


Abstract
Mouse models have been engineered to reveal the biological mechanisms of human diseases based on an assumption. The assumption is that orthologous genes underlie conserved phenotypes across species. However, genetically modified mouse orthologs of human genes do not often recapitulate human disease phenotypes which might be due to the molecular evolution of phenotypic differences across species from the time of the last common ancestor. Here, we systematically investigated the evolutionary divergence of regulatory relationships between transcription factors (TFs) and target genes in functional modules, and found that the rewiring of gene regulatory networks (GRNs) contributes to the phenotypic discrepancies that occur between humans and mice. We confirmed that the rewired regulatory networks of orthologous genes contain a higher proportion of species-specific regulatory elements. Additionally, we verified that the divergence of target gene expression levels, which was triggered by network rewiring, could lead to phenotypic differences. Taken together, a careful consideration of evolutionary divergence in regulatory networks could be a novel strategy to understand the failure or success of mouse models to mimic human diseases. To help interpret mouse phenotypes in human disease studies, we provide quantitative comparisons of gene expression profiles on our website (http://sbi.postech.ac.kr/w/RN).
INTRODUCTION
Mice are very useful model organisms for studying human disease biology, considering the common anatomical features and physiological processes among mammals (1,2). Human phenotyping using mice has been possible based on the assumption that orthologues may be associated with similar phenotypes (3,4). Using a reverse-genetics approach with CRISPR-Cas9 engineering facilitates the design of disease models by knocking out mouse orthologues of human disease gene candidates (5). Currently, numerous mouse models are available to identify the molecular mechanisms of human diseases and are rapidly applicable to the development of therapeutic strategies and prognostic markers of diseases (2).
Unfortunately, due to the ∼100-million-year divergence between humans and mice, knockout mouse models of human diseases often fail to recapitulate the human phenotypes of interest (1,6). Despite their highly conserved sequences, functional divergence between orthologous gene products has frequently emerged during evolution (7). A plausible hypothesis for this observation is that the expression of orthologous genes has changed and given rise to phenotypic differences between species. Owing to the efforts of systematic phenotyping and semantic comparison, it is now possible to directly test this hypothesis in a comprehensive manner (8). Thus, taking advantage of systematic phenotype comparisons between humans and mice, our previous study recently demonstrated that orthologous genes with greater levels of phenotypic divergence convey highly diverged cis-regulatory elements (REs) and altered transcription across tissues. (9)
However, it remains largely unknown how the evolution of cis-regulatory regions impacts phenotypic divergence, since changes in cis-regulatory regions often have no impact on gene regulatory networks (GRNs). Although cis-regulation diverged extensively in terms of nucleotide sequences and tissue location, its significance in phenotypic divergence via varied gene expression is only speculative (10–12). The trans-regulatory circuitry is highly conserved between humans and mice, despite the substantial plasticity of the cis-regulatory regions (13). With transcription factors (TFs) connected by shared regulatory targets, the TF-to-TF networks are nearly identical between humans and mice. These findings strongly indicate that changes in cis-regulatory regions may only slightly affect gene expression, hindering our ability to assess phenotypic divergence in orthologous genes that rely on TF conservation.
We hypothesize that the rewiring of GRNs leads to phenotypic divergence of orthologous genes by altering functional modules composed of many regulatory targets, rather than a single target. It was previously shown that genotype–phenotype relationships tend to be modular. Indeed, bipartite networks connecting gene knockouts and their functional traits have revealed highly modular structures in both mice (14) and humans, and genes associated with similar diseases often share physical interactions and similar expression profiles (15). Genotypic and gene expression relationships usually display modular behaviors. Bipartite networks comprising expression quantitative trait loci exhibit a highly modular structure, where the gene modules are likely associated with similar biological processes (16). Importantly, genes within these functional modules have co-evolved (17,18). Therefore, comprehensively understanding the modular structure among regulatory target genes would provide insights into the evolution of GRNs and their subsequent phenotypic divergence.
Here, we introduce a computational framework to quantify the evolutionary rewiring of GRNs. For semantic comparisons between the descriptions of human diseases and mouse phenotypic outcomes (9), we utilized the phenotype similarity (PS) score, a quantitative measure of the phenotypic similarity of orthologous genes between the two species. Taking advantage of the PS score, we found that phenotypic discrepancies can be explained by the rewiring of regulatory network connections between two species. Furthermore, we show that species-specific REs, such as promoters and enhancers, contribute to rewired regulatory connections (RCs) and phenotypic differences between humans and mice. We validated these correlations by transcriptomic profiling using multiple transcriptome databases, revealing that the divergence of gene expression is triggered by rewired RCs and leads to phenotypic differences between species. We provide quantitative comparisons of orthologous gene expression profiles between humans and mice on our website (http://sbi.postech.ac.kr/w/RN), which can be utilized to interpret phenotypic differences in mouse models of human diseases.
MATERIALS AND METHODS
Calculating PS score
We collected human gene–phenotype relationships using the OMIM and HPO databases (19,20). OMIM provides manually curated relationships between genetic variants and Mendelian disorders (21). HPOs associate a disorder with a standard phenotype term. We compiled links between 2380 genes and 6506 HPOs. Next, we exploited mouse gene–phenotype relationships in the MGI database, which houses gene–phenotype relationship data obtained from mouse gene knockout experiments used for phenotyping (22). We downloaded and compiled ‘MGI GenePheno rpt’ and ‘MPK_ENSEMBL.rpt’ files from the MGI database. We used phenotypes from mouse models with only one MGI accession number because these phenotypes are associated with perturbation of single genes. If multiple mouse models were available for a single gene, we used all phenotypic information from the models. We collected links between 5737 genes and 7839 MPOs. Thus, ‘associated phenotypes of the gene’ in this study indicates (i) recorded disease symptoms when the gene is mapped to the disease, or (ii) observed phenotypic outcomes when mouse models showed genetic perturbation of the gene. PS scores of orthologous genes were calculated based on semantic comparisons utilizing PhenoDigm (8). Normalization of PS scores was conducted by computing Z-scores with SR, which carry similar numbers of associated phenotype ontologies. Consequently, PS scores of 2142 genes were calculated with 642 HPGs and 642 LPGs (Supplementary Figure S1; Supplementary Figure S2; Supplementary data S1). The detailed methods and validation of the PS scores were described previously (9). Importantly, the critical difference between the PS score and the IMPC measurement is that PS scores are calculated for orthologous genes, while IMPC focuses on the relationship between a mouse gene and a human disease. We compared our PS scores with IMPC similarities by classifying genes into two groups, those mimicking any human diseases versus the rest, based on IMPC database. LPGs are significantly enriched with a set of mouse genes that fail to mimic human disease phenotypes based on the data of IMPC (Supplementary Figure S3) (23). Notably, to investigate characteristics of orthologous genes of phenotypic differences, we analyzed enrichment of developmental (and late-onset) phenotypes in orthologous genes of phenotypic differences. LPGs and HPGs may not be simply classified into gene groups of early or late-onset phenotypes (Supplementary Figure S4). We downloaded phenotype onset data from Orphanet. (https://www.orpha.net/consor/cgi-bin/index.php) Genes with age of onset, including ‘Antenatal’ and ‘Neonatal’, were sorted into a gene group of early onset phenotypes, and genes with age of onset, including ‘Adult’ and ‘Elderly’, were sorted into a gene group of late onset phenotypes. The analysis shows LPGs and HPGs have similar proportions in gene groups of early and late onset phenotypes.
Constructing regulatory networks
The construction of regulatory networks comprised three steps: building a functional module of a gene in humans, transferring the functional module for the mouse orthologous gene, and connecting transcription factors to functional modules in each species. (i) Functional module in humans: we first selected genes that are involved in the same biological process with a human gene. The gene set was defined as a functional module and designated as target genes in the regulatory network of the gene. Annotated biological processes associated with genes were downloaded from GSEA msigdb [https://www.gsea-msigdb.org/gsea/index.jsp, c5: gene ontology gene sets and biological processes]. To delete general functional terms, we only used gene ontology biological processes with fewer than 50 genes. (ii) Functional module in mice: mouse functional modules were transferred from human functional modules based on one-to-one orthologous relationships.
(iii) Connecting TFs to functional modules: to generate species-specific regulatory networks, we gathered TF–target gene relationships in humans and mice. The Regulatory Network Repository (RegNetwork) (http://www.regnetworkweb.org/home.jsp) provides integrated data concerning RCs between TFs and target genes in humans and mice for 391 human TFs and 215 mouse TFs (24). To exploit RCs of high confidence, we analyzed TF–target gene associations validated by experimental evidence and removed all predicted connections. Regulatory networks were built by linking RCs between TFs and target genes in the functional modules, in which RCs were filtered by enrichment testing. We used the hypergeometric distribution for the enrichment testing of RCs and corrected for multiple hypothesis testing (25). If the target genes of a certain TF were enriched in a functional module of a human orthologous gene with an adjusted P value lower than 0.01, the RCs between the TF and functional module were used to construct the regulatory network. The same procedure is applied for mouse regulatory networks. Finally, for an orthologous relationship, one regulatory network was built in humans, and one regulatory network was constructed in mice. Additionally, we analyzed the RCs from the literature-based database TRRUST, which provides TF–target gene regulatory relationships for 800 human TFs and 828 mouse TFs, (https://www.grnpedia.org/trrust/) (26).
Validating the construction of the regulatory network
Validation of the regulatory networks was conducted in three steps. First, to test whether a set of genes in a functional module was co-regulated as a unit, we measured co-expression within the functional modules (Supplementary Figure S5). Co-expression within a functional module was calculated by measuring the Pearson coefficient (ρ) of tissue transcriptomes between the human orthologous gene and other genes in the functional module. 10 000 random modules were generated for each regulatory network. Each random module contains the same number of genes as that of the functional module, and genes were selected from among genes that have transcriptomic data in ENCODE and orthologous relationships with mouse genes. The statistical significance of co-expression levels within functional modules was tested against random functional modules. Transcriptomic data from both species were downloaded from ENCODE (27).
Second, the co-expression levels of TFs and functional modules were evaluated. For a functional module, TFs were divided into two segments: TFs with RCs and TFs without RCs (Supplementary Figure S6). TFs with RCs have connections with functional modules in both species. TFs without RCs have connections in only one species. Co-expression levels in the functional modules were measured using the Pearson coefficient of tissue transcriptomes of the TF and genes in the functional module.
Finally, to test whether functional modules of regulatory networks represent disease phenotypes of human orthologous genes, we calculated the similarity of disease symptoms within the functional modules (Supplementary Figure S7). Phenotypic similarity within functional modules was measured using the overlap of disease symptom terms between the human orthologous gene and other genes in the functional module. Similar to the first validation step, for each regulatory network, 10 000 random functional modules were generated by collecting 10 000 random gene sets. The statistical significance of the phenotypic similarity was tested using random functional modules. Disease symptoms were obtained from HPOs and OMIM, and the annotation of the gene in OMIM was conducted using BioMart data, which provides information on the relationships between an Ensembl gene name and MIM morbid accession number (21,28).
Calculating the conservation of the regulatory network
To measure the evolution of the regulatory network of an orthologous gene, we calculated the conservation of RCs between the two species. Based on the constructed regulatory networks of each species, we used the Jaccard similarity coefficient, which is frequently used to measure the evolutionary rewiring of biological networks (29), to examine the RCs in humans and mice. The measurement is defined as the size of the intersection divided by the size of the union of the groups.
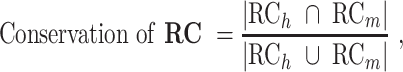 |
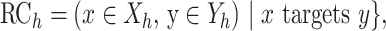 |
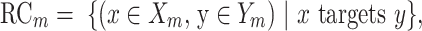 |
where Yh and Ym are the functional modules of the orthologous genes in human and mouse, respectively, and Xh and Xm are the TFs whose targets are significantly enriched in Yh and Ym, respectively. The phrase ‘x targets y’ was used to identify the RCs between ‘x’ and ‘y’ in the RegNetwork (24) and TRRUST (26) databases. Additionally, we analyzed the rewiring of the regulatory networks of orthologous genes using a different similarity measurement, the overlap coefficient (OC), which is defined as the size of the intersection divided by the smaller size of the two groups. OC can capture high similarity when one group is almost included in the other group, and this condition could be regarded as conserved regulatory networks in the evolutionary lineage.
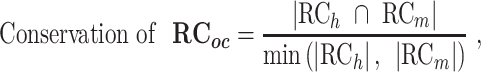 |
where all conditions are the same as described above.
To calculate the conservation of the co-regulation by TFs, we analyzed the RCs linked to the same target gene in a functional module of the regulatory networks. Co-regulatory relationships are defined as the connections of one target gene in a functional module to two or more TFs. If a co-regulatory relationship is detected in both species, the relationship is classified as conserved. Conversely, when one or more changes are observed in the RCs of a co-regulatory relationship, the relationship is classified as rewired. The ‘conservation of the co-regulation by TFs’ was calculated using the proportion of conserved co-regulatory relationships in a functional module of the regulatory network. Furthermore, to calculate the conservation of the regulation of a single gene, we analyzed RCs only linked to a single gene without functional modules in the regulatory network.
Construction of co-regulation network
We constructed the co-regulation networks for each module by assigning co-regulation links to the genes sharing one or more TFs. This procedure changes the bipartite networks (TF–target) into unipartite ones (target – target), investigating another facet of the network structure. Here, we explored k-core because it can characterize sub-modular structure given a module. (30) Within the co-regulation networks, k-cores were identified by collecting the connected components after discarding nodes with the degree smaller than k. To quantify the conservation of cores, we followed the approach proposed in (31), the maximum-matching ratio, which was designed for the comparison between protein complexes. Between a mouse core A and a human core B, their overlap was calculated by the overlap score, |A∩B|2/|A||B|, and the core overlap is the average of the overlap scores. Since a co-regulation networks might have two or more cores, the pairs with the greatest overlap score were taken. Each core was allowed to participate in the pair only once. The link overlap is the Jaccard index of the sets of links.
Calculating conservation of REs in regulatory networks
We leveraged species-specific REs used in our previous works (32). Human and mouse RE (promoter and enhancers) candidates predicted by human and mouse ENCODE projects (http://promoter.bx.psu.edu/ENCODE/download.html) were used. Next, REs were classified as species-specific or conserved by conducting BLASTZ chain alignments of human and mouse genomes and using BnMapper to align mouse cis-REs with the human genome (33). Specifically, ‘mm9.hg19.rBest.chain’ was used to conduct one-to-one mapping of sequence chains between the mouse and human genomes (‘mm9’ and ‘hg19’). One-to-many orthologous sequences were excluded from the mapping analysis.
To quantitatively represent the species specificity of the REs in regulatory networks, we measured the ‘conservation of REs’ by calculating the ratio of conserved REs to all detected REs in the functional module of each regulatory network. ‘Species-specific RE in regulatory network’ was measured by calculating the ratio of species-specific REs to all detected REs in the functional module of each regulatory network. All processes were conducted in promoters and enhancers.
Calculating conservation of TFBSs in regulatory networks
To obtain the genomic locations of TFBSs in the regulatory network, we used a curated collection of sequence-binding motifs for 662 TFs, and each was assigned a confidence score based on its evolutionary conservation across mammals (34–36). To use reliable associations between TFBSs and their target genes, TFBSs were assigned to the genes when the sites were localized within 5000 bps from the TSSs. A TSS of a gene is defined as the first 5′ base of the gene sequence deposited in the Ensembl genome annotation system.
To examine the evolutionary divergence of TFBSs in the regulatory network, we aligned human and mouse genomes and calculated the sequence identities of the TFBSs in the regulatory network of each HPG and LPG and then found the average. As an alignment method, we used BLASTZ (37,38), specifically designed to align two long genomic sequences. This alignment method has been used in studies of the evolutionary conservation of sequences, such as miRNAs (39) and TFBSs (40). The TFBSs of HPGs and LPGs were mapped to the human genome (hg19) and aligned with the mouse genome (mm10). We obtained 105 HPGs and 113 LPGs comprising at least one aligned TFBS in their regulatory networks. The calculation of sequence identities of the TFBSs was conducted except for 2 bp at both ends of the TFBSs (4 bp in total) since sequence mismatches in peripheral positions have less impact on TF binding than those in core positions (41).
Validating the transcriptomic differences between human and mouse
Validating the rewired RCs was conducted by calculating the changes in target gene expression in the regulatory networks between humans and mice. We utilized the expression datasets of both species from ENCODE and FANTOM5, which provide expression levels for each gene in human and mouse homologous tissues (27). Specifically, in the ENCODE dataset, transcriptomes of both species were available in 13 tissues (brain, lung, heart, liver, spleen, adrenal gland, adipose tissue, kidney, pancreas, small intestine, sigmoid colon, testis and ovary). In FANTOM5, transcriptomes of the both species were available in 21 tissues (lymph node, artery, appendix, cerebellum, colon, diencephalon, epididymis, hippocampus, lung, medulla oblongata, ovary, pancreas, prostate, skin, spinal cord, spleen, submandibular gland, testis, tongue, uterus and vagina). To compare gene expression data from each species, we performed quantile normalization of the FPKM values. Covariance- and rank-based correlations of the gene expression profiles were calculated using the Pearson coefficient and Kendall coefficient (τ). The expression conservation of the regulatory network is defined as the average value of the correlation of target gene expression levels in the functional module.
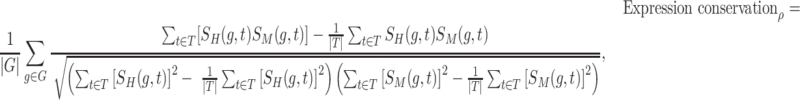 |
where G is a gene group in a functional module within the regulatory network, and T is a homologous tissue used for measuring transcriptomic profiling. SH(g, t) and SM (g, t) are the expression levels (FPKM) of gene g in a homologous tissue t of human and mouse, respectively.
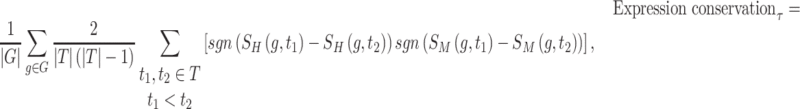 |
where all the conditions are the same as described above.
Furthermore, module boundaries to shape the phenotypic outcomes could be diverse, so we processed all transcriptomic analyses using different boundaries for the functional modules as follows: gene function (gene ontology bioprocess), protein complex (CORUM), and biological pathway (Reactome) (42–44).
RESULTS
Measurements of the PS scores of orthologous genes in humans and mice
To create a normalized PS score, all So were transformed into Z-scores by comparisons with random gene pair semantic similarity scores (SR) (Supplementary Figure S1C). So could have bias due to the number of phenotype ontologies associated with orthologous genes. To delete term number bias in each orthologous gene pair, we processed the normalization step using SR with similar numbers of phenotype terms in both human and mouse species. For the normalization of NEXN, SR were chosen with the same number of HPO terms for human genes and MPO terms for mouse orthologues. The PS score was calculated by transforming SO into Z-scores by term number normalization. (SR of NEXN is 4.72.) Finally, we obtained 2,142 PS scores for orthologous relationships (Supplementary Figure S1D; Supplementary data S1); 642 high phenotype similarity genes (HPGs) represented the set of orthologous genes ranked in the top 30% of PS score and showed significantly high phenotype similarity with SR (P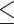 1.33 × 10–2). In contrast, 642 low phenotype similarity genes (LPGs) represented the orthologous gene group ranked in the bottom 30% of PS score. The presence of LPGs, which show no statistical difference in phenotypic similarity distribution compared with SR (P ≥ 0.2486), indicates that a certain proportion of orthologous genes do not share the same or similar phenotypes across species. Notably, we randomly deleted x% of phenotype terms in genotype–phenotype mapping in both species and then newly calculated the PS scores for each orthologous relationship (10 ≤ x ≤ 90). The original PS score and the x%-deleted-version PS score show high correlation, and the convergent tendency was detected using a small proportion of deleted phenotype terms (Supplementary Figure S8).
1.33 × 10–2). In contrast, 642 low phenotype similarity genes (LPGs) represented the orthologous gene group ranked in the bottom 30% of PS score. The presence of LPGs, which show no statistical difference in phenotypic similarity distribution compared with SR (P ≥ 0.2486), indicates that a certain proportion of orthologous genes do not share the same or similar phenotypes across species. Notably, we randomly deleted x% of phenotype terms in genotype–phenotype mapping in both species and then newly calculated the PS scores for each orthologous relationship (10 ≤ x ≤ 90). The original PS score and the x%-deleted-version PS score show high correlation, and the convergent tendency was detected using a small proportion of deleted phenotype terms (Supplementary Figure S8).
Constructing GRNs for orthologous genes based on functional modules
To analyze whether observed phenotypic differences can be explained by the rewiring of regulatory networks between species, we first constructed regulatory networks for each orthologous gene. The regulatory network of an orthologous gene consists of a functional module, TFs, and RCs between TFs and target genes. A functional module is defined as genes sharing biological processes with an orthologous gene and is regarded as a gene regulation target of a regulatory network. Gene sets in functional modules showed significantly high co-expression levels compared to random gene modules (Supplementary Figure S5; P = 8.91 × 10–150, Mann–Whitney U test;), indicating that the expression levels of genes in a functional module are regulated together. TFs are used when RCs exist between a TF and a gene in a functional module (24,26). TFs that are not significantly enriched in a functional module are filtered because those TFs are not likely to be considered regulators of the functional module (25).
Whether regulatory networks of orthologous genes can be built on a collection of false-positive connections during immunoprecipitation in ChIP-seq may be speculative (45). To validate this idea, we analyzed the co-expression of TFs and target genes in the functional module. We compared TF–target gene co-expression levels with and without RCs (Supplementary Figure S6). TFs with RCs were highly co-expressed in the functional module compared with TFs without RCs (P = 1.11 × 10–41; Mann–Whitney U test). Furthermore, we validated that the regulatory network of an orthologous gene can represent phenotypes of the orthologous gene; thus, we analyzed the phenotypic similarity between genes in a functional module of the regulatory network. Specifically, the phenotypic similarity between a human orthologous gene and the gene set of the functional module was calculated using the overlap of annotated HPO terms to genes. We found that the functional modules of the regulatory network showed significantly high similarity to HPOs of the human orthologous gene compared to sets of random gene modules (Supplementary Figure S7; P = 8.10 × 10–182, Mann–Whitney U test;). Taken together, we constructed regulatory networks for orthologous genes that were potentially generated by the regulatory relationship between TFs and target gene sets, representing the phenotypes associated with the orthologous genes.
Analysis of the conservation of gene regulation
Based on the constructed regulatory networks, we analyzed the conservation of RCs, REs, and transcriptomic patterns in tissues. First, to quantitatively measure the rewiring of regulatory networks, we compared the RCs of regulatory networks between human and mouse species (Figure 1A). Evolutionary explanations for the discrepancies in RCs between species could be found in noncoding REs (46). Thus, to measure the conservation of REs, we classified all REs into species-specific or conserved REs (Figure 1B). After aligning the human and mouse noncoding sequences, promoters and enhancers that were detected in only one species were regarded as species-specific REs. For the validation of the conservation of the regulatory network, we proceeded to analyze the conservation of transcriptomic patterns across 21 tissues ((Figure 1C). The following results show the comparison of LPGs and HPGs to identify the correlation between phenotypic differences and the rewiring of regulatory networks between human and mouse species occurring during evolution.
Figure 1.

Conservation of regulatory network connections, regulatory elements, and transcriptomic tissue patterns. (A) The conservation of regulatory connections (RCs) represents the conservation of regulatory networks between humans and mice. (B) The conservation of regulatory elements (REs) was measured by detecting REs including enhancers and promoters in both species. If an enhancer was detected in one species, it was mapped as a species-specific enhancer. If an enhancer was found in both species, it was considered a conserved enhancer. (C) The conservation of gene expression was calculated by comparing tissue transcriptomes in humans and mice using ENCODE and FANTOM5.
Rewiring of regulatory networks indicates phenotypic differences in orthologous genes
We observed that the evolutionary rewiring of regulatory networks contributes to phenotypic discrepancies in orthologous genes between humans and mice. Importantly, this observation was only shown when we used regulatory networks that we based on functional modules, not on the direct regulation of a single gene. The regulatory networks of LPGs showed low conservation scores of RCs, whereas those of HPGs had relatively high conservation scores (Figure 2A, P = 1.93 × 10–4, Mann–Whitney U test; Supplementary Figure S9; Supplementary data S2). Specifically, we quantified the conservation of RCs between humans and mice using the Jaccard index for the regulatory networks of each orthologous gene. When we exploited a database of regulatory connections, TRRUST, which uses literature-based data mining, significant differences in the conservation of RCs were also observed between the regulatory networks of LPGs and HPGs (Supplementary Figure S10) (26). We validated the conservation of RCs using a different similarity coefficient method, the overlap coefficient, and discovered that regulatory networks of LPGs are rewired more frequently than HPGs (Supplementary Figure S11A). In contrast, without regulatory networks for orthologous genes, the regulation of single genes for TFs does not explain the phenotypic differences between species (Figure 2B, P = 1.58 × 10–1, Mann–Whitney U test; Supplementary Figure S11B). Additionally, evolutionary rewiring of the regulatory networks in varied module sizes and diverse PG class cutoffs captured phenotypic differences between orthologous genes between the species. (Supplementary Figure S12; Supplementary Figure S13). These results suggest that genes that confer different phenotypes tend to be regulated by distinct TFs between humans and mice.
Figure 2.

Regulatory network conservation and phenotypic similarity. (A) The conservation of regulatory networks in low phenotype similarity genes (LPGs) and high phenotype similarity genes (HPGs). (B) The conservation of the regulation of a single gene in LPGs and HPGs. (C) The conservation of transcription factor (TF) co-regulation of networks in LPGs and HPGs. (D) The conservation of TF co-regulation of single genes in LPGs and HPGs. The number of LPGs and HPGs used in each test is written under the x-axis. An LPG, phosphorylase kinase catalytic subunit gamma 2 (PHKG2), was used to illustrate (E) the phenotypic similarity matrix and (F) regulatory network conservation, and an HPG, tetratricopeptide repeat domain 21B (TTC21B), was used to illustrate (G) the phenotypic similarity matrix and (H) regulatory network conservation. Filtered-out TF indicates a TF that was filtered out during the enrichment test because of its regulatory links to the functional module.
TFs often cooperate with each other to regulate the expression of target genes (47,48). Based on this biological mechanism, we scrutinized the conservation of the co-regulation by TFs of the regulatory networks for each orthologous gene. The conservation of the co-regulatory relationship between human and mouse was significantly lower in the regulatory networks of LPGs than in those of HPGs (Figure 2C; P = 2.68 × 10–3, Mann–Whitney U test;). Similar to results shown in Figure 2B, the conservation of TF co-regulation could not explain the phenotypic differences in orthologous genes between human and mouse (Figure 2D; P = 2.67 × 10–1, Mann–Whitney U test). Additionally, we tested whether LPGs were under different developmental trajectories between the species. Orthologous genes of phenotypic differences showed different expression trajectories in organ development (Supplementary Figure S14).
PHKG2, an LPG, showed low phenotypic similarity with a PS score 0.15 (Figure 2E), and high network rewiring was observed between humans and mice (Figure 2F). HPOs associated with PHKG2 usually mapped to liver physiology and morphological dysfunctions such as ‘cirrhosis’, ‘hepatomegaly’, and ‘elevated hepatic transaminases’. Unlike the human gene, Phkg2, the orthologous gene in mouse, showed erythrocyte-related phenotypes from MPOs like “increased erythrocyte cell number.” The regulatory network for PHKG2 exhibited a low conservation of RCs, and FOXO3 and CREB1 were the main contributors to this low conservation (Figure 2F). FOXO3 has RCs in the human regulatory network and was reported as a regulator of liver growth (49), whereas Creb1, which has RCs in the mouse network, was reported as a regulator of hemoglobin differentiation in mice (50). Next, we analyzed the rewiring of regulatory networks of genes related to burn, an inflammatory disease, which was previously analyzed in terms of mimicking the human disease with gene expression changed between the two species. Regulatory networks of the burn-related genes were highly rewired between humans and mice (Supplementary Figure S15; P = 5.9 × 10−3, Mann–Whitney U test). This result indicates a challenge exists to engineer mouse models of inflammatory diseases, including burn, consistent with previous study findings (51).
Conversely, as examples of HPGs, TTC21B and Ttc21b (which have an orthologous relationship) map to similar phenotypic traits from HPOs and MPOs, mostly to limb bone and chest rib morphology (Figure 2G). The PS score (2.66) indicated a high phenotypic similarity for this orthologous relationship, and a high conservation of network connections was observed with consistent regulation by GLI1, GLI2 and GLI3 across species (Figure 2H). Previous studies have reported that GLI1, GLI2 and GLI3 play key roles as osteogenic progenitors for bone formation and fracture repair both in humans and mice (52–54).
The structure of co-regulation networks impacts on phenotypic differences
We next investigated how the rewiring of RCs collectively impacts on the regulation of the functional modules based on network topology (Figure 3A). We constructed co-regulation networks, in which target genes were connected by shared TFs, and investigated their core structures within the functional modules. The cores were defined as k-cores, which are the subnetworks comprising only the nodes with the network degree greater than or equal to k. It was shown that the k-cores are effective decomposition for examining the structural diversity embedded in the networks. (30)
Figure 3.

The structure of co-regulation and phenotypic differences. (A) LPGs have lower overlap of co-regulation networks between humans and mice compared with HPGs. (B and C) Core overlap and link overlap of co-regulatory networks in low phenotype similarity genes (LPGs) and high phenotype similarity genes (HPGs). (D) Comparison of core overlap between HPGs and LPGs along with link overlap. (E and F) Examples of co-regulatory networks in LPGs (Glucokinase, GCK) and in HPGs (Bone Morphogenetic Protein Receptor Type 1B, BMPR1B).
We observed that co-regulatory networks of LPGs were less conserved for the core structure of the co-regulation networks than those of HPGs. With k = 3, co-regulatory networks of LPG showed lower overlap of core structures between human and mouse than those of HPGs. (Figure 3B; P = 3.03 × 10–2, Mann–Whitney U test) To quantify the overlap, we calculated the maximum matching ratio that takes the average overlap score with 1-to-1 pairs of the best matching. (see Methods) We also observed similar results with varying k = [1, 4] (Supplementary Figure S16A). The only exception was the case of k = 2, in which the lower overlap of LPGs was not statistically significant (P = N.S., 5.69 × 10–2).
One would expect that the dissimilarity of core structures is derived from the rewiring of co-regulation links. As expected, co-regulatory networks of LPGs exhibited lower overlap of co-regulation links than co-regulatory networks of HPGs. (Figure 3C; P = 4.76 × 10–3, Mann–Whitney U test) Interestingly, we found that the core structures play important role for the distinction between co-regulatory networks of LPGs and HPGs with controlled levels of the rewiring. Divided into three bins with the link overlap, the low and medium group showed differences in the core structure between the co-regulatory networks of LPGs and HPGs (Figure 3D; k = 3). This finding indicates that the core structure further characterizes the conservation of co-regulation networks relevant to the phenotypic conservation, accompanied with that of links. Of note, in case of k = 2, co-regulatory networks of LPGs exhibited a lower core overlap in the medium group. (Supplementary Figure S16B) Examples of LPGs and HPGs are Glucokinase (GCK) and Bone Morphogenetic Protein Receptor Type 1B (BMPR1B), respectively. (Figure 3E and F)
Species-specific REs are related to network rewiring and phenotypic differences
REs that control the expression of target genes are expected to undergo faster evolution than TFs (46); therefore, their sequence alterations could be indicative of regulatory network rewiring across evolution in many cases (55). We found that species-specific REs, including promoters and enhancers, can bring about the rewiring of regulatory networks across species. More specifically, the conservation of RCs in a regulatory network is positively correlated with promoter and enhancer conservation between the two species (Figure 4A and B; P = 1.58 × 10–13 and ρ = 0.403 (promoter), P = 3.97 × 10–7 and ρ = 0.204 (enhancer)). The conservation of REs can be expressed as a quantitative score of the ratio of conserved REs to all REs in a regulatory network. Next, to test whether species-specific REs could contribute to the evolution of associated phenotypes, we analyzed the proportion of species-specific REs in regulatory networks according to LPGs and HPGs. We found that regulatory networks of LPGs have a higher proportion of species-specific REs compared to those of HPGs (Figure 4C and D; P = 5.74 × 10–8 (promoter), P = 1.15 × 10–24 (enhancer), Mann–Whitney U test). Additionally, the discrepancy of the TF sequence could not explain the rewiring of the regulatory networks of orthologous genes (Supplementary Figure S17) (56). These data reveal that the evolutionary divergence of REs potentially leads to the rewiring of RCs and brings about associated phenotypic differences in orthologous genes between humans and mice.
Figure 4.

Conservation of regulatory elements, the regulatory network, and associated phenotypes. (A and B) Correlation between the conservation of regulatory connections and regulatory elements (REs): promoters (A) and enhancers (B). (C and D) Species-specific REs [promoters (C) and enhancers (D)] in regulatory networks for low phenotype similarity genes (LPGs) and high phenotype similarity genes (HPGs). (E and F) Examples of species-specific enhancers in regulatory networks of PHKG2 (LPGs) (E) and PKP2 (HPGs) (F).
Species-specific REs in regulatory networks could explain the phenotypic differences in orthologous genes. For example, the regulatory network of PHKG2, an LPG, has multiple species-specific enhancer candidates in the upstream regions of target genes such as Ppara and Nfkb1 (Figure 4E). Specifically, the mouse histone modification marker H3K4me1, which is a hallmark of enhancer activity, was not aligned with similar markers found on human regulatory noncoding sequences. Conversely, the regulatory network of plakophilin 2 (PKP2), an HPG, showed multiple conserved enhancer candidates in the upstream regions of Tnni1 and Sgk3 (Figure 4F).
Next, for a more in-depth analysis, the sequence identity of transcription factor-binding sites (TFBSs) were analyzed according to genes with phenotypic differences. We found that target genes in the regulatory networks of genes with phenotypic differences show a low sequence identity of TFBSs between humans and mice (Supplementary Figure S18A, P = 4.23 × 10–2, Mann–Whitney U test). Specifically, we processed the sequence alignments by utilizing a BLASTZ search between human and mouse chromosomes (37) and measured the sequence similarities between human TFBSs with the aligned mouse chromosome. In the regulatory network of PHKG2, an LPG, the MYB proto-oncogene (MYB, a TF) was found to regulate high-mobility group protein 1 (HMBG1), and the TFBS of MYB showed sequence differences between humans and mice (Supplementary Figure S18B). Meanwhile, for the regulatory networks of genes with similar phenotypes, the TFBSs of target genes showed high sequence conservation between species. In the regulatory network of the ATP-binding cassette subfamily D member 3 (ABCD3), an HPG, the peroxisome proliferator-activated receptor gamma TF regulated acyl-CoA synthetase long chain family member 1 (ACSL1), and its TFBS showed significantly high identity between humans and mice (Supplementary Figure S18C). Furthermore, we validated the correlation between the conservation of RCs and TFBS sequence identities in the regulatory networks. We found that the conservation of RCs was substantially lower in the regulatory networks of low TFBS sequence identity than those of high TFBS sequence identity (Supplementary Figure S19, P = 3.51 × 10–4, Mann–Whitney U test). Thus, we conclude that the evolution of TFBS sequence differences may induce phenotypic differences through the rewiring of regulatory networks. Additionally, we analyzed the predictive power of the features (conservation of RC and conservation of RE) and found that those features inferred the phenotypic similarity of orthologous genes between humans and mice (Supplementary Figure S20).
Validation of phenotypic differences between human and mouse transcriptomes
Regulatory networks control target gene expression; thus, regulatory changes across species can lead to transcriptomic divergence during evolution (46). We observed that highly conserved regulatory networks of orthologous genes resulted in high expression conservation between humans and mice. Specifically, regulatory networks of highly conserved RCs showed significantly high levels of expression conservation between species compared to those with low conserved RCs (Figure 5A; P = 1.01 × 10–5, Mann–Whitney U test). Expression conservation was measured by calculating the similarity of tissue expression patterns obtained from the Encyclopedia of DNA Elements (ENCODE) and Functional Annotation of the Mouse/Mammalian Genome (FANTOM5) datasets. We validated phenotypic differences by analyzing transcriptomic divergence in the functional modules of regulatory networks between humans and mice. Target genes in the regulatory networks of LPGs showed lower expression conservation than those of HPGs (Figure 5B; P = 1.03 × 10–4, Mann–Whitney U test; supplementary data S3).
Figure 5.

Transcriptomic validation of regulatory network rewiring and phenotypic differences. (A) Comparison of the conservation of gene expression in regulatory networks with low and high conservation of regulatory connections (RCs). (B) The conservation of gene expression in regulatory networks of low phenotype similarity genes (LPGs) and high phenotype similarity genes (HPGs). (C) The PS score and gene set in functional modules of the regulatory networks of TRPM7, an LPG. (D) The conservation of gene expression in the regulatory network of ALAS2, with a conservation score of all genes in the functional module of the regulatory network. Examples of genes representing tissue transcriptomes in humans and mice. (E) The PS score and gene set in the functional modules of the regulatory networks of ALAS2, an HPG. (F) The conservation of gene expression in the regulatory network of ALAS2, with a conservation score of all genes in the functional module of the regulatory network. Examples of genes representing tissue transcriptomes in humans and mice.
For example, the transient receptor potential cation channel subfamily M member 7 (TRPM7), an LPG, exhibited different human disease phenotypes compared with the mouse orthologous gene, Trpm7 (Figure 5C). Genomic alterations in human TRPM7 are known to associate with Parkinson's disease via the phenotype terms of “Parkinsonism” and “muscle weakness.” However, either the knockout or mutation of mouse Trpm7 exhibited different phenotypic symptoms, such as “abnormal cell physiology” and “embryonic growth arrest.” Moreover, the orthologous relationship between human TRPM7 and mouse Trpm7 showed low expression conservation in the functional module of the regulatory network (Figure 5D). In the tissue transcriptome obtained from ENCODE, different gene expression patterns were detected in the brain, for instance, for PGAM family member 5 (PGAM5), transmembrane protein 123 (TMEM123), and baculoviral IAP repeat-containing 2 (BIRC2), which are target genes of the regulatory network of TRPM7.
Conversely, 5′-aminolevulinate synthase 2 (ALAS2), an HPG, mapped to the human disease term “sideroblastic anemia,” and the mouse orthologue Alas2 is associated with the mouse phenotype terms ‘pancytopenia’ and ‘anemia’, a near-identical finding (Figure 5E). Expression conservation of the ALAS2 functional module was relatively higher than that of TRPM7 (Figure 5F). Target genes in the regulatory network of ALAS2 exhibited similar expression patterns between human and mouse as shown by the tissue transcriptomic data, for instance, in biliverdin reductase B (BLVRB), protoporphyrinogen oxidase (PPOX), and heme oxygenase 1 (HMOX1).
Our approach is valid, not merely using functional modules as defined by gene ontology but also using different types of molecular interactions. One might ask whether such a functional module boundary might influence the analyses; thus, we applied diverse biological boundaries of functional modules, such as bioprocess, mammalian protein complex, and pathway modules (42–44). We found that the conservation of expression was higher in functional modules of HPGs than those of LPGs in the examined tests of available functional boundaries (Supplementary Figure S21). For robust validation, we measured the conservation of expression using different similarity coefficient measurements of independent transcriptomic databases (i.e. ENCODE and FANTOM5). All of the results indicated that the divergent expression of target genes in regulatory networks contributed to the phenotypic differences of orthologous genes between humans and mice.
DISCUSSION
Our analysis will be useful for developing mouse models and the interpretation of biological results from mouse genetics studies. Mouse phenologues, orthologous gene representing identical or similar phenotypes as human molecular or disease symptoms, should be considered candidate genes in experiments using mouse models (1). When it comes to biological interpretation, unexpected phenotypic readouts from mouse studies could be due to different evolutionary trajectories of regulatory mechanisms and discordance of tissue gene expression levels between the two species. Here, we showed that rewiring of GRNs and the divergence of modular gene expression across species correlated with the phenotypic differences of orthologous genes (Figure 2 and (Figure 5). We provide quantitative counting of a tissue transcriptomic conservation to examine the conservation of phenotypic differences of orthologous genes (http://sbi.postech.ac.kr/w/RN). We demonstrated that orthologous genes with high expression conservation, which are controlled by conserved regulatory networks between humans and mice, are more likely to be suitable for the analysis of human diseases using mouse models.
By reducing the high complexity of GRNs using the functional modules of regulatory targets, we demonstrated that the rewiring of RCs derived from varied cis-regulatory regions contributes to the phenotypic divergence of orthologous genes between humans and mice. GRNs are complicated because TFs usually have many regulatory targets with various molecular functions and exert their activity in a combinatorial manner (48). Indeed, we observed that we could not characterize phenotypic divergence by investigating each regulatory target alone (Figure 2), possibly because RCs linked to a single gene are unable to capture the functional divergence associated with polygenic traits across species. However, the investigation of all regulatory targets may also not be a plausible approach, since the roles of TFs in GRNs were almost identical between humans and mice (13,32). Therefore, we anticipate that the functional modules, as the unit of evolutionary divergence, effectively connects the cis-REs and phenotypes. We indeed observed that the core structures of co-regulation networks within functional modules further improves our ability to comprehend phenotypic divergence between human and mouse (Figure 3), indicating the modular structure at many different hierarchies may play a pivotal role to comprehend the phenotypic consequences emerged from the GRN’s divergence. Importantly, previous studies support the hypothesis that the functional module is the unit of evolutionary divergence (17,18,57). Notably, the rewiring of regulatory networks with predicted regulatory connections could not explain the phenotypic differences of orthologous genes between humans and mice (Supplementary Figure S22), indicating the need for sophisticated prediction of regulatory connections.
We expanded the boundaries of phenotypes to physiological traits in phenotypic evolution analysis, and quantitatively measured phenotypic similarity by comparing phenotype terms between humans and mice (supplementary Figure S1). Previous analyses of gene regulation have revealed that different developmental processes lead to distinct morphological phenotypes across species. Specifically, Those studies analyzed changes in TF activity related to the loss of specific morphological phenotypes (58,59). Due to previously uncovered associations between human genes and diseases, we can take advantage of HPOs, which include ontology terms associated with disease symptoms (19). MPOs could also be used to examine phenotype terms associated with gene–phenotype relationships in mice (20). Genotype–phenotype mapping is still limited to precisely compare phenotype terms because all the detectable symptoms are not tested in whole genes in all tissues. The PS score is expected to be the more explicit measurement with the accumulation of phenotype annotations to genes across species. Additionally, phenotypes that may be associated with certain physiological systems affected in humans have not been thoroughly investigated in mouse models. In this case, the PS scores of orthologous genes related to those phenotypes could have low PS scores due to study bias. With the improvement in the MGI database for physiological phenotypes of human diseases, the PS score will be updated to overcome the limitation.
The integration of biological omics data within the genotype–phenotype relationship is needed to better understand phenotypic discrepancies that arise during species evolution (60–62). In our analysis, we explain phenotypic evolution by analyzing the regulation of gene expression, which is a part of the central dogma for the shaping of phenotypes. A part of genotype–phenotype relationships may not perfectly explain phenotypic differences, and some LPGs still show high conservation scores of the regulatory networks in our results (Figure 2). The molecular evolution of other biological processes such as post-transcriptional and translational regulation may also contribute to the shaping of distinct phenotypes (63,64); thus, their molecular evolution may further explain phenotypic differences between species. Moreover, comparative analyses of proteomes across species have revealed conserved and unique biological processes (65). With advances in proteomics techniques for the detection of multiple proteins and biomarkers of post-translational regulation, cross-species comparisons of proteomes could also be used to explain phenotypic differences of orthologous genes between species in the near future. The integration of gene expression and proteome profiling is expected to provide a clearer understanding of the phenotypic evolution of orthologous genes between humans and mice.
DATA AVAILABILITY
The code and data to reproduce the results and all the figures are available through GitHub (https://github.com/doyeon94/regulatory_network). The main data underlying this article are also available in the online supplementary materials or on our companion website (http://sbi.postech.ac.kr/w/RN). Additional data underlying this article will be shared on reasonable request by the corresponding author.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the Kim laboratory for the helpful discussions. D.H. and S.K. conceived and designed the experiments. D.H., I.K., Y.O. and D.K. performed the experiments. D.H., D.K., I.K., J.K., S.K.H. and S.K. analysed the data. D.H., D.K., I.K., J.K. and S.K. wrote the paper.
Contributor Information
Doyeon Ha, Department of Life Sciences, Pohang University of Science and Technology, Pohang, Korea.
Donghyo Kim, Department of Life Sciences, Pohang University of Science and Technology, Pohang, Korea.
Inhae Kim, ImmunoBiome Inc., Pohang, Korea.
Youngchul Oh, Department of Life Sciences, Pohang University of Science and Technology, Pohang, Korea.
JungHo Kong, Department of Life Sciences, Pohang University of Science and Technology, Pohang, Korea.
Seong Kyu Han, Department of Life Sciences, Pohang University of Science and Technology, Pohang, Korea.
Sanguk Kim, Department of Life Sciences, Pohang University of Science and Technology, Pohang, Korea; Graduate School of Artificial Intelligence, Pohang University of Science and Technology, Pohang, Korea; Institute of Convergence Research and Education in Advanced Technology, Yonsei University, Seoul, Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Korean National Research Foundation [2021R1A2B5B01001903, 2020R1A6A1A03047902, 2017M3C9A604765, in part]; Ministry of Oceans and Fisheries (“Omics based on fishery disease control technology development and industrialization” [20150242]); IITP [2019-0-01906, Artificial Intelligence Graduate School Program, POSTECH]. Funding for open access charge: Korean National Research Foundation [2021R1A2B5B01001903, 2020R1A6A1A03047902, 2017M3C9A604765]; Ministry of Oceans and Fisheries (“Omics based on fishery disease control technology development and industrialization” [20150242]); IITP [2019-0-01906, Artificial Intelligence Graduate School Program, POSTECH].
Conflict of interest statement. None declared.
REFERENCES
- 1. Perlman R.L. Mouse models of human disease: an evolutionary perspective. Evol. Med. Public Heal. 2016; 2016:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gould S.E., Junttila M.R., De Sauvage F.J.. Translational value of mouse models in oncology drug development. Nat. Med. 2015; 21:431–439. [DOI] [PubMed] [Google Scholar]
- 3. Aitman T.J., Boone C., Churchill G.A., Hengartner M.O., MacKay T.F.C., Stemple D.L.. The future of model organisms in human disease research. Nat. Rev. Genet. 2011; 12:575–582. [DOI] [PubMed] [Google Scholar]
- 4. Schughart K., Libert C., Kas M.J.. Controlling complexity: the clinical relevance of mouse complex genetics. Eur. J. Hum. Genet. 2013; 21:1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rockman M. V. Reverse engineering the genotype–phenotype map with natural genetic variation. Nature. 2008; 456:738–744. [DOI] [PubMed] [Google Scholar]
- 6. Benton M.J., Donoghue P.C.J.. Paleontological evidence to date the tree of life. Mol. Biol. Evol. 2007; 24:26–53. [DOI] [PubMed] [Google Scholar]
- 7. King M., Wilson a C. Humans and Chimpanze es. Science. 1975; 188:107–116. [DOI] [PubMed] [Google Scholar]
- 8. Smedley D., Oellrich A., Köhler S., Ruef B., Westerfield M., Robinson P., Lewis S., Mungall C.. PhenoDigm: analyzing curated annotations to associate animal models with human diseases. Database. 2013; 2013:bat025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han S.K., Kim D., Lee H., Kim I., Kim S. Divergence of noncoding regulatory elements explains gene–phenotype differences between human and mouse orthologous genes. Mol. Biol. Evol. 2018; 35:1653–1667. [DOI] [PubMed] [Google Scholar]
- 10. Balmer J.E., Blomhoff R.. Evolution of transcription factor binding sites in mammalian gene regulatory regions: Handling counterintuitive results. J. Mol. Evol. 2009; 68:654–664. [DOI] [PubMed] [Google Scholar]
- 11. Schmidt D., Wilson M.D., Ballester B., Schwalie P.C., Brown G.D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C.P., Mackay S.et al.. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science (80-.). 2010; 328:1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vierstra J., Rynes E., Sandstrom R., Zhang M., Canfield T., Scott Hansen R., Stehling-Sun S., Sabo P.J., Byron R., Humbert R.et al.. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science. 2014; 346:1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stergachis A.B., Neph S., Sandstrom R., Haugen E., Reynolds A.P., Zhang M., Byron R., Canfield T., Stelhing-Sun S., Lee K.et al.. Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature. 2014; 515:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Z., Liao B.Y., Zhang J.. Genomic patterns of pleiotropy and the evolution of complexity. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:18034–18039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goh K. Il, Cusick M.E., Valle D., Childs B., Vidal M., Barabási A.L. The human disease network. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:8685–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fagny M., Paulson J.N., Kuijjer M.L., Sonawane A.R., Chen C.Y., Lopes-Ramos C.M., Glass K., Quackenbush J., Platig J.. Exploring regulation in tissues with eQTL networks. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E7841–E7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dey G., Jaimovich A., Collins S.R., Seki A., Meyer T.. Systematic discovery of human gene function and principles of modular organization through phylogenetic profiling. Cell Rep. 2015; 10:993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kachroo A.H., Laurent J.M., Yellman C.M., Meyer A.G., Wilke C.O., Marcotte E.M.. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science. 2015; 348:921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Köhler S., Carmody L., Vasilevsky N., Jacobsen J.O.B., Danis D., Gourdine J.P., Gargano M., Harris N.L., Matentzoglu N., McMurry J.A.et al.. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019; 47:D1018–D1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jupp S., Burdett T., Malone J., Leroy C., Pearce M., McMurry J., Parkinson H.. A new ontology lookup service at EMBL-EBI. CEUR Workshop Proc. 2015; 1546:118–119. [Google Scholar]
- 21. Amberger J.S., Bocchini C.A., Scott A.F., Hamosh A.. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019; 47:D1038–D1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kolishovski G., Lamoureux A., Hale P., Richardson J.E., Recla J.M., Adesanya O., Simons A., Kunde-Ramamoorthy G., Bult C.J.. The JAX Synteny Browser for mouse-human comparative genomics. Mamm. Genome. 2019; 30:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muñoz-Fuentes V., Cacheiro P., Meehan T.F., Aguilar-Pimentel J.A., Brown S.D.M., Flenniken A.M., Flicek P., Galli A., Mashhadi H.H., Hrabě de Angelis M.et al.. The International Mouse Phenotyping Consortium (IMPC): a functional catalogue of the mammalian genome that informs conservation. Conserv. Genet. 2018; 19:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Z.P., Wu C., Miao H., Wu H.. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database. 2015; 2015:bav095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karczewski K.J., Snyder M., Altman R.B., Tatonetti N.P.. Coherent tfunctional modules improve transcription factor target identification, cooperativity prediction, and disease association. PLoS Genet. 2014; 10:e1004122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han H., Cho J.W., Lee S., Yun A., Kim H., Bae D., Yang S., Kim C.Y., Lee M., Kim E.et al.. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018; 46:D380–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forrest A.R.R., Kawaji H., Rehli M., Baillie J.K., De Hoon M.J.L., Haberle V., Lassmann T., Kulakovskiy I. V., Lizio M., Itoh M.et al.. A promoter-level mammalian expression atlas. Nature. 2014; 507:462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smedley D., Haider S., Ballester B., Holland R., London D., Thorisson G., Kasprzyk A.. BioMart - biological queries made easy. BMC Genomics. 2009; 10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shou C., Bhardwaj N., Lam H.Y.K., Yan K.K., Kim P.M., Snyder M., Gerstein M.B.. Measuring the evolutionary rewiring of biological networks. PLoS Comput. Biol. 2011; 7:e1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ugander J., Backstrom L., Marlow C., Kleinberg J.. Structural diversity in social contagion. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:5962–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nepusz T., Yu H., Paccanaro A.. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods. 2012; 9:471–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D.et al.. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014; 515:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Denas O., Sandstrom R., Cheng Y., Beal K., Herrero J., Hardison R.C., Taylor J.. Genome-wide comparative analysis reveals human-mouse regulatory landscape and evolution. BMC Genomics. 2015; 16:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kheradpour P., Ernst J., Melnikov A., Rogov P., Wang L., Zhang X., Alston J., Mikkelsen T.S., Kellis M.. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013; 23:800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kheradpour P., Kellis M.. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014; 42:2976–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kheradpour P., Stark A., Roy S., Kellis M.. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res. 2007; 17:1919–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwartz S., Kent W.J., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W.. Human-mouse alignments with BLASTZ. Genome Res. 2003; 13:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schwartz S., Zhang Z., Frazer K.A., Smit A., Riemer C., Bouck J., Gibbs R., Hardison R., Miller W.. PipMaker - a web server for aligning two genomic DNA sequences. Genome Res. 2000; 10:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bentwich I., Avniel A., Karov Y., Aharonov R., Gilad S., Barad O., Barzilai A., Einat P., Einav U., Meiri E.et al.. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005; 37:766–770. [DOI] [PubMed] [Google Scholar]
- 40. Cawley S., Bekiranov S., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J.et al.. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004; 116:499–509. [DOI] [PubMed] [Google Scholar]
- 41. Chekmenev D.S., Haid C., Kel A.E.. P-Match: transcription factor binding site search by combining patterns and weight matrices. Nucleic Acids Res. 2005; 33:432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carbon S., Douglass E., Dunn N., Good B., Harris N.L., Lewis S.E., Mungall C.J., Basu S., Chisholm R.L., Dodson R.J.et al.. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019; 47:D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Giurgiu M., Reinhard J., Brauner B., Dunger-Kaltenbach I., Fobo G., Frishman G., Montrone C., Ruepp A.. CORUM: the comprehensive resource of mammalian protein complexes - 2019. Nucleic Acids Res. 2019; 47:D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fabregat A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B.et al.. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018; 46:D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Newell R., Pienaar R., Balderson B., Piper M., Essebier A., Bodén M.. ChIP-R: assembling reproducible sets of ChIP-seq and ATAC-seq peaks from multiple replicates. Genomics. 2021; 113:1855–1866. [DOI] [PubMed] [Google Scholar]
- 46. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 172:650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J., Choi M., Kim J.R., Jin H., Kim V.N., Cho K.H.. The co-regulation mechanism of transcription factors in the human gene regulatory network. Nucleic Acids Res. 2012; 40:8849–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R.et al.. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012; 489:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cox J., Weinman S.. Mechanisms of doxorubicin resistance in hepatocellular carcinoma. Hepatic Oncol. 2016; 3:57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sangerman J., Lee M.S., Yao X., Oteng E., Hsiao C.-H., Li W., Zein S., Ofori-Acquah S.F., Pace B.S.. Mechanism for fetal hemoglobin induction by histone deacetylase inhibitors involves γ-globin activation by CREB1 and ATF-2. Blood. 2006; 108:3590–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seok Junhee, Warren H. Shaw, Alex G.C., Michael N.M., Henry V.B., Xu W., Richards D.R., McDonald-Smith G.P., Gao H., Hennessy L.et al.. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Salem O., Wang H.T., Alaseem A.M., Ciobanu O., Hadjab I., Gawri R., Antoniou J., Mwale F.. Naproxen affects osteogenesis of human mesenchymal stem cells via regulation of Indian hedgehog signaling molecules. Arthritis Res. Ther. 2014; 16:R152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shi Y., He G., Lee W.C., McKenzie J.A., Silva M.J., Long F.. Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat. Commun. 2017; 8:2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hojo H., Ohba S., Taniguchi K., Shirai M., Yano F., Saito T., Ikeda T., Nakajima K., Komiyama Y., Nakagata N.et al.. Hedgehog-Gli activators direct osteo-chondrogenic function of bone morphogenetic protein toward osteogenesis in the perichondrium. J. Biol. Chem. 2013; 288:9924–9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wittkopp P.J., Kalay G.. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2012; 13:59–69. [DOI] [PubMed] [Google Scholar]
- 56. Needleman S.B., Wunsch C.D.. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970; 48:443–453. [DOI] [PubMed] [Google Scholar]
- 57. Ryan C.J., Krogan N.J., Cunningham P., Cagney G.. All or nothing: protein complexes flip essentiality between distantly related eukaryotes. Genome Biol. Evol. 2013; 5:1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Langer B.E., Roscito J.G., Hiller M.. Reforge associates transcription factor binding site divergence in regulatory elements with phenotypic differences between species. Mol. Biol. Evol. 2018; 35:3027–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roscito J.G., Sameith K., Parra G., Langer B.E., Petzold A., Moebius C., Bickle M., Rodrigues M.T., Hiller M.. Phenotype loss is associated with widespread divergence of the gene regulatory landscape in evolution. Nat. Commun. 2018; 9:4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maher B. The human encyclopaedia. Nature. 2012; 489:46–48. [DOI] [PubMed] [Google Scholar]
- 61. Breschi A., Gingeras T.R., Guigó R.. Comparative transcriptomics in human and mouse. Nat. Rev. Genet. 2017; 18:425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abascal F., Acosta R., Addleman N.J., Adrian J., Afzal V., Aken B., Akiyama J.A., Jammal O. Al, Amrhein H., Anderson S.M.et al.. Perspectives on ENCODE. Nature. 2020; 583:693–698.32728248 [Google Scholar]
- 63. Payne J.L., Wagner A.. The causes of evolvability and their evolution. Nat. Rev. Genet. 2019; 20:24–38. [DOI] [PubMed] [Google Scholar]
- 64. Yang E.W., Bahn J.H., Hsiao E.Y.H., Tan B.X., Sun Y., Fu T., Zhou B., Van Nostrand E.L., Pratt G.A., Freese P.et al.. Allele-specific binding of RNA-binding proteins reveals functional genetic variants in the RNA. Nat. Commun. 2019; 10:1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bayés À., Collins M.O., Croning M.D.R., van de Lagemaat L.N., Choudhary J.S., Grant S.G.N.. Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PLoS One. 2012; 7:e46683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code and data to reproduce the results and all the figures are available through GitHub (https://github.com/doyeon94/regulatory_network). The main data underlying this article are also available in the online supplementary materials or on our companion website (http://sbi.postech.ac.kr/w/RN). Additional data underlying this article will be shared on reasonable request by the corresponding author.