

In this study, Dolce et al. investigated connections between Ctf4-mediated processes involved in drug resistance, and conducted a suppressor screen of ctf4Δ sensitivity to the methylating agent MMS. Their findings demonstrate a chromatin-based drug resistance mechanism in which defects in parental histone transfer after replication fork passage impair error-free recombination bypass and lead to up-regulation of TLS-mediated mutagenesis and drug resistance.
Keywords: DNA damage tolerance, histone deposition, replication fork, recombination, mutagenesis, Mcm2–Ctf4–Polα, Dpb3–Dpb4
Abstract
Ctf4 is a conserved replisome component with multiple roles in DNA metabolism. To investigate connections between Ctf4-mediated processes involved in drug resistance, we conducted a suppressor screen of ctf4Δ sensitivity to the methylating agent MMS. We uncovered that mutations in Dpb3 and Dpb4 components of polymerase ε result in the development of drug resistance in ctf4Δ via their histone-binding function. Alleviated sensitivity to MMS of the double mutants was not associated with rescue of ctf4Δ defects in sister chromatid cohesion, replication fork architecture, or template switching, which ensures error-free replication in the presence of genotoxic stress. Strikingly, the improved viability depended on translesion synthesis (TLS) polymerase-mediated mutagenesis, which was drastically increased in ctf4 dpb3 double mutants. Importantly, mutations in Mcm2–Ctf4–Polα and Dpb3–Dpb4 axes of parental (H3–H4)2 deposition on lagging and leading strands invariably resulted in reduced error-free DNA damage tolerance through gap filling by template switch recombination. Overall, we uncovered a chromatin-based drug resistance mechanism in which defects in parental histone transfer after replication fork passage impair error-free recombination bypass and lead to up-regulation of TLS-mediated mutagenesis and drug resistance.
The DNA replication process is constantly challenged by endogenous and exogenous sources of genotoxic stress (Branzei and Foiani 2010). If not properly handled, these lesions can lead to genome instability and tumorigenesis and can negatively impact organism development (Jackson and Bartek 2009). To manage replication-associated DNA lesions, eukaryotic cells are endowed with DNA damage tolerance (DDT) mechanisms. There are two main modes of DDT (Branzei and Psakhye 2016). One mode relies on specialized translesion synthesis (TLS) DNA polymerases that can bypass lesions by incorporating nonspecific nucleotides and is generally considered to be error-prone and mutagenic (Waters et al. 2009). The other mode involves a replication-dependent recombination-mediated switch of templates to the newly synthesized sister chromatid (Branzei and Psakhye 2016). This replication-associated recombination process is known as template switching and is generally considered to be error-free, although, depending on the genomic context, it can cause genomic rearrangements (Carr and Lambert 2013; Branzei and Szakal 2016). Several factors associated with the replisome and influencing the DNA damage response are involved in promoting one or both modes of DDT, indicating a complex cross-talk between DNA metabolism pathways to ensure accurate genomic replication (Branzei and Foiani 2010).
Ctf4 is a replisome component conserved through evolution with key structural roles and the ability to recruit proteins with different functions at the replication fork (Simon et al. 2014; Samora et al. 2016; Villa et al. 2016). Ctf4 links the replicative helicase complex with DNA polymerase α, coupling CMG (Cdc45–MCM–GINS) helicase progression with DNA synthesis (Gambus et al. 2009). Recent work delineated that together with Mcm2 and Polα, Ctf4 promotes the transfer of parental (H3–H4)2 histones onto the lagging strand (Gan et al. 2018; Li et al. 2020a). Moreover, Ctf4 is important for error-free DNA damage tolerance via template switching and supports a normal replication fork architecture (Fumasoni et al. 2015), in addition to roles in sister chromatid cohesion (Mayer et al. 2004; Lengronne et al. 2006; Borges et al. 2013; Samora et al. 2016; Srinivasan et al. 2020). Yeast genetic studies and CRISPR/Cas9 screens in human cells identified Ctf4 as a promising cancer therapeutic target (van Pel et al. 2013; Behan et al. 2019); however, how its various functions are interconnected and relevant for drug resistance and DNA repair is not known.
While Ctf4 plays key roles in the DNA synthesis and parental histone transfer onto the lagging strand, leading strand synthesis is primarily carried out by DNA polymerase ε, with a switch to polymerase δ upon DNA damage (Guilliam and Yeeles 2021). Polε consists of the catalytic subunit Pol2 and three auxiliary subunits: Dpb2, Dpb3, and Dpb4 (Tsubota et al. 2006; Chilkova et al. 2007; Aksenova et al. 2010). Dpb3 and Dpb4 are two nonessential small histone-fold proteins that contribute to normal replication fork progression by stabilizing the interaction between Polε and template DNA (Aksenova et al. 2010). Dpb3 and Dpb4 fold together, forming a heterodimer that resembles H2A–H2B (Tsubota et al. 2006). A similar heterodimer between Dpb4 and Dls1 (Dpb3-like subunit 1) is present in the ISW2/yCHRAC chromatin remodeler, which counteracts Dpb3–Dpb4-mediated silencing at telomeres (Iida and Araki 2004). Recent work demonstrated that Dpb3–Dpb4 bind H3–H4 in vitro, participate in the inheritance of heterochromatin, and facilitate the transfer of parental (H3–H4)2 histones on the leading strand (He et al. 2017; Yu et al. 2018).
Here, based on studies suggesting Ctf4 as a promising cancer therapeutic target (van Pel et al. 2013; Behan et al. 2019) with potential roles in tumorigenesis of certain cancers (Sato et al. 2010; Liu et al. 2019), we aimed to investigate Ctf4 functions relevant for replication and resistance to genotoxic agents. We conducted a robot-assisted genetic screen to isolate suppressors of the ctf4Δ hypersensitivity to the DNA-damaging agent methyl methane sulfonate (MMS), which is known to induce DDT and DNA damage response activation (Branzei and Psakhye 2016). We isolated dpb3Δ as a suppressor of ctf4Δ hypersensitivity and uncovered that this suppression takes place in the context of the Dpb3–Dpb4 heterodimer. Dpb3 loss did not affect sister chromatid cohesion (SCC) or replication fork architecture in either WT or ctf4Δ contexts. Strikingly, in trends similar to Ctf4 loss, dpb3Δ cells had a reduced ability to engage in error-free postreplicative gap filling via template switching. We uncovered that the decreased DNA damage sensitivity of ctf4Δ dpb3Δ was mediated by hyperactivation of a mutagenic pathway strongly relying on polymerase ζ that contributed to a synergistic increase in mutagenesis and to drug resistance in the double mutants. Importantly, using engineered mutations in the Dpb3 histone-fold domain and in Mcm2–Ctf4–Polα, we uncovered that parental H3–H4 histone deposition on both leading and lagging strands are important to promote error-free template switching. Defects in Dpb3–Dpb4 arise as compensatory mechanisms to mediate drug resistance in ctf4Δ cells and cause a synergistic increase in mutagenesis. We propose that this chromatin-based mechanism of drug resistance and defective error-free recombination is relevant for the replication stress response and chemotherapeutic resistance of tumor cells harboring mutations in certain replisome components.
Results
An unbiased genetic screen identifies dpb3Δ as a suppressor of ctf4Δ MMS sensitivity
Ctf4 is a replisome-associated factor connecting replicative helicase progression with recombination-mediated DDT by template switching and sister chromatid cohesion (SCC) (Hanna et al. 2001; Fumasoni et al. 2015; Samora et al. 2016; Srinivasan et al. 2020). To gain insights into Ctf4 functional networks and Ctf4-mediated drug resistance processes, we conducted a robot-assisted synthetic genetic array (SGA)-based screen to identify suppressors of its MMS and CPT hypersensitivity. We chose these drugs because they interfere with replication fork progression and we wanted to increase the chances to identify suppressors. We crossed ctf4Δ cells with the yeast gene deletion library (Winzeler et al. 1999), selected double mutants, and subsequently replicated them on plates containing MMS or CPT (Fig. 1A). Among the candidate suppressors found at least once in the screens, overall repeated three times, we isolated and validated dpb3Δ. Next, we validated this result using the W303 yeast background, where we reproduced the finding that deletion of DPB3 reduces the hypersensitivity of ctf4Δ cells to MMS (Fig. 1B) and to CPT (Supplemental Fig. S1A). In this study, we focused on the MMS resistance. Dpb3 and Dpb4, the two nonessential subunits of the polymerase ε holoenzyme, fold together and form a heterodimer (Tsubota et al. 2006), while Dpb4 has functions also in the context of the ISW2/yCHRAC complex (Iida and Araki 2004; Casari et al. 2021). We found that single DPB3 or DPB4 deletions caused the same phenotype as loss of both Dpb3 and Dpb4 regarding their effect on ctf4Δ’s sensitivity to MMS (Fig. 1B), indicating that they function together in modulating the damage sensitivity in the absence of Ctf4.
Figure 1.

Dpb3 loss suppresses the MMS sensitivity but not the cohesion defects of ctf4Δ cells. (A) Schematic representation of the suppressor screen conducted to find suppressors of the MMS sensitivity of ctf4Δ cells. (B) Cells with the indicated genotypes were serially diluted and plated on YPD plates supplemented with the indicated concentrations of MMS to validate the outcome of the suppressor screen in the W303 background in two independent experiments. (C) Cohesion assay. WT, ctf4Δ, dpb3Δ, and ctf4Δ dpb3Δ cells were arrested G1 with α factor and released in medium supplemented with nocodazole for 3 h. Samples were then collected, fixed with ethanol, and processed to be analyzed with a fluorescence microscope. The histogram reports the percentage of cells (with mean and standard deviation) that show two dots from two independent experiments. The two dots indicate the separation of the two sister chromatids. P-values were obtained by using an ordinary one-way ANOVA test with Tukey's multiple comparison test. (*) P < 0.05. Compared with WT, the P-value for ctf4Δ = 0.029, and the P-value for ctf4Δ dpb3Δ = 0.012. See also Supplemental Figure S1.
Several mutants of Ctf4 have been reported in which Ctf4 interaction with specific binding partners is diminished or abolished (Villa et al. 2016). Here we used ctf4-4E, which is defective in interaction with polymerase α and is poorly associated with replication forks, and ctf4-3E, which is proficient in interaction with polymerase α but is defective in interaction with Tof2 and Dpb2 and is characterized by ribosomal DNA instability (Villa et al. 2016). We found that ctf4-4E mutations, but not ctf4-3E, cause hypersensitivity to MMS, which is suppressed by dpb3Δ (Supplemental Fig. S1B). Upon replication stress, Ctf4 interacts with Mms22 in the context of the Mms22–Cul8–Mms1 ubiquitin ligase complex to regulate the replicative function of Mrc1 (Buser et al. 2016). Notably, loss of Mms22 also renders cells sensitive to MMS, and this sensitivity is suppressed by DPB3 deletion (Supplemental Fig. S1C). Moreover, truncation of the N-terminal of Ctf4 that abolishes its interaction with Mms22 resulted in severe damage sensitivity that was suppressed by DPB3 deletion (Supplemental Fig. S1D). Thus, functions of Ctf4 and Mms22 performed in the context of the replisome and impacting the ability of cells to tolerate genotoxic stress are affected by Dpb3–Dpb4.
Suppression of ctf4Δ MMS sensitivity is not linked to restoration of sister chromatid cohesion defects
The absence of Ctf4 leads to impaired sister chromatid cohesion (Srinivasan et al. 2020). We examined whether the DNA damage sensitivity suppression conferred by DPB3 deletion in ctf4Δ correlates with restoration of the cohesion defects. To this end, we used a cohesion assay strain containing a tandem array of Tet operators at the URA3 locus on chromosome V while expressing a GFP-TetR fusion integrated at the HIS3 locus. This configuration allows visualization under the microscope of the SCC state, where transition from one to two GFP signals indicates premature sister chromatid separation (Michaelis et al. 1997). The cohesion defect is usually analyzed during metaphase arrest, using nocodazole as a microtubule-disrupting drug. Upon nocodazole treatment, wild-type cells arrested with largely unseparated chromatids, and only ∼13% of cells showed premature separation (Fig. 1C). This assay revealed that differently from Ctf4, loss of which causes ∼30% premature separation, Dbp3 is not required for SCC. Moreover, the SCC defects of the double mutant ctf4Δ dpb3Δ were equal to those of ctf4Δ cells (Fig. 1C), revealing that the damage sensitivity suppression is independent of SCC restoration.
Histone-fold motif of Dpb3 in the context of DNA polymerase ε modulates the DNA damage sensitivity of ctf4Δ cells
We observed that Dpb3 and Dpb4 have similar effects in suppressing the MMS sensitivity of cells deleted for CTF4 (Fig. 1B). Dpb4 functions are also manifested in the context of a chromatin-remodeling complex named ISW2/yCHRAC, in which it forms a heterodimer with a Dpb3-like protein called Dls1, which contains a histone-fold domain (Iida and Araki 2004). The ISW2 complex participates in heterochromatin inheritance, counteracting Dpb3–Dpb4's role in silencing at telomeres and mating type loci. We investigated whether the drug resistance phenotype conferred by DPB3 and DPB4 deletion in ctf4Δ pertains to ISW2-dependent transcriptional silencing regulation at specific genomic loci. We reasoned that, in such a case, Dls1 loss will likely have an effect similar to that of dpb4Δ. However, different from dpb3Δ and dpb4Δ, dls1Δ did not suppress the damage sensitivity of ctf4Δ cells (Fig. 2A). Thus, Dpb3's effect on ctf4Δ sensitivity is manifested in a polymerase ε context.
Figure 2.

Dpb3 histone-fold functions in the context of polymerase ε mediate damage sensitivity in ctf4 cells. (A,C) MMS sensitivity assay. Cells of the indicated genotypes were grown overnight at 28°C, serially diluted, and spotted on YPD plates containing MMS at the indicated concentrations. Cells were allowed to grow for 3 d at 28°C before images were taken. Three independent experiments were performed, showing similar results. (B) Schematic representation of the histone-fold motifs of the Dpb3–Dpb4 heterodimer (Dpb3 in green, Dpb4 in gray). The highlighted Dpb3 lysines represented by red dots (16K, 18K, 19K, 62K, and 64K) were mutated into alanine (A) or aspartate (D) to impair the histone-fold motif properties of Dpb3.
Dpb3 and Dpb4 subunits contain histone-fold domains that can interact with histones and DNA (Tsubota et al. 2006). Notably, point mutations in the histone-fold domain of Dpb3 have been isolated that affect its histone-binding ability in vitro and reduce chromatin silencing at subtelomeric regions (Tsubota et al. 2006). We asked whether the Dpb3 histone-fold motif plays a role in the rescue of ctf4Δ cells’ hypersensitivity to DNA-damaging drugs. We generated de novo a histone-fold motif-deficient dpb3 mutant (referred to here as dpb3-hfm) by substituting five critical lysine residues involved in histone binding to alanine and aspartate (K16A, K18D, K19A, K62A, and K64A) (Fig. 2B), as reported previously (Tsubota et al. 2006). The dpb3-hfm mutant behaved identically to WT regarding MMS sensitivity but improved ctf4Δ growth in the presence of MMS, like dpb3Δ (Fig. 2C). Thus, the histone-binding ability of the Dpb3–Dpb4 subcomplex modulates the drug sensitivity of ctf4Δ cells.
Dpb3 loss does not alter replication fork architecture in WT or ctf4Δ cells
Histones and nucleosome dynamics have roles in the regulation of DNA replication and DNA repair (Hauer and Gasser 2017). Both Ctf4 and Dpb3 have been associated with different axes of parental (H3–H4)2 deposition on the newly synthesized strands (He et al. 2017; Gan et al. 2018; Yu et al. 2018). We investigated whether this process affects replication fork structure, which is altered in Ctf4 and AND-1 mutants in budding yeast and vertebrate cells (Fumasoni et al. 2015; Abe et al. 2018). Using in vivo psoralen-mediated DNA interstrand cross-linking combined with low-angle rotary shadowing and transmission electron microscopy, we analyzed the fine ultrastructure of DNA replication intermediates (Neelsen et al. 2014). We performed this experiment starting from cells synchronously released in MMS from G2/M arrest (Fig. 3). In line with previous reports (Fumasoni et al. 2015), we observed an increase in reversed forks in ctf4Δ (Fig. 3). Differently from ctf4Δ, dpb3Δ cells had a WT pattern of DNA replication intermediates. Moreover, the ctf4Δ dpb3Δ cells had a trend in the overall abundance of reversed forks similar to that of ctf4Δ, indicating that the improved drug resistance in the double mutant is not related to further alterations or restoration of replication fork structure and functionality.
Figure 3.

Dpb3 does not affect replication fork architecture in WT and ctf4Δ cells. (A,B) Representative electron microscopy images of normal and reversed replication forks, with their relative schematic representations. (P) Parental strands, (D) daughter strands, (R) reversed strands. (C) Electron microscopy (EM) analysis of normal and reversed forks. WT (FY1296), ctf4Δ (HY2194), dpb3Δ (HY3373), and ctf4Δ dpb3Δ (HY7259) were synchronized in G2/M using nocodazole at 25°C and released at 30°C in YPD supplemented with 0.033% MMS. Cells collected at 105 min were in vivo psoralen cross-linked, genomic DNA was extracted, and replication intermediates were enriched via BND cellulose prior to EM analysis. The histogram reports the percentage of reversed forks (with mean and standard deviation) of two independent experiments. P-values were obtained by using an ordinary one-way ANOVA test with Dunnett's multiple comparison test. (**) P < 0.01. Compared with WT, the P-value for ctf4Δ = 0.007, and the P-value for ctf4Δ dpb3Δ = 0.007. The number (n) of analyzed DNA replication intermediates for each genotype and condition is shown below the plots.
Ctf4 and Dpb3 facilitate template switching and synergize in suppressing mutagenesis
We aimed to understand whether the growth advantage of ctf4Δ dpb3Δ cells exposed to MMS is related to rewiring of DDT in ctf4Δ. Previous work in our laboratory uncovered that upon exposure to MMS, ctf4Δ cells are defective in forming replication-associated recombination structures as visualized by neutral–neutral 2D gel electrophoresis of replication intermediates (Fumasoni et al. 2015). Recombination by template switching is mediated by the formation of sister chromatid junctions composed of pseudo-double Holliday junction-like structures, which are subsequently processed by the Sgs1–Top3–Rmi1 (STR) complex (Branzei et al. 2008; Giannattasio et al. 2014). To address potential defects in the formation/stability of template switch intermediates, we used a Tc-sgs1 mutant background in which Sgs1 can be conditionally depleted upon addition of tetracycline (Agashe et al. 2021). This strategy avoids potential growth defects of double-knockout mutants, already noted for sgs1Δ ctf4Δ (Fumasoni et al. 2015), while enabling stabilization of recombination structures arising in the course of the experiment. WT, ctf4Δ, dpb3Δ, and ctf4Δ dpb3Δ strains expressing Tc-sgs1 were arrested in G1 with α factor and released in medium supplemented with MMS and tetracycline. After in vivo psoralen-mediated DNA interstrand cross-linking, genomic DNA extracted from cells collected at the indicated time points was subjected to 2D gel analysis with a probe specific for ARS305, an early and efficient origin of replication located on chromosome 3 (Fig. 4A; Supplemental Fig. S2). In this setup, we reproduced the previously reported defect of ctf4Δ in accumulating DNA replication-associated template switching intermediates (Fumasoni et al. 2015) and found that ctf4Δ dpb3Δ cells are also similarly defective in the generation of template switch intermediates (Fig. 4B). Notably, we also observed a reduction in the level of replication-associated recombination structures arising in dpb3Δ cells. Thus, the results uncover a role for polymerase ε in generating error-free template switch intermediates, which was not very pronounced in hypomorphic pol2 mutants previously used in our laboratory (Vanoli et al. 2010).
Figure 4.

Ctf4 and Dpb3 mediate error-free DNA damage tolerance via template switching. (A) Schematic representation of the major 2D gel signals and of the ARS305 region recognized by the ARS305 probe. (B) Neutral–neutral 2D gel analysis of DNA replication and recombination intermediates extracted from cells with the indicated genotypes. The strains with the indicated backgrounds were synchronized in G1 with α factor at 25°C and released at 30°C in YPD supplemented with 0.033% MMS. Depletion of Sgs1 was achieved by adding 1 mM tetracycline to YPD medium during G1 arrest and release. Cells were collected at the indicated time points, and genomic DNA was extracted and digested with NcoI restriction enzyme for 2D gel analysis of the DNA replication intermediates in the ARS305 region. Depletion of HA-Sgs1 was monitored by Western blot using tubulin as a loading control. Cell cycle progression was monitored by FACS analysis. (1N) G1 cell cycle phase, (2N) G2/M cell cycle phase. Quantification of the X- molecule signals is reported in the histogram. The intensity of the signals was normalized to the monomer spot and is shown in the histograms relative to the highest signal, assigned as 100%. The experiment was performed in duplicate, with a different restriction fragment being analyzed via EcoRV and HindIII digestion, and qualitatively identical results were obtained (see Supplemental Fig. S2). (C) Spontaneous mutation rates at the trp1 and CAN1 loci (×10−7) in cells with the indicated genotypes. Mutation rates with 95% confidence intervals were calculated using the generating function (GF) estimator software bz-rates. See also Supplemental Figure S3.
The above result highlights that the improved viability of ctf4Δ dpb3Δ cells in the presence of DNA damage is not due to a rescue of the ctf4Δ defect in generating replication-associated error-free DNA recombination intermediates or restoration of replication fork architecture. We next investigated whether salvage pathways, potentially error-prone, are instead induced. To this end, we performed spontaneous mutagenesis assays, measuring the rate of reversion of the trp1-1 mutation, which makes the cells capable of growing in the absence of tryptophane, and the rate of forward mutation at the CAN1 gene, encoding for the plasma membrane arginine permease, which makes the cells resistant to canavanine. Of interest, we found that deletions of CTF4 and DPB3 have synergistic effects in spontaneous and DNA damage-induced mutagenesis (Fig. 4C; Supplemental Fig. S3). This increased mutation rate was largely dependent on Rev3, a subunit of polymerase ζ (Fig. 4C). Overall, our data suggest that the simultaneous absence of Ctf4 and Dpb3 causes a defect in the utilization of the error-free branch of DDT and induces increased usage of error-prone translesion polymerases and, in particular, of Rev3/Polζ (Fig. 4C).
TLS activities contribute to the MMS resistance of ctf4 dpb3 cells
The strong sensitivity to MMS of ctf4Δ is partially rescued by deletion of DPB3 (Fig. 1A) or by mutations in the histone-fold motif of Dpb3 (Fig. 2C). This improved viability of ctf4Δ dpb3Δ in the presence of MMS does not associate with a rescue in template switch defects of ctf4Δ but with strongly up-regulated mutagenesis (Fig. 5A). One possibility is that the increased usage of translesion synthesis in ctf4 dpb3 cells accounts for the increased resistance of these cells to MMS (Fig. 4C). To evaluate this hypothesis, we examined the contribution of different TLS polymerases to the viability of ctf4Δ dpb3Δ and ctf4Δ dpb3-hfm cells exposed to MMS-induced DNA damage.
Figure 5.
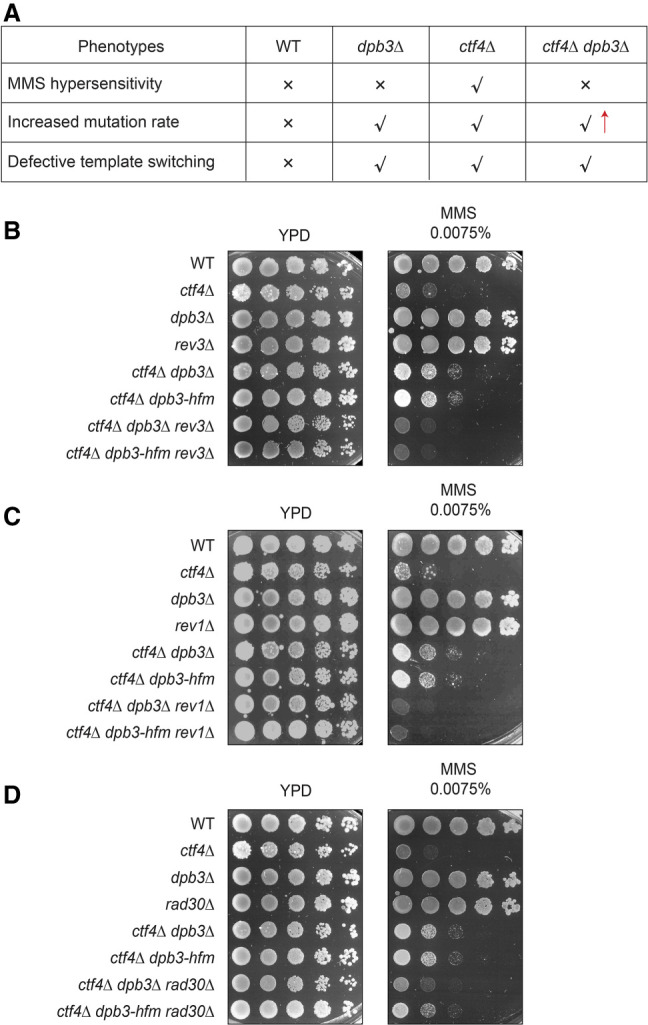
Translesion synthesis polymerases are required for MMS resistance of ctf4Δ dpb3 mutants. (A) Table summarizing the phenotypes of ctf4Δ dpb3Δ cells versus single mutants and WT. (B–D) MMS sensitivity assay of cells with the indicated genotypes. Cells were grown overnight at 28°C, and then serially diluted and spotted on YPD plates supplemented with the indicated MMS concentration to test their sensitivity to the drug. Images were taken after 3 d of incubation at 28°C.
We tested the contribution of Polη (encoded by RAD30) and of Polζ (comprising Rev3–Rev7) associating with Rev1 to the viability of ctf4Δ dpb3Δ and ctf4Δ dpb3-hfm mutants grown in the presence of MMS. We found that both REV3 and REV1 deletions abolished the acquired growth advantage of ctf4Δ dpb3Δ cells and ctf4Δ dpb3-hfm mutant cells in the presence of MMS (Fig. 5B,C). Moreover, we uncovered that although Rad30 contributed to MMS resistance in ctf4Δ dpb3Δ cells, it contributed much less to the MMS resistance of ctf4Δ dpb3-hfm cells (Fig. 5D). These results suggest potential competition between Rad30 and Dpb3, independently of Dpb3 binding to H3–H4. Thus, both Polζ and Polη contribute to the drug resistance phenotype of ctf4 dpb3 mutants.
Template switching relies on efficient deposition of parental histones on both strands
Our results revealed that in the absence of Dpb3, template switching is impaired (Fig. 4B). It was recently shown that the Dpb3–Dpb4 heterodimer is involved in the deposition of parental (H3–H4)2 histones on the leading strand (Yu et al. 2018). We examined whether the histone-fold motif structure function of Dpb3 plays a role in promoting efficient template switch recombination upon genotoxic stress during replication. We performed 2D gel analysis in dpb3Δ and the histone-fold motif-deficient dpb3 (dpb3-hfm) cells upon MMS-induced DNA damage in sgs1 mutant backgrounds. Because dpb3Δ and dpb3-hfm mutations did not show negative genetic interactions with sgs1Δ, we could next address the role of these mutations in the formation or stability of template switch intermediates that accumulate in sgs1Δ cells (Liberi et al. 2005; Branzei et al. 2008). We found that in the sgs1Δ background, both dpb3Δ and dpb3-hfm mutations reduced the levels of X molecules mediating template switching (Fig. 6A). Thus, the ability of Dpb3 to bind and potentially transfer (H3–H4)2 parental histones on the leading strand promotes efficient damage-dependent replication-associated recombination proximal to replication forks. Because relatively little is known about Dpb3 histone transfer functions, we investigated the potential role of parental histone transfer in error-free template switching using mutant alleles specifically defective in the histone transfer process from the parental strand to the lagging strand.
Figure 6.

Replication-coupled recombination requires efficient deposition of parental histones on both newly synthesized strands. (A) 2D gel analysis of cells with the indicated genotypes. Cells were synchronized in G1 with α factor at 25°C and released at 30°C in YPD supplemented with 0.033% MMS. Cells were collected at the indicated time points and subjected to in vivo psoralen-mediated DNA interstrand cross-linking. Genomic DNA was extracted and digested with NcoI restriction enzyme for 2D gel analysis of the replication intermediates accumulating in the ARS305 region. Cell cycle progression was monitored by FACS analysis. (1N) G1 cell cycle phase, (2N) G2/M cell cycle phase. Quantification of the X-molecules signals was performed as described in Figure 4 and is reported next to the corresponding experiment. Experiments were conducted twice with similar results. (B) 2D gel analysis of the DNA replication intermediates accumulating in the ARS305 region of the cells with the indicated genotypes is shown. The experiment was set up as in A, with the addition of 1 mM tetracycline during G1 arrest and release to deplete Sgs1. Depletion of HA-Sgs1 was monitored by Western blot, using tubulin as a loading control. FACS analysis of the cellular DNA content during the experiment is also shown. See also Supplemental Figure S4.
Recent studies have uncovered a role for Mcm2–Ctf4–Polα in interacting with histones and promoting (H3–H4)2 transfer from the parental strand to the lagging strand (Evrin et al. 2018; Gan et al. 2018; Li et al. 2020b). We used pol1-2A2 and mcm2-3A mutants in which histone-fold motifs of Pol1 and Mcm2, respectively, are mutated. These mutants were previously shown to specifically impair the histone-binding and deposition functions of these two proteins (Evrin et al. 2018; Gan et al. 2018; Li et al. 2020b). 2D gel analysis of replication intermediates revealed a significant decrease in template switch intermediates when transfer of parental (H3–H4)2 on the lagging strand is affected (Fig. 6B; Supplemental Fig. S4A). Notably, although pol1-2A2 and mcm2-3A mutants had only mild MMS sensitivity (Supplemental Fig. S4B), we observed a synergistic increase in MMS sensitivity when mcm2-3A was combined with rev3Δ (Supplemental Fig. S4C), supporting also in a genetic manner the key role of the histone transfer to the lagging strand in the DDT pathway. Moreover, pol1-2A2 and mcm2-3A mutations did not suppress or aggravate the MMS sensitivity of ctf4Δ (Supplemental Fig. S4D), in line with an epistatic function of these factors in histone transfer and DNA repair. Altogether, our data indicate that deposition of parental histones in the wake of the replication fork facilitates error-free mediated bypass of lesions.
Discussion
DNA lesions induced by exogenous or endogenous sources impose the frequent utilization of DDT mechanisms in the context of chromatin. While the role of chromatin in the repair of double-strand breaks is significantly advanced (Hauer and Gasser 2017; Clouaire et al. 2018), much less is known about the function of chromatin pathways in the repair or tolerance of DNA damage during replication. Certain replisome components have multiple roles in maintaining genome and chromatin integrity. The replisome factor Ctf4 is a genome integrity factor par excellence, promoting error-free DNA damage tolerance, facilitating a normal replication fork architecture, shaping chromatin assembly, and mediating cohesion between sister chromatids. How these functions are interconnected and possibly linked to the severe chemosensitivity of ctf4Δ cells remains largely unknown.
Starting with an unbiased genetic screen approach, we identified that mutations in Dpb3–Dpb4 histone-fold components of polymerase ε associate with drug resistance in mutants lacking Ctf4 (ctf4Δ) or characterized by poor Ctf4 enrichment (ctf4-4E) to the replisome. Mechanistically, the suppression is linked to the ability of Dpb3–Dpb4 to bind histones and potentially transfer parental (H3–H4)2 histones onto leading strands in the wake of replication forks (Fig. 7). Moreover, we found that the improved viability of ctf4 dpb3 mutants relates to a rewiring of DNA repair pathways engaged at damaged replication forks rather than replication fork architecture and stability.
Figure 7.

Model representing the regulating roles of histone transfer pathways on the usage of error-free versus error-prone pathways of DNA damage tolerance. (Top) Schematics showing that Dpb3–Dpb4 and Mcm2–Ctf4–Polα facilitate parental (H3–H4)2 transfer to the leading and lagging strands of the replication fork. (Bottom) In WT cells, error-free and error-prone pathways are correctly balanced to favor error-free template switching and exclude TLS polymerases from the nascent strands. When Dbp3–Dpb4 and Mcm2–Ctf4–Polα pathways of parental histone transfer are defective, the error-free branch of DDT is impaired and TLS polymerases have increased access to the nascent strands, leading to increased mutagenesis and drug resistance.
Previous work reported that mutations in chromatin components at damaged replication forks can restore replication fork stability and, as consequence, cause chemoresistance (Ray Chaudhuri et al. 2016; Rondinelli et al. 2017; Berti et al. 2020; Kim et al. 2020). In a different view, recent work from the Cantor laboratory (Cong et al. 2021; Panzarino et al. 2021) indicated that daughter strand gaps rather than fork stability align best with chemosensitivity in tumor cells. Ctf4-defective cells have features of increased replication fork degradation and reduced gap filling by recombination-mediated template switching (Fumasoni et al. 2015; Abe et al. 2018). Here we found that the drug sensitivity rescue provided by Dpb3 and Dpb4 mutations is not associated with restoration in replication fork architecture or error-free damage bypass. Indeed, the relative percentage of reversed forks, considered to be the entry point of different nucleases for nascent strand degradation (Kolinjivadi et al. 2017a,b; Berti et al. 2020), remained the same in ctf4Δ and ctf4Δ dpb3Δ double mutants in different experimental conditions. Interestingly, the improved viability of ctf4Δ dpb3Δ cells in the presence of MMS was related to increased usage of TLS polymerases, which became important for viability and strongly contributed to the mutation burden in the double mutants.
Importantly, our study led us to discover that parental histone transfer onto the newly synthesized strands promotes the error-free branch of DNA damage tolerance through template switching and contributes to suppressing mutagenic TLS-mediated gap filling (Fig. 7). Mutations in a subset of factors implicated in parental histone transfer have been reported in congenital diseases in which replication defects were postulated to underlie complex outcomes (Schmit and Bielinsky 2021). Our findings highlight that perturbed or defective histone distribution during replication can severely reduce error-free damage bypass through template switching and can unleash the mutagenic branch of the DNA damage tolerance pathway, thus negatively affecting genome integrity and, potentially, development.
Our present findings are also relevant for chemotherapy. Certain aggressive cancers, such as high-grade glioma and triple-negative breast cancers, often display alterations in genes encoding for chromatin components, such as remodelers and histones (The Cancer Genome Atlas Research Network 2008; Nik-Zainal et al. 2012; Stephens et al. 2012). These tumors are prone to develop chemoresistance (Osuka and Van Meir 2017; Nedeljković and Damjanović 2019), a trait that contributes to their dismal prognosis. The etiology of chemoresistance in gliomas treated with tezolomide, a methylating agent similar to MMS, is likely compounded by hypermutation in mismatch repair (MMR) and Polε genes (Touat et al. 2020), although the mechanism remains to be deciphered. We propose a model of replication stress-induced MMS resistance in ctf4 cells in which mutations in DPB3 and DPB4 inactivate parental histone transfer to the leading strand and unleash mutagenic DNA synthesis dependent on TLS polymerases. These results indicate that TLS polymerase inhibitors affecting Polζ function may be best suited to deal with refractory and aggressive tumors carrying mutations in replisome and chromatin components.
Overall, our study uncovers that parental histone transfer during replication contributes to the establishment of an optimum environment for the error-free bypass of replication-associated lesions with an impact on the normal cellular physiology and drug resistance in cancer cells exposed to chemotherapies.
Materials and methods
Yeast strains
The Saccharomyces cerevisiae strains used in this study are primarily derivatives of the W303 background and are listed in Supplemental Table S1. The yeast strains and mutants were constructed by a PCR-based strategy and by genetic crosses. The dpb3-hfm (K16A, K18D, K19A, K62A, and K64A) mutant was generated using several rounds of polymerase chain reaction (PCR) to introduce mutations leading to the indicated amino acid substitutions in the DPB3 ORF DNA, and this PCR cassette was introduced to replace the dpb3Δ locus. All of the strains were verified by PCR, sequencing, and phenotype.
DNA damage sensitivity suppressor screen
The query strain was grown in YPD overnight, pin-spotted on a 768-format plate, and incubated overnight at 30°C. Mating was performed by pin replica plating the query strain ctf4Δ and the YKO (yeast knockout) mutants on the same medium. Zygotes were pin replica plated on double-selective medium to allow the survival of only the diploids, which were subsequently sporulated. Haploids underwent two consecutive rounds of selection, and double mutants were obtained by consecutive pin replica on double selection plates. This double mutant library was then replica plated on 0.01% methyl methane sulphonate (MMS) and 10 μg/mL camptotechin (CPT) plates. Suppressors were identified by larger colonies growing on MMS or CPT plates. The screen was repeated three times, and selected suppressors were tested manually on different yeast backgrounds for validation.
Yeast growing conditions, cell cycle arrests, and drug treatments
Yeast strains were grown in YPD medium at 25°C unless otherwise indicated. For cell cycle synchronization, logarithmic cells grown at 25°C were arrested in G1 using α factor to a final concentration of 3 μg/mL for 2–2.5 h. Arrest in G2/M was performed by using nocodazole to a final concentration of 20 μg/mL and dimethyl sulfoxide (Sigma) to a final concentration of 1% for 2.5 h at 25°C. G1 or G2/M arrest was verified microscopically and by FACS analysis. Upon synchronization, cells were released by two washes in YP medium, upon which they were resuspended in YPD medium supplemented with MMS (Sigma) and grown at 30°C. For 2D gel analysis and EM analysis, MMS was used at a concentration of 0.033%. For 2D gel analysis of Tc-HA-Sgs1 cells, YPD medium during the arrest and the release was supplemented with tetracycline at 1 mM in order to conditionally deplete HA-tagged Sgs1.
Drug sensitivity assay
Cells were inoculated in YPD and grown overnight at 28°C. The next day, cells were counted and serially diluted before being spotted on YPD plates containing the indicated concentrations of MMS. The plates were incubated at 28°C, and images were taken after 3 d in order to allow cell growth. All of the experiments were performed at least twice independently.
2D gel electrophoresis and electron microscopy sample preparation
Yeast cultures (2 × 109 to 4 × 109 cells) were synchronized in G1 with α factor at 25°C and released at 30°C in YPD media containing 0.033% MMS. In the case of conditionally depleted HA-Sgs1, tetracycline was added to the YPD media during the synchronization and the release at a final concentration of 1 mM. Samples were collected at the indicated time points and incubated with 0.1% sodium azide for 30 min on ice. Replication intermediates were stabilized by in vivo psoralen-mediated interstrand DNA cross-linking and DNA was extracted with CTAB as described in Giannattasio et al. (2014). DNA was digested with NcoI or EcoRV and HindIII restriction endonucleases. In order to separate the replication structures, the digested DNA was run on a one-dimension agarose gel to separate the intermediates by size and then orthogonally on a two-dimension gel in the presence of EtBr to separate the intermediates by size and shape. The separated DNA molecules were transferred onto GeneScreen membranes via Southern blotting following standard procedures. Signals were detected using radioactive-labeled probes against ARS305 (chromosome III 39,026–41,647). Radiolabeling was performed using the Prime-A gene labeling system and purified with ProbeQuant G-50 microcolumns. The 2D gel signals were acquired using Amersham Typhoon scanner software V1.0, and images were retrieved with ImageJ 1.50i software. Quantification of 2D gel signals was performed as in Fumasoni et al. (2015). Experiments were performed independently twice, with either Tc-sgs1 or sgs1Δ alleles and either NcoI or EcoRV and HindIII digestion strategies.
FACS analysis
Approximately 7 × 106 cells were collected and resuspended in 70% ethanol overnight at 4°C. Cells were then washed with 10 mM Tris-HCl (pH 7.5), and RNA and proteins were removed by 0.4 mg/mL RNase A and 1 mg/mL proteinase K treatment. Cells were stained with 1 μM Sytox Green (Invitrogen). Samples were briefly sonicated and analyzed using a Becton Dickinson FACSCalibur system.
Protein techniques
Proteins were analyzed from denatured yeast crude extracts as previously described (Liberi et al. 2000). Briefly, 108 cells/mL were harvested, resuspended in 2 mL of 20% TCA, and transferred to 2-mL Eppendorf tubes. The pellet was then resuspended in 200 μL of 20% TCA, and an equal volume of zyrconium beads (425–600 μM; Sigma) was added. Cells were broken by continuous vortexing for 10 min, and 400 μL of 5% TCA was added to have a final concentration of 10% TCA. The lysates were transferred to new 1.5-mL tubes and centrifuged at 3000 rpm for 10 min at room temperature. The pellet was resuspended in 100 μL of 2× Laemmli buffer. The pH was then adjusted with 50 μL of 1 M Tris base. The protein extracts were boiled for 5 min at 95°C and centrifuged at 13,000 rpm for 2 min at room temperature. The supernatant was collected and analyzed by SDS-PAGE. Western blots were analyzed with α-HA (mouse monoclonal; dilution 1:2000; Thermo Fisher 12CA5) and α-Tubulin (mouse monoclonal; dilution 1:7000; Sigma T5168).
Replication intermediate enrichment and electron microscopy analysis
Genomic DNA was extracted using the CTAB-Psoralen procedure and enriched for replication intermediates as described by Neelsen et al. (2014). Briefly, 15 μg of DNA for each strain was digested with PvuI for 3 h following the manufacturer's instructions and additionally treated with RNase III to avoid dsRNA contamination of the samples. The digestion mix was adjusted to 300 mM NaCl and was then loaded onto a chromatography column containing 1 mL of BND cellulose stock (0.1 g/column; Sigma B-6385) pre-equilibrated with 10 mM Tris-HCl (pH 8) and 300 mM NaCl. DNA was incubated with the BND cellulose for 30 min with resuspension every 10 min to allow full binding of the DNA molecules, and the flow-through was collected by gravity flow. One milliliter of 10 mM Tris-HCl (pH 8) containing 1 M NaCl was added twice to the column to elute linear double-stranded molecules (salt elution, 70%–90% of total DNA). Six-hundred milliliters of 10 mM Tris-HCl (pH 8) and 1 M NaCl containing 1.8% (w/v) caffeine were finally added and incubated for 10 min in order to induce elution of the replication intermediates (RIs). DNA was then purified and concentrated using conical Amicon Ultra centrifugal filters (0.5 mL of 100K-MWCO 100K) following the manufacturer's instructions. Fractions of the samples were then spread onto carbon-coated EM grids, and the DNA intermediates adsorbed on the carbon surface were positively stained with uranyl acetate in the presence of ethanol followed by low-angle platinum-based rotatory shadowing and analyzed as described by Neelsen et al. (2014). The assignment criteria for single-stranded regions on the DNA molecules analyzed in this work were recently described (Neelsen et al. 2014). We note that in order to assign a ssDNA region on a DNA filament it is necessary to identify two points on the DNA molecule that define the borders of the ssDNA region, in which the thickness of the DNA filament (in our experimental conditions, ∼10 nm) decreases close to one-half. We note that the observed thickness of the molecules is largely determined by the amount of deposited heavy atoms during the shadowing procedure. In this experimental condition, the thickness of the DNA fibers is distributed ∼10 nm. The length measurements were performed using a conversion factor expressed in nanometers per base pair obtained using a plasmid of known length used as an internal standard (Neelsen et al. 2014). The EM pictures were acquired using a FEI Tecnai12 G2 Bio twin microscope operated at 120 KV and a side-mounted Gatan Orius SC-1000 camera. The raw files of the EM pictures were generated using the Gatan microscopy suite digital micrograph, saved in the dm3 format, and analyzed using the open source software ImageJ. The pixel size was automatically corrected at each magnification used.
Sister chromatid cohesion
Logarithmically growing cells were adjusted to 8 × 106 cells/mL concentration and treated with 3 μg/mL α factor to induce G1 arrest. Cells were then washed using YP and released in YPD containing 20 μg/mL nocodazole (0.1% DMSO total) in order to allow one round of replication. After 2 h and half of nocodazole treatment, G2 arrest was checked under the microscope. Two milliliters of cells was collected and spun, and the pellet was fixed in 1 mL of 100% cold ethanol. Samples were vortexed and stored overnight at −20°C. The next day, samples were vortexed to eliminate possible clumps, and then 200 μL of cells was diluted with 800 μL of 50 mM Tris HCl (pH 7.6) and sonicated 8–10 sec prior to microscope analysis. Cells were imaged on a DeltaVision microscope (Applied Precision) using a 100× oil immersion lens. Fluorescence was visualized with a conventional FITC excitation filter and a long pass emission filter. Images were analyzed using ImageJ software. Data from two independent replicates are presented as mean ± standard deviation. Statistical analysis was performed by one-way analysis of variance (ANOVA) test (P < 0.05 [*] and not significant [ns]) with Tukey's multiple comparison test using GraphPad Prism 9.0 software.
Mutagenesis assays
Spontaneous mutagenesis at the CAN1 and trp1 loci was assessed by measuring the canavanine-resistant fraction and the fraction of cells that reverted their auxotrophy for tryptophane of parallel saturated populations, respectively. Individual YPD cultures were set up with a 1:20,000 inoculum from an overnight culture that should contain the smallest number of additional mutations possible at the trp1 and CAN1 loci. Cultures were incubated with constant shaking for 36 h at 30°C in order to promote the acquisition of spontaneous mutations. Appropriate dilutions were made, after which cells were pelleted, washed with sterile water, and plated on YPD plates or SC plates lacking arginine and supplemented with 80 μg/mL canavanine or lacking only tryptophane. After 3–5 d of incubation on plates at 30°C, colonies were counted. Three independent experiments were performed for each strain. MMS-induced mutagenesis assay at the CAN1 locus was performed as described above, with the addition of 0.005% MMS 4 h before cell collection. Two independent experiments were performed for each strain. Spontaneous mutation rates were estimated using the generating function (GF) estimator software bz-rates (http://www.lcqb.upmc.fr/bzrates).
Data availability
The raw data associated with this study are available at the Mendeley data set (doi:10.17632/vf7z4c6698.1).
Supplementary Material
Acknowledgments
We thank K. Labib and Z. Zhang for sharing strains, Branzei team members for sharing unpublished reagents and critical discussions, C. Lucca for help in operating the robot for the SGA screens, and B. Szakal for help with the artistic work. We thank the Imaging Facility and the Electron Microscopy Technological Development Unit of Istituto FIRC (Fondazione Italiana per la Ricerca sul Cancro) di Oncologia Molecolare for technical support. This study was supported by the Italian Association for Cancer Research (AIRC IG 18976 and IG 23710) and European Research Council (Consolidator Grant 682190) grants to D.B. C.R.J. was partly supported by AIRC fellowship 23998.
Author contributions: D.B. conceived the study. D.B. and V.D. designed the experiments. V.D. performed most of the experiments with some assistance from C.R.J. M.F. conducted the suppressor screens and the initial validation of the suppressor. S.D. and M.G. performed the electron microscopy experiments, from enrichment of replication intermediates to acquisition and analysis of the data. V.D. and D.B. constructed the figures. D.B. wrote the manuscript with input from V.D. and feedback from all coauthors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.349207.121.
Competing interest statement
The authors declare no competing interests.
References
- Abe T, Kawasumi R, Giannattasio M, Dusi S, Yoshimoto Y, Miyata K, Umemura K, Hirota K, Branzei D. 2018. AND-1 fork protection function prevents fork resection and is essential for proliferation. Nat Commun 9: 3091. 10.1038/s41467-018-05586-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agashe S, Joseph CR, Reyes TAC, Menolfi D, Giannattasio M, Waizenegger A, Szakal B, Branzei D. 2021. Smc5/6 functions with Sgs1–Top3–Rmi1 to complete chromosome replication at natural pause sites. Nat Commun 12: 2111. 10.1038/s41467-021-22217-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenova A, Volkov K, Maceluch J, Pursell ZF, Rogozin IB, Kunkel TA, Pavlov YI, Johansson E. 2010. Mismatch repair-independent increase in spontaneous mutagenesis in yeast lacking non-essential subunits of DNA polymerase ε. PLoS Genet 6: e1001209. 10.1371/journal.pgen.1001209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. 2019. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568: 511–516. 10.1038/s41586-019-1103-9 [DOI] [PubMed] [Google Scholar]
- Berti M, Cortez D, Lopes M. 2020. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol 21: 633–651. 10.1038/s41580-020-0257-5 [DOI] [PubMed] [Google Scholar]
- Borges V, Smith DJ, Whitehouse I, Uhlmann F. 2013. An Eco1-independent sister chromatid cohesion establishment pathway in S. cerevisiae. Chromosoma 122: 121–134. 10.1007/s00412-013-0396-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M. 2010. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11: 208–219. 10.1038/nrm2852 [DOI] [PubMed] [Google Scholar]
- Branzei D, Psakhye I. 2016. DNA damage tolerance. Curr Opin Cell Biol 40: 137–144. 10.1016/j.ceb.2016.03.015 [DOI] [PubMed] [Google Scholar]
- Branzei D, Szakal B. 2016. DNA damage tolerance by recombination: molecular pathways and DNA structures. DNA Repair 44: 68–75. 10.1016/j.dnarep.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Vanoli F, Foiani M. 2008. SUMOylation regulates Rad18-mediated template switch. Nature 456: 915–920. 10.1038/nature07587 [DOI] [PubMed] [Google Scholar]
- Buser R, Kellner V, Melnik A, Wilson-Zbinden C, Schellhaas R, Kastner L, Piwko W, Dees M, Picotti P, Maric M, et al. 2016. The replisome-coupled E3 ubiquitin ligase Rtt101Mms22 counteracts Mrc1 function to tolerate genotoxic stress. PLoS Genet 12: e1005843. 10.1371/journal.pgen.1005843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068. 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AM, Lambert S. 2013. Replication stress-induced genome instability: the dark side of replication maintenance by homologous recombination. J Mol Biol 425: 4733–4744. 10.1016/j.jmb.2013.04.023 [DOI] [PubMed] [Google Scholar]
- Casari E, Gobbini E, Gnugnoli M, Mangiagalli M, Clerici M, Longhese MP. 2021. Dpb4 promotes resection of DNA double-strand breaks and checkpoint activation by acting in two different protein complexes. Nat Commun 12: 4750. 10.1038/s41467-021-25090-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilkova O, Stenlund P, Isoz I, Stith CM, Grabowski P, Lundstrom EB, Burgers PM, Johansson E. 2007. The eukaryotic leading and lagging strand DNA polymerases are loaded onto primer-ends via separate mechanisms but have comparable processivity in the presence of PCNA. Nucleic Acids Res 35: 6588–6597. 10.1093/nar/gkm741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouaire T, Rocher V, Lashgari A, Arnould C, Aguirrebengoa M, Biernacka A, Skrzypczak M, Aymard F, Fongang B, Dojer N, et al. 2018. Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Mol Cell 72: 250–262.e6. 10.1016/j.molcel.2018.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S, Krais J, VanderVere-Carozza PS, Pawelczak KS, Calvo J, et al. 2021. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol Cell 81: 3128–3144.e7. 10.1016/j.molcel.2021.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrin C, Maman JD, Diamante A, Pellegrini L, Labib K. 2018. Histone H2A-H2B binding by Polα in the eukaryotic replisome contributes to the maintenance of repressive chromatin. EMBO J 37: e99021. 10.15252/embj.201899021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D. 2015. Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 complex. Mol Cell 57: 812–823. 10.1016/j.molcel.2014.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambus A, van Deursen F, Polychronopoulos D, Foltman M, Jones RC, Edmondson RD, Calzada A, Labib K. 2009. A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase α within the eukaryotic replisome. EMBO J 28: 2992–3004. 10.1038/emboj.2009.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H, Serra-Cardona A, Hua X, Zhou H, Labib K, Yu C, Zhang Z. 2018. The Mcm2–Ctf4–Polα axis facilitates parental histone H3–H4 transfer to lagging strands. Mol Cell 72: 140–151.e3. 10.1016/j.molcel.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Zwicky K, Follonier C, Foiani M, Lopes M, Branzei D. 2014. Visualization of recombination-mediated damage bypass by template switching. Nat Struct Mol Cell Biol 21: 884–892. 10.1038/nsmb.2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Yeeles JT. 2021. The eukaryotic replisome tolerates leading-strand base damage by replicase switching. EMBO J 40: e107037. 10.15252/embj.2020107037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna JS, Kroll ES, Lundblad V, Spencer FA. 2001. Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol 21: 3144–3158. 10.1128/MCB.21.9.3144-3158.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer MH, Gasser SM. 2017. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev 31: 2204–2221. 10.1101/gad.307702.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Li Y, Dong Q, Chang AY, Gao F, Chi Z, Su M, Zhang F, Ban H, Martienssen R, et al. 2017. Coordinated regulation of heterochromatin inheritance by Dpb3–Dpb4 complex. Proc Natl Acad Sci 114: 12524–12529. 10.1073/pnas.1712961114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida T, Araki H. 2004. Noncompetitive counteractions of DNA polymerase ε and ISW2/yCHRAC for epigenetic inheritance of telomere position effect in Saccharomyces cerevisiae. Mol Cell Biol 24: 217–227. 10.1128/MCB.24.1.217-227.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461: 1071–1078. 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Lee SY, Choi JH, Woo HG, Xhemalce B, Miller KM. 2020. PCAF-mediated histone acetylation promotes replication fork degradation by MRE11 and EXO1 in BRCA-deficient cells. Mol Cell 80: 327–344.e8. 10.1016/j.molcel.2020.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, de Antoni A, Técher H, Baldi G, Costanzo V. 2017a. Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett 591: 1083–1100. 10.1002/1873-3468.12556 [DOI] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. 2017b. Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol Cell 67: 867–881.e7. 10.1016/j.molcel.2017.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengronne A, McIntyre J, Katou Y, Kanoh Y, Hopfner KP, Shirahige K, Uhlmann F. 2006. Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol Cell 23: 787–799. 10.1016/j.molcel.2006.08.018 [DOI] [PubMed] [Google Scholar]
- Li H, Yao NY, O'Donnell ME. 2020a. Anatomy of a twin DNA replication factory. Biochem Soc Trans 48: 2769–2778. 10.1042/BST20200640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hua X, Serra-Cardona A, Xu X, Gan S, Zhou H, Yang WS, Chen CL, Xu RM, Zhang Z. 2020b. DNA polymerase α interacts with H3–H4 and facilitates the transfer of parental histones to lagging strands. Sci Adv 6: eabb5820. 10.1126/sciadv.abb5820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi G, Chiolo I, Pellicioli A, Lopes M, Plevani P, Muzi-Falconi M, Foiani M. 2000. Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and Cdk1 activity. EMBO J 19: 5027–5038. 10.1093/emboj/19.18.5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M. 2005. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19: 339–350. 10.1101/gad.322605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Hu Y, Qin L, Peng XB, Huang YX. 2019. MicroRNA-494-dependent WDHDI inhibition suppresses epithelial-mesenchymal transition, tumor growth and metastasis in cholangiocarcinoma. Dig Liver Dis 51: 397–411. 10.1016/j.dld.2018.08.021 [DOI] [PubMed] [Google Scholar]
- Mayer ML, Pot I, Chang M, Xu H, Aneliunas V, Kwok T, Newitt R, Aebersold R, Boone C, Brown GW, et al. 2004. Identification of protein complexes required for efficient sister chromatid cohesion. Mol Biol Cell 15: 1736–1745. 10.1091/mbc.e03-08-0619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K. 1997. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91: 35–45. 10.1016/S0092-8674(01)80007-6 [DOI] [PubMed] [Google Scholar]
- Nedeljković M, Damjanović A. 2019. Mechanisms of chemotherapy resistance in triple-negative breast cancer—how we can rise to the challenge. Cells 8: 957. 10.3390/cells8090957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Chaudhuri AR, Follonier C, Herrador R, Lopes M. 2014. Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol Biol 1094: 177–208. 10.1007/978-1-62703-706-8_15 [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. 2012. Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993. 10.1016/j.cell.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuka S, Van Meir EG. 2017. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Invest 127: 415–426. 10.1172/JCI89587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzarino NJ, Krais JJ, Cong K, Peng M, Mosqueda M, Nayak SU, Bond SM, Calvo JA, Doshi MB, Bere M, et al. 2021. Replication gaps underlie BRCA deficiency and therapy response. Cancer Res 81: 1388–1397. 10.1158/0008-5472.CAN-20-1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. 2016. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535: 382–387. 10.1038/nature18325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, et al. 2017. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol 19: 1371–1378. 10.1038/ncb3626 [DOI] [PubMed] [Google Scholar]
- Samora CP, Saksouk J, Goswami P, Wade BO, Singleton MR, Bates PA, Lengronne A, Costa A, Uhlmann F. 2016. Ctf4 links DNA replication with sister chromatid cohesion establishment by recruiting the Chl1 helicase to the replisome. Mol Cell 63: 371–384. 10.1016/j.molcel.2016.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Koinuma J, Fujita M, Hosokawa M, Ito T, Tsuchiya E, Kondo S, Nakamura Y, Daigo Y. 2010. Activation of WD repeat and high-mobility group box DNA binding protein 1 in pulmonary and esophageal carcinogenesis. Clin Cancer Res 16: 226–239. 10.1158/1078-0432.CCR-09-1405 [DOI] [PubMed] [Google Scholar]
- Schmit M, Bielinsky AK. 2021. Congenital diseases of DNA replication: clinical phenotypes and molecular mechanisms. Int J Mol Sci 22: 911. 10.3390/ijms22020911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AC, Zhou JC, Perera RL, van Deursen F, Evrin C, Ivanova ME, Kilkenny ML, Renault L, Kjaer S, Matak-Vinković D, et al. 2014. A Ctf4 trimer couples the CMG helicase to DNA polymerase α in the eukaryotic replisome. Nature 510: 293–297. 10.1038/nature13234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M, Fumasoni M, Petela NJ, Murray A, Nasmyth KA. 2020. Cohesion is established during DNA replication utilising chromosome associated cohesin rings as well as those loaded de novo onto nascent DNAs. Elife 9: e56611. 10.7554/eLife.56611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. 2012. The landscape of cancer genes and mutational processes in breast cancer. Nature 486: 400–404. 10.1038/nature11017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, Cortes-Ciriano I, Birzu C, Geduldig JE, Pelton K, et al. 2020. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580: 517–523. 10.1038/s41586-020-2209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubota T, Tajima R, Ode K, Kubota H, Fukuhara N, Kawabata T, Maki S, Maki H. 2006. Double-stranded DNA binding, an unusual property of DNA polymerase ɛ, promotes epigenetic silencing in Saccharomyces cerevisiae. J Biol Chem 281: 32898–32908. 10.1074/jbc.M606637200 [DOI] [PubMed] [Google Scholar]
- Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D. 2010. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet 6: e1001205. 10.1371/journal.pgen.1001205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Pel DM, Stirling PC, Minaker SW, Sipahimalani P, Hieter P. 2013. Saccharomyces cerevisiae genetics predicts candidate therapeutic genetic interactions at the mammalian replication fork. G3 (Bethesda) 3: 273–282. 10.1534/g3.112.004754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa F, Simon AC, Ortiz Bazan MA, Kilkenny ML, Wirthensohn D, Wightman M, Matak-Vinkovíc D, Pellegrini L, Labib K. 2016. Ctf4 is a hub in the eukaryotic replisome that links multiple CIP-box proteins to the CMG helicase. Mol Cell 63: 385–396. 10.1016/j.molcel.2016.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC. 2009. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microb Mol Biol Rev 73: 134–154. 10.1128/MMBR.00034-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. 10.1126/science.285.5429.901 [DOI] [PubMed] [Google Scholar]
- Yu C, Gan H, Serra-Cardona A, Zhang L, Gan S, Sharma S, Johansson E, Chabes A, Xu RM, Zhang Z. 2018. A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science 361: 1386–1389. 10.1126/science.aat8849 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data associated with this study are available at the Mendeley data set (doi:10.17632/vf7z4c6698.1).