

Abstract
Defects in mitochondrial oxidative phosphorylation (OXPHOS) have been reported in COVID-19 patients, but the timing and organs affected vary among reports. Here, we reveal the dynamics of COVID-19 through transcription profiles in nasopharyngeal and autopsy samples from patients and infected rodent models. While mitochondrial bioenergetics is repressed in the viral nasopharyngeal portal of entry, it is up regulated in autopsy lung tissues from deceased patients. In most disease stages and organs, discrete OXPHOS functions are blocked by the virus, and this is countered by the host broadly up regulating unblocked OXPHOS functions. No such rebound is seen in autopsy heart, results in severe repression of genes across all OXPHOS modules. Hence, targeted enhancement of mitochondrial gene expression may mitigate the pathogenesis of COVID-19.
Graphical Abstract
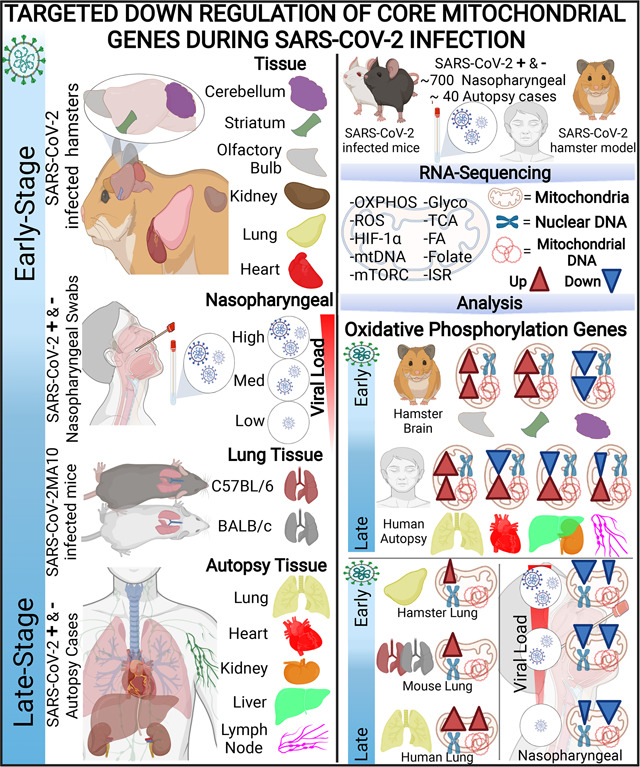
One-Sentence Summary:
Covid-19 is associated with targeted inhibition of mitochondrial gene transcription.
COVID-19 is currently responsible for more than 6 million deaths and has infected close to 375 million people. While generally considered an inflammatory disease, recent evidence suggests that SARS-CoV-2 infection also inhibits mitochondrial bioenergetics, and that mitochondrial inhibition can activate of the inflammasome. Thus, in infected individuals, mitochondrial inhibition may not only contribute to excessive cytokine production but may be particularly impactful for the heart and brain, since these organs are highly reliant on mitochondrial energy production (1–5).
SARS-CoV-2 Infection Modulates Mitochondrial Bioenergetics.
SARS-CoV-2 infection has been found to markedly alter mitochondrial morphology in multiple human and monkey organs and cell lines (6–8). Infected Calu-3 cells showed mitochondria matrix condensation, swollen cristae, and decreased mitochondrial ATP synthase (complex V) subunit 5B (7). Lung-related microvascular endothelial cells (HULEC) and Human Pulmonary Alveolar Epithelial Cells (HPAEpiC) infected with SARS-CoV-2 demonstrated fragmented mitochondria with swollen cristae, increased mitochondrial reactive oxygen species (mROS) production, decreased oxidative phosphorylation (OXPHOS) complex I subunit NDUFA4, and decreased mitochondrial inner membrane protein TIMM23 (6). SARS-CoV-2 infection decreased both protein and transcript levels of the OXPHOS complex IV assembly factors COA4 and COA6 and the inner mitochondrial membrane protein TIMM8B, but increased glycolysis proteins including hexokinase (HK) isoforms 1, 2, and 3, and tricarboxylic acid (TCA) cycle malic enzymes 2 and 3 (6, 9, 10) (Fig. S1).
Shifting the carbon source from glucose to galactose inhibited SARS-CoV-2 replication in TMPRSS-Vero E6 monkey and A549-hACE2 human cells (11). Treatment of CaCo-2 and Calu-3 cells with the HK inhibitor 2-deoxy-D-glucose (2-DG) suppressed viral replication (9, 12). Infected A549-hACE2 cells, human iPSC-derived airway organoids (hPSC-AO), and BALF and lung autopsy samples showed increased transcription of both HIF-1α and HIF-1α target genes involved in glycolytic metabolism (10, 13, 14). Treatment of hPSC-AO, human monocytes, and CaCo-2 cells with the HIF-1α inhibitors, chetomin or BAY87–2243, inhibited viral replication (14–16), and expressing inhibitory RNAs specific for HIF-1α in infected hPSC-AOs reduced viral replication, transcription of HIF-1α, and glycolysis gene expression (14).
SARS-CoV-2 infected human monocytes exhibited elevated glycolysis, suppressed OXPHOS, induced mROS production, increased hypoxia inducing factor 1-α (HIF-1α), up regulated HIF-1α target genes, increased viral load, and mRNA expression of IL-1β, IL-6, TNF and interferons. Treatment with 2-DG reduced viral load; and treatment with the OXPHOS complex V (ATP synthase) inhibitor, oligomycin, increased viral load and cytokine mRNA levels. Treatment with ROS scavengers N-acetyl cysteine (NAC) and MitoQ decreased HIF-1α protein levels, mRNA expression of pro-inflammatory cytokines, glycolytic and mitochondrial proteins, and viral load (16).
SARS-CoV-2 infected human peripheral blood mononuclear cells (PMBC) and CaCo-2 cells also increased transcription of the HIF-1α (12). SARS-CoV-2 infection of PBMCs showed down regulation of mtDNA transcripts, mitochondrial function, and increased glycolysis (17) in association with elevated mitokines, fibroblast growth factor 21 (FGF21)(18) and growth differentiation factor 15 (GDF-15)(19). Expression of FGF-21 and GDF-15 are increased when the mitochondrial unfolded protein response (UPRMT) and the cytosolic unfolded protein response (UPRCT) are activated causing an imbalance between the nDNA and mtDNA mitochondrial translation products (20).
Mitochondrial function is modulated by mTORC1 activity and mTORC1 is increased in COVID-19 lung tissue, upon SARS-CoV-2 infection of Vero cells, and in infected human bronchial epithelial cells. Activation of mTORC1 is known to increase glycolytic metabolism by activating c-Myc and stabilizing HIF-1α (21, 22), which impairs mitochondrial biogenesis (22). Conversely, the mTORC1 inhibitor rapamycin, which increases mitochondrial function, decreased viral replication in cell lines (23). Thus, SARS-CoV-2 infection impairs mitochondrial function, increases mROS production, and stabilizes HIF-1α which facilitates viral replication.
SARS-CoV-2 Polypeptides Bind Mitochondrial Proteins.
One mechanism by which SARS-CoV-2 manipulates mitochondrial function is by direct binding of viral polypeptides to host proteins. Up to 16% of host proteins bound to individual SARS-CoV-2 polypeptides in human HEK293T and A549 cells were mitochondrial (13, 24, 25) (Table S1). For example, the SARS-CoV-2 M protein interacted with the mitochondrial protein synthesis proteins leucyl-tRNA synthase (TARS2) and asparaginyl-tRNA synthase (NARS2); the coenzyme Q synthesis protein COQ8B; the N terminal imported protein peptidases PMPCA and PMPCB; and complex I subunits NDUFS2, NDUFB8 and NDUFB10. The SARS-CoV-2 non-structural protein, Nsp6, bound OXPHOS complex V (ATP synthase) subunits; SARS-CoV-2 open reading frame 10 (Orf10) bound the mitochondrial inner membrane protein import polypeptide, TIMM8; and Orf9b bound mitochondrial outer membrane protein import polypeptide, TOMM70 (13, 24–27) (Table S1).
While binding of viral polypeptides to host proteins could account for the mitochondrial effects of acute infection, SARS-CoV-2 infected K18-hACE2 mice showed broadly reduced nDNA and mtDNA encoded mitochondrial OXPHOS proteins in the heart, lung, kidney, and spleen (28). This implies a more global inhibition of mitochondrial biogenesis, which could occur at the transcriptional level throughout the infection cycle. To address this potentiality, we investigated the effects of SARS-CoV-2 infection on the transcript levels of mitochondrial OXPHOS, glycolysis, nutrient sensing, and stress response genes. We examined human early-stage infection nasopharyngeal swabs and late-stage infection autopsy samples of SARS-CoV-2 positive and negative individuals collected early in the pandemic in New York City (29). We then examined SARS-CoV-2 infected hamsters and mice to analyze changes in bioenergetic gene expression at early-stage and mid-stage infection.
These analyses revealed major changes in mitochondrial gene expression as the SARS-CoV-2 infection progressed. These changes proved to be due to two interacting factors, the concerted down regulation of groups of mitochondrial genes by the virus which is opposed by the host cell compensatory induction of mitochondrial gene expression. In the end, while the host compensatory response prevailed in the autopsy lung, it failed dramatically in the autopsy heart.
RESULTS
We calculated the relative expression levels of host genes in RNA-seq data from ~700 nasopharyngeal samples and ~40 autopsy cases from SARS-CoV-2 positive and negative individuals, using the curated cellular bioenergetics genes listed in Table S2 plus the gene and pathway lists from MitoCarta and MitoPathway (30). These results were validated and extended using hamsters and mouse models.
Early-Stage SARS-CoV-2 Infection: Transcript Changes in Nasopharyngeal and Blood Samples.
Mitochondrial Gene Expression Analysis of SARS-CoV-2 Infected Nasopharyngeal Samples.
Analysis of the nasopharyngeal samples, revealed the interaction between viral suppression of mitochondrial gene expression and host compensatory mitochondrial gene induction (Fig. 1). Samples were binned into no, low, mediate, and high SARS-CoV-2 viral loads, and infections by other pathogens. The expression in the nDNA-coded genes for OXPHOS complexes I, II, III, IV, and V was significantly perturbed in the intermediate and high viral load samples exhibiting a striking dichotomy in responses-some OXPHOS genes being strongly down regulated (blue) and others strongly up regulated (red)(Fig. 1A).
Fig. 1. Mitochondrial OXPHOS complex module gene expression in nasopharyngeal samples from COVID-19 patients.

A) Heatmaps of t-score statistics for specific mitochondrial complex genes expression. Correlation plots associated with each heatmap are shown for all the samples. Circle size is proportional to the correlation coefficient. Two-tailed Student’s t-test *p<0.05, **p<0.01, ***p<0.001. B) Schematic of the human mtDNA with the position that miR-2392 will interact. C) Volcano plots for differentially expressed genes related to mitochondrial processed (MitoCarta). D) Lollipop plots custom-curated mitochondria gene sets determined by fGSEA. NES, nominal enrichment score.
Each mitochondrial OXPHOS complex is composed of both nDNA and mtDNA coded proteins assembled in stoichiometric ratios via subassembly modules. Analysis of the differentially expressed genes indicated that SARS-CoV-2 infection coordinately suppressed the expression of genes associated within mitochondrial OPXPHOS complex assembly modules. This contrasted with the up regulation of mitochondrial genes not inhibited by the virus.
The mtDNA codes for 13 OXPHOS polypeptides plus the tRNAs and rRNAs necessary for the expression of these genes via mitochondrial protein synthesis. The remaining mitochondrially-localized proteins are nuclear coded, with the genes dispersed across the chromosomes. The nDNA transcripts are translated on cytosolic ribosomes and the polypeptides imported into the mitochondrion via transport through the outer mitochondrial membrane (TOMM) and inner mitochondrial membrane (TIMM) import complexes.
The multi-subunit OXPHOS complexes are assembled via intermediate stage modules, often requiring specific assembly factors (31–33). To obtain a better understanding as to how SARS-CoV-2 intervenes in mitochondrial bioenergetics, we analyzed the mRNA levels of the structural genes and assembly factors for each of the OXPHOS complex modules (Table S2).
Complex I is composed of six modules. Four modules nucleate around mtDNA coded polypeptides, MT-ND1, MT-ND2, MT-ND4, or MT-ND5. The remaining two modules, Q and N, contain only nDNA subunits (Fig. 1A, Table S2). The nDNA coded subunit genes incorporated into the mtDNA polypeptide nucleated modules revealed a striking result. Four of the five structural genes of the MT-ND2 module were down regulated by viral infection. In contrast, five of the six assembly factors were up regulated. All six of the structural subunit genes of the MT-ND4 module were down regulated, but assembly genes FOXRED1 and TMEM70 were up regulated. All of MT-ND5 module’s nDNA coded genes were down regulated, and seven of the eleven N module nDNA genes were down regulated. By contrast, two of the six nDNA genes of the MT-ND1 module were strongly up regulated and half of the nDNA genes of the Q module were up regulated and half down regulated.
Complex II subunits and assembly factors are all nDNA coded. Five of the eight were down regulated in the high viral load samples (Fig. 1A).
Complex III encompasses one mtDNA subunit, MT-CYB, and has a more linear assembly. Most complex III nDNA genes were down regulated except for the intermediate assembly steps encompassing UQCC1, UQCC2, UQCC3, and CYC1 which were strongly up regulated.
Complex IV is assembled around mtDNA genes. All four nDNA genes of the MT-CO3 module were down regulated. A cluster of five genes of the MT-CO2 module were strongly down regulated, while assembly factor SCO2 was strongly up regulated. MT-CO1 module gene expression was minimally affected.
Complex V transcript analysis revealed that all of the nDNA genes in modules ATP5MC1, ATP5PB, MT-ATP6/8, as well as the assembly factors DMAC2L and TMEM70, were down regulated, with the ATP5PB gene being especially suppressed. Expression of ATP5F1A module genes was variable.
Finally, the expression of the mitochondrial ribosomal genes was variable. The MRPS10, MRPS16, MRPS34, MRPL36 genes were strongly down regulated while MRPL44 was strongly up regulated (Fig. 1A).
This parallel down regulation of multiple nDNA genes in individual OXPHOS modules suggests that these genes may be coordinately transcribed. If true, SARS-CoV-2 would only need to intervene in the common transcriptional factor(s) of one of the modules for each complex to disrupt OXPHOS biogenesis. In response, the host cell appears to counter by up regulating all the OXPHOS genes, however only those not blocked by the virus are induced.
This interaction between viral inhibition of the transcription of certain OXPHOS genes versus the host induction of all mitochondrial genes is corroborated by the unique expression profile of the mtDNA coded OXPHOS genes (Fig. 1B). In the samples with other viral infections or SARS-CoV-2 negative samples, all the mtDNA transcripts were proportionately down regulated. By contrast, in the samples with the highest SARS-CoV-2 RNAs, most mtDNA mRNAs were either unchanged or down regulated, with the exception of two mtDNA mRNAs, MT-ND1 and MT-ND6, which were strongly up regulated. The mtDNA polypeptide genes are transcribed from two promoters as long polycistronic transcripts for each mtDNA strand, with the polypeptide genes punctuated by tRNA gene sequences. As the transcripts are synthesized, the tRNAs are cleaved out generating the mature mRNAs. For the G-rich “heavy” (H), one promoter transcribes 12 polypeptide genes. For the C-rich “light” (L) strand the other promoter transcribes only one polypeptide gene, MT-ND6 (Fig. 1B).
In our companion paper (34), we reported that SARS-CoV-2 up regulates microRNA-2392 (miR-2392). It has been proposed that miR-2392 enters the mitochondrion and binds to the mtDNA tRNA MT-TQ gene sequence, impeding further progression of the mitochondrial RNA polymerase (35). We would thus anticipate that in COVID-19 the genes between the H strand promoter and MT-TQ would be induced while those downstream from MT-TQ to be reduced. This proves to be the case. The MT-ND1 mRNA upstream of MT-TQ is strongly up-regulated relative to uninfected samples while the next three genes MT-ND2, MT-CO1, and MT-CO2 have lower mRNA levels and the farther downstream polypeptide genes MT-CO3 and MT-CYB were strongly downregulated. The only other strongly up-regulated mtDNA gene is MT-ND6 which is transcribed from the independent L-stand promoter.
To further clarify the effects of SARS-CoV-2 infection on mitochondrial function, we used volcano plots to broaden our analysis to the transcript levels of all MitoCarta identified mitochondrial proteins (Fig. 1C). In the high viral load samples, several mRNAs were strongly up regulated. Most striking was cytosine monophosphate kinase 2 (CMPK2). CMPK2 is the rate limiting step for mtDNA replication, being central to the creation of oxidized mtDNA (Ox-mtDNA) which is released from the mitochondrion and binds to the inflammasome (36). PNPT1 (polyribonucleotide nucleotidyltransferase 1), also highly induced, is thought to be involved in processing mitochondrial double-stranded RNA released into the cytosol to engage MDA5-driven anti-viral signaling and trigger the type I interferon response (37). Also, up regulated was IFI27 (Interferon Alpha Inducible Protein 27), which in blood is the transcript that provides the greatest discrimination between SARS-CoV-2 positive and negative patients (38), and ACOD1(Aconitate Decarboxylase 1 or IRG1), which regulates itaconate production and the metabolic adaptation of inflammatory myeloid cells (39). Additional up regulated genes included central energy metabolism genes including SCO2 (cytochrome c oxidase, COX assembly), SLC25A3 (phosphate carrier) critical for ATP synthesis, and genes for metabolism and for regulating apoptosis (Table S3).
Down-regulated genes include ATP5PB (ATP Synthase Peripheral Stalk-Membrane Subunit B); PDK3 (Pyruvate Dehydrogenase Kinase 3); and additional metabolic genes (Table S3). Hence, SARS-CoV-2 infection shows a dose-response regulation of mitochondrial gene expression. In addition to the general inhibition of OXPHOS gene expression and mitochondrial metabolism, SARS-CoV-2 activates the mitochondrial innate immunity response (CMPK2, IFI27 and IRG1) genes.
Pathway Analysis of Gene Expression Changes in SARS-CoV-2 Infected Nasopharyngeal Samples.
To broaden our analysis of the transcriptional changes of cellular bioenergetic pathways during early nasopharyngeal infections, we performed Gene Set Enrichment Analysis (GSEA) on custom mitochondria pathways and MitoPathway created pathways (Fig. 1D, Fig. S2). At high viral loads, gene expression levels of the OXPHOS complexes I, II, III, and V were strongly down regulated along with mitochondrial cytosolic protein import complexes, mitochondrial TCA cycle and metabolic genes, fatty acid oxidation, glutathione synthesis, HIF-1α regulated mitochondrial genes, and the pentose phosphate shunt. By contrast, genes for fatty acid synthesis, nucleotide synthesis, and the integrated stress response (ISR) were up-regulated (Fig. 1D). Since pharmacological inhibition of fatty acid synthesis in SARS-CoV-2 infected hPSC-AO, HEK293T-hACE2 cells and K18-hACE2 mice inhibits viral replication (14, 40), induction of fatty acid biosynthesis must be required for viral replication, perhaps for the formation of the viral double membrane assembly structures (41).
The overall down regulation of OXPHOS complex IV genes is contrasted by the strong induction of the complex IV assembly gene, SCO2, (Fig.1D), consistent with a cellular compensatory effort to offset the viral reduction in complex IV gene transcription. The mitochondrial inner membrane nucleotide transport proteins (SLC25A3 and SLC25A4) required for ATP synthesis were also down regulated, as were the mtDNA’s replication and transcription genes, other transmembrane solute carriers (SLC25A1), coenzyme Q (CoQ) synthesis genes which bind M, the inner and outer mitochondrial membrane polypeptide transport carrier genes including TOMM70 which binds Orf9b, and mitochondrial fission and fusion genes (Fig. S2).
By contrast, the hypoxia initiation factor regulatory genes were strongly up regulated, though on average the large number of HIF-1α target genes mitigates this effect. A number of the mitochondrial bioenergetic and biogenesis genes that were down-regulated at high viral loads were up-regulated at lower SARS-CoV-2 loads (Fig. 1A), perhaps again reflecting a cellular compensatory response to the early SARS-CoV-2 inhibition of mitochondrial function.
In summary, SARS-CoV-2 inhibits the transcription of a wide range of genes critical for the bioenergetic machinery, thus inhibiting cellular oxidation of substrates in favor of activation of HIF-1α. This likely directs substrates through the early stages of glycolysis to increase fatty acid synthesis to promote viral replication. In response, the cell up regulates transcription of nDNA and mtDNA bioenergetic genes as well as mitochondrial innate immunity genes (CMPK2, IFI27 and IRG1) (36, 42).
Interaction Between SARS-CoV-2 Protein Inhibition and Transcript Inductions in Nasopharyngeal and Peripheral Blood Samples.
To assess the potential functional effects of the changes in nasopharyngeal gene expression with functional protein changes, we compared changes in the acutely infected patient nasopharyngeal transcript levels with changes in the acutely SARS-CoV-2 infected human colon epithelial carcinoma cell line, CaCo-2 (9) (Fig. S3A&B). This revealed the strong transcriptional down regulation of the cytosolic ribosomal protein mRNAs, which would activate UPRMT, UPRCT, and ISR, though the ribosomal protein levels were not significantly changed in this short-term CaCo-1 infection experiment. Consistent with a marked down regulation of multiple OXPHOS mRNAs, CaCo-2 infected cells down regulated OXPHOS complex I (NADH dehydrogenase) in association with the strong increase in pyruvate dehydrogenase kinase, which negatively regulates pyruvate dehydrogenase, and proteins involved in increasing mitochondrial inner membrane permeability.
mtDNA transcripts and mitochondrial RNA polymerase (POLRMT) have been reported to be down regulated in blood cells while interferon response genes were induced (17) with the mitochondrial IFI27 transcript being the most discriminatory factor in SARS-CoV-2 blood (38, 43). We observed in COVID19 positive cases, the blood mRNA levels for FOXRED1 (complex I assembly) and SCO2 (complex IV assembly) were up regulated (Fig. S4A) as we observed in complex I module MT-ND4 and in complex IV MT-CO2 module, SCO2 induction also being observed by others (17) (Fig. S4B). The mRNA levels for the antioxidant enzyme, catalase (CAT) and for ferritin heavy chain (FTH1) for iron homeostasis, were reduced, though their protein levels in COVID19 positive case plasma were increased (43). The reduction in CAT transcription is consistent with the virus inhibiting OXPHOS and antioxidant enzymes to increase mROS levels (Fig. S4B)(17). Finally, mRNA and protein levels were up regulated for nDNA replication (TYMS, TK1, PAICS), folate metabolism (DHFR), and stress response (DDIT4 regulation of mTOR).
Late-Stage SARS-CoV-2 Infection and Lethality: Transcript Changes in COVID-19 Human Autopsy Samples.
To determine the changes in bioenergetic gene expression late in infection, we analyzed the expression profile of patients who died of COVID-19. The autopsy samples from 35 deceased COVID-19 patients encompassing high and low SARS-CoV-2 levels were compared to five uninfected controls. Samples were collected from the heart, kidney, liver, lung, and lymph nodes (44). We also included publicly available RNA-seq data on monocytes from COVID-19 patients (45).
Perturbation of OXPHOS Gene Expression in Autopsy Tissue of SARS-CoV-2 Infected Patients.
Analysis of the transcriptional changes of mitochondrial OXPHOS bioenergetics and protein synthesis genes revealed a striking down regulation of virtually all OXPHOS genes in the heart (Fig. 2A). This included OXPHOS structural genes and assembly factors for complexes I, II, III, IV, and V as well as mitochondrial ribosomal proteins. This was not simply the product of terminal destruction of heart cells, since assembly genes for complex IV (COX16, COX20, COA6, SCO2, and PET100) were up regulated.
Fig. 2. Mitochondrial OXPHOS complex module gene expression in COVID-19 patients from autopsy tissues.

A) and B) Heatmaps display the t-score statistics when comparing viral load versus negative patient samples for nasopharyngeal swab and autopsy samples for specific mitochondrial complex genes. Correlation plots associated with each heatmap are shown for all the samples. Circle size is proportional to the correlation coefficient. Statistical significance determined using a two-tailed Student’s t-test *p<0.05, **p<0.01, ***p<0.001. C) Volcano plots for differentially expressed MitoCarta genes from autopsy COVID-19 patient samples. D) Lollipop plots for statistically significant custom mitochondria and MitoPathway gene sets determined by fGSEA. NES, nominal enrichment score.
The kidney and liver of SARS-CoV-2 infected patients showed down regulation of remarkably similar subsets of the OXPHOS genes, though many fewer genes than in the heart. These included the down regulation of selected complex I structure genes in the MT-ND2, MT-ND4 and NDUFV1 modules, complex II SDHA, and complex III structural and assembly genes CYC1, UQCRC1, BCS1L and TTC19. This further supports the possibility that SARS-CoVC-2 may interfere with specific transcriptional elements for OXPHOS genes.
Changes in the transcriptional profiles of the lymph nodes revealed individual complex genes being down regulated: NDUFA5, NDUFA9, NDUFC2 in complex I modules NDUFS2, MT-ND1, and MT-ND2; SDHC and SDHD in complex II; and CMC1 and COX20 in complex IV modules MT-CO1 and MTCO2. Only modest changes were seen in OXPHOS gene expression in the monocytes (Fig. 2A).
Mitochondrial OXPHOS gene expression was almost uniformly up regulated in lung autopsy samples in striking contrast to other organs, as previously reported (10). Notably induced OXPHOS genes in lung included SDHB in complex II; UQCRB, UQCRQ, UQCC2, UQCRH, and UQCRFS1 in complex III; ATP5F1C and all of the genes in the ATP5PB and the MT-ATP6/8 modules of complex V; as well as the majority of the genes for the mitochondrial ribosomal proteins (Fig. 2A). Possibly by the time the COVID-19 patients die from organ failure, the lungs have cleared the virus and the up regulation of OXPHOS gene expression comes to predominate.
In contrast to the variable expression of the nDNA coded OXPHOS genes between tissues, all the mtDNA coded genes were strongly up regulated in all the autopsy tissues (Fig. 2B). This implies that miR-2392 is no longer inhibiting mtDNA transcription consistent with lower viral RNA levels, and confirms that the down regulation of OXPHOS genes in the heart, kidney and liver is not the result of generalized tissue and organ destruction.
The differential regulated mitochondrial genes in autopsy tissues is further exemplified by analysis of the MitoCarta genes relative to uninfected controls plotted in volcano plots (Fig. 2C). There was a clear progression in the extent of down regulated mitochondrial gene expression from most severe in the heart, followed by kidney and liver, and then lymph node. The lung showed the reverse bias with predominantly up regulation of mitochondrial genes (Fig. 2C). The reduced effects on mitochondrial gene expression between high and low viral loads of each tissue confirm that these gene expression changes are the result of viral infection.
To further elucidate the functional implications of the differential mitochondrial gene expression among the different tissues, we selected some of the most striking up and down regulated genes of each tissue and determined their functions (Fig. 2C, Table S3). In the COVID-19 heart the most strongly up regulate gene was MUCB, a component of the mitochondrial Ca++ uniporter, which coincides with induction of CYP24A1 which regulates vitamin D and Ca++ and PO4− levels. This implies that mitochondrial Ca++ regulation is an important aspect of SARS-CoV-2 pathogenesis. The rate limiting gene for mtDNA replication, CMPK2, was up regulated along with other mtDNA replication genes (PIF-1 and ANGEL2). Additional up regulated heart genes included C15orf48, a COX protein that on viral infection displaces NDUFA4 plus generates miR-147b that inhibits NDUFA4 translation and enhances RIG-1/MDA-5 mediated viral immunity (46), as well as genes associated with apoptosis, mitochondrial metabolism, and mtDNA coded genes (Fig. 2C, Table S3).
The most strongly down regulated gene in the COVID-19 heart was GPAM (glycerol-3-phosphate acyltransferase), the commitment step of glycerolipid synthesis. An array of other mitochondrial OXPHOS, peroxisomal, and mitochondrial metabolism genes were also down regulated, including complex II subunit SDHC; complex III assembly factor BCSIL; and complex IV subunit COX8A (Fig. 2C, Table S3).
In the COVID-19 kidney, MCUB was up regulated as in heart. Specific mtDNA replication, transcription and repair genes; apoptosis genes, metabolic genes, regulatory genes, and mtDNA transcripts (MT-ND6, MT-CO1, MT-ATP6) were also up regulated (Fig. 2C).
The strongest COVID-19 down regulated gene in kidney was GLDC for glycine cleavage, a gene which is up regulated in the autopsy heart. Multiple mitochondrial genes were also down regulated including SDHA, a subunit of complex II parallel to the down regulation of SDHC in the heart; ADCK1, which regulates coenzyme Q biosynthesis; mitochondrial fatty acid synthesis; fatty acid oxidation genes; mitochondrial metabolic genes; and genes involved in mitochondrial biogenesis including PDF, mitochondrial polypeptide deformylase, and GATB, which converts the miss addition of glutamate on tRNAGln to glutamine (Fig. 2C Table S3).
In the COVID-19 autopsy liver, up regulated genes included the OXPHOS regulatory genes C2orf69 and HIGD1A and mtDNA transcripts MT-CO3, MT-ATP6, and MT-ND6, as well as regulators of apoptosis and metabolism. Down regulated genes included cytosolic fatty acid synthesis (FASN), mitochondrial fatty acid synthesis, mitochondrial metabolism, and a mitochondrial fission protein (Fig. 2C, Table S3).
For the COVID-19 autopsy lymph nodes, up regulated genes included mtDNA nucleoid genes (ATAD3A and RECQL4); fatty acid oxidation genes; and metabolism genes. Down regulated genes include the Ca++ uniporter (MCUB), which was the opposite of heart and kidney, and the Sirtuin that regulates glutamine metabolism (SIRT4) (Fig. 2C, Table S2).
For the COVID-19 autopsy lungs, OXPHOS genes C2orf69 and COA6 for complex IV assembly; single carbon metabolism genes MTHFD1L and MTHFD2; heme and iron-sulfur center enzymes; heat shock and apoptosis genes; peptidases; DNA repair genes; and fatty acid metabolism were up regulated. In contrast, fatty acid synthetase (FASN), fatty acid oxidation, peroxisomal, metabolic and mitochondrial regulatory genes were repressed (Fig. 2C, Table S3).
Hence, COVID-19 inhibited transcription of bioenergetic genes in the autopsy heart, kidney, liver, and lymph nodes. Selected genes participating in mitochondrial OXPHOS, fatty acid and amino acid metabolism, protein import, mitochondrial dynamics, and molecular transport were repressed several log-fold in the heart, as they were in the nasopharyngeal samples (Fig. 2D, Fig. S2), and to a lesser extent progressively in the kidney, liver, and lymph node. By contrast, in the lung OXPHOS genes were coordinately induced.
This human COVID-19 gene expression data indicates that SARS-CoV-2 infection initially impairs mitochondrial gene expression in the nasopharyngeal tissues and then progresses to down regulation of mitochondrial gene expression in the heart, kidney, and liver leading to death. However, in the lung the virus appears to be cleared before death resulting in the surviving lung cells up regulating inducing mitochondrial gene expression.
Expression of OXPHOS-Associated Genes in COVID19.
We next focused on transcriptional changes in processes that complement mitochondrial respiration: mitochondrial ATP/ADP-Pi exchange, TCA cycle, coenzyme Q biosynthesis, mitochondrial fatty acid synthesis (mtFASII), fatty acid oxidation, and the cytosolic protein import system (Fig. 3). As with the OXPHOS genes, the COVID-19 autopsy heart showed a systematic down regulation of transcripts for multiple mitochondrial functions, while the kidney and liver showed less robust inhibition of mitochondrial gene expression, which paralleled the down regulation of mitochondrial gene expression in the infected nasopharyngeal samples. Again, the COVID-19 autopsy lung showed the converse with the general induction of mitochondrial genes (Fig. 3).
Fig. 3. Analysis on mitochondrial ATP/ADP-Pi exchange, TCA cycle, coenzyme Q biosynthesis, mitochondrial fatty acid synthesis (mtFASII), fatty acid oxidation, and cytosolic protein import system.

A) The circular heatmap displays the t-score statistics when comparing viral load versus negative patient samples for nasopharyngeal swab and autopsy samples for specific mitochondrial genes. B) Correlation plots associated with each group are shown for all the samples. Circle size is proportional to the correlation coefficient. Statistical significance determined using a two-tailed Student’s t-test *p<0.05, **p<0.01, ***p<0.001.
The differential expression of the mitochondrial inner membrane solute carrier genes is illustrative of the above dynamics. In the heart the phosphate carrier (SLC25A3), the adenine nucleotide translocases (ADP/ATP carries) (SLC25A4, ANT1) and SLC25A6, ANT3), the citrate carrier (SLC25A1), and the Ca++-binding aspartate-glutamate carrier (SLC25A12) were down regulated. By contrast, the ATP-Mg++/Pi transporters (SLC25A24 and SLC25A25) were up regulated. SLC25A3, SLC25A4, SLC25A6, SLC25A1, SLC25A12 are critical for mitochondrial energy production and ATP export, while SLC25A24 and SLC25A25 import ATP into the mitochondria without energetic benefit. The parallelism between the nasopharyngeal and heart samples is striking for these transport systems, with both tissues showing a pronounced down regulation of the phosphate carrier (SLC25A3) as well as ANT3 (SLC25A6) and up regulation of the ATP-Mg++/Pi transporter (SLC25A25). Thus, as the virus inhibits mitochondrial energetics, the cell responds by attempting to sustain intramitochondrial adenine nucleotide levels by importing ATP (Fig. 3).
Regulation of Mitochondrial Allied Bioenergetic Pathways
Changes in the expression of genes for fatty acid synthesis, NADPH synthesis, glutathione synthesis, folate metabolism, glycolysis, gluconeogenesis, and nucleotide metabolism exhibited a number of down regulated transcripts in heart and nasopharyngeal tissue, and generally the opposite in autopsy lung. However, the differences are much less pronounced than seen for the mitochondrial OXPHOS genes (Fig. 4 A&B).
Fig. 4. Glycolysis pathways, HIF pathways, ISR, and mTOR genes show differential expression.

A), C), E), and G) Circular and tabular heatmaps display the t-score statistics when comparing viral load versus negative patient samples for nasopharyngeal swab and autopsy samples for genes related to specific pathways. The outer dendrogram in C) visualizes similar clusters as identified by complete-linkage clustering using Euclidean distance within each segment. B), D), F), and H) Correlation plots associated with each group are shown for all the samples. Circle size is proportional to the correlation coefficient. Statistical significance determined using a two-tailed Student’s t-test *p<0.05, **p<0.01, ***p<0.001.
Of particular interest was the expression of the single carbon metabolism genes which are metabolically interconnected between the mitochondrion and the nucleus-cytosol. We saw a general up regulation of all the mitochondrial folate enzyme genes, except dihydrofolate reductase 2 (DHFR2), in the heart, kidney, liver, lymph node verses the down regulation of the cytosolic and nuclear folate enzyme genes in the heart and progressively less so in kidney and liver. The lung pattern shows these trends but is more variable (Fig. 4 A&B). The up regulation of the mitochodnrial folate genes and the down regulation of the cytosolic and nuclear folate genes in the autopsy heart, kidney and liver (Fig. 4A&B) parallels the up regulation of mitochondrial single carbon metabolism seen in the cells and tissues of humans and mice with genetic defects in mitochondrial bioenergetics and biogenesis linked to activation of the UPRMT and the ISR (47).
In the nasopharyngeal tissue, we see the variable regulation of folate genes. However, we observed a striking induction of mtDNA replication gene, CMPK2, which has been linked to activation of the inflammasome (36).
For glycolysis in the heart, we observed the up regulation of lactate dehydrogenases A (LDHA) which converts pyruvate to lactate and favors glycolysis, but the down regulation of lactate dehydrogenase B (LDHB) which converts lactate to pyruvate and favors OXPHOS, a pattern also reported in peripheral blood (17). The glucose carriers (SLC2A1 and SLC2A4) were down regulated as was the monocarboxylate carrier 1 (SLC16A1), which transports lactate across the mitochondrial inner membrane. However, monocarboxylate carrier 5 (SLC16A4) was strongly up regulated, as it is in cancer cells, being responsive to HIF-1α activation (48) (Fig. 4 A&B).
The down regulation of the glycolysis genes, GAPDH and PGK1, in the nasopharyngeal samples may seem anomalous, since we might have expected glycolysis to be induced to compensate for the mitochondrial ATP deficiency. However, inhibition of the later steps of glycolysis would drive carbon from the earlier steps of glycolysis into fueling fatty acid and NADPH synthesis which would generate the lipids critical for the production of the viral RNA replication centers.
In stark contrast to the down regulation of mitochondrial genes, the HIF-1α target genes were generally up regulated in autopsy tissues. This proved to be even more true for the autopsy lung in which HIF-1α was one of the most up regulated genes. This corresponds to the down regulation of COX4i1 and the induction of COX4i2 along with the mitochondrial LON protease which degrades COX4i1. This change shifts complex IV from a high oxygen to low oxygen optimal configuration (49). Perhaps, in the recovering lung cells, respiration is still impaired and maintaining the pseudohypoxic state is necessary to repair viral damage. This is again the opposite of the early infection nasopharyngeal tissues in which there is a variable expression of HIF-1α regulatory and target genes (Fig. 4 C&D).
Alterations in the ISR and mTOR Pathways in SARS-CoV-2 Infected Tissues.
The differential inhibition of the cytosolic (Fig. S3A) and the mitochondrial (Figs. 2 & 3) ribosomes, plus increased mROS (50) would activate the UPRMT and UPRCT and the ISR via endoplasmic reticulum (ER) kinases HRI (EIF2AK1), PKR (EIF2AK2), PERK (EIF2AK3), and GCN2 (EIK2AK4)]. These kinases phosphorylate the cytosolic elongation factor, eIF2α, and phosphorylation of eIF2α impairs cytosolic protein synthesis and activates expression of the ISR transcription factors ATF4 and ATF5. Increased levels of ATF4 and ATF5 induce the expression of the downstream genes GDF15, GADD34 and CHOP (51, 52). The ISR also induces the expression of the mitochondrial OXPHOS supercomplex assembly factor SCAF1 (53) and the stress hormones FGF21 and GDF15 (47). While GDF15 mRNA was up-regulated in nasopharyngeal, lung, and heart tissues, FGF21 expression was not affected (Fig. 4A).
PKR, which binds double standed RNA, was strongly up regulated in the nasopharyngeal samples along with ATF3, ATF4, ATF5, CHOP, GADD34, GDF15, while eIF2α was strongly down regulated. The autopsy heart, kidney, liver and lymph nodes showed a milder induction of the of the ISR genes, while the autopsy lung showed a mild up regulation of all of the ISR genes including eIF2α. Hence, during initial infection with high levels of SARS-CoV-2 replication intermediates, PKR is activated and induces the ISR. As the initial viral insult subsides, eIF2α is induced in lung, kidney, liver, and lymph node, presumably to return to normal cytosolic translation, though this does not happen in the heart (Fig. 4E&F).
SARS-CoV-2 infection is also associated with altered mTOR function, which also relates to mitochondrial dysfunction (47, 53). mTOR target genes were up regulated in the autopsy lung and lymph nodes, and to a lesser extent in the naropharyngeal samples (Fig. 4G&H). However, they were markedly down regulated in the autopsy heart, kidney, and liver. The induction of mTOR gene expression in the nasopharyngeal, lung, and lymph node tissues did not correlate with the induction of the AMPK genes (PRKAA2, PRKAB1, PRKAG1), but with the STK11 kinase as well as AKT1 and AKT2. By contrast, the down regulation of the mTOR genes in the heart, kidney, and liver correlated with the strong up regulation of AKT3. Presumably, in the heart, kidney, and liver, SARS-CoV-2 impairs mTOR gene expression which induces AKT3.
In summary, with the strong induction of PKR (EIF2AK2) in the acute infection nasopharyngeal samples down stream ISR genes were activated. ISR transcription was also activated in the other COVID-19 tissues. However, what distinguished the heart, kidney, and liver from the nasopharyngeal, lung and lymph node samples was the expression of the mTOR target genes which were down regulated in the former but up regulated in the latter. This corresponds to the differential induction of STK11 plus AKT1 and AKT2 versus AKT3 (Fig. 4G&H).
Utilization of Changes in COVID-19 Patient mRNA Levels to Investigate Viral-Induced Metabolic Fluxes.
To further clarify the metabolic effects of SARS-CoV-2 acute nasopharyngeal and late-stage systemic infection we used the observed alterations in the host mRNA levels to estimate the flux rates of metabolites through bioenergetics via our computational model of cellular pathways (54). Analysis of mitochondrial bioenergetic and antioxidant defenses supported the conclusion that mitochondria function was down regulated in the nasopharyngeal samples and autopsy heart, kidney, liver and lymph node but strikingly up regulation in autopsy lung (Fig. 5, Fig. S4).
Fig. 5. Metabolic flux analysis on RNA-seq data from nasopharyngeal and autopsy patient.

A) Upset plot of the overlapping up regulated and down regulation metabolic fluxes with the nasopharyngeal and autopsy RNA-seq patient samples. The red color dots represent the up regulated metabolic fluxes and the blue color dots represent the down regulated fluxes. The bar chart on top are the number of overlapping metabolic fluxes for each intersect. The set size bar plot represents the total number of metabolic fluxes contained in each row. B), C), and D) Heatmaps of the of the log2 fold-change values for the significantly regulated fluxes in the OXPHOS, ROS detoxification, Vitamin D, and Mitochondrial Transport, major group flux pathways. The color bar represents the log2 fold-change values. The color of low but significant values are faint and may not be apparent in these heat maps.
In high and medium viral load nasopharyngeal samples, down regulation was observed for OXPHOS function; heme biosynthesis; ROS detoxification; and glutathione, cysteine, and pentose-phosphate metabolism. Similarly, autopsy heart samples showed a predominantly down regulation of mitochondrial function including 24 mitochondrial transport functions, far more than any other tissue, as well as reduced cysteine metabolism. Liver showed down regulation of six mitochondrial transport functions as well as glutathione and cysteine metabolism, while kidney exhibited down regulation five mitochondrial transport functions as well as glutathione and cysteine metabolism. Lymph nodes manifested down regulation of eight mitochondrial transporters and NAD, cysteine, and pentose phosphate metabolism.
The autopsy lung, by contrast, exhibited marked up regulation of OXPHOS; heme biogenesis and degradation; ROS detoxification; glutathione, cysteine and pentose-phosphate pathways; plus vitamin D metabolism. The liver and kidney also experienced increased heme biogenesis, ROS detoxification, glutathione metabolism, while liver exhibited increased heme metabolism, a few mitochondrial transport reactions, and cysteine and pentose phosphate pathways. The lymph nodes exhibited increased NAD metabolism and mitochondrial transport. For ROS detoxification, catalase was up regulated in liver and kidney, but down regulated in nasopharyngeal tissue. The only up regulated functions in the heart involved glutathione and vitamin D metabolism (Fig. 5).
Analysis of the modulation of the flux rates of the OXPHOS complexes is problematic due to their multiple subunit composition and the differential inhibition of the specific subunit transcripts by the virus versus induction of the remaining subunit genes by the host. Hence, high expression of some subunits by the host can overshadow the inhibition of others by the virus. Taking this into account, the heart had the lowest gene expression derived OXPHOS flux, while the kidney and liver, with increased calculated OXPHOS flux, had less severe viral inhibition of OXPHOS.
Consequently, calculated relative metabolic fluxes showed the same pattern as that observed for the individual mitochondrial genes. Nasopharyngeal and heart mitochondrial gene expression was strongly impaired; autopsy lung strongly induced; and liver, kidney and lymph nodes arrayed between.
Early-Stage SARS-CoV-2 Infection in the Hamster Model: Analysis of Bioenergetic Gene Expression in Visceral and Brain Tissues
Analysis of the nasopharyngeal and autopsy samples revealed that mitochondrial gene expression is down regulated during early viral infection but is up regulated in later stages of infection when the virus is being cleared. To further elucidate the effects on mitochondrial function during early infection, we infected hamsters by nasopharyngeal administered SARS-CoV-2. When the virus reached its maximum titer in the hamster lung at four days post infection, we analyzed the mitochondrial gene transcript levels (Fig. 6). At this early time in infection, transcription of bioenergetics genes was not markedly perturbed in the lung, heart, kidney, and brain olfactory bulb. There were minimal changes in mitochondrial OXPHOS, mtFASII, coenzyme Q, protein import, TCA cycle, fatty acid oxidation, glutathione synthesis, and fatty acid synthesis gene expression. A subset of mitochondrial ribosomal genes were induced in the heart, lung and kidney, and mild increases were seen in heart and lung folate metabolism, NADP synthesis, nucleotide metabolism, HIF-1α target genes, and ISR genes.
Fig. 6. Mitochondrial specific analysis of RNA-seq data from SARS-CoV-2 infected hamster heart, lung, kidney, olfactory bulb, cerebellum, and striatum.

A) Heatmaps display of mitochondrial specific genes with t-score statistics comparing SARS-CoV-2 infected hamsters vs. controls. B) and C) Lollipop plots for statistically significant custom mitochondria and MitoPathway gene sets determined by fGSEA. NES, nominal enrichment score. D) Volcano plots for differential MitoCarta genes from hamster tissues.
In stark contrast to the minimal effects of acute SARS-CoV-2 infection on the viscera, brain tissues showed striking changes in mitochondrial-related gene expression (Fig. 6A,B,&C). The cerebellum exhibited coordinate down regulation of bioenergetic processes including OXPHOS, mitochondrial ribosome, mtFASII, and cytosolic fatty acid synthesis and glycolytic genes. Conversely, the striatum showed a robust up regulation of OXPHOS, mitochondrial ribosomal, cytosolic protein import, glutathione synthesis genes, and a strong down regulation of glycolysis and cytosolic fatty acid synthesis genes
Volcano plot analysis of (Fig. 6D) revealed a bias toward increased mitochondrial gene expression in heart and kidney, presumably reflecting the high reliance of these organs on mitochondrial energy production; little perturbation of lung mitochondrial gene expression; general down regulation of olfactory bulb and cerebellum mitochondrial gene expression; and a marked increase in striatal mitochondrial gene expression. The functions of the genes highlighted in the volcano plots are listed in Table S3. Notable up regulated genes in the heart and kidney include the branched chain amino acid transporter (Slc25a44) in the heart and the complex I subunit (Ndufs7) and mitochondrial fusion protein (Miga2) in the kidney. Notable down regulated genes in the lung included the mitochondrial translational factor gene (Tsfm) and the mitochondrial superoxide dismutase (Sod2) gene.
The mitochondrial Ca++ uniporter gene (Mcu) was up regulated in the olfactory bulb, which parallels the induction of the mitochondrial Ca++ uniporter gene MCUB in the autopsy heart and kidney. Down regulated mitochondrial genes included the mitochondrial ATP synthase subunit 5 (Atp5p), mitochondrial alanyl-tRNA synthetase (Aars2), and citrate carrier (Slc25a1). In the cerebellum, the mitochondrial antioxidant gene Mpv171 was up regulated while several neurological and mitochondrial genes were down regulated including the citrate carrier (Slc25a1).
Finally, in the striatum, an array of mitochondrial genes was up regulated including complex IV subunit (Cox7a2), complex I subunits (Ndufa9 and Ndufb6), TCA cycle fumarate hydratase-1 (Fh1), and mtDNA complex I subunit (MT-ND3). Notable striatal down regulated genes included citrate carrier (Slc125a1), mitochondrial folate serine hydroxymethyltransferase 2 (Shmt2), coenzyme Q synthesis (Co8b), and lactate dehydrogenase D (Ldhd).
These hamster results indicate that early in viral infection, when SARS-CoV-2 gene expression in the lung peaks and the viral polypeptides are binding to mitochondrial proteins to disrupt mitochondrial OXPHOS, the virus has yet to strongly influence host lung or visceral mitochondrial gene transcription. Surprisingly, however, we observed a striking differential expression of the brain bioenergetic genes, presumably responding to diffusible factors elaborated in response to viral infection, possibly impacting neurological function.
Mid-Stage SARS-CoV-2 Infection in BALB/c and C57BL/6 Mouse Models: Analysis of Bioenergetic Gene Expression in the Lungs.
To investigate the effects on mitochondrial function during a more advance stage of SARS-CoV-2 infection in the lung, we analyzed the mitochondrial transcripts of the lungs of BALB/c and C57BL/6 mice infected with mouse-adapted SARS-CoV-2MA10 virus at four days post-infection (55). BALB/c mice are more susceptible to lung pathologies and dysfunction caused by SARS-CoV-2 MA10 compared to C57BL/6 mice (55), and examining tissue in this time point provided data on the lung’s induction of mitochondrial gene expression as viral load declines.
We observed marked differences in the up and down regulated MitoCarta genes when comparing the two infected mouse strains (Fig. 7A&B). We found a more robust induction of OXPHOS complex I, II, III, IV, and V genes in the BALB/c compared to the C57BL/6 lungs. Similar patterns emerged for genes involved in mitochondrial protein import, TCA cycle, fatty acid oxidation, and inner membrane transport (including the ADP/ATP translocases), mitochondrial ribosomal genes, and ISR genes. Up regulation of genes involved in glycolysis, glutathione synthesis, folate metabolism, pentose-phosphate shunt, nucleotide synthesis, and HIF-1α target genes which were more intense in the BALB/c mice than the C57BL/6 mice. Finally, the cytosolic fatty acid synthesis genes, particularly genes that encode fatty acid synthetase (Fasn), acetyl CoA-carboxylase alpha (Acaca), and long-chain fatty acid ligase 1 (Acsl1), were more down regulated in the BALB/c than the C57BL/6 mice (Fig. 7B).
Fig. 7. Mitochondrial specific analysis on RNA-seq data from SARS-CoV-2 infected mouse lungs.

A) Upset plot of the overlapping MitoCarta genes comparing lungs from BALB/c and C57BL/6 mice infected with SARS-CoV-2. B) Heatmaps display of mitochondrial specific genes with t-score statistics comparing SARS-CoV-2 infected mice vs. controls. C) Volcano plots for differential MitoCarta genes from C57BL/6 and BALB/c tissues.
Volcano plots (Fig. 7C) and gene-level analysis of the mouse expression data revealed similar expression changes to the human autopsy lung. Among the top three to four induced genes of both mouse strains were Cmpk2 and Acod1, involved in the mitochondrial modulation of inflammation, and Gatm which is required for creatin synthesis. Both strains experienced an up regulation of a range of mitochondrial metabolic and biogenesis genes, but the down regulation of Alas2 involved in porphyrin synthesis (Fig. 7C, Table S3).
Assuming that the C57BL/6 and BALB/c mice represent different stages of recovery from the viral infection of the lung, the C57BL/6 mice may be caught in the transition as the viral effects decline and the host induction is initiated. The BALB/c mouse may represent further disease progression with greater viral reduction and host gene induction, analogous to the human autopsy lung tissues.
DISCUSSION
While previous studies have demonstrated that SARS-CoV-2 can inhibit mitochondrial function by the binding of viral structural, Nsp, and Orf proteins to critical host mitochondrial proteins (13, 24, 25) our current results demonstrate that a much more global mitochondrial inhibitory effect occurs at the transcriptional level. Nasopharyngeal and autopsy heart, kidney, and liver samples with high SARS-CoV-2 RNA levels manifest inhibition of transcription of mitochondrial genes associated with OXPHOS complexes I, II, III, IV, and V; fatty acid oxidation; antioxidant defenses; mitochondrial translational machinery; cytosolic protein import; mtFASII; mtDNA biogenesis; and intermediate metabolism. This contrasts with the autopsy lung which shows the opposite transcriptional profile, a general up regulation of mitochondrial gene expression. This striking difference demonstrates that the mitochondrial gene expression profiles changed during the course of the viral infection.
The relationship between viral load inhibition of mitochondrial transcriptional versus host compensatory induction is demonstrated by the differential regulation of nDNA OXPHOS genes in the nasopharyngeal tissues. The OXPHOS enzyme complexes are assembled from multiple nDNA and mtDNA coded subunits assembled through sub-enzyme modules which are then combined into the final enzyme. To assemble the polypeptides for each nodule in stoichiometric ratio, the modular genes would need to be coordinately regulated. In such a model, SARS-CoV-2 would need only to compromise the master transcription regulatory factor(s) for one enzyme complex module to block assembly of the enzyme. The transcriptional profiles of the OXPHOS modules in the SARS-CoV-2 infected nasopharyngeal samples supports the hypothesis of the coordinate regulation of the OXPHOS module genes and that SARS-CoV-2 blocks modular assembly by impacting the master transcriptional regulator of the modules nDNA genes. The host cells counter the viral inhibition by the coordinated up regulation of nDNA mitochondrial gene expression, demonstrated by the striking up regulation of the complex IV assembly gene, SCO2. Other pathway genes show potential coordinate modulation, such as the down regulation of the cytosolic-nuclear folate genes in association with the up regulation of the mitochondria folate genes.
SARS-CoV-2 manipulation of transcriptional pathways to impair mitochondrial bioenergetics is exemplified by the viral regulation of expression of the nasopharyngeal mtDNA transcripts. The SARS-CoV-2 genome codes for three sequences that are homologous to the seed sequences of miR-2392 (34). If these generate RNAs that mimic miR-2392, then at high SARS-CoV-2 viral loads, there would be increased inhibition of the genes and RNAs that bind to the miR-2392 seed sequence. It was previously proposed that miR-2392 binds to the mtDNA MT-TQ gene resulting in inhibition of mtDNA transcription. At high SARS-CoV-2 RNA levels, sufficient miR-2392 mimic RNA would be generated to enter the mitochondrion and block mtDNA transcription. This conjecture was supported by the high expression of the mtDNA H strand MT-ND1 gene upstream from MT-TQ, and the reduced expression of the downstream mtDNA H strand mRNAs. SARS-CoV-2 manipulation of regulatory genes was further supported by the altered gene expression of the mTOR nutrient sensing pathway genes in association with the energy sensing kinases.
In the nasopharyngeal and heart, kidney, and liver samples, inhibition of OXPHOS and limitation of antioxidant defenses would result in increased mROS which stabilizes HIF-1α. This redirects metabolites away from the mitochondrion and toward glycolysis to generate lipid precursors. The imbalance in nDNA and mtDNA mitochondrial polypeptides also activates the UPRMT and UPRER, which activates the ISR resulting in bias of protein synthesis away from cellular maintenance and toward vial biogenesis.
The human autopsy data confirms that, as viral titer declines, normal mitochondrial function resurges repairing tissue damage. However, if the virally-induced inhibition has been too severe, as in the case of the autopsy heart, kidney, and liver, then organ failure ensues resulting in death.
While the early and late phases of COVID-19 modulation of bioenergetic gene transcription were established from the human nasopharyngeal and autopsy studies, the relationship between the initial viral protein inhibition of host mitochondrial proteins and the viral inhibition of bioenergetic gene transcription was not clear. To address this discrepancy, we analyzed the transcriptional effects of early hamster nasopharyngeal infection. This revealed that at peak lung viral titer, mitochondrial gene expression was not markedly impaired in the lung, heart, and kidney. Hence, the viral regulation of host mitochondrial transcription occurs after the initial viral protein inhibition of mitochondrial protein function. Surprisingly, however, brain mitochondrial gene expression was affected possibly accounting for the commonly experienced “brain fog” (1).
Late in the course of SARS-CoV-2 lung infection, an upsurge of bioenergetic gene expression occurred in the autopsy lung, likely associated with the clearance of the virus from the autopsy lung. To further clarify the host cell response to the progressive lung clearance of the virus, we studied the mid phase response of the lungs of C57BL/6 and BALB/c mice, C57BL/6 being more resistant than BALB/c mice to SARS-CoV-2MA10. The BALB/c lung showed the strong up regulation of mitochondrial gene expression seen in the human autopsy lung. However, the C57BL/6 mice appeared to be at an earlier disease stage, with the OXPHOS genes beginning to be induced, but mtDNA genes, OXPHOS assembly genes, and fatty acid oxidation genes still being down regulated.
Therefore, the most immediate bioenergetic effects of SARS-CoV-2 infection occur through the binding of viral polypeptides to critical host cell mitochondrial proteins disrupting mitochondrial metabolism and energetics. Next, the virus initiates a more global attack on mitochondrial bioenergetics by inhibiting the transcription of critical OXPHOS genes, a broad array of other mitochondrial genes, but at the same time preserving functions such as mitochondrial folate metabolism important for viral biogenesis. The global inhibition of mitochondrial OXPHOS would increase mROS which stabilizes HIF-1α resulting in the redirection of substrates away from mitochondrial oxidation and towards glycolysis to generate viral precursors. The host responds by the coordinate induction of bioenergetic gene expression but is foiled by viral blockage at critical transcriptional control nodes. The host also up regulates the mitochondrial innate immune genes (CMPK2, IFI27, ACOD1/IRG1), which contributes to the ultimate elimination of the virus, but exacerbating the host inflammatory response.
The host also responds by the coordinate induction of bioenergetic gene expression in other tissues but is foiled by viral blockage at critical transcriptional control nodes. While in the final stages of infection the lung succeeds in fully inducing mitochondrial transcription, this fails in the heart resulting in the comprehensive suppression of virtually all cardiac mitochondrial gene expression. This may explain why cardiac dysfunction is a prominent finding in COVID-19 (2–5). Since the heart is highly reliant on mitochondrial bioenergetics, protracted inhibition of mitochondrial bioenergetics could be an important factor on COVID-19 associated deaths.
The pivotal role of the mitochondrion in these processes suggests that an effective approach to mitigating the adverse effects of SARS-CoV-2 might be stimulation of mitochondrial function in combination with inhibition of mROS production. The beneficial effects of reducing mROS have been shown by the reduction of the HIF-1α protein levels, pro-inflammatory mRNA levels, and viral load by treating SARS-CoV-2 infected monocytes with the antioxidants N-acetyl-cysteine and MitoQ (16). The potential beneficial effects of up regulating mitochondrial function have been suggested by the rapamycin inhibition of mTORC1 which decreased viral replication (23). Viral pathology might also be mitigated by neutralizing miR-2393 (34) and activation of mitochondrial biogenesis with bezafibrate (56).
Limitations of the Study
This study has a number of limitations. First, some of the clinical components of the study were designed retrospectively. Consequently, COVID-19 and control groups are not matched for demographic variables, comorbidities, or in-hospital treatments.
Supplementary Material
Acknowledgments:
This work was supported by supplemental funds for COVID-19 research from Translational Research Institute of Space Health through NASA Cooperative Agreement NNX16AO69A (T-0404 to A.B.) and further funding from KBR, Inc provided to A.B.
This work used resources services, and support provided via the COVID-19 HPC Consortium (https://covid19-hpc-consortium.org/), provided specifically by the NASA High-End Computing (HEC) Program through the NASA Advanced Supercomputing (NAS) Division at Ames Research Center which was awarded to A.B.
Additional support came from DOD grant W81XWH-21-1-0128 awarded to D.C.W.
Competing interests:
R.E.S. is on the scientific advisory of Miromatrix, Inc and is a consultant for Alnylam, Inc. D.C.W. is on the scientific advisory boards of Pano Therapeutics, Inc. and Medical Excellent Capital, and has a grant from March, Therapeutics, Inc.
Footnotes
Data and material availability:
The published article includes all datasets generated and analyzed during this study. Processed bulk RNA-seq data for the human related data from the nasopharyngeal and autopsy data is available online with dbGaP Study Accession number: phs002258.v1.p1 and online here at: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002258.v1.p1 and also https://covidgenes.weill.cornell.edu/. The hamster and murine RNA-seq data is deposited on SRA (https://www.ncbi.nim.gov/sra), IDs pending. This study did not generate new unique reagents.
References
- 1.Kingery J. R. et al. , Health status, persistent symptoms, and effort intolerance one year after acute COVID-19 Iinfection. Journal of General Internal Medicine ePub ahead of print, 10.1007/s11606-021-07379-z (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nambiar L. et al. , Acute COVID-19-associated decrements in left and right ventricular function predict all-cause mortality. Journal of the American Society of Echocardiography 35, 229–234 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puntmann V. O. et al. , Outcomes of cardiovascular magnetic resonance imaging in patients recently recovered from coronavirus disease 2019 (COVID-19). JAMA Cardiology 5, 1265–1273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szekely Y. et al. , Spectrum of cardiac manifestations in COVID-19: a systematic echocardiographic study. Circulation 142, 342–353 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie Y., Xu E., Bowe B., Al-Aly Z., Long-term cardiovascular outcomes of COVID-19. Nat. Med. ePub ahead of print, 10.1038/s41591-022-01689-3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang P. et al. , A cross-talk between epithelium and endothelium mediates human alveolar-capillary injury during SARS-CoV-2 infection. Cell Death Dis. 11, 1042 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cortese M. et al. , Integrative imaging reveals SARS-CoV-2-induced reshaping of subcellular morphologies. Cell Host Microbe 28, 853–866.e855 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nardacci R. et al. , Evidences for lipid involvement in SARS-CoV-2 cytopathogenesis. Cell Death Dis. 12, 263 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bojkova D. et al. , Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583, 469–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller B. et al. , Host mitochondrial transcriptome response to SARS-CoV-2 in multiple cell models and clinical samples. Sci. Rep. 11, 3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y. et al. , SARS-CoV-2 hijacks folate and one-carbon metabolism for viral replication. Nat. Commun. 12, 1676 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishnan S. et al. , Metabolic perturbation associated with COVID-19 disease severity and SARS-CoV-2 replication. Mol. Cell. Proteomics 20, 100159 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stukalov A. et al. , Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 594, 246–252 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Duan X. et al. , An airway organoid-based screen identifies a role for the HIF1alpha-glycolysis axis in SARS-CoV-2 infection. Cell Rep 37, 109920 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian M. et al. , HIF-1α promotes SARS-CoV-2 infection and aggravates inflammatory responses to COVID-19. Signal Transduct. Target. Ther. 6, 308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Codo A. C. et al. , Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab 32, 437–446 e435 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medini H., Zirman A., Mishmar D., Immune system cells from COVID-19 patients display compromised mitochondrial-nuclear expression co-regulation and rewiring toward glycolysis. iScience 24, 103471 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ajaz S. et al. , Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. American Journal of Physiology. Cell Physiology 320, C57–C65 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luis R. de Guadiana Garcia et al. , Circulating levels of GDF-15 and calprotectin for prediction of in-hospital mortality in COVID-19 patients: A case series. The Journal of Infection 82, e40–e42 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houtkooper R. H. et al. , Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dodd K. M., Yang J., Shen M. H., Sampson J. R., Tee A. R., mTORC1 drives HIF-1alpha and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 34, 2239–2250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de la Cruz Lopez K. G., Toledo Guzman M. E., Sanchez E. O., Garcia Carranca A., mTORC1 as a regulator of mitochondrial functions and a therapeutic target in cancer. Frontiers in Oncology 9, 1373 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mullen P. J. et al. , SARS-CoV-2 infection rewires host cell metabolism and is potentially susceptible to mTORC1 inhibition. Nat. Commun. 12, 1876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon D. et al. , Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 370, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gordon D. E. et al. , A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang H. W. et al. , SARS-CoV-2 proteome microarray for global profiling of COVID-19 specific IgG and IgM responses. Nat. Commun. 11, 3581 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J. et al. , SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep 34, 108761 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S. et al. , Metabolic reprogramming and epigenetic changes of vital organs in SARS-CoV-2 induced systemic toxicity. JCI Insight 6, e145027 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butler D. et al. , Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat. Commun. 12, 1660 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rath S. et al. , MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Signes A., Fernandez-Vizarra E., Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 62, 255–270 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guerrero-Castillo S. et al. , The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 25, 128–139 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Cuperfain A. B., Zhang Z. L., Kennedy J. L., Goncalves V. F., The complex interaction of mitochondrial genetics and mitochondrial pathways in psychiatric disease. Mol. Neuropsychiatry 4, 52–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald J. T. et al. , The great deceiver: miR-2392’s hidden role in driving SARS-CoV-2 infection. bioRxiv preprint, 2021.2004.2023.441024 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan S. et al. , Mitochondrial miRNA determines chemoresistance by reprogramming metabolism and regulating mitochondrial transcription. Cancer Res. 79, 1069–1084 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong Z. et al. , New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhir A. et al. , Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta R. K. et al. , Blood transcriptional biomarkers of acute viral infection for detection of pre-symptomatic SARS-CoV-2 infection: a nested, case-control diagnostic accuracy study. The Lancet Microbe 2, e508–e517 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu R., Chen F., Wang N., Tang D., Kang R., ACOD1 in immunometabolism and disease. Cell Mol Immunol 17, 822–833 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu J. et al. , Pharmacological inhibition of fatty acid synthesis blocks SARS-CoV-2 replication. Nat. Metab. 3, 1466–1475 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams C. G. et al. , Inhibitors of VPS34 and fatty-acid metabolism suppress SARS-CoV-2 replication. Cell Rep. 36, 109479 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.West A. P., Shadel G. S., Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17, 363–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan K. D. et al. , The COVIDome Explorer researcher portal. Cell Rep 36, 109527 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park J. et al. , System-wide transcriptome damage and tissue identity loss in COVID-19 patients. Cell Rep Med 3 100522, ePub ahead of print, https://www.cell.com/cell-reports-medicine/fulltext/S102666-103791(100522)100022-100522 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rother N. et al. , Hydroxychloroquine inhibits the trained innate immune response to interferons. Cell Rep. Med. 1, 100146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee C. Q. E. et al. , Coding and non-coding roles of MOCCI (C15ORF48) coordinate to regulate host inflammation and immunity. Nat. Commun. 12, 2130 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khan N. A. et al. , mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 26, 419–428 e415 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Fisel P., Schaeffeler E., Schwab M., Clinical and functional relevance of the monocarboxylate transporter family in disease pathophysiology and drug therapy. Clin. Transl. Sci. 11, 352–364 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fukuda R. et al. , HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Liu L., Wise D. R., Diehl J. A., Simon M. C., Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J. Biol. Chem. 283, 31153–31162 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong Y. L. et al. , The small molecule ISRIB rescues the stability and activity of Vanishing White Matter Disease eIF2B mutant complexes. eLife 7, e32733 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong Y. L. et al. , eIF2B activator prevents neurological defects caused by a chronic integrated stress response. eLife 8, e42940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balsa E. et al. , ER and nutrient stress promote assembly of respiratory chain supercomplexes through the PERK-eIF2alpha axis. Mol. Cell 74, 877–890 e876 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y., Kim M. S., Nguyen E., Taylor D. M., Modeling metabolic variation with single-cell expression data. bioRxiv preprint available at 10.1101/2020.01.28.923680 (2020). [DOI] [Google Scholar]
- 55.Leist S. et al. , A mouse-adapted SARS-CoV-2 induces acute lung injury and mortality in standard laboratory mice. Cell 183, 1070–1085.e1012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J. et al. , Association of mitochondrial biogenesis with variable penetrance of schizophrenia. JAMA Psychiatry 78, 911–921 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated and analyzed during this study. Processed bulk RNA-seq data for the human related data from the nasopharyngeal and autopsy data is available online with dbGaP Study Accession number: phs002258.v1.p1 and online here at: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002258.v1.p1 and also https://covidgenes.weill.cornell.edu/. The hamster and murine RNA-seq data is deposited on SRA (https://www.ncbi.nim.gov/sra), IDs pending. This study did not generate new unique reagents.