

Abstract
The mechanisms whereby neutrophils respond differentially to live and dead organisms are unknown. We show here that neutrophils produce 5‐ to 30‐fold higher levels of the Cxcl2 chemokine in response to live bacteria, compared with killed bacteria or isolated bacterial components, despite producing similar levels of Cxcl1 or pro‐inflammatory cytokines. Secretion of high levels of Cxcl2, which potently activates neutrophils by an autocrine mechanism, requires three signals. The first two signals are provided by two different sets of signal peptides released by live bacteria, which selectively activate formylated peptide receptor 1 (Fpr1) and Fpr2, respectively. Signal 3 originates from Toll‐like receptor activation by microbial components present in both live and killed bacteria. Mechanistically, these signaling pathways converge at the level of the p38 MAP kinase, leading to activation of the AP‐1 transcription factor and to Cxcl2 induction. Collectively, our data demonstrate that the simultaneous presence of agonists for Fpr1, Fpr2, and Toll‐like receptors represents a unique signature associated with viable bacteria, which is sensed by neutrophils and induces Cxcl2‐dependent autocrine cell activation.
Keywords: formylated peptide receptors, innate immunity, live/dead discrimination, neutrophils, toll‐like receptors
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Live bacteria concomitantly activate Fpr1, Frp2 and Toll‐like receptors to induce p38 MAP kinase signalling, leading to higher levels of the Cxcl2 chemokine and ROS.
Introduction
Activation of host defenses during infection is triggered by specialized pathogen recognition receptors (PRRs), such as Toll‐like receptors (TLRs), expressed by cells of the innate immune system. Each PRR is activated upon binding to one or few cognate ligands, consisting of evolutionary conserved microbial molecules designated as pathogen‐associated molecular patterns or PAMPs. In order to avoid excessive or prolonged activation that may lead to serious pathology, the defense system regulates the magnitude of its responses according to the actual danger associated with the presence of microbial products. This is accomplished in two ways. First, the presence of self‐components associated with tissue damage (damage‐associated molecular patterns or DAMPs) is sensed by some PRRs and other specialized receptors, leading to enhanced inflammatory responses (Zindel & Kubes, 2020). Second, immune cells selectively detect the presence of molecules associated with viable organisms, which enables them to respond more vigorously to live than to dead microbes. The importance of this form of live/dead discrimination has long been recognized in regard to the superior ability of live vaccines, compared to inactivated ones, to induce protective immunity (Rauh & Schmidt, 1965; Lauvau et al, 2001), although the underlying molecular mechanisms are still incompletely understood (Ugolini & Sander, 2019). It has been proposed that the presence of products from dead bacteria (such as DNA, RNA, or polymeric peptidoglycan) in the extracellular space signals the end of the infection process, leading to downregulation of inflammatory responses and tissue repair (Vance et al, 2009; Neyen et al, 2016). Others have postulated, in addition, the existence of a class of PAMPs, called vita‐PAMPs, that are specifically associated with viable bacteria and are capable of triggering distinctive responses in macrophages and dendritic cells, including robust inflammasome activation and production of interferon‐β (Sander et al, 2011; Blander & Sander, 2012). Importantly, these studies have shown that procariotic RNA, which is more abundant in live than in killed bacteria, can act as a vita‐PAMP by triggering TLR signaling in macrophages and dendritic cells. Convincing evidence that other bacterial products, such as quorum sensing molecules and cyclic di‐nucleotides, act as vita‐PAMPs is not available yet (Ugolini & Sander, 2019). Little is known of the live/dead pathogen discrimination capabilities of neutrophils. This kind of knowledge seems important, since neutrophils are the first cells to reach infection sites and potently shape subsequent events, including the recruitment of other cell types, pathogen clearance, and tissue repair or damage (Kobayashi et al, 2005; Deniset & Kubes, 2016; Kang et al, 2020). Neutrophils migrate from the blood into sites of infection and/or tissue damage by moving toward increasing concentrations of different, locally presented chemoattractants released from a variety of sources (Majumdar et al, 2014). Primary attractants, such as formylated peptides, originate from bacteria or damaged cells and provide short range guidance cues toward these “end targets”. Secondary or intermediate attractants, such as leukotriene B4 (LTB4) and chemokines of the CXCL8 family, are released from endothelial cells and other resident cells (Kolaczkowska & Kubes, 2013). Secondary attractants can be also produced by neutrophils themselves during their migration, thereby relaying to other neutrophils chemotactic signals in a feed‐forward amplification loop. This relay of signals between neutrophils choreographs dynamic cell aggregations, as visualized by intravital microscopy, and extends the recruitment range of these cells over long distances from primary attractants (Kienle & Lammermann, 2016). In the present study, we focused on neutrophil‐derived Cxcl2, since this chemokine plays non‐redundant roles in relaying chemotactic signals to other neutrophils and in promoting autocrionous cell activation (Li et al, 2016; Girbl et al, 2018; Lentini et al, 2020). Cxcl2 (or Mip2) is a member of the CXCL8 family of cytokines (Russo et al, 2014), which, together with Cxcl1 (or KC) performs in the mouse similar functions as human CXCL8 in recruiting neutrophils to inflammation sites (Zlotnik & Yoshie, 2012). We show here that neutrophils produce high levels of Cxcl2 in response to live, but not killed, bacteria in a manner involving the simultaneous activation of TLRs and of receptors for formylated peptides (Fpr). This form of live/dead discrimination requires signaling from both Fpr1 and Fpr2, each of which plays a non‐redundant role in recognizing a specific set of secretory signal peptides released by live bacteria. TLRs and Fprs strongly synergize in determining the sequential activation of the p38 MAP kinase and the AP‐1 transcription factor, leading to Cxcl2 induction. Our data indicate that formylated peptides function as a novel class of vita‐PAMPs in driving high‐level Cxcl2 production and neutrophil activation.
Results
Live, but not killed, bacteria induce high‐level Cxl2 responses in neutrophils
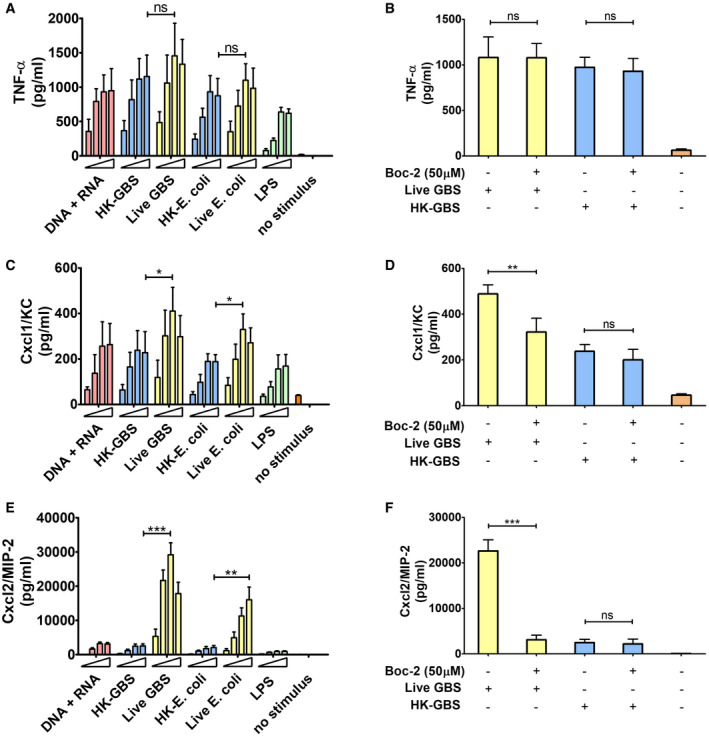
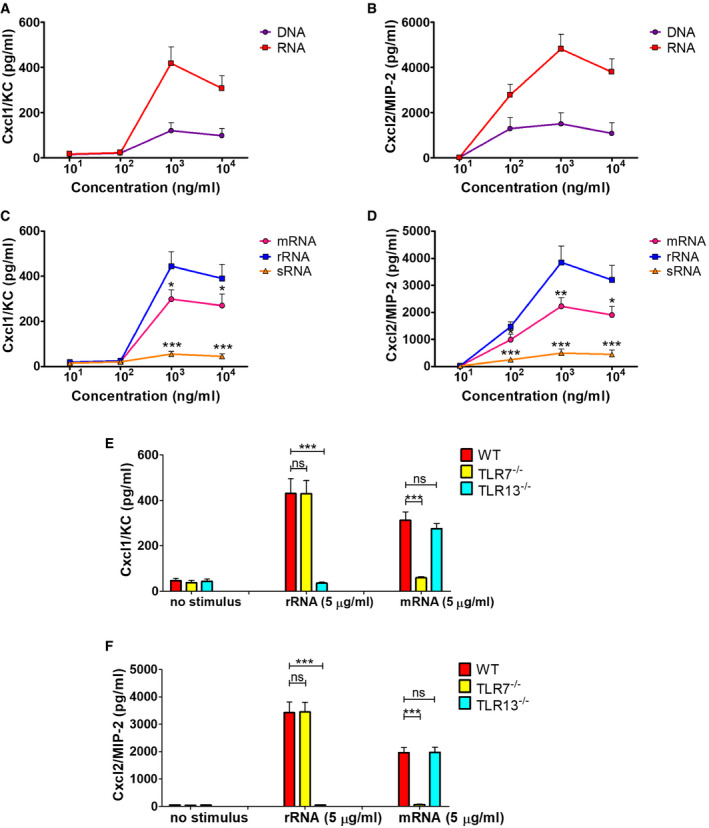
In view of the crucial role of neutrophil‐derived Cxcl2 in host defenses (Girbl et al, 2018; Lentini et al, 2020), we sought to identify the bacterial products responsible for induction of this chemokine in bone marrow‐derived murine neutrophils. We initially used mixtures of prokaryotic DNA + RNA, since it was previously found that innate antibacterial responses require the endosomal receptors TLR7, TLR9, and TLR13 (Signorino et al, 2014; Mohammadi et al, 2016; Fama et al, 2020; Lentini et al, 2020), which are known to recognize bacterial RNA (TLR7 and 13) and DNA (TLR9). Accordingly, a 1:1 DNA + RNA mixture potently induced TNF‐α and Cxcl1 production in neutrophils at levels that were similar or slightly lower than those induced by whole bacteria, such as group B streptococcus (gram‐positive) or Escherichia coli (gram‐negative; Fig 1A and C). Surprisingly, however, DNA+RNA, as well as killed bacteria or LPS, induced Cxcl2 elevations that were 10‐ to 30‐fold lower than those induced by live bacteria (Fig 1E). Bacteria killed with ethanol, gentamycin, or ultraviolet light were similarly unable to induce high‐level Cxcl2 responses (data not shown). We also tested purified mRNA, which was previously found to act as a vita‐PAMP in macrophages (Sander et al, 2011), in comparison with other nucleic acid species. However, only modest Cxcl2 responses were elicited by mRNA, rRNA, or DNA, although these stimuli induced robust Cxcl1 production in a fashion that was totally dependent on their respective cognate TLR (TLR7, 13 and 9, respectively; Fig EV1). These data indicate that live bacteria have a unique ability to stimulate neutrophils for the production of high levels of Cxcl2, which cannot be mimicked using other stimuli, including killed bacteria, LPS, or previously described vita‐PAMPS, such as mRNA. Therefore, other microbial components produced by live, but not killed, bacteria seemed to be required, in addition to nucleic acids, for high‐level Cxcl2 responses in neutrophils.
Figure 1. Live, but not killed, bacteria induce high‐level Cxcl2 production.
-
A–FTNF‐α, Cxcl1, and Cxcl2 (A, C, and E, respectively) were measured at 24 h after stimulation of bone marrow‐derived neutrophils (5 × 105 cells) with increasing concentrations of bacterial nucleic acids (1, 5, 10, and 20 µg of DNA + RNA in equal proportions), HK‐bacteria (1, 5, 10 or 20 μg/ml) or live bacteria (MOIs of 2, 5, 10, and 20). LPS (1, 10, 100, and 1,000 ng/ml) was used as a positive control stimulus. (B, D and F) Effect of FPR antagonist Boc‐2 on GBS‐induced TNF‐α, Cxcl1, and Cxcl2 production, respectively. Bone marrow‐derived neutrophils were exposed to Boc‐2 (50 µM) for 1 h and then stimulated with HK‐GBS (10 µg/ml) or live GBS (MOI of 5). Data are expressed as means + standard deviations of measurements from three independent experiments conducted in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001, as determined by unpaired t‐test; ns, not significant.
Figure EV1. Production of Cxcl1 and Cxcl2 in neutrophils stimulated with bacterial nucleic acids.
-
A–DConcentrations of Cxcl1 (A, and C) and Cxcl2 (B, and D) in supernatants of neutrophil cultures stimulated with increasing concentrations (101, 102, 103, and 104 ng/ml) of the indicated nucleic acids. mRNA, messenger RNA; rRNA, ribosomal RNA; sRNA small RNA.
-
E, FCxcl1 (E) and Cxcl2 (F) concentrations in supernatants of bone marrow‐derived neutrophils from mice with genetic deficiency in TLR7 or TLR13 after stimulation with rRNA or mRNA. Chemokine levels in supernatants were measured at 24 h after stimulation.
Data information: Data are expressed as means + SD of three independent experiments conducted in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001 versus RNA (A and B), rRNA (C and D) or WT mice (E and F), determined by unpaired t‐test.
High‐level Cxcl2 responses require formyl peptide receptors
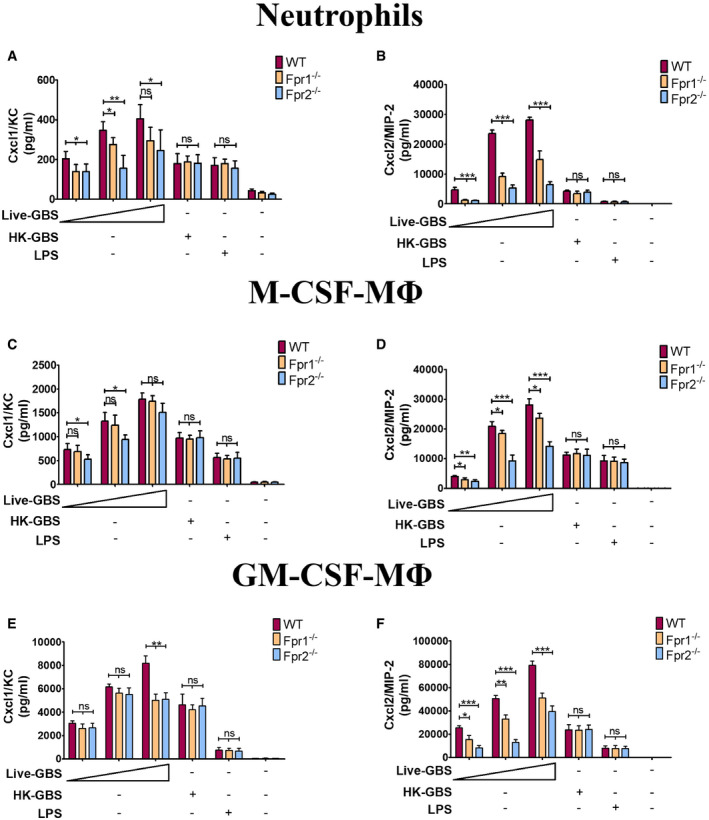
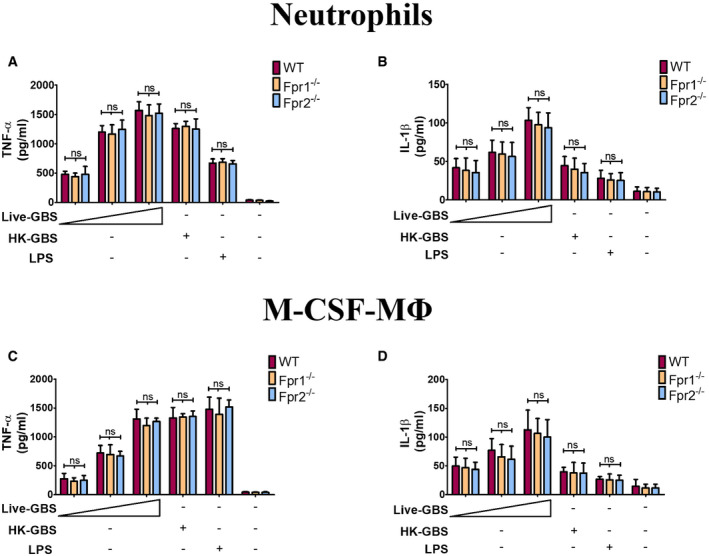
An obvious difference between live and dead bacteria is that protein synthesis is inactive in the latter. Since protein synthesis in prokaryotes is uniquely initiated by N‐formylated methionine (fMet) and fMet‐bearing peptides can function as PAMPs (Le et al, 2002; He & Ye, 2017), we hypothesized that such peptides are used by neutrophils for discrimination of live from dead bacteria. To verify our hypothesis, we focused on group B streptococcus (GBS) because it potently induced Cxcl2 release in our previous experiments (Fig 1). Moreover, this is a frequent pathogen (Raabe & Shane, 2019), which has been extensively used to model innate anti‐bacterial responses (Costa et al, 2012; Landwehr‐Kenzel & Henneke, 2014). To explore the role of receptors for fMet‐bearing peptides in Cxcl2 induction neutrophils were pre‐treated with 50 µM t‐Boc‐FLFLF (also termed Boc‐2), which, at this concentration, blocks both Fpr1 and Fpr2 signaling (Stenfeldt et al, 2007). Strikingly, treatment with 50 µM Boc‐2 almost completely abrogated Cxcl2 responses to live GBS but was totally ineffective when using killed bacteria as a stimulus (Fig 1F). Moreover, TNF‐α and Cxcl1 responses to live organisms were not affected or only moderately decreased, respectively, by Boc‐2 treatment (Fig 1B and D). When we used the Fpr2‐specific antagonist WRW4 (Bae et al, 2004), or 10 µM Boc‐2 (which, at this concentration, specifically blocks Fpr1, but not Fpr2; Stenfeldt et al, 2007), we found that both treatments significantly reduced Cxcl2 responses induced by stimulation with live bacteria (Fig 2). Fpr involvement was further confirmed using cells from KO mice lacking Fpr1 or Fpr2, since marked reductions in Cxl2 responses to live bacteria were observed in the absence of either receptor, while TNF‐α, IL‐1β, and Cxcl1 responses were unaffected or slightly reduced (Figs 3A and B, and Fig EV2). Likewise, selective reduction in Cxcl2 levels was observed using M‐CSF‐ or GM‐CSF‐polarized macrophages lacking Fpr1 or Fpr2, although these differences were not as marked as those observed in neutrophils (Fig 3C–F). These data indicated that both Fpr1 and Fpr2 have non‐redundant roles in Cxcl2 responses to live bacteria in neutrophils and macrophages, and cannot compensate for the absence of each other.
Figure 2. Effect of FPR antagonists on GBS‐induced Cxcl2 production.
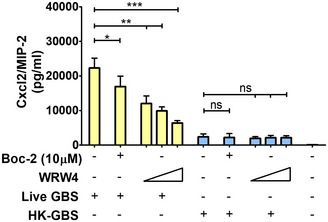
Bone marrow‐derived neutrophils were exposed to Boc‐2 (10 µM) or WRW4 (1, 5, and 10 µM) for 1 h and then stimulated with HK‐GBS (10 µg/ml) or live GBS (MOI of 5). Data are expressed as means + standard deviations of measurements from three independent experiments conducted in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001, as determined by unpaired t‐test; ns, not significant.
Figure 3. FPRs are required for Cxcl2 responses to live streptococci.
-
A–FCxcl1 and Cxcl2 concentrations in 24‐h culture supernatants of neutrophils (A and B), CSF‐polarized (C and D) or GM‐CSF‐polarized (E and F) BMDMs (ΜΦ) from WT or Fpr‐deficient mice stimulated with live GBS (MOI of 2, 5, and 10) or HK‐GBS (10 µg/ml). LPS (100 ng/ml) was used as a positive control stimulus. Means + SD of data from three independent experiments conducted in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001 vs. WT mice, determined by unpaired t‐test.
Figure EV2. Production of pro‐inflammatory cytokines in Fpr‐defective phagocytes.
-
A–DConcentrations of TNF‐α (A and C) and IL‐1β (B and D) in 24‐h culture supernatants of neutrophils (A and B) or M‐CSF‐polarized BMDMs (C and D) from WT or Fpr‐deficient mice stimulated with live GBS (MOI of 2, 5, and 10) or HK‐GBS (10 µg/ml). LPS (100 ng/ml) was used as a positive control stimulus. Means + SD of data from three independent experiments conducted in duplicate. ns, non‐significant, as determined by unpaired t‐test.
Fpr signaling is required for in vivo Cxcl2 production and host defenses
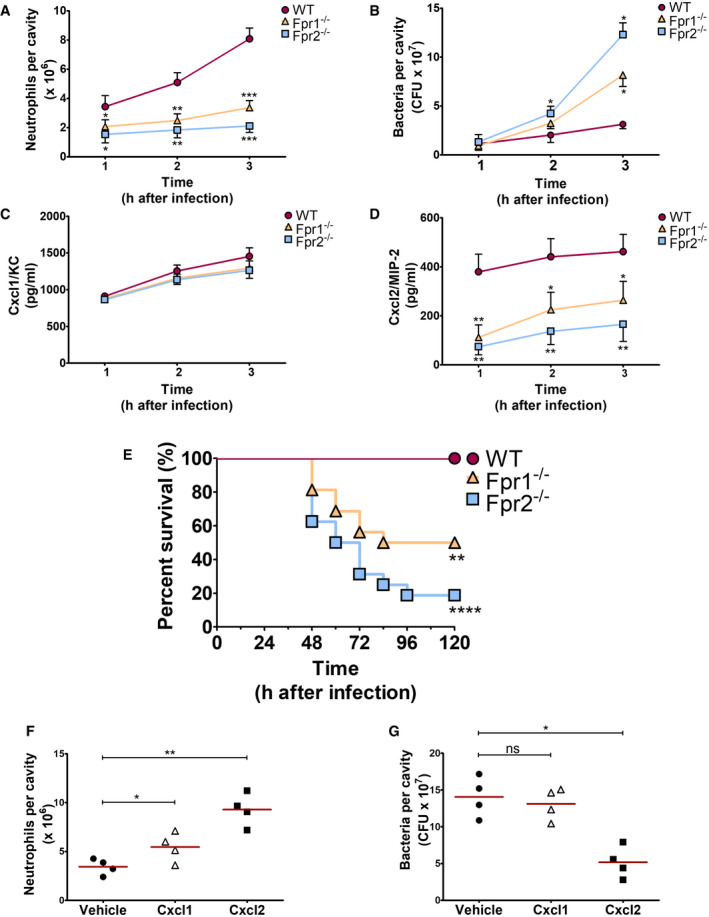
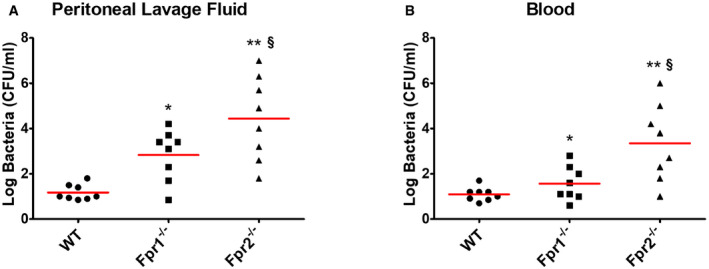
To ascertain whether Fpr signaling is involved in Cxcl2 responses not only in vitro, but also in vivo, Fpr1‐ and Fpr2‐defective mice were infected i.p. with the GBS strain H36B under conditions resulting in robust neutrophil influx into the peritoneal cavity (Lentini et al, 2020). Significant Cxcl1/2 elevations and neutrophil recruitment were already detected at 1 h after infection and increased over the next two hours (Fig 4A, C and D). Cxcl2, but not Cxcl1, elevations were decreased in peritoneal exudates obtained from either Fpr1 or Fpr2 mice at all time points, relative to wild‐type (WT) animals, concomitantly with decreased neutrophil influx and increased bacterial burden (Fig 4A–D). To further analyze the role of Fpr signaling in host defenses, mice were infected i.p. with a low GBS dose and observed for signs of disease. All WT animals remained in good health under these conditions, while 50 and 88% of Fpr‐1 and Fpr‐2 KO mice, respectively, showed persistent signs of disease, such as reduced mobility or hunching, and were humanely euthanized (Fig 4E). Uncontrolled bacterial growth was confirmed as the cause of illness in these animals (Fig EV3). To investigate whether decreased Cxcl2 responses were responsible for the impaired ability of immune‐deficient animals to control bacterial infection, Fpr2−/− mice were treated with recombinant Cxcl2 at 1 h post‐challenge. Under these conditions, administration of Cxcl2 rescued neutrophil recruitment and bactericidal activity to wild‐type levels (Fig 4F and G). In contrast, exogenous administration of Cxcl1 did not decrease bacterial burden, despite producing a moderate increase in neutrophil recruitment (Fig 4F and G). Collectively, these data indicate that Fpr1/2 signaling is crucially required for robust Cxcl2 responses, which lead to effective control of bacterial infection.
Figure 4. Fpr1 or Fpr2 are required for in vivo neutrophil recruitment, Cxcl2 production, and antibacterial defenses during GBS infection.
-
A–DKinetics of neutrophil influx (A), bacterial burden (B), and Cxcl1/2 production (C and D) in peritoneal lavage fluid samples after i.p. challenge with live GBS (2 × 107 CFU).
-
EKaplan–Meier analysis of survival rates in WT, Fpr1, and Fpr2 KO mice after i.p. challenge with 2 × 105 bacteria. Data are the cumulative results from two experiments, each involving 8 animals per group. **P < 0.01, ****P < 0.0001 vs. WT mice, as determined by Kaplan–Meier analysis.
-
F, GNeutrophil influx (F) and bacterial burden (G) in peritoneal lavage fluid samples collected from Fpr2−/− mice at 3 h after i.p. challenge with live GBS (2 × 107 CFU). At 1 h post‐challenge mice were inoculated i.p. with 1 μg of recombinant Cxcl1 or Cxcl2 or with vehicle.
Data information: In (A–D), means + SDs of three duplicate determinations, each conducted in a different animal. In (F and G), individual data points and means. *P < 0.05, **P < 0.01, ***P < 0.001 versus WT mice (A, C, and D) or versus vehicle‐treated mice (F and G), determined by unpaired t‐test (A, C, D and F) or Mann–Whitney U‐test (B and G).
Figure EV3. Bacterial burden in Fpr‐defective mice after challenge with GBS.
-
A, BCFU numbers in peritoneal lavage fluid (A) and blood (B) samples from wild type (WT), Fpr1, and Fpr2 KO mice at 24 h after i.p. infection with live GBS (2 × 105 CFU). Horizontal red bars indicate mean values. Each determination was conducted on a different animal in the course of two experiments, each involving 4 animals per group. *P < 0.05, **P < 0.01 vs. WT mice, § P < 0.05 vs. Fpr1‐deficient mice, as determined by the Wilcoxon test.
Bacterial signal peptides synergize with TLR agonists in inducing the production of Cxcl2 and reactive oxygen species
Next, we sought to identify the Fpr agonists involved in Cxcl2 responses to GBS. Since previous studies have indicated that N‐terminal, 3–6 amino acid long, sequences from some bacterial signal peptides can activate FPRs (Bufe et al, 2015), we investigated whether such peptides are also involved in Fpr‐dependent live/dead pathogen discrimination. Interrogation of a signal peptide database (http://www.signalpeptide.de) led to the identification of 15 unique sequences in GBS (Table 1). The corresponding hexapeptides, all bearing an N‐terminal fMet, were synthesized and tested for their ability to activate neutrophils in comparison with the prototypical Fpr1 and 2 agonists f‐MIFL and WKYMVM, respectively. Out of the 15 GBS peptides, 4 promoted strong in vitro chemotactic responses, 7 induced moderate, but significant, chemotaxis and the remaining 4 were inactive. Notably, for each peptide, chemotactic activity was totally dependent on either Fpr1 or Fpr2, as determined using neutrophils from Fpr‐deficient mice (Fig 5A). Moreover, N‐formyl substitution at the N‐terminal methionine residue was required for these effects, since de‐formylated peptides were totally inactive. The peptides identified as Fpr agonists also induced intracellular calcium increases in an Fpr‐dependent manner (Fig 5B). Despite their ability to promote chemotaxis and calcium increase, only modest Cxcl2 responses were induced by these fMet peptides or by the prototypical Fpr agonists f‐MIFL and WKYMVM when used alone as stimuli over a wide dose range (Fig 5C). However, Fpr1‐ and/or Fpr2‐ specific peptides in combination with HK‐GBS produced considerably higher Cxcl2 elevations than those observed with HK‐GBS alone (Fig 5C). Notably, a combination of three stimuli (HK‐GBS, Fpr1‐ and Fpr‐2‐specific peptides) induced even higher Cxcl2 elevations than those induced by a combination of two. Similar synergistic activities were observed when using bacterial RNA or other TLR ligands in place of killed bacteria (Fig 5D). In contrast, TLR agonists, including HK‐GBS, did not significantly synergize with Fpr agonists in the induction of Cxcl1 or TNF‐α (Appendix Fig S1).
Table 1.
Group B streptococcal signal peptides examined in this study.
| Signal peptide | Protein | UniProtKB/Swiss‐Prot Entry Name | Status |
Sequence (6 aa) |
Receptor Dependency |
|---|---|---|---|---|---|
| GBS fPep1 | Membrane protein insertase YidC 1 (oxaA 1) | OXAA1_STRA3 | (hypothetical) | fMKKKLK a | Fpr2 |
| GBS fPep2 | Foldase protein prsA | PRSA_STRA3 | (hypothetical) | fMKTRSK b | Inactive |
| GBS fPep3 | Phosphate‐binding protein PstS 1 | PSTS1_STRA3 | (hypothetical) | fMKMKKN | Fpr2 |
| GBS fPep4 | Phosphate‐binding protein PstS 2 | PSTS2_STRA3 | (hypothetical) | fMKKHKM a | Fpr2 |
| GBS fPep5 | Membrane protein insertase YidC 2 (oxaA 2) | OXAA2_STRA5 | (hypothetical) | fMKKTLK a | Fpr2 |
| GBS fPep6 | Plasminogen binding surface Protein (PbsP) | Q8E6Y0_STRA3 | Confirmed (Buscetta et al, 2016) | fMKISQY | Fpr2 |
| GBS fPep7 | PcsB protein | Q9AKA4_STRAG | Confirmed (Reinscheid et al, 2001; Papasergi et al, 2013) | fMKKRIL a | Fpr2 |
| GBS fPep8 | cAMP factor | Q53651_STRAG | Confirmed (Podbielski et al, 1994; Papasergi et al, 2013) | fMNVTHM | Fpr1 |
| GBS fPep9 | Hyaluronate lyase | O86478_STRAG | Confirmed (Gase et al, 1998; Papasergi et al, 2013) | fMEIKKK | Inactive |
| GBS fPep10 | Fibrinogen‐binding adhesin FbsA | Q84EL7_STRAG | Confirmed (Schubert et al, 2002; Papasergi et al, 2013) | fMFNKIG | Fpr2 |
| GBS fPep11 | IgA FC receptor | BAG_STRAG | Confirmed (Jerlstrom et al, 1991) | fMFKSNY | Fpr1 |
| GBS fPep12 | BPS protein | Q9XAS7_STRAG | Confirmed (Erdogan et al, 2002) | fMFRQYN | Fpr1 |
| GBS fPep13 | Bsp protein | Q8VLP7_STRAG | Confirmed (Reinscheid et al, 2002; Papasergi et al, 2013) | fMMKKGQ | Inactive |
| GBS fPep14 | Hyaluronate lyase | HYSA_STRA3 | Confirmed (Gase et al, 1998; Papasergi et al, 2013) | fMKQVVD | Inactive |
| GBS fPep15 | C protein alpha‐antigen | BCA_STRA1 | Confirmed (Auperin et al, 2005; Papasergi et al, 2013) | fMFRRSK | Fpr2 |
Figure 5. Bacterial signal peptides activate neutrophils and synergize with TLRs in Cxcl2 induction.
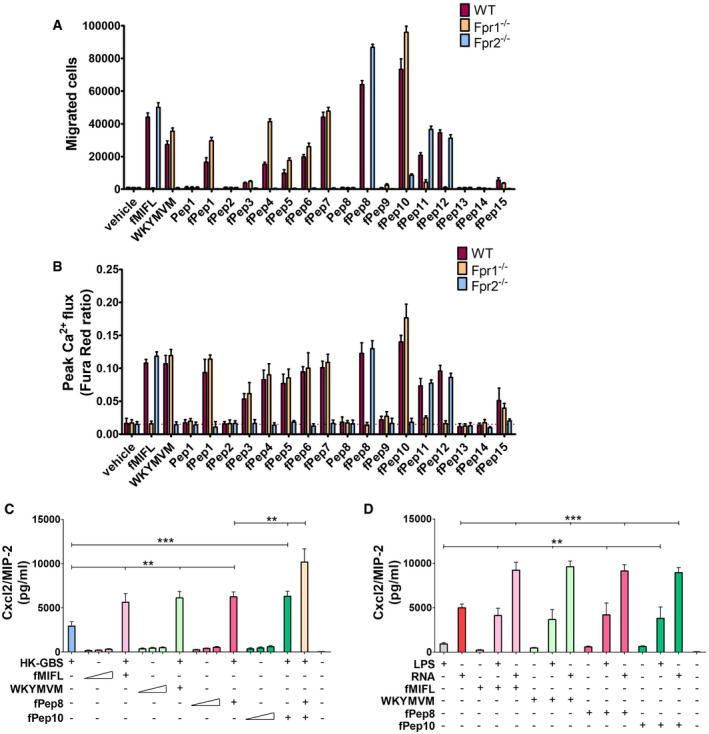
-
ANeutrophils from WT or Fpr‐deficient mice were allowed to migrate toward GBS signal peptide sequences (fPep1‐15, 1 µM), positive control peptides (f‐MIFL or WKYMVM, 1 µM) or vehicle (0.2% DMSO).
-
BAnalysis of intracellular calcium flux in neutrophils stimulated with signal peptides as monitored by flow cytometry.
-
C, DCxcl2 release in neutrophil cultures stimulated with combinations of HK‐bacteria, GBS fMet peptides, or TLR agonists. Neutrophils were exposed to a 50 µM concentration of Fpr agonists (fMIFL, WKYMVM or fPep8 and fPep10) for 1 h before the addition of the indicated TLR agonists, which included HK‐GBS (10 μg/ml), LPS (1 µg/ml), and RNA (1 µg/ml). In some cases, neutrophils were exposed to graded doses (10, 20, and 50 µM) of the indicated Fpr agonists alone.
Data information: Data are expressed as means + SD from three independent experiments, each conducted in duplicate. **P < 0.01, ***P < 0.001, as determined by unpaired t‐test.
To prove that N‐formylated peptides are involved in Cxcl2 induction in the context of whole bacteria, GBS were grown in the presence of a sub‐inhibitory concentration of actinonin, a peptide deformylase inhibitor, that increases N‐formylated peptide levels in bacteria (Fu et al, 2003). Strikingly, actinonin‐grown bacteria induced Cxcl2 levels that were approximately 2‐fold higher than those induced by bacteria cultured in actinonin‐free medium (Fig 6A). Next, we found that TLR and Fpr agonists synergized also in inducing the production of reactive oxygen species (ROS), as shown in Fig EV4A and B. Moreover, live bacteria induced markedly higher ROS responses than killed organisms (Fig EV4C). Notably, responses to live GBS or E. coli required the TLR adaptor MyD88, Fpr1, and Fpr2, each of which played a non‐redundant role in ROS induction, as shown using neutrophils lacking these factors (Fig 6B and C). Since ROS can function as signaling molecules (Nguyen et al, 2017), we investigated whether increased Cxcl2 responses to live bacteria are secondary to ROS release. However, this was not the case, since stimulation of neutrophils in the presence of ROS quenchers had minimal or no effects on GBS‐induced Cxcl2 production (Fig 6D). Collectively, these data indicated that combinations of Fpr1, Fpr2, and TLR agonists, including signal peptide sequences, can largely recapitulate Cxcl2 and ROS responses to live bacteria. Accordingly, the latter responses required Fpr1, Fpr2, and TLR signaling.
Figure 6. ROS production requires the synergistic activation of Fprs and TLRs.
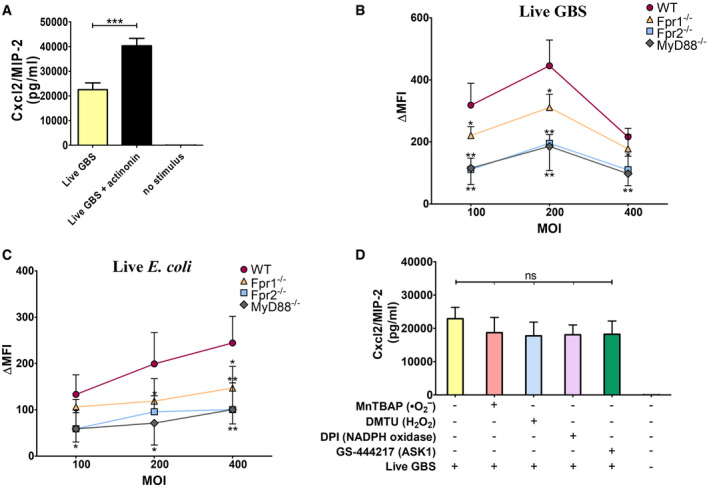
-
AEffect of actinonin on Cxcl2 production by neutrophils. Neutrophils were stimulated with live GBS (MOI 5) in the presence or absence of the formyl peptidase inhibitor actinonin (32 µg/ml).
-
B, CROS production in neutrophils from WT, MyD88, or Fpr1/2‐deficient mice stimulated with different doses of live bacteria.
-
DEffect of treatment with ROS scavengers/inhibitors on Cxcl2 production. Neutrophils were treated with the singlet oxygen scavenger MnTBAP (100 μM), the hydrogen peroxide scavenger DMTU (10 mM), the NADPH oxidase inhibitor DPI (20 μM) or the ASK1 inhibitor GS‐444217 (1 μM) for 1 h before the addition of live GBS (MOI of 5).
Data information: Data are means + SD from three independent experiments conducted in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001, as determined by unpaired t‐test.
Figure EV4. ROS production in neutrophils stimulated with Fpr and TLR agonists.
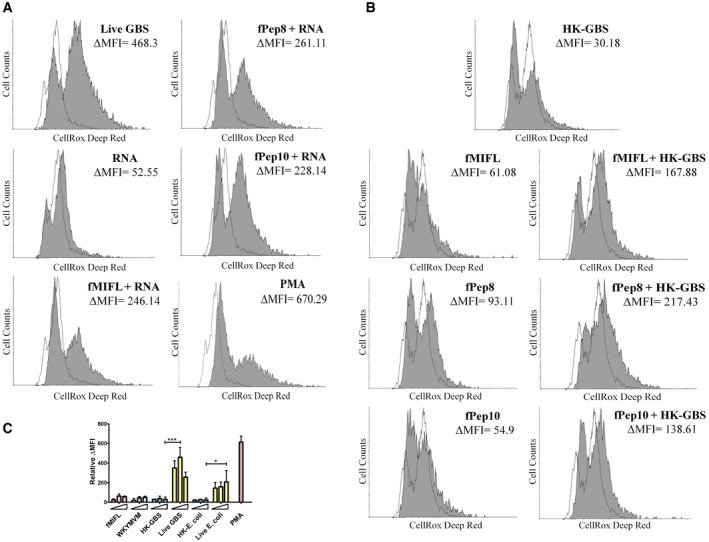
-
A, BROS production in neutrophils stimulated with live GBS (MOI 200) or combinations of Fpr agonists (fMIFL, WKYMVM, or GBS formylated peptides, 50 µM) and TLR agonists (HK‐GBS, 10 μg/ml or bacterial RNA, 1 µg/ml). Phorbol‐12‐myristate‐13‐acetate (PMA, 25 ng/ml) was used as a positive control stimulus. Data from one representative experiment of three producing similar results. Cells were stained with the CellROX fluorescent reagent. The shadowed area indicates stimulated cells.
-
CROS production in neutrophils stimulated with increasing concentrations of fMIFL or WKYMVM (10, 20, and 50 µM), HK‐bacteria (GBS or E. coli, 10, 20, and 40 μg/ml) or increasing numbers of live bacteria (MOIs of 100, 200, and 400). PMA (25 ng/ml) was used as a positive control stimulus. Data are expressed as means + SD from three experiments conducted in duplicate. *P < 0.05, ***P < 0.001, as determined by unpaired t‐test.
Cxcl2 responses to live bacteria are driven by the Tak1 and p38 kinases and by the AP‐1 and NF‐κB transcription factors
Next, we sought to analyze the mechanisms underlying the synergistic effects of TLR/FPR signaling in Cxcl2 responses to live bacteria. Since such responses are transcriptionally regulated (De Filippo et al, 2008; Lentini et al, 2020), we focused on signaling kinases, such as TAK1, MAPKs, and Akt, which are known to be involved in TLR/FPR‐dependent transcription factor activation (Mancuso et al, 2002; Ear et al, 2010). Strikingly, Cxcl2 responses to live GBS were almost completely abrogated by the p38 MAPK‐specific inhibitor SB203580 or the Tak1‐specific inhibitor 5Z‐7‐oxozeaenol (Fig 7A). Moderate, but significant, reductions were observed with inhibitors of the MEK1/ERK pathway, such as UO126, FR18, and PD09, while JNK, Akt or TBK1 inhibition was ineffective (Fig 7A). We next focused on the role of the NF‐κB, STAT3 and AP‐1, since these transcription factors have putative binding sites in the Cxcl2 promoter (Fig EV5). Notably, while STAT3 inhibition had no effects, AP‐1 or NF‐κB blockade markedly reduced Cxcl2 responses to live bacteria (Fig 7B). To determine whether TAK1, p38, AP‐1 and NF‐κB inhibition selectively affects Cxcl2 production, we also measured Cxcl1 and TNF‐α levels after stimulation with live bacteria. Appendix Fig S2A and B show that TAK1 and NF‐κB inhibitors markedly reduced the production of these mediators, which was similar to the results previously obtained with Cxcl2. In contrast, despite their ability to almost completely abrogate Cxcl2 production (Fig 7), p38 or AP‐1 blockade only slightly reduced the release of Cxcl1 or TNF‐α (Appendix Fig S2A and B). Moreover, p38 or AP‐1 blockade had modest effects on the production of all cytokines when using killed bacteria or LPS as stimuli (Appendix Fig S2C–H). Collectively, these data suggest that p38 and AP1 are selectively involved in high‐level Cxcl2 induction by live bacteria. To obtain further insights into the involvement of this signaling pathway, we analyzed the activation of AP1 components and p38 after stimulation with live GBS. Live GBS induced more robust and lasting p38 phosphorylation than heat‐killed bacteria (confront Fig 8A with Appendix Fig S3). Activated FosB and c‐Jun, in forms that were capable of binding to the TPA response element (TRE) sequence, could be detected in nuclear extracts of GBS‐stimulated neutrophils shortly after p38 phosporylation (Fig 8A and B). Activation of FosB and c‐Jun was almost totally prevented by p38 blockade, indicating that this MAPK functions upstream of AP‐1 (Fig 8C). Notably, both AP‐1 and p38 activation were markedly reduced in GBS‐stimulated neutrophils lacking Fpr1, Fpr2, or MyD88 (Fig 8D and F), or in neutrophils treated with 5–7 oxo, an inhibitor of the TAK1 kinase, which is known to be downstream of TLRs (Fig 8E). Collectively, these data suggest that stimulation with live bacteria activates two parallel signaling pathways, initiated by TLR and Fprs, respectively, which converge at the level of the p38 MAPK, whose phosphorylation leads to activation of the AP‐1 transcription factor and to high‐level Cxcl2 production.
Figure 7. Effects of signaling molecule or transcription factor inhibitors on Cxcl2 production.
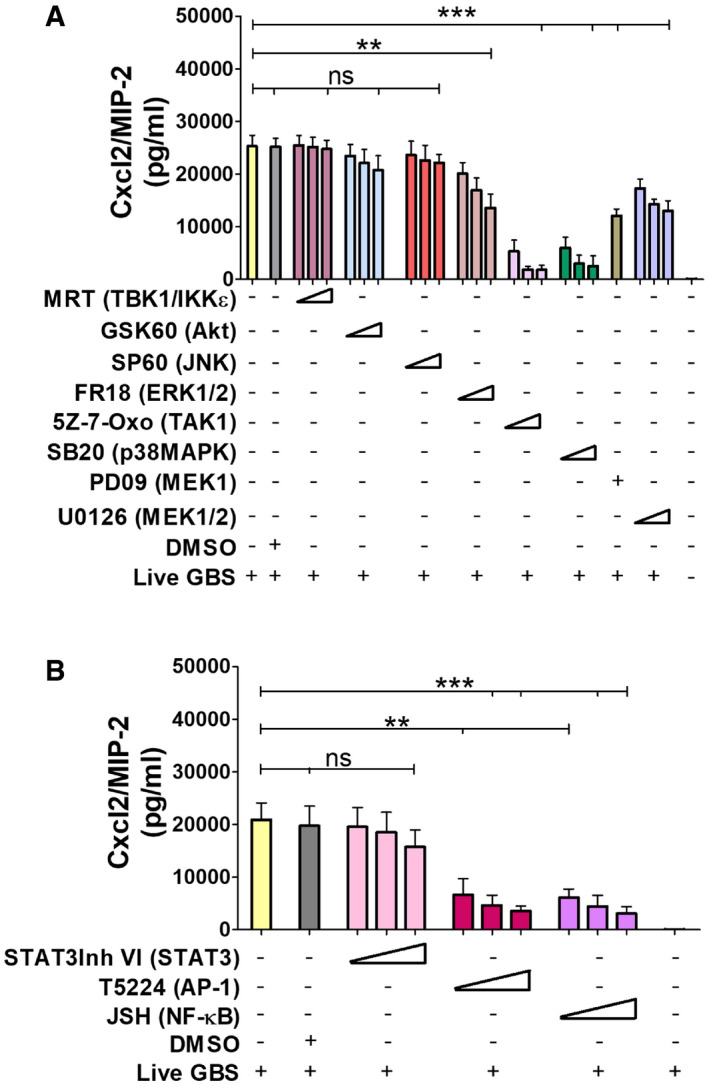
-
A, BBone marrow‐derived neutrophils were exposed to different concentrations of signaling molecule (A) or transcription factors inhibitors (B) for 1 h and then stimulated with live GBS (MOI 5). Target molecules are indicated in parentheses. The concentrations used for each inhibitor and their abbreviations are indicated under the “Materials and Methods” section. Data are means + SD from three independent experiments conducted in duplicate. **P < 0.01, ***P < 0.001, as determined by unpaired t‐test.
Figure EV5. Schematic representation of the Cxcl2 promoter.
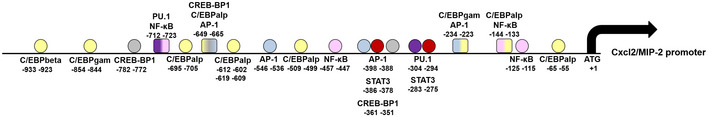
The sequence 1,000 nt upstream of the start site was analyzed or the presence of transcription factor‐binding sites using the Alibaba2.1 software (Grabe, 2002).
Figure 8. The Tak1 and p38 kinases and the AP‐1 and NF‐κB transcription factors are involved in Cxcl2 responses to live GBS.
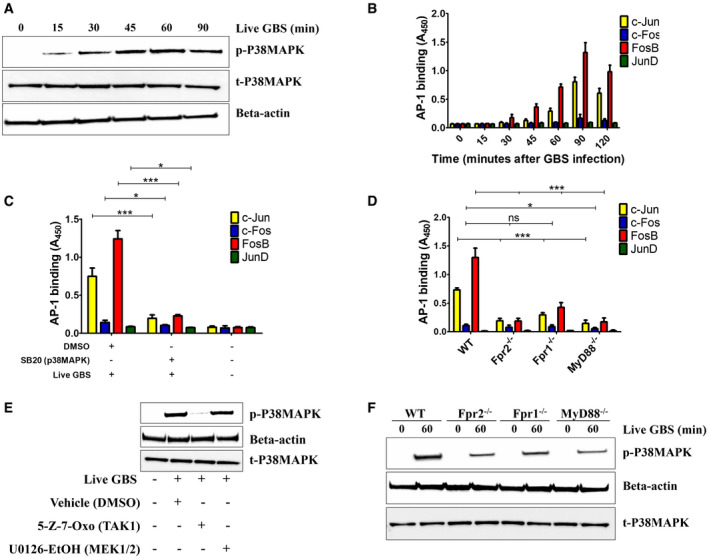
- Time course of p38‐MAPK activation in lysates of neutrophils stimulated with live GBS, as detected by phospho‐specific antibodies using Western blot analysis. Data are from one representative experiment of three showing similar results.
- Time course of AP‐1 DNA‐binding activity in neutrophils stimulated with live GBS (MOI 5). Data are means + SD from three independent experiments conducted in duplicate.
- Effect of p38MAPK inhibition on the activation of AP‐1 family members in neutrophils stimulated with live GBS. Neutrophils were pretreated with SB202190 (5 μM), stimulated for 90 min with live GBS (MOI 5), lysed and assayed for AP‐1 activation. Data are means + SD from three independent experiments conducted in duplicate. *P < 0.05, ***P < 0.001, as determined by unpaired t‐test.
- Effect of the absence of Fpr1, Fpr2, or the TLR adaptor MyD88 on the activation of AP‐1 family members in neutrophils. Neutrophils from WT, Fpr‐, or MyD88‐deficient mice were stimulated for 90 min with live GBS (MOI 5), lysed and assayed for AP‐1 activation. Data are means + SD from three independent experiments conducted in duplicate. *P < 0.05, ***P < 0.001 vs. WT mice, as determined by unpaired t‐test.
- Effect of TAK1 inhibition on p38 MAPK activation in neutrophils stimulated with live GBS (MOI 5).
- Effect of the absence of Fpr1, Fpr2, or the TLR adaptor MyD88 on p38MAPK activation in neutrophils stimulated with live GBS (MOI 5). Shown is a blot representative of three separate experiments.
Discussion
The vast majority of bacterial infections are rapidly dealt with by the recruitment of neutrophils, which become activated and proceed to kill microbes. Pathogen death signals the beginning of a new phase in host responses, characterized by cell de‐activation and repair of the tissue damage caused by neutrophils and microbial toxins. During the entire process, neutrophils and tissue‐resident cell accurately assess microbial viability in order to avoid inappropriate inflammatory responses and maintain homeostasis. Accordingly, neutrophils react vigorously in vitro to the presence of live, but not killed, bacteria by releasing ROS, although the mechanism underlying these differential effects are unclear (Rodriguez‐Rodrigues et al, 2017; Lentini et al, 2020). We show here that, in order to discriminate live from dead bacteria, neutrophils employ a multiple signal system that differs from the single, RNA‐based mechanism used by mononuclear phagocytes for the same purpose (Sander et al, 2011; Blander & Sander, 2012). Stimulation with bacterial RNA or other TLR agonists is necessary, but insufficient, to fully activate crucial antibacterial functions in neutrophils, such as the production of high levels of Cxcl2 and ROS. Rather, concomitant activation of both Fpr1 and Fpr2 is also required. This occurs in the presence of a variety of Fpr1 and Fpr2 agonists released by live bacteria during protein synthesis. We speculate that a multiple signal system of this kind operates as a fail‐safe mechanism that prevents full neutrophil activation and the accompanying tissue damage in the absence of an immediate threat, such as that posed by the presence of live bacteria in sterile tissues. While much has been learned in the past 50 years by using isolated microbial compounds, such as synthetic N‐formyl peptides (Schiffmann et al, 1975) and TLR agonists (Zindel & Kubes, 2020), there is now a need to integrate this knowledge into the context of host responses to whole pathogens, as they occur during natural infection.
Here, we find that neutrophils produce very high Cxcl2 levels in the presence of live, but not killed, bacteria or isolated microbial products. This contrasts with the production of other mediators, including Cxcl1, which was similar after stimulation with viable and dead organisms and did not require Fpr signaling. Our data suggest that this differential induction is linked to the presence in the Cxcl2, but not Cxcl1, promoter of AP‐1 binding sites (Fig EV5). Indeed, we show here that AP‐1 is selectively required for Cxcl2 induction by live bacteria and is downstream to the p38 MAP kinase, which is targeted by both TLR and Fpr signaling pathways. Recent studies have shed new light on the distinctive, non‐redundant role of Cxcl2 in neutrophil responses. Neutrophil‐derived Cxcl2 can potently amplify its own production by autocrine and paracrine signaling in the presence of other activating stimuli (Li et al, 2016) and orchestrate self‐guided, abluminal migration through venular walls at endothelial cell junctions (Girbl et al, 2018). Moreover, neutrophils make a major contribution to the in vivo production of Cxcl2, which drives self‐regulated chemotaxis toward sites of immune complex deposition (Li et al, 2016) or bacterial infection (Lentini et al, 2020). Neutrophil‐derived Cxcl2 (but not Cxcl1) plays an important role in bacterial clearance not only by promoting the recruitment of other neutrophils, but also by increasing their antibacterial activities, including ROS production (Lentini et al, 2020). Accordingly, in the present study, exogenous administration of Cxcl2, but not Cxcl1, rescued the phenotype of Fpr‐defective mice by restoring Cxcl2 production and containment of bacterial infection to wild‐type levels. Studies are underway to examine the role of in vivo Cxcl2 production by cell types other than neutrophils, including mast cells (De Filippo et al, 2008, 2013) in bacterial infection.
Since formylated peptides induce the release of pre‐formed lipid mediators, such as LTB4, but not of chemokines (Afonso et al, 2012; Majumdar et al, 2016), it has been postulated that the latter are indirectly induced through a lipid/cytokine/chemokine cascade (Sadik & Luster, 2012). Our data, instead, suggest a direct link between stimulation with formylated peptides and Cxcl2 production, provided that TLR agonists are also present. This is in agreement with observations that LTB4 was required for the production of Cxcl1 and other chemokines, but not Cxcl2 (Brandt et al, 2018), in the context of bacterial infection. Therefore, it appears that formylated peptides, a class of primary attractants, might directly establish secondary Cxcl2 gradients during infection, without the intervention of other mediators, such as lipids or pro‐inflammatory cytokines, in the presence of TLR agonists. Another major finding of the present study is that Fpr1 and Fpr2 recognize distinctive sets of formylated signal peptides released by live bacteria. Signal peptides are cleaved off from variety of proteins (including extracellular, cell membrane, outer membrane, and cell wall proteins) after they passage through the cell membrane. Although they are likely to represent a significant portion of the formylated peptides produced during bacterial growth, our data do not exclude that other Fpr ligands also play a role in the ability of neutrophils to discriminate between live and dead bacteria. It should be noted, in this respect, that Fprs are highly promiscuous receptors that recognize a large variety of N‐formylated and non‐formylated compounds (Dahlgren et al, 2016), and further research is needed to establish the contribution of different classes of Fpr agonists to the overall ability of various species of pathogenic bacteria to stimulate Fprs. We identified here eleven novel N‐terminal hexapeptide sequences in GBS, which could induce neutrophil chemotaxis and synergize with TLR agonists in inducing Cxcl2 production. Fpr2 was required for the neutrophil‐stimulating activities of the majority of these sequences, and interestingly, this was associated with a more prominent role of Fpr2 than Fpr1 in promoting anti‐GBS host defenses (Fig 4). This finding was unexpected, since the majority of the f‐Met peptides studied thus far preferentially target Fpr1, rather than Fpr2, or target both receptors with similar potency (Southgate et al, 2008; Bufe et al, 2015; Dahlgren et al, 2016).
Therefore, our newly identified Fpr agonist sequences may be useful to obtain further insights into the structural requirements involved in Fpr1/2 activation (Bufe et al, 2015; Raabe et al, 2019) and into the role of these receptors in the pathogenesis of GBS infections. Taken together, our data extend previous studies showing that secretory signal peptides represent a large class of evolutionary conserved molecules that can serve as ligands for Fprs and function as PAMPs (Bufe et al, 2015; Bufe & Zufall, 2016). In conclusion, we propose that neutrophils sense the presence of live bacteria by integrating signals originating from Fpr1, Fpr2, and TLRs. The simultaneous presence of bacterial agonists for these receptors leads to neutrophil activation and increased bactericidal activities by triggering a self‐regulated, Cxcl2‐centered amplification mechanism.
Materials and Methods
Ethics statement
All studies were performed in strict accordance with the European Union guidelines for the use of laboratory animals. The procedures were approved by the Animal Welfare Committee of the University of Messina (OPBA) and by the Ministero della Salute of Italy (permit n° 665/2015‐PR).
Mice and bacterial strains
C57BL/6 wild‐type (WT) mice were purchased from Charles River Laboratories. Fpr1−/− and Fpr2−/− mice were developed by Dr. Ji Ming Wang (National Institute of Health, Bethesda, MD) (Gao et al, 1999; Chen et al, 2010) and kindly provided by Dr. Lars‐Ove Brandenburg, Aachen University, Aachen, Germany. MyD88 and TLR7−/− knockout mice were obtained from Shizuo Akira (Osaka University, Japan). Heterozygous TLR13−/+ mice were provided by the KOMP Repository (www.komp.org) and the Mouse Biology Program (www.mousebiology.org) at the University of California Davis. Subsequently, TLR13−/− mice were bred in the Animal Facility of the Department of Pathology of the University of Messina, Messina, Italy, as described (Mohammadi et al, 2016). All KO mice, bred on a C57BL/6J background, were born and developed normally. All mice used in the present study were housed under specific pathogen‐free conditions in individually ventilated cages. GBS WT strain H36B serotype Ib was used for most experiments. An encapsulated Escherichia coli strain K1 E‐R8 was also used in selected experiments. Bacteria were grown at 37°C with 5% CO2 to the mid‐log phase in Todd‐Hewitt broth (THB, Oxoid), washed twice in nonpyrogenic PBS (0.01 M phosphate, 0.15 M NaCl [pH 7.4]; Euroclone), and resuspended to the desired concentration in PBS. To obtain preparations of heat‐killed bacteria (HK‐GBS), GBS were grown in Carey’s chemically defined medium (Carey et al, 1980) to the late log phase, washed three times, and resuspended in nonpyrogenic PBS. Bacteria were killed by heating at 80°C for 45 min, followed by extensive washing with distilled water and lyophilization. One μg of the heat‐killed bacteria preparation corresponds to approximately 1 × 106 bacteria.
Murine infection model
Six‐week‐old female WT or Fpr1/2‐deficient mice were injected intraperitoneally (i.p.) with the indicated CFUs of live H36B GBS grown to the mid‐log phase. In each experiment, the actual number of injected bacteria was determined by colony counts. Clinical conditions and lethality rates were evaluated every 12 h for 10 days after inoculation. Mice showing signs of disease, such as lethargy, reduced mobility or hunching, were humanely euthanized. In some experiments, peritoneal lavage fluids (PLF) were collected at various times after challenge to measure host cell numbers by flow cytometry, bacterial CFUs, and cytokine concentrations by ELISA. In other experiments, Fpr2‐defective mice were inoculated with 1 μg of recombinant Cxcl1 or Cxcl2 (both from R&D Systems) at 1 h post‐challenge and PLF was collected at 3 h post‐challenge to measure neutrophils numbers and bacterial CFU. In selected experiments, mice were sacrificed at 24 h after infection to measure bacterial burden in PLFs and blood.
GBS signal peptides
Fifteen GBS signal peptide sequences were identified in the Signal Peptide Database (http://www.signalpeptide.de). Custom‐made peptides were synthesized by Genscript Corp. with a purity of ≥ 95%. Details on sequence and source are given in Table 1.
Extraction and purification of bacterial nucleic acids
Bacterial pellets (GBS grown to mid‐log phase in THB) were suspended in 350 μl of Tris–HCl (pH 8, 10 mM) in 1.5 ml microcentrifuge tubes, to which 34 mg of glass beads (106 μm, Sigma‐Aldrich) were added. The tubes were placed in a RETSH MM30 homogenizer and shaken at 30 Hz for 20 min. To isolate RNA in the homogenized samples, RNA purification columns (RNeasy minikit, Qiagen) were used, according to the manufacturer’s protocols. Purified mRNA was obtained from total RNA by sequential removal of rRNA and small RNA with a Microbe Express kit and a Megaclear kit, respectively, according to the manufacturer’s protocols (Invitrogen™). Electrophoresis on agarose gels, followed by slicing of 23S and 16S bands and electroelution, was used to isolate rRNA. Small RNA (under 200 nucleotides; enriched in tRNA and 5S rRNA) was obtained from total RNA by use of a microRNA purification kit according to the manufacturer’s protocol (Norgen). Purification of GBS genomic DNA was performed with the DNeasy Blood and Tissue kit (Qiagen). The quantity and purity of all preparations were determined by Nanodrop 2000 spectrophotometry (Thermo Fisher Scientific) using the manufacturer’s instructions and by electrophoresis on agarose gels. DNA and RNA preparations were “complexed” with DOTAP {N‐[1‐(2,3‐dioleoyloxyl)propyl]‐N,N,N‐trimethylammonium methyl‐sulfate; Sigma‐Aldrich} as described previously (Mancuso et al, 2009) before cell stimulation. Control cultures received DOTAP alone.
Isolation and stimulation of bone marrow‐derived cells
Bone marrow‐derived neutrophils were isolated from the bone marrow of WT and KO mice using Percoll density gradient centrifugation as described (Lentini et al, 2020). Staining with May/Grunwald/Giemsa showed that ~90% of isolated cells (15–20 ± 0.6 × 106 cells per mouse) were morphologically mature neutrophils (bands and segmented). Moreover, purity of these neutrophil populations was > 95%, as determined by flow cytometry using the neutrophil‐specific marker Ly‐6G. To obtain bone marrow‐derived macrophages, marrow cells were cultured for 6–7 days in RPMI 1640 supplemented with 10% FCS, penicillin (50 IU/ml), and streptomycin (50 μg/ml). Medium was supplemented with either 100 ng/ml macrophage colony‐stimulating factor (M‐CSF) or 20 ng/ml granulocyte–macrophage colony‐stimulating factor (GM‐CSF) (both from PeproTech) to obtain bone marrow‐derived, M‐CSF‐polarized (M‐CSF‐MΦ) or GM‐CSF‐polarized (GM‐CSF‐MΦ) macrophages, respectively, as previously described (Fama et al, 2020). Isolated bone marrow‐derived cells (5 × 105 per well in 0.2 ml of RPMI supplemented with 10% FCS) were seeded in microtiter plates and stimulated with HK or live bacteria grown to the mid‐log phase at the indicated concentrations or multiplicities of infection (MOI), respectively. In selected experiments, bacteria were grown in THB containing a sub‐inhibitory concentration of the formyl peptidase inhibitor actinonin (32 µg/ml; Sigma‐Aldrich). All stimulations were carried out by centrifuging cell suspensions for 10 min at 400 × g to facilitate bacteria/neutrophil interactions. After incubation for 1 h at 37°C with 5% CO2, penicillin (250 IU/ml), and streptomycin (250 μg/ml) were added to kill extracellular bacteria. In preliminary tests, we determined the number of colony forming units at various times following antibiotic exposure and observed complete bacterial killing. Control wells were stimulated with Escherichia coli K12 ultrapure LPS (InvivoGen). In selected experiments, neutrophils (5 × 105/well) were preincubated for 1 h at 37°C with 5% CO2 with the selective Fpr2 antagonist WRW4 (Abcam) and the pan‐FPR inhibitor Boc‐2 (GenScript) at the indicated concentrations before stimulation with HK or live GBS. In another set of experiments, neutrophil stimulation with HK‐GBS, LPS, or bacterial RNA was preceded by a 1 h preincubation at 37°C in 5% CO2 with the indicated concentrations of the selective FPR agonists f‐MIFL (GenScript) or WKYMVM (Abcam) or with different concentrations of formylated peptides (fPep8 and fPep10) from GBS. The contribution of ROS as signaling molecules to GBS‐induced Cxcl2 production was evaluated by pretreating neutrophils (for 1 h before stimulation with live GBS) with the following molecules: (i) the ROS scavengers dimethylthiourea (DMTU) and Mn(III) tetrakis (4‐benzoic acid) porphyrin (MnTBAP) (both from Sigma‐Aldrich); (ii) the NADPH oxidase inhibitor diphenyleneiodonium chloride (DPI) (from Calbiochem); (iii) Apoptosis Signal‐regulating Kinase 1 (ASK1) inhibitor GS‐444217 (from MedChemExpress). In preliminary experiments we tested the ability of DMTU, MnTBAP and DPI to inhibit ROS production in GBS‐stimulated neutrophils using the CellROX kit.
For the inhibitor studies, neutrophils were pretreated for 1 h in the presence or absence of intracellular kinase inhibitors: 5Z‐7‐Oxozeaenol (TAK1 inhibitor, 500 nM, 1 and 5 μM), SB202190 (p38MAPK inhibitor, 500 nM, 5 and 10 μM), PD98059 (MEK1 inhibitor, 50 μM), FR180204 (ERK1/2 inhibitor, 500 nM, 1 and 10 μM), SP600125 (JNK inhibitor, 5, 10 and 20 μM), GSK690693 (AKT inhibitor, 500 nM, 5 and 10 μM), JSH‐23 (NF‐kB inhibitor, 10, 50, and 100 μM) (all purchased from Sigma‐Aldrich), U0126‐EtOH (MEK1/2 inhibitor, 500 nM, 1 and 10 μM) and T5224 (AP‐1 inhibitor, 10, 50 and 100 μM) (both from MedChemExpress), MRT67307 (TBK1 and IKKε inhibitor, 1, 2 and 3 μM; InvivoGen) and STAT3 Inhibitor VI (STAT3 inhibitor; Calbiochem, 10, 50, and 100 μM). Cell culture supernatants were collected at 24 h and stored at −80°C for cytokine measurements.
Reactive oxygen species measurement
Bacteria‐induced reactive oxygen species (ROS) production by neutrophils was measured using the CellROX Deep Red Flow Cytometry Assay Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. Briefly, neutrophils (5 × 105 per well in 0.2 ml of RPMI supplemented with 10% FCS) were seeded in microtiter plates and stimulated for 30 min with live GBS or combinations of the indicated fMet peptides with TLR agonists (HK‐GBS or bacterial RNA). In some experiments, cells were preincubated with the TLR agonists for 1 h before stimulation with f‐Met peptides. In another set of experiments, neutrophils from WT, MyD88, or Fpr1/2‐deficient mice were stimulated with live GBS and E. coli at the indicated MOIs. After stimulation, samples then stained with the CellROX fluorescent reagent at a final concentration of 5 μM for 30 min at 37˚C. Cells were washed, fixed with 3.7% formaldehyde for 15 min, and analyzed with a BD FACSCanto II instrument.
Western blot analysis of neutrophil lysates
Bone marrow‐derived neutrophils were seeded on 6‐well plates (5 × 106/well in 2 ml of RPMI 1640 supplemented with 10% FCS) and collected at various times after the addition of live GBS or plain medium as a control. For the inhibitor studies, neutrophils were pretreated with selected inhibitors (5Z‐7‐Oxozeaenol or U0126‐EtOH, 1 μM) or control vehicle (0.2% DMSO) for 1 h prior to stimulation with live GBS (MOI 5) for 60 min. Cells were washed three times with ice‐cold PBS and lysed by vigorous vortexing in RIPA lysis buffer [50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1% Triton X‐100, 20% glycerol, 1× protease inhibitor cocktail (Promega)]. Lysates were then centrifuged at 13,000 g for 15 min at 4°C. Protein concentration in each sample was determined using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific). Protein samples (30 μg of protein per lane) were run on precast Bolt Bis‐Tris 4–12% gels (Invitrogen) with 1× MOPS buffer (Invitrogen) and transferred on PVDF (polyvinylidene difluoride) membranes (Bio‐Rad). Membranes were washed in TBS‐T (Tris‐buffered saline with 0.1% Tween‐20) and blocked with TBS‐T containing 5% bovine serum albumin (BSA, Sigma‐Aldrich) for 2 h. Membranes were subsequently incubated with primary antibodies in TBS‐T containing 5 or 1% BSA at 4°C overnight. The following primary antibodies were used: anti‐phospho p38MAPK (T180/Y182; Cell Signaling Technology), anti‐total p38 MAPK Antibody (9212; Cell Signaling Technology), and anti‐beta actin (ab8229; Abcam). After overnight incubation, membranes were washed with TBS‐T and incubated with secondary antibodies (swine anti‐rabbit immunoglobulins HRP‐conjugated, DAKO, or goat IgG HRP‐conjugated, R&D Systems Sciences) for 2 h at room temperature in TBS‐T containing 1% milk or BSA. Membranes were then washed with TBS‐T and developed using chemiluminescence reagent (Immobilon Forte Western HRP substrate, Merck Millipore). Images were captured with Bio‐Rad's ChemiDoc XRS system. Beta‐actin was used as loading control.
Neutrophil migration assay
Neutrophil chemotaxis was determined using ChemoTX multiwell chambers (NeuroProbe) with 3 µm pore size filters according to the manufacturer’s protocol. Briefly, peptides or vehicle were diluted to the desired concentration in RPMI medium supplemented with 0.1% BSA (wt/vol) and 320 µl were added to the lower chamber of each well. Neutrophil suspensions (1 × 105 per well in 57 µl of RPMI/0.1% BSA) were placed on top of the polycarbonate filter above each well and allowed to migrate for 90 min at 37°C with 5% CO2. The assay was terminated by detaching and wiping the filter to remove non‐migrated cells from the filter top. Thereafter, the number of transmigrated cells was determined using BD TruCount tubes (BD Biosciences).
Determination of intracellular calcium levels in neutrophils
Intracellular calcium was measured using the Fura Red (Thermo Fisher Scientific), a calcium‐sensitive dye using ratiometric analysis, as previously described (Wendt et al, 2015). Neutrophils were resuspended at 1 × 107 cells/ml in pre‐warmed Hanks balanced salt solution (HBSS; Euroclone) and incubated with 1 μM Fura Red dye at 37°C for 30 min. After incubation, cells were washed three times in HEPES buffered saline solution (HBSS supplemented with 1 mM CaCl2, 0.5 mM MgCl2, 0.1% BSA, 10 mM HEPES), resuspended in HEPES buffer and stimulated with GBS fMet peptides. Ratiometric calcium flux was measured by flow cytometry as described (Wendt et al, 2015).
Cytokine and AP‐1 transcription factor activation measurement
Keratinocyte‐derived chemokine (KC; CXCL1), macrophage inflammatory protein 2 (MIP‐2; CXCL2), tumor necrosis factor alpha (TNF‐α), and interleukin 1 beta (IL‐1β) concentrations were determined in duplicate using the murine enzyme‐linked immunosorbent assay (ELISA) kits CXCL1/KC Quantikine, CXCL2/MIP‐2 DuoSet, TNF‐α DuoSet and Mouse IL‐1 beta/IL‐1F2 DuoSet, according to the manufacturer’s recommendations (R&D Systems). The lower detection limits of these assays were 15.6, 15.6, 31.3, and 15.6 pg/ml, respectively. For AP‐1 transcription factor activation measurements, nuclear extracts (15 μg) from neutrophils stimulated for different time points with live GBS (MOI of 5) were assayed for binding of the activated AP‐1 family members c‐Jun, c‐Fos, FosB, and JunD to a TPA‐responsive element (TRE) oligonucleotide immobilized onto a 96‐well plate using the AP1 Transcription Factor Assay Kit (ab207196; Abcam) according to the manufacturer’s protocols. The AP‐1 transcription factor assay was performed in the presence of soluble wild‐type or mutated consensus binding oligonucleotides. Results are expressed as specific AP‐1 binding activity (i.e., absorbance measured in the presence of the mutated oligonucleotide minus that measured in the presence of the wild‐type oligonucleotide).
Flow cytometry
The evaluation of neutrophil influx into the peritoneal cavity was performed on a FACS Canto II flow cytometer (BD Biosciences) as previously described (Cardaci et al, 2012; Biondo et al, 2014; Lentini et al, 2020). Briefly, all PLF cells were blocked with 0.5 μg Fc Block (BD Biosciences) for 20 min at room temperature and incubated in the dark for 20 min with the rat anti‐mouse Ly‐6G monoclonal antibody (clone 1A8, Thermo Fisher Scientific), which is highly specific for neutrophils (Daley et al, 2008). The respective isotype Abs were used as control. The quantification of neutrophil numbers in PLF samples and in neutrophil migration assay were determined using BD Trucount™ Absolute Counting Tubes (BD Biosciences). Data analysis was performed using Flowing Software 2.5.1.
Statistical analyses
Differences in cytokine and transcription factor activation levels were assessed by Student's t‐test. The same statistic test was used for the analysis of neutrophil counts and ROS production. Survival data were analyzed by Kaplan–Meier survival plots, and differences in bacterial CFU counts were assessed by the Mann–Whitney U‐test or the Wilcoxon test. Differences were considered statistically significant when P values were less than 0.05 (P < 0.05). Statistical analyses were performed with GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA).
Author contributions
Giuseppe Teti: Conceptualization; Supervision; Investigation; Writing—original draft; Writing—review and editing. Germana Lentini: Data curation; Investigation; Methodology; Writing—review and editing. Giuseppe Valerio De Gaetano: Methodology. Agata Famà: Investigation; Methodology. Roberta Galbo: Methodology. Francesco Coppolino: Methodology. Giuseppe Mancuso: Methodology. Concetta Beninati: Conceptualization; Investigation; Methodology; Writing—original draft; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
GT and CB conceived the study. GL designed and performed most of the experiments and analyzed the data. GVG, AF, RG, GM, and FC performed several experiments and contributed to experimental design. GL, GT, and CB wrote the paper. All authors approved the final version of the manuscript and agree to be held accountable for the content therein.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgments
The work was funded in part by grant PRIN 2017, research Projects of National Relevance, Prot. 2017M8R7N9. Heartfelt thanks to Dr. Mariia Yurchenko from the Department of Clinical and Molecular Medicine, Norwegian University of Science and Technology, Trondheim, Norway, for providing advice and reagents for the analysis of intracellular signaling. We thank Dr. Andrew Waller, Intervacc AB, Hägersten, Sweden for his suggestion to use bacteria treated with peptide deformylase inhibitors as a stimulus and Prof. Lars Brandenburg, Aachen University, Aachen, Germany for sending Fpr‐defective mice. We also thank Dr. Angelina Midiri from our University for help with the in vivo experiments.
Disclosure and competing interests statement
Concetta Beninati and Giuseppe Teti act as scientific advisors for, respectively, Scylla Biotech Srl. and Charybdis Vaccines Srl. without receiving any compensation for these activities. Charybdis Vaccines S.r.l. and Scylla Biotech S.r.l. did not provide funding for this study and had no role in its conduction. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The EMBO Journal (2022) 41: e109386.
Data availability
This study includes no data deposited in external repositories.
References
- Afonso PV, Janka‐Junttila M, Lee YJ, McCann CP, Oliver CM, Aamer KA, Losert W, Cicerone MT, Parent CA (2012) LTB4 is a signal‐relay molecule during neutrophil chemotaxis. Dev Cell 22: 1079–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auperin TC, Bolduc GR, Baron MJ, Heroux A, Filman DJ, Madoff LC, Hogle JM (2005) Crystal structure of the N‐terminal domain of the group B streptococcus alpha C protein. J Biol Chem 280: 18245–18252 [DOI] [PubMed] [Google Scholar]
- Bae YS, Lee HY, Jo EJ, Kim JI, Kang HK, Ye RD, Kwak JY, Ryu SH (2004) Identification of peptides that antagonize formyl peptide receptor‐like 1‐mediated signaling. Journal of Immunology 173: 607–614 [DOI] [PubMed] [Google Scholar]
- Biondo C, Mancuso G, Midiri A, Signorino G, Domina M, Lanza Cariccio V, Mohammadi N, Venza M, Venza I, Teti G et al (2014) The interleukin‐1beta/CXCL1/2/neutrophil axis mediates host protection against group B streptococcal infection. Infect Immun 82: 4508–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Sander LE (2012) Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol 12: 215–225 [DOI] [PubMed] [Google Scholar]
- Brandt SL, Klopfenstein N, Wang S, Winfree S, McCarthy BP, Territo PR, Miller L, Serezani CH (2018) Macrophage‐derived LTB4 promotes abscess formation and clearance of Staphylococcus aureus skin infection in mice. PLoS Pathog 14: e1007244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bufe B, Schumann T, Kappl R, Bogeski I, Kummerow C, Podgorska M, Smola S, Hoth M, Zufall F (2015) Recognition of bacterial signal peptides by mammalian formyl peptide receptors: a new mechanism for sensing pathogens. J Biol Chem 290: 7369–7387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bufe B, Zufall F (2016) The sensing of bacteria: emerging principles for the detection of signal sequences by formyl peptide receptors. Biomol Concepts 7: 205–214 [DOI] [PubMed] [Google Scholar]
- Buscetta M, Firon A, Pietrocola G, Biondo C, Mancuso G, Midiri A, Romeo L, Galbo R, Venza M, Venza I et al (2016) PbsP, a cell wall‐anchored protein that binds plasminogen to promote hematogenous dissemination of group B Streptococcus. Mol Microbiol 101: 27–41 [DOI] [PubMed] [Google Scholar]
- Cardaci A, Papasergi S, Midiri A, Mancuso G, Domina M, Cariccio VL, Mandanici F, Galbo R, Passo CL, Pernice I et al (2012) Protective activity of Streptococcus pneumoniae Spr 1875 protein fragments identified using a phage displayed genomic library. PLoS One 7: e36588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey RB, Eisenstein TK, Shockman GD, Greber TF, Swenson RM (1980) Soluble group‐ and type‐specific antigens from type III group B Streptococcus. Infect Immun 28: 195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Le Y, Liu Y, Gong W, Ying G, Huang J, Yoshimura T, Tessarollo L, Wang JM (2010) A critical role for the g protein‐coupled receptor mFPR2 in airway inflammation and immune responses. Journal of Immunology 184: 3331–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, Midiri A, Galbo R, Trieu‐Cuot P, Papasergi S et al (2012) Activation of the NLRP3 inflammasome by group B streptococci. Journal of Immunology 188: 1953–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren C, Gabl M, Holdfeldt A, Winther M, Forsman H (2016) Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger‐associated molecular pattern generated by bacteria and mitochondria. Biochem Pharmacol 114: 22–39 [DOI] [PubMed] [Google Scholar]
- Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE (2008) Use of Ly6G‐specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 83: 64–70 [DOI] [PubMed] [Google Scholar]
- Deniset JF, Kubes P (2016) Recent advances in understanding neutrophils. F1000Research 5: 2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ear T, Fortin CF, Simard FA, McDonald PP (2010) Constitutive association of TGF‐beta‐activated kinase 1 with the IkappaB kinase complex in the nucleus and cytoplasm of human neutrophils and its impact on downstream processes. Journal of Immunology 184: 3897–3906 [DOI] [PubMed] [Google Scholar]
- Erdogan S, Fagan PK, Talay SR, Rohde M, Ferrieri P, Flores AE, Guzman CA, Walker MJ, Chhatwal GS (2002) Molecular analysis of group B protective surface protein, a new cell surface protective antigen of group B streptococci. Infect Immun 70: 803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fama A, Midiri A, Mancuso G, Biondo C, Lentini G, Galbo R, Giardina MM, De Gaetano GV, Romeo L, Teti G, et al (2020) Nucleic acid‐sensing toll‐like receptors play a dominant role in innate immune recognition of pneumococci. MBio 11: e00415‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, Hogg N (2013) Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 121: 4930–4937 [DOI] [PubMed] [Google Scholar]
- De Filippo K, Henderson RB, Laschinger M, Hogg N (2008) Neutrophil chemokines KC and macrophage‐inflammatory protein‐2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. Journal of Immunology 180: 4308–4315 [DOI] [PubMed] [Google Scholar]
- Fu H, Dahlgren C, Bylund J (2003) Subinhibitory concentrations of the deformylase inhibitor actinonin increase bacterial release of neutrophil‐activating peptides: a new approach to antimicrobial chemotherapy. Antimicrob Agents Chemother 47: 2545–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao JL, Lee EJ, Murphy PM (1999) Impaired antibacterial host defense in mice lacking the N‐formylpeptide receptor. J Exp Med 189: 657–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gase K, Ozegowski J, Malke H (1998) The Streptococcus agalactiae hylB gene encoding hyaluronate lyase: completion of the sequence and expression analysis. Biochem Biophys Acta 1398: 86–98 [DOI] [PubMed] [Google Scholar]
- Girbl T, Lenn T, Perez L, Rolas L, Barkaway A, Thiriot A, del Fresno C, Lynam E, Hub E, Thelen M et al (2018) Distinct compartmentalization of the chemokines CXCL1 and CXCL2 and the atypical receptor ACKR1 determine discrete stages of neutrophil diapedesis. Immunity 49: 1062–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabe N (2002) AliBaba2: context specific identification of transcription factor binding sites. In Silico Biol 2: S1–15 [PubMed] [Google Scholar]
- He HQ, Ye RD (2017) The formyl peptide receptors: diversity of ligands and mechanism for recognition. Molecules 22: 455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlstrom PG, Chhatwal GS, Timmis KN (1991) The IgA‐binding beta antigen of the c protein complex of Group B streptococci: sequence determination of its gene and detection of two binding regions. Mol Microbiol 5: 843–849 [DOI] [PubMed] [Google Scholar]
- Kang L, Yu H, Yang X, Zhu Y, Bai X, Wang R, Cao Y, Xu H, Luo H, Lu LU et al (2020) Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat Commun 11: 2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienle K, Lammermann T (2016) Neutrophil swarming: an essential process of the neutrophil tissue response. Immunol Rev 273: 76–93 [DOI] [PubMed] [Google Scholar]
- Kobayashi SD, Voyich JM, Burlak C, DeLeo FR (2005) Neutrophils in the innate immune response. Arch Immunol Ther Exp 53: 505–517 [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13: 159–175 [DOI] [PubMed] [Google Scholar]
- Landwehr‐Kenzel S, Henneke P (2014) Interaction of Streptococcus agalactiae and cellular innate immunity in colonization and disease. Front Immunol 5: 519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauvau G, Vijh S, Kong P, Horng T, Kerksiek K, Serbina N, Tuma RA, Pamer EG (2001) Priming of memory but not effector CD8 T cells by a killed bacterial vaccine. Science 294: 1735–1739 [DOI] [PubMed] [Google Scholar]
- Le Y, Murphy PM, Wang JM (2002) Formyl‐peptide receptors revisited. Trends Immunol 23: 541–548 [DOI] [PubMed] [Google Scholar]
- Lentini G, Famà A, Biondo C, Mohammadi N, Galbo R, Mancuso G, Iannello D, Zummo S, Giardina M, De Gaetano GV et al (2020) Neutrophils enhance their own influx to sites of bacterial infection via endosomal TLR‐dependent Cxcl2 production. Journal of Immunology 204: 660–670 [DOI] [PubMed] [Google Scholar]
- Li JL, Lim CH, Tay FW, Goh CC, Devi S, Malleret B, Lee B, Bakocevic N, Chong SZ, Evrard M et al (2016) Neutrophils self‐regulate immune complex‐mediated cutaneous inflammation through CXCL2. J Invest Dermatol 136: 416–424 [DOI] [PubMed] [Google Scholar]
- Majumdar R, Sixt M, Parent CA (2014) New paradigms in the establishment and maintenance of gradients during directed cell migration. Curr Opin Cell Biol 30: 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar R, Tavakoli Tameh A, Parent CA (2016) Exosomes mediate LTB4 release during neutrophil chemotaxis. PLoS Biol 14: e1002336 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, Teti G, Beninati C (2009) Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol 10: 587–594 [DOI] [PubMed] [Google Scholar]
- Mancuso G, Midiri A, Beninati C, Piraino G, Valenti A, Nicocia G, Teti D, Cook J, Teti G (2002) Mitogen‐activated protein kinases and NF‐kappa B are involved in TNF‐alpha responses to group B streptococci. Journal of Immunology 169: 1401–1409 [DOI] [PubMed] [Google Scholar]
- Mohammadi N, Midiri A, Mancuso G, Patane F, Venza M, Venza I, Passantino A, Galbo R, Teti G, Beninati C et al (2016) Neutrophils directly recognize group B streptococci and contribute to interleukin‐1beta production during infection. PLoS One 11: e0160249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyen C, Runchel C, Schupfer F, Meier P, Lemaitre B (2016) The regulatory isoform rPGRP‐LC induces immune resolution via endosomal degradation of receptors. Nat Immunol 17: 1150–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen GT, Green ER, Mecsas J (2017) Neutrophils to the ROScue: mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol 7: 373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasergi S, Galbo R, Lanza‐Cariccio V, Domina M, Signorino G, Biondo C, Pernice I, Poyart C, Trieu‐Cuot P, Teti G et al (2013) Analysis of the Streptococcus agalactiae exoproteome. J Proteomics 89: 154–164 [DOI] [PubMed] [Google Scholar]
- Podbielski A, Blankenstein O, Lutticken R (1994) Molecular characterization of the cfb gene encoding group B streptococcal CAMP‐factor. Med Microbiol Immunol 183: 239–256 [DOI] [PubMed] [Google Scholar]
- Raabe CA, Groper J, Rescher U (2019) Biased perspectives on formyl peptide receptors. Biochim Biophys Acta 1866: 305–316 [DOI] [PubMed] [Google Scholar]
- Raabe VN, Shane AL (2019) Group B Streptococcus (Streptococcus agalactiae). Microbiol Spectr 7: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh LW, Schmidt R (1965) Measles immunization with killed virus vaccine. serum antibody titers and experience with exposure to measles epidemic. Am J Dis Child 109: 232–237 [DOI] [PubMed] [Google Scholar]
- Reinscheid DJ, Gottschalk B, Schubert A, Eikmanns BJ, Chhatwal GS (2001) Identification and molecular analysis of PcsB, a protein required for cell wall separation of group B streptococcus. J Bacteriol 183: 1175–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinscheid DJ, Stosser C, Ehlert K, Jack RW, Moller K, Eikmanns BJ, Chhatwal GS (2002) Influence of proteins Bsp and FemH on cell shape and peptidoglycan composition in group B streptococcus. Microbiology 148: 3245–3254 [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Rodrigues N, Castillo LA, Landoni VI, Martire‐Greco D, Milillo MA, Barrionuevo P, Fernandez GC (2017) Prokaryotic RNA associated to bacterial viability induces polymorphonuclear neutrophil activation. Front Cell Infect Microbiol 7: 306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo RC, Garcia CC, Teixeira MM, Amaral FA (2014) The CXCL8/IL‐8 chemokine family and its receptors in inflammatory diseases. Exp Rev Clin Immunol 10: 593–619 [DOI] [PubMed] [Google Scholar]
- Sadik CD, Luster AD (2012) Lipid‐cytokine‐chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol 91: 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, Swanson JA, Muller M, Blander JM (2011) Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature 474: 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann E, Corcoran BA, Wahl SM (1975) N‐formylmethionyl peptides as chemoattractants for leucocytes. Proc Natl Acad Sci USA 72: 1059–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert A, Zakikhany K, Schreiner M, Frank R, Spellerberg B, Eikmanns BJ, Reinscheid DJ (2002) A fibrinogen receptor from group B Streptococcus interacts with fibrinogen by repetitive units with novel ligand binding sites. Mol Microbiol 46: 557–569 [DOI] [PubMed] [Google Scholar]
- Signorino G, Mohammadi N, Patanè F, Buscetta M, Venza M, Venza I, Mancuso G, Midiri A, Alexopoulou L, Teti G et al (2014) Role of Toll‐like receptor 13 in innate immune recognition of group B streptococci. Infect Immun 82: 5013–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southgate EL, He RL, Gao JL, Murphy PM, Nanamori M, Ye RD (2008) Identification of formyl peptides from Listeria monocytogenes and Staphylococcus aureus as potent chemoattractants for mouse neutrophils. Journal of Immunology 181: 1429–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenfeldt AL, Karlsson J, Wenneras C, Bylund J, Fu H, Dahlgren C (2007) Cyclosporin H, Boc‐MLF and Boc‐FLFLF are antagonists that preferentially inhibit activity triggered through the formyl peptide receptor. Inflammation 30: 224–229 [DOI] [PubMed] [Google Scholar]
- Ugolini M, Sander LE (2019) Dead or alive: how the immune system detects microbial viability. Curr Opin Immunol 56: 60–66 [DOI] [PubMed] [Google Scholar]
- Vance RE, Isberg RR, Portnoy DA (2009) Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6: 10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt ER, Ferry H, Greaves DR, Keshav S (2015) Ratiometric analysis of fura red by flow cytometry: a technique for monitoring intracellular calcium flux in primary cell subsets. PLoS One 10: e0119532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindel J, Kubes P (2020) DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annual Review of Pathology 15: 493–518 [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O (2012) The chemokine superfamily revisited. Immunity 36: 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]