


Abstract
The CRISPR-Cas9 genome editing tool is used to study genomic variants and gene knockouts, and can be combined with transcriptomic analyses to measure the effects of such alterations on gene expression. But how can one be sure that differential gene expression is due to a successful intended edit and not to an off-target event, without performing an often resource-demanding genome-wide sequencing of the edited cell or strain? To address this question we developed CRISPRroots: CRISPR–Cas9-mediated edits with accompanying RNA-seq data assessed for on-target and off-target sites. Our method combines Cas9 and guide RNA binding properties, gene expression changes, and sequence variants between edited and non-edited cells to discover potential off-targets. Applied on seven public datasets, CRISPRroots identified critical off-target candidates that were overlooked in all of the corresponding previous studies. CRISPRroots is available via https://rth.dk/resources/crispr.
INTRODUCTION
The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas9 system is an RNA-guided antiviral defense complex capable of cleaving foreign DNA complementary to a short segment of a guide RNA (gRNA) molecule at a DNA site juxtaposed to a motif known as PAM (Protospacer Adjacent Motif) (1). This machinery, originally discovered in prokaryotes, has recently been transformed into a multipurpose genome engineering and visualization technology (2). Among the main applications of CRISPR–Cas9 there are (i) gene knockouts, used to investigate the effects of single or multiple allele losses and (ii) knockins of sequence variants, in which endogenous genes are altered to study genetic disorders. In the former case, gene loss is achieved by mutagenic errors at the cleavage site introduced by error-prone DNA repair pathways such as the non-homologous end-joining (NHEJ) or the microhomology-mediated end joining (MMEJ) (3). In the latter case, a DNA template carrying the genomic variant of interest is delivered to the cell and integrated via homology-directed repair (HDR) after cleavage at a nearby location (4). Following Cas9-mediated editing, cells are sequenced at the targeted locus to examine if the editing was successful. Additionally, few off-target sites predicted by bioinformatics tools based on a gRNA–target sequence-similarity search are typically sequenced to verify the absence of unwanted cleavage events (5).
Genome engineering can be combined with RNA sequencing (RNA-seq) to identify genes whose expression levels are altered as a consequence of the edit (knockin or knockout) (6–13). RNA-seq data can additionally be used to evaluate the presence and abundance of the modified transcript and its possible down-regulation, or total absence, after monoallelic or multiallelic knockout (8). In this regard, RNA-seq can also highlight unwanted editing effects that remain hidden in the sequencing of a short DNA region overlapping the target cleavage site, such as extended loss of heterozygosity and partial or complete loss of a chromosome, all events that have been previously observed in Cas9-edited cells (14–18). In the past few years, RNA-seq data was used in the analysis of potential Cas9 off-target effects either by comparing variants discovered in the transcriptome of edited and non-edited cells (9), or by incorporating off-target predictions with gene expression changes to identify downregulated genes overlapping potential off-targets (7). Although neither of these methods provide a complete assessment, the combination of both allows to prioritize predicted off-targets for validation by pointing to scenarios presenting tangible transcriptome variations. This procedure exploits fully the RNA-seq data, which is instead ignored by generic off-target prediction tools that are based solely on the search for gRNA targets in a given genome while ignoring the transcriptional activity.
Although the literature currently contains a modest number of studies applying CRISPR editing in combination with RNA-seq of at least three replicates of edited and wild-type samples (required for statistical significance), we anticipate that the number of such studies will grow rapidly in the future. In 2011, there were only two papers in PubMed (19) combining CRISPR and RNA-seq, while this number has increased to about 300 per year, with a current total of 843 (Supplementary Table S1). Automatizing the screening of such datasets is currently hindered by the lack of details on the gRNA(s) and the edited site(s) in the data repositories. These are usually provided separately (e.g. in a related article), and need to be found manually.
To better exploit the potential of RNA-seq data we developed CRISPRroots, a tool that compares RNA-seq reads from Cas9-edited cells and corresponding isogenic controls to evaluate potential off-targets and verify on-target editing outcomes. We assess CRISPRroots on seven published RNA-seq datasets with at least three replicates of edited and control samples and show that there are multiple potential off-targets of high relevance that were not taken into account by the corresponding studies. The pipeline around CRISPRroots integrates pre-processing, mapping, gene quantification, differential expression, off-target prediction, variant discovery, Cas9-gRNA binding properties, and assessment of genome integrity with cutting-edge tools. The CRISPRroots pipeline is made in a user-friendly Snakemake (20) workflow that optimizes the handling of computing resources, parallelises tasks, and minimizes software prerequisites via the definition of Conda environments (https://docs.anaconda.com/), facilitating re-usability and reproducibility.
MATERIALS AND METHODS
Implementation
CRISPRroots is implemented as a pipeline consisting of a number of key modules: (1) RNA-seq read processing and mapping; (2) Somatic variant calling; (3) Variant-based off-target screening; (4) Differential gene expression; (5) Assessment of on-target knockins and knockouts; (6) gRNA off-target prediction; (7) Expression-based off-target screening. Combining these modules as depicted in Figure 1 results in an on/off-target report elucidating whether the on-target edit was successful or not and highlighting possible off-target events found in the RNA-seq data or in promoter regions, which are therefore potentially involved in gene expression regulation. In the following we describe the content of these modules.
Figure 1.
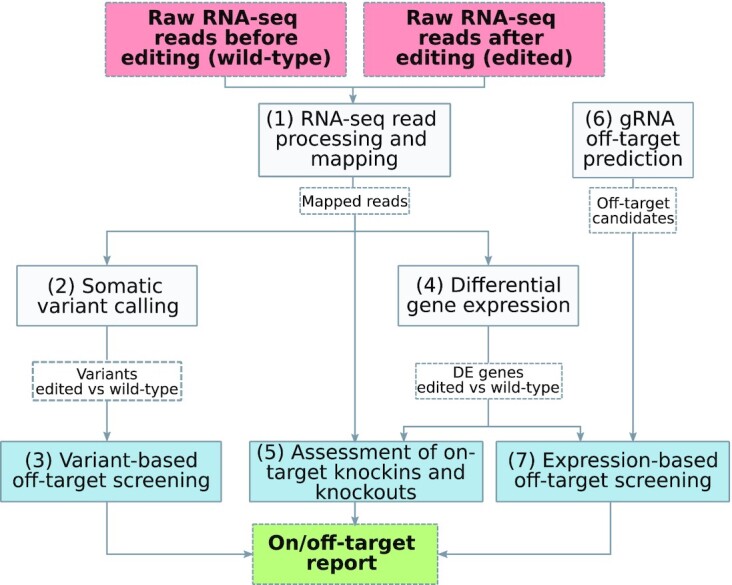
Overview of the CRISPRroots pipeline. We implemented the following main external tools in the seven modules: (1) Cutadapt, Bbduk, FastQC, MultiQC, STAR; (2) Mutect2; (3) RIsearch1; (4) featureCounts, DESeq2; (5) SAMtools; (6) RIsearch2, CRISPRoff; and (7) BEDtools, RIsearch1. The CRISPRroots specific modules are colored in blue. Key input/output files are displayed in dashed boxes. As an option, the off-target search and evaluation (modules 3, 6, 7) can run on a variant-aware version of the genome, generated after discovering germline variants with HaplotypeCaller.
RNA-seq read processing and mapping. The quality of raw reads is assessed with FastQC v.0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and summarized with MultiQC v.1.9 (21). This process is repeated after each of the subsequent filtering steps. The removal of adapters (provided as FASTA files) is performed with Cutadapt v.2.10 (22), which also filters out short reads and low-quality reads. Additional filters can be defined in the configuration file. Reads are cleaned from residual ribosomal RNAs with Bbduk v.37.62 (https://sourceforge.net/projects/bbmap/). Clean reads are mapped to the genome with STAR v.2.6.1a (2-pass mode) (23), and the resulting mapping files are sorted and indexed with SAMtools v.1.9 (24).
Somatic variant-calling. Somatic variants between multiple edited and wild-type samples are discovered with the Mutect2 (25) tool from GATK v.4.2.0.0 (26) after processing the reads as follows: (i) mapped reads are sorted by query name with SortSam (Picard v.2.23.0; http://broadinstitute.github.io/picard); (ii) duplicated read pairs are marked and sorted by coordinates with MarkDuplicates (GATK); (iii) split reads are separated with SplitNCigarReads (GATK); (iv) short variants are called with Mutect2 (min base quality=30; minimum callable depth=10); (v) results are filtered with FilterMutectCalls (GATK). Step (iii) is specific and necessary to call variants in RNA-seq data, as the splicing of introns results in Ns in the CIGAR string describing the mapping. Mutect2 is used with default options, and learns unknown parameters in the filter models from the unfiltered data (25). Because step (iv) is highly demanding in terms of computational resources, reads are first grouped by chromosome, and separate instances of Mutect2 are executed in parallel with GNUparallel (27).
Variant-based off-target screening. Possible cleaved loci are derived from the coordinates and pattern of somatic short variants (SNVs and indels) as follows (Figure 2A): (i) SNVs: the phospho-diester bonds immediately before or after the variated nucleotide; (ii) insertions: the phospho-diester bond linking the nucleotides in the reference between which the insertion is located; (iii) deletions: the phospho-diester bonds immediately before and after any of the removed bases. Knowing that the cut site is three nucleotides upstream from the PAM, all possible related PAM sites, on any strand, are identified. The search for a PAM can be extended up to n nucleotides (default n = 2), to account for possible bulges between the PAM and the cut site. Then, possible gRNA binding regions are defined as the complementary sequences upstream of the cut site that have the same length as the gRNA plus an arbitrary number of m nucleotides (default m = 2) to account for possible bulges on the DNA. Bulges on the gRNA are allowed as well. Interactions between the gRNA and its possible targets are evaluated in terms of resulting gRNA-DNA binding energy, ΔGB, and complementarity in the seed region. The ΔGB is computed following the CRISPRoff v.1.1 (28) binding energy model: ΔGB = δPAM(ΔGH − ΔGO − ΔGU), where ΔGH is the gRNA-DNA binding energy, ΔGO is the energy penalty for opening the target DNA, ΔGU is the penalty for opening up possible gRNA structures, and δPAM is a PAM weight (NGG = 1, NAG = 0.9, NGA = 0.8) (28). The weighted gRNA-DNA binding energy, ΔGH, is computed by RIsearch1 v.1.2 (29), which allows to force interactions to start at the 3′ end of the target (DNA) and end at the 3′ and 5′ ends of the query (gRNA) and the target, respectively. This is done to penalize interactions with PAM-proximal mismatches more severely compared to CRISPRoff (a positive energy is added for mismatches instead of not adding any cost) and to enable the evaluation of potential off-targets to which the gRNA binds forming bulges on the DNA or on the gRNA itself. A positive energy, defined in the RIsearch1 scoring matrix, is added in the presence of bulges and thus the binding is penalized. In RIsearch1, gRNA-target bindings are evaluated using the scoring matrix ‘su_95’, option ‘-f’ to force the start and end of the interactions, and the same array of Cas9 positional weights defined in CRISPRoff. The gRNA minimum free energy, ΔGU, is obtained with RNAfold v.2.2.5 (30). The DNA–DNA opening energy, ΔGO, is computed with the same function as in CRISPRoff, but limiting the calculation to the DNA segment involved in the optimal gRNA–DNA binding to avoid adding energy penalties for unused bases (e.g. Figure 4B shows examples with only a portion of the target DNA being involved in the binding). For each variant, among all possible gRNA bindings starting at any position on the DNA and ending at the PAM site, the one with lowest ΔGB is retained. Flags are added to the final results to signal repeat-masked regions and known SNPs that were intersected with the variant coordinates using BEDtoolsintersectBed v.2.29.2 (31).
Differential gene expression. featureCounts from the Subread package v.2.0.1 (32) is used to quantify the genes present in a merged set of annotations derived from both GENCODE v.33 (33) and FANTOM-CAT v.1.0.0 (34). Only non-chimeric reads are counted (both mate reads for paired-end sequencing). If known, the library type can be specified directly in the config file. If unknown, the strand specificity can be discovered with RSeQC v.4.0.0 (35) within the pipeline’s environment. Differential expression analysis is performed on the gene read counts with DESeq2 from Bioconductor v.3.8 (36). The comparison is done between the conditions ‘Edited’ and ‘Original’ (the non-edited wild-type) which are defined in a sample table (see Supplementary Table S2 for an example). A gene is considered differentially expressed (DEG) if its absolute log2 fold change is >0.5 and the related Benjamini-Hochberg (37) adjusted Wald-test P-value is <0.01. Genes with mean normalized read count across samples <10 are considered as not expressed.
Assessment of on-target knockins and knockouts. Expected knockin mutations are defined in the configuration file by their sequence pattern, genomic coordinates, and possible role in splicing (splice donor, splice acceptor, intron). For every edited position CRISPRroots summarizes the number of reads reporting the reference nucleotides, variants, skips (which symbolize spliced introns), and other events (insertions or deletions) from a pileup of the mapped reads generated with SAMtools. Numerical summaries of the read counts for the different alleles and the genotype interpretation are provided. Special attention is given to variations affecting splice sites and introns, which can alter not only the sequence of a transcript but also the way it is spliced (Figure 2B). For instance, if a splice acceptor is disrupted, splicing can terminate at a downstream splice acceptor (skipping continuation) or not take place at all (intron retention). Because of this, while SNVs affecting coding loci are assessed at a single genomic position, neighboring nucleotides are included in the evaluation of splice donors, acceptors, and introns. Expression changes at the on-target gene are evaluated with DESeq2. Read counts normalized by size factors, the log2 fold change, and the adjusted P-value are summarized in the output.
gRNA off-target prediction. To examine potential off-targets that impair expression or that are located in untranscribed regions, and that hence might not be captured by the analysis in (3), we perform a genome-wide search for off-targets with CRISPRoff. Following the CRISPRoff guidelines, off-targets with up to 6 mismatches to the gRNA are searched with RIsearch2 v.2.1 (38). This tool enables fast searching of gRNA binding via a suffix arrays approach, but does not allow to constrain and weight gRNA-DNA interactions with any number of bulges as RIsearch1. The search is carried out either in the reference genome or, optionally, in its variant-aware version. Potential off-target locations are evaluated with CRISPRoff, supplied with RNAfold v.2.2.5 (30). Predicted off-targets are filtered to eliminate non-spontaneous bindings (ΔGB > 0).
Expression-based off-target screening. Gene expression changes analyzed with DESeq2 are employed in concert with off-target predictions to identify candidate off-targets overlapping differentially expressed genes or their promoter regions. The genomic coordinates of DEGs and their promoter regions (by default, 1 kb upstream of the transcription start site) are intersected with the cleavage coordinates of the predicted off-targets with BEDtools. The ΔGB, initially calculated by CRISPRoff, is re-evaluated with RIsearch1 with the same strategy described above for the variant-based screening, to obtain a more precise evaluation of the binding. An exception are potential cleavage events inside an expressed gene or its promoter that is localized on a hemizygous chromosome (e.g. chrX and chrY in male human). If those events are linked to a variant, they are already reported in the output of module (3) and, hence, they are removed from the list of potential expression-based off-targets.
Figure 2.

Analysis of sequence variations at possible on-/off-targets. (A) Strategy for variant-based off-target screening. Short genomic variants discovered from RNA-seq are screened to find Cas9 binding sites proximal to the possible ‘cut’ positions associated to the variants. All gRNA–DNA interactions ending at one of the identified binding sites are evaluated, and the energetically most favourable one is retained as most likely off-target for each variant. (B) Patterns of on-target single nucleotide variations. Four different types of on-target editing events are shown. For each of them, the reference pileup and examples of other possible mutant pileups (in red) are given. The positions analyzed to evaluate on-target edits are highlighted with grey boxes.
Figure 4.

Predicted Cas9 off-target criticalities discovered in test datasets. (A) Number of predicted off-targets identified in each dataset split by degree of severity (critical or major) and by discovery method (variant or expression-based screening). Major predicted off-targets related to the expression-based screening are divided in type 1 (T1), type 2 (T2) and type 3 (T3). All major predicted off-targets related to variants are of type 2. (B) For each dataset the most favourable (lowest ΔGB) predicted off-target is reported (preference is given to the critical ones with canonical NGG PAM and not overlapping repeat-masked regions). The gRNA-DNA binding pattern is represented with the following symbols: |, canonical base pair; W, wobble base pair; M, mismatch. The portion of the gRNA-DNA interaction with lowest resulting binding energy ΔGB is highlighted in yellow, i.e. the region comprising the segment of the DNA target involved in the most energetically favourable binding interaction with the gRNA. Information on the associated downregulated gene(s) is provided (right). Log2 FC, log2 fold change; P-adj, Benjamini-Hochberg adjusted Wald test P-value. (C) Correlation between the number of Cas9 binding sites in the differentially expressed genes and the number of potential off-targets discovered by the expression-based CRISPRroots analysis. Major type 3 off-targets are excluded because they overlap non-expressed genes or intergenic regions. (D) Correlation between the number of short variants discovered from the RNA-seq in Cas9-edited vs controls cells and the number of variant-based potential off-targets.
(Optional) Variant-aware reference genome. The search and evaluation of potential off-targets (modules 3, 6 and 7) can be carried out on either the reference genome or on a variant-aware version of it. The variant-aware reference genome includes short genomic variants discovered from RNA-seq with HaplotypeCaller (GATK) (39). This tool performs local reassembly of haplotypes in regions that differ from a given reference sequence. In contrast to Mutect2, which tolerates differences in the ploidy profiles of the detected somatic variants, HaplotypeCaller assumes a fixed ploidy as it is designed to call germline variants. The variant-aware genome is generated as follows: (i) split reads are used to call short variants to the reference with HaplotypeCaller (minimum phred-scaled confidence for variant calling=20); (ii) results are filtered with VariantFiltration (GATK) following the GATK recommendations (as of 2019, firstly defined in (40)) to remove clusters of SNVs (window size=35, number of SNVs to define a cluster=3) and any variant with either phred-scaled probability of strand bias (FS) > 30 or variance confidence normalized by depth (QD) < 2. Additionally, variants with approximate read depth (DP) <10 are removed. (iii) variants called between non-edited samples and the reference genome are intersected with the BCFtools v.1.9 (41) isec function keeping only instances carrying identical alleles to produce a solid set of variants to the reference, supported by all samples. (iv) a variant-aware version of the reference genome is generated with BCFtoolsconsensus, which also provides a chain file to lift annotation coordinates. The pipeline can be configured to take either the reference (REF) or the alternative (ALT) allele in the presence of heterozygous variants. Although this procedure only provides the union of different haplotypes (non-reference alleles), to our knowledge there is no tool that can insert germline variants in a reference genome while preserving the haplotypes assembled during variant calling. For the test cases presented here, the pipeline was run twice, using in turn the REF and the ALT allele for heterozygous variants. By using the variant-aware genome it may be possible to find potential off-targets that would remain hidden in a reference-based analysis. However, the generation of a variant-aware genome requires significant time and resources, and it did not provide any relevant benefit in the definition of major or critical candidate off-targets concerning our test cases. Thus, the procedure is set as an option in the pipeline.
Datasets
To test the pipeline, seven RNA-seq datasets were retrieved from five public studies. Because one study employed two gRNAs for a single knockout, there are a total number of eight test cases of Cas9-gRNA activity (Table 1).
Table 1.
List of test cases and the respective gRNAs and PAMs
| Test case | Study | gRNA | PAM |
|---|---|---|---|
| QPRT-INS | (7) | GCAGCGGGCCAGCGTGTTGA | GGG |
| QPRT-DEL | (7) | GCAGTTGAGTTGGGTAAATA | TGG |
| GRIN2B-FW | (8) | GATGGCAATGCCATAGCCAG | TGG |
| GRIN2B-REV | (8) | AGATTCTGGGTGGAAGCGCC | AGG |
| APOE | (9) | CCTCGCCGCGGTACTGCACC | AGG |
| PIK3CA-HET | (10) | ATGAATGATGCACATCATGG | TGG |
| PIK3CA-HOMO | (10) | ATGAATGATGCACATCATGG | TGG |
| OGFOD1 | (11) | GGCAGGACGCCGTTCAGTCA | CGG |
1-2: QPRT. Haslinger et al. generated QPRT-homozygous knockout cells by means of two gRNAs targeting different loci, which generated an insertion (QPRT-INS395A) and a deletion (QPRT-DEL268T) at the target sites in SH-SY5Y cells (7). RNA-seq data produced via MACE (massive analysis of cDNA ends) (42) was downloaded in 3 replicates for 3 experimental settings (QPRT-INS395A, QPRT-DEL268T and wild-type empty control (eCtrl)). In the off-target assessment included in the study, no predicted off-target with up to 4 mismatches is reported to overlap a gene downregulated in knockout compared to eCtrl and not downregulated between additionally sequenced wild-type cells and eCtrl.
3-4: GRIN2B. Bell et al. generated biallelic GRIN2B knockouts with a two-gRNA Cas9-mediated double nickase system, with two gRNAs (GRIN2B-FW and GRIN2B-REV) and differentiated the cells in cortical neurons (8). Of note, the usage of a double nickase system is expected to importantly reduce, but not abolish, off-target activity (43). RNA-seq data was downloaded in 4 replicates for both knockout and control cells.
5: APOE. APOE3 to APOE4 induced pluripotent stem cells (iPSCs) were generated by Lin et al. (9) and RNA-seq data was sequenced in 3 replicates for both edited and non-edited cells. The study also presents an off-target analysis based on exonic variants between edited APOE4 iPSCs and parental APOE3 iPSC, which did not highlight any variation possibly related to off-targets.
6-7: PIK3CA. Heterozygous and homozygous knockins of PIK3CAH1047R in iPSCs were obtained by Madsen et al. (10). RNA-seq data in 3 replicates was downloaded for heterozygous (PIK3CA-HET), homozygous (PIK3CA-HOMO), and wild-type iPSCs. The authors confirmed the absence of unwanted edits at 17 off-target locations predicted with http://crispr.mit.edu from the Zhang Lab or Cas-OFFinder (44) by Sanger sequencing.
8: OGFOD1. The effect of Cas9-mediated homozygous knockout of OGFOD1 in cardiomyocytes was investigated by Stoehr et al. (11). The top 20 off-targets predicted by CRISPOR (45) were sequenced, without finding mutations attributable to off-target effects (11). RNA-seq data was downloaded in four replicates for both knockout and wild-type cells.
Data pre-processing
As part of the CRISPRroots pipeline, raw RNA-seq reads were pre-processed by removing low quality 3′ ends (min phred score = 30), adapters, dangling Ns, and reads shorter than 90% of their original length after cleaning.
RESULTS
Assessment of CRISPR–Cas9 on-target editing activity
We applied the CRISPRroots pipeline (version 1.1) on seven public RNA-seq datasets from both Cas9 knockout (QPRT-INS, QPRT-DEL, GRIN2B-FW/REV, OGFOD1) (7,8,11) and knockin (APOE, PIK3CA-HET, PIK3CA-HOMO) (9,10) experiments. As mentioned above these seven datasets constitute eight test cases, as two gRNAs (FW and REV) were employed for the knockout of GRIN2B in the GRIN2B-FW/REV dataset (Table 1). The datasets are highly heterogeneous in terms of cell types, library preparation, sequencing strategy, and sequencing depth (Table 2). The amount of sequenced reads varies from 5.5–10.5 M in the MACE-sequenced samples (QPRT-INS and QPRT-DEL datasets) to 12.4–83.3 M reads (or paired-end reads) in other samples. The heterogeneity of these datasets allows us to assess the stability of CRISPRroots in the presence of input data with different properties.
Table 2.
Properties of the RNA-seq datasets selected for testing the pipeline. The sequencing strategies, the approximate number of reads (or pairs of reads in paired-end sequencing) before and after pre-processing, mapping to the human genome (hg38), and feature-assignment to a set of merged GENCODE (33) and FANTOM-CAT (34) annotations are reported for each of the seven datasets
| Dataset | Sequencing protocol, read length (nt) | Raw reads min–max (M) | Pre-proc. reads min–max (M) | Uniquely mapped reads mean ± std (M) | Mapped reads assigned to a feature mean ± std (M) |
|---|---|---|---|---|---|
| QPRT-INS (7) | single, <69 | 5.5–10.5 | 4.5–8.2 | 5.2 ± 1.0 | 4.6 ± 0.9 |
| QPRT-DEL (7) | single, <69 | 6.0–8.2 | 4.5–6.4 | 4.7 ± 0.6 | 4.2 ± 0.6 |
| GRIN2B-FW/REV (8) | paired, 125 | 35.1–44.62 | 27.1–34.0 | 29.6 ± 2.3 | 26.4 ± 2.0 |
| APOE (9) | single, 50 | 12.4–14.5 | 9.4–12.3 | 8.5 ± 1.1 | 7.2 ± 0.8 |
| PIK3CA-HET (10) | single, 50 | 22.9–32.0 | 22.5–31.5 | 20.7 ± 2.5 | 18.8 ± 2.3 |
| PIK3CA-HOMO (10) | single, 50 | 23.1–32 | 22.7–31.5 | 21.7 ± 3.1 | 19.7 ± 2.8 |
| OGFOD1 (11) | paired, 50 | 55.6–83.3 | 47.4–71.8 | 51.9 ± 6.8 | 43.9 ± 5.8 |
Distinct strategies are applied on knockout and knockin experiments to assess on-target editing activities, as explained below. Depending on the settings employed for editing, a successful knockout is indicated by a significant loss or complete absence of the target gene in the transcriptome and/or by the presence of loss of function indels at the cleavage locus in aberrant transcripts. We evaluate the knockout effectiveness by comparing the expression level of the target gene in the edited and non-edited cells, and by genotyping target locations on the DNA from mapped RNA-seq reads. We find that three of the four homozygous knockout datasets show a significant downregulation of the respective target genes (Figure 3A): QPRT-INS log2 fold-change (l2fc) = –3.27, Benjamini-Hochberg adjusted Wald test P-value (P-adj) = 7.8e-187; QPRT-DEL l2fc = –2.50, P-adj = 6.9e-132; and OGFOD1 l2fc = –1.68, P-adj = 1.6e-52. In the dataset GRIN2B-FW/REV the expression of the target gene is not downregulated (l2fc = –0.02, P-adj = 0.979).
Figure 3.

On-target Cas9-mediated edits in test datasets. (A) Transcript expression log2 fold change of genes targeted for Cas9-directed knockout or knockin computed by comparing expression levels in edited and non-edited cells with DESeq2. Significance determined by the Benjamini-Hochberg adjusted Wald test. (B) Fraction of reads mapping to edited nucleotides carrying the reference allele (REF), the alternative one (the intended edit) (ALT) or anything else (variant/indel/skip) (OTHER) in the test datasets GRIN2B, APOE, and PIK3CA-HET. The genomic coordinates of the edit in the human genome (hg38) are reported on the X-axis. Cell replicates are represented with different symbols. Note that only two of the three edited replicates of PIK3CA-HET have reads overlapping the loci described.
RNA-seq reads mapping at the target cleavage sites of the two gRNAs employed for the knockout of GRIN2B (FW and REV) reveal that the edited cells bear in-frame deletions of variable length. These deletions generate ‘skips’ in the mapping of RNA-seq reads to the genome at the target cleavage sites (Figure 3B). The presence of deletions was also substantiated by Sanger sequencing in the original publication (8), in which these deletions were characterized as frame-shifting. For the QPRT-INS and QPRT-DEL datasets, the status of the on-target edits cannot be assessed from the mapped reads, as the applied MACE sequencing protocol only sequences the 3′ ends of the RNA (7,42). In the OGFOD1 dataset, all the reads fully overlapping the edited locus have deletions of 4 nt, as previously validated by Sanger sequencing in the related study (11).
On-target edits in Cas9-directed knockin datasets are inspected using mapped RNA-seq reads in edited and non-edited lines. Silent mutations introduced to avoid successive Cas9 cleavage are also assessed. Our screening shows that in the APOE, PIK3CA-HET and PIK3CA-HOMO dataset almost all reads (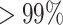 ) at the editing loci map to the wild-type allele in the non-edited cells (Figure 3B). In the APOE edited lines the wild-type allele is substituted with the designed one, and the latter is present in
) at the editing loci map to the wild-type allele in the non-edited cells (Figure 3B). In the APOE edited lines the wild-type allele is substituted with the designed one, and the latter is present in 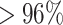 of the reads covering the edited positions in two of the replicates and in
of the reads covering the edited positions in two of the replicates and in 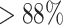 in a third replicate. The remaining mapped reads contain skips in correspondence to the editing site. In replicate 3, which has the lowest percentage of edited reads, there are 2 skip reads out of 17 total reads covering one edited site (chr19:44908684) and 2 out of 19 at the other (chr19:44908692) (Figure 3B). Reads mapping to the three edits in PIK3CA-HET knockin carry homozygous silent mutations at positions chr3:179234289 and chr3:179234292. The reads covering the third editing site (A>G H1047R target edit at chr3:179234297) are heterozygous in only one replicate, while a second replicate exclusively possesses the reference nucleotide at this locus. However, this is supported by only three reads. No read mapping to the edited loci is found in a third replicate. The homozygous editing at the same coordinates in PIK3CA-HOMO is supported by the exclusive presence of the designed nucleotides at all the edited locations in two of the three replicates, while no read maps to these sites in a third replicate. Of note, the read coverage at these genomic coordinates is low in both wild-type and edited cells (six reads in one replicate and two in the other for all three editing sites). The expression levels of edited genes are also evaluated, as significant downregulation of a gene targeted for editing may signal the partial or complete loss of a chromosome due to Cas9 cleavage. In the analyzed knockin datasets the expression of the edited genes does not change (Figure 3A): APOE l2fc = −0.03, P-adj = 1; PIK3CA-HET l2fc = 0.11, P-adj = 1; PIK3CA-HOMO l2fc = −0.21, P-adj = 0.46.
in a third replicate. The remaining mapped reads contain skips in correspondence to the editing site. In replicate 3, which has the lowest percentage of edited reads, there are 2 skip reads out of 17 total reads covering one edited site (chr19:44908684) and 2 out of 19 at the other (chr19:44908692) (Figure 3B). Reads mapping to the three edits in PIK3CA-HET knockin carry homozygous silent mutations at positions chr3:179234289 and chr3:179234292. The reads covering the third editing site (A>G H1047R target edit at chr3:179234297) are heterozygous in only one replicate, while a second replicate exclusively possesses the reference nucleotide at this locus. However, this is supported by only three reads. No read mapping to the edited loci is found in a third replicate. The homozygous editing at the same coordinates in PIK3CA-HOMO is supported by the exclusive presence of the designed nucleotides at all the edited locations in two of the three replicates, while no read maps to these sites in a third replicate. Of note, the read coverage at these genomic coordinates is low in both wild-type and edited cells (six reads in one replicate and two in the other for all three editing sites). The expression levels of edited genes are also evaluated, as significant downregulation of a gene targeted for editing may signal the partial or complete loss of a chromosome due to Cas9 cleavage. In the analyzed knockin datasets the expression of the edited genes does not change (Figure 3A): APOE l2fc = −0.03, P-adj = 1; PIK3CA-HET l2fc = 0.11, P-adj = 1; PIK3CA-HOMO l2fc = −0.21, P-adj = 0.46.
Identification of potential CRISPR–Cas9 off-target sites
Cas9 off-target activity at sites located within a gene or any genomic feature that affect transcription, such as promoters and enhancers, can produce sequence variations that are conveyed to the transcriptome or alter gene expression. To discover potential off-targets from RNA-seq data, we propose a combination of two strategies: the analysis of Cas9 binding sites linked to genomic variants, and the identification of differentially expressed genes harboring predicted off-target sequences. A well supported variant discovered between edited and non-edited cells can delineate off-target events and the corresponding sequence changes introduced by the DNA repair process. Instead, predicted off-targets linked to a differentially expressed gene but without a well supported variant need to be validated by additional DNA sequencing to account for possible silenced alleles (that carry the variant) in an homologous chromosome, whose sequence is unknown in the RNA-seq. An exception for this are predicted off-targets in hemizygous chromosomes (e.g. the Y chromosome; see Methods). The gRNA-DNA interactions of potential off-targets are evaluated in terms of complementarity and the resulting binding free-energy ΔGB with a modified version of the CRISPRoff (28) energy model (see Methods). Given n as the maximum number of mismatches or bulges in the seed, possible off-target interactions are classified into five categories as follows: (i) critical: binding with fully complementary gRNA−DNA seed and linked to a downregulated gene or a variant; (ii) major type 1: binding with fully complementary gRNA-DNA seed and linked to an upregulated gene; (iii) major type 2: binding with ≤n mismatches or bulges in the seed and linked to a variant or to a differentially expressed gene; (iv) major type 3: predicted off-target with perfect complementarity to the gRNA but overlapping a not expressed gene or an intergenic region; (v) minor: any other potential variant-based or expression-based off-target. Possible off-target interactions that after the re-evaluation of the binding energy with RIsearch1 (see Methods) are energetically unfavourable (ΔGB > 0) or that have more than n mismatches or bulges in the seed are flagged. The RNA-DNA base pairs dG·rU and dT·rG, whose contribution to the gRNA−DNA binding energy is limited compared to that of canonical base pairs, are regarded as matches in the binding pattern.
The off-target screening on the test datasets was carried out by searching for either of the PAMs: NGG, NAG and NGA. Up to n = 1 mismatches or bulges were tolerated in the seed region (10 nucleotides from the PAM start position (46)). For each dataset, the off-target analysis was performed on a dedicated variant-aware genome in which short variants to the reference discovered from the RNA-seq data of the wild-type samples were introduced. The procedure was repeated twice, by selecting either the reference or the alternative allele in case of heterozygous background mutations.
Our method identifies critical or major predicted off-targets in all of seven test datasets used for testing (eight test cases, Figure 4A), none of which is among those sequenced in the related studies. In seven test cases, CRISPRroots identified at least one potential off-target with up to four total mismatches or bulges to the gRNA (Figure 4B). The only exception is the dataset APOE, whose single candidate off-target has a total of five mismatches and one bulge in its binding pattern to the gRNA. Predicted off-targets classified as critical and overlapping the promoter or sequence of a downregulated gene are detected in six of eight test cases (Figure 4A, B). Particularly many critical predicted off-target sequences appear in PIK2CA-HOMO, and 12 of those have high similarity of the gRNA with repeatmasked sequences followed by an AGA non-canonical PAM site (Supplementary Table S3). Three test datasets have one predicted off-target sequence that is fully complementary (with wobble base pairs counted as matches) to the entire length of the applied gRNA. These off-targets are intergenic in PIK3CA-HET (Figure 4B) and PIK3CA-HOMO, or overlap a non-expressed gene in QPRT-DEL (Supplementary Table S3). Potential off-targets linked to variants discovered between edited and non-edited lines are observed in four of eight test cases (Figure 4A). Of these, the only critical one (no mismatch in the seed) is a C>T variant found in the GRIN2B dataset and related to the gRNA GRIN2B-REV. This variant is located on chromosome 19 at position 44908822 in hg38, and it corresponds to a missense SNP in the APOE gene (dbSNP (47): rs7412). The co-occurrence of T at position 44908822 and 44908684 in chromosome 19 is referred to as the APOE2 allele and it is associated with reduced risk of Alzheimer’s disease, while the C variant at chr19:44908822 makes a ‘neutral’ APOE (48). All samples have a T at position chr19:44908684, thus the C>T variant at chr19:44908,822 changes the APOE of the cortical neurons in the GRIN2B dataset from neutral to protective. This variant is found in all four GRIN2B loss of function samples and in two of four controls (Supplementary Figure S1). Although this variant is relevant in the study of cortical neurons, the fact that it is also present in half of the controls makes Cas9 off-target editing unlikely at this position.
The only dataset with no predicted off-target linked to differential expression is APOE, which has also the lowest amount of DEGs and variants (DEGs n=18, variants n = 1689), excluding the MACE-sequenced samples. Hence, we checked for the possibility that our pipeline detects possible off-targets in transcriptomic data just by chance based on the size of the search space, i.e. if the number of genomic variants or of binding sites within DEGs correlates with the number of possible off-targets. We did not find a correlation between the number of expression-based potential off-targets (critical, major type 1, or major type 2) and the number of binding sites in DEGs that were identified by searching for the NGG PAM and its reverse complement (Pearson’s r = 0.31, P-value = 0.450; Figure 4C). Also, the number of potential off-targets identified from the variant-based screening is not correlated with the total number of variants discovered between edited and non-edited cells (Pearson’s r = 0.58, P-value = 0.132; Figure 4D).
Running time
The time required to execute the full CRISPRroots pipeline (starting from raw reads) launched on a cluster of standard Linux nodes (Intel® Xeon® CPU E5-2650, 60G RAM and 16 cores) varied from 6 to 8 h for test cases with three replicates per condition (QPRT-INS/DEL, APOE and PIK3CA-HET/HOM) to 15-20 h for test cases with four replicates per condition (GRIN2B and OGFOD1). Up to half of the computing time was consumed by the somatic and germline variant calling.
DISCUSSION
Selecting gRNAs with high on-target effectiveness and low off-target potential is the main objective in the design of Cas9-mediated genome engineering. Following the editing, intended on-target modifications and a restricted number of predicted off-targets are usually validated by DNA sequencing. Given the designed gRNA and a reference genome, off-target predictions are identified and scored by computational tools based on the gRNA sequence similarity and/or other binding properties in relation to DNA sites flanked by valid PAMs. Within this process, the information present in eventual RNA-seq data associated with the experiments remains unused. Previous attempts in exploiting RNA-seq to discover off-targets are rather incomplete, as they employed exclusively either variant discovery (9) or expression changes (7). The latter strategy was additionally limited by the parameters employed for off-target prediction, which allowed up to four mismatches and did not account for wobble base-pairs.
Here, we introduced a comprehensive method, CRISPRroots, to analyse on- and off-targets from RNA-seq data. CRISPRroots allows to (i) verify on-target edits intended to affect the transcriptome, (ii) detect off-target events directly visible in RNA-seq reads and (iii) prioritize other potential off-target events based on the evidence provided by gene expression changes.
CRISPRroots incorporates knowledge of called variants into a relaxed calculation of gRNA−DNA binding energies to increase off-target sensitivity. The related gRNA binding site might not be found in the RIsearch2 search that is provided to CRISPRoff, for instance because of the presence of a bulge or because of the limit of up to six mismatch/wobble base pairs. Thus, we evaluate possible off-targets related to genomic variants with RIsearch1, that also allows for bulges and any number of mismatches. Furthermore, CRISPRroots allows to search for off-targets either in the reference genome or, optionally, in a variant-aware genome in which short variants discovered from RNA-seq are introduced in the reference. Despite the chance of finding off-targets overlapping germline variants discovered in the transcriptome is undoubtedly limited, their potential occurrence remains a major safety concern in gene therapies.
CRISPRroots prioritizes potential off-targets that affect the transcriptome whereas predicted off-targets without expression- or variant-based support are downgraded. Despite the limitations that come from using RNA-seq for off-target ranking, the prioritized potential off-targets in CRISPRroots are the most interesting ones due to the evidence of related consequences on the transcriptome. Both the potential off-targets related to variants in the transcriptome and those on DEGs are more likely to have direct functional consequences, while those in a non-transcribed region may not result in concrete issues. A limitation of the software is that it cannot distinguish between DEGs altered by the activity of off-targets and those altered by the effects of the intended on-target edit. A downstream analysis with, e.g., the STRING (49) database of protein-protein interactions could provide the information necessary to filter potential off-targets related to DEGs functionally linked to the on-target. However, we believe this practice to be hazardous, in particular for potential off-targets with high gRNA affinity, and in contradiction to the primary goal of the software to select potential off-targets for validation prioritization.
CRISPRroots identified in all of seven test datasets potential off-target criticalities that were not addressed by the original studies. No difference was found in the total critical or major candidate off-targets after running CRISPRroots using either the reference or the alternative allele in heterozygous mutations. Potential off-targets were hidden in previous investigations because of the limited search ability of the chosen off-target prediction tools that did not account for at least one of the following elements: (i) dG·rU and dT·rG wobble base pairs, that are disguised as mismatches in similarity-based off-target searches; (ii) alternative (non-canonical) PAM sites such as NAG and NGA; (iii) high number of mismatches tolerated in gRNA-DNA binding, which is often limited in the off-target searches to 3 or 4. Additionally, some of the potential off-targets highlighted by our method were not selected for validation despite being detected as possible off-targets because of the non-identical sorting of the predictions, ruled by scores differing between off-target prediction tools. The analysis performed by CRISPRroots includes wobble base pairs and up to an arbitrary number of mismatches in a user-defined seed region and adjacent to both canonical and non-canonical PAMs. The scoring system we define is also optimized, as it is based on evidence provided by the sequencing data (variations in the sequence or expression level of genes) which is not accounted for by other off-target predictors. The underlying binding energy model employed in CRISPRroots has high off-target prediction performance (28) and is applicable to any genome.
CRISPRroots evaluates gRNA binding energies and transcriptome changes in RNA-seq data to drastically shorten the list of potential Cas9-gRNA off-target events, making their validation more feasible and impactful. In our test cases, the number of critical off-targets overlapping genes or promoters predicted by CRISPRoff has mean(±std) of 24.25(±15.31), while after the careful re-evaluation of the CRISPRoff results and the inclusion of gene expression change evidence in CRISPRroots the critical potential off-targets are reduced to 3.9(±5.91) (Supplementary Table S4). The filters based on binding patterns and energies also allow to better classify, or rule out, an important number of unfavourable interactions at potential off-target sites related to sequence variants discovered between edited and non-edited cells. Counting potential off-targets with up to 6 mismatches or bulges in the binding to the gRNA and linked to a sequence variant without including binding energy consideration leads to a mean(±std) of 15.75(±17.37) sites, while the additional binding analysis of CRISPRroots allows to highlight the 1.25(±1.50) most suitable events (Supplementary Table S4). Due to the lack of experimentally supported true positive off-targets, we cannot evaluate the false positive rate of our high-scoring candidates. Therefore we emphasize that the putative off-targets should be treated as ranked candidates for experimental validation.
An alternative strategy for off-target control was proposed by Haslinger et al. (7). Their study provides RNA-seq data for both a non-targeting empty control vector eCtrl and the wild-type line, and genes differentially expressed between eCtrl and wild-type are excluded from the expression-based off-target analysis presented in the study. The CRISPRroots method currently supports only comparisons between two conditions, edited and wild-type. Thus, only the eCtrl RNA-seq was used in our analysis as non-edited data. The filtering step proposed by Haslinger et al. is attractive, but requires the additional sequencing of non-targeting controls, which is not a common practice. Also, while some potential off-targets could be reasonably excluded based on this criterion, others are not as straightforward. For instance, in QPRT-DEL we detect a potential off-target overlapping the gene RPH3A, downregulated in the edited cells compared to controls. The gene was also reported to be downregulated in wild-type compared to eCtrl in the original study, but to a lower extent (l2fc = −0.95, P-adj=3.2e-4 in wild-type versus eCtrl; l2fc = −1.31, P-adj = 1.8e-6 QPRT-DEL knockout vs eCtrl). Excluding this potential off-target would be incautious, as the change related to QPRT-DEL knockout versus eCtrl is stronger and more significant than that recorded in the wild-type versus eCtrl. Genes harboring predicted off-targets and presenting an increase of expression were also not investigated by Haslinger et al. Although we agree that upregulation is a less likely off-target outcome, other types of mRNA misregulation rather than knockdown cannot be excluded (50). In CRISPRroots predicted off-targets related to upregulated genes are classified as major rather than critical, but not eliminated.
In regard to the on-target editing events, we did not observe any evident inconsistency to the reported knockout and knockin events. This is to a certain extent expected, given that all of the edited sites were verified by sequencing in the original studies. For mapping reads originating from the knockin sequence, partial matches to the reference genome are tolerated with the default parameter settings of STAR. In case of knockins of exogenous genes, it is necessary to introduce the knockin sequence in the reference genome before running the pipeline. Even though Cas9 edits are commonly verified by Sanger sequencing in genome engineering experiments, the validation of such edits in RNA-seq is of relevance as it provides the expression levels of the edited alleles. This procedure also excludes possible errors, e.g. in the labeling or sequencing of samples.
In conclusion, we demonstrate our method to be very useful, as it allows for the identification of possible off-target criticalities that were not investigated in published datasets. The method is included in the first comprehensive pipeline for the analysis of RNA-seq data from CRISPR-Cas9 editing experiments, CRISPRroots. The pipeline can also be applied in studies involving other RNA-directed endonucleases by adjusting the configuration parameters. We believe that this tool will help saving time and resources in the analysis of genome engineered data, facilitating the advancement in this field.
DATA AVAILABILITY
The datasets analysed in this study are available in GEO with accession numbers: GSE113734 (7), GSE114685 (8), GSE102956 (9), GSE126562 (10), GSE130521 (11). These datasets were derived from the following public domain resource: https://www.ncbi.nlm.nih.gov/geo. The analyses were performed with CRISPRroots version 1.1. The CRISPRroots software is freely available via https://rth.dk/resources/crispr and on GitHub via https://github.com/RTH-tools/crisprroots. The software comes with a tutorial on how to reproduce the results presented in this article.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Christian Anthon for developing the CRISPRroots webpage.
Author contributions: G.I.C. implemented the on-target and off-target assessment method. G.I.C. and V.G. developed and tested the pipeline. G.I.C. drafted the manuscript. All authors edited and reviewed the manuscript. S.E.S. and J.G. supervised the study.
Contributor Information
Giulia I Corsi, Center for non-coding RNA in Technology and Health, Department of Veterinary and Animal Sciences, University of Copenhagen, Thorvaldsensvej 57, 1871 Frederiksberg, Denmark.
Veerendra P Gadekar, Center for non-coding RNA in Technology and Health, Department of Veterinary and Animal Sciences, University of Copenhagen, Thorvaldsensvej 57, 1871 Frederiksberg, Denmark.
Jan Gorodkin, Center for non-coding RNA in Technology and Health, Department of Veterinary and Animal Sciences, University of Copenhagen, Thorvaldsensvej 57, 1871 Frederiksberg, Denmark.
Stefan E Seemann, Center for non-coding RNA in Technology and Health, Department of Veterinary and Animal Sciences, University of Copenhagen, Thorvaldsensvej 57, 1871 Frederiksberg, Denmark.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work (including publication costs) was supported by Innovation Fund Denmark [4108-00008B and 4096-00001B to J.G.] and the Danish Research Council [9041-00317B to J.G.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doudna J.A., Charpentier E.. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 3. Chuai G., Wang Q.-L., Liu Q.. In silico meets in vivo : towards computational CRISPR-based sgRNA design. Trends Biotechnol. 2017; 35:12–21. [DOI] [PubMed] [Google Scholar]
- 4. Lin S., Staahl B.T., Alla R.K., Doudna J.A.. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 2014; 3:e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zischewski J., Fischer R., Bortesi L.. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 2017; 35:95–104. [DOI] [PubMed] [Google Scholar]
- 6. Zhang H., Shi J., Hachet M.A., Xue C., Bauer R.C., Jiang H., Li W., Tohyama J., Millar J., Billheimer J.et al.. CRISPR/Cas9-mediated gene editing in human iPSC-derived macrophage reveals lysosomal acid lipase function in human macrophages—brief report. Arteriosclerosis Thrombosis Vasc. Biol. 2017; 37:2156–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haslinger D., Waltes R., Yousaf A., Lindlar S., Schneider I., Lim C.K., Tsai M.-M., Garvalov B.K., Acker-Palmer A., Krezdorn N.et al.. Loss of the Chr16p11.2 ASD candidate gene QPRT leads to aberrant neuronal differentiation in the SH-SY5Y neuronal cell model. Mol. Autism. 2018; 9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bell S., Maussion G., Jefri M., Peng H., Theroux J.-F., Silveira H., Soubannier V., Wu H., Hu P., Galat E.et al.. Disruption of GRIN2B Impairs Differentiation in Human Neurons. Stem Cell Rep. 2018; 11:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin Y.-T., Seo J., Gao F., Feldman H.M., Wen H.-L., Penney J., Cam H.P., Gjoneska E., Raja W.K., Cheng J.et al.. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018; 98:1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Madsen R.R., Knox R.G., Pearce W., Lopez S., Mahler-Araujo B., McGranahan N., Vanhaesebroeck B., Semple R.K.. Oncogenic PIK3CA promotes cellular stemness in an allele dose-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:8380–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stoehr A., Kennedy L., Yang Y., Patel S., Lin Y., Linask K.L., Fergusson M., Zhu J., Gucek M., Zou J.et al.. The ribosomal prolyl-hydroxylase OGFOD1 decreases during cardiac differentiation and modulates translation and splicing. JCI Insight. 2019; 5:e128496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Wel T., Hilhorst R., den Dulk H., van den Hooven T., Prins N.M., Wijnakker J.A.P.M., Florea B.I., Lenselink E.B., van Westen G. J.P., Ruijtenbeek R.et al.. Chemical genetics strategy to profile kinase target engagement reveals role of FES in neutrophil phagocytosis. Nat. Commun. 2020; 11:3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chandrasekaran A., Dittlau K.S., Corsi G.I., Haukedal H., Doncheva N.T., Ramakrishna S., Ambardar S., Salcedo C., Schmidt S., Zhang Y.et al.. Astrocytic reactivity triggered by defective autophagy and metabolic failure causes neurotoxicity in frontotemporal dementia type 3. Stem cell reports. 2021; 16:2736–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee H., Kim J.-S.. Unexpected CRISPR on-target effects. Nat. Biotechnol. 2018; 36:703–704. [DOI] [PubMed] [Google Scholar]
- 15. Ledford H. CRISPR gene editing in human embryos wreaks chromosomal mayhem. Nature. 2020; 583:17–18. [DOI] [PubMed] [Google Scholar]
- 16. Liang D., Gutierrez N.M., Chen T., Lee Y., Park S.-W., Ma H., Koski A., Ahmed R., Darby H., Li Y.et al.. Frequent gene conversion in human embryos induced by double strand breaks. 2020; bioRxiv doi:20 June 2020, preprint: not peer reviewed 10.1101/2020.06.19.162214. [DOI]
- 17. Zuccaro M.V., Xu J., Mitchell C., Marin D., Zimmerman R., Rana B., Weinstein E., King R.T., Palmerola K.L., Smith M.E.et al.. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell. 2020; 183:1650–1664. [DOI] [PubMed] [Google Scholar]
- 18. Alanis-Lobato G., Zohren J., McCarthy A., Fogarty N.M.E., Kubikova N., Hardman E., Greco M., Wells D., Turner J.M.A., Niakan K.K.. Frequent loss of heterozygosity in CRISPR-Cas9–edited early human embryos. Proc. Natl/ Acad. Sci. U.S.A. 2021; 118:e2004832117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coordinators N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017; 46:D8–D13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Köster J., Rahmann S.. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012; 28:2520–2522. [DOI] [PubMed] [Google Scholar]
- 21. Ewels P., Magnusson M., Lundin S., Käller M.. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016; 32:3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011; 17:10. [Google Scholar]
- 23. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The Sequence Alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benjamin D., Sato T., Cibulskis K., Getz G., Stewart C., Lichtenstein L.. Calling somatic SNVs and indels with Mutect2. 2019; bioRxiv doi:02 December 2019, preprint: not peer reviewed 10.1101/861054. [DOI]
- 26. der Auwera G.A., O’Connor BD V.. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. 2020; 1st edn.O’Reilly Media, Inc, City. [Google Scholar]
- 27. Tange O. GNU parallel - the command-line power tool. The USENIX Magazine. 2011; 36:42–47. [Google Scholar]
- 28. Alkan F., Wenzel A., Anthon C., Havgaard J.H., Gorodkin J.. CRISPR-Cas9 off-targeting assessment with nucleic acid duplex energy parameters. Genome Biol. 2018; 19:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wenzel A., Akbaşli E., Gorodkin J.. RIsearch: fast RNA–RNA interaction search using a simplified nearest-neighbor energy model. Bioinformatics. 2012; 28:2738–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lorenz R., Bernhart S.H., zu Siederdissen C.H., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2013; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 33. Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2018; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hon C.-C., Ramilowski J.A., Harshbarger J., Bertin N., Rackham O.J.L., Gough J., Denisenko E., Schmeier S., Poulsen T.M., Severin J.et al.. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature. 2017; 543:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang L., Wang S., Li W.. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012; 28:2184–2185. [DOI] [PubMed] [Google Scholar]
- 36. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B (Methodological). 1995; 57:289–300. [Google Scholar]
- 38. Alkan F., Wenzel A., Palasca O., Kerpedjiev P., Rudebeck A., Stadler P.F., Hofacker I.L., Gorodkin J.. RIsearch2: suffix array-based large-scale prediction of RNA–RNA interactions and siRNA off-targets. Nucleic Acids Res. 2017; 45:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Poplin R., Ruano-Rubio V., DePristo M.A., Fennell T.J., Carneiro M.O., der Auwera G.A.V., Kling D.E., Gauthier L.D., Levy-Moonshine A., Roazen D.et al.. Scaling accurate genetic variant discovery to tens of thousands of samples. 2017; bioRxiv:24 July 2018, preprint: not peer reviewed 10.1101/201178. [DOI]
- 40. DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M.et al.. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011; 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M.et al.. Twelve years of SAMtools and BCFtools. GigaScience. 2021; 10:giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhernakov A., Rotter B., Winter P., Borisov A., Tikhonovich I., Zhukov V.. Massive analysis of cDNA ends (MACE) for transcript-based marker design in pea (Pisum sativum L.). Genomics Data. 2017; 11:75–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ran F., Hsu P., Lin C.-Y., Gootenberg J., Konermann S., Trevino A.E., Scott D., Inoue A., Matoba S., Zhang Y.et al.. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013; 154:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bae S., Park J., Kim J.-S.. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014; 30:1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.-B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J.et al.. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016; 17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang F., Doudna J.A.. CRISPR–Cas9 structures and mechanisms. Ann. Rev. Biophys. 2017; 46:505–529. [DOI] [PubMed] [Google Scholar]
- 47. Sherry S.T., Ward M.-H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K.. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Z., Shue F., Zhao N., Shinohara M., Bu G.. APOE2: protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020; 15:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Szklarczyk D., Gable A.L., Nastou K.C., Lyon D., Kirsch R., Pyysalo S., Doncheva N.T., Legeay M., Fang T., Bork P.et al.. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020; 49:D605–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tuladhar R., Yeu Y., Piazza J.T., Tan Z., Clemenceau J.R., Wu X., Barrett Q., Herbert J., Mathews D.H., Kim J.et al.. CRISPR-Cas9-based mutagenesis frequently provokes on-target mRNA misregulation. Nat. Commun. 2019; 10:4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analysed in this study are available in GEO with accession numbers: GSE113734 (7), GSE114685 (8), GSE102956 (9), GSE126562 (10), GSE130521 (11). These datasets were derived from the following public domain resource: https://www.ncbi.nlm.nih.gov/geo. The analyses were performed with CRISPRroots version 1.1. The CRISPRroots software is freely available via https://rth.dk/resources/crispr and on GitHub via https://github.com/RTH-tools/crisprroots. The software comes with a tutorial on how to reproduce the results presented in this article.