

Summary Paragraph
Apicomplexa are unicellular eukaryotes and obligate intracellular parasites, including Plasmodium, the causative agent of malaria and Toxoplasma, one of the most widespread zoonotic pathogens. They possess specialized secretory organelles, the rhoptries, that undergo regulated exocytosis during invasion1. Rhoptry proteins are injected directly into the host cell to support invasion and subversion of host immune function2. The mechanism by which they are discharged is unclear and appears distinct from those in bacteria, yeast, animals or plants. Here we show that rhoptry secretion in Apicomplexa shares structural and genetic elements with the exocytic machinery of ciliates, their free-living relatives. Rhoptry exocytosis depends on intramembranous particles in the shape of a rosette embedded into the plasma membrane of the parasite apex. Formation of this rosette requires multiple Non-discharge (Nd) proteins conserved and restricted to ciliates, dinoflagellates, and Apicomplexa, that together constitute the superphylum Alveolata. We identified Nd6 in association with an apical vesicle at the site of exocytosis. Sandwiched between the rosette and the tip of the rhoptry, this vesicle appears as a central element of the rhoptry secretion machine. Our results describe a conserved secretion system that was adapted to provide defense for free-living unicellular eukaryotes and host cell injection in intracellular parasites.
Apicomplexan parasites are invasive and defined by the presence of an apical complex used to recognize and gain entry into host cells. It includes two secretory organelles: micronemes and rhoptries3. Microneme proteins are secreted to the parasite surface and mediate motility, host cell recognition and invasion4. Rhoptry proteins are injected directly into the host cell2, where they anchor the machinery propelling the parasite into the host cell5, facilitate nutrient import6–8, interfere with the immune response, and modulate gene expression to promote parasite infection9. Rhoptry secretion requires a trigger in the form of microneme proteins binding to host cell receptors9,10. Importantly, rhoptry proteins not only cross the plasma membrane of the parasite (exocytosis) but also that of the host1,11. Rhoptry exocytosis factors identified to date10,12,13 are mostly specific to Apicomplexa, suggesting that these cells depend on unique lineage-restricted secretory mechanisms. Consistent with this, no eukaryotic SNAREs, the main drivers for fusing vesicles to target membrane in eukaryotic system, have so far been associated with rhoptry exocytosis. Moreover, Apicomplexa lack genes encoding prokaryotic secretion systems14. Thus, how rhoptry effectors are delivered into the host cytoplasm remains unclear.
To explore the mechanisms of rhoptry secretion, we searched for examples of regulated secretion in organisms phylogenetically closely related to Apicomplexa. Apicomplexa are grouped together with ciliates and dinoflagellates, in the superphylum Alveolata15. All Alveolata bear alveolar sacs beneath the plasma membrane, which give the superphylum its name, and contain elaborate membrane-bounded secretory organelles whose wide range of different morphologies and functions may belie a shared evolutionary origin15. Known as trichocysts in the ciliate Paramecium, they function in defense, docking at the plasma membrane and discharging in response to predation16. Exocytic membrane fusion occurs at plasma membrane sites consisting of rosettes of 8–9 intramembranous particles (IMPs)17,18. Previous electron microscopy (EM) studies have described similar rosettes at the apex of several apicomplexan parasites19–22. Though suggested to be involved in exocytic fusion19, the function of the IMP rosette has never been experimentally addressed in Apicomplexa. Analysis of Paramecium tetraurelia mutants defective in both trichocyst exocytosis and rosette assembly23,24 led to identification of nd (non-discharge) genes25–28 (Supplementary Table 1). To determine if similar factors could be involved in exocytosis of secretory organelles in Apicomplexa, we first searched for Nd homologs in the tree of life. Genome mining for nd genes and phylogenetic analyses revealed that nd6 and nd9 are found in ciliates, but also in dinoflagellates, chromerids and apicomplexans (Extended data Fig. 1 and Extended Data figure 1). We also found Nd9 homolog in Perkinsids, a sister group of Dinoflagellata. Altogether, this analysis suggested a conserved evolution and common function of nd genes across Alveolata.
To define the parasite localization of Nd proteins, we tagged nd6 (TGGT1_248640) and nd9 (TGGT1_249730) at the endogenous loci in Toxoplasma gondii, an experimentally tractable apicomplexan (Extended data Fig. 2). Both proteins displayed a punctate signal throughout the cytoplasm, but in addition TgNd6 accumulated at the apical tip of the parasite (Fig. 1a, b). More precise localization by immuno-Electron Microscopy (EM) localized TgNd6 at the site of rhoptry exocytosis, in association with the parasite plasma membrane and an underlying membranous spheroid known as the “apical vesicle” (Fig. 1c). Previously observed by EM in Toxoplasma20,29, the composition and function of apical vesicles remain unknown, although links to rhoptries have been suggested21,29.
Fig. 1 |. Rhoptry secretion is dependent on rosette formation.

a, Immunofluorescence assay (IFA) of control untagged line and endogenously HA3-tagged TgNd6 and HA3-tagged TgNd9 tachyzoites. The green arrow points to TgNd6-HA3 apical puncta. ARO is a marker for rhoptries. b, Super-resolution microscopy of the apical dot of TgNd6. Schematic of apical end of a tachyzoite. Maximum intensity projections of z-stacks of TgNd6-HA3 parasites transiently expressing RNG1_GFP (top) and of TgNd6-HA3 parasites expressing centrin 2-Ty3 (bottom). The protein RNG1_GFP marks the apical polar ring (APR) and CEN2_GFP marks pre-conoidal rings (PCR). Higher magnifications show that TgNd6-HA3 (red arrow) localizes above the apical polar ring (green arrow) and co-localizes partially with CEN2. DIC: differential interference contrast. c, Immunogold labelling of TgNd6-HA3. Right panel shows TgNd6-HA3 on the apical vesicle. Insert panel: higher magnification of the apical vesicle. Micronemes (m) and rhoptries (Rh) are visible in transverse section of the conoid (Co). Bar is 200 nm. d, Quantification of invasion after depletion of TgNd6 and TgNd9. Mean ± SD of n=3 independent experiments. e, Immunoblot showing microneme secretion (arrow=processed/secreted TgAMA1) in Tgnd6-iKD ± IAA 24h (top) and Tgnd9-iKD ± ATC 72h (bottom). P = pellet, Sup: supernatant, Ind: propanolol-induced secretion. GRA3, loading control. f, Rhoptry secretion assay by Secreted Cre, epitope-tagged (SeCrEt). Rhoptry secretion quantification of Tgnd6i-KD (left) ± IAA and Tgnd9-iKD (right) ± ATC. Mean ± SD of n=3 independent experiments. f, Left: Freeze-fracture electron microscopy of a T. gondii tachyzoite (P face) showing a rosette of intramembranous particles (white arrow). Middle: Higher magnification of the right panel. The white arrows point to the eight IMPs of the rosette. Right: Quantification of rosettes of IMPs in Tgnd9-iKD ± ATc 72h and Tgnd6-iKD ± IAA 24h using freeze fracture. (d, f) Unpaired two tail student’s t test: **** p-value < 0.0001, *** p-value < 0.001, ** p-value < 0.01, * p-value < 0.05.
Because TgNd6 and TgNd9 were predicted to be fitness-conferring genes30, in order to investigate their function we generated inducible knock-down mutants using an auxin-inducible degron for TgNd631 and tetracycline-induced repression for TgNd932,33 (Extended data Fig. 2). Parasites conditionally depleted of TgNd6 or TgNd9 showed reduced plaque formation on fibroblast monolayers, indicating the inability of both mutants to efficiently complete the lytic cycle (Extended Data Fig. 3a). Tgnd6-iKD and Tgnd9-iKD mutants showed no detectible defects in their intracellular replication, egress, conoid extrusion, motility, or host cell attachment (Extended Data Fig. 3b–g. In contrast, invasion was severely impaired for both mutants (Fig. 1d).
To understand the mechanistic basis of this invasion defect, we evaluated microneme and rhoptry secretion in the Tgnd6-iKD and Tgnd9-iKD mutants. Release of the micronemal protein AMA1 into the supernatant was unimpeded by the absence of TgNd6 or TgNd9 (Fig. 1e). In contrast, rhoptry secretion was significantly impaired by loss of TgNd6 or TgNd9 as revealed by quantifying the release of rhoptry protein ROP1 into host cells using immunofluorescence assays (IFA)34 (Extended Data Fig. 3h), or the delivery of Cre recombinase fused to the rhoptry protein toxofilin into the nucleus of a suitable reporter cell35 (Fig. 1f). This was not due to defects in rhoptry biogenesis, as the mutants showed normal organelle formation and apical positioning, as judged by IFA and EM (Extended Data Fig. 4). Analysis by freeze fracture EM of the apex of TgNd6-iKD mutants showed no significant reduction in the presence of the apical rosette. In contrast T. gondii depleted of TgNd9 showed a strong decrease in cells with a rosette at the parasite apex (Fig. 1f). All together, these results link rosette formation with rhoptry secretion and support an evolutionarily conserved mechanism of regulated exocytosis in Alveolata.
To determine whether other Apicomplexa require nd genes for rhoptry function, we analysed Nd9 in Plasmodium falciparum, the causative agent of the deadliest form of malaria. We confirm that P. falciparum intracellular merozoites possess a fusion rosette of 8 IMPs (Fig. 2a). As nd9 is also predicted to be essential in P. falciparum36, we used the rapamycin-inducible dimerizable Cre recombinase (DiCre) system36 to conditionally excise the Pfnd9 gene (PF3D7_1232700) (Extended Data Fig. 6a–c) while also adding a triple HA tag at the C-terminal end. We could not detect the protein by IFA and observed only a faint band at the expected size in late schizonts (Extended Data Fig. 5d), both consistent with the very low transcript level of PfNd9 (Plasmodb.org). DiCre mediated ablation of Pfnd9 resulted in substantial reduction in parasite proliferation (Fig. 2b), which was due to the inability of Pfnd9-iKO mutants to reinvade host cells (Fig. 2b), while their intracellular development and egress were unaffected (Fig. 2b, c; Extended Data Fig. 5e). As in T. gondii, microneme secretion was unaltered in Pfnd9-iKO parasites (Fig. 2d) but rhoptry secretion was affected (Fig. 2e).
Fig. 2 |. PfNd9 is essential for rhoptry secretion in P. falciparum.
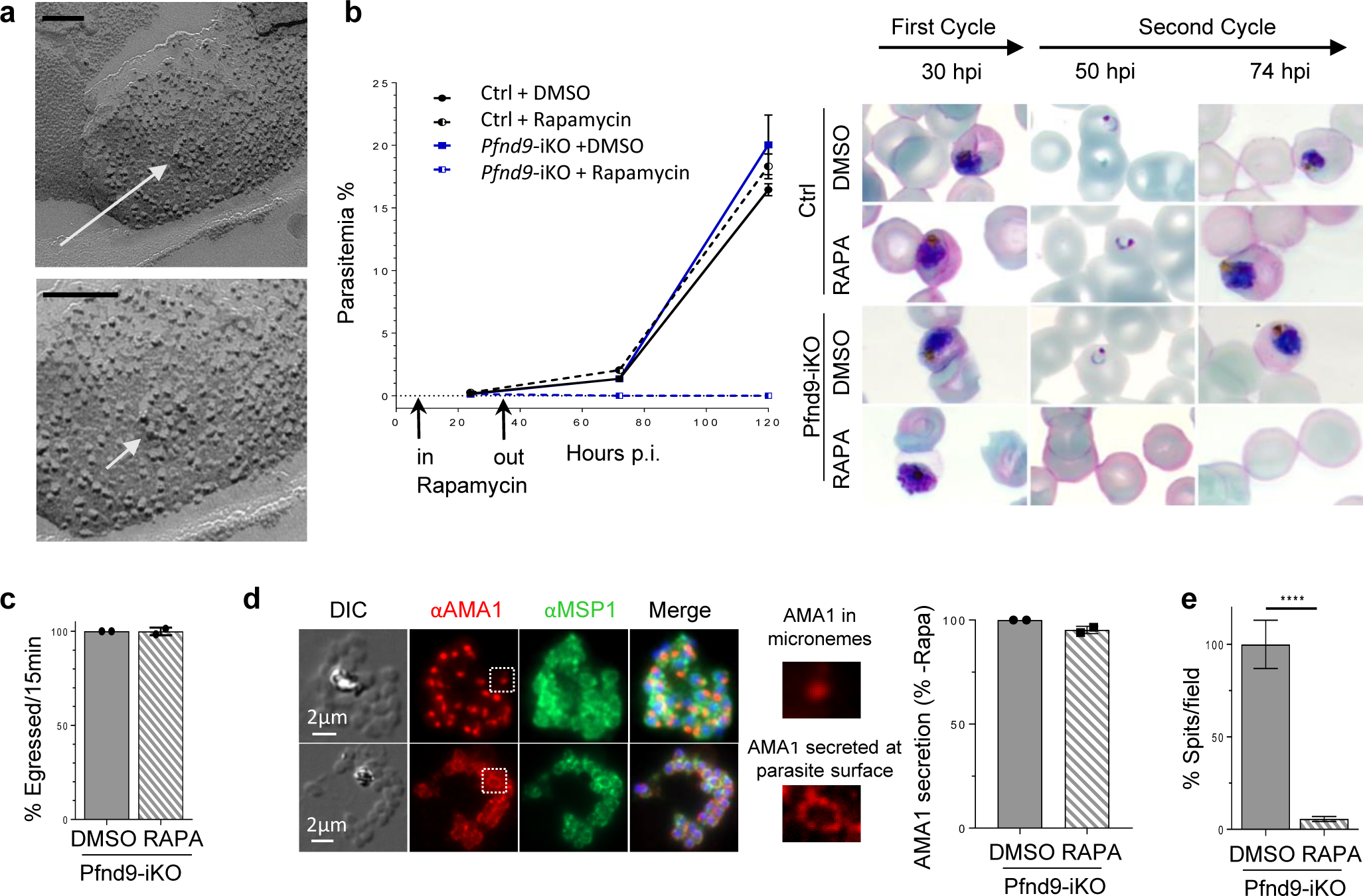
a, Freeze-fracture electron microscopy of a P. falciparum merozoite (P face) showing a rosette of intramembranous particles (white arrow). Higher magnification at the bottom. Bar is 100 nm. b, Growth curves (parasitaemias) of p230p DiCre (Ctrl) and Pfnd9-iKO mutant ± rapamycin shows that PfNd9-depleted parasites have a growth defect. On the right: Giemsa staining of the growth experiment illustrating development and reinvasion of p230p DiCre (Ctrl) and Pfnd9-iKO merozoites (along 2 cycles) ± rapamycin treatment. c, Quantification of egress of Pfnd9iKO ± rapamycin schizonts. Data collected from 8 movies of Pfnd9-iKO ± rapamycin. d, Left: IFA illustrating AMA1 protein stored in micronemes (top) or secreted and translocated at the surface of the parasite prior to egress (bottom). MSP1: surface marker. Right: Proportion of infected cells (± rapamycin) exhibiting AMA1 secretion.. e, Quantification of rhoptry secretion events in Pfnd9-iKO ± rapamycin-treated schizonts using anti-PfRAP2 antibodies to visualise rhoptry secretion events (‘spits’ of RAP2 export into the RBC).
To gain a more complete understanding of the molecular composition of the rhoptry secretion machine, we searched for Nd interacting proteins in T. gondii. Nd9 displays two Armadillo repeats while Nd6 shows homology with GDP/GTP exchange factors (GEF) known to activate GTPases (Fig. 3a). We used Nd9 for immunoprecipitation (IP) experiments as we found Nd6 to be largely insoluble (Extended data Fig. 2g). Mass spectrometry analysis revealed robust interaction of TgNd9 with TgNd6 and with TgFER2 (TGGT1_260470), a member of the ferlin calcium sensor family, known to be essential for Toxoplasma rhoptry secretion12 (Fig. 3a and Supplementary Table 2). The Nd9 IP also enriched TGGT1_222660, a protein harboring Armadillo repeats and Leucine Rich Repeats, named hereafter TgNdP1 (Nd Partner 1) and TGGT1_316730 (TgNdP2: Nd Partner 2), a protein with a C2 calcium lipid binding domain (Fig. 3a). Both genes are broadly shared among Alveolata (Extended Data Fig. 1c, d and Supplementary Data 1) and predicted to be fitness score-conferring in Toxoplasma30 and Plasmodium37. In contrast, Nd9-IP interactor TGGT1_277840, a GTPase, is restricted to apicomplexans and dinoflagellates and TGGT1_253570 is only found in the apicomplexan subgroup Coccidia. Importantly, all these proteins are also recovered when using reverse IP with tagged-TgNdP1 protein (Supplementary Table 3).
Fig. 3 |. Nd6 and Nd9 are part of an Alveolate complex essential for organellar secretion in T. gondii (Apicomplexa) and T. thermophila (Ciliate).
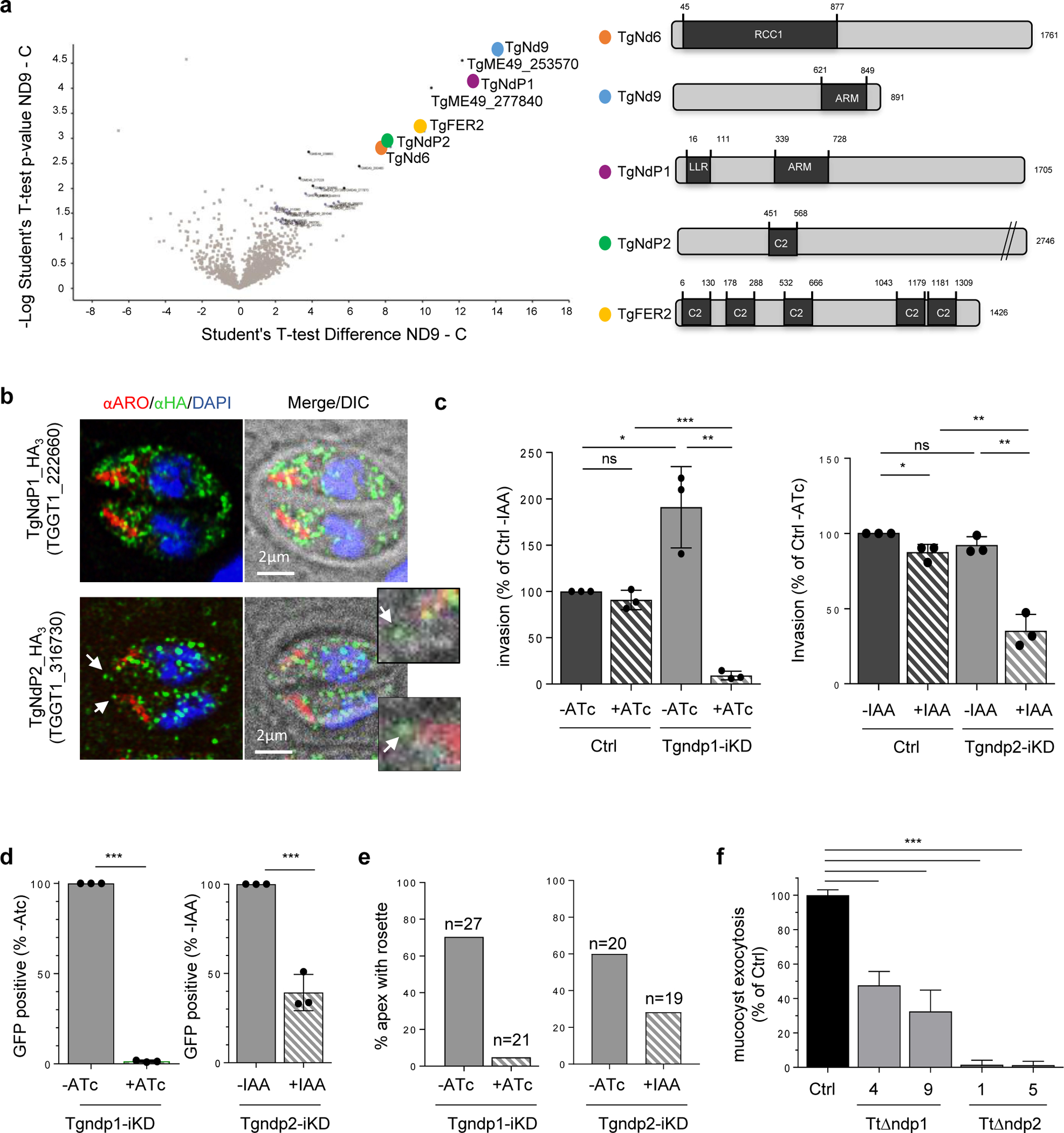
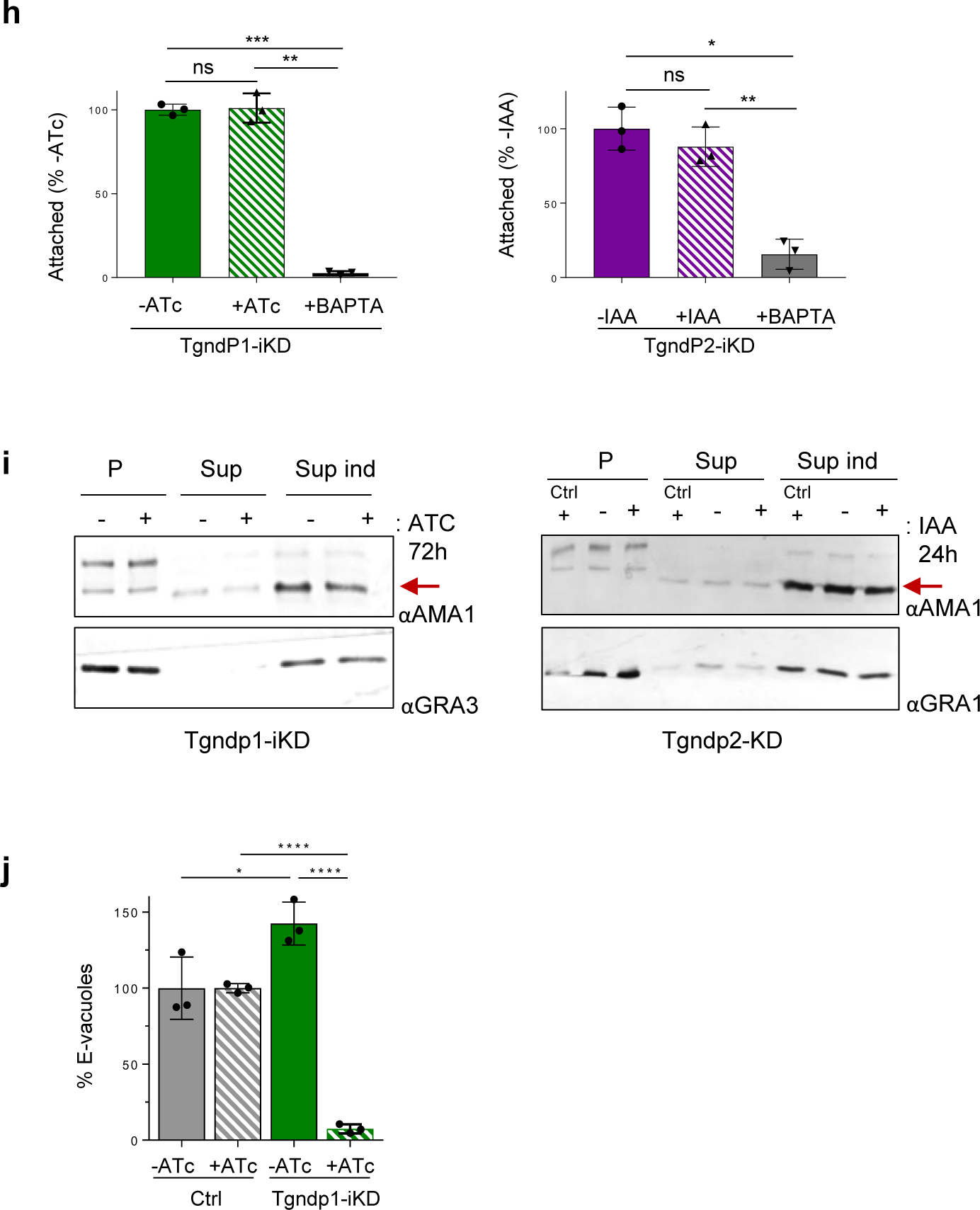
a, Mass spectrometry analysis of immuno-isolated Nd9-HA. Left: Volcano Plot of proteins differentially enriched in Nd9 vs control IP. This plot presents the fold change (Difference) and significance (-Log p) obtained from a t-test of three independent IPs using LFQ intensity values. Right: Schematic representation of TgNds proteins using SUPERFAMILY58. RCC1: regulator of chromosome condensation 1-like domains (RLDs), a versatile domain that performs many different functions, including guanine nucleotide exchange on small GTP-binding proteins59. LRR: Leucine Rich Repeat domain. ARM: Armadillo Repeat domain. C2: lipid-calcium binding domain. b, Immunofluorescence (IF) of endogenously HA3-tagged TgNdP1 and TgNdP2 tachyzoites. The white arrow points to TgNdP1-HA3 apical dots of the two parasites, which are magnified on the right. ARO: rhoptry marker. c, Quantification of invasion after depletion of TgNdP1 (left) and TgNdP2 (right). Mean ± SD of n=3 independent experiments. d, Rhoptry secretion quantification of TgndP1i-KD (left) ± ATc and TgndP2-iKD (right) ± IAA. Mean ± SD of n=3 independent experiments. e, Quantification of rosettes of IMPs in TgndP1-iKD ± ATC 72h and in TgndP2-iKD ± IAA 24h. f, Quantification of mucocyst exocytosis by dibucaine assay. Data collected from three experiments. (c, e) Unpaired two tail student’s t test: **** p-value < 0.0001, *** p-value < 0.001, ** p-value < 0.01, * p-value < 0.05.
To learn more about the new conserved partners of TgNd9, we next generated T. gondii lines in which TgNdP1 or TgNdP2 was tagged by an epitope and could be ablated conditionally (Extended Data Fig. 6 and 7). Both proteins appear as punctate cytoplasmic staining (Fig. 3b) but TgNdP2—like TgNd6—also appears as a dot at the apical tip of the parasite, although consistently with lower intensity. Depletion of TgNdP1 or TgNdP2 resulted in a profound growth defect (Extended Data Fig. 7a) that we linked to impairment of host cell invasion and rhoptry secretion (Fig. 3c, d and Extended Data Fig. 7). Again, loss of rhoptry secretion went hand-in-hand with loss of the rosette in Tgndp1-iKD and Tgndp2-iKD parasites (Fig. 3e).
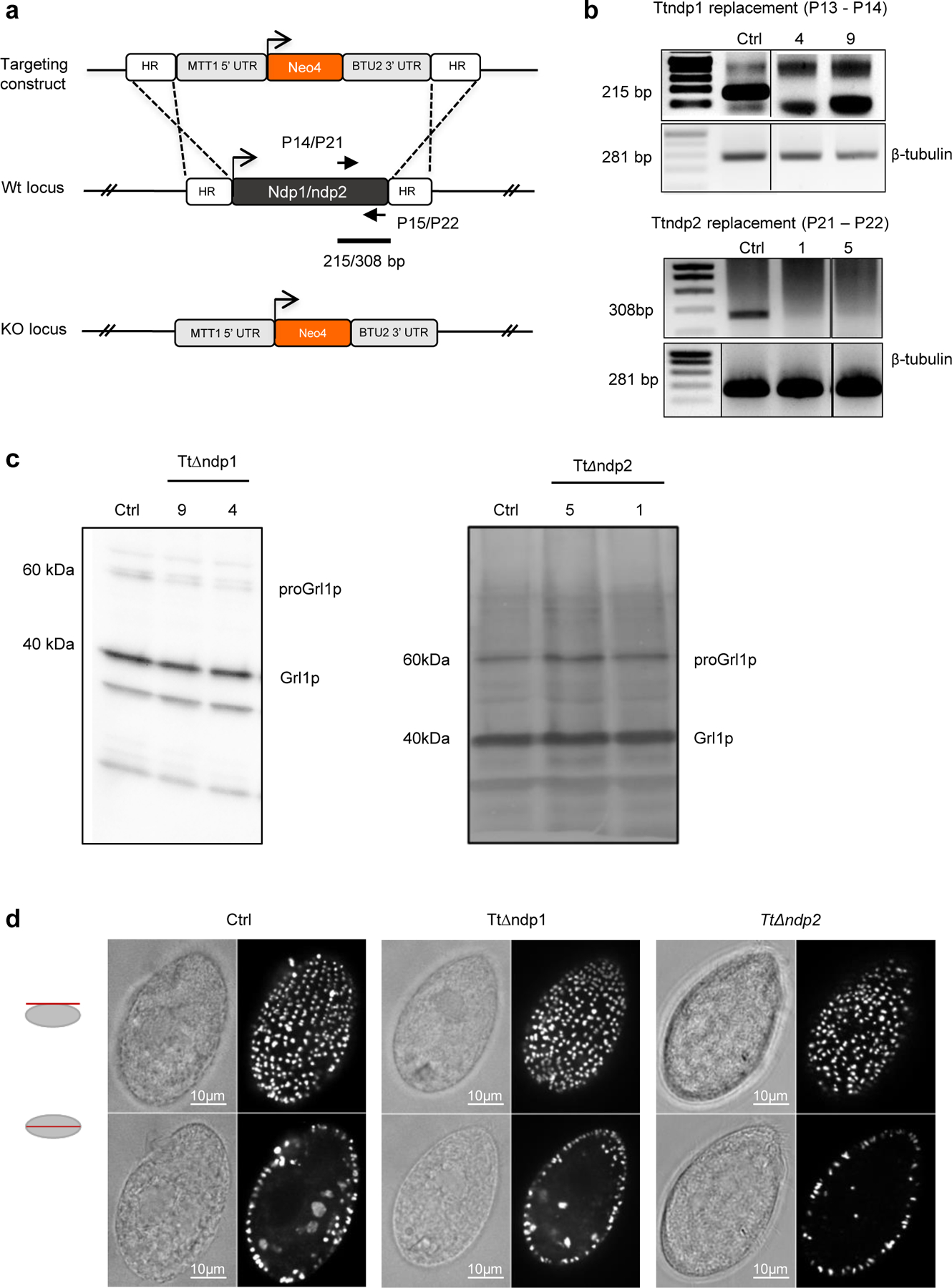
To further validate our conservation data and broaden our discoveries back to Ciliate, we generated knock-outs in Tetrahymena thermophila for orthologues of both NdP1 (TTHERM_01287970, TtnΔndp1) and NdP2 (TTHERM_00498010; TtnΔndp2) (Extended Data Fig. 7a, b). The homologous organelles to Paramecium trichocysts in Tetrahymena are called mucocysts, and they are non-essential for laboratory growth. We found that all TtnΔndp1 and TtnΔndp2 clones were defective in mucocyst secretion, which was triggered by exposure to dibucaine38 (Fig. 3f). We further showed that the impairment of exocystosis was not due to defects in mucocyst biogenesis, given that mucocyst maturation (as measured by processing of mucocyst pro-proteins) and trafficking (monitored by IFA) remained unaltered (Extended Data Fig. 8c, d). Taken together, we identified a complex of proteins essential for organellar exocytosis and rosette assembly, conserved across Alveolata.
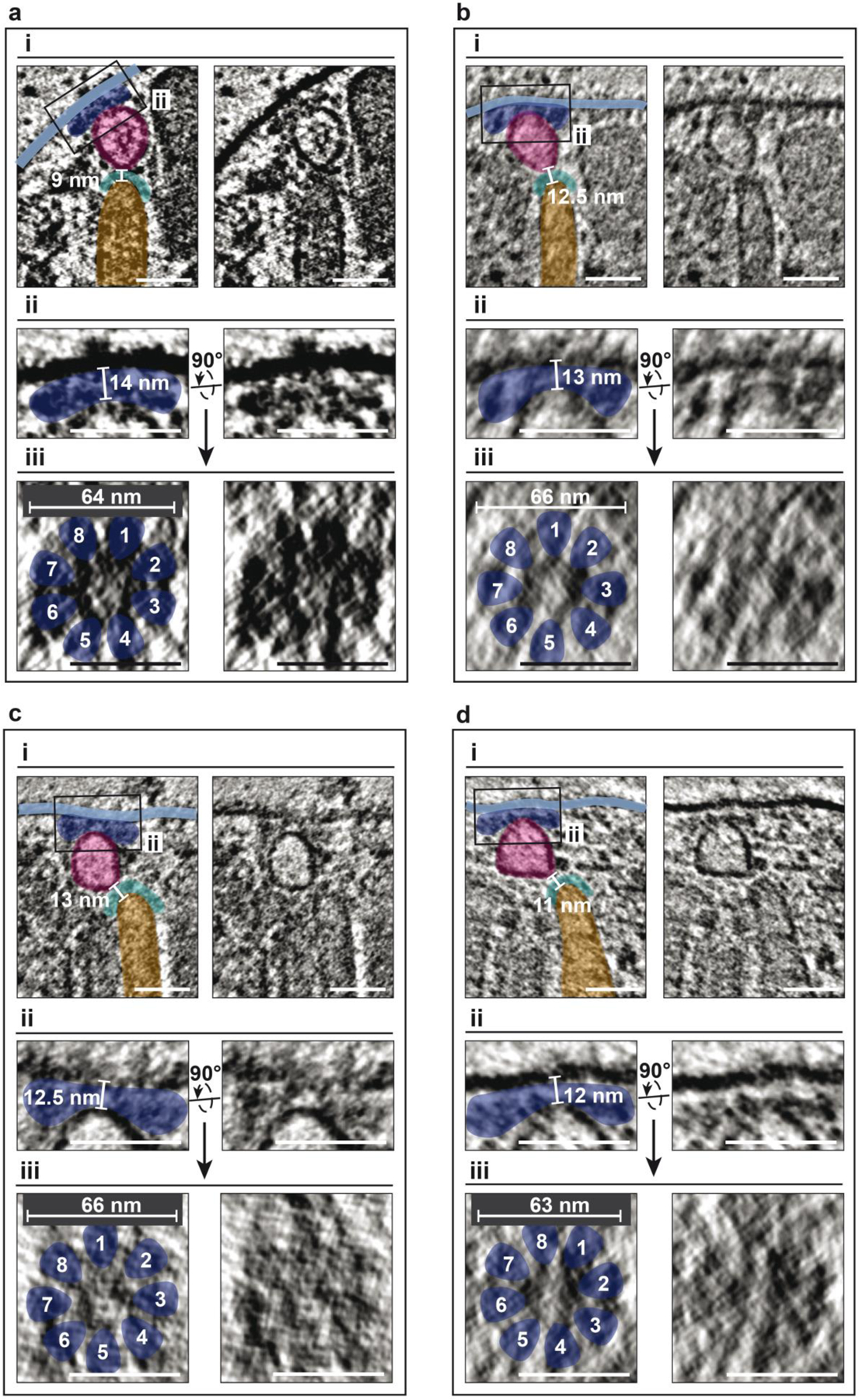
The exact position of the rosette relative to the apical tip of the rhoptry and the enigmatic apical vesicle remained elusive, since the freeze-fracture technique—used to image the rosette on the membrane—does not capture the internal structures at the same time. To understand how the rosette is connected to the rhoptry tip, we imaged the Toxoplasma apex by cryo-electron tomography (cryo-ET)—a technique combining the advantages of 3D imaging with molecular resolution, to reveal ultrastructure in situ in its native biological context. We were able to define three linked elements (Fig. 4a and b, Extended Fig. 9): 1) the rosette (dark blue), 2) the apical vesicle (magenta), and 3) the apical tip of the rhoptry organelle (cyan). Electron density showed an 8-fold symmetry around a central axis (Fig. 4c and d), that extended under the parasite plasma membrane (light blue) and interacted with the apical vesicle (Fig. 4c, top). The rosette is tightly sandwiched between the apical vesicle and the plasma membrane and extensively interacts with both membranes. The apical vesicle in turn sits over the rhoptry tip (Fig. 4a).The contiguity of all elements from the rhoptry tip to the plasma membrane supports the idea that the rosette and the apical vesicle are fundamental to the rhoptry secretion machine. Interestingly, the connection between apical vesicle and rosette is sometimes observed even in the absence of a docked rhoptry (Fig. 4e) suggesting that they assemble independently of rhoptry docking. To test this hypothesis, we took advantage of the Toxoplasma ARO mutant, in which rhoptries fail to dock and are dispersed in the cytoplasm39. In these mutant cells, both the rosette and the apical vesicle were present at the apex of the parasite (Fig 4e, f). Altogether, these results positioned the rosette and apical vesicle at the heart of the exocytic machinery in Apicomplexa and revealed that rhoptries do not make contact and fuse directly with the plasma membrane.
Fig. 4 |. The rhoptry secretion machine includes an apical vesicle tightly connected with the rosette.

a, A slice through a tomogram showing a side view of the apical complex – conoid (brown), micronemes (yellow), plasma membrane (PM; light blue) and the rhoptry secretion system consisting of the rosette (dark blue), apical vesicle (AV; magenta), rhoptry (orange) and rhoptry tip density (cyan). Original image (right) is annotated with color overlays (left). b, Left: magnified image of the boxed region in (a) showing the connections between the rhoptry, tip density, AV, rosette and the PM. The rhoptry tip is 9 nm distant from the AV. Right: 3-dimensional (3D) segmentation of the scene above. The PM is rendered transparent on the right to reveal the rosette. c, Magnified image of the boxed region in (b) showing the side view of the rosette. The AV is 14 nm distant from the PM. d) Top view of the rosette from a horizontal tomogram section, perpendicular to the plane in (c), showing 8-fold rotational symmetry and a diameter of ~67 nm. e) AV connected with the PM via a rosette in the absence of a rhoptry. All measurements are made in 3D. Images in (b-e) are low pass filtered to boost contrast. The images in (c) and (d) are from two different cells that were initially oriented perpendicular to each other on the EM grid. f, Quantification of apical rosettes in TgARO-iKD mutants ± ATc 72h. g, Left: Ultrastructure of wild type (RH strain type I) tachyzoites with the apical vesicle positioned beneath the plasma membrane, where the rosette is located, and above the tip of the rhoptry neck. Right: In TgARO-iKD ATc-treated tachyzoites (72h), the apical vesicle is still properly positioned under the rosette, while rhoptries are not engaged in the conoid. co: conoid; m: micronemes; Rh: rhoptry; APR: apical polar ring; PCR: pre-conoidal apical rings; AV: apical vesicle; CM: pair of intraconoidal microtubules. h, Schematic of similarities and differences of the exocytic machinery between ciliates and Apicomplexa. Exocytosis in Alveolata (Ciliate, Dinoflagellate and Apicomplexa) is outlined by the presence of a rosette of particles embedded in the outer membrane, defining the site of exocytosis. In ciliates, organelles discharge can have a defensive or predatory function. Rhoptry exocytosis in Apicomplexa is one of the critical steps of host cell invasion and therefore fundamental for parasitism. In Apicomplexa, but not in ciliates, an apical vesicle (Av) of unknown function is present between the tip of the rhoptry and the plasmalemma, plausibly involved in the injection of rhoptry proteins into the host cell. PM, plasma membrane, PVM, parasitophorous vacuole membrane, AS, alveolar sac, which is homologous to the IMC (inner membrane complex) of Apicomplexa.
Our work breaks ground on the molecular and structural mechanisms for rhoptry exocytosis in Apicomplexa. We define the apical rosette of IMPs as the site for rhoptry exocytosis in Apicomplexa, and we characterize an Alveolata-specific Nd complex necessary for the assembly of the rosette. In Paramecium, physiological studies predicted PtNd6 to be active at the plasma membrane23 while PtNd9 appeared to be a diffusible cytoplasmic component interacting with both trichocyst and plasma membranes40,41. However, both the localization and identity of partners in Ciliata remained unknown. We localized TgNd9 to the cytoplasm and TgNd6 to the site of exocytosis in Toxoplasma. We found both proteins to form a complex that included TgNdP1 and TgNdP2, which we demonstrated to be essential for organelle exocytosis in both Ciliata and Apicomplexa. This complex further includes proteins with C2 domains (TgFER2 and TgNdP2), a homolog of the membrane fusion Ferlin family (FER2)12, a GTPase (TGME49_277840), and a putative GEF protein (TgNd6). These proteins and their domains yield a regulatory model in which calcium signaling and nucleotide binding and hydrolysis constitute key steps that control rosette assembly and/or organelle discharge (More discussion in Supplementary text).
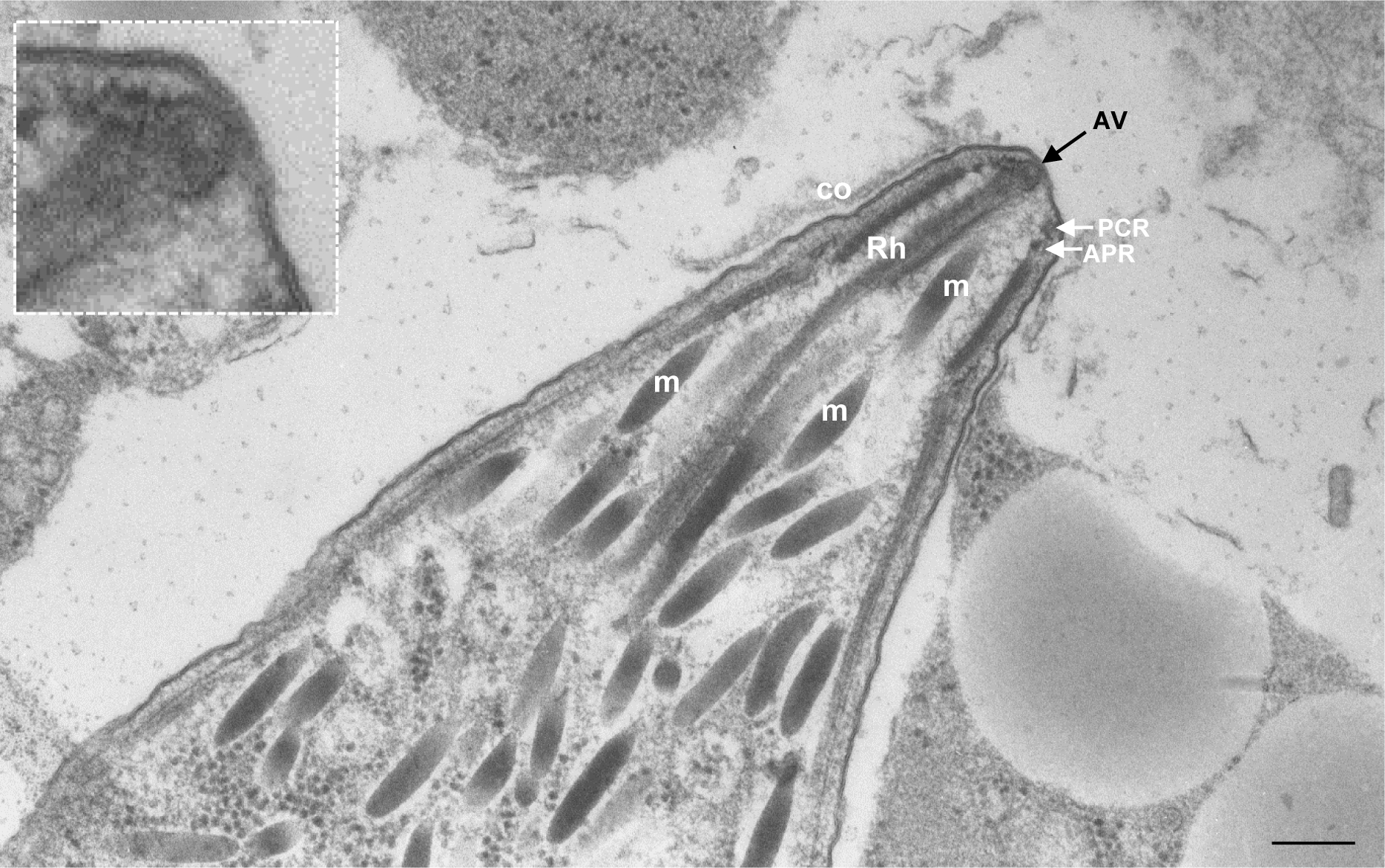
In this study, we also shed new light on the enigmatic apical vesicle. Described in Toxoplasma21,29, it is also visible in early micrographs from other apicomplexan parasites like Eimeria19,42 and Besnoitia43, and we found it in Sarcocystis (Extended Data Fig. 10). We show that the apical vesicle is on one side tightly connected with the plasma membrane via the rosette, and from the other sits on the tip of the rhoptry, precluding a direct link and fusion between the rhoptry and the plasma membrane. In contrast, no comparable apical vesicle is present in Paramecium or Tetrahymena, where trichocysts and mucocysts fuse directly at the plasma membrane17,24. In Paramecium, the rosette is formed at the time of docking of trichocysts to the plasma membrane40. In contrast, we showed that the docking of the rhoptries in Toxoplasma is dispensable for rosette formation (Fig. 4e–g). This suggests that the assembly of the rosette in Toxoplasma might be instead induced by docking of the apical vesicle (Supplementary Text).
The rosette and the set of Nd proteins highlight a common ancestry for the fusion machinery linked to secretory organelles in two groups of protists that diverged hundreds of millions of years ago and which have adopted radically different lifestyles. While Paramecium trichocysts are discharged into the environment to thwart predators, apicomplexan rhoptries translocate their contents directly into the cytoplasm of host organisms to infect and parasitize (Fig. 4h). The differences suggest that the apical vesicle is an adaptation in Apicomplexa for parasitism and cell invasion. Its presence may reflect additional complexity of the secretory machinery in apicomplexans, in which exocytosis must be coupled with injection of rhoptry content through the barrier of a host cell membrane. In support of this hypothesis, a similar vesicle is present at the apex of Perkinsus marinus (previously named Dermocystidium marinum)44. This organism possesses rhoptries and invades and parasitizes the cells of oysters. Phylogenetically, Perkinsus falls between dinoflagellates and apicomplexans and is viewed as a basal taxon and example of early adaptation to parasitism.
The architecture and molecular composition of the rhoptry system that enables the intracellular parasitism of Apicomplexa is now revealed. Future studies combining high resolution structural biology approaches with the genetics and cell biology of Toxoplasma will allow a full understanding of how this fascinating mechanism unfolds and is regulated. Such an understanding may find application in designing strategies against malaria, cryptosporidiosis and toxoplasmosis.
Methods
Parasite Immunofluorescence microscopy
Immunofluorescence assays (IFAs) on intracellular T. gondii parasites were conducted as previously described45. Briefly, cell monolayers were washed and fixed in 4% paraformaldehyde (PFA) in Phosphate Saline Buffer (PBS) for 20 min. After three washes with PBS, cells were permeabilized with 0.1% (v/v) Triton X-100 or with saponin 0.1% (v/v) (for invading parasites) in PBS for 5 min, blocked with 10% fetal bovine serum in PBS (PFBS) for 45 min, incubated with primary Abs diluted in 2% PFBS, washed three times, and then incubated with secondary antibody. The coverslips were mounted onto microscope slides using Immunomount (Calbiochem).
For IFA on P. falciparum, thin blood smears of highly synchronized DMSO and rapamycin-treated PfNd9-iKO schizonts were air-dried, fixed in 4% (w/v) PFA and permeabilized in 0.1% (v/v) Triton X-100. After blocking in 1.5% BSA, samples were probed for 1 h with the suitable antibodies: rabbit anti-PfAMA146 antibody (1:1000); mouse anti-PfMSP146 antibody (1:1000); mouse anti-PfRAP247 antibodies (1:500). The secondary antibodies used were Alexa Fluor 488 and 594-conjugated antibodies against mouse or rabbit IgG (highly cross-adsorbed) both diluted 1:4000 (Molecular Probes). Samples were then counterstained with Hoechst and slides mounted with Vectashield® antifade mounting medium.
Excepted when specified, observations were performed with a Zeiss Axioimager Z1 epifluorescence microscope equipped with a Zeiss Axiocam MRm CCD camera and 100X/1.4 Oil Plan Apochromat objective. Images were processed using Zen Blue 2.3 pro (Zeiss) software. Imaging of TgNd6, TgNd9, TgNdP1 and TgNdP2 co-localization (Fig. 1 and Fig. 3) was performed on a ZEISS confocal LSM880, equipped with an Airyscan detector and a 63X/1.4 Oil Plan Apochromat objective. Zen Black (Zeiss) was used for image airyscan processing. The frontal orthogonal projection shown in Fig. 1a was obtained using the weighted average orthogonal projection method, and the 3D surface reconstruction was made using Zen 3D Visualization module.
Adjustments for brightness and contrast were applied uniformly on the entire images and when required, matching pairs of images were recorded with the same exposure time and processed identically. All optical images were collected at the Montpellier Ressources Imagerie facility of the University of Montpellier (MRI, www.mri.cnrs.fr).
Invasion assays
Toxoplasma Plaque assays, replication assays, and two-color invasion assays were performed as described previously34,48. Briefly, to synchronize invasion, freshly egressed tachyzoites (5 × 106), pre-treated for 48 h ± ATc (Tgnd9-iKD and Tgndp1-iKD) or 24h ± IAA (Tgnd6-iKD and Tgndp2-iKD), were harvested and settled on ice for 20 min on HFF monolayer grown on coverslips in 24-well plates. Invasion was allowed for 5 min at 38 °C and stopped by fixation with 4% PFA in Hank’s Balanced Salt Solution (HBSS) for 20 min at room temperature. To detect extracellular parasites, immuno-detection was performed with the mouse mAb T4 1E5 anti-SAG1 antibody (dilution 1:1000) in 2% FCS/HBSS, without previous permeabilization. After permeabilization with 0.01% saponin for 15 min, a second IFA was performed using rabbit anti-ROP1 antibody (dilution 1:1000) to label the parasitophorous vacuole of intracellular parasites. Extracellular and intracellular parasites were counted by microscopic examination of at least 20 fields per coverslip (n=3 coverslips). Graphs show the mean of three independent invasion assays.
Microneme secretion assay
Microneme secretion in T. gondii was assayed by monitoring the release into the culture medium. Freshly egressed T. gondii tachyzoites, pre-treated for 48 h ± ATc (Tgnd9-iKD and Tgndp1-iKD) or 24h ± IAA (Tgnd6-iKD and Tgndp2-iKD), were harvested by centrifugation at 600 g, and washed twice in intracellular buffer (5 mM NaCl, 142 mM KCl, 1 mM MgCl2, 2 mM EGTA, 5.6 mM glucose and 25 mM HEPES, pH 7.2), prewarmed to 37°C. Parasites were resuspended in DMEM (supplemented with 2 mM glutamine) ± propranolol 500 μM, and incubated at 37°C for 20 min to induce microneme secretion. Parasites were pelleted at 1000 g for 5 min at 4°C, washed once in PBS and stored at −20°C. Supernatants were centrifuged at 2000 g for 5 min, at 4°C and used as ESA (excreted/secreted antigen). Pellets and ESA samples were analyzed for micronemal protein (AMA1) by Western blot. Microneme secretion in P. falciparum was assayed by monitoring the surface translocation of AMA1. Highly synchronized DMSO and rapamycin-treated Pfnd9-iKO schizonts were exposed to 10 μM E64 to block egress. IFAs were then performed with anti-PfAMA1 antibodies to analyze AMA1 translocation to the merozoite surface.
Rhoptry secretion assays
To evaluate the efficiency of rhoptry secretion in T. gondii, we expressed in the Tgnd-iKD and Tgndp-iKD lines a rhoptry secretion reporter protein consisting of the rhoptry protein toxofilin fused with Cre-recombinase, a nuclear localization signal (NLS), and a myc tag, collectively called Secreted Cre, Epitope-tagged (SeCrEt)35. The reporter strains generated were called Tgnd9-iKD_SeCrEtUPRT, Tgndp1-iKD_SeCrEtUPRT, and Tgnd6-iKD_SeCrEtHXGPRT. Ds Red cells, constitutively expressing DsRed and able to switch to eGFP expression upon Cre-mediated recombination, were used as reporter cells35. DsRed Cells were maintained at < 50% confluence (regardless of the flask size) and plated the day before parasite infection, at a density of 2 ×105 cells /ml (in T25 flasks). Tachyzoites pre-treated for 48 h ± ATc (Tgnd9-iKD and Tgndp1-iKD) or 24h ± IAA (Tgnd6-iKD) were collected and used to infect the DsRed cells at a multiplicity of infection (MOI) of 3. 24hpi infected DsRed cells were trypsinized and examined by fluorescence-activated cell sorting (FACS) to assess the percentage of Ds-Red (rhoptry secretion impairment) and GFP (successful rhoptry secretion) expressing cells. Number of GFP positive cells were normalized as the percentage compared to 100% in the control (-IAA or -ATc).
Additionally, rhoptry secretion has been also quantified by classical e-vacuole assay34. Freshly egressed parasites, pre-treated for 48 h ± ATc (Tgnd9-iKD and Tgndp1-iKD) or 24h ± IAA (Tgnd6-iKD), were preincubated with 1 μM of cytochalasin D (cytD) for 10 min and then incubated with HFF cells in the presence of cytD for 15 min. IFA were then performed with anti-ROP1 (rhoptry secretion) and anti-SAG1 (parasite surface), and the number of ROP1 stainings per field was determined by microscopic examination of at least 20 fields per coverslip (n = 3 coverslips).
To quantify rhoptry secretion in P. falciparum, the same amount of DMSO and rapamycin-treated Pfnd9-iKO purified schizonts were arrested with 1.5 μM C2 for 4 hours before being washed twice in order to allow them to egress. Parasites were then incubated for 30 min in complete medium with 1 μM cytD in the presence of red blood cells (RBCs). IFAs were then performed with anti-PfRAP2 antibodies to visualise rhoptry secretion events (‘spits’ of RAP2 export into the RBC). The number of secretion events were counted over the total of RBCSs by microscopic examination of 11 fields (~3000 events analyzed).
Freeze-fracture
Cells used for the freeze-fracture were harvested, pelleted and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer at room temperature for 2 hours. Following primary fixation, the samples were rinsed in the same buffer, and kept overnight in 30% glycerol 0.1 M phosphate solution. The cells were then quickly frozen by immersion in liquid nitrogen under vacuum. The frozen samples were fractured in a BAL-TEC BAF 060 apparatus; subsequently the fracture face was shadowed by evaporating platinum 45° (3.2nm) and Carbon 90° (25nm). The replicas were then washed in 6.5% sodium hypochlorite, rinsed in a chloroform (2/3) - (1/3) solution, rinsed in distilled water and mounted on copper grids. In order to interpret the replicas obtained in freeze-fracture technique one must keep in mind that the portion of the lipid bilayer associated with the exterior of the cell is termed the E-face, or extracellular face. The portion associated with the interior of the cell is termed the P-face, or protoplasmic face49.
In vitro growth assay of P. falciparum parasite asexual development
The growth capability of Pfnd9-iKO mutant parasites was assessed by comparing it with the Pf3D7diCre parental line ± rapamycin in biological triplicates. DMSO (vehicle control) and rapamycin-treated cultures were synchronized at ring stages by sorbitol synchronization and parasitaemia adjusted to 0.2% to follow growth over 3 cycles. Parasites around 30 hours post-invasion were collected at each cycle to determine parasitaemia by counting Giemsa-stained parasites on at least 1000 RBCs per culture.
P. falciparum induced egress and time-lapse microscopy
P. falciparum egress was imaged using C2 compound to tightly synchronize egress. DMSO and rapamycin-treated Pfnd9-iKO parasites were highly synchronized and blocked 40 hpi (mature schizonts) with 1.5 μM C2 compound to prevent egress. Four hours later parasites were washed twice with warm RPMI 1640 medium (Gibco), to remove C2, and placed into a 35 mm Dish (MatTek). The parasites suspension was sealed by adhering a 22×22 mm square coverslip to the Dish and the preparation was introduced on a temperature-controlled microscope stage at 37 °C. Bright field images were collected 5 min after washing off the C2 at 1 s intervals over a total of 15 min using a ZEISS AxioObserver Microscope fitted with a coolsnap HQ2 digital camera and 63X/1.4 Oil Plan Apochromat objective. Images were exported to MOV movies using ZEN 2 (blue edition) software. Number of egress events in 15mn were normalized as the percentage compared to 100% in the control.
Tetrahymena dibucaine assay to quantify mucocyst secretion
Tetrahymena cells were grown to stationary phase (106 cells/ml) in 25 ml SPP (2% proteose peptone, 0.1% yeast extract, 0.2% dextrose and 0.003% ferric-EDTA supplemented with 250 μg/ml penicillin G, 250 μg/ml streptomycin sulfate, and 0.25 μg/ml amphotericin B fungizone) for 48 h, and then concentrated by centrifugation into a lose 1.5ml pellet. Cells were stimulated with 2.5 mM dibucaine, vigorously mixed for 30 s and diluted to 15 ml with 10 mM Hepes, pH 7.5, and 5 mM CaCl2. After gently mixing, the culture was centrifuged at 1,200 g for 2 min, resulting in the formation of a cell pellet/flocculent bilayer. Quantification of mucocysts secretion has been done by weighing the flocculent layer overlying the pellet of cells.
Transmission electron microscopy
Tgnd9-iKD and Tgnd6-iKD parasites, together with the Δku80-TATi parental strain, were treated with ATc for 72 hours. Extracellular parasites were collected in the culture medium after natural egress and fixed by adding and equal volume of phosphate buffer 0.1 M containing 5% of glutaraldehyde for one hour at RT. Parasites were then centrifuged and resuspended in 1 ml of fresh buffer with 2.5% glutaraldehyde for 2h before being kept at 4°C until further processing. Each of the following steps were performed in suspension followed by centrifugation steps in a tabletop microcentrifuge. Sample were post-fixed with 1% OsO4 and 1.5% potassium ferricyanide in 0.1 M phosphate buffer for 1 hour at RT, washed in water and then incubated in 2% uranyl acetate in water overnight at 4°C. Tachyzoites were then dehydrated in growing concentration of acetonitrile, followed by impregnation in Epon118: acetonitrile 50:50 for 2 hours, 90:10 for 2 additional hours and then overnight in pure epon. Pellets were polymerized in fresh Epon for 48h at 60°C. 70 nm ultrathin sections were cut with a Leica ultracut (Leica microsystems), counterstained with uranyl acetate and lead citrate.
Immunoelectron microscopy
Tgnd6-HA3 parasites, infected fibroblast monolayers were trypsinized and fixed with equal volume of 8% formaldehyde (FA) in phosphate buffer overnight at 4°C and resuspended in 4% fresh FA until further processing. Cells were then incubated with 0.1 % glycine in phosphate buffer, pelleted and embedded in 12% gelatin, cut in small blocks (< 1 mm) and infused in 2.3 M sucrose on a rotating wheel for 24 hours at 4°C. Gelatin blocks were mounted on specimen pins and frozen in liquid nitrogen. Cryo-sectioning was performed on a Leica UC7 cryo-ultramicrotome, 70 nm cryosections were picked-up in a 1:1 mixture of 2.3 M sucrose and 2% methylcellulose in water and stored at 4°C. For on-grid immunodetection, grids were floated on 2% gelatin in PBS for 30 min at 37°C to remove methylcellulose/sucrose mixture, then blocked with 1% skin-fish gelatin (SFG, Sigma) in PBS for 5 min. Successive immunolabeling steps were performed on drops as follows: 1) rat monoclonal anti-HA antibodies (clone 3F10, Roche) in 1% BSA, 2) rabbit polyclonal anti-rat IgG antibody (Sigma) in 1% BSA, 3) Protein A-gold (UMC Utrecht) in 1% BSA. Four washes (2 min each) with 0.1% BSA were performed between steps. After Protein A treatment, grids were washed four times for 2 min each with PBS, fixed 5 min in 1% glutaraldehyde in water, and washed six times for 2 min each with distilled water. Grids were then incubated with 2% methylcellulose: 4% uranyl acetate 9:1 on ice in the dark for 15 min, picked-up on a wire loop and air-dried.
All chemicals were from Electron Microscopy Sciences (USA), solvents were from Sigma. Observations and image acquisition were performed on a Jeol 1200 EXII transmission electron microscope at the Electron Microscopy Platform of the University of Montpellier (MEA; http://mea.edu.umontpellier.fr). Transmission electron microscopy images were processed with Fiji for contrast optimization and the Image J plugin was used to make the EM panels.
Cryo-Electron-Tomography (Cryo-ET)
Freshly isolated Toxoplasma gondii cells were suspended in HBSS along with fiducials (10 nm Au fiducials from Ted Pella for alignment of tilt series). A ~4 μL drop of the suspension was applied onto EM grids, excess liquid blotted away, and plunge frozen in a liquid ethane/propane mixture using a EM GP2 automatic plunger (Leica Microsystems, Wetzlar, Germany)50. The blotting chamber was set to 95–100% relative humidity at 37 °C and blotting was done either from the sample side of the grid or from the back using Whatman filter paper. Plunge-frozen grids were subsequently loaded into autogrid cartridges (ThermoFisher). EM cartridges containing frozen grids were stored in liquid nitrogen and maintained at ≤−170 °C throughout storage, transfer and cryo-EM imaging.
Cryo-ET was performed on plunge frozen cells using a ThermoFisher Krios G3i 300 keV FEG cryo-TEM equipped with a 6k x 4k K3 direct electron detector (Gatan, Inc.) at the Singh Center for Nanotechnology (University of Pennsylvania). The camera was operated in electron counting mode to enable motion correction. An energy filter (Gatan Imaging Filter, Gatan, Inc.) with a slit width of 20 eV was used to increase the contrast of the projection images. Additionally, a Volta phase plate and a defocus of negative 2–3 μm were used to boost the contrast. A magnification of 33000X was used for tilt series corresponding to pixel sizes of 2.65 Å. SerialEM software51 was used for all imaging. Cells were first assessed at lower magnifications for suitability of ice thickness and outer membrane integrity (the parasites sometimes tend to vesiculate their outer membrane possibly in response to blotting). Once the target cells were identified and marked, anchor maps were used to revisit these locations and collect tilt-series in an automated fashion. Each tilt-series was collected from negative 60° to positive 60° with an increment of 2° in an automated fashion using the low dose functions of tracking and focusing. The cumulative dose of each tilt-series ranged between 100 and 150 e−/Å2. Once acquired, tilt-series were binned twice or four times before alignment and reconstruction into 3D tomograms with the IMOD software package52 Tilt series were aligned using the 10 nm Au fiducials using our in-house automated pipeline and tomogram reconstructions were performed using SIRT-like filter in IMOD. For presentation purposes, tomograms were favorably oriented in three dimensions before averaging a few slices around the slice of interest (to enhance contrast) using the slicer window in IMOD. In a few cases (Figs. 4a, right panel, to 4c), contrast was further enhanced using non-linear anisotropic diffusion filter in IMOD.
Immunopurification and mass spectrometry analysis
Immunopurification was performed using anti HA magnetic beads (Pierce R88836) as previously described53. Purified proteins were loaded on a SDS-PAGE and digested in gel (2 bands per sample) as previously described54. Samples were loaded onto a 25 cm reversed phase column (75 mm inner diameter, Acclaim Pepmap 100® C18, Thermo Fisher Scientific) and separated with an Ultimate 3000 RSLC system (Thermo Fisher Scientific) coupled to a Q Exactive HFX (Thermo Fisher Scientific). MS/MS analyses were performed in a data-dependent mode. Full scans (375 – 1,500 m/z) were acquired in the Orbitrap mass analyzer with a resolution of 60,000 at 200 m/z. For the full scans, 3e6 ions were accumulated within a maximum injection time of 60 ms. The twelve most intense ions with charge states ≥ 2 were sequentially isolated (1e5) with a maximum injection time of 45 ms and fragmented by HCD (Higher-energy collisional dissociation) in the collision cell (normalized collision energy of 28%) and detected in the Orbitrap analyzer at a resolution of 30,000.
Raw spectra were processed using the MaxQuant55 using standard parameters with label-free quantification (LFQ) and match between runs56. MS/MS spectra were matched against the UniProt Reference proteomes of T. gondii and Human (respectively Proteome ID UP000001529 and UP000005640) and 250 frequently observed contaminants as well as reversed sequences of all entries (MaxQuant contaminant database). Statistical analysis were done using Perseus on LFQ data57.
Amino acid sequence alignments and phylogenetic analysis, cell and parasite culture, parasite cloning strategies, parasite transfections, parasite immunoblots, T. gondii plaque assays, T. gondii intracellular growth assay, T. gondii immunofluorescence-based induced egress assay, T. gondii conoid extrusion assay, T. gondii gliding assays, T. gondii attachment assay, Ciliate culture conditions, Ciliate biolistic transformation, generation of Tetrahymena knockout strain, Ciliate immunoblots, Ciliate immunofluorescence microscopy, statistical analysis, reagents and antibodies are found in Supplementary Methods.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon request.
Extended Data
Extended Data Fig. 1 |.
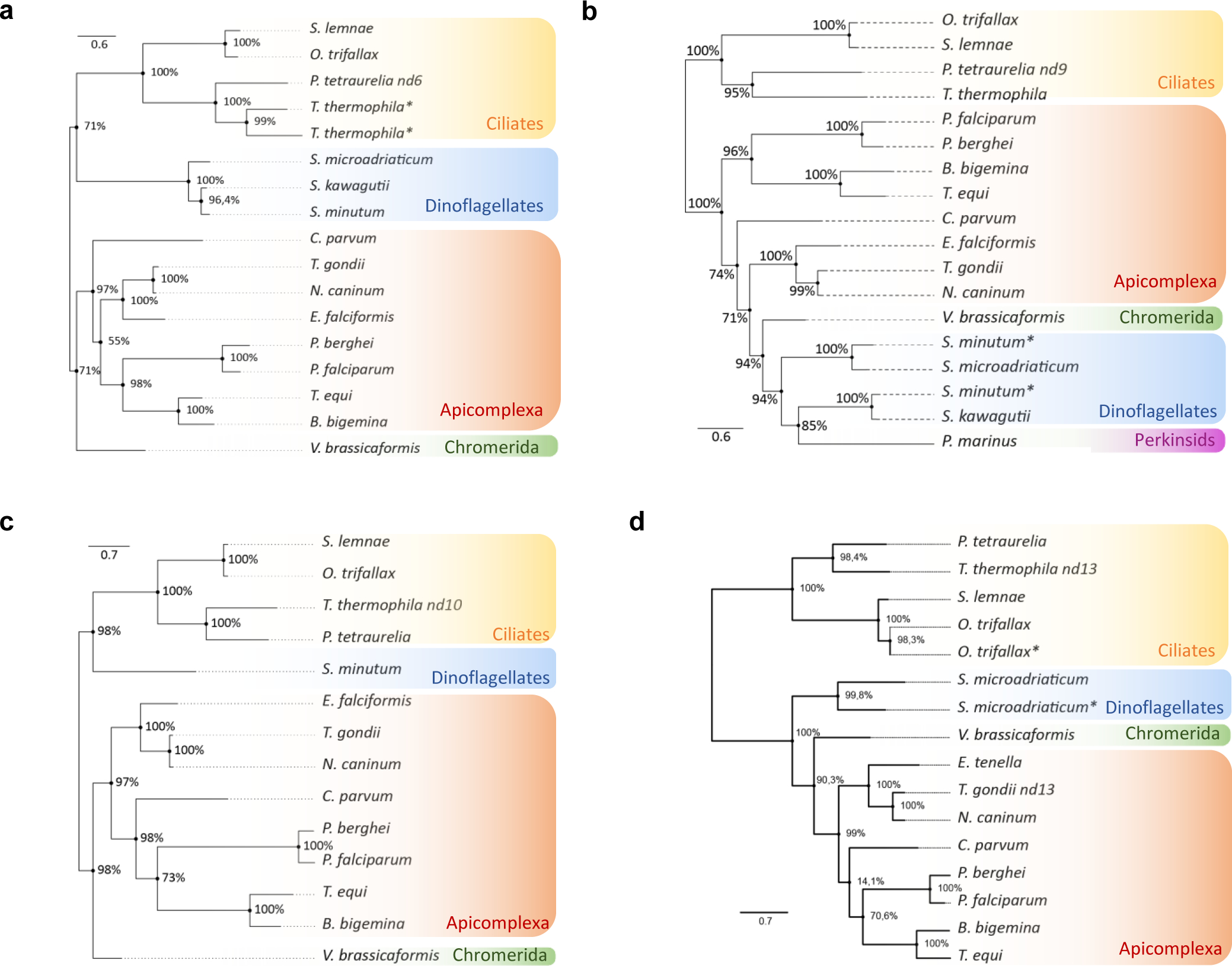
Amino acid sequence alignments and phylogenetic analysis
Extended Data Fig. 2 |.

Generation of TgNd6-KD and TgNd9-KD
Extended Data Fig. 3 |.
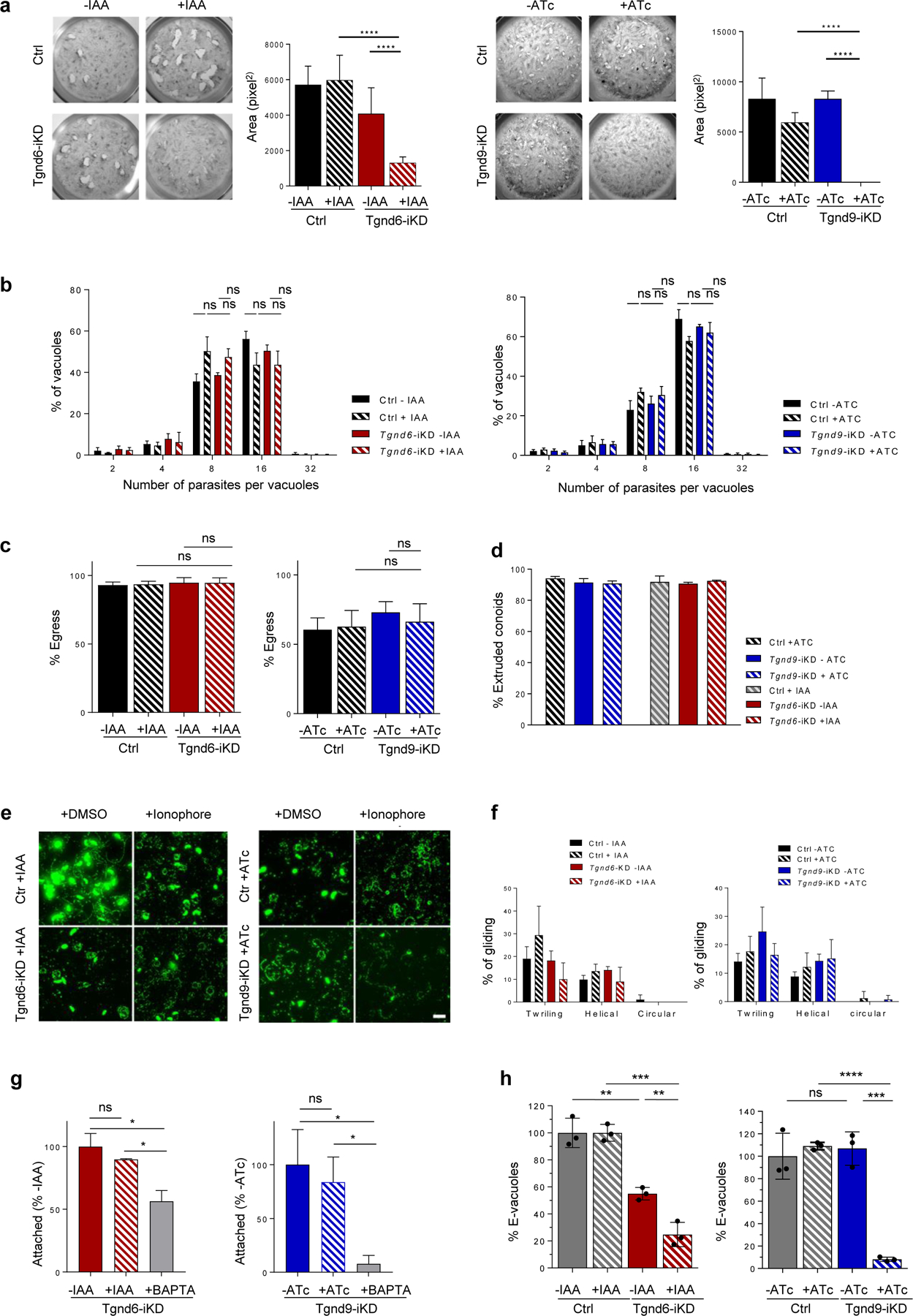
Depletion of TgNd6 or TgNd9 in T.gondii parasites does not affect replication, egress, nor gliding motility.
Extended Data Fig. 4 |.
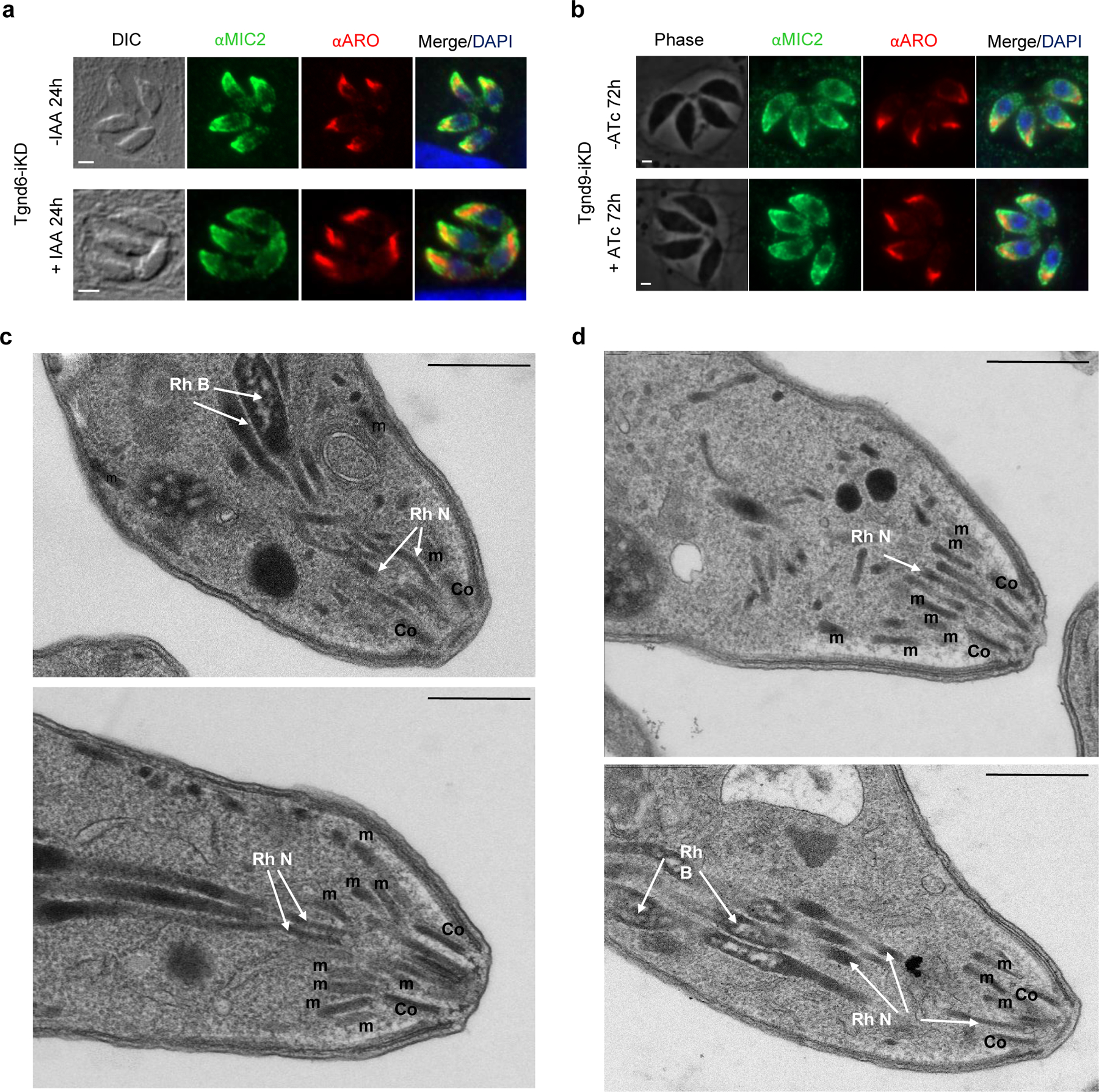
Secretory organelles biogenesis and positioning is not affected in Tgnd6iKO and Tgnd9iKO.
Extended Data Fig. 5 |.
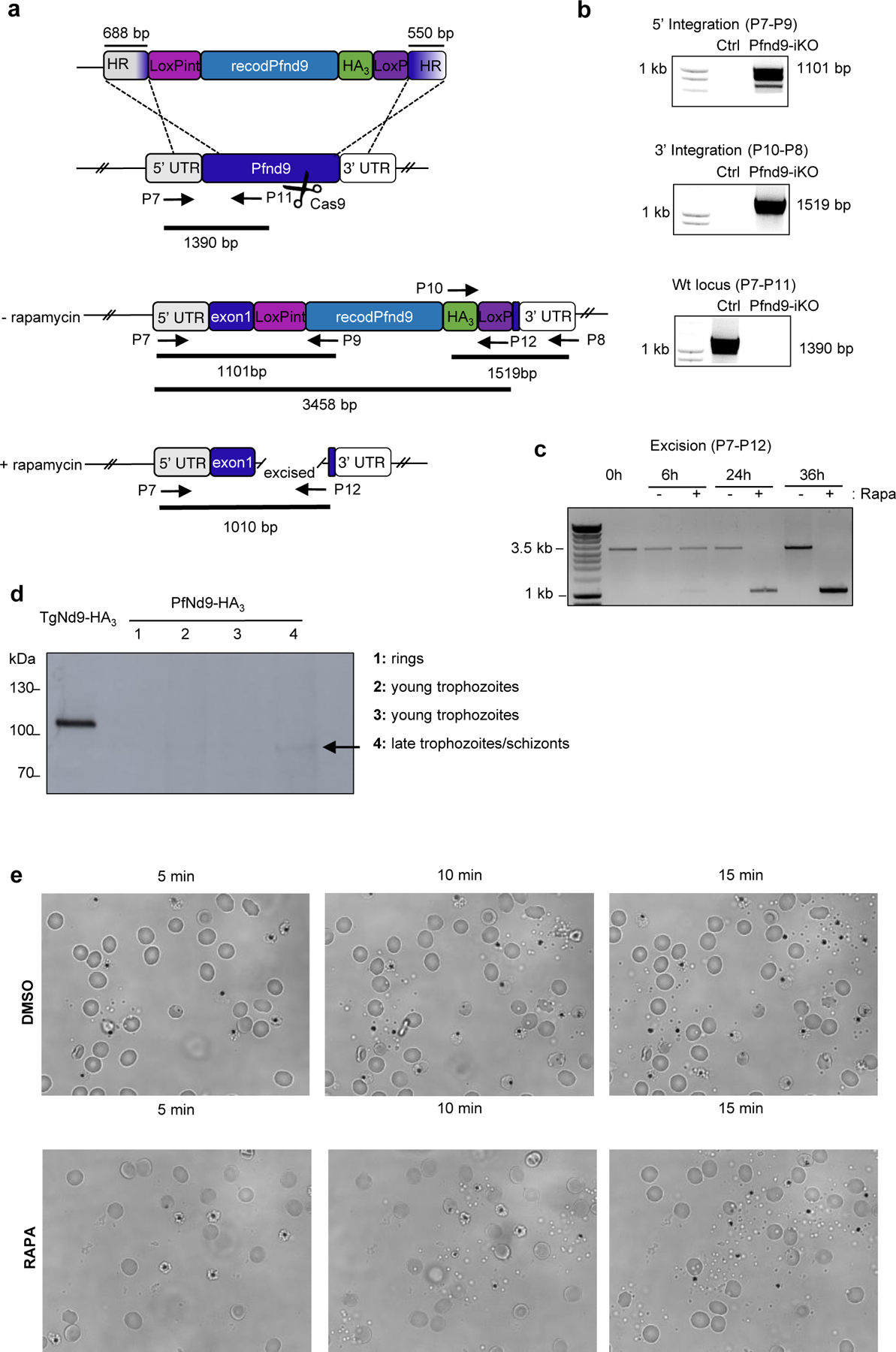
Generation of Pfnd9iKO
Extended Data Fig. 6 |.
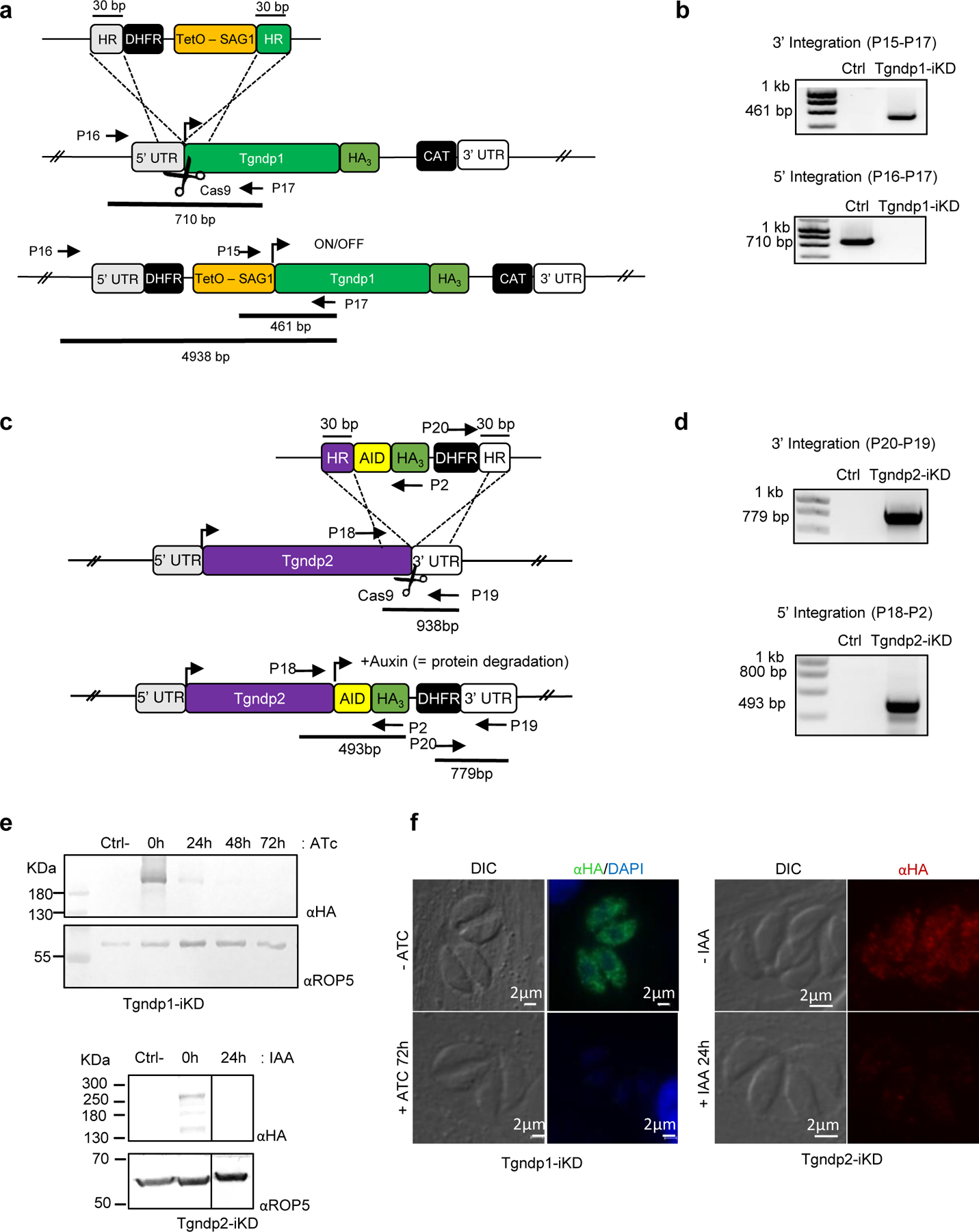
Generation of Tgnd6-iKD and Tgnd9-iKD
Extended Data Fig. 7 |.
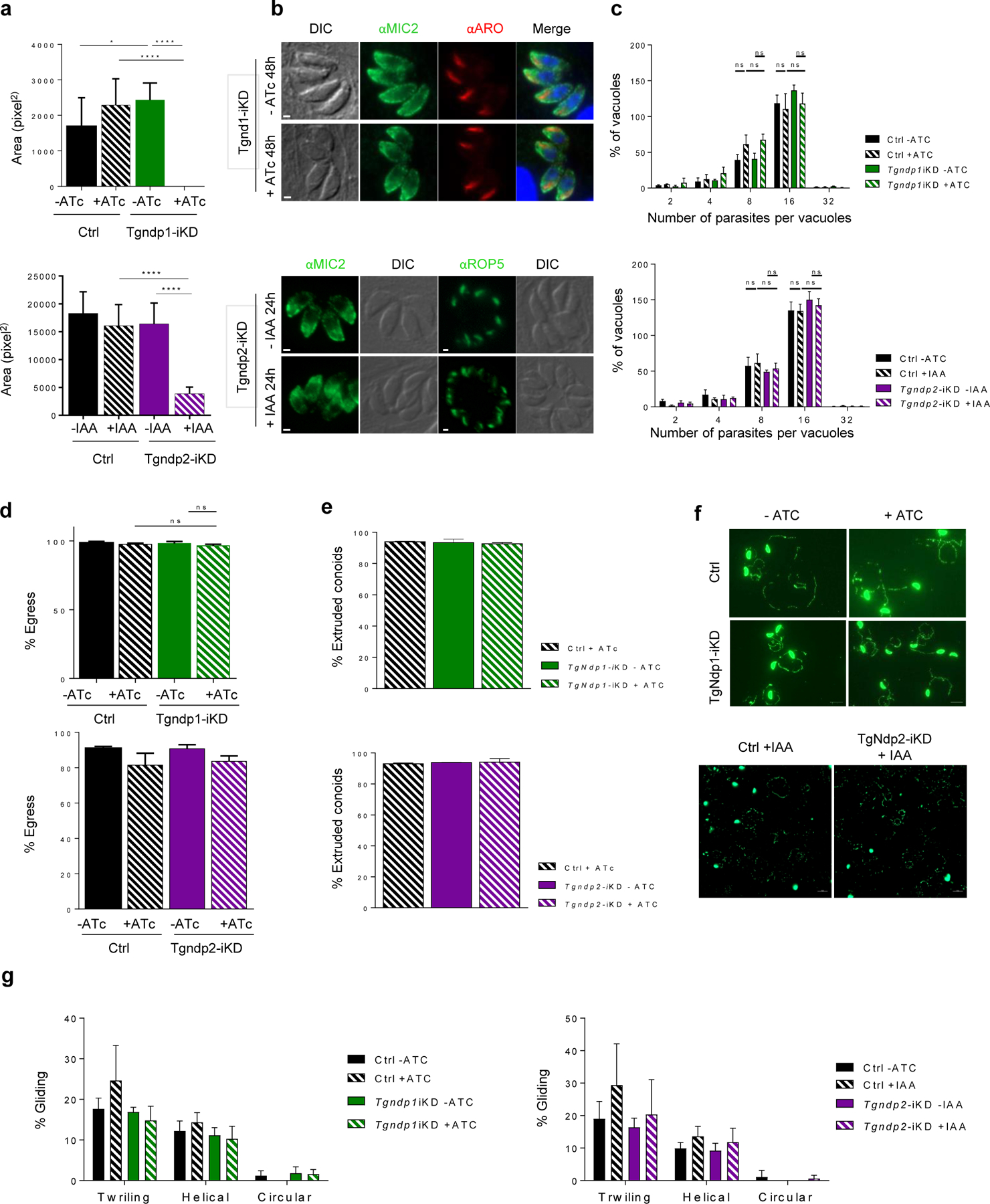
Depletion of NdP1 or NdP2 in T. gondii parasites does not affect replication, egress, gliding motility, nor attachment
Extended Data Fig. 8 |.
Mucocyst biogenesis and positioning is not affected in TtΔnd10
Extended Data Fig. 9 |.
Cryo-ET of rhoptry secretion system.
Extended Data Fig. 10 |.
Apical area of a Sarcocystis tenella cystozoite.
Extended Data Fig. 11 |.
Unedited full-length gels/blots.
Supplementary Material
Acknowledgements
We thank Sebastian Lourido for the pU6-Universal plasmid, Artur Scherf for anti-PfRAP2 antibodies, Soldati-Favre for providing us the anti-ARO antibodies and TgARO-iKD strain; Michael Blackman for anti-PfAMA1, anti-MSP1 and vector and strain for DiCRE, John Boothroyd for mAb CCL2 anti-TgAMA1; Ross Waller for RNG1-GFP plasmid, Anita Koshy for the toxofilin-Cre plasmid and Helen Blau’s lab for the Cre reporter DSred cell line. We thank Oliver Billker for providing us with Compound 2. We thank Veronique Richard and Frank Godiard for their technical assistance on the electron microscopy service of the University of Montpellier. We are also grateful to Elodie Jublanc and the imaging facility MRI at the University of Montpellier, part of the national infrastructure France-BioImaging supported by the French National Research Agency (ANR-10-INBS-04, «Investments for the future»), and Christophe Duperray of the MRI-Cytometry at the Institute for Regenerative Medicine and Biotherapy for their assistance and technical support. We would like to thank Julien Marcetteau for the graphical design of Fig. 1b and Fig. 4. Dr Maryse Lebrun is an INSERM researcher. This work was supported by the Laboratoire d’Excellence (LabEx) (ParaFrap ANR-11-LABX-0024), by the ANR (ANR-16-CE15-0010-01), by the Fondation pour la Recherche Medicale (Equipe FRM DEQ20170336725), by the FACCTS (France and Chicago Collaborating in the Sciences) and by a David and Lucile Packard Fellowship for Science and Engineering N°2019-69645.
Footnotes
Competing interests
The authors declare no competing financial interests.
Supplementary Information is available for this paper.
References
- 1.Dubremetz JF Rhoptries are major players in Toxoplasma gondii invasion and host cell interaction. Cell Microbiol 9, 841–8 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Boothroyd JC & Dubremetz JF Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat Rev Microbiol 6, 79–88 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Carruthers VB & Sibley LD Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur J Cell Biol 73, 114–23 (1997). [PubMed] [Google Scholar]
- 4.Frenal K, Dubremetz JF, Lebrun M & Soldati-Favre D Gliding motility powers invasion and egress in Apicomplexa. Nat Rev Microbiol 15, 645–660 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Besteiro S, Dubremetz JF & Lebrun M The moving junction of apicomplexan parasites: a key structure for invasion. Cell Microbiol 13, 797–805 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Ito D, Schureck MA & Desai SA An essential dual-function complex mediates erythrocyte invasion and channel-mediated nutrient uptake in malaria parasites. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Counihan NA et al. Plasmodium falciparum parasites deploy RhopH2 into the host erythrocyte to obtain nutrients, grow and replicate. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguitragool W et al. Malaria parasite clag3 genes determine channel-mediated nutrient uptake by infected red blood cells. Cell 145, 665–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh S, Alam MM, Pal-Bhowmick I, Brzostowski JA & Chitnis CE Distinct external signals trigger sequential release of apical organelles during erythrocyte invasion by malaria parasites. PLoS Pathog 6, e1000746 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kessler H et al. Microneme protein 8--a new essential invasion factor in Toxoplasma gondii. J Cell Sci 121, 947–56 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Nichols BA, Chiappino ML & O’Connor GR Secretion from the rhoptries of Toxoplasma gondii during host-cell invasion. J Ultrastruct Res 83, 85–98 (1983). [DOI] [PubMed] [Google Scholar]
- 12.Coleman BI et al. A Member of the Ferlin Calcium Sensor Family Is Essential for Toxoplasma gondii Rhoptry Secretion. MBio 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suarez C et al. A lipid-binding protein mediates rhoptry discharge and invasion in Plasmodium falciparum and Toxoplasma gondii parasites. Nat Commun 10, 4041 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tseng TT, Tyler BM & Setubal JC Protein secretion systems in bacterial-host associations, and their description in the Gene Ontology. BMC Microbiol 9 Suppl 1, S2 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gubbels M-J & Duraisingh MT Evolution of apicomplexan secretory organelles. Int. J. Parasitol. 42, 1071–1081 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plattner H Trichocysts-Paramecium’s Projectile-like Secretory Organelles: Reappraisal of their Biogenesis, Composition, Intracellular Transport, and Possible Functions. J Eukaryot Microbiol 64, 106–133 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Plattner H, Miller F & Bachmann L Membrane specializations in the form of regular membrane-to-membrane attachment sites in Paramecium. A correlated freeze-etching and ultrathin-sectioning analysis. J Cell Sci 13, 687–719 (1973). [DOI] [PubMed] [Google Scholar]
- 18.Knoll G, Braun C & Plattner H Quenched flow analysis of exocytosis in Paramecium cells: time course, changes in membrane structure, and calcium requirements revealed after rapid mixing and rapid freezing of intact cells. J Cell Biol 113, 1295–304 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubremetz JF & Torpier G Freeze fracture study of the pellicle of an eimerian sporozoite (Protozoa, Coccidia). J Ultrastruct Res 62, 94–109 (1978). [DOI] [PubMed] [Google Scholar]
- 20.Porchet E & Torpier G [Freeze fracture study of Toxoplasma and Sarcocystis infective stages (author’s transl)]. Z Parasitenkd 54, 101–24 (1977). [DOI] [PubMed] [Google Scholar]
- 21.Porchet-Hennere E & Nicolas G Are rhoptries of Coccidia really extrusomes? J Ultrastruct Res 84, 194–203 (1983). [DOI] [PubMed] [Google Scholar]
- 22.Dubremetz JF & Entzeroth R Exocytic events during cell invasion by Apicomplexa. Adv. Cell Mol. Biol. Membr. Membr. Traffic Protozoa Ed AM Tartakoff H Plattner 2A, 83–98 (1993). [Google Scholar]
- 23.Lefort-Tran M, Aufderheide K, Pouphile M, Rossignol M & Beisson J Control of exocytotic processes: cytological and physiological studies of trichocyst mutants in Paramecium tetraurelia. J Cell Biol 88, 301–11 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beisson J, Lefort-Tran M, Pouphile M, Rossignol M & Satir B Genetic analysis of membrane differentiation in Paramecium. Freeze-fracture study of the trichocyst cycle in wild-type and mutant strains. J Cell Biol 69, 126–43 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gogendeau D, Keller AM, Yanagi A, Cohen J & Koll F Nd6p, a novel protein with RCC1-like domains involved in exocytosis in Paramecium tetraurelia. Eukaryot Cell 4, 2129–39 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Froissard M, Keller AM & Cohen J ND9P, a novel protein with armadillo-like repeats involved in exocytosis: physiological studies using allelic mutants in paramecium. Genetics 157, 611–20 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skouri F & Cohen J Genetic approach to regulated exocytosis using functional complementation in Paramecium: identification of the ND7 gene required for membrane fusion. Mol Biol Cell 8, 1063–71 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Froissard M, Keller AM, Dedieu JC & Cohen J Novel secretory vesicle proteins essential for membrane fusion display extracellular-matrix domains. Traffic 5, 493–502 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Paredes-Santos TC, de Souza W & Attias M Dynamics and 3D organization of secretory organelles of Toxoplasma gondii. J Struct Biol 177, 420–30 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Sidik SM et al. A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 166, 1423–1435 e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long S et al. Calmodulin-like proteins localized to the conoid regulate motility and cell invasion by Toxoplasma gondii. PLoS Pathog 13, e1006379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meissner M, Schluter D & Soldati D Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science 298, 837–40 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Sheiner L et al. A systematic screen to discover and analyze apicoplast proteins identifies a conserved and essential protein import factor. PLoS Pathog 7, e1002392 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamarque MH et al. Plasticity and redundancy among AMA-RON pairs ensure host cell entry of Toxoplasma parasites. Nat Commun 5, 4098 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Koshy AA et al. Toxoplasma secreting Cre recombinase for analysis of host-parasite interactions. Nat Methods 7, 307–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collins CR et al. Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol Microbiol 88, 687–701 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang M et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Briguglio JS, Kumar S & Turkewitz AP Lysosomal sorting receptors are essential for secretory granule biogenesis in Tetrahymena. J Cell Biol 203, 537–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mueller C et al. The Toxoplasma protein ARO mediates the apical positioning of rhoptry organelles, a prerequisite for host cell invasion. Cell Host Microbe 13, 289–301 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Beisson J, Cohen J, Lefort-Tran M, Pouphile M & Rossignol M Control of membrane fusion in exocytosis. Physiological studies on a Paramecium mutant blocked in the final step of the trichocyst extrusion process. J Cell Biol 85, 213–27 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonnemain H, Gulik-Krzywicki T, Grandchamp C & Cohen J Interactions between genes involved in exocytotic membrane fusion in paramecium. Genetics 130, 461–70 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varghese T Fine structure of the endogenous stages of Eimeria labbeana. I. The first generation merozoites. J Protozool 22, 66–71 (1975). [DOI] [PubMed] [Google Scholar]
- 43.Sheffield HG Electron microscope study of the proliferative form of Besnoitia jellisoni. J Parasitol 52, 583–94 (1966). [PubMed] [Google Scholar]
- 44.Perkins FO Zoospores of the Oyster Pathogen, Dermocystidium marinum. I. Fine Structure of the Conoid and Other Sporozoan-Like Organelles. J. Parasitol. 62, 959–974 (1976). [Google Scholar]
- 45.El Hajj H et al. Molecular signals in the trafficking of Toxoplasma gondii protein MIC3 to the micronemes. Eukaryot Cell 7, 1019–28 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burghaus PA & Holder AA Expression of the 19-kilodalton carboxy-terminal fragment of the Plasmodium falciparum merozoite surface protein-1 in Escherichia coli as a correctly folded protein. Mol Biochem Parasitol 64, 165–9 (1994). [DOI] [PubMed] [Google Scholar]
- 47.Douki JB et al. Adhesion of normal and Plasmodium falciparum ring-infected erythrocytes to endothelial cells and the placenta involves the rhoptry-derived ring surface protein-2. Blood 101, 5025–32 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Cerede O et al. Synergistic role of micronemal proteins in Toxoplasma gondii virulence. J Exp Med 201, 453–63 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Branton D et al. Freeze-etching nomenclature. Science 190, 54–6 (1975). [DOI] [PubMed] [Google Scholar]
- 50.Iancu CV et al. Electron cryotomography sample preparation using the Vitrobot. Nat. Protoc. 1, 2813–2819 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Kremer JR, Mastronarde DN & McIntosh JR Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116, 71–6 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Lebrun M et al. The rhoptry neck protein RON4 relocalizes at the moving junction during Toxoplasma gondii invasion. Cell Microbiol 7, 1823–33 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Skorupa A et al. Angiogenin induces modifications in the astrocyte secretome: relevance to amyotrophic lateral sclerosis. J Proteomics 91, 274–85 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Cox J & Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–72 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Cox J et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10, 1794–805 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Tyanova S et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–40 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Wilson D, Madera M, Vogel C, Chothia C & Gough J The SUPERFAMILY database in 2007: families and functions. Nucleic Acids Res 35, D308–13 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hadjebi O, Casas-Terradellas E, Garcia-Gonzalo FR & Rosa JL The RCC1 superfamily: from genes, to function, to disease. Biochim Biophys Acta 1783, 1467–79 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon request.