

In a recent article in RPTH, Gangnus and Burckhardt present the preanalytic variables that need be considered for the reliable measurement of bradykinin (BK). 1 This topic has been the holy grail of the kinin field. In 1949, Roche E Silva et al. published a manuscript describing a snake venom and trypsin releasable agent that slowed gut contractions of smooth muscle that they named bradykinin (BK) due to its property to slow activity. 2 It was derived from a globulin precursor in blood and serum that they called bradykininogen. This peptide‐like entity degrades quickly and when infused into animals produces arterial hypotension. 2 BK is a nine amino acid peptide (RPPGFSPFR) that is derived from plasma high and low molecular weight kininogens (HK and LK) respectively (Figure 1A). Once formed it binds to one of two G‐protein coupled receptors, the bradykinin B2 receptor (B2R) that is constitutively present or the bradykinin B1 receptor (B1R) that arises in inflammatory states. Removal of the 9th amino acid of BK, which results in Des‐Arg‐9 BK or BK‐(1–8), is the optimal ligand for the B1R. BK and derivatives stimulate endothelial cell NO formation (endothelial cell relaxing factor) leading to cyclic guanosine monophosphate production to produce smooth muscle relaxation and vasodilation. 3 , 4 , 5 In most vascular beds, BK induces vasodilation; in coronary and renal arterial circulation, BK causes vasoconstriction. BK is also the most potent stimulus for tissue plasminogen activator release from endothelial cells and is essential for endothelial cell barrier function. 6 , 7
FIGURE 1.
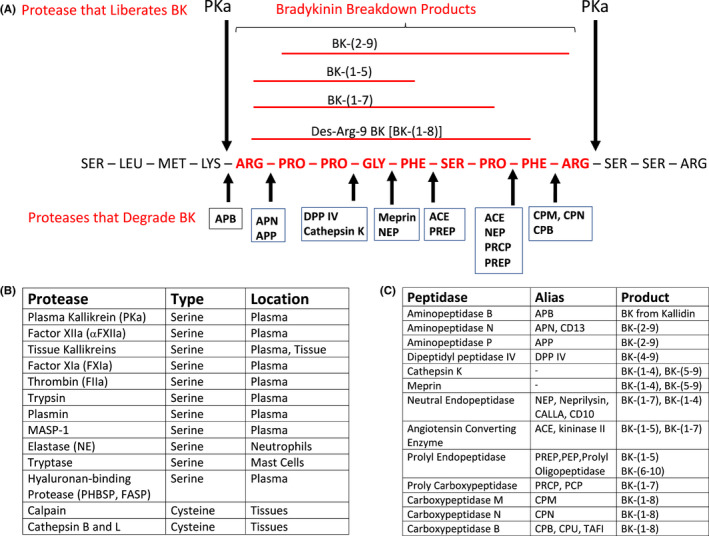
Bradykinin formation and degradation. Panel A. Bradykinin (in red lettering) is a 9 amino acid peptide in kininogens. This panel shows where plasma kallikrein (PKa) cleaves kininogen (HK) to liberate bradykinin (BK). The peptidases (kininases) that degrade BK are shown as abbreviations below the red sequence of BK. The peptide bond that each of these kininases cleave are indicated by the thick small arrows. The full name of the abbreviated kininases is shown in Panel C. The BK breakdown products from the various kininases are delineated in red above the sequence of bradykinin. Panel B lists the many human serine and cysteine plasma and tissue proteases that cleave kininogens to liberation BK. Panel C presents the full name and alias of 13 plasma and membrane kininases. The BK degradation peptides produced by these kininases are shown in the table
How is BK released from its plasma precursors HK and LK? BK is liberated by plasma kallikrein (PKa) and factor XIIa when HK or LK are cleaved between a Lys‐Arg at the N‐terminus and Arg‐Ser at the C‐terminus. The Km of plasma kallikrein and factor XIIa to liberate BK are similar, but PKa is considered the dominant producer in vivo. Bradykinin in plasma is degraded in seconds; therefore, to have biologic influence, it must be released in sequestered locations. HK saturates all vessels to serve as a receptor of prekallikrein. Prekallikrein bound to HK on vessels is activated to PKa by membrane‐expressed proly carboxypeptidase (PRCP). Vessel wall bound PKa then hydrolyzes its receptor, HK, to liberate BK. 8 , 9 Also, the constitutive plasma concentration of HK alone blocks factor XII from binding to vessel wall. Ambient BK formed on the vessel wall at a minimum is modulated by the local concentration of bound prekallikrein, PRCP, and C1 inhibitor, the major serpin inhibitor of PKa and factor XIIa. In addition to PKa, there is a large family of tissue kallikreins that are defined by their ability to liberate Lys‐BK (Kallidin) from LK and HK. In Figure 1B, the listed serine proteases liberate BK from kininogens in a catalytic manner. Most are trypsin‐based serine proteases of the blood coagulation and related systems. The human cysteine proteases, calpains and cathepsins, cleave kininogen in stoichiometric concentrations since kininogens are the major plasma inhibitor of cysteine proteases. Of note, infectious parasites produce cysteine proteases like cruzipain and falcipain and bacterial proteases staphopain, streptopain, and gingipain that cleave HK liberating BK in disease states.
In plasma, forty percent of formed BK binds to its receptors initiating metabolism. 10 The remaining peptide is metabolized by ambient plasma and membrane‐associated serine or metalloproteases called kininases. Figure 1C lists 13 kininases that cleave BK at almost every peptide bond as shown in Figure 1A. Angiotensin converting enzyme (ACE, kininase II) is the major plasma and endothelial cell bradykininase. 11 It is important to realize that almost no breakdown product of BK is functionally inactive. We recognized the BK‐(1–5) (RPPGF) is a direct retro binding inhibitor to the active site of α‐thrombin. 12 Souza‐Silva et al. recently have recognized that BK‐(1–3), BK‐(1–5), and BK‐(1–7) induce NO formation independent of the B2R and B1R. 13 The functions of all BK peptidases is not known. It is recognized that inhibition of angiotensin converting enzyme (kininase II), neutral endopeptidase (NEP), and dipeptidyl peptidase IV is associated with secondary angioedema. This ill effect arises as a consequence of their important roles in management of hypertension, heart failure, and diabetes. Aminopeptidase N (CD13) and neutral endopeptidase (CD10, CALLA) are markers of acute myelogenous and lymphocyte leukemia, respectively. PRCP not only degrades BK‐(1–8) into BK‐(1–7), but as mentioned above, is a plasma prekallikrein activator. 9 Carboxypeptidase B, also called carboxypeptidase U, and in the hemostasis/fibrinolysis field as thrombin‐activable fibrinolysis inhibitor (TAFI), cleaves the C‐terminal arginine residue from bradykinin. Thus, the influence of BK on vascular biology is much more sophisticated than just this single peptide. At any instant, its effect is based on its concentration, rate of formation and degradation of it and all its biologic breakdown products.
Measurement of bradykinin in plasma and other biologic fluids is difficult. The challenges include collection and transport of the sample without contact activation and inhibition of BK forming and degrading enzymes in vitro. The combined concentration of human HK and LK is 1.5 to 2 μM allowing for enormous potential for BK formation in vivo and ex vivo. The actual peptide of bradykinin was not synthesized and characterized until about 1960. 14 With the development of immunologic assays, precise measurement of BK was an early goal. In O’Donnell et al. BK levels were measured by radioimmunoassay. 15 Samples were collected using polypropylene tubing into chilled tubes containing EDTA and hexadimethrine bromide to inhibit factor XII and contact activation and metalloproteases. After processing the plasma at 4°C, the normal and septic patient BK levels were in ng/ml quantities. However, the 4°C processing inactivates C1 inhibitor to promote contact activation resulting in increased BK levels. 16 Scicli et al. by carefully dripping 6 ml of human blood into 100% ethanol into siliconized hardware followed by several extractions, separations, and assaying the same day were able to measure 25.2 ± 2.6 pg/ml BK in 22 normal subjects by radioimmunoassay (RIA). 17 We observed that collection of samples into an ACE inhibitor from cultured cells' supernatant is essential to measure maximal formed BK levels from PKa cleavage of HK. 8 Iwaki and Castellino also observed that the addition of corn trypsin inhibitor that prevents contact activation during the preparatory procedures and aprotinin that inhibits plasmin and PKa to EDTA was essential to collect and measure ng/ml plasma BK levels by RIA and EIA. 18 More recently, Marceau et al. showed that the addition of enalaprilat to previously frozen plasma samples in sodium citrate or EDTA is sufficient to obtain low ng/ml BK levels in normals using BK EIA platforms. 19 More recently developments in mass spectrometric detection have enabled the simultaneous quantification of multiple kinins by overcoming the immunoassay cross‐reactivity. 20
Nevertheless, the heterogeneity of sample collection, stabilization, transport, processing, and assay, makes its measurement less reproducible and, perhaps, reliable even at 70 years since its discovery. Into this conundrum of variable sample collection and assay performance, Drs. Gangnus and Burckhardt present a critical methodological study of the preanalytical variables for the reliable determination of BK and related BK kinin peptides. 1 These investigators combine their knowledge of clinical laboratory testing metrics and bradykinin biology. They determined suitable protease inhibitors, blood sampling conditions, and specimen handling for optimal BK and 6 kinin peptides [Lys‐BK, BK‐(1–8), Lys‐BK‐(1–8), BK‐(1–7), BK‐(2–9), and BK‐(1–5)] measurement using an established LC‐MS/MS platform. 20 These kinin peptides encompass the products of most plasma and membrane peptidases such as aminopeptidase B, aminopeptidases N and P, ACE, prolyl endopeptidase, prolyl carboxypeptidase, neutral endopeptidase, and carboxypeptidases M, N, and B (Figure 1C).
The determination of suitable protease inhibitors was a crucial first step in the success of this investigation. Starting with 17 candidate inhibitors, the investigators empirically reduced the inhibitor pool to a 7‐member cocktail, each with a specific target. Preliminary studies showed that EDTA plasma alone was associated with high pg/ml to low ng/ml BK levels in normal controls. Additionally, the values increase over 30 min after sample collection and further increase at 4.5 h even in the presence of leupeptin or chicken‐egg trypsin inhibitor. The final collection cocktail includes EDTA and sodium citrate, inhibitors of carboxypeptidase N, proly carboxypeptidase, ACE, and aminopeptidase P. It also includes omapatrilat that inhibits ACE and neutral endopeptidase. Tube contact activation is prevented by hexadimethrine bromide (Polybrene) that blocks factor XIIa and nafamostat that inhibits both factor XIIa and PKa. Added formic acid contributes to carboxypeptidase N, ACE, neutral endopeptidase, aminopeptidase P, and prolyl endopeptidase inhibition. Last, chloroquine was needed as an additional inhibitor of prolyl carboxypeptidase.
Subsequent studies examined best practice blood collection devices. 1 A 21 G butterfly needle with minimal tubing was best for blood aspiration directly into collection tubes containing protease inhibitor was optimal. Further minimal time in the collection device and centrifugation within 30 minutes are important for optimal results since time delays of 60 minutes or more lead to more degradation products.
These efforts reveal that BK and its related peptides have baseline values in the single digit pg/ml to undetectable. Using such assays on clinical samples should obtain a stable snapshot of BK and 6 kinin peptides in normal and disease states with contact and kallikrein/kinin systems' activation. In sum, the work of Gangnus and Burckhardt has great promise to measure reliable BK results in angioedema and sepsis and may initiate a new era where BK measurement becomes a marker of health and disease.
RELATIONSHIP DISCLOSURE
All authors have no conflict of interest in relation to the present topic being discussed. Dr. Schmaier has received consulting fees from Intellia and Cardinal Health and research support from Takeda.
AUTHOR CONTRIBUTIONS
ASP, SS, and AHS each contributed to the writing and preparation of the figure in this manuscript.
ACKNOWLEDGEMENTS
This work is funded in part by NIH grants AI130131, HL144113, HL143402, and CA223301 to Dr. Schmaier.
Pinheiro AS, Silbak S, Schmaier AH. Bradykinin – An elusive peptide in measuring and understanding. Res Pract Thromb Haemost. 2022;6:e12673. doi: 10.1002/rth2.12673
Handling Editor: Prof. Yotis Senis
REFERENCES
- 1. Gangnus T, Burckhardt BB. Reliable measurement of plasma kinin peptides: Importance of preanalytical variables. Res Pract Thromb Haemost. 2022;6:e12646. doi: 10.1002/rth2.12646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rocha E, Silva M, Beraldo WT, Rosenfeld G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am J Physiol. 1949;156:261‐273. [DOI] [PubMed] [Google Scholar]
- 3. Cherry PD, Furchgott RF, Zawadzki JV, Jothianandan D. Role of endothelial cells in relaxation of isolated arteries by bradykinin. Proc Natl Acad Sci U S A. 1982;79:2106‐2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rapoport RM, Murad F. Agonist‐induced endothelium‐dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ Res. 1983;52:352‐357. [DOI] [PubMed] [Google Scholar]
- 5. Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium‐derived relaxing factor. Nature. 1987;327:524‐526. [DOI] [PubMed] [Google Scholar]
- 6. Smith D, Gilbert M, Owen WG. Tissue plasminogen activator release in vivo in response to vasoactive agents. Blood. 1985;66:835‐839. [PubMed] [Google Scholar]
- 7. Silva LS, Pinheiro AS, Teixeira DE, et al. Kinins released by erythrocytic stages of Plasmodium falciparum enhance adhesion of infected erythrocytes to endothelial cells and increase blood brain barrier permeability via activation of bradykinin receptors. Front Med (Lausanne). 2019;6:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao Y, Qiu Q, Mahdi F, Shariat‐Madar Z, Røjkjaer R, Schmaier AH. Assembly and activation of HK‐PK complex on endothelial cells results in bradykinin liberation and NO formation. Am J Physiol Heart Circ Physiol. 2001;280:H1821‐H1829. [DOI] [PubMed] [Google Scholar]
- 9. Shariat‐Madar Z, Mahdi F, Schmaier AH. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J Biol Chem. 2002;277:17962‐17969. [DOI] [PubMed] [Google Scholar]
- 10. Munoz CM, Leeb‐Lundberg LM. Receptor‐mediated internalization of bradykinin. DDT1 MF‐2 smooth muscle cells process internalized bradykinin via multiple degradative pathways. J Biol Chem. 1992;267:303‐309. [PubMed] [Google Scholar]
- 11. Alhenc‐Gelas F, Bouby N, Girolami JP. Kallikrein/K1, kinins, and ACE/Kininase II in homeostasis and in disease insight from human and experimental genetic studies, therapeutic implication. Front Med (Lausanne). 2019;6:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hasan AA, Amenta S, Schmaier AH. Bradykinin and its metabolite, Arg‐Pro‐Pro‐Gly‐Phe, are selective inhibitors of alpha‐thrombin‐induced platelet activation. Circulation. 1996;94:517‐528. [DOI] [PubMed] [Google Scholar]
- 13. Souza‐Silva IM, Amorim de Paula C, Santos AK, et al. The kallikrein‐kinin system is falling into pieces: bradykinin fragments are biological active peptides. bioRxiv. doi: 10.1101/2020.09.14.296004 [DOI] [Google Scholar]
- 14. Elliott DF. The discovery and characterization of bradykinin. In: Erdos EG, ed. Handbook of Experimental Pharmacology, Bradykinin, Kallidin and Kallikrein (vol 25). Springer; 1970:7‐13. [Google Scholar]
- 15. O'Donnell TF Jr, Clowes GH Jr, Talamo RC, Colman RW. Kinin activation in the blood of patients with sepsis. Surg Gynecol Obstet. 1976;143:539‐545. [PubMed] [Google Scholar]
- 16. Weiss R, Silverberg M, Kaplan AP. The effect of C1 inhibitor upon Hageman factor autoactivation. Blood. 1986;68:239‐243. [PubMed] [Google Scholar]
- 17. Scicli AG, Mindroiu T, Scicli G, Carretero OA. Blood kinins, their concentration in normal subjects and in patients with congenital deficiency in plasma prekallikrein and kininogen. J Lab Clin Med. 1982;100:81‐93. [PubMed] [Google Scholar]
- 18. Iwaki T, Castellino FJ. Plasma levels of bradykinin are suppressed in factor XII‐deficient mice. Thromb Haemost. 2006;95:1003‐1010. [DOI] [PubMed] [Google Scholar]
- 19. Marceau F, Bachelard H, Rivard GÉ, Hébert J. Increased fibrinolysis‐induced bradykinin formation in hereditary angioedema confirmed using stored plasma and biotechnological inhibitors. BMC Res Notes. 2019;12:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gangnus T, Burckhardt BB. Targeted LC‐MS/MS platform for the comprehensive determination of peptides in the kallikrein‐kinin system. Anal Bioanal Chem. 2021;413:2971‐2984. [DOI] [PMC free article] [PubMed] [Google Scholar]