

Abstract
The kinesin-4 motor KIF7 is a conserved regulator of the Hedgehog signaling pathway. In vertebrates, Hedgehog signaling requires the primary cilium, and KIF7 and Gli transcription factors accumulate at the cilium tip in response to Hedgehog activation. Unlike conventional kinesins, KIF7 is an immotile kinesin and its mechanism of ciliary accumulation is unknown. We generated KIF7 variants with altered microtubule binding or motility. We demonstrate that microtubule binding of KIF7 is not required for the increase in KIF7 or Gli localization at the cilium tip in response to Hedgehog signaling. In addition, we show that the immotile behavior of KIF7 is required to prevent ciliary localization of Gli transcription factors in the absence of Hedgehog signaling. Using an engineered kinesin-2 motor that enables acute inhibition of intraflagellar transport, we demonstrate that kinesin-2 KIF3A/KIF3B/KAP mediates the translocation of KIF7 to the cilium tip in response to Hedgehog pathway activation. Together, these results suggest that KIF7’s role at the tip of the cilium is unrelated to its ability to bind to microtubules.
INTRODUCTION
Hedgehog (Hh) signaling is an evolutionarily conserved pathway that plays essential roles during embryonic development and adult tissue homeostasis (Kong et al., 2019; Qi and Li, 2020). The kinesin-4 family member Drosophila Costal2 (Cos2) and its vertebrate homologue KIF7 are conserved regulators of Hh signaling (He et al., 2017) and mutations in human Kif7 are associated with several diseases including hydrolethalus, acrocallosal syndrome, and Joubert syndrome (He et al., 2017).
Hh signaling in vertebrates requires the primary cilium, a microtubule-based organelle that protrudes from the cell surface (Bangs and Anderson, 2017; He et al., 2017). In the absence of Hh signaling, KIF7 and suppressor of fused (SUFU) restrain Gli transcription factors in the cytosol and promote Gli3 processing into its transcriptional repressor form. In the presence of Hh signaling, KIF7, SUFU, and Gli transcription factors are enriched at the tip of cilium and Gli2 is converted into its transcriptional activator form (Corbit et al., 2005; Haycraft et al., 2005; Endoh-Yamagami et al., 2009; Liem et al., 2009; Humke et al., 2010; Zeng et al., 2010; He et al., 2014; Liu et al., 2014). KIF7 has also been suggested to regulate the length of the primary cilium and organize the cilium tip based on its ability to bind selectively to the plus ends of microtubules in in vitro assays (He et al., 2014; Jiang et al., 2019).
How Hh pathway activation results in accumulation of KIF7 and Gli proteins at the tip of the primary cilium is not known. As a kinesin, it seemed possible that KIF7 could convey itself, and perhaps SUFU and Gli proteins, to the tip of the cilium. However, the kinesin motor domain of KIF7 only binds statically to microtubules (He et al., 2014; Yue et al., 2018). Even though KIF7 is an immotile kinesin, it is possible that KIF7’s ability to bind to, or even move along, microtubules is altered during Hh signaling to facilitate its localization to the ciliary tip. An alternative possibility is that another kinesin motor is required for the anterograde transport of KIF7 to the tip of the primary cilium. Intraflagellar transport (IFT) is driven by microtubule-based motors during cilium assembly and maintenance. While IFT is primarily driven by the kinesin-2 KIF3A/KIF3B/KAP and cytoplasmic dynein-2 motors, several other kinesins have been localized to primary and motile cilia and may function in ciliary transport (Lechtreck, 2015; He et al., 2017; Prevo et al., 2017; Reilly and Benmerah, 2019; Webb et al., 2020).
Here, we consider four models for how Hh stimulation results in an accumulation of KIF7 at the tip of the primary cilium (Figure 1): 1) increased access of KIF7 to the ciliary compartment; 2) increased KIF7 loading onto IFT trains; 3) increased ability of KIF7 to bind to microtubule plus ends at the tip of the primary cilium; and 4) increased interaction of KIF7 with an anchoring protein present at the tip of the primary cilium. It is important to note that these models are not mutually exclusive, and several mechanisms may cooperate to control KIF7 localization at the cilium tip. We find that IFT but not KIF7’s microtubule binding is required for Hh-induced accumulation of KIF7 at the tip of the primary cilium. We suggest that Hh stimulation increases the loading of KIF7 and associated Gli proteins onto IFT trains.
FIGURE 1:
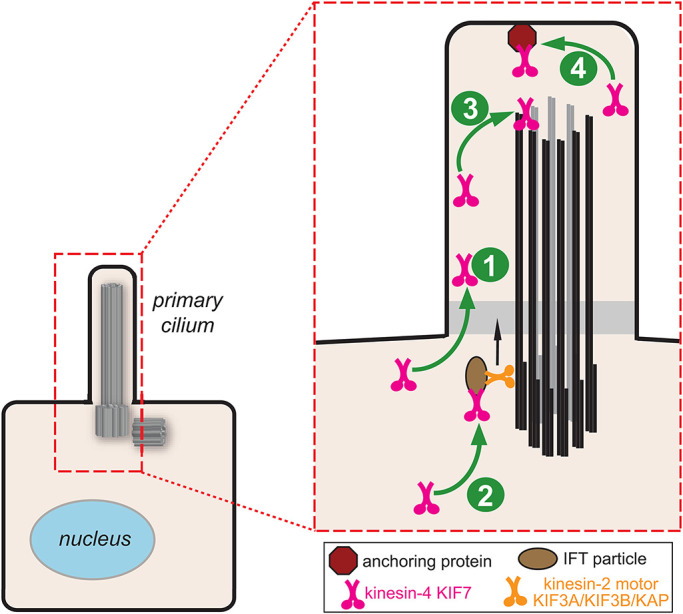
Models for Hh-induced accumulation of KIF7 in the primary cilium. Hh signaling increases 1) the ability of KIF7 to access the ciliary compartment, 2) KIF7 loading onto IFT trains, 3) the ability of KIF7 to bind to microtubule plus ends at the tip of the primary cilium, and/or 4) the interaction of KIF7 with an anchoring protein present at the tip of the primary cilium.
RESULTS AND DISCUSSION
Characterization of KIF7 mutants with altered microtubule binding affinity and motility
We first set out to create a KIF7 variant that lacks microtubule-binding ability. Previous work demonstrated that mutation of residues in loop12 and α-helix5 (L12/α5) of the motor domain of kinesin-1 abolish microtubule binding (Woehlke et al., 1997; Marx et al., 2009). We thus mutated the equivalent residues of the KIF7 motor domain (Supplemental Figure S1B) and verified that the mutant protein, KIF7noMT, does not bind to microtubules using a fluorescence-based microtubule binding assay (Supplemental Figure S1E). When expressed in mammalian cells, KIF7noMT (Figure 2A) localized diffusely throughout the cell and to cytoplasmic puncta in COS-7, NIH 3T3, and mouse embryonic fibroblasts (MEF) cells (Figure 2B and Supplemental Figure S2), suggesting that it does not interact with cytosolic microtubules.
FIGURE 2:

Intracellular localization of KIF7-mCit WT and mutant versions. (A) Schematic of the domain organization of KIF7 WT, mutant, and chimeric proteins. X denotes the position of mutations that ablate (noMT) or cause constitutive (rigor) microtubule binding. (B) Representative images of mCit-tagged KIF7 WT and variant proteins expressed in COS-7 cells. White dashed lines indicate the periphery of transfected cells. Scale bars, 10 µm.
We next set out to create a KIF7 variant that binds strongly to microtubules, that is, a “rigor” mutant. We mutated the last amino acid in the nucleotide-binding P-loop motif (T101; Supplemental Figure S3B) as this results in a rigor motor for kinesin-1 (Nakata and Hirokawa, 1995; Crevel et al., 1996); however, KIF7T101N showed a decreased ability to bind to microtubules (Supplemental Figure S3, C, E, and G). We thus took advantage of recent structural work demonstrating that loop 5 (L5), a segment important for opening the nucleotide pocket in response to microtubule binding (Cochran, 2015), has limited conformational flexibility in KIF7 (Jiang et al., 2019). We surmised that further reductions in L5 flexibility could result in stronger KIF7 microtubule binding. We thus replaced L5 in KIF7 with the corresponding sequences from the kinesin-1 motor KIF5C (Supplemental Figure S3B). As the mutant protein displayed increased microtubule binding (Supplemental Figure S3, C, F, and G), we thus refer to this variant as KIF7rigor (Figure 2A). When expressed in mammalian cells, KIF7rigor bound strongly to microtubules and, in some cells, caused their bundling and collapse around the nucleus (Figure 2B and Supplemental Figure S2).
To create a motile version of KIF7, we replaced the motor domain of KIF7 with the motor domains of either the kinesin-4 member KIF21A (hereafter referred to as KIF7motile_21A) or the kinesin-1 member KIF5C (hereafter referred to as KIF7motile_5C; Figure 2A). KIF21A’s motor domain is closely related to that of KIF7 (50.6% identical), yet KIF21A is a motile kinesin (van der Vaart et al., 2013; Cheng et al., 2014). However, because KIF21A does not localize to the primary cilium in basal or Hh-stimulated states (Supplemental Figure S4) and thus may be incapable of motility along axonemal microtubules, we created a second chimeric protein using the motor domain of the kinesin-1 KIF5C (40.3% identical) as KIF5C localizes to the primary cilium in the basal and Hh-stimulated states (Supplemental Figure S4). While the KIF5C and KIF21A motor domains provide the chimeric proteins with an ability to walk along microtubules, the coiled-coil and tail regions of KIF7 provide sequences for localization to the primary cilium, interaction with Gli transcription factors, and dephosphorylation by liprin-α1 (PPFIA1) and phosphatase PP2A (Cheung et al., 2009; Endoh-Yamagami et al., 2009; Liu et al., 2014; Blasius et al., 2021). When expressed in cells, both KIF7motile_21A and KIF7motile_5C chimeric proteins accumulated at the cell periphery, indicative of directed motion to the microtubule plus ends (Figure 2B and Supplemental Figure S2). Overall, we conclude that the KIF7 variants display the intended altered microtubule-binding affinity and/or motility.
KIF7’s microtubule binding is not required for its Hh-stimulated ciliary accumulation
We tested whether altered microtubule binding and/or motility of KIF7 impacts its ability to accumulate at the tip of the primary cilium in the basal and Hh-stimulated states. To avoid potential complications due to heterodimerization of the KIF7 variants with the endogenous protein, we expressed wild-type (WT) or mutant KIF7 proteins in Kif7−/− MEFs (Liu et al., 2014). We note that the expression level of KIF7 does not influence its localization to cytosolic microtubules or to the cilium tip (Blasius et al., 2021). Hh pathway activation was accomplished by treatment with Smoothened agonist (SAG; Chen et al., 2002).
Hh stimulation resulted in a significant increase in the number of cells with KIF7 at the tip of the cilium (29.6% of unstimulated cells and 82.8% of SAG-treated cells) (Figure 3), consistent with previous work (Liu et al., 2014). Hh stimulation also resulted in a significant increase in the number of cells with the KIF7noMT variant at the tip of the cilium (14.1% of unstimulated cells and 51.4% of SAG-treated cells; Figure 3), indicating that KIF7’s microtubule binding is dispensable for its Hh-induced increase in ciliary localization. Similar results were recently reported for a different KIF7noMT variant which was found to localize to the cilium tip in ∼30% of Hh-stimulated cells (Jiang et al., 2019), however, this finding was interpreted by the authors as KIF7’s cilium entry or tip localization being dependent on its own microtubule binding. We believe the different interpretations of these similar results can be reconciled by the fact that Jiang et al did not examine KIF7noMT’s ciliary localization in the absence of Hh stimulation. Our finding that the magnitude of KIF7noMT’s Hh-induced increase in ciliary tip localization is similar to that of the WT protein indicates that KIF7’s microtubule binding is dispensable for its Hh-induced increase in ciliary localization. These findings do not support model 3 (Figure 1) as the primary mechanism by which Hh stimulation causes an increase in KIF7 localization to the cilium tip. Further evidence against model 3 comes from our recent work demonstrating that Hh stimulation does not induce KIF7 to bind to the plus ends of microtubules in cells (Blasius et al., 2021).
FIGURE 3:

KIF7’s microtubule binding is dispensable for its Hh-induced accumulation at the tip of the cilium. (A) Representative images of the subcellular region containing the primary cilium in Kif7−/− MEFs expressing mCit-tagged KIF7 WT or variant proteins (green) and either untreated (−SAG) or treated with SAG for 4 h (+SAG). The cells were fixed and stained with antibodies against acetylated tubulin (cilium; red), pericentrin (basal body; magenta), and with DAPI (nucleus; blue). Arrowheads indicate tips of cilia. Scale bar, 5 µm. (B–D) Quantification of the percent of cells exhibiting ciliary tip localization of KIF7 WT or variant proteins. The data are plotted to display statistical comparisons (B) between −SAG and +SAG conditions for each expressed protein (two-tailed t test) or (C, D) for the −SAG or +SAG conditions across KIF7 WT and variants (one-way ANOVA with Dunnet’s post hoc test). n.s., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001. Each spot indicates the mean of one independent experiment. Error bars, SEM across more than 30 cilia and more than or equal to three independent experiments.
Hh stimulation also resulted in a significant increase in the number of cells with the KIF7rigor variant at the tip of the cilium (68.3% of unstimulated cells and 94.3% of SAG-treated cells; Figure 3B). This result also does not support model 3 (Figure 1) being the primary mechanism for Hh-induced accumulation of KIF7 in the primary cilium. Because the KIF7rigor variant localized along cytosolic microtubules rather than at their plus ends (Figure 2 and Supplemental Figure S2), we were surprised that this variant localized at the cilium tip rather than along the shaft of the cilium (Figure 3A). It appears that this variant’s loss of autoinhibition (Figure 2B) alters its ability to enter and/or exit the cilium compartment, similar to the behavior of a pathogenic mutant that results in loss of autoinhibition (Blasius et al., 2021).
Interestingly, both motile KIF7motile_21A and KIF7motile_5C variants showed strong localization to the tip of the primary cilium without Hh stimulation (86.6% and 92.5% of cells, respectively) and modest increases upon SAG treatment (to 94.2% and 96.6% of cells, respectively; Figure 3B). Thus, access of KIF7 to the ciliary compartment is not restricted in the unstimulated state. These results suggest that a Hh-induced change in the ability of KIF7 to access the ciliary compartment (model 1; Figure 1) is unlikely to be the primary mechanism by which Hh stimulation causes an increase in KIF7 localization to the cilium tip. Further evidence against model 1 comes from our recent work demonstrating that the truncated version KIF7(1-558) localizes along the shaft of the primary cilium (Blasius et al., 2021), suggesting that the motor domain of KIF7 can access the cilium in the unstimulated state.
Microtubule-binding and motile mutants of KIF7 enable Gli transcription factors to localize at the tip of the cilium in the absence of Hh signaling
We investigated the effects of altering KIF7’s ability to bind to and/or move along microtubules on the ciliary localization of Gli2 and Gli3. We coexpressed 6xMyc-Gli2 or 6xMyc-Gli3 with WT or mutant KIF7 proteins in Kif7−/− MEFs and determined the ciliary tip localization of Gli2 and Gli3 without or with SAG treatment. In the control situation, Kif7−/− MEFs expressing the fluorescent protein mCitrine (mCit) were unable to accumulate Gli2 or Gli3 at the tip of the cilium in response to SAG treatment (Figure 4), consistent with previous work (Endoh-Yamagami et al., 2009; Liu et al., 2014).
FIGURE 4:

Increased microtubule binding or motility of KIF7 results in Gli accumulation at the cilium tip in the absence of Hh stimulation. (A, B) Representative images of the subcellular region containing the primary cilium in Kif7−/− MEFs coexpressing mCit-tagged KIF7 WT or variants (green) together with either (A) 6xMyc-Gli2 or (B) 6xMyc-Gli3. The cells were untreated (−SAG) or treated with SAG for 4 h (+SAG) and then the cells were fixed and stained with antibodies against the Myc tag (Gli proteins; red) and Arl13b (primary cilium; magenta). Each fluorescence channel is offset by 10 pixels for clarity. Arrowheads indicate tips of cilia. Scale bar, 2 μm. (C, D) Quantification of the percent of Kif7−/− MEF cells with ciliary tip localization of (C) 6xMyc-Gli2 or (D) 6xMyc-Gli3 when coexpressed with KIF7-mCit WT or variant proteins and either untreated (−SAG) or treated with SAG (+SAG). Each spot indicates the mean of one independent experiment. Error bars, SEM for more than 30 cilia across three independent experiments. n.s., not significant; **, p < 0.01; ***, p < 0.001 (two-tailed t test).
Expression of KIF7 rescued the Hh-stimulated increase in tip localization of Gli2 and Gli3. For Gli2, the ciliary tip localization increased from 36.0% to 75.0% of cells upon SAG stimulation (Figure 4, A and C) and for Gli3, the ciliary tip localization increased from 11.8% to 68.3% of cells upon SAG stimulation (Figure 4, B and D). Interestingly, expression of KIF7noMT had a similar rescuing effect on Hh-induced ciliary tip localization of Gli2 (from 37.6% to 77.6% of cells; Figure 4, A and C) and Gli3 (from 15.7% to 70.9% of cells; Figure 4, B and D). This result indicates that the ability of KIF7 to bind to microtubules is dispensable for the Hh-induced increase in ciliary localization of both Gli2 and Gli3. This finding also does not support model 3 (Figure 1) as the primary mechanism for Hh-induced regulation of KIF7 or Gli protein localization.
In contrast, expression of the rigor or motile KIF7 variants had a dramatic effect on Gli localization in the absence of Hh pathway activation. Coexpression with the KIF7rigor mutant resulted in localization of Gli2 to the tip of the cilium in 66.1% of cells and Gli3 in 67.2% cells in the unstimulated state (Figure 4). SAG treatment resulted in a modest increase in ciliary tip localization for Gli2 (to 84.1% of cells) and Gli3 (74.4% of cells). Similar results were obtained upon coexpression of Gli2 and Gli3 with the active motors KIF7motile_21A and KIF7motile_5C as both Gli2 and Gli3 showed a high percentage of ciliary tip localization (>69.6%) in the absence or presence of SAG treatment (Figure 4). We note that this is not simply due to overexpression since overexpression of WT KIF7 was not sufficient to promote localization of Gli proteins to the tips of cilia (Figure 4 and Liu et al., 2014).
Overall, these results demonstrate that the accumulation of Gli transcription factors at the tip of the primary cilium parallels that of KIF7. This work extends previous findings demonstrating that KIF7 can bind to both Gli2 and Gli3 proteins (Cheung et al., 2009; Endoh-Yamagami et al., 2009) by further delineating the sequences necessary for interaction with the Gli proteins to outside of the KIF7 motor domain. This work also extends previous findings demonstrating that KIF7 is necessary for Gli localization to the primary cilium (Endoh-Yamagami et al., 2009; Liu et al., 2014) by demonstrating that the interaction between KIF7 and the Gli proteins is sufficient for localization of the latter to the cilium. As the Gli proteins interact with the IFT kinesin-2 motor through synergistic interactions with KAP3 and KIF3A (Carpenter et al., 2015), our results suggest that these interactions may be mediated or facilitated by KIF7.
IFT kinesin-2 is required for Hh-induced accumulation of KIF7 in the primary cilium
The ability of KIF7noMT to accumulate at the tip of the primary cilium in response to Hh signaling suggests that another kinesin motor drives the ciliary trafficking of KIF7. Although the kinesin-2 KIF3A/KIF3B/KAP complex is the main anterograde motor, several other kinesins have been identified to play a role in ciliary transport and/or assembly (Lechtreck, 2015; He et al., 2017; Reilly and Benmerah, 2019). We recently developed inhibitable forms of the kinesin-2 motor KIF3A/KIF3B/KAP by appending homodimerizing DmrB domains to the motor domains of KIF3A and KIF3B. The inhibitable KIF3A/KIF3B/KAP motor (hereafter referred as to i3Ai3B) is capable of rescuing cilium assembly and Hh signaling in Kif3a/Kif3b−/− cells (Engelke et al., 2019). However, in the presence of B/B homodimerizer, the i3A and i3B motor domains become crosslinked, the stepping motion of i3Ai3B along the microtubule is inhibited, and IFT is rapidly (within 2 min) stopped (Figure 5A; Engelke et al., 2019).
FIGURE 5:

Kinesin-2 inhibition blocks Hh-induced accumulation of KIF7 at the tip of the primary cilium. (A) Schematic of the engineered inhibitable KIF3A/KIF3B/KAP (i3Ai3B) and inhibition of its motility by B/B homodimerizer. (B) Kif3a−/−; Kif3b−/− cells were serum-starved and transfected with i3A, i3B, and KIF7-mCit WT or variants (green). Two days later, cells were either unstimulated (no SAG), or stimulated with SAG in combination with EtOH vehicle (+SAG) or 50 nM B/B inhibitor (+SAG, +B/B) for 45 min and then fixed and stained with an antibody against Arl13b (cilium; magenta). Arrowheads indicate the tips of cilia. Scale bar, 2 µm. (C) Quantification of the percentage of transfected and ciliated cells in which KIF7-mCit WT and variants accumulate at the tip of the cilium. Each spot indicates the mean of one independent experiment. Error bars, SEM for more than or equal to 30 cilia across three independent experiments. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 as determined by one-way ANOVA with post hoc Dunnet’s test.
The ability to rapidly and specifically block IFT driven by KIF3A/KIF3B/KAP enabled us to directly test this motor’s role in localization of KIF7 to the tip of the primary cilium. Kif3a/Kif3b−/− cells were cotransfected with plasmids for expression of i3Ai3B and KIF7. In the control condition (no SAG treatment, no kinesin-2 inhibition), KIF7 localized to the tip of the cilium in 25.6% of cells (Figure 5, B and C). Addition of SAG resulted in increased ciliary tip localization for KIF7 (to 70.7% of cells; Figure 5, B and C). However, upon inhibition of KIF3A/KIF3B/KAP (+B/B homodimerizer), only 2.8% of cells showed KIF7 ciliary tip localization in response to SAG treatment (Figure 5, B and C). These results indicate that IFT is responsible for the Hh-induced localization of KIF7 to the tip of the primary cilium.
To test whether KIF7’s ability to bind to or move along microtubules could play a synergistic role with its transport by IFT, Kif3a/Kif3b−/− cells were cotransfected with plasmids for expression of i3Ai3B with the KIF7 variants. SAG treatment resulted in a significant increase in KIF7noMT localized at the tip of the cilium (from 10.2% to 33.2% of cells), however this Hh-induced tip localization was completely blocked by inhibition of the kinesin-2 IFT motor (+B/B; Figure 5, B and C). In a similar manner, SAG treatment resulted in a significant increase in KIF7rigor localized at the tip of the cilium (from 67.0% to 82.4% of cells), however this Hh-induced tip localization was completely blocked by inhibition of the kinesin-2 IFT motor (+B/B; Figure 5, B and C). In contrast, inhibition of KIF3A/KIF3B/KAP did not decrease the strong ciliary tip localization of KIF7motile_5C and KIF7motile_21A (>83.3% in all conditions; Figure 5, B and C).
These results provide strong support for model 2 (Figure 1) by demonstrating that the Hh-induced increase in KIF7 at the tip of the primary cilium requires the canonical IFT motor, the kinesin-2 KIF3A/KIF3B/KAP. This finding is consistent with recent work demonstrating that KIF7’s ciliary tip localization does not require the homodimeric kinesin-2 motor KIF17 in hTERT-RPE cells (Schwarz et al., 2017). We propose that Hh stimulation increases the loading of KIF7 and Gli proteins onto IFT trains. The premise for this proposal is pioneering work in Chlamydomonas where different cellular conditions dictate the differential loading of ciliary proteins onto IFT trains (Pan and Snell, 2005; Wren et al., 2013; Craft et al., 2015; Hunter et al., 2018). The loading of KIF7 onto IFT trains does not appear to be hindered or facilitated by KIF7’s ability to bind to microtubules as the KIF7noMT and KIF7rigor variants show similar Hh-stimulated increases in tip localization as KIF7 (Figure 3).
IFT-dependent transport of KIF7 and Gli proteins can explain why the KIF7rigor variant and associated Gli proteins accumulate at the cilium tip rather than along the axonemal shaft (Figures 3 – 5). That is, KIF7 may be carried to the cilium tip in a conformation where its motor domain, even in the rigor state, cannot access the microtubule surface. Alternatively, the microtubule binding of KIF7 may be weaker than that of KIF3A/KIF3B/KAP and thus unable to act as a “brake” on IFT motility during transport.
CONCLUSIONS
We demonstrate that KIF7’s ability to bind to microtubules is dispensable for its own Hh-induced localization at the tip of the primary cilium as well as for Hh-induced ciliary localization of Gli2 and Gli3 transcription factors. Rather, the IFT kinesin-2 motor KIF3A/KIF3B/KAP is required for KIF7’s localization to the cilium tip upon Hh pathway activation, thus providing strong support for model 2 (Figure 1). Further work is needed to determine how Hh pathway activation increases the loading of KIF7 and Gli proteins onto IFT trains. A direct test of model 4 (Figure 1) requires identification of KIF7 binding partners at the cilium tip and it remains possible that anchoring of KIF7 at the cilium tip (model 4) may work synergistically with a Hh-induced increase in IFT of KIF7 (model 2). Motile variants of KIF7 presumably bypass IFT to drive their own and Gli accumulation at the plus ends of axonemal microtubules, suggesting that the immotile behavior of KIF7 is critical for restricting cilium tip localization of KIF7 and Gli proteins in the absence of Hh stimulation.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Plasmids
6xmyc-Gli2, 6xmyc-Gli3 and MmKIF7 (FL) plasmids were gifts from Benjamin L. Allen (University of Michigan, MI). Mouse KIF7 was amplified by PCR and subcloned in mCitrine-N1 vector (based on Takara Bio Inc.’s EYFP-N1 vectors). KIF7noMT-mCit (H298A/R302A/K305A) was generated using QuickChange site-directed mutagenesis. KIF7motile_5C [KIF5C(1-379)-KIF7(379-1348)-mCit] and KIF7motile_21A [KIF21A (1-409)-KIF7(379-1348)-mCit] chimeric proteins were generated using overlap extension PCR. Rat KIF5C(1-379) fragment was amplified from KIF5C(1-560) plasmid (Cai et al., 2009) and human KIF21A(1-409) fragment was amplified from KIF21A plasmid (Huang and Banker, 2012), which was a gift from G. Banker (Oregon Health Sciences, Portland, OR). KIF7rigor mutant was generated using overlap extension PCR, in which a DNA fragment containing the L5 sequences from KIF5C was synthesized and inserted into KIF7. The inhibitable kinesin-2 motor (i3Ai3B) constructs, DmrB-Δ12KIF3A and DmrB-Δ6KIF3B, have been described (Engelke et al., 2019). The truncated KIF7(1-558)noMT-mCit mutant was amplified by PCR from KIF7noMT-mCit and subcloned in mNeonGreen-N1 vector. All plasmids were verified by DNA sequencing.
Cell culture and transfection
COS-7 (monkey kidney fibroblast) cells obtained from ATCC (RRID: CVCL_0224) were cultured in DMEM (Life Technologies) with 10% (vol/vol) Fetal Clone III (HyClone) and 1% GlutaMAX (Life Technologies) at 37°C with 5% CO2. COS-7 cells were transfected with Trans-IT LT1 (Mirus), according to the manufacturer’s instructions. Male NIH 3T3 cells (MEFs), purchased from ATCC (Cat#CRL-1658 RRID: CVCL_0594), were cultured in DMEM (Life Technologies) supplemented with 10% fetal clone III (Hyclone) and 1% GlutaMAX (Life Technologies) at 37°C and 5% CO2. Kif3a;Kif3b−/− NIH 3T3 cells have been described previously (Engelke et al., 2019). Both NIH-3T3 cell lines were transfected using Lipofectamine 2000 (Life Technologies), according to the manufacturer’s instructions. Kif7−/− MEF cells were gifts from Stephane Angers (University of Toronto) (Liu et al., 2014). MEF cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1% GlutaMAX (Life Technologies) at 37°C and 5% CO2. MEF cells were transfected using a combination of Fugene HD (Promega; Cat. No. E2311) and Lipofectamine LTX (ThermoFisher; Cat. No. 15338100) according to Ishiguro et al. (2017). After transfection, confluent MEF cells were starved with 0.5% FBS in DMEM for 24 h to induce ciliation. 20 h posttransfection, the cells were treated with 400 nM SAG (Enzo Life Sciences) for 4 h to activate the Hedgehog signaling pathway. All cell lines are tested annually for Mycoplasma contamination.
Inhibition of i3Ai3B in Kif3a/Kif3b −/− NIH 3T3 cells
Kif3a;Kif3b−/− NIH 3T3 cells were seeded on cover glasses and 12 h later, the culture medium was switched to 1% Fetal Clone III (serum-starvation) and cells were co-transfected with i3Ai3B motors and KIF-mCit WT or variants. Two days later, cells were either treated with vehicle (0.1% ethanol final) or with 500 nM SAG (Enzo Life Sciences) in combination with vehicle or 50 nM B/B homodimerizer (Clontech). After 45 min of treatment, cells were fixed and stained.
Immunofluorescence
Cells were fixed with 3.7% (vol/vol) paraformaldehyde (Thermo Fisher Scientific) in PBS and permeabilized with 0.2% Triton X-100 in PBS and then blocked in blocking solution (0.2% fish skin gelatin in PBS). Primary and secondary antibodies were applied in blocking solution at room temperature for 1 h each. Nuclei were stained with DAPI (final concentration 10.9 µM). The glass coverslips were mounted in ProlongGold (Life Technologies). Primary antibodies: Mouse anti-β-tubulin (1:2000, E7; Developmental Studies Hybridoma Bank); Mouse anti-acetylated tubulin (1:10,000, T6793, Sigma); Rabbit anti-pericentrin (1:500, ab4448, Abcam); Rabbit anti-Arl13B (1:1000, 17711-1-AP; Proteintech); Mouse anti-myc (1:500, 9E10; Invitrogen); Mouse anti-polyglutamylated tubulin (1:1000, GT335; Life Sciences). Secondary antibodies used were 594 nm anti-rabbit, 594 nm anti-mouse and 680 nm anti-rabbit (1:500; Jackson ImmunoResearch Laboratories).
Fluorescence microscopy
Images were acquired on an inverted epifluorescence microscope (Nikon TE2000E) equipped with a 100× 1.40 numerical aperture (NA) or a 60×, 1.40 NA oil-immersion objective and a Photometrics CoolSnapHQ camera driven by NIS-Elements (Nikon) software. Image analysis was performed using ImageJ (National Institutes of Health [NIH]). Although the level of KIF7 expression does not influence its ability to localize to the tip of the primary cilium (Blasius et al., 2021), images were only acquired of ciliated cells expressing the indicated constructs at low levels. The cilium tip was distinguished from the base of the cilium by staining for polyglutamylated tubulin of the centrioles or pericentrin of the pericentriolar material. A region of interest (ROI) at the tip of the cilium was selected in the Arl13b (cilium membrane marker) channel. The same ROI was then applied to the KIF7 and/or Gli images and the fluorescence intensity was measured at the cilium tip. The same ROI was applied to a region of the cell lacking the primary cilium and this background fluorescence was subtracted from the cilium tip fluorescence. If the resulting background-subtracted fluorescence was >0, then the KIF7 or Gli protein was designated as positive for cilium tip localization.
Cell lysates
To prepare cell lysates for the fluorescence-based in vitro microtubule-binding assay, COS-7 cells were collected 16 h posttransfection. The cells were harvested by low-speed centrifugation at 3000 × g for 3 min at 4°C. The pellet was rinsed once in PBS and resuspended in ice-cold lysis buffer (25 mM HEPES/KOH, 115 mM potassium acetate, 5 mM sodium acetate, 5 mM MgCl2, 0.5 mM ethylene glycol tetraacetic acid (EGTA), and 1% Triton X-100, pH 7.4) freshly supplemented with 1 mM ATP, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitors (P8340; Sigma-Aldrich). After the lysate was clarified by centrifugation at 20,000 × g for 10 min at 4°C, aliquots of the supernatant were snap-frozen in liquid nitrogen and stored at −80°C until further use. The amount of motor in the COS-7 lysates was normalized across constructs by a dot-blot, in which the same volumes of COS-7 lysates were spotted onto a nitrocellulose membrane that was air-dried for 1 h and immunoblotted with a monoclonal primary antibody to GFP (66002-1; Proteintech) and secondary antibody 680 nm–anti-mouse (Jackson ImmunoResearch Laboratories) at room temperature for 1 h each. The fluorescence intensity of the spots on the nitrocellulose membrane was detected by Azure c600 (Azure Biosystems) and quantified to normalize the motor concentration across lysates using Fiji/ImageJ (NIH).
Fluorescence-based in vitro microtubule-binding assay
The assays were performed at room temperature in a flow cell (∼10 µl volume) assembled by attaching a clean no. 1.5 coverslip to a glass slide with two strips of double-sided tape. HiLyte647-labeled microtubules were polymerized from purified tubulin including 10% Hily647-labeled tubulin (Cytoskeleton) in BRB80 buffer (80 mM Pipes/KOH, pH 6.8, 1 mM MgCl2, and 1 mM EGTA) supplemented with 1 mM GTP at 37°C for 30 min. Polymerized microtubules were stored at room temperature in the dark for further use after the addition of five volumes of prewarmed BRB80 containing 20 μM taxol and an additional 1 h incubation at 37°C. Polymerized microtubules were diluted in BRB80 buffer containing 10 μM taxol and added into a flow cell and incubated for 5 min for nonspecific adsorption onto the coverslip. Subsequently, blocking buffer (15 mg/ml bovine serum albumin [BSA] and 10 μM taxol in P12 buffer [12 mM Pipes/KOH, pH 6.8, 2 mM MgCl2, and 1 mM EGTA]) was added into the flow cell and incubated for 5 min to prevent nonspecific binding of kinesin motors onto the coverslip surface. Finally, kinesin motors in the motility mixture (0.5–1 μl of cell lysate, 2 mM ATP or AMPPNP, 0.4 mg/ml casein, 6 mg/ml BSA, 10 μM taxol, and oxygen scavenging [1 mM DTT, 1 mM MgCl2, 10 mM glucose, 0.2 mg/ml glucose oxidase, and 0.08 mg/ml catalase] in P12 buffer) was added to the flow cell. The flow cell was sealed with molten paraffin wax and imaged by TIRF microscopy using an inverted microscope Ti-E/B (Nikon) equipped with the perfect focus system (Nikon), a 100× 1.49 NA oil-immersion total internal reflection fluorescence (TIRF) objective (Nikon), three 20-mW diode lasers (488 nm, 561 nm, and 640 nm), and an electron-multiplying charge-coupled device detector (iXon X3DU897; Andor Technology). The fluorescence intensities of motors along the microtubules were measured using Fiji/ImageJ (NIH), and the fluorescence intensity of an adjacent region was subtracted as background.
Protein sequence analysis
Sequence alignments and percent amino acid identity between the core motor domains of rat kinesin-1 KIF5C (amino acids 1–324), mouse kinesin-4 KIF7 (amino acids 1–347), and human kinesin-4 KIF21A (amino acids 1–368) were determined using Clustal Omega (1.2.4; EMBL-EBI).
Statistical analysis
Statistical analyses were performed and graphs were generated using Prism software (GraphPad). Comparisons between −SAG and +SAG conditions for each expressed protein were carried out using a two-tailed t test (Figures 3B, and 4, C and D). Multiple comparisons across WT and variant proteins (Figure 3, C and D) or across treatments for the same protein (Figure 5B) were carried out using one-way ANOVAs with post hoc Dunnett’s test.
Supplementary Material
Acknowledgments
We thank members of the Verhey laboratory for helpful discussions and Stephane Angers for Kif7 –/– MEFs. The work was funded by grants no. R35GM-131744 and R01GM-116204 to K.J.V. and no. R01GM-131744 to K.J.V. and Benjamin Allen from the National Institutes of Health.
Abbreviations used:
- Hh
hedgehog
- i3Ai3B
inhibitable KIF3A/KIF3B kinesin
- IFT
intraflagellar transport
- mCit
monomeric Citrine
- SAG
smoothened agonist
- SUFU
suppressor of fused
- WT
wild-type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E21-04-0215) on October 27, 2021.
REFERENCES
- Bangs F, Anderson KV (2017). Primary cilia and mammalian hedgehog signaling. Cold Spring Harb Perspect Biol 9, a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius TL, Yue Y, Prasad R, Liu X, Gennerich A, Verhey KJ (2021). Sequences in the stalk domain regulate auto-inhibition and ciliary tip localization of the immotile kinesin-4 KIF7. J Cell Sci 134, jcs258464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, McEwen DP, Martens JR, Meyhofer E, Verhey KJ (2009). Single molecule imaging reveals differences in microtubule track selection between Kinesin motors. PLoS Biol 7, e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter BS, Barry RL, Verhey KJ, Allen BL (2015). The heterotrimeric kinesin-2 complex interacts with and regulates GLI protein function. J Cell Sci 128, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, Beachy PA (2002). Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA 99, 14071–14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Desai J, Miranda CJ, Duncan JS, Qiu W, Nugent AA, Kolpak AL, Wu CC, Drokhlyansky E, Delisle MM, et al. (2014). Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron 82, 334–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HO, Zhang X, Ribeiro A, Mo R, Makino S, Puviindran V, Law KK, Briscoe J, Hui CC (2009). The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci Signal 2, ra29. [DOI] [PubMed] [Google Scholar]
- Cochran JC (2015). Kinesin motor enzymology: chemistry, structure, and physics of nanoscale molecular machines. Biophys Rev 7, 269–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021. [DOI] [PubMed] [Google Scholar]
- Craft JM, Harris JA, Hyman S, Kner P, Lechtreck KF (2015). Tubulin transport by IFT is upregulated during ciliary growth by a cilium-autonomous mechanism. J Cell Biol 208, 223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevel IM, Lockhart A, Cross RA (1996). Weak and strong states of kinesin and ncd. J Mol Biol 257, 66–76. [DOI] [PubMed] [Google Scholar]
- Endoh-Yamagami S, Evangelista M, Wilson D, Wen X, Theunissen JW, Phamluong K, Davis M, Scales SJ, Solloway MJ, de Sauvage FJ, Peterson AS (2009). The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr Biol 19, 1320–1326. [DOI] [PubMed] [Google Scholar]
- Engelke MF, Waas B, Kearns SE, Suber A, Boss A, Allen BL, Verhey KJ (2019). Acute inhibition of heterotrimeric kinesin-2 function reveals mechanisms of intraflagellar transport in mammalian cilia. Curr Biol 29, 1137–1148. e1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK (2005). Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 1, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Agbu S, Anderson KV (2017). Microtubule motors drive hedgehog signaling in primary cilia. Trends Cell Biol 27, 110–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Subramanian R, Bangs F, Omelchenko T, Liem KF Jr, Kapoor TM, Anderson KV (2014). The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat Cell Biol 16, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CF, Banker G (2012). The translocation selectivity of the kinesins that mediate neuronal organelle transport. Traffic 13, 549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R (2010). The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 24, 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter EL, Lechtreck K, Fu G, Hwang J, Lin H, Gokhale A, Alford LM, Lewis B, Yamamoto R, Kamiya R, et al. (2018). The IDA3 adapter, required for intraflagellar transport of I1 dynein, is regulated by ciliary length. Mol Biol Cell 29, 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro K, Watanabe O, Nakamura M, Yamamura T, Matsushita M, Goto H, Hirooka Y (2017). Combinational use of lipid-based reagents for efficient transfection of primary fibroblasts and hepatoblasts. Biotechniques 63, 37–39. [DOI] [PubMed] [Google Scholar]
- Jiang S, Mani N, Wilson-Kubalek EM, Ku PI, Milligan RA, Subramanian R (2019). Interplay between the kinesin and tubulin mechanochemical cycles underlies microtubule tip tracking by the non-motile ciliary kinesin Kif7. Dev Cell 49, 711–730.e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong JH, Siebold C, Rohatgi R (2019). Biochemical mechanisms of vertebrate hedgehog signaling. Development 146, dev166892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF (2015). IFT-cargo interactions and protein transport in cilia. Trends Biochem Sci 40, 765–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem KF Jr, He M, Ocbina PJ, Anderson KV (2009). Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci USA 106, 13377–13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Couzens AL, Deshwar AR, McBroom-Cerajewski LDB, Zhang X, Puviindran V, Scott IC, Gingras AC, Hui CC, Angers S (2014). The PPFIA1-PP2A protein complex promotes trafficking of Kif7 to the ciliary tip and Hedgehog signaling. Sci Signal 7, ra117. [DOI] [PubMed] [Google Scholar]
- Marx A, Hoenger A, Mandelkow E (2009). Structures of kinesin motor proteins. Cell Motil Cytoskeleton 66, 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N (1995). Point mutation of adenosine triphosphate-binding motif generated rigor kinesin that selectively blocks anterograde lysosome membrane transport. J Cell Biol 131, 1039–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Snell WJ (2005). Chlamydomonas shortens its flagella by activating axonemal disassembly, stimulating IFT particle trafficking, and blocking anterograde cargo loading. Dev Cell 9, 431–438. [DOI] [PubMed] [Google Scholar]
- Prevo B, Scholey JM, Peterman EJG (2017). Intraflagellar transport: mechanisms of motor action, cooperation, and cargo delivery. FEBS J 284, 2905–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Li X (2020). Mechanistic insights into the generation and transduction of Hedgehog Signaling. Trends Biochem Sci 45, 397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly ML, Benmerah A (2019). Ciliary kinesins beyond IFT: Cilium length, disassembly, cargo transport and signalling. Biol Cell 111, 79–94. [DOI] [PubMed] [Google Scholar]
- Schwarz N, Lane A, Jovanovic K, Parfitt DA, Aguila M, Thompson CL, da Cruz L, Coffey PJ, Chapple JP, Hardcastle AJ, Cheetham ME (2017). Arl3 and RP2 regulate the trafficking of ciliary tip kinesins. Hum Mol Genet 26, 2480–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vaart B, van Riel WE, Doodhi H, Kevenaar JT, Katrukha EA, Gumy L, Bouchet BP, Grigoriev I, Spangler SA, Yu KL, et al. (2013). CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev Cell 27, 145–160. [DOI] [PubMed] [Google Scholar]
- Webb S, Mukhopadhyay AG, Roberts AJ (2020). Intraflagellar transport trains and motors: insights from structure. Semin Cell Dev Biol 107, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehlke G, Ruby AK, Hart CL, Ly B, Hom-Booher N, Vale RD (1997). Microtubule interaction site of the kinesin motor. Cell 90, 207–216. [DOI] [PubMed] [Google Scholar]
- Wren KN, Craft JM, Tritschler D, Schauer A, Patel DK, Smith EF, Porter ME, Kner P, Lechtreck KF (2013). A differential cargo-loading model of ciliary length regulation by IFT. Curr Biol 23, 2463–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Blasius TL, Zhang S, Jariwala S, Walker B, Grant BJ, Cochran JC, Verhey KJ (2018). Altered chemomechanical coupling causes impaired motility of the kinesin-4 motors KIF27 and KIF7. J Cell Biol 217, 1319–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Jia J, Liu A (2010). Coordinated translocation of mammalian Gli proteins and suppressor of fused to the primary cilium. PLoS One 5, e15900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.