

Abstract
Triangulene, known as Clar's hydrocarbon, is a prototypical non-Kekulé diradical comprised of six benzenoid rings fused in a triangular shape. We synthesized and characterized its trimesityl derivative, illustrating that three bulky substituents installed in the centers of the zigzag edges suffice to protect all reactive positions. This work brings prospects to use triangulene and its open-shell analogs in spintronic materials via solution-phase synthesis.
Three substituents shield the reactive core of Clar’s hydrocarbon triangulene and make it persistent at room temperature in solution. Image of Erich Clar from Dr. Winfried Willicks (1926–2010) is courtesy of Glasgow University, School of Chemistry.
Open-shell graphene fragments1–4 have recently emerged as promising materials for spintronics.5,6 It is envisioned that the spins of unpaired electrons in these molecules can function as information carriers and spin-based devices made of these materials can meet the increasing demand for speed and miniaturization in information processing.7,8 The main advantage of graphene-based materials over other materials like metals is the small spin–orbit coupling of carbon, a pre-requisite for achieving long spin-state lifetimes at room temperature.9,10
Graphene fragments with two or more unpaired π-electrons belong to the family of non-Kekulé conjugated hydrocarbons,11 which have fewer π-bonds than permitted by the standard rule of valence. They are characterized by zigzag edges and ground states of highest possible multiplicity. A prototypical textbook example is the smallest non-Kekulé graphene fragment triangulene,12,13 also known as Clar's hydrocarbon, comprised of six benzenoid rings fused in a triangular shape with three zigzag edges (Fig. 1, left). Due to its symmetry, triangulene has two non-bonding molecular orbitals, each occupied by one electron, and a triplet ground state that conforms to Ovchinnikov's rule,14 or Lieb's theorem.15 The triplet ground state of triangulene is also predicted by multi-configurational calculations,16 which estimate the energy difference between the triplet ground state and the lowest singlet state to be in the range 13–16 kcal mol−1. The majority of the spin density in triangulene is localized at its zigzag edges,17 which results in its high reactivity.
Fig. 1. Triangulene derivatives prepared since the pioneering work of Erich Clar.

The chemistry of non-Kekulé hydrocarbons was pioneered by Erich Clar, who attempted the first synthesis of pristine triangulene in the early 1950s.12 Although he successfully prepared several direct precursors of triangulene, he could not isolate and characterize it. A derivative, stable in oxygen-free solution at room temperature, was achieved by Bushby et al. via instalment of oxy groups at the centers of the edges (Fig. 1, red arrows).18,19 The triplet ground state of the obtained diradical trianion was confirmed by the detection of a half-field line in the continuous wave (cw) electron paramagnetic resonance (EPR) spectrum in a frozen matrix at 13 K. Kinetic stabilization via introduction of steric hindrance was demonstrated by Nakasuji et al. in 2001 who installed bulky tert-butyl groups at the vertices of triangulene (Fig. 1, black arrows).20 Blocking the vertices was only partially successful because the most reactive positions (red arrows) remained exposed and oligomerization occurred even at temperatures below 0 °C. The EPR characterization of the mixture was performed in frozen solution. Due to the presence of a monoradical impurity, however, a fully resolved and characterized EPR spectrum could not be acquired and the half-field line expected for a triplet state was too weak to be detected on account of a small D value.
Pristine triangulene was synthesized in 2017 by Pavliček et al. on various surfaces under ultra-high vacuum conditions.21 Although it was shown that triangulene was formed and does not bind to the surface, using current technology it is not possible to determine the ground-state multiplicity of this molecule. Following this seminal work, the on-surface synthesis of π-extended [4]-22, [5]-23 and [7]triangulene24 homologs as well as π-extended triangulene structures, namely, dimer,25 bowtie9 and a rhombus-shaped graphene fragment,10 was accomplished. Most recently, Fasel et al. achieved the on-surface synthesis of a spin chain made of triangulene units as a model system to study correlations in quantum spin-liquid phases, demonstrating the possibility to implement measurement-based quantum computation.26
Simultaneously to this work,27 Shintani et al.28 reported the synthesis of a persistent triangulene derivative kinetically stabilized by six substituents (Fig. 1, middle), which allowed its isolation in the solid state and structure confirmation by X-ray crystallography. The triplet ground state was confirmed by means of SQUID magnetometry and EPR spectroscopy. We envisioned that three bulky substituents installed in the centers of the edges would result in a sufficient steric protection of all nine reactive positions: the most reactive central (Fig. 1, red arrows) as well as their neighboring positions.
We explored several approaches to achieve the synthesis of such a trisubstituted triangulene (Fig. S129,30 and S231, ESI†). The approach based on the method of Wu et al.23 was successfully applied in the synthesis of dihydro-precursors 1a,b equipped with three mesityl substituents (Scheme 1). Its preparation involved two key building blocks: 9-bromo-10-mesitylanthracene (3) and (2,6-bis(methoxymethyl)phenyl)boronic acid.32,33 Anthracene derivative 3 was synthesized via a nucleophilic addition of mesitylmagnesium bromide to anthrone (2; step a), followed by the in situ elimination of water and electrophilic bromination (step b). The boronic acid and 3 were combined in a Suzuki cross-coupling reaction yielding intermediate 4 (step c). Dialdehyde intermediate 7 was prepared in three steps from 4, including the substitution of the methoxy groups with bromide (d), substitution of bromides with acetate followed by ester hydrolysis (e) and Swern oxidation (f). Upon the addition of mesitylmagnesium bromide (step g), the formed diol underwent a Lewis acid-catalyzed cyclization to provide a mixture of dihydro-triangulenes 1a and 1b (step h), which could not be separated and with 1a being the major isomer. Based on DFT which predicts 1b to be more stable by ∼7 kcal mol−1, the composition of the isomeric mixture suggests that the final cyclization step proceeds under kinetic control. The final oxidation to generate Mes3-Tr was performed using p-chloranil. The precise addition of oxidizing agent allows the oxidation to be performed in a stepwise fashion; first a mixture of monoradicals 8a + 8b is formed, then diradical Mes3-Tr. This differs from the procedure reported by Shintani et al.,28 where the last step must be performed under reductive conditions. The oxidation method allows formation of pristine triangulene monoradical and its analysis by X-ray diffraction (Fig. S5, ESI†). The solid-state structure of 8a confirmed that the mesityl groups are oriented perpendicularly to the triangulene plane. Therefore, spin delocalization to the substituents as well as perturbation of the electronic structure of triangulene should be negligible.
Scheme 1. Reaction conditions: (a) (i) MesMgBr, THF, 65 °C, (ii) aq. HCl, 65 °C, (b) NBS, CHCl3, 60 °C, (c) (2,6-bis(methoxymethyl)phenyl)boronic acid, K2CO3, Pd2dba3, DPEPhos, toluene/ethanol/water, 100 °C, (d) 33% HBr in CH3COOH, DCM, r.t., (e) (i) CH3COOK, TBAB, DMF, 100 °C, (ii) KOH, THF, 80 °C, (f) (COCl)2, DMSO, NEt3, DCM, –78 °C to r.t., (g) MesMgBr, THF, 0 °C, (h) BF3·Et2O, DCM, r.t.

The oxidation process was followed by UV-vis spectroscopy (Fig. 2). The sample was sufficiently stable to be handled outside the glovebox using deoxygenated solvents and routine Schlenk techniques. The UV-vis spectrum of 1 shows two distinct absorption bands, which can be assigned to 1a and 1b (Fig. 2 and Fig. S4, ESI†) based on TD-DFT calculations (Tables S5 and S6, ESI†). Isomer 1a shows the characteristic vibronic progression of an anthracene subunit (Fig. S3, ESI†) shifted bathochromically due to an additional coplanar benzene ring in the triangulene core. The oxidation of 1 provided initially a mixture of two monoradical species 8 (Fig. 2). While 8a can be formed from both 1a and 1b, monoradical 8b ensues only from oxidation of 1b.
Fig. 2. UV-vis spectra recorded during the oxidation process for toluene solutions of a dihydro-precursor mixture 1a + 1b (black trace) with 5 equivalents of p-chloranil (CA), providing, after 2 hours, a mixture of monoradical species 8a + 8b (red trace) and, after 3 days, the diradical species Mes3-Tr (yellow trace) together with the excess CA and formed reduced CA. The oxidation was performed at a concentration of 2.5 mM. For the UV-vis measurements, an aliquot sample of this solution was diluted to 2.5 × 10−5 M.

According to the maximum intensity of the characteristic absorption bands (490 nm, 8a and 527 nm, 8b) that match the TD-DFT calculations (Tables S7 and S8, ESI†) well, the concentration of both monoradicals 8 reached a maximum in 2 hours and then decreased as Mes3-Tr was formed. The oxidation process is clean; the disappearance of signals in the 1H NMR spectrum demonstrates full consumption of 1 with no side products and EPR spectroscopy (vide infra) shows clean transformation of 8 to Mes3-Tr. The presence of Mes3-Tr in solution was also confirmed by (+)ESI-HRMS (ESI†).
The magnetic properties of the species formed after chemical oxidation of 1a + 1b were characterized by EPR spectroscopy. Fig. 3A and B shows the continuous wave EPR spectra measured at the X-band (9.75 GHz) at room temperature at different times after addition of one equivalent of p-chloranil to a 0.5 mM solution of 1a + 1b in toluene. Both spectra exhibit a width of roughly 3.5 mT and are characterized by a pronounced hyperfine structure originating from the coupling of unpaired electron spin(s) to the protons. The isotropic g value is very similar in both cases and amounts to 2.0025 and 2.0027, respectively.
Fig. 3. EPR characterization of the mono- and diradical species. Monoradical mixture 8a + 8b and diradical Mes3-Tr formed after chemical oxidation of solutions of a dihydro-precursor mixture (1a + 1b) in toluene using p-chloranil. (A and B) Room temperature cw EPR spectra of a 0.5 mM solution in toluene after addition of 1 equivalent of the oxidant recorded at the X-band about 2 hours (A) and 6 hours (B) after oxidant addition together with a numerical simulation of the data using hyperfine couplings obtained from DFT calculations. (C and D) Transient nutation data recorded at the Q-band on frozen solutions (120 K) of a 2.5 mM solution in toluene shortly after addition of either 0.1 (C) or 5 (D) equivalents of the oxidant. The horizontal dotted lines indicate the expected normalized frequencies of 1.0 for a doublet state (monoradical) and 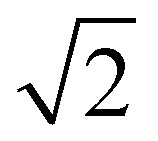 for a triplet state (diradical).
for a triplet state (diradical).

Shortly after the addition of oxidant or when small amounts of the oxidant were added (Fig. 3A), the cw EPR spectrum was found to consist of a range of very narrow lines with an average peak separation of ∼0.04 mT. This spectrum shows close resemblance with the cw EPR spectrum reported previously for the triangulene monoradical with three tert-butyl substituents20 and is therefore assigned to the monoradical species 8a + 8b. Addition of a large excess of p-chloranil or prolonged oxidation time resulted in a symmetric cw EPR spectrum (Fig. 3B) characterized by far fewer well-resolved lines with an average peak separation of approximately 0.12 mT, assigned to Mes3-Tr.
To support the assignment of the two measured EPR spectra to the monoradical and triplet diradical species, DFT calculations of the corresponding spin densities and hyperfine coupling constants (see the ESI†) were carried out using the ORCA program package (Version 4.0).34 We then simulated the expected EPR spectra based only on the computed isotropic hyperfine coupling constants using the ‘pepper’ routine35 of EasySpin.36 All calculated couplings smaller than 0.5 MHz were neglected as they are not resolved in the experimental spectra and only contribute to the linewidth. The resulting simulations are shown in Fig. 3A and B and the ESI.† When applying a global scaling factor of 0.93 to all calculated proton couplings for the triplet diradical, a near perfect match with the experimental spectrum was obtained (Fig. 3B), strongly supporting the triplet nature of Mes3-Tr. The simulation of the monoradical spectrum is less straightforward since two different isomers are expected to contribute and it is difficult to determine their ratio. For the simulation of the mixture of monoradicals 8a + 8b, a ratio of 50 : 50 was assumed. Although the agreement is not perfect, the size of the hyperfine couplings and main features of the monoradical spectrum are well reproduced (Fig. 3A).
As an additional proof for the assignment of the spectrum in Fig. 3B to the triplet diradical Mes3-Tr, transient nutation experiments were performed on two different samples of an oxidized mixture of 1a + 1b in frozen toluene at 120 K. The measurements were performed at Q-band frequencies (34.0 GHz) using the microwave pulse sequence ξ–τ–π–τ–echo, with τ = 140 ns and π = 40 ns, where the flip angle ξ was gradually increased by increasing the corresponding microwave pulse length in steps of 2 ns, starting at 20 ns, that is, 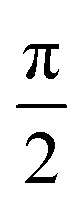 . For every magnetic field position, the obtained time traces were subjected to a Fourier transform to yield the frequency maps for 0.1 (Fig. 3C) and 5 equivalents (Fig. 3D) of the oxidant. The sample with 5 equivalents of p-chloranil was frozen shortly after oxidant addition to be sure that a mixture of 8 and the putative Mes3-Tr is obtained. The contribution of the signal of 8 (pure doublet species) is used here as an internal standard to obtain the reference frequency ω0, needed for a correct normalization of the frequency axis for determination of the spin multiplicity of Mes3-Tr. For a triplet state, a frequency of
. For every magnetic field position, the obtained time traces were subjected to a Fourier transform to yield the frequency maps for 0.1 (Fig. 3C) and 5 equivalents (Fig. 3D) of the oxidant. The sample with 5 equivalents of p-chloranil was frozen shortly after oxidant addition to be sure that a mixture of 8 and the putative Mes3-Tr is obtained. The contribution of the signal of 8 (pure doublet species) is used here as an internal standard to obtain the reference frequency ω0, needed for a correct normalization of the frequency axis for determination of the spin multiplicity of Mes3-Tr. For a triplet state, a frequency of 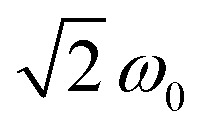 would be expected according to
would be expected according to 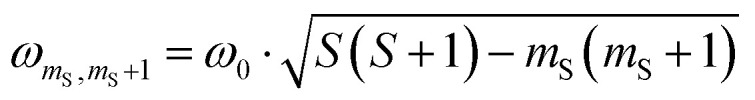 ,37–39 which is in perfect agreement with our experimental observations, as graphically illustrated in Fig. 3D (horizontal dotted lines).
,37–39 which is in perfect agreement with our experimental observations, as graphically illustrated in Fig. 3D (horizontal dotted lines).
In addition, variable-temperature cw EPR measurements of Mes3-Tr in frozen toluene in the range 120–170 K show that the signal intensity decreases with increasing temperature (Fig. S9, ESI†), strongly suggesting that the triplet is the ground state.
Room temperature X-band cw EPR spectra were recorded after 3 hours, 2 weeks and 3 weeks after oxidant addition. No significant changes in the shape or intensity of the signal were observed after 3 weeks (Fig. S10, ESI†), showing the outstanding stability of Mes3-Tr in deoxygenated solution. It is noteworthy that Mes3-Tr together with the hexasubstituted derivative28 are rare examples20,40 of purely organic triplet diradicals, where the triplet state could be detected in isotropic solution at room temperature. The reason might be the scarcity of persistent non-Kekulé diradicals prepared until now.
Due to high reactivity, the chemistry of non-Kekulé open-shell graphene fragments has until recently27,28 been limited to ultra-high vacuum and low-temperature conditions. In this work, we presented a persistent derivative of an iconic member of this class of compounds—triangulene—kinetically stabilized by only three substituents, which sterically shield all nine reactive positions. The persistence effect of the substituents was demonstrated by the kinetic stability of this compound for several weeks in solution at room temperature. The triplet ground state was validated by means of continuous wave, pulse and variable-temperature EPR spectroscopy. The diradical species were generated by means of stepwise oxidation, which allows formation of pristine monoradical species, inaccessible via the alternative reduction28 protocol. The realization of persistent derivatives of triangulene will motivate the synthesis of its analogs, enabling implementation of open-shell graphene fragments into solution-processed and solid materials.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Electronic supplementary information (ESI) available: Synthetic procedures and characterization data for all new compounds, UV-vis, EPR and crystallographic data, results of DFT calculations, NMR and HRMS spectra, and Cartesian coordinates. CCDC 2104838. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2cc00352j
References
- Morita Y. Suzuki S. Sato K. Takui T. Nat. Chem. 2011;3:197–204. doi: 10.1038/nchem.985. [DOI] [PubMed] [Google Scholar]
- Zeng W. Wu J. Chemistry. 2021;7:358–386. doi: 10.1016/j.chempr.2020.10.009. [DOI] [Google Scholar]
- Liu J. Feng X. Angew. Chem., Int. Ed. 2020;59:23386–23401. doi: 10.1002/anie.202008838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y. and Nishida S., in Stable Radicals, ed. R. G. Hicks, John Wiley & Sons, Ltd, Chichester, UK, 2010, pp. 81–145 [Google Scholar]
- Žutić I. Fabian J. Das Sarma S. Rev. Mod. Phys. 2004;76:323–410. doi: 10.1103/RevModPhys.76.323. [DOI] [Google Scholar]
- Bullard Z. Girão E. C. Owens J. R. Shelton W. A. Meunier V. Sci. Rep. 2015;5:7634. doi: 10.1038/srep07634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K. Nakazawa S. Rahimi R. Ise T. Nishida S. Yoshino T. Mori N. Toyota K. Shiomi D. Yakiyama Y. Morita Y. Kitagawa M. Nakasuji K. Nakahara M. Hara H. Carl P. Höfer P. Takui T. J. Mater. Chem. 2009;19:3739–3754. doi: 10.1039/B819556K. [DOI] [Google Scholar]
- Chappert C. Fert A. Van Dau F. N. Nat. Mater. 2007;6:813–823. doi: 10.1038/nmat2024. [DOI] [PubMed] [Google Scholar]
- Mishra S. Beyer D. Eimre K. Kezilebieke S. Berger R. Gröning O. Pignedoli C. A. Müllen K. Liljeroth P. Ruffieux P. Feng X. Fasel R. Nat. Nanotechnol. 2020;15:22–28. doi: 10.1038/s41565-019-0577-9. [DOI] [PubMed] [Google Scholar]
- Mishra S. Yao X. Chen Q. Eimre K. Gröning O. Ortiz R. Di Giovannantonio M. Sancho-García J. C. Fernández-Rossier J. Pignedoli C. A. Müllen K. Ruffieux P. Narita A. Fasel R. Nat. Chem. 2021;13:581–586. doi: 10.1038/s41557-021-00678-2. [DOI] [PubMed] [Google Scholar]
- Minkin V. I. Pure Appl. Chem. 1999;71:1919–1981. [Google Scholar]
- Clar E. Stewart D. G. J. Am. Chem. Soc. 1953;75:2667–2672. doi: 10.1021/ja01107a035. [DOI] [Google Scholar]
- Clar E. Stewart D. G. J. Am. Chem. Soc. 1954;76:3504–3507. doi: 10.1021/ja01642a044. [DOI] [Google Scholar]
- Ovchinnikov A. A. Theor. Chim. Acta. 1978;47:297–304. doi: 10.1007/BF00549259. [DOI] [Google Scholar]
- Lieb E. H. Phys. Rev. Lett. 1989;62:1201. doi: 10.1103/PhysRevLett.62.1201. [DOI] [PubMed] [Google Scholar]
- Das A. Müller T. Plasser F. Lischka H. J. Phys. Chem. A. 2016;120:1625–1636. doi: 10.1021/acs.jpca.5b12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz R. Boto R. A. García-Martínez N. Sancho-García J. C. Melle-Franco M. Fernández-Rossier J. Nano Lett. 2019;19:5991–5997. doi: 10.1021/acs.nanolett.9b01773. [DOI] [PubMed] [Google Scholar]
- Allinson G. Bushby R. J. Paillaud J. L. Oduwole D. Sales K. J. Am. Chem. Soc. 1993;115:2062–2064. doi: 10.1021/ja00058a076. [DOI] [Google Scholar]
- Allinson G. Bushby R. J. Paillaud J.-L. Thornton-Pett M. J. Chem. Soc., Perkin Trans. 1. 1995:385–390. doi: 10.1039/P19950000385. [DOI] [Google Scholar]
- Inoue J. Fukui K. Kubo T. Nakazawa S. Sato K. Shiomi D. Morita Y. Yamamoto K. Takui T. Nakasuji K. J. Am. Chem. Soc. 2001;123:12702–12703. doi: 10.1021/ja016751y. [DOI] [PubMed] [Google Scholar]
- Pavliček N. Mistry A. Majzik Z. Moll N. Meyer G. Fox D. J. Gross L. Nat. Nanotechnol. 2017;12:308–311. doi: 10.1038/nnano.2016.305. [DOI] [PubMed] [Google Scholar]
- Mishra S. Beyer D. Eimre K. Liu J. Berger R. Gröning O. Pignedoli C. A. Müllen K. Fasel R. Feng X. Ruffieux P. J. Am. Chem. Soc. 2019;141:10621–10625. doi: 10.1021/jacs.9b05319. [DOI] [PubMed] [Google Scholar]
- Su J. Telychko M. Hu P. Macam G. Mutombo P. Zhang H. Bao Y. Cheng F. Huang Z.-Q. Qiu Z. Tan S. J. R. Lin H. Jelínek P. Chuang F.-C. Wu J. Lu J. Sci. Adv. 2019;5:eaav7717. doi: 10.1126/sciadv.aav7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S. Xu K. Eimre K. Komber H. Ma J. Pignedoli C. A. Fasel R. Feng X. Ruffieux P. Nanoscale. 2021;13:1624–1628. doi: 10.1039/D0NR08181G. [DOI] [PubMed] [Google Scholar]
- Mishra S. Beyer D. Eimre K. Ortiz R. Fernández-Rossier J. Berger R. Gröning O. Pignedoli C. A. Fasel R. Feng X. Ruffieux P. Angew. Chem., Int. Ed. 2020;59:12041–12047. doi: 10.1002/anie.202002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S. Catarina G. Wu F. Ortiz R. Jacob D. Eimre K. Ma J. Pignedoli C. A. Feng X. Ruffieux P. Fernández-Rossier J. Fasel R. Nature. 2021;598:287–292. doi: 10.1038/s41586-021-03842-3. [DOI] [PubMed] [Google Scholar]
- This content is a preprint and has not been peer-reviewed, Aug 26, 2021,; Valenta L. Mayländer M. Kappeler P. Blacque O. Šolomek T. Richert S. Juríček M. ChemRxiv. 2021 doi: 10.33774/chemrxiv-2021-gsrtp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arikawa S. Shimizu A. Shiomi D. Sato K. Shintani R. J. Am. Chem. Soc. 2021;143:19599–19605. doi: 10.1021/jacs.1c10151. [DOI] [PubMed] [Google Scholar]
- Ribar P. Valenta L. Šolomek T. Juríček M. Angew. Chem., Int. Ed. 2021;60:13521–13528. doi: 10.1002/anie.202016437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribar P. Šolomek T. Juríček M. Org. Lett. 2019;21:7124–7128. doi: 10.1021/acs.orglett.9b02683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C. J. Wentworth K. J. Johnson R. P. Angew. Chem., Int. Ed. 2019;58:15793–15796. doi: 10.1002/anie.201907226. [DOI] [PubMed] [Google Scholar]
- Gu Y. Wu X. Gopalakrishna T. Y. Phan H. Wu J. Angew. Chem., Int. Ed. 2018;57:6541–6545. doi: 10.1002/anie.201802818. [DOI] [PubMed] [Google Scholar]
- van de Kuil L. A. Luitjes H. Grove D. M. Zwikker J. W. van der Linden J. G. M. Roelofsen A. M. Jenneskens L. W. Drenth W. van Koten G. Organometallics. 1994;13:468–477. doi: 10.1021/om00014a020. [DOI] [Google Scholar]
- Neese F. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2018;8:e1327. [Google Scholar]
- The ‘pepper’ routine was used here since routines typically used for cw EPR simulations in the fast motion regime cannot account for species with S = 1
- Stoll S. Schweiger A. J. Magn. Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Astashkin A. V. Schweiger A. Chem. Phys. Lett. 1990;174:595–602. doi: 10.1016/0009-2614(90)85493-V. [DOI] [Google Scholar]
- Mizuochi N. Ohba Y. Yamauchi S. J. Phys. Chem. A. 1997;101:5966–5968. doi: 10.1021/jp971569y. [DOI] [Google Scholar]
- Mizuochi N. Ohba Y. Yamauchi S. J. Phys. Chem. A. 1999;103:7749–7752. doi: 10.1021/jp9922559. [DOI] [Google Scholar]
- Li Y. Huang K.-W. Sun Z. Webster R. D. Zeng Z. Zeng W. Chi C. Furukawa K. Wu J. Chem. Sci. 2014;5:1908–1914. doi: 10.1039/C3SC53015A. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.