


Abstract
Subdomain 5BSL3.2 of hepatitis C virus RNA lies at the core of a network of distal RNA–RNA contacts that connect the 5′ and 3′ regions of the viral genome and regulate the translation and replication stages of the viral cycle. Using small-angle X-ray scattering and NMR spectroscopy experiments, we have determined at low resolution the structural models of this subdomain and its distal complex with domain 3′X, located at the 3′-terminus of the viral RNA chain. 5BSL3.2 adopts a characteristic ‘L’ shape in solution, whereas the 5BSL3.2–3′X distal complex forms a highly unusual ‘Y’-shaped kissing junction that blocks the dimer linkage sequence of domain 3′X and promotes translation. The structure of this complex may impede an effective association of the viral polymerase with 5BSL3.2 and 3′X to start negative-strand RNA synthesis, contributing to explain the likely mechanism used by these sequences to regulate viral replication and translation. In addition, sequence and shape features of 5BSL3.2 are present in functional RNA motifs of flaviviruses, suggesting conserved regulatory processes within the Flaviviridae family.
INTRODUCTION
RNA structures play essential functions in the replication cycles of RNA viruses, compensating in this way the limited protein-coding capacity of their relatively short genomes. As several of these viruses are important human pathogens with the potential of generating world-wide pandemics (1), it is important to study these structures and the mechanism by which they regulate viral activity.
A specific subset of these functional RNA motifs is represented by the complexes formed by RNA structures separated by hundreds or even thousands of bases in the viral genomes. These distal RNA–RNA complexes are relatively common in positive-sense RNA viruses and are particularly interesting because they help determine the overall spatial organization and long-range interactions of the viral genomes and have often been linked to essential viral replication steps (2).
A network of these remote RNA–RNA contacts has been detected in the genomic RNA of the hepatitis C virus (HCV). This virus affects approximately 58 million people worldwide and, despite current treatments, remains a concern due to limited access to drugs, appearance of resistances and lack of an effective vaccine (3). The RNA–RNA interactions of HCV connect the 5′ and 3′ ends of the viral genome and likely serve to coordinate the translation, replication and packaging processes along the HCV cycle. At the core of this interaction network is subdomain 5BSL3.2 (also termed SL9266) (4), which binds the internal ribosome entry site (IRES) located at the 5′ untranslated region (UTR) of the viral genome (5,6), domain 3′X at the 3′ UTR (6–11) and an additional structure (SL9005) in the open reading frame (ORF) (6,12). Subdomain 5BSL3.2 (designated cre46) is a conserved 46-nt hairpin formed by the NS5B polymerase-coding region of the ORF and comprises an extended apical loop of 12 nucleotides (nt) and two double-helical stems separated by a large 8-nt bulge (7,13) (Figure 1). A 5-nt cytosine-rich GCCCG tract within this bulge has been reported to base-pair with a UGGGU segment contained in the apical loop of subdomain IIId of the IRES (5,6) or alternatively with a CGGGC tract of SL9005 in the ORF (6,12). The latter contact has been reported to be important for replication (12). The IRES interaction likely functions as a translational repressor (14), since the IIId GGG residues form Watson–Crick base pairs with residues C1116CC1118 of the small ribosomal subunit (15) and are essential for IRES activity (16). On the other hand, the apical loop of 5BSL3.2 contains a 7-nt UCACAGC sequence (identified as k′) that contacts a complementary GCUGUGA (k) segment within domain 3′X (6–11). This 3′X k sequence is part of a self-complementary 16-nt segment termed dimer linkage sequence (DLS) that has been shown to promote the dimerization of virus genomes in vitro (17–21) (Figure 1). The 5SL3.2–3′X k′-k contact has been reported to be essential for virus replication (7,8) and translation (11,14,22). Not surprisingly, all of the components of this RNA–RNA interaction network, subdomain 5BSL3.2, IRES subdomain IIId, SL9005 and domain 3′X, are evolutionarily conserved within the extremely variable genome of HCV. In fact, the overlapped DLS and k sequences are absolutely conserved among HCV genotypes and isolates (23).
Figure 1.
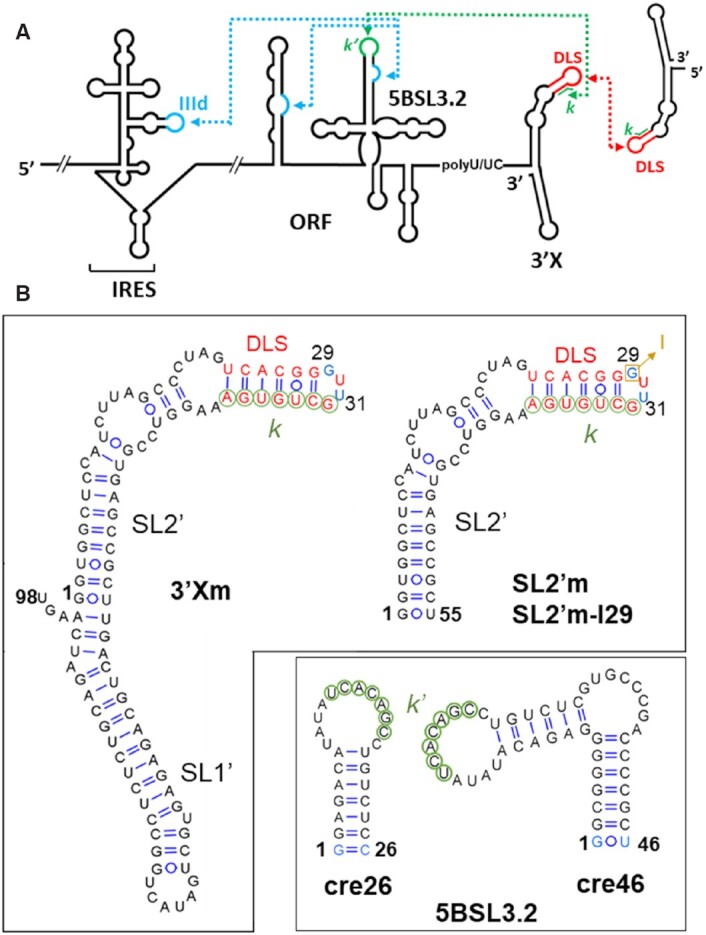
Secondary structures of the hepatitis C virus RNA sequences analyzed in this study. (A) Secondary structure representation of the HCV genomic RNA. Distal RNA–RNA interactions with functional significance are indicated with blue and green dotted arrows; the red dotted arrow represents an intermolecular contact between palindromic DLS sequences that promotes 3′X RNA homo-dimerization. (B) Secondary structures of the 98-nt full-length domain 3′X, 55-nt 3′X subdomain SL2′, 46-nt full-length subdomain 5BSL3.2 (cre46) and 26-nt apical 5BSL3.2 hairpin (cre26). 3′Xm and SL2′m are dimerization-defective variants of the wild-type sequences 3′X and SL2′, respectively, containing a double C29G/A31U mutation in the apical palindromic tetraloop of SL2′ (indicated with blue nt). In the SL2′m-I29 sequence, G29 is replaced with inosine (depicted in brown). The blue-colored nucleotides in cre26 and cre46 indicate changes relative to the wild-type sequence, introduced to increase transcription yield. 3′X sequence k (indicated with green circles) has been shown to establish a distal interaction involving Watson–Crick pairs with a complementary k′ sequence (green circles) in the apical loop of 5BSL3.2. The 16-nt palindromic DLS sequence (depicted with red nt) overlaps with the k tract and is absolutely conserved among all HCV isolates.
We have previously studied the interaction between subdomain 5BSL3.2 and domain 3′X using NMR spectroscopy (10). The vast majority of remote RNA–RNA interactions involve the unpaired nt of two distally located loops, which form antiparallel base pairs in so-called kissing-loop complexes. In contrast, the 5BSL3.2–3′X interaction was found to involve unpaired nt in the 5BSL3.2 apical loop and base-paired nt within a double-helical stem of domain 3′X. This interaction mode was proposed to result in an unusual kissing junction that could have functional consequences (10). Here we describe at low resolution the global structure of this 5BSL3.2–3′X distal complex, together with the 3D shape of the isolated 5BSL3.2 subdomain playing a pivotal role in the RNA–RNA contact network of HCV.
MATERIALS AND METHODS
Sequences
The sequences analyzed in this study correspond to HCV genotype 1b and were obtained from vector pFK-I389FLUCNS3-3′ET (24).
Secondary structure predictions
These calculations were carried out with the MC-Fold (25) web server using as restraints the base-pairs observed by NMR spectroscopy. The structures were drawn using VARNA (http://varna.lri.fr/) (26).
Preparation of RNA samples
The 46 nt-long full-length 5BSL3.2 subdomain sequence (cre46), the 26 nt-long molecule comprising the upper portion of 5BSL3.2 (cre26), the 55 nt-long dimerization-defective C29G/A31U mutant SL2′ subdomain of 3′X (SL2′m), and the specificity controls SL2′m-k’ and cre26-k (Supplementary Figure S1) were prepared by T7-polymerase in vitro transcription using synthetic oligonucleotide DNA templates. Construct 3′Xm, comprising the full-length dimerization-defective C29G/A31U mutant 3′X domain (98 nt), was transcribed from a pUC19 plasmid containing a ScaI restriction site for linearization and run-off transcription (10). We also generated uniformly 13C/15N-labeled transcripts of cre46 using NTPs obtained from CortecNet. All transcription products were separated overnight on large-scale denaturing gels containing 20% acrylamide and 8 M urea and using Tris-borate as running buffer. The RNA transcripts aimed for SAXS experiments were eluted from the gel by passive diffusion into a 50 mM sodium acetate, pH 5.2, 2 mM EDTA solution, concentrated by diafiltration after washing with excess elution buffer, and subsequently transferred by diafiltration into aqueous solutions containing 10 mM Tris-HCl, pH 7.0, and 0.1 mM EDTA. The RNA products used in NMR experiments were electroeluted from the gel, ethanol-precipitated twice, desalted with Sephadex G-25 cartridges and transferred by diafiltration into aqueous solutions containing 10 mM sodium phosphate, pH 6.0, and 0.1 mM EDTA. SL2′m-I29, the 55-nt dimerization-defective SL2′ subdomain containing inosine instead of guanine at position 29, was obtained HPLC-purified from Integral DNA Technologies, and transferred by diafiltration into the same solution. Before SAXS or NMR data acquisition, all samples were heated at 95°C for 5 min and immediately placed on ice. When needed, 6 mM MgCl2 was added after this thermal treatment, followed by diafiltration. The 3′Xm-cre46 and SL2′m-I29-cre26 complexes were obtained by incubating the two interacting species for 150 min at 25°C in the presence of 6 mM MgCl2, each previously subjected to thermal treatment in the absence of the cation. To obtain a homogeneous sample for SAXS, the 3′Xm-cre46 complex was subsequently purified by size exclusion chromatography using a HiLoad 16/60 Superdex 75 column (GE Healthcare) with 10 mM Tris-HCl, pH 7.0, and 6 mM MgCl2 as running buffer and a flow rate of 0.05 ml/min, and subsequently concentrated by diafiltration. The purity of all samples was assessed with denaturing electrophoresis experiments. Each SAXS sample was prepared at three different RNA concentrations, ranging from 0.5 to 2 mg ml–1 (approximately 12.5–240 μM). The RNA concentration in the NMR samples ranged from 77 to 185 μM.
Gel electrophoresis experiments
Native gels were run at 4°C for 12 h under constant voltage (80 V). We used 20% 19:1 acrylamide:bisacrylamide gels and 89 mM Tris-borate, 6 mM MgCl2 (TBM) as running buffer. These experiments involved 10–40 μM RNA samples, prepared as specified above. All gels were stained with methylene blue and distained in water. These experiments were used to measure the homodimerization and 5BSL3.2 (cre26 and cre46) binding capacities of 3′Xm, SL2′m and SL2′m-I29 molecules (Supplementary Figures S2 and S3). Further assays involving SL2′m-k’ and cre26-k sequences were carried to evaluate the specificity of the 3′X–5BSL3.2 interaction in the ionic conditions used for the SAXS and NMR analyses (Supplementary Figure S4).
SAXS data acquisition
SAXS experiments involved the isolated cre46 and 3′Xm species, which were analyzed in the absence and presence of magnesium, and the 3′Xm–cre46 complex, studied in the presence of 6 mM MgCl2, X-ray scattering measurements were carried out at room temperature using a Rigaku BIOSAXS 2000 instrument at the SAXS Core Facility of the National Cancer Institute at Frederick (MD, USA), and beamline 12D-ID-B of the Advanced Photon Source (APS) at the Argonne National Laboratory (IL, USA). The samples were initially screened and examined at the SAXS Core Facility using a photon energy of 8 keV and total exposure time of 2 h, refreshed each 15 min for an image frame. Selected samples were measured at the 12-ID-B beamline with a photon energy of 13.3 keV and an off-center Pilatus 2M detector. Simultaneous WAXS data were recorded, and the q range examined was ∼0.005 < q < 0.84 Å−1. To evaluate the magnitude of possible interparticle interactions, each RNA was measured at three different concentrations. A total of 45 sequential data frames were recorded for each RNA sample and matching buffer using a flow cell and an exposure time of 1 s to minimize radiation damage and obtain good signal-to-noise ratios. The 2D scattering patterns were corrected and reduced to 1D scattering profiles using Rigaku SAXSLab software, or Matlab scripts developed at the APS12-ID-B beamline. No radiation damage was observed as evidenced by the absence of systematic signal changes in sequential SAXS profiles; this was subsequently confirmed by gel electrophoresis after the X-ray measurements. The scattering profiles of RNAs were calculated by subtracting the background buffer contribution from the sample solution profile. The WAXS data were used to guide accurate background subtraction for the SAXS data using NCI IGOR Pro macros.
SAXS data analysis
For each RNA sequence and ionic strength condition, the radius of gyration (Rg) was obtained across the three different RNA concentrations with PRIMUS (27) by using the Guinier approximation at the low scattering angle region of the profiles (Figure 2 and Supplementary Table S1). We checked the aggregation state of the samples by evaluating the linearity of the Guinier plots. The concentration effects were eliminated by extrapolating data to infinite dilution using PRIMUS (27) or an NCI SAXS Core in-house Matlab script. The program GNOM (28) was then applied using a maximum q value of 0.3 Å−1 to calculate the pair distance distribution function (PDDF) and the maximum intramolecular distance (Dmax). Dmax was monitored in steps of 2 Å until a good fit to the experimental data was obtained and the PDDF curve fell smoothly to zero. The molecular weight of the RNA molecules was estimated from the scattering data using three different methods based on apparent volume, correlation volume and Porod volume (29–31) (Supplementary Table S2).
Figure 2.

Small-angle X-ray scattering data of the 5BSL3.2 subdomain (top), dimerization-defective 3′Xm domain (middle) and complex between 3′Xm and 5BSL3.2 (bottom) of HCV RNA. (A, D, G) Scattering intensities versus momentum transfer q, obtained in the absence (blue) and presence (red) of magnesium. The insets show, for each ionic condition, the Guinier region of the scattering curve with a linear fit line. (B, E, H) Kratky plots in the absence (blue) and presence (red) of magnesium. (C, F, I) Pair distance distribution functions (PDDF) in the absence (blue) and presence (red) of magnesium. Experimental conditions: 0.5–2 mg ml–1; 10 mM Tris-HCl, pH 7.0, 0 mM MgCl2 (all sequences, low ionic strength) or 10 mM Tris-HCl, pH 7.0, and 6 mM MgCl2 (all sequences, higher ionic strength). 5BSL3.2 and 3′Xm were studied in both ionic conditions and the 3′Xm–5BSL3.2 complex in the latter one, since magnesium is required for the interaction. The Guinier region of the scattering curves (insets in Figure 2A, D and G) was linear in all cases, indicating that none of the samples was aggregated.
Ab initio 3D modeling of SAXS data
Twenty low-resolution envelope models were calculated for each sequence and condition from the GNOM output using DAMMIM (32) in ‘slow’ mode. The resulting ab initio shape models were superimposed with SUPCOMB (33), averaged with DAMAVER (34) based on normalized spatial discrepancy (NSD) criteria, and filtered with DAMFILT (34). For each RNA sequence and condition, the resulting DAMFILT envelope was compared with the most representative envelope (NSD = 0) and with a refined envelope generated with DAMSTART (34). The shapes were approximately similar for all systems (data not shown).
NMR spectroscopy
These experiments primarily involved exchangeable protons and nitrogens and were utilized to study the secondary structure of cre46 in the absence and presence of 6 mM magnesium chloride, as well as SL2′m-I29 in the absence and presence of 6 mM MgCl2 and cre26. NMR spectra were acquired on a cryoprobe-equipped, 600 MHz Bruker Avance II spectrometer, and analyzed using Topspin 3.6 (Bruker Biospin) and NMRFARM-Sparky (35). All systems were studied using unlabeled samples and homonuclear watergate-NOESY (with 150 ms mixing time) and watergate-TOCSY experiments (60 ms mixing time) recorded in 90% H2O/10% D2O, typically at two temperatures (16 and 27°C). The recycle delays were 1.6 and 2 s for all homonuclear TOCSY and NOESY experiments, respectively. A 13C/15N-labeled cre46 molecule was additionally analyzed with heteronuclear 1H-15N HSQC and HNN-COSY experiments recorded at 27 °C. For 1H-15N HSQC experiments, we acquired 256 indirect experiments with 64 scans per experiment. For HNN-COSY experiments, the delay for evolution of the 2JNN coupling was set to 15 ms, and we collected 128 complex points in the t1 dimension with 240 scans for each t1 increment. The recycle delays were between 1.0 and 1.3 s for the HSQC and HNN-COSY experiments.
Secondary structure restraints
(i) Isolated species. We determined the secondary structure of the full-length 5BSL3.2 subdomain cre46 sequence through a combination of 1H-15N HSQC and HNN-COSY together with homonuclear NOESY and TOCSY data. These analyses, supported by previous cre26 assignments (10), allowed detection and identification of the base pairs contained in the lower and upper double-helical stems of 5BSL3.2. The secondary structure of the isolated SL2′m-I29 subdomain was obtained from analyses of homonuclear NOESY and TOCSY data obtained in the absence and presence of magnesium, supported by previous assignments of subdomain SL2′m obtained in similar solution conditions (10). The secondary structures adopted by the homodimerization-defective 3′Xm domain and the 5BSL3.2 cre26 apical hairpin were previously determined by heteronuclear NMR spectroscopy experiments using the same sequences and similar solution conditions and assignment strategies (10). (ii) 3′Xm-cre46 complex. Previous NMR analyses of the wild-type SL2′-cre26 complex (20) and homodimerization-defective 3′Xm-cre26 and SL2′m-cre26 complexes (10) indicated that within the 3′X domain, subdomain SL1′ and the lower double-helical stem of subdomain SL2′ remained undisturbed upon complex formation. In contrast, a disruption of the upper region of subdomain SL2′, where the k nt are base-paired, was detected, together with the establishment of new Watson–Crick pairs with the k′ nt of the cre26 apical loop (10). On the other hand, chemical shift perturbations in cre26 were mostly restricted to the 12-nt apical loop (10). Further secondary structure organization of the upper region of subdomain SL2′ in contact with 5BSL3.2 was deduced from NMR analyses of the inosine-containing SL2′m-I29 sequence, which detected the formation of a diagnostic I:C pair in the presence of magnesium and cre26.
Building and analysis of atomic models
The SimRNA web server (36) was used to generate all-atom models of the 3′Xm and cre46 molecules and the 3′Xm–cre46 complex with secondary structure restrictions obtained from NMR analyses. Specifically, we used as restraints the base-pairing interactions indicated in Figures 3C, 4C and 6D. 10 000 models were generated for each system, with 1000 simulation steps per model. Theoretical scattering profiles were generated for each model and compared with the experimental SAXS data at q < 0.5 Å–1 using the FoXS server (37). We selected the 20 best models (with the lowest χ2 fit values) of each species. These models were subsequently refined by progressively minimizing their potential energy with the ff99-OL3 force field of AMBER 20 (38) and a generalized Born model for solvent simulation. The refined models were evaluated with MolProbity (39), and the best model was overlaid with the average ab initio envelope with SUPCOMB (33). The selection of the best model was based on dispersion fit χ2 values and Molprobity results (Supplementary Table S3). The NMR analyses supplied restraints for G:C, A:U and G:U pairs within double-helical stems but did not provide information about other, non-canonical base pairs. This limited the accuracy of the atomic models. Images of SAXS envelopes and atomic models were generated with PyMOL (Schrödinger, LLC), and RNA 3D models were analyzed with UCSF Chimera (40) and RNApdbee (41).
Figure 3.
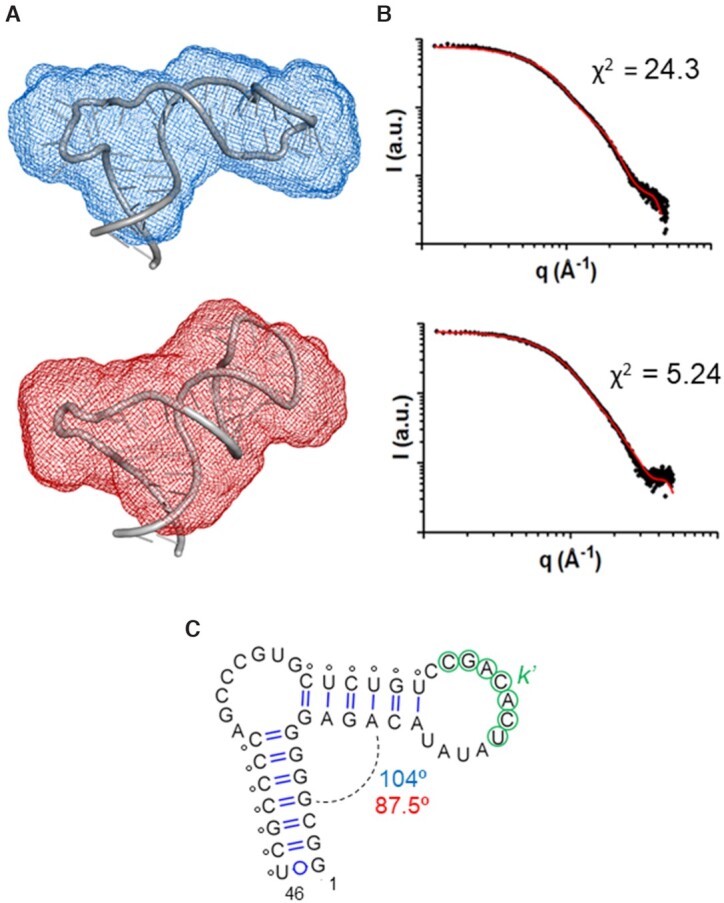
Low-resolution models of subdomain 5BSL3.2 of HCV determined by NMR spectroscopy and SAXS. (A) Molecular envelopes of cre46 calculated from the SAXS profiles in the absence (blue) and presence (red) of magnesium, superposed with the best energy-minimized atomic models, selected by fitting the theoretical SAXS profiles to the experimental curve. The normalized spatial discrepancy (NSD) between scattering envelopes was 0.734 and 0.631 for low and higher ionic strength, respectively. (B) Theoretical dispersion profiles calculated from the best energy-minimized 5BSL3.2 atomic models (red line) in the absence (top) and presence (bottom) of magnesium, overlaid with the experimental SAXS profiles (black dots). (C) Experimental secondary structure of 5BSL3.2. The average angle between the two double helical stems in the absence and presence of magnesium is indicated in blue and red, respectively. The small circles indicate base-pairing restraints derived from NMR spectroscopy and used for model building. The k′ nt are indicated with green color.
Figure 4.

Low-resolution models of the dimerization-defective 3′Xm domain of HCV determined by NMR and SAXS. (A) Molecular envelopes of 3′Xm calculated from the dispersion profiles in the absence (blue) and presence (red) of magnesium, superposed with the best energy-minimized atomic models. The normalized spatial discrepancy (NSD) between scattering envelopes was 0.769 and 0.642 for low and higher ionic strength, respectively. (B) Theoretical dispersion profiles calculated from the best energy-minimized 3′Xm atomic models (red line) in the absence (top) and presence (bottom) of magnesium, overlaid with the experimental SAXS profiles (black dots). (C) Experimental secondary structure of 3′Xm. The C29G and A31U mutations blocking homodimerization are indicated in green. The average angle between the lower and upper stems of subdomain SL2′ in the absence and presence of magnesium is indicated in blue and red, respectively. The small circles indicate base-pairing restraints derived from NMR spectroscopy and used for model building. DLS nt are indicated in red and k nt are marked with green circles.
Figure 6.

Low-resolution model of the 3′Xm-5BSL3.2 complex of HCV determined by NMR and SAXS. (A) Molecular envelope of 3′Xm–5BSL3.2 calculated from the dispersion profile, superposed with the best energy-minimized atomic model. The normalized spatial discrepancy (NSD) between scattering envelopes was 0.754. (B) Theoretical dispersion profile calculated from the best energy-minimized 3′Xm-cre46 atomic model (red line) overlaid with the experimental SAXS profile (black dots). (C) Low-resolution model of the 3′Xm-5BSL3.2 complex shown in two different orientations. 3′Xm DLS nt, 3′Xm k nt and other 3′Xm nt are, respectively, indicated in red, green and blue, whereas 5BSL3.2 k′ nt and other 5BSL3.2 nt are, respectively, depicted in light green and orange. (D) Experimental secondary structure of the 3′Xm-5BSL3.2 complex. The C29G and A31U mutations blocking 3′Xm homodimerization are indicated in green, DLS nt are depicted in red, and k and k′ nt are represented with green circles. Average angles between double-helical stems are indicated, and the small circles denote NMR-derived base-pairing restraints utilized for model building.
NS5B complex modeling
We used the dispersion atomic models of cre46 and 3′Xm, the X-ray structure of the HCV NS5B initiation complex (PDB 4WTL (42)) and the MOE 2020 package (CCG Inc.) to build a model of a putative 5BSL3.2–NS5B–3′X ternary complex. The 5BSL3.2 apical loop was placed near a conserved arginine-rich patch in the thumb domain based on similarities with flaviviral 3′-SL and SLA hairpins interactions with NS5 (43,44). The unpaired 3′-terminal nt of the 3′X domain, which acts as a template for negative-sense RNA synthesis initiation, was superposed with the template strand present in the active site of the polymerase (42). The conservation of NS5B residues present at the NS5B–5BSL3.2 model interface was assessed with ClustalW (45) alignments.
RESULTS
Experimental strategy and sequence design
To obtain the low-resolution structural models of subdomain 5BSL3.2 and the complex formed between 5BSL3.2 and domain 3′X, we used a strategy based on NMR spectroscopy and SAXS analyses. Each of the isolated RNA species was examined first in the absence and presence of magnesium using these two techniques, before studying the 3′X–5BSL3.2 interaction in the presence of the cation, which is required for complex formation.
We have shown before that 3′X and 5BSL3.2 form a stable complex in the presence of magnesium. However, in these conditions, domain 3′X also forms homodimers (10,20,21). In order to obtain homogeneous samples, we inserted a double C29G/A31U mutation in domain 3′X (Figure 1 and Supplementary Figure S1): this mutation disrupts the DLS palindrome and blocks 3′X homodimerization without affecting the structure of the domain or its interaction with 5BSL3.2 ((10) and Supplementary Figure S2). Thus, our analyses included a 98 nt-long molecule comprising the full-length C29G/A31U dimerization-defective 3′X domain (hereafter identified as 3′Xm), and a 46 nt-long molecule comprising the full-length 5BSL3.2 subdomain (designated cre46) (Figure 1). Atomic models of the isolated 3′Xm and cre46 molecules and the 3′Xm–cre46 complex were generated using secondary structure restraints based on NMR spectroscopy observations. The best models were then selected by comparing the theoretical and experimental dispersion curves and superposed on the envelopes generated from the dispersion data.
Low-resolution model of subdomain 5BSL3.2
NMR analyses of the full-length 5BSL3.2 sequence (cre46) in the absence and presence of magnesium (Supplementary Figure S5) confirmed that this RNA formed a hairpin closed by a 12-nt apical loop and comprising two double-helical stems separated by an 8-nt bulge (Figure 1). These results were in agreement with previous studies of the smaller cre26 construct (10) and earlier homonuclear NMR analyses of a similar full-length 5BSL3.2 sequence (7).
The experimental SAXS curves obtained for cre46 in the absence and presence of magnesium are shown in Figure 2A. The shape of the Porod-Debye and Kratky plots indicated that cre46 was folded in both conditions, with only slightly more flexibility in the absence of the cation (Figure 2B and Supplementary Figure S6A). The pair distance distribution functions (PDDF) exhibited in both cases one main distance population at about 20 Å, coinciding with the diameter of an A-form RNA helix, but the maximum intramolecular distance (Dmax) was shorter in the presence of magnesium (70 versus 76 Å in the absence of the cation), in agreement with the smaller radius of gyration (Rg) registered in this ionic condition (Figure 2C and Supplementary Table S1). Concurring with these observations, the 3D envelopes calculated from the dispersion data at each ionic strength were different, with a more compact shape detected in the presence of magnesium (Figure 3).
An ensemble of 10 000 cre46 atomic models was generated using as restraints the base pairs detected by NMR spectroscopy (Supplementary Figure S5), and the models giving rise to the best fit with the experimental scattering curves in either the absence or presence of magnesium were selected and energy-minimized. The best energy-minimized atomic models are shown in Figure 3A, superposed with the molecular envelopes determined from the SAXS data; Figure 3B shows the fit between the theoretical dispersion profiles of the best models and the experimental SAXS curves. cre46 adopted a monomeric hairpin structure in both ionic conditions, as confirmed by molecular weight calculations based on the dispersion data (Supplementary Table S2). Both hairpins were closed by an extended 12-nt apical loop and comprised two A-form double-helical segments separated by the 8-nt bulge. The presence of this large bulge induced a strong curvature between the two double-helical stems, and the interhelical angle became more acute in the presence of magnesium (87.5o versus 104o in the absence of the cation); this likely contributed to the compaction of the structure observed in this ionic condition (Figure 3).
Low-resolution model of homodimerization-defective 3′Xm domain
The secondary structure of domain 3′Xm was previously determined by NMR spectroscopy experiments using similar solution conditions. As observed for the wild-type 3′X domain (20), it was found to comprise two SL1′ and SL2′ double-helical subdomains. The DLS sequence was folded in the upper hairpin of the SL2′ subdomain, with the k nt base-paired in this double-helical stem (10) (Figure 1).
An analysis of the 3′Xm dispersion data indicated that this sequence was folded in both ionic conditions but was more flexible in the absence of magnesium (Figure 2E and Supplementary Figure S6B). The Dmax values obtained in the absence and presence of magnesium (147 and 142 Å, respectively) were comparable and similar to the Dmax measured for the wild-type sequence at low ionic strength, 148 Å (21). However, the PDDF curves and Rg values in the presence and absence of magnesium were different, indicating dissimilar overall conformations (Figure 2F and Supplementary Table S1). Molecular weight calculations based on the scattering profiles indicated that 3′Xm retained a monomeric state under both conditions (Supplementary Table S2), unlike the wild-type domain that homodimerized at higher ionic strength (21). This confirmed our 3′Xm electrophoretic observations (Supplementary Figure S2) and previous findings (10). An inspection of the 3D envelopes generated from the dispersion data indicated that 3′Xm adopted the characteristic elongated shape observed for the wild-type 3′X sequence (21) in both solution conditions, although a more compact shape was again observed in the presence of the cation (Figure 4A).
As with the 5BSL3.2 subdomain, 3′Xm atomic models were generated using as restraints the base pairs previously observed by NMR spectroscopy for this molecule (10), and the best models were selected by comparing the theoretical scattering profiles with the experimental dispersion curves obtained in the absence and presence of magnesium. The best energy-minimized atomic models fitted well into the SAXS envelopes, and in both cases the atomic models giving rise to the best fits comprised two coaxially stacked SL1′ and SL2′ stems (Figure 4A). A similar structure was also observed for the wild-type sequence at low ionic strength (21). All 3′X models shared this overall structure, but in agreement with the dispersion data the conformation of the 3′Xm models was not the same in the absence and presence of magnesium. Despite the differences, we detected a 115o average curvature between the lower and upper double-helical stems of subdomain SL2′ shared by all selected models in either solution condition (Figure 4C). This curvature was also detected in the monomeric models of the wild-type 3′X domain (21).
Secondary structure of 3′Xm bound to 5BSL3.2
Previous NMR analyses of wild-type and dimerization-defective 3′X sequences in contact with the 5BSL3.2 cre26 hairpin indicated that within the 3′X domain, subdomain SL1′ and the lower double-helical stem of subdomain SL2′ remained undisturbed upon complex formation (10,11,20). The changes only affected the upper region of subdomain SL2′ where the k nt are base-paired, and involved the establishment of new Watson-Crick pairs between these nt and the k′ nt of the 5BSL3.2 apical loop (10,11). On the other hand, chemical shift perturbations in cre26 were mostly restricted to the apical loop (10).
In silico predictions indicated that a secondary structure reorganization of the upper region of subdomain SL2′ was likely after the formation of the intermolecular k-k′ duplex (Figure 5A). However, the exact base-pairing arrangement was difficult to determine from heteronuclear NMR analyses of complexes between 3′X and 5BSL3.2 sequences (data not shown). To solve this problem, we substituted guanine in position 29 of a dimerization-defective SL2′m subdomain molecule with its analog inosine (Figure 1). The imino proton of inosine has the significant advantages of exhibiting a characteristic NOE pattern together with distinctive chemical shift values that depend on base-pairing (46) (Figure 5B). Furthermore, the chemical environment of I29 would be different in the isolated subdomain and the two main secondary structures predicted for SL2′m bound to 5BSL3.2 (Figure 5A). Gel electrophoresis experiments indicated that the new SL2′m-I29 sequence maintained 5BSL3.2 binding while still blocking homodimerization, as observed for the C29G/A31U 3′Xm and SL2′m mutants (Supplementary Figure S3). NMR analyses of the isolated SL2′m-I29 molecule confirmed the presence of the expected SL2′ subdomain hairpin structure (Supplementary Figure S7). Upon addition of the 5BSL3.2 cre26 hairpin in the presence of magnesium, a characteristic low-field imino proton peak and HN1-H2 NOE pattern were detected, revealing the formation of a diagnostic I:C pair (Figure 5C). This indicated that the establishment of the k-k′ duplex in the SL2′m-I29-cre26 complex implied the folding of the nt forming the 5′-half of the DLS, which are released upon 5BSL3.2 binding, into a short hairpin (Figure 5A, top structure). This result allowed us to complete the list of base-pair restrictions for generating atomic models of the 3′Xm–5BSL3.2 complex for SAXS analyses.
Figure 5.

Secondary structure analysis of the dimerization-defective SL2′m subdomain of HCV in complex with 5BSL3.2. (A) MC-Fold-predicted secondary structures of SL2′m, isolated (left) and in complex with 5BSL3.2 (right). C29G and A31U mutations blocking homodimerization are indicated in blue, DLS nt are depicted in red, the G29I mutation present in the SL2′m-I29 sequence is marked with a square, and k nt are circled in green. For predicting the secondary structure of SL2′m bound to 5BSL3.2, the k nt pairing with 5BSL3.2 were impeded to form base pairs with SL2′m nt, whereas the base pairs marked with black circles were imposed pairing restraints. The base-pairing restraints were derived from NMR analyses of the SL2′m-5BSL3.2 complex (10). (B) Comparison of I:C and I:U pairs with G:C and G:U pairs, indicating the characteristic HN1 chemical shift and NOE pattern of inosine-containing pairs allowing to use this base as a secondary structure probe. (C) NMR spectroscopy analysis of the interaction between SL2′m-I29 and 5BSL3.2 hairpin cre26. (Left) 1D and watergate-NOESY spectra (150 ms mixing time) of unbound SL2′m-I29 (blue) superposed with those of a SL2′m-I29:cre26 mixture at 1:1 molar ratio (red). The spectra were acquired in the presence of 6 mM MgCl2. Sequential cross-peaks between imino protons are indicated with dotted lines, sequential contacts between A H2 and G H1 protons are marked with dashed lines, and NOE interactions across base pairs or within the same nt are labelled with colons. Assignments of C amino protons have been omitted for clarity. The green dotted line indicates the imino peak detected in the presence of cre26, whose chemical shift and NOE pattern indicated the formation of a diagnostic I:C pair in the complex. (Right) Secondary structures of bound SL2′m-I29 and cre26 consistent with the NMR data. Assigned nt are numbered. DLS nt are depicted in red, k and k′ nt are indicated with green circles, SL2′ C29I and A31U mutations blocking dimerization and replacing guanine with inosine are identified in blue, and cre26 mutations introduced to increase transcription yield are represented in light blue. Conditions: 77 μM SL2′m-I29, 0 μM (blue) or 77 μM (red) cre26, 6 mM MgCl2, 16°C.
Low-resolution model of the 3′Xm–5BSL3.2 complex
3′Xm–cre46 complexes were prepared in the presence of magnesium and purified by size-exclusion chromatography (Supplementary Figure S8). The Guinier dispersion plots indicated that the samples were homogeneous (Figure 2G), and the presence of a heterodimeric complex comprising 3′Xm and cre46 was confirmed by molecular weight calculations based on the dispersion data (Supplementary Table S2). The Porod-Debye and Kratky representations revealed that the complex adopted a well-defined structure with limited flexibility (Figure 2H and Supplementary Figure S6C). Two main distance populations were detected in the PDDF curve (Figure 2I): the first distance, 20 Å, is generated by the presence of A-form double-helices and was also detected in the isolated species. However, the 40 Å distance was only detected in the complex and pointed toward the presence of a wider structure. The Dmax and Rg magnitudes were larger than those of the individual components but below the sum of the corresponding 3′Xm and cre46 values (Supplementary Table S1). This is likely due to a conformational change in one or both species and is consistent with the second peak detected in the PDDF graph. The 3D envelopes generated from the dispersion were elongated, resembling those of the isolated 3′X domain, but exhibited a 42 Å-wide lateral protrusion that was not present in the unbound domain (Figures 6A and 4A).
Following the procedure used for the isolated species, 3′Xm-cre46 atomic models were generated using intramolecular and intermolecular secondary structure restraints based on NMR observations. The best model was selected by comparing the theoretical scattering profiles with the experimental dispersion curves, energy-minimized and superposed on the scattering envelope (Figure 6A and B). Globally, the complex adopted a Y-shaped structure comprising three double-helical domains and a three-way junction formed by the 3′Xm k-5BSL3.2 k′ pairing region. The first domain (designated I in Figure 6C) is formed by 3′Xm subdomain SL1′ and the lower stem of subdomain SL2′, which are coaxially stacked; this domain forms the lower branch of the ‘Y’ and is the larger one. The second domain (II) comprises the new hairpin formed by the apical region of SL2′, and the third one (III) corresponds to the 5BSL3.2 subdomain (Figure 6C and D). The 3′Xm k and 5BSL3.2 k′ nt form a 7-nt intermolecular double helix stabilized by coaxial stacking with the upper double-helical stem of 5BSL3.2 as well as a new middle stem formed by subdomain SL2′ (Figure 6C and D). The new SL2′ hairpin contains the first half of the DLS sequence, occupies the protuberance detected in the dispersion envelope and is located opposite the 5BSL3.2 subdomain, forming with it the two upper branches of the ‘Y’.
Subdomain 5BSL3.2 maintained in the complex the sharp bend at the 8-nt bulge observed in the unbound species (Figures 3, 6 and Supplementary Figure S9). Subdomain SL1′ and the lower double-helical stem of SL2′ did not undergo significant changes relative to the isolated 3′Xm domain and remained coaxially stacked upon complex formation (Figure 6 and Supplementary Figure S9). In contrast, the apical part of SL2′ underwent substantial changes with 5BSL3.2 binding. First, as explained above a new 5-base pair stem (A10-U14:A39-U43) spontaneously formed in the middle region of the subdomain, which was coaxially stacked with the intermolecular k-k′ helix (Figure 6 and Supplementary Figure S9). Second, the SL2′ DLS nt that did not pair with 5BSL3.2 generated a new 5-base pair (A15-C19:G27-U31) hairpin closed by a 7-nt apical loop. This hairpin was detected by NMR spectroscopy using the SL2′m-I29 mutant sequence (Figure 5).
We also generated 3′Xm-5BSL3.2 complex models where domain 3′Xm formed three SL1, SL2 and SL3 double-helical subdomains. This secondary structure arrangement was proposed by earlier chemical modification experiments (18,47–50) and exposes the k sequence in the SL2 loop, where it would be available for pairing with the 5BSL3.2 loop k′ nt. However, these models gave rise to poorer fitting with the experimental SAXS curve (χ2 = 26.7 relative to 7.91), less overlap with the dispersion envelopes and significantly poorer inter-domain stacking (Supplementary Figure S10). This result confirmed previous studies based on NMR spectroscopy, gel electrophoresis, thermal melting and functional experiments (10,11,20), indicating that domain 3′X does not require a conformational transition to a SL1, SL2 and SL3 hairpin structure to interact with 5BLS3.2 or regulate the virus replication cycle.
DISCUSSION
Subdomain 5BSL3.2 plays a pivotal role in the network of distal RNA–RNA interactions detected in the HCV genome: it binds domain 3′X in the 3′UTR through its apical loop (6–11) and can also contact subdomain IIId in the IRES (5,6), or alternatively subdomain SL9005 in the ORF (6,12), with its bulge nt. Our combined NMR spectroscopy and SAXS experiments indicate that subdomain 5BSL3.2 adopts a distinctive ‘L’ 3D shape due to the presence of the large 8-nt bulge, which imposes an angle close to 90o between the upper and lower double-helical stems (Figure 3). The scattering data also indicated remarkable stability in the absence or presence of magnesium (Figure 2B and Supplementary Figure S6A), pointing to a significant degree of local structure within the 12-nt apical loop, as previously observed by NMR spectroscopy (10). The ‘L’-shaped structure of 5BSL3.2 is reminiscent of that of stem-loop A (SLA) of the dengue and Zika flaviviruses, which also contains a large bulge comprising 9 or 11 nt, respectively (44).
The SAXS and NMR results indicate that domain 3′X adopts an elongated two-stem conformation in the absence and presence of magnesium, characterized by coaxial stacking between the SL1′ and SL2′ helices, the exposure of the central 4-nt DLS tract in the apical loop of subdomain SL2′, and the presence of an unpaired 3′-tail that protrudes laterally from the domain and contains the three terminal nt of the viral genome (Figure 4). A slight increase in flexibility was detected in the absence of magnesium, but in agreement with previous NMR observations (10) the SAXS data were consistent with the presence of a stable structure, as opposed to a mixture of conformations, under both ionic conditions (Figure 2E and Supplementary Figure S6B). The 3′X domain sequence analyzed in this report contained two DSL mutations abrogating homodimerization (Figure 1). This did not change the structure of the domain (10) and allowed us to study the monomeric form of the domain and its complex with 5BSL3.2 in the presence of magnesium. We previously found that the wild-type 3′X sequence adopted a similar two-stem structure in solution but homodimerized at higher ionic strength due to the exposure of the DLS palindrome in the SL2′ terminal loop (20,21). The two-stem structure of domain 3′X is compatible with replication, since the presence of at least three nt after a stable secondary structure has been reported to promote primer-independent initiation of RNA synthesis by NS5B (51).
The NMR and dispersion data showed that the distal complex formed by 5BSL3.2 and domain 3′X adopts a characteristic ‘Y’ shape in solution. As previously predicted on the basis of NMR evidence (10), the complex is an unusual kissing junction; the lower branch of the ‘Y’ is formed by the coaxially stacked 3′X subdomain SL1′ and lower stem of subdomain SL2′. The two upper branches, on the other hand, comprise subdomain 5BSL3.2 and a new hairpin formed by SL2′ DLS nt previously paired to the k nt (Figure 6). The kissing junction is formed by an intermolecular helix where the k′ nt of 5BSL3.2 and k nt of 3′X are base-paired. This helix is coaxially stacked with the upper stem of 5BSL3.2 and the lower stem of subdomain SL2′. Although this structure was obtained with a dimerization-defective 3′X domain (Figure 1), previous NMR evidence indicated that the wild-type sequence is capable of forming a similar structure ((11,20) and Supplementary Figure S9B). As observed for 5BSL3.2 and 3′X, the SAXS data revealed that the complex adopted a well-defined structure with limited flexibility (Figure 2H and Supplementary Figure S6C). This means that the establishment of the interaction freezes the spatial distance and relative orientation of the two RNA species forming the complex.
In order to base-pair with the 5BSL3.2 loop, the upper stem of subdomain SL2′ must transiently adopt an open conformation. In agreement with this fact, we detected substantially increased flexibility for the isolated SL2′ subdomain relative to the full-length 3′X domain or subdomain SL1 in previous SAXS studies (21). In this report we observed a small decrease in flexibility in the isolated 5BSL3.2 and 3′Xm species when adding MgCl2 (Figure 2B, E, Supplementary Figure S6A and S6B). The Dmax and Rg values also became smaller in the presence of the cation (Figure 2C and F and Supplementary Table S1). The 3′X–5BSL3.2 complex formed a remarkably stable fold in the presence of magnesium (Figure 2H and Supplementary Figure S6C).
In addition to forming a distal complex with domain 3′X as described in this report (Figure 6), 5BSL3.2 has been shown to bind specifically to the HCV NS5B polymerase, and this interaction requires the presence of the 8-nt bulge (52,53) imposing the ‘L’-shape to the subdomain. On the other hand, the CACAG segment of the UCACAGC k′ sequence located in the apical loop of 5BSL3.2 is conserved within the Flaviviridae family (54), which comprises hepaciviruses like HCV as well as flaviviruses as dengue and Zika. In flaviviruses, this segment is also located in the apical loop of a 3′UTR stem-loop (called 3′-SL) and contacts the flaviviral NS5 polymerase via an arginine-rich patch in the thumb domain (43), playing a role in replication (43,55). On the other hand, the SLAs of the dengue and Zika flaviviruses, situated at the 5′UTR of the viral genomes, act as promoters for negative-sense RNA synthesis and comprise approximately 70 nt. These stem-loops have been recently reported to also adopt an L-shape and to recognize the thumb domain of NS5 (44); in this case, the apical loop was proposed to bind to the arginine-patch recognized by 3′-SL, whereas the bottom stem, which is considerably longer than that of 5BSL3.2, would interact with the MTase domain of the flavivirus NS5 (44), absent in the NS5B polymerase of HCV. Guided by the experimental 5BLS3.2-NS5B binding evidence and the observed similarities with flaviviruses, we built a hypothetical model of the ‘L’-shaped 5BSL3.2 structure bound to the thumb domain of the polymerase. In this model, the size complementary of the protein and RNA structures turned out to be excellent and, in line with the interactions observed in flaviviruses, the apical loop of 5BSL3.2 was placed near an arginine-rich loop at the thumb surface that is conserved in hepaciviruses (Figure 7A). Formation of this complex would not block the template binding channel of the polymerase nor the double-stranded RNA exit channel, and would allow the unpaired 3′-terminal tail of the 3′X domain to enter the active site of the polymerase. In fact, since this 3′-terminal tail protrudes from the continuous stem of domain 3′X, it is possible to model the domain laterally bound to NS5B with the terminal strand entering the template binding channel of the protein (Figure 7A). This binding mode agrees with the lesser affinity reported for NS5B-3′X binding relative to 5BSL3.2 or the 3′UTR polyU/C stretch (56), and is likewise consistent with the reduction of HCV replication induced by 3′X mutations stabilizing the lower stem of SL2′, located next to the unpaired 3′-tail (11).
Figure 7.
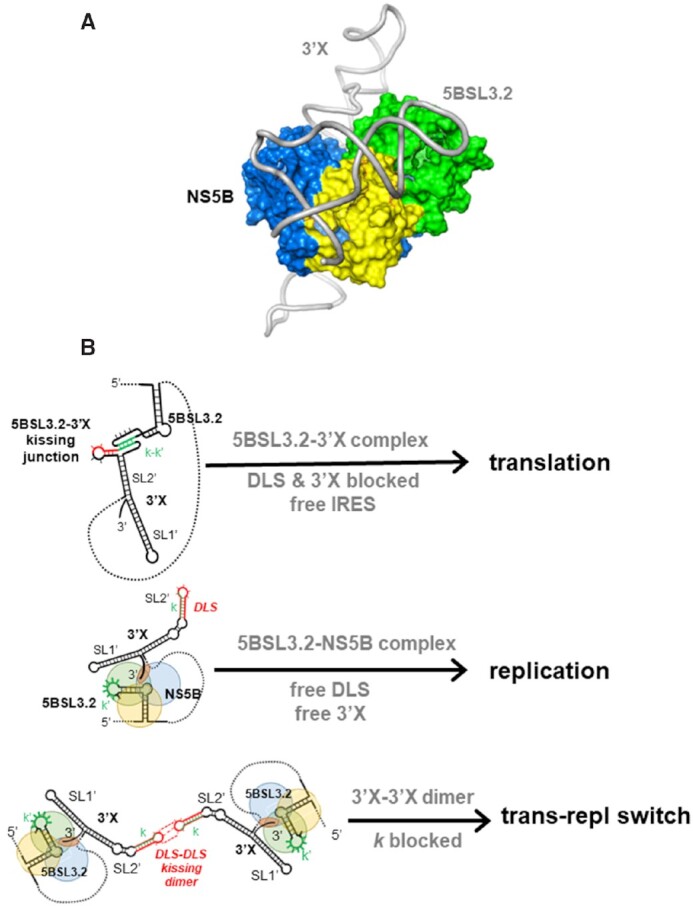
Model of putative NS5B-5BSL3.2–3′X complex and scheme summarizing the possible functions of the 5BSL3.2, 3″X and 5BSL3.2–3′X structures. (A) Hypothetical model of a putative NS5B-5BSL3.2–3′X ternary complex, with 5BSL3.2 bound to the thumb domain and the 3′-terminal tail of 3′X accessing the template binding channel of the polymerase. The model was generated based on sequence and structural similarities between 5BSL3.2 of HCV and the 3′-SL and SLA RNA motifs of flaviviruses, which recognize the thumb surface of the NS5 polymerase (43,44). (B) Model of HCV replication and translation regulation by 5BSL3.2, 3′X and their distal complexes. (Top) Formation of the 5BSL3.2–3′X distal contact favors polyprotein translation, while simultaneously obstructing the formation of NS5B-5BSL3.2–3′X replication complexes and blocking the DLS palindrome. (Middle) After viral protein translation, recognition of 5BSL3.2 by the NS5B polymerase favors 3′X biding to the template channel and the start of negative-sense RNA synthesis. (Bottom) At higher viral RNA concentration, replication-competent 3′X-3′X homodimers are formed through kissing loop contacts between palindromic DLS loops. These homodimers inhibit 5BSL3.2–3′X complex formation by blocking the k sequence and stabilize the replication stage of the HCV cycle. In (A) and (B), the thumb, palm and fingers domains of NS5B are colored green, yellow and blue, respectively.
The distal contact between 5BSL3.2 and 3′X has been reported to be required for either replication (7,8) or translation (14,22). We recently conducted a mutagenesis analysis of domain 3′X that supported the latter view, since 3′X mutants that adopted the wild-type conformation and bound 5BSL3.2 exhibited better translation activities than mutants that were unable to recognize 5BSL3.2 (11). The models described in this report provide a rationale explaining why formation of the 5BSL3.2–3′X complex may disfavor negative-sense RNA synthesis. When not bound to domain 3′X, the ‘L’-shaped 5BSL3.2 subdomain can recognize the thumb domain of NS5B and in this state there is enough flexibility for the 3′X domain to reach the template entry channel of the polymerase and start negative sense RNA synthesis (Figure 7B). In contrast, when the relatively rigid 5BSL3.2–3′X complex forms, a hypothetical binding of the ‘Y’ junction to the thumb domain would not physically allow the 3′-terminal tail to reach the template channel and vice versa, binding of the 3′-terminal tail to the template channel would likely be disfavored by the solvent exposure of the bulky upper region of the ‘Y’ (Figure 7).
Another important consequence of 5BSL3.2–3′X complex formation is the concealment of the palindromic DLS sequence of the 3′X domain (Figure 6). This sequence, absolutely conserved in all viral isolates (23), is exposed in the apical loop of subdomain SL2′ ((10,20,21) and Figure 4) and promotes 3′X domain homodimerization (17–21) through the formation of an intermolecular kissing loop contact between two palindromic SL2′ loops (19). The mechanism by which 3′X dimerization influences the viral cycle is not clear, but we and others have proposed that it likely contributes to control the switch between the translation and replication processes of the virus (11,18,19,57). This hypothesis is compatible with the available structural evidence and the mechanistic model described above. Formation of DLS–DLS homodimers would impede 5BSL3.2–3′X complex formation by blocking the k sequence of domain 3′X, and would therefore promote 5BSL3.2–NS5B recognition and the concomitant entry of the 3′X terminal tail in the NS5B template channel, stabilizing the replication stage of the HCV cycle (Figure 7B). In this regard, the 3′X–3′X homodimers have been reported to be optimal templates for NS5B (57) and may contribute to increase template selectivity and to ensure that only full-length RNA molecules are used for negative-sense RNA synthesis. Conversely, the establishment of the 5BSL3.2–3′X contact necessarily blocks homodimerization by concealing the DLS palindrome (Figure 6) and would disfavour negative sense RNA synthesis as explained above. Intriguingly, palindromic sequences promoting dimerization have been detected in the SLA promoters of flaviviruses (44), pointing to common regulatory mechanisms within the Flaviviridae family.
It is important to consider that the network of RNA–RNA and RNA–protein interactions reported to regulate the HCV cycle is likely dynamic in a cellular context. For example, after contacting subdomain 5BSL3.2, NS5B may shuffle downstream to recognize the flexible polyU/C segment (56), keeping the 3′X tail in the template channel and allowing 5BSL3.2 to contact subdomain IIId of the IRES and block translation. Another important question that remains unanswered is whether formation of the 3′X–5BSL3.2 complex has a direct effect on translation initiation through the recognition of the unusual kissing junction by a protein or RNA factor. Further experiments are needed to verify this hypothesis, as well as to validate the proposed 5BSL3.2–NS5B–3′X interaction model.
In conclusion, the low-resolution models of two crucial RNA domains of the HCV genome and their distal contact have shed light on the mechanisms likely used by the virus to regulate replication and translation. Furthermore, the detection of conserved RNA and RNA–protein motifs shared with flaviviruses suggests common regulatory mechanisms within the Flaviviridae family.
DATA AVAILABILITY
Experimental SAXS data and 3D models of full-length subdomain 5BSL3.2 and dimerization-defective full-length domain 3′Xm in the absence and presence of magnesium, as well as the 3′Xm–5BSL3.2 complex in the presence of magnesium, have been deposited in the Small Angle Scattering Biological Data Bank (SASBDB) with accession codes SASDMM4, SASDMN4, SASDMP4, SASDMQ4 and SASDMR4.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the use of the SAXS Core facility of Center for Cancer Research (CCR), National Cancer Institute (NCI). The SAXS core has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract 75N91019D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. NCI-SAXS Core is supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This research used 12-ID-B beamline of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The authors thank Dr Xiaobing Zuo (ANL) for his expert support.
Contributor Information
Jesús Castillo-Martínez, Centro de Investigación Traslacional San Alberto Magno, Universidad Católica de Valencia, 46001, Valencia, Spain; Escuela de Doctorado, Universidad Católica de Valencia, 46001, Valencia, Spain.
Lixin Fan, Basic Science Program, Frederick National Laboratory for Cancer Research, Small-Angle X-ray Scattering Core Facility of National Cancer Institute, National Institutes of Health, Frederick, MD 21702, USA.
Mateusz P Szewczyk, Centro de Investigación Traslacional San Alberto Magno, Universidad Católica de Valencia, 46001, Valencia, Spain; Escuela de Doctorado, Universidad Católica de Valencia, 46001, Valencia, Spain.
Yun-Xing Wang, Protein-Nucleic Acid Interaction Section, Center for Structural Biology, National Cancer Institute, National Institutes of Health, Frederick, MD 21702, USA.
José Gallego, Centro de Investigación Traslacional San Alberto Magno, Universidad Católica de Valencia, 46001, Valencia, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministerio de Ciencia e Innovación [BFU2015-65103-R, RTI2018-093935-B-I00 to J.G]; Fundación Universidad Católica de Valencia San Vicente Mártir [E-46–2017-0118740 to J.C.-M.]. Funding for open access charge: Ministerio de Ciencia e Innovación [RTI2018-093935-B-I00].
Conflict of interest statement. None declared.
REFERENCES
- 1. Petersen E., Koopmans M., Go U., Hamer D.H., Petrosillo N., Castelli F., Storgaard M., Al Khalili S., Simonsen L.. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 2020; 20:e238–e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nicholson B.L., White K.A.. Functional long-range RNA-RNA interactions in positive-strand RNA viruses. Nat. Rev. Microbiol. 2014; 12:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cox A.L. Challenges and promise of a hepatitis c virus vaccine. Cold Spring Harb. Perspect. Med. 2020; 10:a036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Romero-López C., Berzal-Herranz A.. The 5BSL3.2 functional RNA domain connects distant regions in the hepatitis c virus genome. Front Microbiol. 2017; 8:2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Romero-López C., Berzal-Herranz A.. A long-range RNA-RNA interaction between the 5′ and 3′ ends of the HCV genome. RNA. 2009; 15:1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shetty S., Stefanovic S., Mihailescu M.R.. Hepatitis c virus RNA: molecular switches mediated by long-range RNA-RNA interactions?. Nucleic Acids Res. 2013; 41:2526–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Friebe P., Boudet J., Simorre J.P., Bartenschlager R.. Kissing-loop interaction in the 3′ end of the hepatitis c virus genome essential for RNA replication. J. Virol. 2005; 79:380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. You S., Rice C.M.. 3′ RNA elements in hepatitis c virus replication: kissing partners and long poly(U). J. Virol. 2008; 82:184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tuplin A., Struthers M., Simmonds P., Evans D.J.. A twist in the tail: SHAPE mapping of long-range interactions and structural rearrangements of RNA elements involved in HCV replication. Nucleic Acids Res. 2012; 40:6908–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cantero-Camacho Á., Gallego J.. An unexpected RNA distal interaction mode found in an essential region of the hepatitis c virus genome. Nucleic Acids Res. 2018; 46:4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castillo-Martínez J., Ovejero T., Romero-López C., Sanmartín I., Berzal-Herranz B., Oltra E., Berzal-Herranz A., Gallego J.. Structure and function analysis of the essential 3′X domain of hepatitis c virus. RNA. 2020; 26:186–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diviney S., Tuplin A., Struthers M., Armstrong V., Elliott R.M., Simmonds P., Evans D.J.. A hepatitis c virus cis-acting replication element forms a long-range RNA-RNA interaction with upstream RNA sequences in NS5B. J. Virol. 2008; 82:9008–9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. You S., Stump D.D., Branch A.D., Rice C.M.. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis c virus RNA replication. J. Virol. 2004; 78:1352–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Romero-López C., Berzal-Herranz A.. The functional RNA domain 5BSL3.2 within the NS5B coding sequence influences hepatitis c virus IRES-mediated translation. Cell. Mol. Life Sci. 2012; 69:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Quade N., Boehringer D., Leibundgut M., van den Heuvel J., Ban N.. Cryo-EM structure of hepatitis c virus IRES bound to the human ribosome at 3.9-Å resolution. Nat. Commun. 2015; 6:7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsuda D., Mauro V.P.. Base pairing between hepatitis c virus RNA and 18S rRNA is required for IRES-dependent translation initiation in vivo. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:15385–15389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cristofari G., Ivanyi-Nagy R., Gabus C., Boulant S., Lavergne J.P., Penin F., Darlix J.L.. The hepatitis c virus core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004; 32:2623–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ivanyi-Nagy R., Kanevsky I., Gabus C., Lavergne J.P., Ficheux D., Penin F., Fossé P., Darlix J.L.. Analysis of hepatitis c virus RNA dimerization and core-RNA interactions. Nucleic Acids Res. 2006; 34:2618–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shetty S., Kim S., Shimakami T., Lemon S.M., Mihailescu M.R.. Hepatitis c virus genomic RNA dimerization is mediated via a kissing complex intermediate. RNA. 2010; 16:913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cantero-Camacho Á., Gallego J.. The conserved 3′X terminal domain of hepatitis c virus genomic RNA forms a two-stem structure that promotes viral RNA dimerization. Nucleic Acids Res. 2015; 43:8529–8539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cantero-Camacho A., Fan L., Wang Y.-X., Gallego J.. Three-dimensional structure of the full-length 3′X-tail of hepatitis c virus RNA in monomeric and dimeric states. RNA. 2017; 23:1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tuplin A., Struthers M., Cook J., Bentley K., Evans D.J.. Inhibition of HCV translation by disrupting the structure and interactions of the viral CRE and 3′ X-tail. Nucleic Acids Res. 2015; 43:2914–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamada N., Tanihara K., Takada A., Yorihuzi T., Tsutsumi M., Shimomura H., Tsuji T., Date T.. Genetic organization and diversity of the 3′ noncoding region of the hepatitis c virus genome. Virology. 1996; 223:255–261. [DOI] [PubMed] [Google Scholar]
- 24. Lohmann V., Körner F., Koch J., Herian U., Theilmann L., Bartenschlager R.. Replication of subgenomic hepatitis c virus RNAs in a hepatoma cell line. Science. 1999; 285:110–113. [DOI] [PubMed] [Google Scholar]
- 25. Parisien M., Major F.. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008; 452:51–55. [DOI] [PubMed] [Google Scholar]
- 26. Darty K., Denise A., Ponty Y.. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I.. PRIMUS: a windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003; 36:1277–1282. [Google Scholar]
- 28. Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992; 25:495–503. [Google Scholar]
- 29. Fischer H., de Oliveira Neto M., Napolitano H.B., Polikarpov I., Craievich A.F.. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 2010; 43:101–109. [Google Scholar]
- 30. Rambo R.P., Tainer J.A.. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013; 496:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P.V., Svergun D.I.. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012; 45:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Svergun D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999; 76:2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kozin M.B., Svergun D.I.. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001; 34:33–41. [Google Scholar]
- 34. Volkov V.V., Svergun D.I.. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003; 36:860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee W., Tonelli M., Markley J.L.. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015; 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Magnus M., Boniecki M.J., Dawson W., Bujnicki J.M.. SimRNAweb: a web server for RNA 3D structure modeling with optional restraints. Nucleic Acids Res. 2016; 44:W315–W319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schneidman-Duhovny D., Hammel M., Tainer J.A., Sali A.. FoXS, foxsdock and multifoxs: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016; 44:W424–W429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salomon-Ferrer R., Case D.A., Walker R.C.. An overview of the amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013; 3:198–210. [Google Scholar]
- 39. Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B.et al.. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018; 27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 41. Zok T., Antczak M., Zurkowski M., Popenda M., Blazewicz J., Adamiak R.W., Szachniuk M.. RNApdbee 2.0: multifunctional tool for RNA structure annotation. Nucleic Acids Res. 2018; 46:W30–W35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Appleby T.C., Perry J.K., Murakami E., Barauskas O., Feng J., Cho A., Fox D., Wetmore D.R., McGrath M.E., Ray A.S.et al.. Viral replication. Structural basis for RNA replication by the hepatitis c virus polymerase. Science. 2015; 347:771–775. [DOI] [PubMed] [Google Scholar]
- 43. Hodge K., Tunghirun C., Kamkaew M., Limjindaporn T., Yenchitsomanus P.T., Chimnaronk S.. Identification of a conserved RNA-dependent RNA polymerase (RdRp)-RNA interface required for flaviviral replication. J. Biol. Chem. 2016; 291:17437–17449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee E., Bujalowski P.J., Teramoto T., Gottipati K., Scott S.D., Padmanabhan R., Choi K.H.. Structures of flavivirus RNA promoters suggest two binding modes with NS5 polymerase. Nat. Commun. 2021; 12:2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Madeira F., Park Y.M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A.R.N., Potter S.C., Finn R.D.et al.. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019; 47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Janke E.M., Riechert-Krause F., Weisz K.. Low-temperature NMR studies on inosine wobble base pairs. J. Phys. Chem. B. 2011; 115:8569–8574. [DOI] [PubMed] [Google Scholar]
- 47. Blight K.J., Rice C.M.. Secondary structure determination of the conserved 98-base sequence at the 3′ terminus of hepatitis c virus genome RNA. J. Virol. 1997; 71:7345–7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ito T., Lai M.M.. Determination of the secondary structure of and cellular protein binding to the 3′-untranslated region of the hepatitis c virus RNA genome. J. Virol. 1997; 71:8698–8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dutkiewicz M., Ciesiolka J.. Structural characterization of the highly conserved 98-base sequence at the 3′ end of HCV RNA genome and the complementary sequence located at the 5′ end of the replicative viral strand. Nucleic Acids Res. 2005; 33:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Romero-López C., Barroso-Deljesus A., García-Sacristán A., Briones C., Berzal-Herranz A.. End-to-end crosstalk within the hepatitis c virus genome mediates the conformational switch of the 3′X-tail region. Nucleic Acids Res. 2014; 42:567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kao C.C., Yang X., Kline A., Wang Q.M., Barket D., Heinz B.A.. Template requirements for RNA synthesis by a recombinant hepatitis c virus RNA-dependent RNA polymerase. J. Virol. 2000; 74:11121–11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang J., Yamada O., Sakamoto T., Yoshida H., Araki H., Murata T., Shimotohno K.. Inhibition of hepatitis c virus replication by pol III-directed overexpression of RNA decoys corresponding to stem-loop structures in the NS5B coding region. Virology. 2005; 342:276–285. [DOI] [PubMed] [Google Scholar]
- 53. Kanamori H., Yuhashi K., Ohnishi S., Koike K., Kodama T.. RNA-dependent RNA polymerase of hepatitis c virus binds to its coding region RNA stem-loop structure, 5BSL3.2, and its negative strand. J. Gen. Virol. 2010; 91:1207–1212. [DOI] [PubMed] [Google Scholar]
- 54. Markoff L. 5′- and 3′-noncoding regions in flavivirus rNA. Adv. Virus Res. 2003; 59:177–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tilgner M., Deas T.S., Shi P.Y.. The flavivirus-conserved penta-nucleotide in the 3′ stem-loop of the west nile virus genome requires a specific sequence and structure for RNA synthesis, but not for viral translation. Virology. 2005; 331:375–386. [DOI] [PubMed] [Google Scholar]
- 56. Oh J.W., Sheu G.T., Lai M.M.. Template requirement and initiation site selection by hepatitis c virus polymerase on a minimal viral RNA template. J. Biol. Chem. 2000; 275:17710–17717. [DOI] [PubMed] [Google Scholar]
- 57. Masante C., Jaubert C., Palau W., Plissonneau J., Besnard L., Ventura M., Di Primo C.. Mutations of the SL2 dimerization sequence of the hepatitis c genome abrogate viral replication. Cell. Mol. Life Sci. 2015; 72:3375–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental SAXS data and 3D models of full-length subdomain 5BSL3.2 and dimerization-defective full-length domain 3′Xm in the absence and presence of magnesium, as well as the 3′Xm–5BSL3.2 complex in the presence of magnesium, have been deposited in the Small Angle Scattering Biological Data Bank (SASBDB) with accession codes SASDMM4, SASDMN4, SASDMP4, SASDMQ4 and SASDMR4.