

Abstract
All living organisms adapt their membrane lipid composition in response to changes in their environment or diet. These conserved membrane‐adaptive processes have been studied extensively. However, key concepts of membrane biology linked to regulation of lipid composition including homeoviscous adaptation maintaining stable levels of membrane fluidity, and gel‐fluid phase separation resulting in domain formation, heavily rely upon in vitro studies with model membranes or lipid extracts. Using the bacterial model organisms Escherichia coli and Bacillus subtilis, we now show that inadequate in vivo membrane fluidity interferes with essential complex cellular processes including cytokinesis, envelope expansion, chromosome replication/segregation and maintenance of membrane potential. Furthermore, we demonstrate that very low membrane fluidity is indeed capable of triggering large‐scale lipid phase separation and protein segregation in intact, protein‐crowded membranes of living cells; a process that coincides with the minimal level of fluidity capable of supporting growth. Importantly, the in vivo lipid phase separation is not associated with a breakdown of the membrane diffusion barrier function, thus explaining why the phase separation process induced by low fluidity is biologically reversible.
Keywords: homeoviscous adaptation, lipid domains, lipid phase separation, membrane fluidity, protein partitioning
Subject Categories: Membranes & Trafficking; Microbiology, Virology & Host Pathogen Interaction
Essential cellular processes in bacteria, including cytokinesis, envelope expansion, chromosome replication/segregation and maintenance of membrane potential, are impaired by low membrane fluidity.

Introduction
Biological membranes are complex arrangements predominantly composed of lipids and both integral and surface‐attached proteins (Nicolson, 2014). The primordial function of biological membranes was likely to act as a simple, semipermeable diffusion barrier separating the cell from its environment, and genomes from each other (Chen & Valde, 2010). Later, membranes and membrane proteins evolved to fulfil a multitude of cellular functions including transport, respiration and morphogenesis. Since the physicochemical state of biological membranes is highly sensitive to changes in the environment including temperature, osmolarity, salinity, pH or diet (Razin, 1967; Hazel, 1995; Ernst et al, 2016), careful homeostatic regulation of key membrane parameters such as thickness or fluidity is vital for cell function (Parsons & Rock, 2013; Ernst et al, 2016; Harayama & Riezman, 2018; Levental et al, 2020).
Arguably, the best studied membrane‐adaptive process is homeoviscous adaptation that acts upon changes in temperature (Hazel, 1995; Parsons & Rock, 2013; Ernst et al, 2016). With increasing temperature, lipid bilayers exhibit reduced head group packing, increased fatty acid disorder and increased fluidity in terms of increased rotational and lateral diffusion of molecules (Chapman, 1975; Heimburg, 2007). While all of these membrane parameters are closely interconnected, membrane fluidity is thought to be the key property actively maintained at stable levels that optimally support vital membrane functions through homeoviscous adaptation processes (Hazel, 1995; Parsons & Rock, 2013; Ernst et al, 2016). All living organisms achieve this by actively adapting their lipid composition. Most commonly, this is obtained by altering the content of lipids carrying fluidity‐promoting unsaturated fatty acids (UFA) or branched chain fatty acids (BCFA) and fluidity‐reducing saturated fatty acids (SFA), respectively, thereby counteracting shifts in membrane fluidity (Hazel, 1995; Diomandé et al, 2015; Ernst et al, 2016).
While adaptive changes in lipid fatty acid composition as well as the regulatory processes involved are increasingly well characterised (Mansilla et al, 2004; Ernst et al, 2018; Ballweg et al, 2020), the cellular consequences of inadequate membrane fluidity are significantly less understood. Sufficiently high membrane fluidity has been implicated in promoting folding, catalytic activity and diffusion of membrane proteins (Lee, 2004; Andersen & Koeppe, 2007). Too high membrane fluidity, in turn, has been shown to increase proton permeability in vitro (Rossignol et al, 1982; van de Vossenberg et al, 1999), thus potentially hampering with efficient ion homeostasis and energy conservation (Valentine, 2007), while too low membrane fluidity impedes respiration due to reduced ubiquinone diffusivity (Budin et al, 2018). However, our understanding of the behaviour of biological membranes upon changing fluidity is predominantly based on in vitro and in silico studies with simplified model lipids, or in vitro studies with either natural lipid extracts or isolated membranes (Baumgart et al, 2007; Schäfer et al, 2011; Nickels et al, 2017).
One of the fascinating properties of lipids is their ability to undergo phase transitions between distinct configurations that differ in terms of ability to form bilayers, membrane thickness and degree of lipid packing (Chapman, 1975). The biologically relevant bilayer‐forming lipid phases are: (i) the liquid‐disordered phase characterised by low packing density and high diffusion rates that forms the regular state of biological membranes, (ii) the cholesterol/hopanoid‐dependent liquid‐ordered phase that forms nanodomains (lipid rafts) found in biological membranes, both representing different fluid phases; and (iii) the gel or solid phase characterised by dense lipid packing with little lateral or rotational diffusion, which is generally assumed to be absent in biologically active membranes (Veatch, 2007; Sáenz et al, 2015; Schmid, 2017). In fact, the temperature associated with gel phase formation has been postulated to define the lower end of the temperature range able to support vital cell functions (Drobnis et al, 1993; Ghetler et al, 2005; Burns et al, 2017), and maintaining biological membranes in the correct phase (homeophasic regulation) has been suggested as an alternative rationale behind temperature‐dependent lipid adaptation (Hazel, 1995). Finally, lipid phases can co‐exist, resulting in separated membrane areas exhibiting distinctly different composition and characteristics (Baumgart et al, 2007; Elson et al, 2010; Heberle & Feigenson, 2011; Nickels et al, 2017; Shen et al, 2017). This principal mechanism of lipid domain formation is best studied in the context of lipid rafts (Lingwood & Simons, 2010; Shaw et al, 2021). Here, the co‐existence of fluid liquid‐disordered and liquid‐ordered phases, and the associated protein segregation, has been demonstrated in membranes of living eukaryotic cells (Toulmay & Prinz, 2013; preprint: Shelby et al, 2021). In contrast, comprehensive in vivo studies on gel‐fluid lipid phase separation in live cells have been challenging due to the tendency of cholesterol/hopanoids to suppress gel‐fluid phase transitions (Heberle & Feigenson, 2011), and due to the difficulty to modify the membrane fatty acid composition and, thus, fluidity without inducing lipotoxicity (Shen et al, 2017; Budin et al, 2018).
While in vitro and in silico approaches with simplified lipid models have provided detailed insights into the complex physicochemical behaviour of lipid bilayers, testing the formed hypotheses and models in the context of protein‐rich biological membranes is now crucial. Bacteria tolerate surprisingly drastic changes in their lipid composition and only possess one or two membrane layers as part of their cell envelope. Consequently, bacteria are both a suitable and a more tractable model to study the fundamental biological process linked to membrane fluidity and phase separation in vivo.
We analysed the biological importance of membrane homeoviscous adaptation in Escherichia coli (phylum Proteobacteria) and Bacillus subtilis (phylum Firmicutes), respectively. These organisms were chosen due to their prominence as Gram‐negative and Gram‐positive model organisms, and the different archetypes of membrane fatty acid composition (straight versus branched chain fatty acids) they represent. We have established protocols that allow the fatty acid composition of both organisms to be progressively altered and the cellular consequences to be directly monitored in growing cells. This approach allowed us to address three central questions linked to homeostatic regulation of membrane composition and fluidity: (i) what are the cellular consequences of an inadequate level of membrane fluidity that necessitate the extensive and conserved homeostatic regulatory processes, (ii) how do changes in lipid fatty acid composition translate to changes in membrane fluidity of living cells and (iii) what is the lipid phase behaviour in living cells with protein‐crowded membranes and intact lipid domain organisation?
Our results demonstrate that too low membrane fluidity results in growth arrest in both organisms, which is accompanied by severe disturbances of the cell morphogenesis and ion homeostasis. Furthermore, too low fluidity triggers a striking, large‐scale lipid phase separation into liquid‐disordered and gel phase membranes, accompanied by segregation of otherwise disperse membrane proteins such as ATP synthase and glucose permease. Our results revealed that phase separation between liquid‐disordered and gel state membranes is associated with loss of essential membrane functions, thereby limiting the range of membrane fluidity able to support life. At last, our findings demonstrating that gel‐liquid phase separation and associated membrane protein segregation indeed occurs in protein‐crowded, native plasma membranes of living cells, are fully consistent with the comparable phenomena observed in in vitro and in silico model systems (Baumgart et al, 2007; Veatch, 2007; Lingwood & Simons, 2010; Schäfer et al, 2011; Domański et al, 2012). Thus, the results provide strong in vivo support for the general validity of the respective models.
Results
Depletion of BCFAs in B. subtilis
To modify the fatty acid composition in B. subtilis, we constructed a Δbkd Δdes deletion strain (Appendix Table S1). The bkd operon encodes enzymes catalysing the conversion of branched chain amino acids into intermediates for BCFA synthesis (Debarbouille et al, 1999). The lack of this activity can be complemented by supplementation with precursors 2‐methylbutyric acid (MB) or isobutyric acid (IB) (Kaneda, 1977; Boudreaux et al, 1981). This provides the experimental means to control the lipid iso‐ and anteiso‐BCFA composition (Appendix Fig S1A) normally responsible for the homeostatic adaptation of membrane fluidity in response to environmental changes (Diomandé et al, 2015). In addition, the strain is deficient for the lipid desaturase Des to prevent rapid adaptation of membrane fluidity by converting SFAs or BCFAs into UFAs (Diomandé et al, 2015). In the remaining text, the B. subtilis strain is labelled “Δbkd” for simplicity.
We compared growth of B. subtilis 168 used as wild‐type (WT) and Δbkd cells at 37°C upon supplementation with BCFA precursors MB or IB (Fig 1A). While BCFA precursors had little impact on growth of WT cells, the auxotrophic Δbkd strain only grew in the presence of either of the precursors. Corresponding fatty acid analyses revealed large shifts in the composition of the Δbkd strain depending on the supplied precursor (Fig 1B and Appendix Fig S1B). As expected, cells supplemented with MB exhibited a high content (77%) of anteiso‐BCFAs, whereas cells grown with IB showed a high content of iso‐BCFAs (77%). To obtain cells depleted for both BCFA types, cells were grown in the presence of IB, followed by wash and incubation in precursor‐free (PF) medium. This precursor depletion leads to growth arrest after about 90 min (Appendix Fig S1B), corresponding to an accumulated SFA content of ~50% (Fig 1B).
Figure 1. Membrane fatty acid composition‐dependent growth of B. subtilis and E. coli .
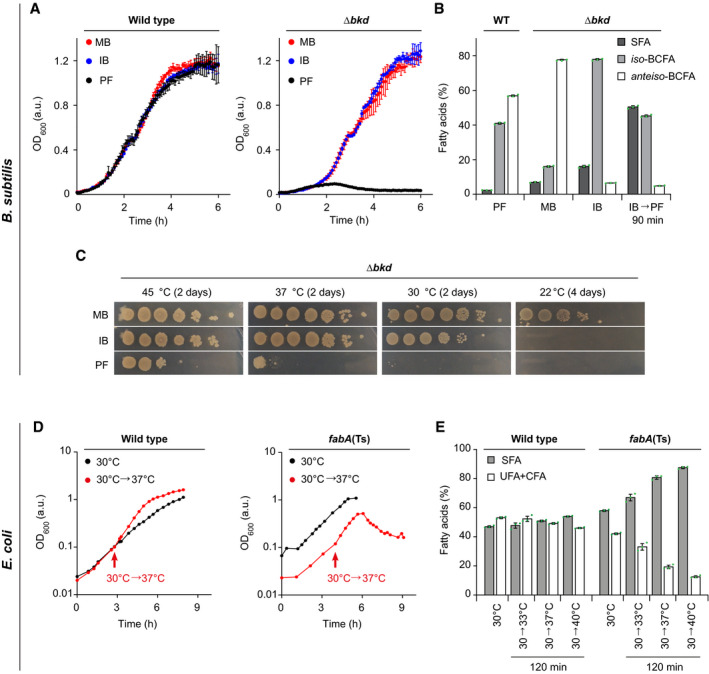
- Growth of B. subtilis WT and fatty acid precursor‐auxotrophic Δbkd cells in medium supplemented with precursor 2‐methyl butyric acid (MB), isobutyric acid (IB) or grown precursor‐free (PF).
- Fatty acid composition of B. subtilis WT cells grown in PF medium, and Δbkd grown with MB, IB or depleted for precursor for 90 min (IB→PF). For detailed analyses, see Appendix Fig S1B.
- Temperature‐dependent growth of B. subtilis Δbkd on solid medium in serial 10‐fold dilutions. For comparison between WT, Δdes and Δbkd Δdes cells, see Appendix Fig S1C.
- Temperature‐dependent growth behaviour of E. coli WT and fabA(Ts), including a shift from 30 to 37°C as non‐permissive temperature of fabA(Ts).
- Fatty acid composition of E. coli WT and fabA(Ts) cells grown at 30°C and shifted to different temperatures for 120 min. For detailed analyses, see Appendix Fig S2B and C.
Data information: (A) The diagram depicts mean and standard deviation (SD) of technical triplicates for each strain. (B, E) The histograms depict mean and SD of biological triplicates. (C, D) The experiments are representative of three independent repeats. CFA, cyclopropane fatty acid. Strains used: (A–C) B. subtilis 168, HS527; (D, E) E. coli MG1, MG4 (strains Y‐Mel and UC1098, respectively, additionally encoding fluorescent ATP synthase (FOF1 a‐mNG)).
Source data are available online for this figure.
Analyses of Δbkd cells grown at different growth temperatures with different BCFA precursors (Fig 1C and Appendix Fig S1C) indicated that only MB, the precursor for anteiso‐BCFAs, is capable for supporting robust growth at low temperatures (22°C). At 30°C and 37°C, growth was comparable in the presence of either MB or IB, while no growth was observed in the absence of precursors, demonstrating that a high BCFA level is essential for growth under these conditions. At 45°C, Δbkd could grow at low dilutions even in the absence of BCFA precursors (Fig 1C and Appendix Fig S1C). For these reasons, we chose growth with IB at 37°C, as the reference condition for B. subtilis Δbkd.
Depletion of UFAs in E. coli
In E. coli, membrane fluidity is modulated by UFAs (Parsons & Rock, 2013) (Appendix Fig S2A). While synthesis of UFAs is essential for E. coli growth, a temperature‐sensitive fabF fabA(Ts) mutant (Appendix Table S1), in which a shift to non‐permissive growth temperatures leads to UFA depletion, has been isolated (Cronan & Gelmann, 1973). DNA sequencing of fabF and fabA from this rather old isolate revealed that FabF (β‐ketoacyl‐ACP synthase II) is non‐functional due to S291N and G262S substitutions. FabA (β‐hydroxyacyl‐ACP‐dehydratase/isomerase) carries a single G101D substitution (Rock et al, 1996). Based on the structure of the FabA head‐to‐tail homodimer (Nguyen et al, 2014), G101 is positioned at the border of the dimerisation interface. Consequently, the G101D substitution could plausibly cause thermosensitivity by destabilising the essential dimer structure of FabA at elevated temperatures, thereby provoking the gradual depletion of UFA (Cronan & Gelmann, 1973). Thus, this strain provides the experimental tool to control the UFA/SFA balance in growing cells. Throughout the text, the strain is labelled “fabA(Ts)” for simplicity.
While the growth of fabA(Ts) at 30°C is comparable to E. coli Y‐Mel used as WT, transfer to non‐permissive temperatures such as 37°C only supported growth for about 120 min, followed by growth arrest and onset of cell lysis (Fig 1D). Corresponding fatty acid analyses confirmed a strong, temperature‐dependent decrease in UFAs (Fig 1E and Appendix Fig S2). In agreement with Cronan and Gelmann (1973), a minimal amount of 10–15% UFAs appeared to be essential to support growth (Fig 1E and Appendix Fig S2B). In comparison, WT cells showed only minor, temperature‐dependent changes in fatty acid composition caused by homeoviscous adaptation towards increased SFA content at higher temperatures (Fig 1E and Appendix Fig S2C).
Reduced membrane fluidity in cells depleted of UFAs or BCFAs
To confirm that changes in fatty acid composition translate to shifts in in vivo membrane fluidity, we monitored changes in steady‐state fluorescence anisotropy of 1,6‐diphenyl‐1,3,5‐hexatriene (DPH), the rotational freedom of which is sensitive to acyl chain disorder and, thus, indirectly to the fluidity of lipid bilayers (Lentz, 1993). DPH anisotropy measurements with B. subtilis Δbkd revealed the highest membrane fluidity for cells with the highest anteiso‐BCFA content (Fig 2A). Cells with high iso‐BCFA content exhibited membrane fluidity levels slightly lower than those found for WT. These results confirm that anteiso‐BCFAs promote higher membrane fluidity than the corresponding iso‐forms in vivo; a difference previously based on in vitro evidence only (Lewis et al, 1987). The changes observed upon depletion of BCFAs, which is accompanied by accumulation of SFAs, were more drastic and resulted in a gradual reduction of membrane fluidity, ultimately leading to growth arrest (Fig 2A and Appendix Fig S1B).
Figure 2. Reduced membrane fluidity in cells depleted for UFAs or BCFAs.
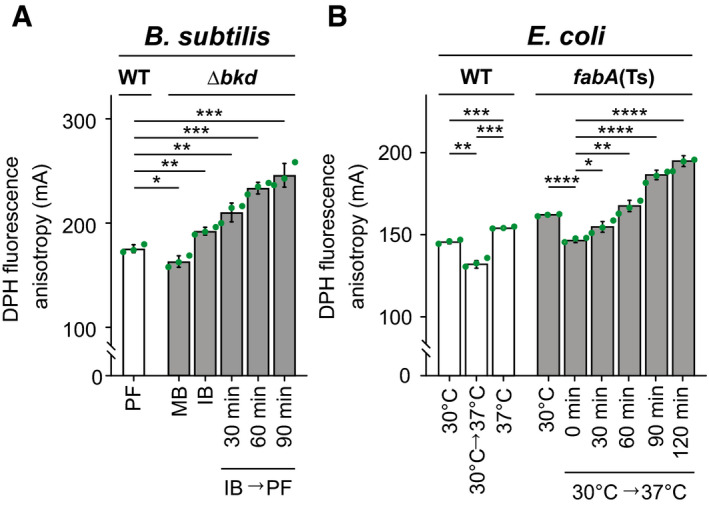
- DPH anisotropy of B. subtilis WT or Δbkd cells supplemented either with MB, IB or depleted for precursor (IB→PF) for the times indicated. High DPH anisotropy indicates low membrane fluidity.
- DPH anisotropy of E. coli WT cells grown steady state at 30, 37°C or shifted from 30 to 37°C followed by immediate measurement. In addition, DPH anisotropy of fabA(Ts) cells grown steady state at 30°C or shifted from 30 to 37°C followed by measurement at the times indicated.
Data information: (A, B) The experiments are representative of three independent repeats. The histograms depict means and SD of technical triplicates, together with P values of an unpaired, two‐sided t‐test. Significance was assumed with ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, n.s., not significant. Strains used: (A) B. subtilis 168, HS527; (B) E. coli Y‐Mel, UC1098.
Source data are available online for this figure.
DPH anisotropy measurements conducted with E. coli followed a similar trend (Fig 2B). Both E. coli WT and fabA(Ts) cells grown at 30°C exhibited an expected, immediate increase in membrane fluidity upon a shift to 37°C; a phenomenon that in WT cells is overtime counteracted by homeoviscous adaptation restoring membrane fluidity close to pre‐shift levels. However, in fabA(Ts) continued growth at 37°C resulted in a gradual increase in DPH anisotropy, thus confirming a substantial reduction of membrane fluidity.
In conclusion, the established fatty acid depletion procedures allow membrane fluidity to be controllably lowered to a point incapable of supporting growth in both organisms. In the following chapters, we use this approach to analyse which cellular processes are impaired by inadequate levels of membrane fluidity.
Consequences of low membrane fluidity on membrane diffusion barrier function
The prevalence of adaptive mechanisms maintaining membrane fluidity (Hazel, 1995) might indicate its importance for preserving the fundamental membrane barrier function. To analyse the consequences of too low membrane fluidity on membrane leakiness, we used the combination of two fluorescent dyes. Sytox Green is a membrane‐impermeable, DNA‐intercalating dye used to assess the integrity of bacterial plasma membranes in terms of permeability (Roth et al, 1997). DiSC3(5), a voltage‐sensitive dye accumulating in cells with high membrane potential (te Winkel et al, 2016), indicates changes in either membrane ion conductivity or respiration.
Growing B. subtilis Δbkd cells, irrespectively of the supplied BCFA precursor, exhibited DiSC3(5) fluorescence signals comparable to those observed for WT (Fig 3A and B, and Appendix Fig S3A–C). This indicates that the corresponding changes in the membrane fatty acid composition and fluidity had surprisingly little impact on membrane potential. In contrast, depletion of BCFAs triggered a gradual membrane depolarisation that was, in a mild form, already detectable after 30 min. A complete membrane depolarisation was observed after 90 min coinciding with growth arrest (Appendix Fig S1B). However, membranes remained impermeable for Sytox Green (Fig 3A), demonstrating that the gradual membrane depolarisation was not caused by a simple disruption of membrane continuity. In contrast, even the severely BCFA‐depleted membranes were fully capable of forming a continuous, tight diffusion barrier.
Figure 3. Consequences of low membrane fluidity on membrane diffusion barrier function.
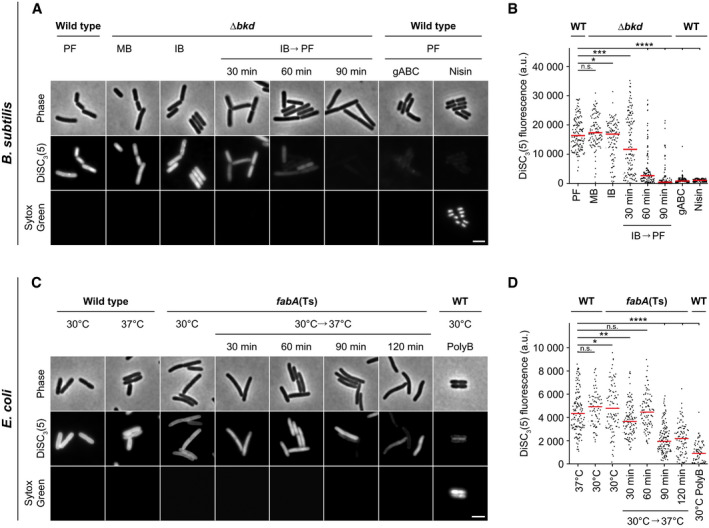
- Images of B. subtilis WT and Δbkd cells co‐labelled with the membrane potential‐sensitive dye DISC3(5) and the membrane permeability indicator Sytox Green. Membrane properties were assessed for Δbkd cells grown in the presence of MB, IB or washed precursor‐free (IB→PF) for the times indicated. As controls, WT cells were measured in the presence of depolarising antimicrobial peptide gramicidin ABC (gABC) or pore‐forming lantibiotic Nisin. For cross‐correlation between membrane depolarisation and membrane permeabilisation, see Appendix Fig S3A–C.
- Quantification of DISC3(5) fluorescence for cells (n = 100–142) depicted in panel A. Median represented by red line.
- Images of E. coli WT and fabA(Ts) cells co‐labelled with the same indicator dyes as in panel A. Membrane properties were assessed for fabA(Ts) at 30°C and upon transfer to non‐permissive 37°C for the times indicated. As controls, WT cells were incubated with the pore‐forming antibiotic Polymyxin B (PolyB). For cross‐correlation between membrane depolarisation and membrane permeabilisation, see Appendix Fig S3D and E. The integrity of the diffusion barrier function was additionally studied via ONPG permeability in a ΔlacY background (see Fig EV1).
- Quantification of DISC3(5) fluorescence for cells (n = 76–141) depicted in panel C. Median represented by red line.
Data information: (A–D) The experiments are representative of three independent repeats. (B, D) Red lines indicate the median. P values represent the results of unpaired, two‐sided t‐tests. Significance was assumed with ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, n.s., not significant. (A, C) Scale bar, 3 µm. Strains used: (A, B) B. subtilis 168, HS527; (C, D) E. coli Y‐Mel, UC1098.
Source data are available online for this figure.
High DiSC3(5) fluorescence signals and, thus, high membrane potential levels were also observed both for E. coli WT and fabA(Ts) grown at the permissive temperature of 30°C (Fig 3C and D, and Appendix Fig S3D and E), whereas depletion of fluidity‐promoting UFA in E. coli fabA(Ts) triggered a gradual loss of membrane potential. However, compared to B. subtilis the loss was delayed and incomplete (Fig 3D). Again, the lack of Sytox Green staining revealed that the membranes were not impaired in their general diffusion barrier function (Fig 3C). To confirm this important finding, membrane permeability was also monitored by following LacZ‐dependent hydrolysis of the chromogenic substrate ONPG (ortho‐nitrophenyl β‐D‐galactopyranoside) in cells deficient for the uptake system LacY. While we were able to detect low level, LacZ‐dependent ONPG hydrolysis in a lacY deletion background, no difference was observed between cells with native fatty acid composition and cells strongly depleted for UFAs undergoing lipid phase separation. Hence, the results provide an independent control for the lack of membrane permeabilisation (Fig EV1).
Figure EV1. Very low membrane fluidity in E. coli does not trigger membrane permeabilisation for ortho‐nitrophenyl β‐D‐galactopyranoside (ONPG).
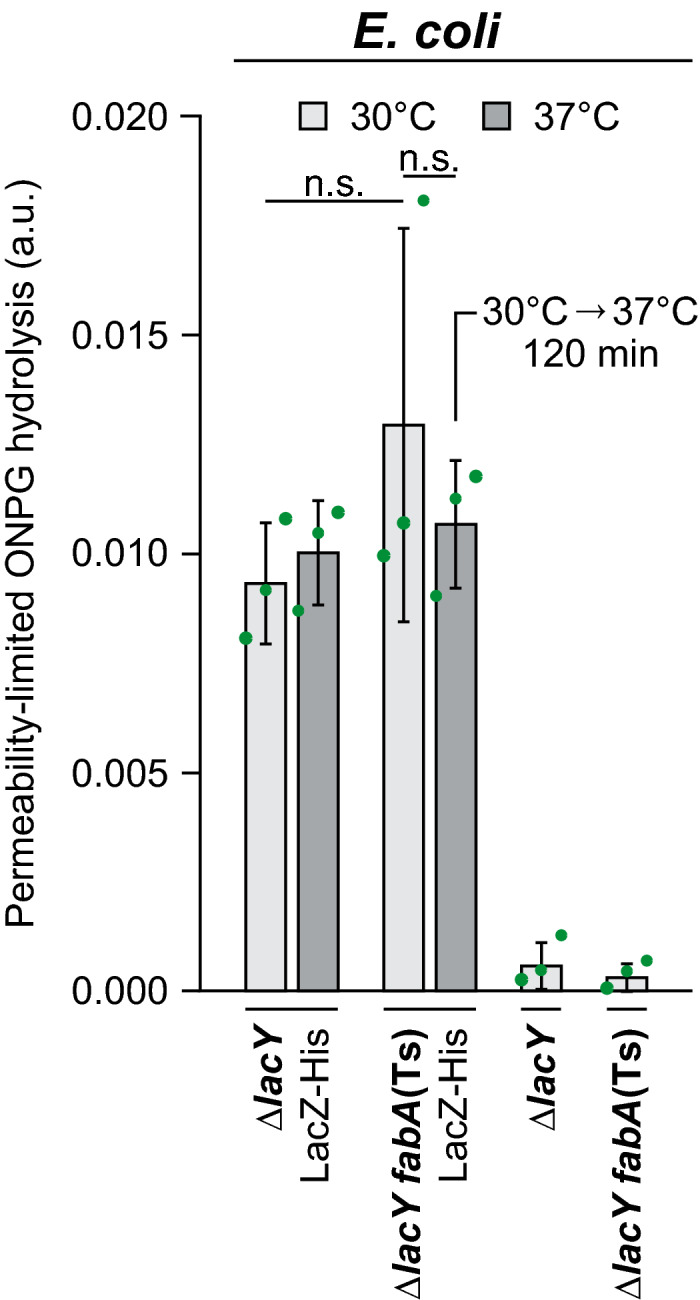
The membrane permeability of ONPG was assessed in strains deficient for the active uptake system LacY simultaneously expressing lacZ from a plasmid‐encoded leaky Ptac promoter (without addition of the inducer IPTG). The graph depicts ONPG hydrolysis rates measured for intact cells upon incubation at 30 and 37°C. In case of the temperature‐sensitive fabA(Ts) strain, growth at the non‐permissive temperature of 37°C was limited to 120 min. As controls, the ONPG conversion rates were measured in strains lacking the lacZ‐expressing plasmid. Note the lack of significant difference in ONPG conversion rates upon strong depletion of unsaturated fatty acids (fabA(Ts) strain at 37°C for 120 min), implying the lack of detectable membrane permeabilisation due to phase separation.
Data information: The graph depicts mean and SD of biological triplicates. The P values represent the results of unpaired, two‐sided t‐tests. Insignificant changes (P > 0.1) are indicated with n.s. Strains used: E. coli Y‐Mel.ΔlacY, UC1098.ΔlacY, Y‐Mel.ΔlacY/pTM30.lacZ‐His2, UC1098.ΔlacY/pTM30.lacZ‐His2.
Source data are available online for this figure.
In summary, while membrane depolarisation is observed due to too low membrane fluidity in both organisms, the core permeability function of the plasma membrane is not compromised even upon conditions unable to support growth. This is consistent with a more subtle effect of low fluidity on membrane‐associated biological processes maintaining ion homeostasis such as respiration.
Consequences of low membrane fluidity on cell morphogenesis
In rod‐shaped bacteria, cell growth and morphogenesis are predominantly driven by two membrane‐associated multiprotein complexes, the elongasome responsible for envelope expansion and rod shape determination (Typas et al, 2012), and the divisome responsible for cytokinesis (Adams & Errington, 2009). The main scaffold proteins for these prominent cellular machineries are the tubulin homolog FtsZ (Adams & Errington, 2009) and the actin homolog MreB (Typas et al, 2012). To assess the functionality of these key cellular machineries, we determined the localisation of corresponding GFP fusions upon depletion of fluidity‐promoting fatty acids. Furthermore, by use of GFP‐fused DNA‐binding protein Hu (B. subtilis) (Köhler & Marahiel, 1997) or DNA staining with intercalating dye DAPI (E. coli), we analysed the cells for potential defects in chromosome replication and segregation.
In B. subtilis, depletion of BCFAs had no effect on nucleoid prevalence or morphology indicating the presence of largely functional DNA replication, segregation and compaction mechanisms (Fig 4A and Appendix Fig S4A). While no DNA‐free cells indicative for defects in DNA replication and segregation were observed in E. coli either, a clear de‐condensation of the nucleoid was evident at later stages of UFA depletion (Fig 4B and Appendix Fig S4B). Intriguingly, this process coincides with partial dissociation of RNA degradosome from the membrane as indicated by an increasingly cytoplasmic localisation of the key scaffold and membrane anchor protein RNase E in cells exhibiting very low fluidity (Fig EV2, EV3, EV4, EV5). These two processes appear to be causally linked since expression of a cytoplasmic variant of RNase E, which lacks an amphipathic helix essential for membrane binding, leads to a comparable decondensation of the nucleoid (Fig EV3A and B).
Figure 4. Consequences of low membrane fluidity on and cell morphogenesis.
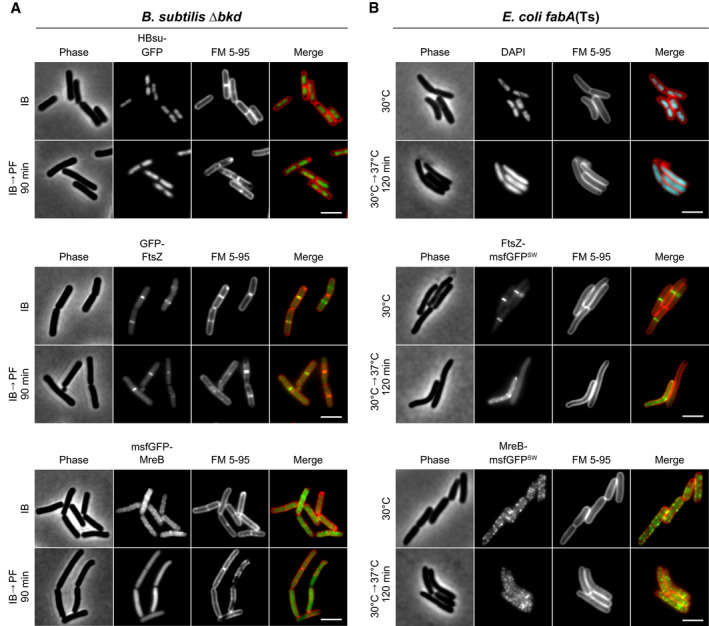
- Images of B. subtilis Δbkd cells stained with membrane dye FM 5‐95 and expressing GFP fusions of DNA‐binding protein HBsu (top), cell division protein FtsZ (middle) or cell elongation protein MreB (bottom). Cells were grown with IB or depleted for precursors for 90 min (IB→PF). For further examples and additional time points, see Appendix Fig S4A.
- Images of E. coli fabA(Ts) cells stained with FM 5–95 for the outer membrane and with DNA‐intercalating dye DAPI (top) or expressing GFP sandwich (SW) fusions to the cell division protein FtsZ (middle) and the cell elongation protein MreB (bottom), respectively. Depicted are cells grown at 30°C or with a temperature shift to 37°C for 120 min. For a more detailed view on the influence of low membrane fluidity on membrane dissociation of RNase E as well as on the cell division machinery, see Figs EV2, EV3, EV4, EV5 and Appendix Fig S5. For further examples and additional time points, see Appendix Fig S4B.
Data information: (A, B) The experiments are representative of biological triplicates. Scale bar, 3 µm. Strains used: (A) B. subtilis HS541, HS548, HS549; (B) E. coli UC1098, BHH500, BHH501.
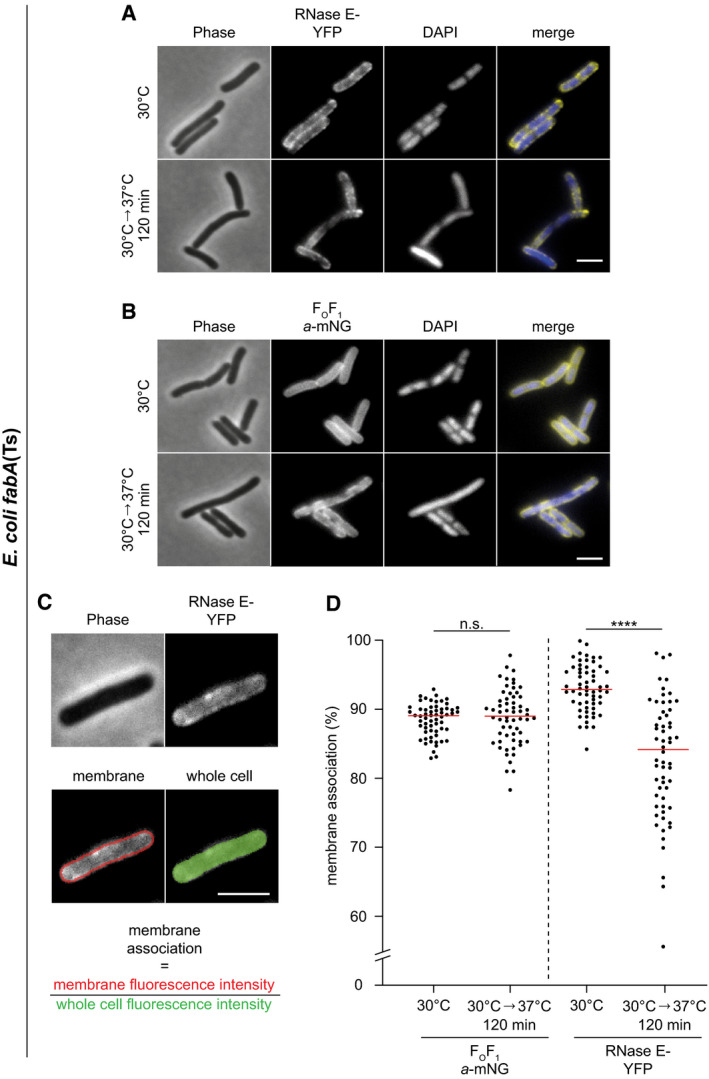
Figure EV2. Very low membrane fluidity triggers partial dissociation of RNase E from the membrane in E. coli fabA(Ts).
-
A, BPhase‐contrast and fluorescence images of E. coli fabA(Ts) strain expressing (A) RNase E‐YFP or (B) FOF1 a‐mNG. Cells were grown in LB at 30°C to an OD600 of 0.3, transferred to the non‐permissive temperature of 37°C for 120 min followed by labelling with DAPI and fluorescence microscopy. Note the increasing cytoplasmic localisation of RNase E‐YFP upon depletion of the membrane for UFA, which coincides with decondensation of the nucleoid (compare Fig 4B).
-
CQuantification of membrane association of RNase E. The degree of membrane association was quantified by automated detection of cells using phase‐contrast images, defining a 3‐pixel wide band around the periphery of the cell, and measuring the relative membrane association as a ratio between the mean peripheral fluorescence signal and the mean fluorescence of the whole cell.
-
DRelative membrane association of FOF1 a‐mNG and RNase E‐YFP at 30°C and 37°C for 120 min in individual cells (n = 60). Red lines indicate the median.
Data information: (A–D) Experiments are representative of independent biological duplicates. (D) Red lines indicate the median. P values represent the results of unpaired, two‐sided t‐tests. Significance was assumed with ****P < 0.0001, n.s., not significant (A–C). Scale bar: 3 µm. Strains used: (A, C, D) E. coli UC1098/pVK207; (B, D) E. coli MG4.
Source data are available online for this figure.
Figure EV3. Expression of cytoplasmic RNase E is sufficient to trigger decondensation of the nucleoid.
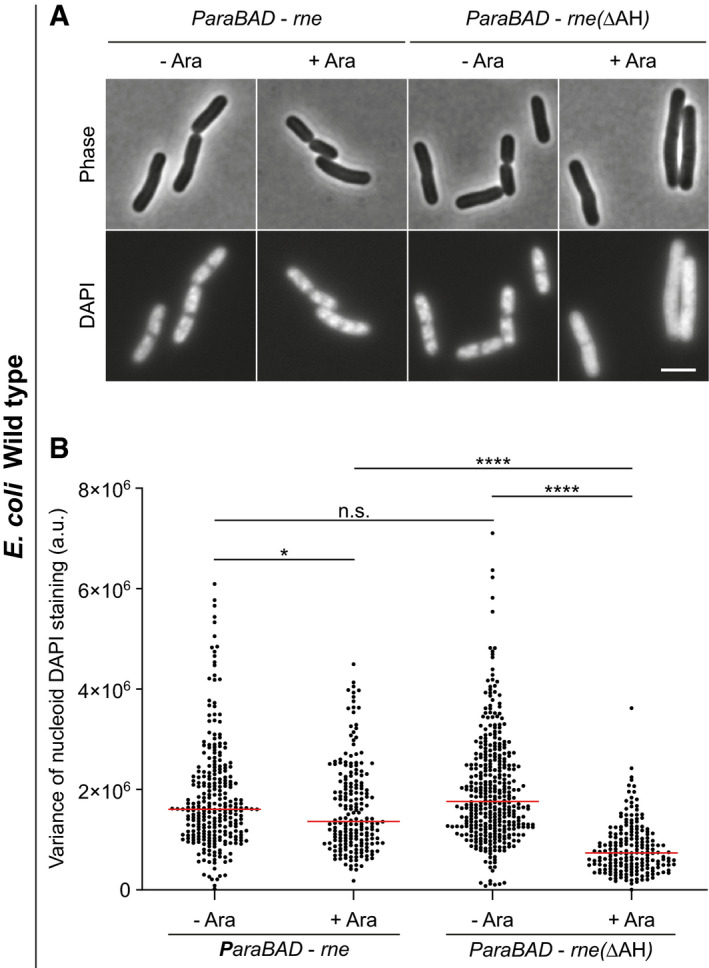
- Phase‐contrast and fluorescence images of E. coli WT cells expressing plasmid‐encoded full‐length membrane‐associated RNase E (Rne) and a corresponding construct encoding RNase E that lacks the membrane‐binding amphipathic helix (RneΔAH), respectively. The cells depicted were grown in LB medium at 37°C to an OD600 of 0.3 followed by induction of rne with 0.2% (w/v) arabinose (Ara) for 60 min, labelling with DAPI for 15 min and fluorescence microscopy. Note the decondensation of the nucleoid observed upon expression of cytoplasmically located RneΔAH, but not in the presence of the native membrane‐associated RNase E.
- Variance of nucleoid staining. The degree of nucleoid condensation was assessed by analysing the variance of DAPI fluorescence within the cell (n = 199–372). In this type of analysis, a more homogeneous fluorescence signal such as that caused by nucleoid decondensation results in a lower variance of the per pixel fluorescence intensity. Red lines indicate the median.
Data information: (A, B) The experiments are representative of biological duplicates. (B) Red lines indicate the median. P values represent results of unpaired, two‐sided t‐tests. Significance was assumed with ****P < 0.0001, *P < 0.05, n.s., not significant. (A) Scale bar: 3 µm. Strains used: (A, B) E. coli MG1655/pJG130, MG1655/pJG131.
Source data are available online for this figure.
Figure EV4. Destabilisation of E. coli divisome by deletion of the division regulator minC triggers hypersensitivity towards low membrane fluidity.
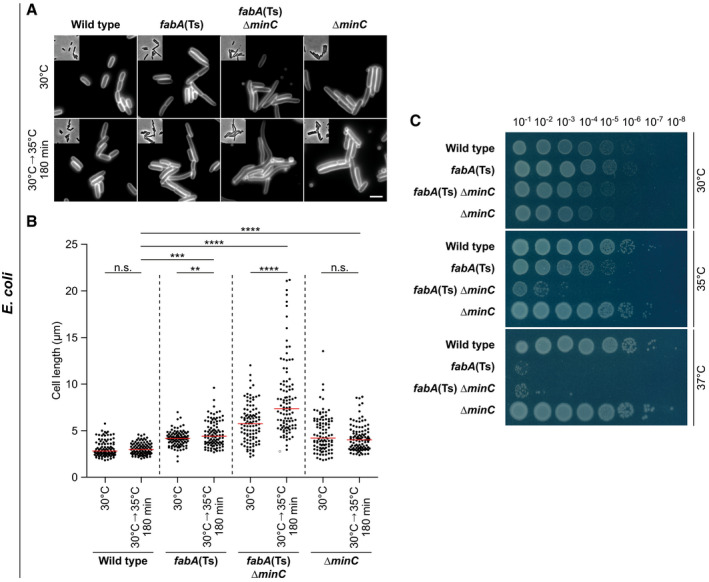
- Images of E. coli WT, fabA(Ts), fabA(Ts) ΔminC and ΔminC cells grown either at the permissive temperature (30°C) or for 180 min at 35°C, which is non‐permissive for the fabA(Ts) ΔminC strain. Cells were stained with the outer membrane dye FM 5–95 prior to microscopy. Note the strong cell elongation of the fabA(Ts) ΔminC strain upon incubation at 35°C, which is indicative of a severe cell division defect.
- Quantification of cell length for cells (n = 100) depicted in panel A. Red lines indicate the median.
- Viability of the strains depicted in panel A upon incubation on agar plates in M9‐glucose minimal medium overnight at different temperatures. The serial dilutions and spot assays were carried out with pre‐cultures grown at 30°C to mid‐log growth phase. Note the temperature hypersensitivity and loss of viability of strain fabA(Ts) ΔminC at 35°C, which indicates that the cell division process has become the limiting factor in tolerance towards low membrane fluidity in this strain. The temperature sensitivity of the strains fabA(Ts) ΔzapA and fabA(Ts) ΔzapB supported this view (see Appendix Fig S5).
Data information: (A–C) The experiments are representative of biological triplicates. (B) Red lines indicate the median, while the P values represent results of unpaired, two‐sided t‐tests. Significance was assumed with ****P < 0.0001, ***P < 0.001, **P < 0.01, n.s., not significant. (A) Scale bar: 3 µm. Strains used: (A–C) E. coli Y‐Mel, UC1098, UC1098.ΔminC, JW1165‐1.
Source data are available online for this figure.
Figure EV5. Protein segregation induced by very low membrane fluidity is limited to the inner cytoplasmic membrane in E. coli .
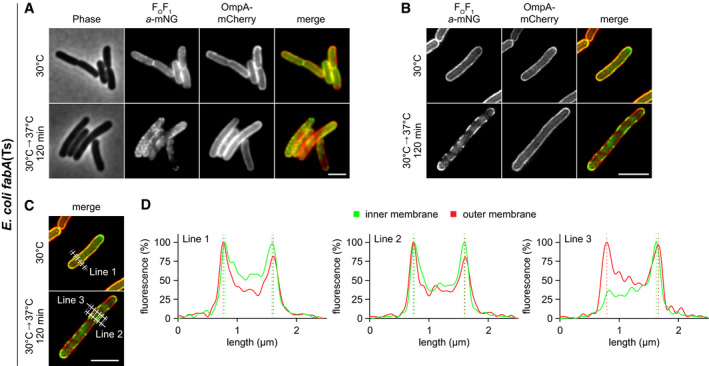
- Membrane protein segregation was monitored in fabA(Ts) cells expressing as inner membrane marker FOF1 a‐mNG and as outer membrane marker OmpA‐mCherry. Widefield microscopy images depict phase‐contrast, fluorescence and overlay images for cells grown at permissive 30°C or at non‐permissive 37°C for 120 min in M9‐glucose minimal medium. Note the transition from disperse localisation into a segregated pattern in case of the inner membrane‐localised FOF1 a‐mNG at 37°C, while the pattern of the outer membrane‐localised OmpA‐mCherry remains homogeneous at both growth temperatures.
- Super‐resolution 2D‐SIM (structured illumination microscopy) images of cells expressing FOF1 a‐mNG and OmpA‐mCherry at both growth temperatures.
- Localisation and orientation of 5‐pixel wide lines used to analyse fluorescence intensity profiles depicted in panel D.
- Fluorescence intensity line scans across the cells imaged with 2D‐SIM microscopy. Note the small, but detectable outward shift between the inner membrane marker FOF1 a‐mNG and outer membrane marker OmpA‐mCherry.
Data information: (A–D) Experiments are representative of biological triplicates (A–C). Scale bar: 3 µm. Strain used: (A–D) E. coli MG4/pGI10.
Source data are available online for this figure.
The cell division machinery, indicated by mid‐cell localisation of FtsZ, turned out to be robust towards changes in membrane fluidity in B. subtilis, with only a weakening of the fluorescent mid‐cell signal observed upon BCFA depletion (Fig 4A and Appendix Fig S4A). In contrast, a clear defect in divisome assembly was observed upon depletion of UFA in E. coli (Fig 4B and Appendix Fig S4B). To confirm that the E. coli cell division machinery indeed is stressed by low membrane fluidity, we combined the UFA depletion with a deletion of cell division regulator MinC (Hu et al, 1999). Indeed, in the absence of MinC, the E. coli cell division process became the growth‐limiting factor upon UFA depletion and even a slight shift in temperature from 30 to 35°C resulted in hypersensitivity towards low fluidity and loss of viability for strain fabA(Ts) ΔminC (Fig EV4, EV5). A clear increased sensitivity towards low membrane fluidity was also observed for strains deficient for the division proteins ZapA or ZapB (Appendix Fig S5) (Galli & Gerdes, 2010).
An inverse sensitivity was observed for the cell elongation machinery using localisation of MreB as proxy. In this case, depletion of BCFA triggered a complete disassembly of the MreB cytoskeleton in B. subtilis (Fig 4A and Appendix Fig S4A), whereas the E. coli counterpart was largely unaffected from UFA depletion in terms of membrane association and filament formation (Fig 4B and Appendix Fig S4B).
In conclusion, very low membrane fluidity incapable to support growth indeed affects membrane‐associated cellular machineries responsible for bacterial growth and division. It is important to note, however, that the changes in membrane fluidity required to disturb cell morphogenesis are rather extreme and go well beyond those observed upon normal changes in growth temperature.
Consequences of low membrane fluidity on membrane homogeneity
In the microscopic experiments described above (Fig 4A and B), cells were stained with FM 5‐95. This hydrophobic fluorescent dye allows visualisation of the plasma membrane of B. subtilis (Sharp & Pogliano, 1999), or the outer membrane in the case of E. coli (Pilizota & Shaevitz, 2012). While no changes in FM 5–95 staining were observed in E. coli cells, the smooth staining observed in Δbkd cells supplemented with IB transitioned into a distinctly irregular pattern upon BCFA depletion (Fig 4A and Appendix Fig S4A). This suggests that the more homogeneous membrane, present under normal growth conditions, segregates into areas with different local physicochemical properties upon low membrane fluidity. Intriguingly, the development of membrane irregularities coincides with growth arrest (Fig 5A and Movie EV1). It is worth emphasising, however, that the changes in lipid fatty acid composition needed to trigger this response are rather extreme compared to changes observed upon normal homeoviscous adaptation (Appendix Fig S6A and B).
Figure 5. Consequences of low membrane fluidity on membrane homogeneity.
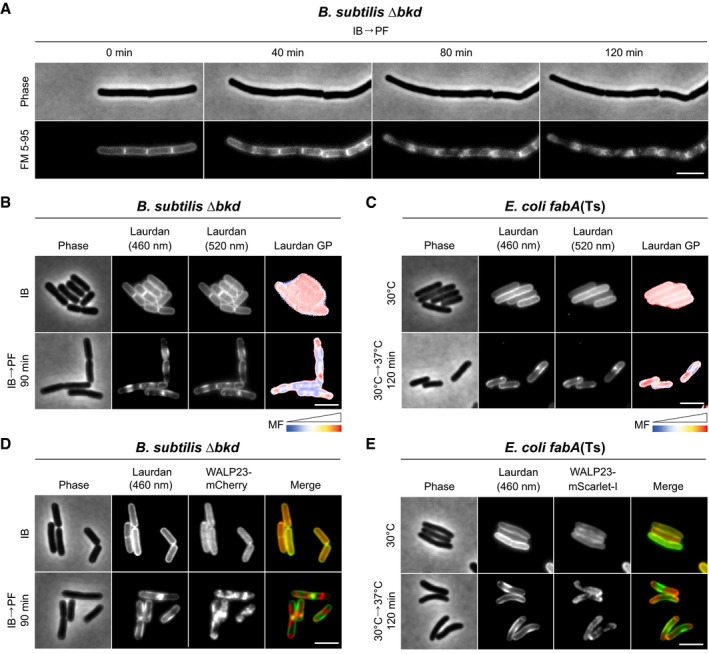
- Images of B. subtilis Δbkd cells grown with IB or without precursor (IB→PF). Cells were stained with membrane fluidity‐sensitive dye Laurdan and imaged at 460 nm, 520 nm and as the corresponding colour‐coded Laurdan GP map.
- Images of E. coli fabA(Ts) cells grown at 30°C or shifted to non‐permissive 37°C for 120 min. Cells were stained and imaged as described in panel B. For a more pronounced view on domain formation associated with local differences in membrane fluidity, see Appendix Fig S7, showing E. coli fabA(Ts) grown in LB instead of M9 minimal medium with glucose/casamino acids.
- Images of B. subtilis Δbkd cells grown and stained with Laurdan (imaged at 460 nm) as in panel B, but additionally expressing WALP23‐mCherry. For corresponding fluorescence intensity correlation between images, see Appendix Fig S8A. Co‐localisation of membrane dye FM 5‐95 and transmembrane peptide WALP23 is shown in Appendix Figs S9 and S8C.
- Images of E. coli fabA(Ts) cells grown and stained as in panel C, but additionally expressing WALP23‐mScarlet‐I. For corresponding fluorescence intensity correlation between images, see Appendix Fig S8B.
Data information: (A–E) The experiments are representative of biological triplicates. Scale bar: 3 µm. MF, membrane fluidity. Strains used: (A, B) B. subtilis HS527; (C) E. coli UC1098; (D) B. subtilis HS547; (E) E. coli UC1098/pBH501.
In vitro, lipid mixtures of low fluidity undergo phase transition into a more tightly packed gel state. We speculated that the lipid de‐mixing observed in B. subtilis (Figs 4A and 5A) could therefore represent large‐scale lipid phase separation between gel and liquid‐disordered phases. Therefore, we analysed the local membrane fluidity of both B. subtilis and E. coli with the fluidity‐sensitive membrane dye Laurdan. Laurdan exhibits shifts in its fluorescence emission spectrum that depends on lipid packing‐linked penetration of H2O to the membrane interior and, thus, the immediate environment surrounding the fluorophore. The interconnected nature of lipid packing and membrane fluidity allows local membrane fluidity to be estimated as Laurdan generalised polarisation (GP) (Parasassi et al, 1990; Wenzel et al, 2018). Indeed, when B. subtilis and E. coli cells were depleted for fluidity‐promoting fatty acids, domain formation associated with both differential staining (Laurdan fluorescence intensity) and differences in local fluidity (Laurdan GP) was observed (Fig 5B and C, and Appendix Fig S7). To verify these findings with an independent dye‐free method, we used cells expressing helical transmembrane peptide WALP23, which preferentially accumulate in liquid‐disordered membrane areas (Schäfer et al, 2011; Scheinpflug et al, 2017b). Indeed, co‐labelling of cells with Laurdan clearly demonstrated that the observed lipid phase separation results in segregation of WALP23 in membrane areas of low Laurdan fluorescence (Fig 5D and E, and Appendix Fig S8A and B). At last, co‐staining with FM 5–95 demonstrated that FM 5–95 and WALP23 share the same preference for higher fluidity areas in de‐mixed membranes (Appendix Figs S8C and S9).
In conclusion, by exhibiting de‐mixing into distinct areas of high and low membrane fluidity, respectively, the observed in vivo domain formation shares the core characteristic of lipid phase separation between fluid and gel state membranes (Baumgart et al, 2007; Domański et al, 2012; Mostofian et al, 2019).
Segregation of membrane proteins into fluid domains of phase‐separated plasma membranes
As indicated by WALP23 (Fig 5D and E), the observed lipid phase separation might have broader consequences on membrane protein localisation. To test this, we focused on E. coli for two reasons. Firstly, the membrane depolarisation caused by low membrane fluidity, which itself can affect membrane protein localisation (Strahl & Hamoen, 2010), is less extensive in E. coli (Fig 3). Secondly, E. coli does not exhibit delocalisation of MreB (Fig 4B and Appendix Fig S4B), which we have previously shown to induce membrane protein clustering (Strahl et al, 2014). As a model protein of choice, we focused on ATP synthase (FOF1), an abundant polytopic membrane protein complex (Junge & Nelson, 2015).
To visualise the localisation of FOF1, fluorescent protein was C‐terminally fused to membrane‐integral FO‐a yielding a stable and active enzyme (Appendix Fig S10A and B). Upon UFA depletion, FOF1 showed clear segregation behaviour (Fig 6A and Movie EV2). When co‐expressed, FOF1 and WALP23 showed clear co‐segregation into the fluid areas of phase‐separated membranes (Fig 6B and Appendix Fig S8D). This property was also confirmed by co‐staining with Laurdan showing an anti‐correlation of the fluorescent signals (Fig 6C and Appendix Fig S8E). In agreement with the smooth outer membrane staining using FM 5–95 (Fig 4B and Appendix Fig S4B), no segregation was observed for the major outer membrane protein OmpA. Co‐labelling of the inner and outer membrane with fluorescent proteins analysed by super resolution structured illumination microscopy demonstrated that the inner membrane marker FOF1 a‐mNG showed segregation into the fluid areas at non‐permissive 37°C, while the pattern of the outer membrane marker OmpA‐mCherry remains homogeneous, thus supporting the view that the outer membrane does not participate in the phase separation process (Fig EV5A–D).
Figure 6. Partitioning of membrane proteins into fluid domains of phase‐separated plasma membranes.
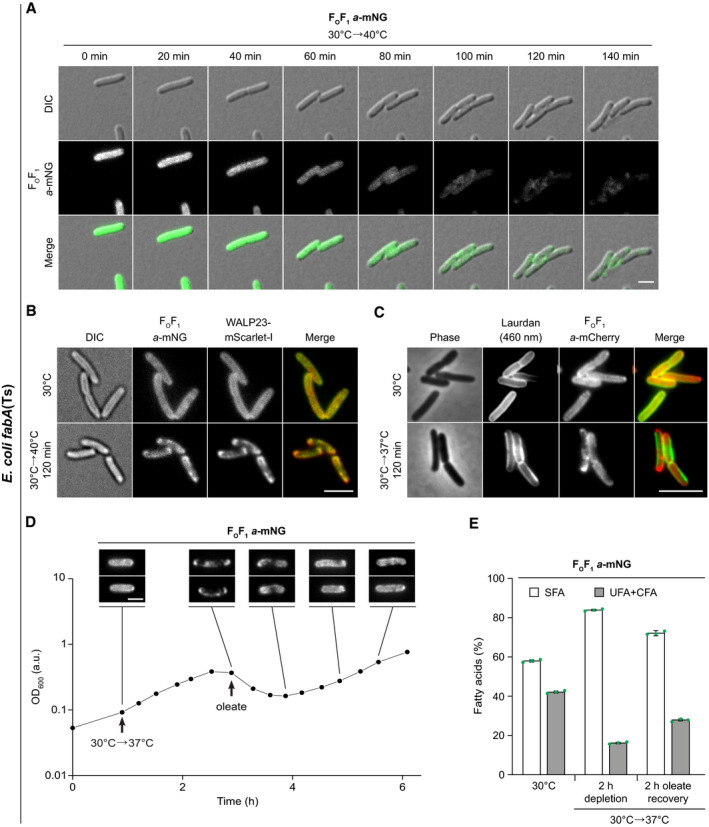
- Images of E. coli fabA(Ts) co‐expressing WALP23‐mScarlet‐I and FOF1 a‐mNG grown at permissive 30°C or shifted to non‐permissive 40°C. For fluorescence intensity correlations, see Appendix Fig S8D. For corresponding images of E. coli fabA(Ts) cells co‐expressing inner membrane marker FOF1 a‐mNG and outer membrane marker OmpA‐mCherry, see Fig EV5.
- Images of FOF1 a‐mCherry‐expressing E. coli fabA(Ts) grown at 30°C or shifted to non‐permissive 37°C and stained with Laurdan. For fluorescence intensity correlations, see Appendix Fig S8E.
- Growth behaviour of FOF1 a‐mNG expressing E. coli fabA(Ts) after shift from 30°C to non‐permissive 37°C and upon recovery through exogenous supplementation with UFA oleate (cis‐Δ9‐C18:1). The corresponding reversible segregation of FOF1 a‐mNG is shown above the growth curve. For further controls, see Appendix Fig S11A. For a comparable fluorescence time lapse analysis of mNG‐labelled glucose permease PtsG, see Appendix Fig S12.
- Fatty acid composition of E. coli fabA(Ts) cells (same cell batch as in D) upon growth at 30°C, upon depletion of UFA by incubation at 37°C for 120 min and upon recovery by oleate supplementation for additional 120 min. For detailed analyses, see Appendix Fig S11B–D.
Data information: (A–D) The experiments are representative of biological triplicates. (E) The histogram depicts mean and SD of biological triplicates. DIC, differential interference contrast. Scale bar: (A, B, D) 2 µm; (C) 3 µm. Strains used: (A, D, E) E. coli MG4; (B), E. coli MG4/pBH501; (C), E. coli LF6.red.
Source data are available online for this figure.
Depletion of UFAs had no substantial influence on the DCCD‐sensitive ATPase activity of FOF1 (Appendix Fig S10B), thus arguing that high viscosity of the surrounding lipids does not significantly hinder FOF1 in its rotation‐based catalytic cycle (Junge & Nelson, 2015). In a wider context, this indicates that the remaining fluid phase, to which FOF1 partitions upon UFA depletion, can retain robust bioactive properties. Motivated by this finding, we analysed whether the lipid phase separation and the associated growth arrest is reversible. Indeed, when severely UFA‐depleted fabA(Ts) cells exhibiting both growth arrest and lipid phase separation were exogenously supplied with oleate (cis‐Δ9‐C18:1), incorporation of oleate into phospholipids, together with growth recovery and restoration of the dispersed distribution of FOF1 was observed (Fig 6D and E, and Appendix Fig S11A–D). Comparable experiments performed with fluorescently labelled glucose permease (PtsG‐mNG) confirmed that the observed protein segregation and its recovery (Appendix Fig S12A–C) is not unique for ATP synthase.
In summary, our results demonstrate that lipid phase separation occurring under conditions of low membrane fluidity has a profound effect on membrane protein distribution, triggering segregation of integral membrane proteins into the remaining liquid‐disordered phase areas.
Restricted diffusion of membrane proteins in UFA‐depleted E. coli membranes
To analyse the consequences of UFA depletion on protein diffusion and, thus, membrane fluidity directly, we followed mNG‐labelled FOF1 (FOF1 a‐mNG) by in vivo single molecule tracking. Consistent with the lack of a specific localisation pattern, FOF1 a‐mNG complexes exhibited free diffusion within the plasma membrane plane of E. coli WT cells (Fig 7A and Movie EV3). The observed lateral mobilities and jump distances were largely independent of the growth temperature (Fig 7B and Appendix Fig S13A), as expected for cells with active homeoviscous adaptation mechanisms. FOF1 a‐mNG expressed in fabA(Ts) cells at 30°C also showed unrestricted lateral mobility comparable to WT. Under conditions of UFA depletion (33–40°C), however, a gradual, temperature‐dependent reduction of lateral displacement and median jump distances was observed (Fig 7A–C, Movie EV4, and Appendix Table S2). This is consistent with either significantly reduced diffusion or a local confinement of FOF1 caused by co‐occurring lipid phase separation (compare Fig 6A–C). Calculation of apparent lateral diffusion coefficients (Dapp) revealed that the lateral mobility was reduced up to 9‐fold (Fig 7D). Comparable results were obtained when the median jump distances and Dapp were analysed for cell‐to‐cell heterogeneity (Appendix Fig S13A and B). The Dapp of 0.0474 ± 0.0015 µm2/s determined here for FOF1‐a‐mNG at 30°C corresponds well with the values obtained previously for mEOS3.2‐FOF1 (0.042 ± 0.011 µm2/s at 22°C and 0.054 ± 0.014 µm2/s at 37°C, respectively) (Renz et al, 2015). These diffusion values are in also in agreement with recent FRAP (fluorescence recovery after photobleaching) and single molecule tracking data for cytoplasmic membrane proteins in E. coli ranging from 0.01 to 0.2 µm2/s (Leake et al, 2008; Kumar et al, 2010; Oswald et al, 2016).
Figure 7. Restricted diffusion of membrane proteins in UFA‐depleted E. coli membranes.
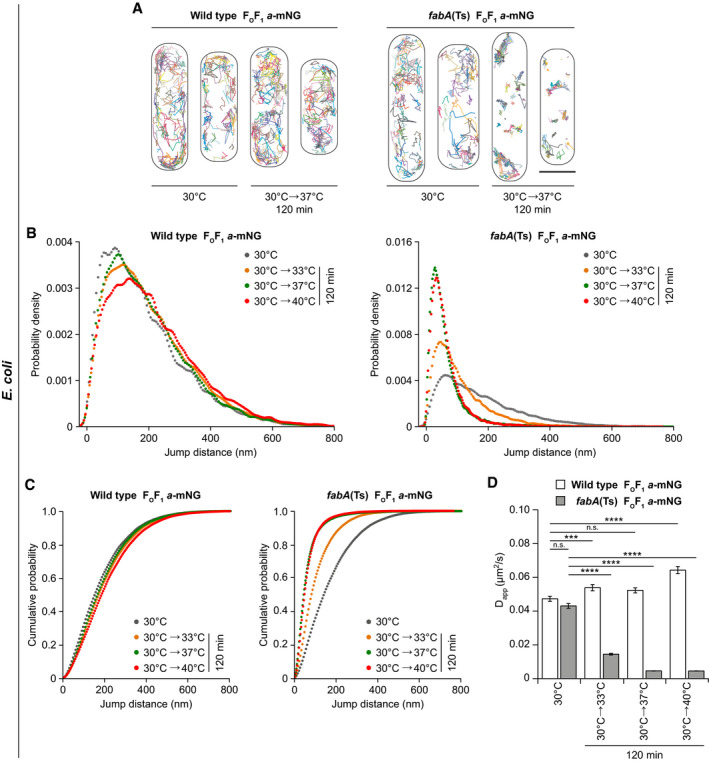
- Probability density plots of FOF1 a‐mNG jump distances in E. coli WT and fabA(Ts) cells.
- Cumulative probability plots of FOF1 a‐mNG jump distances in E. coli WT and fabA(Ts) cells. For an analysis of cell‐to‐cell heterogeneity of median jump distances of FOF1 a‐mNG, see Appendix Fig S13A.
- Apparent lateral diffusion coefficients (Dapp) of FOF1 a‐mNG analysed in panel B. An analysis of cell‐to‐cell heterogeneity of Dapp is shown in Appendix Fig S13B. For a comparable single molecule tracking analysis of transmembrane peptide WALP23‐mNG, see Appendix Fig S14 and Appendix Table S3. A detailed analysis of FOF1 a‐mNG in an osmotically stabilised fabB15(Ts) mutant (2% KCl) at non‐permissive 40°C is shown in Appendix Figs S15 and S16, Appendix Table S4.
Data information: (A) Representative for 3–5 biological replicates. (B, C) Trajectories with ≥ 5 consecutive frames for each growth condition and strain were pooled from 3 to 5 biological replicates (n = 2,345–4,468). See Appendix Table S2 for detailed information on cell numbers and jump distances. (D) Median and SD from 3 to 5 biological replicates, together with P values of a two‐sided Wilcoxon rank sum test. Significance was assumed with ****P < 0.0001, ***P < 0.001, n.s., not significant. (A) Scale bar: 1µm. Strains used: (A–D), E. coli MG1, MG4.
Source data are available online for this figure.
WALP23‐mNG also exhibited rapid, unconfined diffusion when expressed in fabA(Ts) cells at 30°C. As expected due to its single transmembrane helix (Ramadurai et al, 2009; Lucena et al, 2018), the median jump distances and Dapp of WALP23 (Appendix Fig S14A–D and Appendix Table S3) were higher than those observed for FOF1. Upon lipid phase separation caused by UFA depletion, WALP23 exhibited confined mobility comparable to that observed for FOF1, again supporting the notion that both proteins co‐segregate into the remaining fluid phase (Fig 6B and Appendix Fig S8D and E).
Due to the membrane phase separation resulting in protein‐free gel phase regions (Figs 5C and E, and 6B and C), membrane proteins become concentrated in the remaining fluid phase areas, while the membrane plane available for lateral diffusion is strongly reduced (Figs 5, 6, 7). Hence, it is not straightforward to distinguish whether the reduced diffusion dynamics are due to a change in the diffusion coefficient or due to a phase separation‐driven local confinement. While qualitative, the observed displacements appear to be significantly smaller than the remaining fluid phase membrane areas (compare Figs 5C and E, and 6B–D), thus somewhat arguing against confinement as the sole reason for the reduced diffusion dynamics. A reduction of the diffusion coefficient in protein‐overcrowded membrane areas would also be consistent with previous in vitro work demonstrating a linear decrease of both protein and lipid lateral mobility with increasing membrane protein concentrations (Ramadurai et al, 2009).
Osmotic stabilisation has been described for some UFA auxotrophic E. coli strains (Broekman & Steenbakkers, 1973, 1974; Akamatsu, 1974). Whereas osmotic stabilisation does not restore the viability of fabA(Ts) strain UC1098 in non‐permissive temperatures (Akamatsu, 1974), a strain carrying a fabB15(Ts) mutation is able to grow at non‐permissive 40°C as long as an osmotic stabiliser (e.g. 2% KCl) is present (Akamatsu, 1974) (Appendix Fig S15A). Fluorescence microscopy of fabB15(Ts) cells expressing FOF1 a‐mNG revealed that protein partitioning observed in the presence of KCl is detectable but less severe (Appendix Fig S15B), while single molecule tracking of FOF1 a‐mNG showed a lesser reduction of lateral diffusion compared to fabA(Ts) (Appendix Fig S16A and Appendix Table S4). Crucially, fatty acid analyses (Appendix Fig S16B–D) showed that depletion of UFA is significantly less pronounced in this strain upon osmotic stabilisation with KCl. Hence, osmotic stabilisation of this strain does not rescue the cells from low UFA content but rather acts by partially restoring UFA synthesis.
In summary, depletion of UFA in E. coli results in a strong reduction of membrane fluidity that severely restricts lateral diffusion of membrane proteins (summarised in Fig 8). As a complementary, dye‐independent method, the tracking experiments also confirm the lipid phase separation phenomenon, resulting in integral membrane proteins segregated and confined into the remaining fluid membrane areas.
Figure 8. Low membrane fluidity triggers large‐scale gel‐liquid lipid phase separation in vivo .
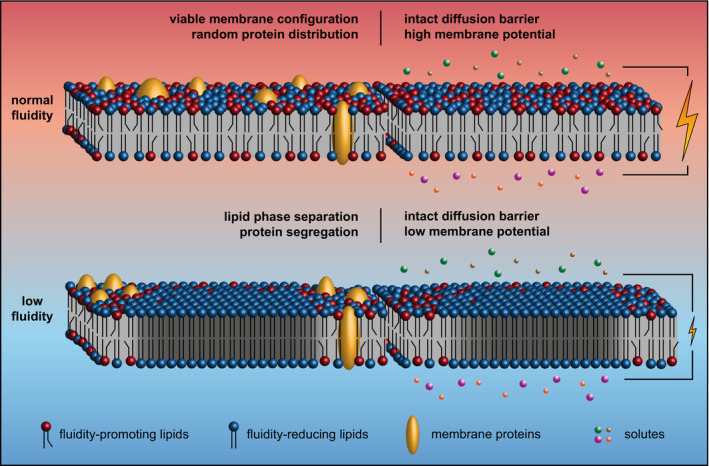
The cartoon illustrates how too low membrane fluidity triggers large‐scale lipid phase separation associated with both segregation and confined diffusion of membrane‐integral proteins (left part). Too low membrane fluidity also results in dissipation of membrane potential likely due to a reduced electron transport chain activity (Budin et al, 2018). The core diffusion barrier function of the membrane, however, is maintained (right part).
Discussion
Consistent with early studies (Willecke & Pardee, 1971; Cronan & Gelmann, 1973; Kaneda, 1977; Boudreaux et al, 1981), the ability to maintain membrane fluidity through synthesis of fluidity‐promoting lipid species is indeed essential for the growth of both E. coli and B. subtilis. However, the magnitude of changes in composition and fluidity which the cells can tolerate is surprisingly large (summarised in Fig 8). While B. subtilis cells finely balance the ratio of iso‐ and anteiso‐BCFA in response to changes in temperature (Suutari & Laakso, 1992; Klein et al, 1999), even the massive changes in the iso/anteiso‐ratio obtained through precursor supplementation had no significant effect on growth behaviour. Similarly, changes in fatty acid composition and membrane fluidity needed to impair growth of E. coli are much more drastic than those observed as part of the normal lipid adaptation upon temperature shifts (Marr & Ingraham, 1962; Sinensky, 1974; Mansilla et al, 2004). Hence, while both E. coli and B. subtilis adapt their membrane composition and fluidity even upon subtle changes in temperature, the failure to do so is not associated with growth inhibitory consequences. This lack of clear phenotypes argues against maintaining membrane fluidity (homeoviscous adaptation) or membrane phase (homeophasic adaptation) as the physiological reason behind the tight temperature‐dependent regulation of membrane lipid composition in these model organisms. While it is possible that the optimal growth conditions applied here suppress phenotypes more evident upon stressful condition such as nutrient and O2 limitation, interspecies competition, or challenge with envelope‐targeting antimicrobial agents, it is important to emphasise that fluidity and phase are not the only parameters relevant for biological membranes. In fact, changes in temperature also induce substantial changes in lipid packing and membrane thickness (Szekely et al, 2011). While speculative, the temperature‐dependent lipid adaptation processes could have evolved to regulate membrane thickness rather than fluidity, thereby carefully maintaining appropriate hydrophobic shielding of integral membrane proteins in a changing environment.
In contrast to the smaller, physiologically more relevant changes discussed above, a more drastic reduction of membrane fluidity indeed has severe, growth inhibitory consequences. Whereas low membrane fluidity associated with lipid phase separation is accompanied by a substantial increase in membrane leakiness in vitro (Papahadjopoulos et al, 1973; Heimburg, 2007; Cordeiro, 2018), our results suggest that, at least in the context of the in vivo plasma membranes of B. subtilis and E. coli, such an effect does not play a significant role. Even the very low fluidity membranes incapable for supporting growth retain a robust diffusion barrier function. Rather than indicating ion leakage, the observed gradual and partial membrane depolarisation is fully consistent with previous reports, demonstrating that the membrane fluidity influences the electron transport chain (ETC) both in E. coli and mitochondria (Budin et al, 2018; Torres et al, 2018). While the enzyme complexes of the ETC maintain their function (as we also observed for FOF1), the diffusivity of ubiquinone is reduced in membranes of low fluidity and thus controls the electron transfer rate in the ETC (Budin et al, 2018). Therefore, maintaining robust ETC activity may well be one of the biological reasons why a fine, homeostatic balance of membrane fluidity is important.
In addition to changes in membrane potential, we also observed severe effects on the machineries responsible for cell morphogenesis. The reduction of membrane fluidity in B. subtilis is associated with rapid delocalisation of MreB indicating disturbance of lateral cell wall synthesis. Conversely, in E. coli both the cell division machinery and the nucleoid morphology were disturbed. However, both cell division and cell wall synthesis machineries are also influenced by membrane depolarisation (Strahl & Hamoen, 2010; Strahl et al, 2014). It therefore remains to be determined whether the observed changes are a direct consequence of low membrane fluidity, or secondarily caused by the gradual membrane depolarisation. It is worth highlighting though that the changes in membrane fluidity required to disturb cell morphogenesis are quite extreme and go well beyond those observed upon normal changes in growth temperature. Consequently, rather than as a sign for sensitivity, these findings provide an indication for the relative robustness of bacterial morphogenetic systems towards normally encountered changes in membrane fluidity.
The most striking phenomenon caused by severe reduction of membrane fluidity is the lipid de‐mixing associated with membrane protein segregation (summarised in Fig 8). Molecular dynamic simulations with membrane models composed of lipids with SFAs and BCFAs revealed increased ordering of the lipid bilayer when the SFA content was systematically increased. In the presence of approximately 20% SFAs (compared to 5–7% in B. subtilis WT cells), a sharp transition representing phase separation between liquid and gel phase was observed (Mostofian et al, 2019). These findings are fully consistent with the in vivo lipid de‐mixing we observe upon depletion of BCFAs in favour of SFAs accumulation in B. subtilis, albeit with a slightly higher SFA content needed. In E. coli, we observe in vivo lipid de‐mixing upon accumulation of SFA to a maximal content of 80% (compared to about 50% in WT (Zhu et al, 2009)), which again is consistent with previous in vitro studies regarding lipid phase separation of SFA/UFA mixtures (Letellier et al, 1977; Morein et al, 1996; Suárez‐Germà et al, 2011). For these reasons, we argue that the observed de‐mixing represents lipid phase separation between liquid‐disordered and gel state membranes, here occurring in intact, living cells. This phenomenon is likely comparable to that observed in eukaryotic endoplasmic reticulum, characterised by low cholesterol levels, upon metabolism of SFA C18:0 (Shen et al, 2017). While the phase separation phenomenon unarguably affects the plasma membranes both in Gram‐positive B. subtilis and Gram‐negative E. coli, we suggest that the process is limited to the plasma membrane and does not encompass the Gram‐negative outer membrane. This notion is based on an even distribution of a major outer membrane protein OmpA and smooth FM 5–95 outer membrane staining in cells with phase‐separated inner membranes.
The specificity of dyes to label different lipid phases is still a matter of debate. Small hydrophobic dyes may themselves alter the composition and ordering of coexisting phases, even when used in trace amounts (Veatch, 2007). However, we are convinced that (i) by using the combination of two chemically distinct fluorescent membrane dyes Laurdan and FM 5–95 exhibiting opposing phase preferences; (ii) by combining dye‐based approaches with localisation of WALP23 peptide previously shown to exhibit liquid‐disordered phase preference in vivo, in vitro and in silico (Ridder et al, 2004; Schäfer et al, 2011; Scheinpflug et al, 2017b); and (iii) by following the reversible phase separation through its consequences on lateral membrane protein diffusion, an approach completely independent of dyes but based on intrinsic chromosomal expression of proteins, we have exhausted the possibility that the observed phase separation is an artefact caused by the labelling techniques used.
Importantly, by demonstrating lipid liquid‐gel phase separation and the associated membrane protein segregation occurring in protein‐crowded, native membranes of living cells, our results are fully consistent with comparable phenomena observed in simplified in vitro and in silico model systems (Picas et al, 2010; Suárez‐Germà et al, 2011; Domański et al, 2012; Shaw et al, 2021), thus providing strong, complementary in vivo support for the general validity of the respective membrane models.
It is perhaps not surprising that the observed lipid phase separation coincides with growth arrest. Transmembrane segments of integral membrane proteins are embedded within the hydrophobic interior of lipid bilayers. Consequently, lipid bilayer thickness, which acutely changes with membrane fluidity and phase, is both important for membrane protein activity and drives partitioning of proteins between different phases (Lenaz, 1987; Lee, 2004; Lorent et al, 2017; Nickels et al, 2019). Peripheral membrane proteins, which establish membrane association through bilayer‐intercalating domains such as amphipathic helices, in turn rely on sufficiently low packing density/high fluidity for efficient membrane association (Drin & Antonny, 2010; Bigay & Antonny, 2012; Strahl & Errington, 2017). At last, the severe restriction of lateral diffusion caused by phase separation is likely interfering with localisation and activity of many membrane‐associated cellular processes relying on diffusion and capture mechanism (Rudner et al, 2002). In conclusion and as suggested earlier based on indirect evidence (Drobnis et al, 1993; Ghetler et al, 2005; Burns et al, 2017), we argue that it is indeed the lipid phase separation process and the formation of gel phase areas that determines the lower end of membrane fluidity capable of supporting viable cell functions.
Materials and Methods
Construction of E. coli strains
All E. coli strains used are listed in Appendix Table S1. E. coli strains MG1 or EB8.1 carrying a C‐terminal mNeonGreen (mNG) or mCherry fusion via a Gly‐Ser linker to the membrane‐integral FO‐a subunit, respectively, were generated using the phage λ Red recombinase to replace a chromosomal sequence (Datsenko & Wanner, 2000). Briefly, the kanamycin resistance cassette of strain EB4 (ΔatpBE::FRT‐kan‐FRT) was exchanged by the HindIII/AseI fragment of plasmids pBH189 (MG1) or pEB21.2 (EB8.1), followed by growth on M9 minimal medium with succinate (0.4% (w/v)) as sole carbon source for selection. As expected, both strains show a Succ+ KanS phenotype. In detail, EB4 cells transformed with temperature‐sensitive plasmid pKD46 encoding λ Red recombinase genes under control of the ParaBAD promoter were grown at 30°C to mid‐logarithmic phase in lysogenic broth (LB) composed of yeast extract (0.5% w/v), tryptone (1% w/v), NaCl (1% w/v) and supplemented with arabinose (0.2% w/v) for induction. Competent cells were prepared by wash with ice‐cold water for removal of salts and medium components, electroporated in the presence of corresponding DNA fragments, grown for 1 h in LB at 37°C for phenotypic expression and plated on selective solid medium.
For generation of E. coli strain UC1098. PtsG‐mNG, a 75 bp linker (encoding the amino acids EFTMVPAAPAPAAAAPAAAPTPASR) from plasmid pBLP2 (coding for a functional PtsG‐GFP fusion protein connected by the same linker region (Kosfeld & Jahreis, 2012)) and the open reading frame (ORF) encoding mNG (pNCS‐mNeonGreen) were inserted into the chromosomally encoded ptsG gene prior to its stop codon using λ Red mutagenesis (Datsenko & Wanner, 2000). Briefly, the kanamycin resistance cassette of strain UC1098.ΔptsG (ΔptsG::FRT‐kan‐FRT) (see below) was exchanged by a PCR product harbouring the ptsG‐linker‐mNG fusion gene flanked by upstream/downstream chromosomal regions of ptsG using growth on M9 minimal medium with glucose (0.4% (w/v)) for selection with ptsG‐mNG‐expressing colonies being significantly larger compared to those of precursor strain ΔptsG. The PCR product was generated by a two‐step PCR overlap extension method. Firstly, four individual PCR products were generated using (i) oligonucleotides 1/2 with lysed cells of E. coli strain Y‐Mel as template, (ii) oligonucleotides 3/4 with pBLP2 as template, (iii) oligonucleotides 5/6 with pNCS‐mNeonGreen as template and (iv) oligonucleotides 7/8 again with lysed cells of Y‐Mel as template. Secondly, the four PCR products and oligonucleotides 1/8 were used for the second amplification step. Oligonucleotides used are listed in Appendix Table S5.
Several E. coli strains were obtained by P1 transduction (Thomason et al, 2007). In detail, P1 liquid lysate was generated by growing the donor strain to optical density at 600 nm (OD600) of 0.1 in LB (3 ml), adding CaCl2 (330 µl 50 mM) and P1 lysate (20 µl of ~10−9 phages/ml) and further growth with good aeration until lysis occurred. 5 drops of chloroform were added to lyse remaining cells, centrifuged twice to pellet debris, and the supernatant was stored with 50 µl of chloroform in the dark at 4°C. For transduction, an overnight culture of the recipient strain (200 µl) was mixed with CaCl2 (28 µl 50 mM) and P1 lysate (50 µl) of the donor strain and incubated for 20 min at 37°C. After addition of LB (0.7 ml) and Na3‐citrate (100 µl 1 M) and further incubation for 40 min, cells were plated on selective solid medium containing Na3‐citrate (20 mM).
Strains LF4 and AB1 were obtained using EB4 as donor, UC1098 and Cy288 as recipients, respectively, using LB with kanamycin (50 µg/ml) for selection leading to a Succ+ KanR phenotype. Subsequently, MG4 and LF6.red were generated by P1 transduction using MG1 and EB8.1 as respective donor strains, LF4 as recipient and M9 minimal medium with succinate (0.4% (w/v)) for selection. Strain BHH87 was generated in the same way as described for strain MG4. For generation of strains BHH100 and BHH101 by P1 transduction, MG1655.mreB‐msfGFP and KC555, respectively, were used as respective donor strains, UC1098 as recipient and LB with kanamycin (50 µg/ml) or chloramphenicol (12.5 µg/ml) for selection. For generation of strain UC1098.ΔptsG, strain JW1087‐2 was used as donor, UC1098 as recipient and LB with kanamycin for selection. For generation of strains Y‐Mel.ΔlacY and UC1098.ΔlacY, strain JW0334‐1 was used as donor, Y‐Mel or UC1098 as recipients and LB medium with kanamycin for selection. For generation of strains UC1098.ΔminC, UC1098.ΔzapA and UC1098.ΔzapB (yiiU being renamed to zapB), strains JW1165‐1, JW2878‐1 and JW3899‐1, respectively, were used as donors, UC1098 as recipient and LB medium with kanamycin for selection. In all strains, the genes of interest were verified by colony PCR and DNA sequencing.
Construction of E. coli plasmids
For construction of plasmids pBH189 and pEB21.2, a BamHI site (encoding a Gly‐Ser linker) and genes encoding mNG (pBH189) and mCherry (pEB21.2), respectively, were inserted into the atp operon of plasmid pBWU13 prior to the stop codon of atpB using a two‐step PCR overlap extension method. Firstly, three individual PCR products were generated using (i) oligonucleotides 9/10 with pSD166 as template, (ii) oligonucleotides 11/12 with pNCS‐mNeonGreen as template for pBH189 or with pQW58 as template for pEB21.2 and (iii) oligonucleotides 13/14 with pSTK3 as template. Secondly, the three PCR products and oligonucleotides 9/14 were used for the second amplification step. HindIII/AseI‐digested PCR products were cloned into correspondingly digested pBH4.
For construction of plasmids pBH500 and pBH501, a linker encoding SGSGSG, and the ORFs encoding mNeonGreen and mScarlet‐I, respectively, were fused with WALP23‐ORF by two‐step PCR. Briefly, for plasmid pBH500, two different PCR products were obtained using oligonucleotides 15/16 with pL030 as template and oligonucleotides 18/19 with pNCS‐mNeonGreen as template. For the second PCR step, oligonucleotides 15/19 were used. For plasmid pBH501, two PCR products were obtained using oligonucleotides 15/17 with pL030 as template and oligonucleotides 20/21 with synthesised mScarlet‐I‐encoding DNA as template. For the second PCR step, oligonucleotides 15/21 were used. In both cases, AvrII/SpeI‐digested PCR products were cloned into correspondingly digested pL030.
For construction of plasmids pJG130 and pJG131, plasmid pBAD322 was linearised with oligonucleotides 26 and 27. Full‐length rne was amplified using oligonucleotides 28/29, while inserts for construction of rne‐ΔAH were amplified using oligonucleotides 28/30 and 29/31. These fragments were fused using NEBuilder® HiFi DNA Assembly Cloning Kit (New England Biolabs).
All constructs were verified by DNA sequencing and are listed in Appendix Table S6. Oligonucleotides used are listed in Appendix Table S5.
E . coli strains and growth conditions
E. coli strains (Appendix Table S1) were grown in M9 minimal medium composed of Na2HPO4•2H2O (0.85% w/v), KH2PO4 (0.3% w/v), NaCl (0.3% w/v), NH4Cl (0.05% w/v), MgSO4•7H2O (0.25% w/v), CaCl2•2H2O (0.015% w/v) and supplemented with thiamine (0.01% w/v), casamino acids (0.1% w/v) and glucose (0.4% w/v) at 30°C, unless stated otherwise. For induction of the temperature‐sensitive phenotype fabA(Ts), pre‐cultures of E. coli strain UC1098 and its derivatives were diluted from an overnight culture grown at 30°C to OD600 of 0.025 and grown to OD600 of 0.5 at 30°C, and again diluted to OD600 of 0.025 in pre‐warmed, fresh medium for complete removal of cells in the stationary growth phase. At OD600 of 0.1–0.2, cells were transferred for 120 min to growth temperatures of 33, 37 or 40°C as indicated. WT strains were handled accordingly. For induction of the thermosensitive phenotype of fabB15(Ts), cells were grown essentially as described (Akamatsu, 1974). Precultures of E. coli strain BHH87 were diluted from an overnight culture, grown at 30°C in the presence or absence of 2% (w/v) KCl, to OD600 of 0.025 and grown at permissive 30°C to OD600 of 0.4–0.5 or grown at non‐permissive 40°C for 180 min, each with or without 2% KCl. The corresponding WT strain was handled accordingly. For recovery from UFA depletion and corresponding phase separation, UC1098 derivatives were subsequently supplemented with potassium oleate (100 µg/ml; Sigma‐Aldrich) dissolved in Brij®58 (0.1% w/v; Sigma‐Aldrich). Fluorescently labelled ATP synthase (FOF1 a‐mNG or FOF1 a‐mCherry; C‐terminal fusion to FO‐a), fluorescently labelled glucose permease (PtsG‐mNG; C‐terminal fusion) and msfGFP sandwich fusions of FtsZ and MreB were expressed from their own locus under control of their native promoter. Fluorescently labelled WALP23 (WALP23‐mScarlet‐I or WALP23‐mNG; amino acid sequence: AWW(LA)8LWWA) was expressed plasmid‐encoded under control of the B. subtilis Pxyl promoter (Appendix Table S6) resulting in constitutive expression in E. coli. Fluorescently labelled RNase E‐YFP (C‐terminal fusion) was expressed plasmid‐encoded under control of its own promoter. RNase E and ΔAH‐RNase E (RNase E lacking the membrane‐binding amphipathic helix) were expressed plasmid‐encoded under control of the ParaBAD‐inducible promoter.
Construction of B. subtilis strains
All B. subtilis strains used are listed in Appendix Table S1. For construction of a B. subtilis strain expressing WALP23 fused to monomeric superfolder GFP (msfGFP), the plasmid pBH500 was linearised with oligonucleotides 22 and 23, msfGFP amplified using oligonucleotides 24 and 25 (Appendix Table S5) and the fragments fused using NEBuilder® HiFi DNA Assembly Cloning Kit (New England Biolabs). The resulting plasmid was transformed into B. subtilis 168, thus resulting in strain JG054. All other B. subtilis strains were constructed by transforming the respective recipient strains with chromosomal DNA from the donor strains or corresponding plasmid DNA. Transformations were carried out as described (Hamoen et al, 2002).
B. subtilis strains and growth conditions
For strain construction, B. subtilis (Appendix Table S1) was grown either in LB (lysogeny broth), Nutrient Broth or Nutrient Agar (Oxoid). If necessary, these media were supplemented with either isobutyric acid (IB) (100 µM; Sigma‐Aldrich) or 2‐methylbutyric acid (MB) (100 µM; Sigma‐Aldrich). All other experiments were carried out with fortified Spizizen minimal medium composed of (NH4)2SO4 (0.2% w/v), K2HPO4 (1.4% w/v), KH2PO4 (0.6% w/v) Na3‐citrate•2H2O (0.1% w/v), MgSO4 (0.09% w/v), ferric ammonium citrate (1.1 µg/ml) supplemented with glucose (0.96% w/v), L‐tryptophan (20 µg/ml) and casamino acids (0.02% w/v). In our hands, the precursor isovaleric acid, which is the primer for the synthesis of iso‐C15:0 and iso‐C17:0, neither supported growth nor resulted in synthesis of the expected iso‐BCFAs, thus implying that this precursor cannot be supplied exogenously in the B. subtilis 168 strain background. All cultures were inoculated by 1:100 dilution of an LB overnight culture supplemented with the corresponding precursor. Depletion of BCFAs was carried out for cells initially grown in the presence of IB (100 µM), followed by washing, pelleting and resuspension in pre‐warmed, precursor‐free medium (PF). Unless stated otherwise, all experiments were carried out at 37°C. Fluorescently labelled WALP23 peptides (WALP23‐mCherry or WALP23‐msfGFP), msfGFP‐MreB and GFP‐FtsZ were expressed ectopically (amyE locus) under control of the Pxyl promoter and induced by xylose (1% w/v in case of WALP23; 0.3% w/v in case of GFP‐FtsZ).
DPH anisotropy measurements
Steady‐state DPH fluorescent anisotropy measurements were carried out with 1,6‐Diphenyl‐1,3,5‐hexatriene (DPH)‐labelled cells using a BMG Clariostar multimode plate reader (BMG Labtech). For B. subtilis, cells taken from cultures at time points of interests were diluted to an OD600 of 0.25 in a pre‐warmed medium, followed by addition of DPH (Sigma Aldrich) dissolved in dimethyl formamide (DMF) to a final concentration of 10 µM DPH and 1% (v/v) DMF. Samples were shaken in dark at 37°C for 5 min, followed by a wash, resuspension in dye‐free medium to an OD600 of 0.5 and transfer to pre‐warmed, black, polystyrene 96‐well microtiter plates (Labsystems) for measurement. Following 1‐min incubation under shaking in the pre‐warmed plate reader to homogenise the sample, the fluorescence anisotropy was measured at 37°C using excitation wavelength of 360–10 nm, emission wavelength of 450–10 nm and a dichroic mirror set at 410 nm. The fluorescence anisotropy (A) was calculated with MARS Data Analysis software (BMG Labtech) using the equation (Iparallel‐Iperpendicular)/(Iparallel+2xIperpendicular).
The corresponding measurements for E. coli were carried out using the same protocol with following modifications. Staining was carried out with cells grown in the presence of non‐growth inhibitory concentrations (30 µg/ml; Sigma‐Aldrich) of the outer membrane‐permeabilising agent Polymyxin B nonapeptide, which is required for good staining of E. coli with DPH. The measurements at 30 and 37°C without temperature shifts were carried out with all media, plastic ware and the plate reader pre‐warmed to the corresponding temperatures. The rapid temperature shift from 30 to 37°C was carried out with cells grown, stained and washed at 30°C, followed by final resuspension in buffer pre‐warmed to 37°C and measurement with microtiter plate as well as plate reader pre‐warmed to 37°C.
Determination of fatty acid composition
The fatty acid composition of E. coli and B. subtilis was determined from 50 to 100 mg (wet weight) of bacterial cells grown as described in the main text. Fatty acids were extracted as methyl esters after saponification and methylation as described (Sasser, 1990). For saponification, cell pellets were mixed with 15% (w/v) NaOH in 50% (v/v) methanol (1 ml), incubated at 100°C for 5 min, vortexed and further incubated for 25 min. After cooling, acid methylation with 6 N HCl in 50% (v/v) methanol (2 ml) was performed for 10 min at 80°C followed by immediate cooling on ice. Methylated fatty acids were extracted by addition of hexane/methyl tert‐butyl ether in a 1:1 ratio (1.25 ml), followed by end‐over‐end incubation for 10 min. After phase separation by centrifugation, the lower phase was discarded. The organic phase was washed with 1.2% (w/v) NaOH (3 ml) by 5‐min end‐over‐end incubation and centrifugation. The upper phase of the phase‐separated sample was used for further analysis.
The fatty acid methyl esters (FAME) were separated and identified by gas chromatography‐mass spectrometry (GC‐MS) with a gas chromatograph (model 7890A; Agilent Technologies) equipped with a 5% phenylmethyl silicone capillary column and a mass spectrometer (model 5975C; Agilent Technologies). Helium was used as carrier gas, injection volume was 1 µl, injector temperature was 250°C, the column temperature was increased from 120 to 240°C at a rate of 5°C/min, and the GC‐MS line transfer temperature was 280°C. FAME were separated by their retention times and identified by their equivalent chain lengths and their mass spectra. Equivalent chain length values were calculated from linear interpolation of unknown peaks’ retention time between two saturated straight chain FAME of a standard.
For the analysis of the fatty acid composition of B. subtilis wild‐type cells grown at different temperatures (Appendix Fig S6), the cells were grown in LB medium and collected when the cultures reached an OD600 of approximately 0.5. The fatty acids were analysed as fatty acid methyl esters with GS‐MS as described above. However, these specific analyses were carried out by the Identification Service of the DSMZ, Braunschweig, Germany.
Glycerophospholipid analysis by MALDI‐TOF mass spectrometry
Extraction of lipids from bacterial cells was performed according to Gidden et al (2009) as follows. 1010 cells (assuming that 1 ml of cell culture with OD600 of 1.0 contains 109 cells (Neidhardt et al, 1990) were harvested, washed twice with cooled water and extracted with 450 µl of dichloromethane:ethanol:water 1:1:1 (v:v:v) overnight at 4°C. 1 μl of the lipid‐containing lower organic phase was spotted on a MALDI target plate (Prespotted AnchorChip 96 Set for Proteomics II; Bruker Daltonics/Eppendorf; washed peptide‐free with 2‐propanol) pre‐spotted with 1 µl of 9‐aminoacridine (10 mg/ml dissolved in acetone:water 9:1 (v/v)) and air‐dried. Mass spectra were obtained on an ultrafleXtreme MALDI‐TOF/TOF mass spectrometer equipped with a smartbeam™ solid‐state laser (Bruker Daltonics) operating in the negative ion mode. The laser was fired with a frequency of 500 Hz with 4 × 250 laser shots per spot. MS/MS spectra were obtained using the “LIFT” technique implemented in the mass spectrometer with an increased laser power. Samples from three independent biological replicates were measured per condition each as technical triplicates, compared with corresponding standard lipids PE (16:0) (18:1) and PG (16:0) (18:1) (Avanti polar lipids) in MS as well as MS/MS spectra and analysed with FlexAnalysis 3.4 (Bruker Daltonics).
ONPG membrane permeability assay
For measuring passive ONPG membrane permeability in E. coli ΔlacY strains carrying plasmid‐encoded lacZ, overnight cultures were grown at 30°C in LB medium followed by 1:100 dilution in M9‐casamino acid medium and growth to an OD600 of 0.3. For maintaining the plasmid, media were supplemented with 50 µg/ml ampicillin. Cells at OD600 of 0.3 were transferred to clear 96‐well microtiter plates followed by addition of 2‐nitrophenyl β‐D‐galactopyranoside (ONPG) dissolved in PBS (Oxoid) to a final concentration of 2 mg/ml. LacZ‐driven hydrolysis of ONPG was monitored by measuring absorbance at 420 nm using a BMG Clariostar multimode plate reader, every 2 min for up to 30 min at 37°C with vigorous shaking in between. Meanwhile, the culture was transferred to 37°C for 120 min in a pre‐warmed water bath. Cells were then adjusted to OD600 0.3 and ONPG measurements were repeated as above. The relative ONPG permeability was calculated as the rate of ONPG conversion, which is permeability‐limited in intact ΔlacY cells.
Fluorescence microscopy
Regular wide‐field fluorescence microscopy was carried out with cells immobilised on Teflon‐coated multi‐spot microscope slides (Hendley‐Essex) with 1.2% (w/v) agarose/H2O (te Winkel et al, 2016). In brief, after the agarose solidified within 10 min at room temperature, 0.5 µl of cell culture was applied to the exposed agarose surface, air‐dried until the liquid‐drop was soaked in, covered with a coverslip and immediately used for microscopy. For staining with various fluorescent dyes, cells were incubated upon shaking at the growth temperature for 5 min with following concentrations: FM 5‐95 (2 µg/ml; Thermo Fisher Scientific), DiSC3(5) (2 µM; Sigma‐Aldrich), Sytox Green (50 ng/ml; Thermo Fisher Scientific) and DAPI (200 ng/ml; Sigma‐Aldrich). Membrane depolarisation of B. subtilis was achieved by 5‐min incubation with small cation specific channel‐forming antimicrobial peptide Gramicidin ABC (10 µM; gABC; Sigma‐Aldrich), and membrane permeabilisation by 5‐min incubation with pore‐forming lantibiotic Nisin (10 μM; Sigma‐Aldrich). In the case of E. coli, membrane depolarisation and permeabilisation were achieved by 15‐min incubation in the presence of pore‐forming antibiotic Polymyxin B (10 µg/ml; Sigma‐Aldrich), and nucleoid staining was achieved by 15‐min incubation with 500 ng/ml DAPI (Severn Biotech). Laurdan microscopy was carried out with cells stained with 100 µM Laurdan (Sigma‐Aldrich; dissolved in 1% (v/v) DMF) as described (Scheinpflug et al, 2017a; Wenzel et al, 2018). Staining of E. coli was carried out with Polymyxin B nonapeptide outer membrane‐permeabilised cells, as was done for DPH. The time lapse microscopy of B. subtilis was carried out with the fortified Spizizen minimal medium (with glucose, tryptophan and casamino acids) diluted to one tenth and supplemented with 1.4% (w/v) low‐melting point agarose. The slide preparation was carried out as described (de Jong et al, 2011).
The fluorescence microscopy of B. subtilis and E. coli cells stained with FM 5‐95, DiSC3(5), Sytox Green, Laurdan or expressing fluorescent protein fusions was performed at 37°C with Nikon Eclipse Ti equipped with either Sutter Instruments Lambda LS Xenon‐arc light source or CoolLed pE‐300white LED light source, CoolLed pE‐4000 LED light source, Photometrics Prime sCMOS camera, Photometrics BSI sCMOS camera and either Nikon Plan Fluor 100×/1.30 NA Oil Ph3, Nikon CFI Plan Apo VC 100×/1.40 NA or Nikon Plan Apo 100×/1.40 NA Oil Ph3 objectives. The used filter sets were Chroma 49000 (for DAPI and Laurdan 460 nm), Chroma 49002 (for GFP and Sytox Green), Chroma 49003 (for YFP), Chroma 49008 (for mCherry, mScarlet‐I and FM5‐95), Semrock Cy5‐4040C (for DiSC3(5)), a custom filter set consisting of Chroma AT350/50x excitation filter, Chroma T400lp beam splitter or Chroma ET525/50m (for Laurdan 520 nm). The microscopy images shown in Appendix Fig S7 were carried out with Applied Precision DeltaVision RT equipped with Photometrics Coolsnap HQ2 camera, Zeiss Plan‐APOCHROMAT 100× objective and the standard DeltaVision filters sets. Dual‐colour 2D‐SIM was carried out with Nikon N‐SIM equipped with Nikon CFI APO TIRF 100×/1.49 oil objective, 488 nm (Coherent Sapphire) and 561 nm (Cobolt Jive 100) solid‐state lasers and Andor Xion X3 EMCCD camera. The used filter sets were Chroma 49904 (for mNG) and 49909 (for mCherry). Image capture and reconstruction of high‐resolution 2D‐SIM images were performed with NIS elements 5.11 (Nikon). All images were analysed using Fiji (Schindelin et al, 2012). Laurdan GP maps were calculated and generated using the ImageJ‐macro as described (Wenzel et al, 2018). The localisation correlation analysis was carried out with the Fiji plugin Coloc 2, using a 3‐pixel wide line following the cell periphery as a region of interest.
For E. coli fabA(Ts), time lapse microscopy and widefield fluorescence microscopy microscope slides were coated with a thin film of 1% (w/v) agarose dissolved in M9 minimal media supplemented with glucose/casamino acids. Cells (1 or 3 µl) were immobilised and imaged with a DeltaVision Elite microscopy system (Applied Precision, GE Healthcare) equipped with an inverted microscope (IX‐71, Olympus), a 100× oil immersion objective (UAPON 100x TIRF, Olympus) or an extended apochromat phase‐contrast objective (UPLXAPO100XOPH, Olympus), solid‐state illumination system (Insight SSI, Applied Precision), a sCMOS camera (pco.edge 4.2, PCO) and acquisition software (softWoRx 5.5, Applied Precision). Z‐Stacks of 300 nm in 5 optical slices were acquired of cells growing on the microscope slide with the microscope tempered to the corresponding temperature and imaged for 2.5 h with 5‐min intervals using an exposure time per frame of 50 ms. Fluorescence of mNeonGreen and mScarlet‐I was excited using a polychromic beamsplitter (405 nm/488 nm/590 nm/650 nm) as well as either a GFP/FITC bandpass filter (461–489 nm) or a mCherry/Alexa594 bandpass filter (562–588 nm). Fluorescence detection was achieved using a GFP/FITC bandpass emission filter (501–559 nm) for mNG and a mCherry/Alexa594 bandpass emission filter (602–648 nm) for mScarlet‐I.
Single molecule imaging of E. coli cells expressing FOF1 a‐mNG or WALP23‐mNG was performed using a total internal reflection fluorescence (TIRF) microscopy system equipped with an inverted microscope (IX‐83, Olympus), a motorised four‐line TIRF condenser (cellTIRF, Olympus), an 150× oil immersion objective (UAPON 150×/1.45 NA TIRF, Olympus), an EMCCD camera (iXON Ultra 897, Andor) and the acquisition software CellSens 2.3 (Olympus). Fluorescence of mNG was excited by a 488 nm laser diode (LuxX 488‐200, Omicron) using a TIRF pentaband polychroic mirror (zt405/488/561/640/730rpc, Chroma). Fluorescence detection and efficient TIRF laser blocking were achieved by a pentabandpass emission filter (BrightLine HC 440/521/607/694/809, Semrock) and an additional single bandpass emission filter (BrightLine HC 525/35, Semrock). All images were analysed using Fiji (Schindelin et al, 2012).
Single molecule tracking
Single molecule imaging of E. coli cells expressing FOF1 a‐mNG was performed using a total internal reflection fluorescence (TIRF) microscope. Cells were pre‐bleached for 2.5 s at 10% laser intensity (approx. 1.5 µW/µm2) to obtain single molecule fluorescence level. Subsequently, single emitter signals were imaged at 30 frames per second for 1,200 frames (40 s) with 5% of laser intensity. All experiments were carried out at room temperature for comparability. Tracking of single molecules and data analysis were carried out with well‐established localisation and tracking algorithms, implemented in a software package called “SLIMfast” (kindly provided by C.P. Richter (Osnabrück)) written in Matlab (Richter et al, 2017; Appelhans et al, 2018). Localisation precision was typically about 20–25 nm. Between 900 and 1,000 frames per image series were used for further step length and diffusion constant analysis. Step length analysis is based on trajectories exhibiting at least five sequential frames (excluding deflation loops and frame gaps). Typically, the population of all trajectories (step size of ≥ 1) is approximately twice as high as those taken into account (step size of ≥ 5).
Analysis of lateral mobility was performed via cumulative probability plots with jump distances of pooled trajectories from 3 to 5 separately grown cell batches and via boxplots to determine the median of all trajectories present within individual cells. Apparent two‐dimensional diffusion coefficients Dapp were estimated by the mean‐squared displacement (MSD) considering a linear free diffusion model. Here, is the lag time defined by multiples of time interval Δt of the image series. For all trajectories with at least ≥5 five sequential frames, MSD was averaged and the diffusion coefficient calculated by the slope of a linear fit based on the first four data points of the MSD. For the determination of standard error, the statistical resampling method of bootstrapping was used, evaluating data sets with N data points 1,000 times (Bradley, 1981).
Membrane vesicles and DCCD‐sensitive ATPase activity
Inverted membrane vesicles were prepared as previously described (Brandt et al, 2013) using 850 ml of cell culture grown in M9 minimal medium with 0.4% (w/v) glucose and 0.1% (w/v) casamino acids. After harvest, cell pellets were resuspended in 25 ml 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 10% (v/v) glycerol and disrupted in the presence of 10 µg/ml DNaseI (Sigma) with a constant cell disruptor system (Daventry) at 4°C and 1.35 kbar. For removal of cell debris, lysates were centrifuged at 35,000 g for 30 min at 4°C. After ultracentrifugation of the supernatant at 250,000 g at 4°C for 60 min, membranes were resuspended with a marten paint brush in a small aliquot (0.3–0.5 ml) of the same buffer and stored in liquid nitrogen.
ATPase activities of inverted membrane vesicles were determined using an automated continuous assay enabling a direct recording of the substrate turnover (Arnold et al, 1976). For inhibition of ATPase activities with 80 µM N,N′‐dicyclohexylcarbodiimide (DCCD; Sigma‐Aldrich; stock solution 40 mM in ethanol), membranes were incubated in 1 ml of 50 mM Tris–HCl, pH 8.0 for 20 min at 37°C prior to measurement (Deckers‐Hebestreit & Altendorf, 1992).
SDS–PAGE and Western blot
Protein concentrations were determined with the BCA assay as recommended by the supplier (Pierce). Proteins (20 µg/lane) were separated by SDS–PAGE using 10% Tris‐Tricine gels (10% T, 3% C) (Schägger & von Jagow, 1987) with PageRulerTM prestained protein ladder (Fermentas) as standard. For immunoblotting, separated proteins were transferred to nitrocellulose membranes (0.45 μm) via wet blotting in carbonate buffer (10 mM NaHCO3, 3 mM Na2CO3, pH 8.9 with NaOH, 20% (v/v) methanol) for 45 min at 1.8 A with cooling (Hilbers et al, 2013). Membranes were blocked with 5% (w/v) skimmed milk powder in TBS buffer (50 mM Tris–HCl pH 7.4, 0.9% (w/v) NaCl), decorated with monoclonal mouse antibodies specific for FO‐a (GDH 14‐5C6 (Jäger et al, 1998)) or mNeonGreen (32F6, ChromoTek) and secondary IRDye™800DX‐labelled goat‐anti‐mouse IgG (H + L) (LI‐COR Biosciences), and detected using a two‐channel Odyssey infrared imaging System (LI‐COR Biosciences).
Quantification and statistical analysis
Statistical analysis was performed using a two‐sided Wilcoxon rank sum test (MATLAB) or an unpaired two‐sided t‐test (GraphPad Prism or Microsoft Excel). Error bars represent SD from three independent biological replicates, unless stated otherwise. Significance was assumed with ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, n.s., not significant.
Author contributions
GD‐H and HS designed research; GD‐H and HS coordinated the collaborative research; MG, AL, JWG, JAB, BH, ZB and HS performed the experiments; MG, AL, JWG, JAB, RK, SW, GD‐H and HS analysed data; HS, GD‐H and MG wrote the paper.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank E. Garner (Harvard), Tanneke den Blaauwen (Amsterdam), S. Lee (Newcastle) and K. Jahreis (Osnabrück) for providing strains/plasmids; E. Becker, L. Fellner, A. Bogdanowski (Osnabrück) and Kenneth H. Seistrup (Newcastle) for aid in generating plasmids/strains and for generating wild‐type fatty acid profiles used in comparisons; H. Winkelmann and H. Arlt for introducing MG into fluorescence microscopy; C.P. Richter and T. Appelhans for technical discussions (Osnabrück). This work was supported by CRC944 (to GD‐H), “Incentive Award” of the faculty of Biology/Chemistry of Osnabrück University (to GD‐H) and Biotechnology and Biological Sciences Research Council New Investigator Award BB/S00257X/1 (to HS). JAB was supported by the Medical Research Council grant MR/N013840/1, JWG by the Biotechnology and Biological Sciences Research Council grant BB/M011186/1, and MG by a stipend of the Hans Mühlenhoff‐Stiftung (Osnabrück).
Disclosure statement and competing interests
The authors declare that they have no conflict of interest.
The EMBO Journal (2022) 41: e109800.
See also: M Bramkamp (March 2022)
Contributor Information
Gabriele Deckers‐Hebestreit, Email: deckers-hebestreit@biologie.uni-osnabrueck.de.
Henrik Strahl, Email: h.strahl@ncl.ac.uk.
Data availability
Additional figures and tables can be found in the Appendix. Correspondence and requests for material should be addressed to H.S. or G.D.‐H. Source data are available online for individual graphs. This study includes no data deposited in external repositories.
References
- Adams DW, Errington J (2009) Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat Rev Microbiol 7: 642–653 [DOI] [PubMed] [Google Scholar]
- Akamatsu Y (1974) Osmotic stabilization of unsaturated fatty acid auxotrophs of Escherichia coli . J Biochem 76: 553–561 [DOI] [PubMed] [Google Scholar]
- Andersen OS, Koeppe RE 2nd (2007) Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct 36: 107–130 [DOI] [PubMed] [Google Scholar]
- Appelhans T, Beinlich FRM, Richter CP, Kurre R, Busch KB (2018) Multi‐color localisation microscopy of single membrane proteins in organelles of live mammalian cells. J Vis Exp 136: e57690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold A, Wolf HU, Ackermann BP, Bader H (1976) An automated continuous assay of membrane‐bound and soluble ATPases and related enzymes. Anal Biochem 71: 209–213 [DOI] [PubMed] [Google Scholar]
- Ballweg S, Sezgin E, Doktorova M, Covino R, Reinhard J, Wunnicke D, Hänelt I, Levental I, Hummer G, Ernst R (2020) Regulation of lipid saturation without sensing membrane fluidity. Nat Commun 11: 756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart T, Hammond AT, Sengupta P, Hess ST, Holowka DA, Baird BA, Webb WW (2007) Large‐scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc Natl Acad Sci USA 104: 3165–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigay J, Antonny B (2012) Curvature, lipid packing, and electrostatics of membrane organelles: defining cellular territories in determining specificity. Dev Cell 23: 886–895 [DOI] [PubMed] [Google Scholar]
- Boudreaux DP, Eisenstadt E, Iijima T, Freese E (1981) Biochemical and genetic characterization of an auxotroph of Bacillus subtilis altered in the acyl‐CoA:acyl‐carrier‐protein transacylase. Eur J Biochem 115: 175–181 [DOI] [PubMed] [Google Scholar]
- Bradley E (1981) Nonparametric estimates of standard error: the jackknife, the bootstrap and other methods. Biometrika 68: 589–599 [Google Scholar]
- Brandt K, Maiwald S, Herkenhoff‐Hesselmann B, Gnirß K, Greie JC, Dunn SD, Deckers‐Hebestreit G (2013) Individual interactions of the b subunits within the stator of the Escherichia coli ATP synthase. J Biol Chem 288: 24465–24479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekman JH, Steenbakkers JF (1973) Growth in high osmotic medium of an unsaturated fatty acid auxotroph of Escherichia coli K‐12. J Bacteriol 116: 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekman JH, Steenbakkers JF (1974) Effect of the osmotic pressure of the growth medium on fabB mutants of Escherichia coli . J Bacteriol 117: 971–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budin I, de Rond T, Chen Y, Chan LJG, Petzold CJ, Keasling JD (2018) Viscous control of cellular respiration by membrane lipid composition. Science 362: 1186–1189 [DOI] [PubMed] [Google Scholar]
- Burns M, Wisser K, Wu J, Levental I, Veatch SL (2017) Miscibility transition temperature scales with growth temperature in a zebrafish cell line. Biophys J 113: 1212–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman D (1975) Phase transitions and fluidity characteristics of lipids and cell membranes. Q Rev Biophys 8: 185–235 [DOI] [PubMed] [Google Scholar]
- Chen IA, Walde P (2010) From self‐assembled vesicles to protocells. Cold Spring Harb Perspect Biol 2: a002170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro RM (2018) Molecular structure and permeability at the interface between phase‐separated membrane domains. J Phys Chem B 122: 6954–6965 [DOI] [PubMed] [Google Scholar]
- Cronan JE Jr, Gelmann EP (1973) An estimate of the minimum amount of unsaturated fatty acid required for growth of Escherichia coli . J Biol Chem 248: 1188–1195 [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debarbouille M, Gardan R, Arnaud M, Rapoport G (1999) Role of BkdR, a transcriptional activator of the SigL‐dependent isoleucine and valine degradation pathway in Bacillus subtilis . J Bacteriol 181: 2059–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckers‐Hebestreit G, Altendorf K (1992) Influence of subunit‐specific antibodies on the activity of the FO complex of the ATP synthase of Escherichia coli. II. Effects of subunit c‐specific polyclonal antibodies. J Biol Chem 267: 12370–12374 [PubMed] [Google Scholar]
- Diomandé SE, Nguyen‐The C, Guinebretière MH, Broussolle V, Brillard J (2015) Role of fatty acids in Bacillus environmental adaptation. Front Microbiol 6: 813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domański J, Marrink SJ, Schäfer LV (2012) Transmembrane helices can induce domain formation in crowded model membranes. Biochim Biophys Acta 1818: 984–994 [DOI] [PubMed] [Google Scholar]
- Drin G, Antonny B (2010) Amphipathic helices and membrane curvature. FEBS Lett 584: 1840–1847 [DOI] [PubMed] [Google Scholar]
- Drobnis EZ, Crowe LM, Berger T, Anchordoguy TJ, Overstreet JW, Crowe JH (1993) Cold shock damage is due to lipid phase transitions in cell membranes: a demonstration using sperm as a model. J Exp Zool 265: 432–437 [DOI] [PubMed] [Google Scholar]
- Elson EL, Fried E, Dolbow JE, Genin GM (2010) Phase separation in biological membranes: integration of theory and experiment. Annu Rev Biophys 39: 207–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Ballweg S, Levental I (2018) Cellular mechanisms of physicochemical membrane homeostasis. Curr Op Cell Biol 53: 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Ejsing CS, Antonny B (2016) Homeoviscous adaptation and the regulation of membrane lipids. J Mol Biol 428: 4776–4791 [DOI] [PubMed] [Google Scholar]
- Galli E, Gerdes K (2010) Spatial resolution of two bacterial cell division proteins: ZapA recruits ZapB to the inner face of the Z‐ring. Mol Microbiol 76: 1514–1526 [DOI] [PubMed] [Google Scholar]
- Ghetler Y, Yavin S, Shalgi R, Arav A (2005) The effect of chilling on membrane lipid phase transition in human oocytes and zygotes. Hum Reprod 20: 3385–3389 [DOI] [PubMed] [Google Scholar]
- Gidden J, Denson J, Liyanage R, Ivey DM, Lay JO (2009) Lipid compositions in Escherichia coli and Bacillus subtilis during growth as determined by MALDI‐TOF and TOF/TOF mass spectrometry. Int J Mass Spectrom 283: 178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamoen LW, Smits WK, de Jong A, Holsappel S, Kuipers OP (2002) Improving the predictive value of the competence transcription factor (ComK) binding site in Bacillus subtilis using a genomic approach. Nucl Acids Res 30: 5517–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harayama T, Riezman H (2018) Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol 19: 281–296 [DOI] [PubMed] [Google Scholar]
- Hazel JR (1995) Thermal adaptation in biological membranes: Is homeoviscous adaptation the explanation? Annu Rev Physiol 57: 19–42 [DOI] [PubMed] [Google Scholar]
- Heberle FA, Feigenson GW (2011) Phase separation in lipid membranes. Cold Spring Harb Perspect Biol 3: a004630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimburg T (2007) Thermal biophysics of membranes. Weinheim, Germany: Wiley‐VCH Verlag; [Google Scholar]
- Hilbers F, Eggers R, Pradela K, Friedrich K, Herkenhoff‐Hesselmann B, Becker E, Deckers‐Hebestreit G (2013) Subunit δ is the key player for assembly of the H+‐translocating unit of Escherichia coli FOF1 ATP synthase. J Biol Chem 288: 25880–25894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Mukherjee A, Pichoff S, Lutkenhaus J (1999) The MinC component of the division site selection system in Escherichia coli interacts with FtsZ to prevent polymerization. Proc Natl Acad Sci USA 96: 14819–14824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger H, Birkenhäger R, Stalz WD, Altendorf K, Deckers‐Hebestreit G (1998) Topology of subunit a of the Escherichia coli ATP synthase. Eur J Biochem 251: 122–132 [DOI] [PubMed] [Google Scholar]
- de Jong IG, Beilharz K, Kuipers OP, Veening JW (2011) Live cell imaging of Bacillus subtilis and Streptococcus pneumoniae using automated time‐lapse microscopy. J Vis Exp 53: e3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge W, Nelson N (2015) ATP synthase. Annu Rev Biochem 84: 631–657 [DOI] [PubMed] [Google Scholar]
- Kaneda T (1977) Fatty acids of the genus Bacillus: an example of branched‐chain preference. Bacteriol Rev 41: 391–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein W, Weber MHW, Marahiel MA (1999) Cold shock response of Bacillus subtilis: Isoleucine‐dependent switch in the fatty acid branching pattern for membrane adaptation to low temperatures. J Bacteriol 181: 5341–5349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler P, Marahiel MA (1997) Association of the histone‐like protein HBsu with the nucleoid of Bacillus subtilis . J Bacteriol 179: 2060–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosfeld A, Jahreis K (2012) Characterization of the interaction between the small regulatory peptide SgrT and the EIICBGlc of the glucose‐phosphotransferase system of E. coli K‐12. Metabolites 2: 756–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Mommer MS, Sourjik V (2010) Mobility of cytoplasmic, membrane, and DNA‐binding proteins in Escherichia coli . Biophys J 98: 552–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leake MC, Greene NP, Godun RM, Granjon T, Buchanan G, Chen S, Berry RM, Palmer T, Berks BC (2008) Variable stoichiometry of the TatA component of the twin‐arginine protein transport system observed by in vivo single‐molecule imaging. Proc Natl Acad Sci USA 105: 15376–15381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AG (2004) How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta 1666: 62–87 [DOI] [PubMed] [Google Scholar]
- Lenaz G (1987) Lipid fluidity and membrane protein dynamics. Biosci Rep 7: 823–837 [DOI] [PubMed] [Google Scholar]
- Lentz BR (1993) Use of fluorescent probes to monitor molecular order and motions within liposome bilayers. Chem Phys Lipids 64: 99–116 [DOI] [PubMed] [Google Scholar]
- Letellier L, Moudden H, Shechter E (1977) Lipid and protein segregation in Escherichia coli membrane: Morphological and structural study of different cytoplasmic membrane fractions. Proc Natl Acad Sci USA 74: 452–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Malmberg E, Symons JL, Fan YY, Chapkin RS, Ernst R, Levental I (2020) Lipidomic and biophysical homeostasis of mammalian membranes counteracts dietary lipid perturbations to maintain cellular fitness. Nat Commun 11: 1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RNAH, Sykes BD, McElhaney RN (1987) Thermotropic phase behavior of model membranes composed of phosphatidylcholines containing dl‐methyl anteisobranched fatty acids. 1. Differential scanning calorimetric and 31P NMR spectroscopic studies. Biochemistry 26: 4036–4044 [DOI] [PubMed] [Google Scholar]
- Lingwood D, Simons K (2010) Lipid rafts as a membrane‐organizing principle. Science 327: 46–50 [DOI] [PubMed] [Google Scholar]
- Lorent JH, Diaz‐Rohrer B, Lin X, Spring K, Gorfe AA, Levental KR, Levental I (2017) Structural determinants and functional consequences of protein affinity for membrane rafts. Nat Commun 8: 1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucena D, Mauri M, Schmidt F, Eckhardt B, Graumann PL (2018) Microdomain formation is a general property of bacterial membrane proteins and induces heterogeneity of diffusion patterns. BMC Biol 16: 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansilla MC, Cybulski LE, Albanesi D, de Mendoza D (2004) Control of membrane lipid fluidity by molecular thermosensors. J Bacteriol 186: 6681–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr AG, Ingraham JL (1962) Effect of temperature on the composition of fatty acids in Escherichia coli . J Bacteriol 84: 1260–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morein S, Andersson AS, Rilfors A, Lindblom G (1996) Wild‐type Escherichia coli regulate the membrane lipid composition in a “window” between gel and non‐lamellar structures. J Biol Chem 271: 6801–6809 [DOI] [PubMed] [Google Scholar]
- Mostofian B, Zhuang T, Cheng X, Nickels JD (2019) Branched‐chain fatty acid content modulates structure, fluidity, and phase in model microbial cell membranes. J Phys Chem B 123: 5814–5821 [DOI] [PubMed] [Google Scholar]
- Neidhardt FC, Ingraham JL, Schaechter M (1990) Physiology of the bacterial cell. A molecular approach. Sunderland, MA: Sinauer Associates; [Google Scholar]
- Nguyen C, Haushalter RW, Lee DJ, Markwick PRL, Bruegger J, Caldara‐Festin G, Finzel K, Jackson DR, Ishikawa F, O’Dowd B et al (2014) Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature 505: 427–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickels JD, Chatterjee S, Mostofian B, Stanley CB, Ohl M, Zolnierczuk P, Schulz R, Myles DAA, Standaert RF, Elkins JG et al (2017) Bacillus subtilis lipid extract, a branched‐chain fatty acid model membrane. J Phys Chem Lett 8: 4214–4217 [DOI] [PubMed] [Google Scholar]
- Nickels JD, Hogg J, Cordner D, Katsaras J (2019) Lipid rafts in bacteria: structure and function. In Health consequences of microbial interactions with hydrocarbons, oil, and lipids, Goldfine H (ed.), pp 1–30. Cham, Switzerland: Springer Nature; [Google Scholar]
- Nicolson GL (2014) The fluid‐mosaic model of membrane structure: Still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim Biophys Acta 1838: 1451–1466 [DOI] [PubMed] [Google Scholar]
- Oswald F, Varadarajan A, Lill H, Peterman EJG, Bollen YEM (2016) MreB‐dependent organization of the E. coli cytoplasmic membrane controls membrane protein diffusion. Biophys J 110: 1139–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papahadjopoulos D, Jacobson K, Nir S, Isac T (1973) Phase transitions in phospholipid vesicles. Fluorescence polarization and permeability measurements concerning the effect of temperature and cholesterol. Biochim Biophys Acta 311: 330–348 [DOI] [PubMed] [Google Scholar]
- Parasassi T, De Stasio G, d'Ubaldo A, Gratton E (1990) Phase fluctuation in phospholipid membranes revealed by Laurdan fluorescence. Biophys J 57: 1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JB, Rock CO (2013) Bacterial lipids: metabolism and membrane homeostasis. Prog Lipid Res 52: 249–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picas L, Carreterro‐Genevrier A, Montero MT, Vázquez‐Ibar JL, Seantier B, Milhiet PE, Hernández‐Borrell J (2010) Preferential insertion of lactose permease in phospholipid domains: AFM observations. Biochim Biophys Acta 1798: 1014–1019 [DOI] [PubMed] [Google Scholar]
- Pilizota T, Shaevitz JW (2012) Fast, multiphase volume adaptation to hyperosmotic shock by Escherichia coli . PLoS One 7: e35205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadurai S, Holt A, Krasnikov V, van den Bogaart G, Killian JA, Poolman B (2009) Lateral diffusion of membrane proteins. J Am Chem Soc 131: 12650–12656 [DOI] [PubMed] [Google Scholar]
- Razin S (1967) The cell membrane of mycoplasma. Ann NY Acad Sci 143: 115–129 [DOI] [PubMed] [Google Scholar]
- Renz A, Renz M, Klütsch D, Deckers‐Hebestreit G, Börsch M (2015) 3D‐localisation microscopy and tracking of FOF1‐ATP synthases in living bacteria. Proc SPIE 9331: 93310D [Google Scholar]
- Richter D, Moraga I, Winkelmann H, Birkholz O, Wilmes S, Schulte M, Kraich M, Kenneweg H, Beutel O, Selenschik P et al (2017) Ligand‐induced type II interleukin‐4 receptor dimers are sustained by rapid re‐association within plasma membrane microcompartments. Nat Commun 8: 15976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridder ANJA, Spelbrink REJ, Demmers JAA, Rijkers DTS, Liskamp RMJ, Brunner J, Heck AJR, de Kruijff B, Killian JA (2004) Photo‐crosslinking analysis of preferential interactions between a transmembrane peptide and matching lipids. Biochemistry 43: 4482–4489 [DOI] [PubMed] [Google Scholar]
- Rock CO, Tsay JT, Heath R, Jackowski S (1996) Increased unsaturated fatty acid production associated with a suppressor of the fabA6(Ts) mutation in Escherichia coli . J Bacteriol 178: 5382–5387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol M, Thomas P, Grignon C (1982) Proton permeability of liposomes from natural phospholipid mixtures. Biochim Biophys Acta 684: 195–199 [DOI] [PubMed] [Google Scholar]
- Roth BL, Poot M, Yue ST, Millard PJ (1997) Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl Environ Microbiol 63: 2421–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner DZ, Pan Q, Losick RM (2002) Evidence that subcellular localization of a bacterial membrane protein is achieved by diffusion and capture. Proc Natl Acad Sci USA 99: 8701–8706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáenz JP, Grosser D, Bradley AS, Lagny TJ, Lavrynenko O, Broda M, Simons K (2015) Hopanoids as functional analogues of cholesterol in bacterial membranes. Proc Natl Acad Sci USA 112: 11971–11976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasser M (1990) Identification of bacteria through fatty acid analysis. In Methods in phyto‐bacteriology 595, Klement Z, Rudolph K, Sands DC (eds.), Hungary: Akadémiai Kiadó Budapest; [Google Scholar]
- Schäfer LV, de Jong DH, Holt A, Rzepiela AJ, de Vries AH, Poolman B, Killian JA, Marrink SJ (2011) Lipid packing drives the segregation of transmembrane helices into disordered lipid domains in model membranes. Proc Natl Acad Sci USA 108: 1343–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger H, von Jagow G (1987) Tricine‐sodium dodecyl sulfate polyacrylamide gel electrophoresis for the separation of proteins in the range of 1 to 100 kDa. Anal Biochem 166: 368–379 Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PubMed] [Google Scholar]
- Scheinpflug K, Krylova O, Strahl H (2017a) Measurement of cell membrane fluidity by Laurdan GP: fluorescence spectroscopy and microscopy. Methods Mol Biol 1520: 159–174 [DOI] [PubMed] [Google Scholar]
- Scheinpflug K, Wenzel M, Krylova O, Bandow JE, Dathe M, Strahl H (2017b) Antimicrobial peptide cWFW kills by combining lipid phase separation with autolysis. Sci Rep 7: 44332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid F (2017) Physical mechanisms of micro‐ and nanodomain formation in multicomponent lipid membranes. Biochim Biophys Acta 1859: 509–528 [DOI] [PubMed] [Google Scholar]
- Sharp MD, Pogliano K (1999) An in vivo membrane fusion assay implicates SpoIIIE in the final stages of engulfment during Bacillus subtilis sporulation. Proc Natl Acad Sci USA 96: 14553–14558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw TR, Ghosh S, Veatch SL (2021) Critical phenomena in plasma membrane organization and function. Annu Rev Phys Chem 72: 51–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelby SA, Castello‐Serrano I, Wisser KC, Levental I, Veatch SL (2021) Membrane phase separation drives organization at B cell receptor clusters. bioRxiv 10.1101/2021.05.12.443834 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Shen Y, Zhao Z, Zhang L, Shi L, Shahriar S, Chan RB, Di Paolo G, Min W (2017) Metabolic activity induces membrane phase separation in endoplasmic reticulum. Proc Natl Acad Sci USA 114: 13394–13399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinensky M (1974) Homeoviscous adaptation ‐ a homeostatic process that regulates the viscosity of membrane lipids in Escherichia coli . Proc Natl Acad Sci USA 71: 522–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl H, Burmann F, Hamoen LW (2014) The actin homologue MreB organizes the bacterial cell membrane. Nat Commun 5: 3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl H, Errington J (2017) Bacterial membranes: structure, domains, and function. Annu Rev Microbiol 71: 519–538 [DOI] [PubMed] [Google Scholar]
- Strahl H, Hamoen LW (2010) Membrane potential is important for bacterial cell division. Proc Natl Acad Sci USA 107: 12281–12286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez‐Germà C, Montero MT, Ignés‐Mullol J, Hernández‐Borrell J, Domènech Ò (2011) Acyl chain differences in phosphatidylethanolamine determine domain formation and LacY distribution in biomimetic model membranes. J Phys Chem B 115: 12778–12784 [DOI] [PubMed] [Google Scholar]
- Suutari M, Laakso S (1992) Unsaturated and branched chain‐fatty acids in temperature adaptation of Bacillus subtilis and Bacillus megaterium . Biochim Biophys Acta 1126: 119–124 [DOI] [PubMed] [Google Scholar]
- Szekely P, Dvir T, Asor R, Resh R, Steiner A, Szekely O, Ginsburg A, Mosenkis J, Guralnick V, Dan Y et al (2011) Effect of temperature on the structure of charged membranes. J Phys Chem B 115: 14501–14506 [DOI] [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Court DL (2007) E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol Chapter 1, Unit 1 17 [DOI] [PubMed] [Google Scholar]
- Torres MJ, Kew KA, Ryan TE, Pennington ER, Lin C‐T, Buddo KA, Fix AM, Smith CA, Gilliam LA, Karvinen S et al (2018) 17β‐Estradiol directly lowers mitochondrial membrane microviscosity and improves bioenergetic function in skeletal muscle. Cell Metab 27: 167–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulmay A, Prinz WA (2013) Direct imaging reveals stable, micrometer‐scale lipid domains that segregate proteins in live cells. J Cell Biol 202: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, Gross CA, Vollmer W (2012) From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10: 123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine DL (2007) Adaptations to energy stress dictate the ecology and evolution of the Archaea. Nat Rev Microbiol 5: 316–323 [DOI] [PubMed] [Google Scholar]
- Veatch SL (2007) From small fluctuations to large‐scale phase separation: Lateral organization in model membranes containing cholesterol. Semin Cell Dev Biol 18: 573–582 [DOI] [PubMed] [Google Scholar]
- van de Vossenberg JLCM, Driessen AJM, da Costa MS, Konings WN (1999) Homeostasis of the membrane proton permeability in Bacillus subtilis grown at different temperatures. Biochim Biophys Acta 1419: 97–104 [DOI] [PubMed] [Google Scholar]
- Wenzel M, Vischer NOE, Strahl H, Hamoen LW (2018) Assessing membrane fluidity and visualizing fluid membrane domains in bacteria using fluorescent membrane dyes. Bio‐protocol 8: e3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willecke K, Pardee AB (1971) Fatty acid‐requiring mutant of Bacillus subtilis defective in branched chain α‐keto acid dehydrogenase. J Biol Chem 246: 5264–5272 [PubMed] [Google Scholar]
- te Winkel JD, Gray DA, Seistrup KH, Hamoen LW, Strahl H (2016) Analysis of antimicrobial‐triggered membrane depolarisation using voltage sensitive dyes. Front Cell Dev Biol 4: 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Zhang YM, Rock CO (2009) Transcriptional regulation of membrane lipid homeostasis in Escherichia coli . J Biol Chem 284: 34880–34888 [DOI] [PMC free article] [PubMed] [Google Scholar]