

Abstract
Lung adenocarcinoma (LUAD) accounts for almost 40% of lung cancers, leading to significant associated morbidity and mortality rates. However, the mechanism of LUAD tumorigenesis remains far from clear. Here, we scanned down‐regulated genes involved in LUAD sourced from The Cancer Genome Atlas and Gene Expression Omnibus data and focused on G protein‐coupled receptor 133 (GPR133). We offer compelling evidence that GPR133 was expressed at low levels in the setting of LUAD, and higher expression was positively related to a better prognosis among patients with LUAD. Functionally, GPR133 inhibited cell proliferation and tumor growth in vitro and in vivo. Regarding the mechanism, flow cytometry assays and western blot assays showed that GPR133 enhanced p21 and decreased cyclin B1 expression, thus triggering LUAD cells at G2/M‐phase arrest. Consistent with this, we evaluated the expression levels of cell‐cycle biomarkers and found that bioinformatics analysis combined with N6‐methyladenosine (methylation at the N6 position in adenosine) RNA immunoprecipitation‐qPCR assay indicated that GPR133 expression was down‐regulated by this modification. Moreover, we observed that methyltransferase‐like 3 was impaired in LUAD, and that it is able to significantly increase levels of GPR133 by enhancing its RNA stability. In conclusion, we found that GPR133 expression was down‐regulated in LUAD via N6‐methyladenosine modification. Increasing GPR133 levels could suppress LUAD cell proliferation and tumor growth.
Keywords: cell cycle, G protein‐coupled receptor 133, lung adenocarcinoma, proliferation
G protein‐coupled receptor 133 (GPR133) is expressed at low levels in lung adenocarcinoma (LUAD). Furthermore, GPR133 inhibits LUAD cell proliferation and tumor growth. Mechanistically, GPR133 induces G2/M‐phase arrest by inhibiting cyclin B1 and enhancing p21 expression. Moreover, we demonstrated that METTL3 could regulate GPR133 in LUAD cells. Together, these results imply that GPR133 affects LUAD progression via regulation of cell proliferation.
Abbreviations
- GAPDH
glyceraldehyde‐3 phosphate dehydrogenase
- GEO
Gene Expression Omnibus
- GEPIA
Gene Expression Profiling Interactive Analysis
- GO
Gene Ontology
- GPCR
G protein‐coupled receptor
- GPR133
G protein‐coupled receptor 133
- GSEA
gene set enrichment analysis
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LUAD
lung adenocarcinoma
- m6A
N6‐methyladenosine
- METTL3
methyltransferase‐like 3
- OS
overall survival
- qRT‐OCR
quantitative RT‐PCR
- SD
standard deviation
- TCGA
The Cancer Genome Atlas
Lung adenocarcinoma (LUAD), which develops along the outer edges of the lungs within glandular cells in the small airways, accounts for approximately 40% of all lung cancer cases and constitutes a major cause of cancer mortality worldwide [1]. Recent efforts have focused on identifying biomarkers for the diagnosis and treatment of LUAD. Several genes, such as epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK) and reactive oxygen species (ROS), were found to exhibit genomic mutations in LUAD [2], while other research found that molecularly targeted therapies directed at receptor tyrosine kinases were clinically successful. Studies are also currently being conducted on other targeted therapies directed against alterations in the KRAS, ERBB2, BRAF, MET, RET, NTRK1 and NTRK2 genes [3]. Encouragingly, immune checkpoint inhibitors targeting programmed cell death 1 receptor/programmed death‐ligand 1 mediated immunosuppression, showing efficacy in up to 30% of patients with LUAD [4]. However, many patients with LUAD have no common genetic mutation or acquire resistance to epidermal growth factor receptor tyrosine kinase inhibitors, even experiencing no response to immune checkpoint inhibition therapy. Thus, it is urgent to advance the understanding of the regulatory mechanisms involved in the development and progression of LUAD. Also, more sensitive novel biomarkers need to be identified for early diagnosis and therapeutic purposes.
The adhesion family forms a large branch of the pharmacologically important superfamily of G protein‐coupled receptors (GPCRs), which play important roles in receptor recognition, signal transduction, the cell cycle and cell differentiation [5]. G protein‐coupled receptor 133 (GPR133), also known as ADGRD1, is an orphan adhesion GPCR member. GPR133 contains a large N‐terminal extracellular domain, with a signal peptide and a pentraxin/concanavalin A domain [6]. Research suggests that the height and length of the R–R interval in the adult cardiac electrical cycle are related to the single‐nucleotide polymorphisms of GPR133 [7, 8]. Notably, GPR133 correlates with altered bone mineral density in mouse knockouts, suggesting that it is a causal genetic driver of such disease in humans [9]. GPR133 expression increased as a function of World Health Organization grade and peaks in glioblastoma [10]. However, the role of GPR133 in LUAD remains unknown.
Here, we conducted a bioinformatics analysis of down‐regulated genes in LUAD using several databases, focusing in particular on GPR133. Our investigation demonstrated that GPR133 expression was decreased in LUAD and positively related to better outcomes among patients with LUAD. Moreover, increased GPR133 expression may significantly suppress the proliferation of LUAD cells in vitro and in vivo.
Materials and methods
Bioinformatics analysis
The mRNA expression profiles and outcomes of patients with LUAD were obtained from The Cancer Genome Atlas (TCGA) data portal (https://tcga‐data.nci.nih.gov/tcga/) and Gene Expression Omnibus (GEO) datasets (https://www.ncbi.nlm.nih.gov/). The down‐regulated genes with fold changes of two or more and P < 0.05 were chosen during subsequent analysis. We merged the earlier down‐regulated genes for intersection and focused on the GPR133 gene. Meanwhile, the r software (R Foundation for Statistical Computing, Vienna, Austria) was used to match GPR133 expression levels with the outcomes of patients with LUAD via TCGA data. Similarly, we analyzed GPR133 expression levels and the prognosis of patients with LUAD using GEO datasets. Moreover, gene set enrichment analysis (GSEA) was implemented via GSEA version 2.2.2 (http://www.broadinstitute.org/gsea) to investigate the biological characteristics of GPR133. The genes correlated with GPR133 were downloaded from the cBioPortal for Cancer Genomics (https://www.cbioportal.org/) and placed into the DAVID Bioinformatics Resources database (https://david.ncifcrf.gov/) to predict their function by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. The N6‐methyladenosine (m6A) sites on the GPR133 transcript were analyzed using the SRAMP online software program (http://www.cuilab.cn/sramp). The expression pattern of methyltransferase‐like 3 (METTL3) and the relationship between METTL3 and GPR133 were estimated using Gene Expression Profiling Interactive Analysis (GEPIA) data (http://gepia.cancer‐pku.cn/).
Cells and clinical samples
The BEAS‐2B human bronchial epithelial cell line and several kinds of LUAD cells (A549, H1299, H1650, H1975 and PC9) were cultured in Roswell Park Memorial Institute (RPMI) 1640 (Gibco Laboratories, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Gibco Laboratories), incubated at 37 °C with 5% CO2 in a humidified incubator. A total of 28 cases of LUAD tissue samples and matched adjacent normal tissues were collected from patients in the People’s Hospital of Wuzhou between December 2011 and March 2019. There were 14 male and 14 female patients. The average age is 53.78 ± 4.67 years. Another cohort of normal tissues (n = 16, 5 female and 11 male patients; average age is 39.41 ± 10.27 years) and LUAD tissues (n = 63, 25 female and 38 male patients; the average age is 64.39 ± 9.17 years) with prognosis information was compiled from the biological resource specimen bank of the People’s Hospital of Wuzhou. The written informed consent was obtained from each patient for use of their tissue samples in research. This study was approved by the ethics committee of the People’s Hospital of Wuzhou.
Transfection, quantitative RT‐PCR and western blot assays
For transfection assay, GPR133‐restored expression plasmid, METTL3 overexpression plasmid and vector plasmid were obtained from GeneCopoeia Biotechnology (Rockville, MD, USA). Lipofectamine 3000 Reagent was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Cells were seeded into a six‐well plate overnight. Per well in a six‐well plate, 3 μg plasmid was incubated with 5 μL P3000 in 250 μL Opti‐MEM. A total of 5 μL Lipofectamine 3000 was mixed with 250 μL Opti‐MEM. Then these two mixtures were mixed together and incubated for 20 min at room temperature. Finally, the mixtures were added into one plate with 1.5 mL complete culture. Forty‐eight hours later, the cells were harvested for other assays.
For quantitative RT‐PCR (qRT‐PCR) assay, cells were treated with TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and total RNAs were extracted and reversely transcribed into cDNA. qRT‐PCR assays were performed to measure the expression levels of target genes. All primers used are listed in Table 1.
Table 1.
Primers in this study. F, forward; R, reverse.
| Primer name | Sequence (5′–3′) |
|---|---|
| GPR133‐F | AAAGTCCCGGAGTGATACTGA |
| GPR133‐R | TTGGTGAGATTCAAGGCTGTC |
| GAPDH‐F | ACAACTTTGGTATCGTGGAAGG |
| GAPDH‐R | GCCATCACGCCACAGTTTC |
| Cyclin D1‐F | GCTGCGAAGTGGAAACCATC |
| Cyclin D1‐R | CCTCCTTCTGCACACATTTGAA |
| Cyclin B1‐F | AATAAGGCGAAGATCAACATGGC |
| Cyclin B1‐R | TTTGTTACCAATGTCCCCAAGAG |
| CDK4‐F | ATGGCTACCTCTCGATATGAGC |
| CDK4‐R | CATTGGGGACTCTCACACTCT |
| p21‐F | TGTCCGTCAGAACCCATGC |
| p21‐R | AAAGTCGAAGTTCCATCGCTC |
| Cdc2‐F | GGATGTGCTTATGCAGGATTCC |
| Cdc2‐R | CATGTACTGACCAGGAGGGATAG |
| IGF2BP3‐F | ACGAAATATCCCGCCTCATTTAC |
| IGF2BP3‐R | GCAGTTTCCGAGTCAGTGTTCA |
For western blot assay, total protein was obtained from cells with lysis buffer and protease inhibitor. Bicinchoninic acid methods (Thermo Fisher Scientific) were used to determine protein concentrations. Proteins were loaded in gel channels to begin SDS/PAGE; transferred into polyvinylidene fluoride or polyvinylidene difluoride membranes; and incubated with 5% BSA, primary antibodies and horseradish peroxidase as a secondary antibody, respectively. All primary antibodies were listed in the manner of name, catalog name, dilution rate: glyceraldehyde‐3 phosphate dehydrogenase (GAPDH; 5174, 1 : 1000; CST), GPR133 (DF4947, 1 : 500; Affinity), p21 (2947, 1 : 500; CST), cyclin B1 (12231, 1 : 500; CST), cyclin D1 (55506, 1 : 500; CST), CDK4 (12790, 1 : 600; CST), Cdc2 (9116, 1 : 500; CST) and METTL3 (86132, 1 : 600; CST). Finally, an ECL chemiluminescence kit (Millipore, Burlington, MA, USA) was used to evaluate protein levels in the samples.
m6A RNA immunoprecipitation‐qPCR assay
The m6A sites of GPR133 were validated using the EZ‐Magna RNA immunoprecipitation kit (Millipore) according to the users’ instructions. Cells were seeded into a 10‐cm cell culture dish for overnight. In brief, cells were collected and lysed by RNA immunoprecipitation lysis buffer for 20 min, then centrifuged at 13 000 rpm for 10 min. The supernatant was incubated with m6A antibody and normal rabbit immunoglobulin G overnight, respectively. RNAs were extracted from magnetic beads, and we analyzed the level of GPR133 in the earlier groups by qRT‐PCR assay.
Immunohistochemistry
Tissues were deparaffinized in xylene, rehydrated with graded alcohol and boiled in 0.01 m citrate buffer (pH 6.0). Subsequently, the tissues were treated with 0.3% hydrogen peroxide, followed by normal goat serum.
All sections were incubated with the primary antibodies (GPR133, 1 : 100) overnight and followed by the biotinylated second antibody, streptavidin alkaline phosphatase, each for 10 min. Then the sections were then counterstained with hematoxylin, dehydrated and mounted. All immunohistochemical staining was evaluated and scored by at least two independent pathologists. The scores were set into four groups: negative (0–3), weak positive (3–6), middle positive (6–9) and strong positive (9–12).
Cell viability assay
Cells were seeded in 96‐well plates at the concentration of 1500 cells per well. MTS was added into wells at 0, 1, 2, 3, 4 and 5 days, respectively, for 3 h. The absolute absorbance value at 490 nm (A 490 nm) was measured using the Varioskan LUX system (Thermo Fisher Scientific).
Colony formation assay and soft agar colony formation assay
For colony formation experiments, the cells were seeded into six‐well plates at a density of 750 cells per well and cultured. After 14 days, cells were fixed with 4% formaldehyde for 15 min and stained with 0.1% crystal violet for 10 min. Finally, the colonies were photographed, and any colonies larger than 1 mm (>50 cells per clone) were counted.
For soft agar colony formation assay, 1500 cells were fully mixed with complete RPMI 1640 medium and 0.75% agarose, then quickly placed on the complete RPMI 1640 medium curing layer with 1.5% agarose. Next, 0.5 mL of RPMI 1640 was added every 5 days to each well to feed cells. After 3 weeks, colonies larger than 50 μm were photographed.
RNA stability assay
A549 cells with decreasing METTL3, H1299 cells with decreasing METTL3 concentrations and control group cells were seeded into six‐well plates, respectively. All cells were treated with 5 μg·mL−1 actinomycin D, and total RNA samples were obtained at 0, 1, 2 and 3 h. The mRNA expression level of GPR133 was estimated by qRT‐PCR assay.
Flow cytometry
A cell‐cycle staining kit was obtained from MultiSciences Biotech (Hangzhou, China), and cell cycles were analyzed by flow cytometry assay using this kit according to the manufacturer’s instructions. Cells were seeded in a six‐well plate, and starvation treatment was deployed overnight. Then cells were harvested, washed with cold phosphate‐buffered saline solution and incubated with DNA staining solution and permeabilization solution for 30 min at room temperature. Finally, cell samples were determined by using fluorescence‐activated cell sorting (Becton, Dickinson & Co., Franklin Lakes, NJ, USA).
Animal experiments
All animal experiments were approved by the ethics committee of the People’s Hospital of Wuzhou. Four‐week‐old immunodeficient mice were purchased from the Guangdong Animal Center (Guangzhou, China). Animals were randomly divided into groups (n = 4). Each group had two male and two female patients. A total of 5 × 105 cells were subcutaneously injected into the nude mice (n = 4 per group). Tumor growth was analyzed by measuring the tumor length (L) and width (W) and calculating the volume (V) using the formula: V = LW 2/2. The tumor tissues were then embedded in paraffin and analyzed.
Statistical analysis
For all statistical tests, a two‐tailed P value <0.05 was considered to be statistically significant. Student’s t‐test and chi‐square test were performed to compare a single gene’s expression levels between two groups. Overall survival (OS) curves were estimated by Kaplan–Meier analysis.
Results
GPR133 was down‐regulated and associated with better prognosis in LUAD
To explore the tumorigenesis and development of LUAD, we searched for down‐regulated genes in LUAD among TCGA and GEO datasets and located a total of 319 genes down‐regulated in LUAD among TCGA, GSE43767 and GSE43458 datasets (Fig. 1A). Notably, the expression level of GPR133 was much lower in 497 samples of LUAD tissue than that in 54 samples of normal tissue. GPR133 expression was also decreased in 80 samples of LUAD tissue as compared with that in 30 samples of normal tissue in GSE43458; similar results were obtained in GSE43767 (Fig. 1B). Consistently, we found that GPR133 expression was more significantly suppressed in 28 samples of LUAD tissue than in paired adjacent tissues (Fig. 1C). Further, we estimated GPR133 expression in LUAD cells, where not only the mRNA but also the protein expression level of GPR133 was much lower in LUAD cells than in normal BEAS‐2B cells (Fig. 1D). Next, we selected A549 and H1299 cells given their low expression of GPR133. To investigate the clinical implications of GPR133 in LUAD, we conducted Kaplan–Meier analysis and found that patients with high GPR133 expression levels had a better OS and disease‐free survival (Fig. 1E). In GSE31210, we performed the chi‐squared test and found that the expression of GPR133 was related to the tumor stage of LUAD, but not with age, sex or smoking history (Table 2). Collectively, these results suggested that GPR133 expression is decreased in LUAD and may be a potential biomarker of a better prognosis in patients with LUAD.
Fig. 1.
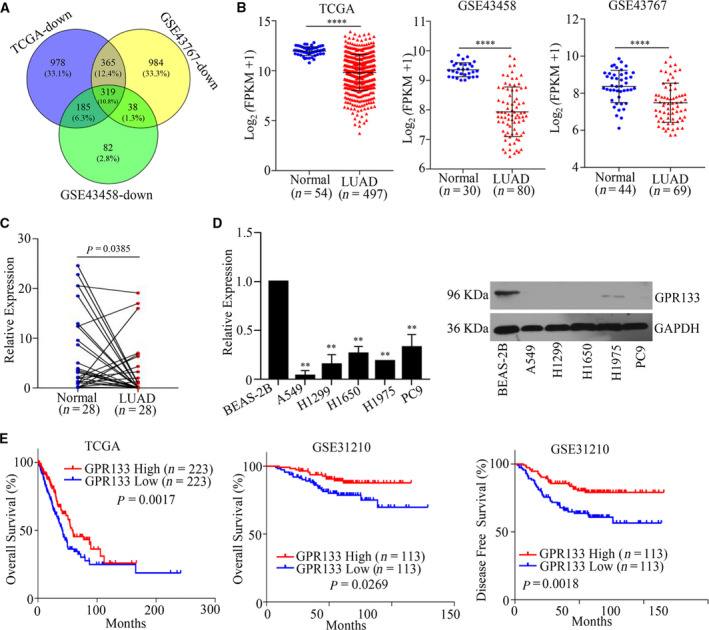
The expression pattern of GPR133 in LUAD. (A) Gene expression information was downloaded from TCGA and GEO databases, and a Venn diagram was used to identify differentially expressed genes. (B) The expression pattern of GPR133 in LUAD was displayed in TCGA and GEO databases vs. normal, ****P < 0.0001. (C) Total RNAs were obtained from 28 samples of LUAD tissue and matched adjacent normal tissues, and qRT‐PCR assays were implemented to detect GPR133 expression in these tissues. Data were reported as mean ± standard deviation (SD) for three independent experiments, and statistical analysis was performed via Student’s t‐test. (D) The mRNA and protein expression levels of GPR133 in LUAD cells were measured by qRT‐PCR and western blot assays, respectively. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. BEAS‐2B, **P < 0.01. (E) Kaplan–Meier analysis was conducted according to GPR133 expression in patients with LUAD from TCGA database and GSE31210 dataset.
Table 2.
The correlation between GPR133 expression and the clinical parameters of LUAD in GSE31210.
| Cases (n) | High | Low | P value | |||
|---|---|---|---|---|---|---|
| Case (n) | Rate (%) | Case (n) | Rate (%) | |||
| Age | ||||||
| ≥60 | 140 | 49 | 35.00 | 91 | 65.00 | 0.297 |
| <60 | 106 | 44 | 41.51 | 62 | 58.49 | |
| Sex | ||||||
| Male | 116 | 39 | 33.62 | 77 | 66.38 | 0.201 |
| Female | 130 | 54 | 41.54 | 76 | 58.46 | |
| Smoking | ||||||
| Yes | 123 | 43 | 34.96 | 80 | 65.04 | 0.357 |
| No | 123 | 50 | 40.65 | 73 | 59.35 | |
| Tumor stage | ||||||
| I | 169 | 68 | 40.24 | 101 | 59.76 | 0.000 |
| II | 58 | 6 | 10.34 | 52 | 89.66 | |
Restored expression of GPR133 inhibited LUAD proliferation
To discover the effects of GPR133 on tumor progression, we first enhanced GPR133 expression in A549 and H1299 cells (Fig. 2A). Interestingly, the cell viability of both A549 and H1299 cells was sharply decreased by accelerating GPR133 in these cells (Fig. 2B). Consistent with this observation, colony formation and soft agar colony formation assays also demonstrated that LUAD cells with restored GPR133 expression formed significantly smaller and fewer colonies than the control group (Fig. 2C,D). These data indicate that GPR133 inhibited the proliferation ability of LUAD cells.
Fig. 2.
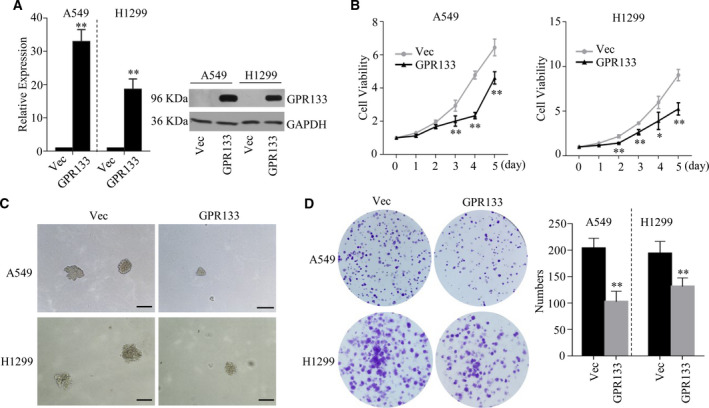
GPR133 suppressed cell proliferation in LUAD. GPR133 overexpression plasmid was transfected into A549 and H1299 cells, respectively. (A) qRT‐PCR assays and western blot assays were performed to verify GPR133 in the mentioned cells. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. Vec, **P < 0.01. (B) The above cells were seeded into 96 wells, and MTS was added into each well at 0, 1, 2, 3, 4 and 5 days. The cell viability of each well was measured at A 490 nm. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. Vec, *P < 0.05, **P < 0.01. Soft agar colony formation assay (C) and colony formation assay (D) were performed using the above cells. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. Vec, **P < 0.01. Scale bars: 100 μm. Vec, Vector.
GPR133‐triggered cell‐cycle arrest in LUAD cells
GSEA analysis was conducted to discover the possible mechanism through which GPR133 is involved in the proliferation of LUAD. The results revealed that GPR133 expression was correlated with the cell cycle in LUAD (Fig. 3A). As shown in Fig. 3B, GO annotations suggested that GPR133 coexpressed genes that were mainly involved in the cell cycle. Thus, we estimated the mRNA and protein expression levels of several biomarkers for the cell cycle. It was noted that GPR133 expression reduced cyclin B1 and enhanced p21 but had no effect on cyclin D1, CDK4 or Cdc2 (Fig. 3C). In addition, a flow cytometry assay was performed to measure the effect of GPR133 expression on the cell cycle of LUAD cells and revealed that overexpressing GPR133 triggered G2/M‐phase arrest in LUAD cells (Fig. 3D). Collectively, these results suggest that GPR133 plays a role in G2/M‐phase arrest in LUAD cells.
Fig. 3.
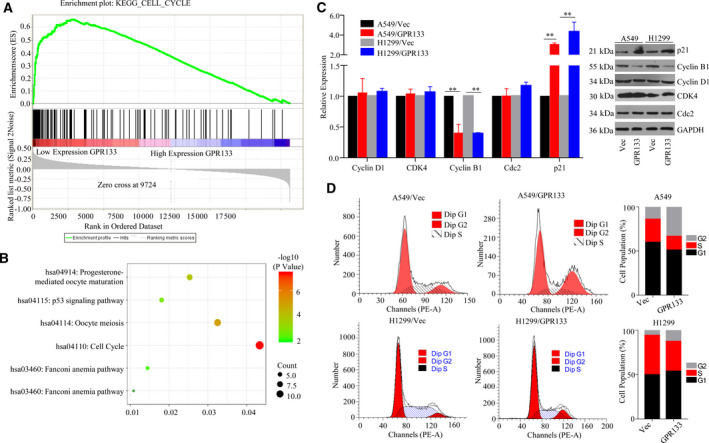
Effect of GPR133 on cell‐cycle distribution in LUAD cells. (A) GSEA assays were performed to explore the mechanism of GPR133 function in LUAD cells. (B) GPR133‐coexpressed genes were analyzed by GO and KEGG using the clusterprofiler software package on the r platform. (C) In LUAD cells with accelerating GPR133 and in control groups, the expression levels of cell‐cycle biomarkers were evaluated by qRT‐PCR and western blot assays. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. Vec, **P < 0.01. (D) Flow cytometry assays were conducted to detect the cell cycle in these cells.
GPR133 expression is enhanced by METTL3‐mediated m6A modification in LUAD
To discover the mechanism of GPR133 down‐regulation in LUAD, we analyzed the m6A sites on the GPR133 transcript using SRAMP and predicted that several m6A sites exist on the GPR133 transcript (Fig. 4A). We next performed m6A RNA immunoprecipitation‐qPCR assays to test this hypothesis, and the results revealed a substantial increase in the m6A level in A549 and H1299 cells (Fig. 4B). Moreover, we examined the correlation between the expression levels of GPR133 and m6A writers in TCGA. As shown in Fig. 4C, METTL3 expression was impaired in 483 samples of LUAD tissue and 347 samples of normal tissue (P < 0.01). Although TCGA and Genotype‐Tissue Expression (GTEx) datasets of LUAD showed weak positive correlation of METTL3 and GPR133 (R = 0.092), further cell line results strengthened it. Notably, qRT‐PCR and western blot assays revealed that the expression levels of METTL3 in A549 and H1299 cells were much lower than those in BEAS‐2B cells (Fig. 4D). On the basis of METTL3 expression, we transfected METTL3 overexpression plasmid into LUAD cells to restore an appropriate level of METTL3 expression (Fig. 4E). Actually, accelerating METTL3 may enhance not only the mRNA but also the protein expression levels of GPR133 (Fig. 4F). To verify whether METTL3 promoted the stability of GPR133 mRNA, we used actinomycin D to observe the mRNA level of GPR133. Upon increasing the actinomycin D treatment time, the decay of GPR133 mRNA was reduced in A549 and H1299 cells with overexpressed METTL3 as compared with in the respective control groups (Fig. 4G). Given these results, we presumed that METTL3 modulates GPR133 in an m6A‐dependent manner.
Fig. 4.
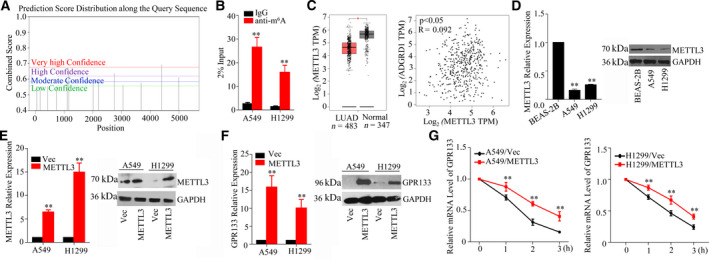
Identification of METTL3 targeting GPR133 in LUAD cells. (A) SRAMP presumed that the m6A was abundant in GPR133 transcripts. (B) The cells were isolated and RNA immunoprecipitation assay was performed. m6A antibody was used to trigger target RNAs, while immunoglobulin G was used as a negative control. The expression of GPR133 was determined in this experiment by qRT‐PCR assay. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. immunoglobulin G, **P < 0.01. (C) The expression pattern of METTL3 in LUAD was exhibited. The relationship between METTL3 and GPR133 was analyzed by Pearson’s correlation coefficient using the GEPIA databank vs. normal, *P < 0.05. (D) The expression levels of METTL3 in BEAS‐2B, A549 and H1299 cells were detected by qRT‐PCR and western blot assays. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test. BEAS‐2B, **P < 0.01. (E) METTL3 overexpression plasmid was transfected into LUAD cells, and the mRNA and protein expression profiles of METTL3 were assessed by qRT‐PCR and western blot assays. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. Vec, **P < 0.01. (F) In LUAD cells with increasing METTL3 expression, the mRNA and protein expression levels of GPR133 were evaluated by qRT‐PCR and western blot assays. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. Vec, **P < 0.01. (G) The decay rate of GPR133’s mRNA at the indicated times after actinomycin D (5 µg·mL−1) treatment in A549 and H1299 cells with restored METTL3 expression was measured. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. Vec, **P < 0.01.
GPR133 inhibited LUAD tumor growth in vivo
In brief, we estimated GPR133 expression patterns in LUAD by immunohistochemistry assay. The results revealed that GPR133 expression was down‐regulated in 63 samples of LUAD tissue and in 17 samples of normal tissue (Fig. 5A). Meanwhile, Kaplan–Meier analysis demonstrated that patients with LUAD with high GPR133 expression levels had a better outcome (Fig. 5A). To further investigate the effects of GPR133 on tumorigenicity in vivo, we subcutaneously injected A549 cells with increasing GPR133 expression into nude mice and assessed the subsequent tumor growth; ultimately, the results showed that overexpression of GPR133 could markedly suppress tumor growth in vivo (Fig. 5B). As compared with in the control group, restored GPR133 expression may significantly suppress both tumor weight and tumor growth (Fig. 5B). All these data support that GPR133 is a tumor inhibitor in LUAD.
Fig. 5.
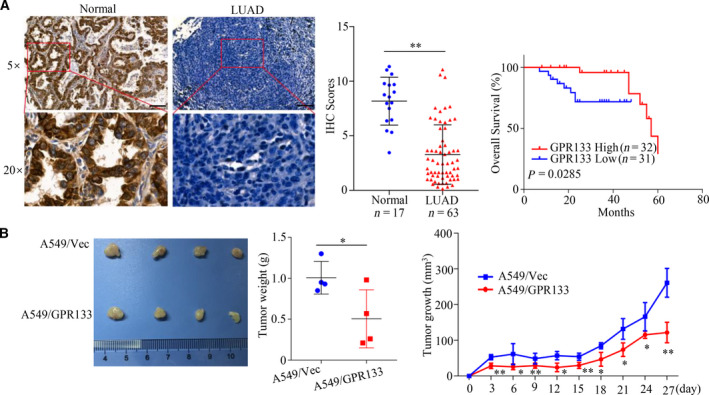
GPR133 inhibited tumor growth in vivo. (A) Immunohistochemistry assays were implemented to determine GPR133 expression in normal tissues and LUAD tissues. Data were reported as mean ± SD for three independent experiments, and statistical analysis was performed via Student’s t‐test vs. normal, **P < 0.01. The relationship between GPR133 expression and the prognosis of patients with LUAD was established by Kaplan–Meier analysis. Scale bars: 100 μm. (B) A total of 5 × 105 A549 cells with overexpressed GPR133 and the control group were subcutaneously injected into nude mice (n = 4 per group), respectively. The tumor size was detected every 2 days; tumor weights were also measured, and the tumor growth curve was drawn. Data were reported as mean ± SD, and statistical analysis was performed via Student’s t‐test vs. Vec, *P < 0.05, **P < 0.01.
Discussion
GPR133, a member of the GPCR superfamily, regulates cell adhesion and cell metabolism. In this study, we provided the first evidence that GPR133 expression is up‐regulated in LUAD by METTL3 in an m6A‐dependent manner. However, METTL3 was impaired in LUAD. Bioinformatics analysis combined with experiments suggested that GPR133 expression is positively related to a better prognosis among patients with LUAD. Functionally, GPR133 suppressed the proliferation of LUAD cells both in vivo and in vitro. Importantly, we showed that GPR133 triggered G2/M‐phase arrest in LUAD cells. Hence targeting GPR133 might represent a novel strategy to treat LUAD.
Recently, m6A has been discovered to be a reversible RNA methylation factor. This dynamic RNA methylation factor is enriched around stop codons, in 3′ untranslated regions and within internal long exons [11]. The act of m6A modification affects fundamental aspects of mRNA metabolism, resulting in posttranscriptional dysregulation of gene expression relating to cell differentiation, cell homeostasis, the cellular response to stress and cancer [12]. In this modification system, m6A ‘writers’ are composed of core catalytic components (METTL3/methyltransferase‐like 14) to install m6A modification [13]. Alkylation repair homolog protein 5 (ALKBH5) and fat mass and obesity‐associated protein, which are termed as m6A erasers, focus on removing m6A modification [14, 15]. The function of m6A is executed by m6A ‘readers’ that bind to m6A directly (YT521‐B homology domain‐containing proteins, Eukaryotic initiation factor 3 and Insulin‐like growth factor 2 mRNA‐binding proteins) or indirectly (HNRNPA2B1) [16, 17, 18].
METTL3 has been recognized as an essential factor in conditions including diabetes, cancers and cardiovascular disease [19, 20]. The METTL3‐mediated m6A modification on AFF4 could promote its expression. In addition, AFF4 is bound to the promoter of MYC. As such, the METTL3/AFF4/MYC axis contributes to bladder cancer tumorigenesis [21]. METTL3 targeted the 3′ untranslated region of HK2 mRNA. Moreover, METTL3 recruited YTHDF1 to enhance HK2 stability, thereby promoting the Warburg effect of cervical cancer [22]. Consistent with the aforementioned study, we found that METTL3 was expressed at low levels in LUAD. METTL3 was positively correlated with GPR133 via the GEPIA databank. Moreover, the overexpression of METTL3 may significantly enhance the mRNA stability of GPR133.
To discover the function of GPR133 expression in LUAD, we performed cell viability and colony formation assays, where the results showed that increasing the GPR133 expression sharply suppressed the proliferation of LUAD cells. Further investigation determined that GPR133 inhibited tumor growth in animal experiments of LUAD. However, it was reported that GPR133 was selectively expressed in hypoxic regions of GBM, while GPR133 knockdown abrogated tumor initiation [10]. Several genes, including GPR133, were found to be up‐regulated in gastrointestinal stromal tumors, but their functions remain unknown [23].
To discover the potential regulatory mechanism of GPR133 in LUAD, we performed GO and KEGG analyses based on GPR133‐related genes. The results indicated that the cell cycle was the most likely possible regulatory mechanism of GPR133 in LUAD. The cell cycle is a key event of cells, and targeting the cell cycle may be an important approach in cancer therapy [24]. It is well known that cell‐cycle machinery is controlled by cyclin‐dependent kinase (CDK), cyclins, and CDK‐inhibitory proteins [25]. Hence we quantified the mRNA and protein expression levels of cell‐cycle biomarkers in LUAD cells with increasing GPR133 expression. Our results indicated that GPR133 inhibited cyclin B1 and enhanced p21 expression in LUAD cells. Notably, cyclin B1 and p21 are famous G2/M‐phase biomarkers. Next, we performed a flow cytometry assay to discern that GPR133 significantly induced G2/M‐phase arrest in LUAD cells. Consistently, PP9 (a natural steroidal saponin) was reported to effectively induce G2/M‐phase arrest by up‐regulating p21 and suppressing cdc25C, cyclin B1 and cdc2 [26]. Evidently, avasimibe dose‐dependently inhibited the proliferation of U251 and U87 human glioblastoma cells. Further research revealed that avasimibe suppressed the expression of CDK2, cyclin E1, CDK4, cyclin D, CDK1, cyclin B1, Aurora A and PLK1, while inducing the expression of p53, p21, p27 and GADD45A [27].
Our encouraging data presented herein lay the foundation for further research of GPR133 in LUAD as a therapeutic target. Indeed, we are currently conducting experiments geared toward further target validation, as well as toward developing GPR133 inhibitors via small biomolecules.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
GW and DZ performed most of the experiments and analyzed results. JX and ZL did the bioinformatics analysis. XL and SZ analyzed the data. ZZ designed the research. GW wrote the paper. ZZ and XL revised the paper.
Acknowledgements
This study was supported by grants from the Scientific Research and Technology Development Project of Wuzhou (Grant No. 201501033).
Guixiong Wu and Dongfeng Zhai contributed equally to this article
Contributor Information
Xin Liu, Email: 229098031@qq.com, Email: eyzhaoziwen@scut.edu.cn.
Ziwen Zhao, Email: eyzhaoziwen@scut.edu.cn.
Data accessibility
All data are included in the manuscript.
References
- 1. Senosain M‐F and Massion PP (2020) Intratumor heterogeneity in early lung adenocarcinoma. Front Oncol 10, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ et al. (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- 3. Kohsaka S, Hayashi T, Nagano M, Ueno T, Kojima S, Kawazu M, Shiraishi Y, Kishikawa S, Suehara Y, Takahashi F et al. (2020) Identification of novel CD74‐NRG2alpha fusion from comprehensive profiling of lung adenocarcinoma in Japanese never or light smokers. J Thorac Oncol 15, 948–961. [DOI] [PubMed] [Google Scholar]
- 4. Herbst RS, Baas P, Kim DW, Felip E, Perez‐Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ et al. (2016) Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): a randomised controlled trial. Lancet 387, 1540–1550. [DOI] [PubMed] [Google Scholar]
- 5. Hamann J, Aust G, Arac D, Engel FB, Formstone C, Fredriksson R, Hall RA, Harty BL, Kirchhoff C, Knapp B et al. (2015) International union of basic and clinical pharmacology. XCIV. Adhesion G protein‐coupled receptors. Pharmacol Rev 67, 338–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bayin NS, Frenster JD, Kane JR, Rubenstein J, Modrek AS, Baitalmal R, Dolgalev I, Rudzenski K, Scarabottolo L, Crespi D et al. (2016) GPR133 (ADGRD1), an adhesion G‐protein‐coupled receptor, is necessary for glioblastoma growth. Oncogenesis 5, e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marroni F, Pfeufer A, Aulchenko YS, Franklin CS, Isaacs A, Pichler I, Wild SH, Oostra BA, Wright AF, Campbell H et al. (2009) A genome‐wide association scan of RR and QT interval duration in 3 European genetically isolated populations: the EUROSPAN project. Circ Cardiovasc Genet 2, 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim JJ, Park YM, Baik KH, Choi HY, Yang GS, Koh I, Hwang JA, Lee J, Lee YS, Rhee H et al. (2012) Exome sequencing and subsequent association studies identify five amino acid‐altering variants influencing human height. Hum Genet 131, 471–478. [DOI] [PubMed] [Google Scholar]
- 9. Sabik OL, Calabrese GM, Taleghani E, Ackert‐Bicknell CL and Farber CR (2020) Identification of a core module for bone mineral density through the integration of a co‐expression network and GWAS data. Cell Rep 32, 108145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frenster JD, Inocencio JF, Xu Z, Dhaliwal J, Alghamdi A, Zagzag D, Bayin NS and Placantonakis DG (2017) GPR133 promotes glioblastoma growth in hypoxia. Neurosurgery 64, 177–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fu Y, Dominissini D, Rechavi G and He C (2014) Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet 15, 293–306. [DOI] [PubMed] [Google Scholar]
- 12. Tong J, Flavell RA and Li HB (2018) RNA m(6)A modification and its function in diseases. Front Med 12, 481–489. [DOI] [PubMed] [Google Scholar]
- 13. Chen XY, Zhang J and Zhu JS (2019) The role of m(6)A RNA methylation in human cancer. Mol Cancer 18, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo X, Li K, Jiang W, Hu Y, Xiao W, Huang Y, Feng Y, Pan Q and Wan R (2020) RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A‐YTHDF2‐dependent manner. Mol Cancer 19, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang L, Song C, Wang N, Li S, Liu Q, Sun Z, Wang K, Yu SC and Yang Q (2020) NADP modulates RNA m(6)A methylation and adipogenesis via enhancing FTO activity. Nat Chem Biol 16, 1394–1402. [DOI] [PubMed] [Google Scholar]
- 16. Zhen D, Wu Y, Zhang Y, Chen K, Song B, Xu H, Tang Y, Wei Z and Meng J (2020) m(6)A reader: epitranscriptome target prediction and functional characterization of N (6)‐methyladenosine (m(6)A) readers. Front Cell Dev Biol 8, 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao Y, Shi Y, Shen H and Xie W (2020) m(6)A‐binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol 13, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu Y, Yang X, Chen Z, Tian L, Jiang G, Chen F, Li J, An P, Lu L, Luo N et al. (2019) m(6)A‐induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer 18, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang J, Liu J, Zhao S and Tian F (2020) N(6)‐methyladenosine METTL3 modulates the proliferation and apoptosis of lens epithelial cells in diabetic cataract. Mol Ther Nucleic Acids 20, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lv J, Zhang Y, Gao S, Zhang C, Chen Y, Li W, Yang YG, Zhou Q and Liu F (2018) Endothelial‐specific m(6)A modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res 28, 249–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, Liang Y, Zhu F, Zhang Y, Zhang X et al. (2019) The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF‐kappaB/MYC signaling network. Oncogene 38, 3667–3680. [DOI] [PubMed] [Google Scholar]
- 22. Wang Q, Guo X, Li L, Gao Z, Su X, Ji M and Liu J (2020) N(6)‐methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell Death Dis 11, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gromova P, Ralea S, Lefort A, Libert F, Rubin BP, Erneux C and Vanderwinden JM (2009) Kit K641E oncogene up‐regulates Sprouty homolog 4 and trophoblast glycoprotein in interstitial cells of Cajal in a murine model of gastrointestinal stromal tumours. J Cell Mol Med 13, 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Urrego D, Tomczak AP, Zahed F, Stuhmer W and Pardo LA (2014) Potassium channels in cell cycle and cell proliferation. Philos Trans R Soc Lond B Biol Sci 369, 20130094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pandey V, Tripathi A, Rani A and Dubey PK (2020) Deoxyelephantopin, a novel naturally occurring phytochemical impairs growth, induces G2/M arrest, ROS‐mediated apoptosis and modulates lncRNA expression against uterine leiomyoma. Biomed Pharmacother 131, 110751. [DOI] [PubMed] [Google Scholar]
- 26. Yao M, Li R, Yang Z, Ding Y, Zhang W, Li W, Liu M, Zhao C, Wang Y, Tang H et al. (2020) PP9, a steroidal saponin, induces G2/M arrest and apoptosis in human colorectal cancer cells by inhibiting the PI3K/Akt/GSK3beta pathway. Chem Biol Interact 331, 109246. [DOI] [PubMed] [Google Scholar]
- 27. Liu JY, Fu WQ, Zheng XJ, Li W, Ren LW, Wang JH, Yang C and Du GH (2020) Avasimibe exerts anticancer effects on human glioblastoma cells via inducing cell apoptosis and cell cycle arrest. Acta Pharmacol Sin 42, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in the manuscript.