

Abstract
The transcription factor ThPOK is critical for homeostasis and differentiation of mature T helper cells. Recent studies have shown that the ThPOK transcriptional silencer element (SilThPOK) can be reactivated in mature CD4 T cell populations to repress ThPOK and thereby promote differentiation to CD4+ IELs in the intestinal mucosa. However, the molecular basis for this dynamic regulation of SilThPOK activity remains unknown. Here, using a combination of reverse genetic and reporter approaches, along with global RNA-, ATAC- and ChIP-seq analyses, we demonstrate an important mechanism of ThPOK regulation by which ThPOK binds to the SilThPOK to drive an auto-feedback loop that maintains its own long-term expression. Disruption of this loop in vivo prevents persistent ThPOK expression in mature peripheral CD4 T cells, leading to genome-wide changes in chromatin accessibility, widespread transcriptional deregulation, and creation of an unusual metastable state, characterized by gain of colonic Treg, cytotoxic T-cell and early DN thymic progenitor specific gene signatures in naïve CD4 T cells. Functionally, abrogation of the ThPOK autoregulatory loop promotes selective differentiation of naive CD4 T cells into GITRlo PD-1lo CD25lo (Triplelo)Tregs in vitro and in vivo, as well as their conversion to CD4+ IELs in the gut, which provide dominant protection from colitis. Thus, the ThPOK autoregulatory loop represents a key mechanism to physiologically control ThPOK expression and T cell differentiation in the gut, with potential therapeutic relevance.
Introduction
The major CD4 and CD8 T cell subsets develop in the thymus and play key roles in orchestrating cell mediated immune responses. CD4 and CD8 lineage commitment is determined in the thymus with remarkable accuracy based on MHC specificity of the TCR expressed by DP (CD4+CD8+) precursors. Thymocytes expressing MHC II-restricted TCRs receive strong and/or persistent TCR signals that promote CD4 commitment, whereas cells expressing MHC I-restricted TCRs do not, and instead undergo commitment to the CD8 lineage in the presence of an appropriate cytokine milieu1. Importantly, the thymic lineage commitment process leads to alternate gene expression programs dictating helper versus cytotoxic functional programs.
The transcription factor ThPOK acts as a “master regulator” of the CD4 commitment process in the thymus, which supports expression of genes related to the T helper lineage program, whilst antagonizing expression of genes associated with the T cytotoxic program2, 3, 4. We and others have previously shown that ThPOK expression in thymocytes is regulated primarily at the transcriptional level via several stage- and lineage-specific cis elements5, 6. Of particular importance is the ~400bp silencer (SilThPOK), whose function has been conserved for 165 million years of evolution, preceding the divergence of marsupial and placental mammals7. The SilThPOK represses ThPOK expression in MHC I-restricted thymocytes, as evidenced by the fact that germline ablation of the SilThPOK causes promiscuous expression of ThPOK and diversion of all thymocytes to the CD4 lineage irrespective of their MHC specificity5, 6. Runx transcription factors are important antagonists of ThPOK that promote CD8 commitment6.
ThPOK continues to be expressed at high levels in peripheral CD4 T cells to stably maintain the helper lineage gene expression program9, for Th1 and TH2, but not TH17 effector differentiation10. ThPOK expression by CD4 T cells is also critical to support effective CD4+ T cell memory11. The mechanisms by which ThPOK expression is maintained in mature extrathymic CD4 T cells remain largely unknown, although a continuing dynamic role for the SilThPOK has recently emerged. Thus, mature CD4 T cells can convert into class II–restricted CTLs13 or into CD4+ CD8αα IELs14 and this conversion is dependent on SilThPOK activity13. Paradoxically, in peripheral CD8 T cells the SilThPOK is no longer required for ThPOK repression, as its deletion at this stage fails to derepress ThPOK15. The ability of the SilThPOK to toggle between ON and OFF states in peripheral CD4 T cells has major implications for T cell function and differentiation. For example, induction of colitis by naive CD4 T cells in an adoptive T cell transfer model requires ThPOK downmodulation in a SilThPOK-dependent manner, implying that SilThPOK actively regulates the fate of naïve T cells arriving at the site of inflammation or infection16. Given the widespread and important roles of ThPOK in CD4 T cell function, understanding the mechanisms that control its induction, maintenance and repression is both fundamentally significant and crucial for designing novel therapeutic strategies against autoimmunity and infectious diseases.
Here we identify an “anti-silencer” element within the SilThPOK, which is essential for persistent ThPOK expression in mature CD4 T cells and consequently for maintenance of mature CD4 T cell identity and function. We show that this element acts as a ThPOK binding site, and drives a positive autoregulatory loop by coordinating enhancer-promoter interactions. Using in vivo colitis and in vitro Treg cell culture models, we further demonstrate that the ThPOK-mediated positive autoregulatory loop is crucial for tissue-specific Treg differentiation, for maintaining Treg integrity in the intestine, and their conversion to the CD4+ IELs. Finally combining ThPOK ChIP-seq, ATAC-seq and RNA-seq data, we dissect how the genome-wide regulatory landscape of naive CD4 T cells is fundamentally dependent on the ThPOK mediated autoregulatory loop.
Results
Identification of an anti-silencer element.
The SilThPOK is reactivated during IEL generation in the gut, indicating that the SilThPOK continues to be dynamically regulated in mature peripheral CD4 T cells, i.e. is not permanently inactivated after CD4 commitment. Here we set out to identify the underlying mechanism for this dynamic control. For this purpose, we generated a series of knockout mouse lines carrying various deletions within the SilThPOK (Fig.1 a–c), and crossed each line to homozygosity. Two lines (ThPOKΔOC74, ΔNR82) showed no or minimal difference in peripheral CD4 T cell frequency compared to WT controls, suggesting that the deleted regions were not important for control of ThPOK expression in peripheral CD4 T cells, while 2 others showed complete absence of peripheral CD8 T cells (ThPOKΔQC48, ΔQK27), suggesting that the deletions blocked SilThPOK function during thymic development (as confirmed by thymic analysis; data not shown) (Fig. 1b,c). Most interestingly, two additional lines with overlapping deletions (ThPOKΔPY3 and ΔOB11) displayed profound alterations in T cell subset representation and T cell surface marker phenotypes (Fig. 1b,c): a) Peripheral CD4 T cells defined by high CD4 levels (CD4hi) were greatly diminished relative to CD8 T cells in ThPOKΔOB11/ΔOB11 mice. b) Two unusual T cell populations appeared, which either expressed both CD4 and CD8 (DP = double positive), or displayed low CD4 levels while lacking CD8 (CD4lo cells). The sum total of peripheral CD4hi, CD4lo and DP cells from ThPOKΔOB11/ΔOB11 mice is comparable to the number of CD4 T cells from WT mice, suggesting that these 3 populations collectively comprise the class II-restricted T cell compartment in ThPOKΔOB11/ ΔOB11 mice.
Fig. 1. Genetic mapping of an anti-silencer element in mature CD4 T cells.
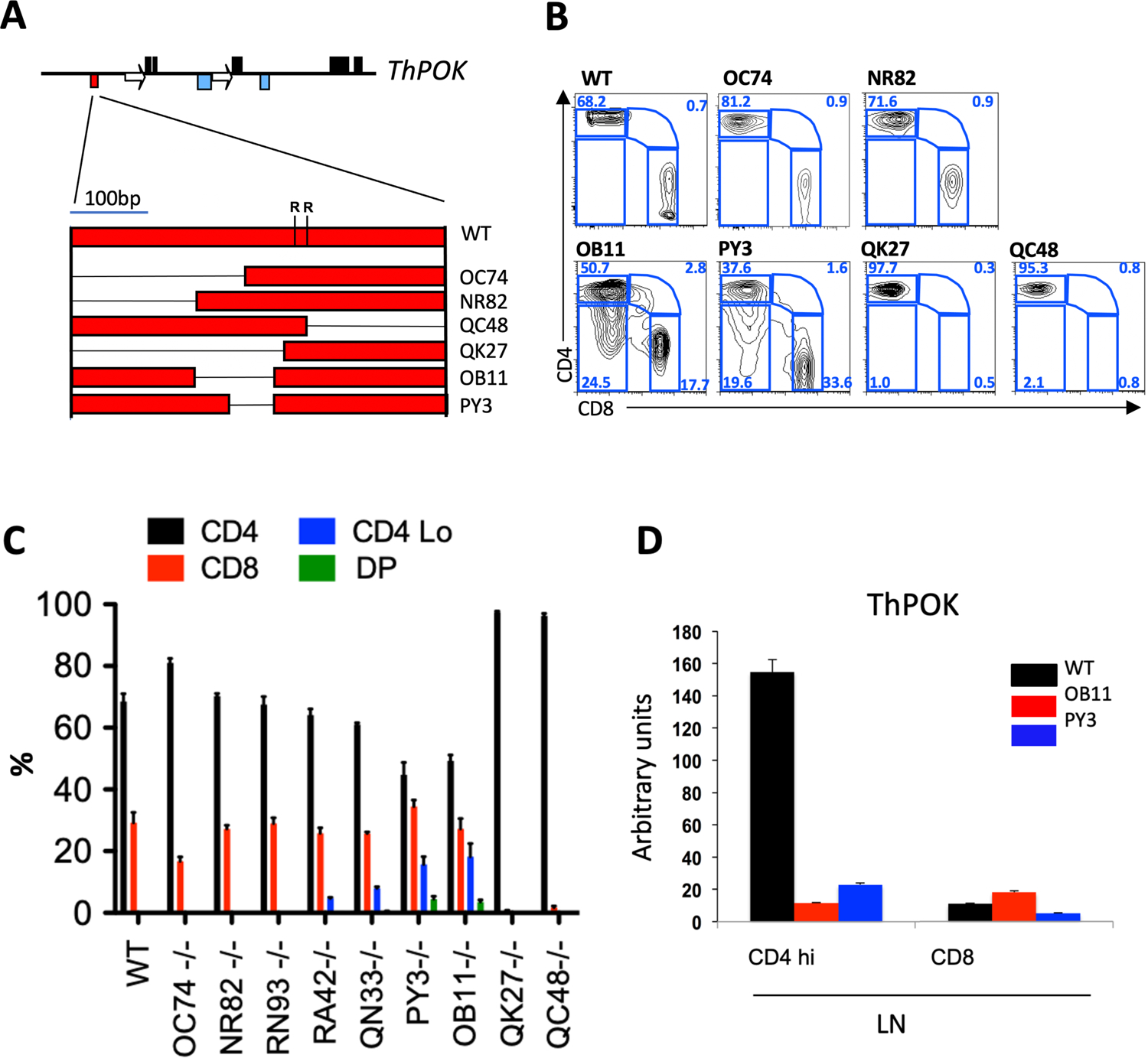
a) Organization of mouse ThPOK gene (top), and diagram of silencer deletion mutants (bottom). Black boxes, blue boxes, arrows, and red boxes indicate exons, enhancers, silencers and promoters, respectively. Deletions within the silencer are indicated by thin black lines. “R” indicates positions of conserved Runx binding sites. b) FACS analysis of CD4, and CD8a expression in gated TCRβ+ PBLs of WT mice and homozygous mutant lines, as designated in panel a. c) Plots indicate % of SP CD4, CD8, CD4+ 8+ (DP) and CD4lo/− cells within gated TCRβ+ PBLs of indicated strains (n = 5, for each strain). Error bars represent standard deviations. d) RNA was collected from freshly isolated cells before probing for ThPOK expression by qPCR.
qRT-PCR analysis showed that ThPOK mRNA is almost absent in peripheral T cells from ThPOKΔOB11/ΔOB11and ThPOKΔPY3/ΔPY3 mice (including SP CD4 cells), consistent with reactivation of the SilThPOK (Fig. 1d). In contrast, thymocytes from both lines showed normal stage-specific expression of ThPOK, except for a reduction at the most mature (CD69−) SP CD4 stage (Suppl. Fig. 1b). However, this has little effect on CD4/CD8 lineage choice, as revealed by normal ratio of SP CD4 to CD8 thymocytes (Suppl. Fig. 1a). Supporting this conclusion, no SP CD8 thymocytes were generated in SilΔOB11/ΔOB11 β2m−/−mice, in which positive selection is restricted to class II-restricted thymocytes (Suppl. Fig. 1c).
In summary, these results indicate that the OB11 region of the SilThPOK is largely dispensable for regulation of SilThPOK activity in thymocytes, but is essential to prevent inappropriate SilThPOK activation in mature peripheral CD4 T cells.
Y1H analysis of ThPOK binding to SilThPOK element.
Bioinformatic (JASPAR) analysis indicates that the 100bp OB11 region contains 4 predicted ThPOK binding sites, 3 of which are shared with the 50bp PY3 deletion region, suggesting potential autoregulatory function (Fig. 2a). To directly assess capacity of these motifs to bind ThPOK, we first utilized a yeast-1-hybrid (Y1H) approach17, 18, 19. For completeness, we examined the entire SilThPOK, subdivided into 15 overlapping 40–100 bp fragments (Fig. 2a). This showed that functional ThPOK binding sites map to a minimal 100bp region (between fragments 5 and 14), which closely coincides with the OB11 region (Fig. 2a). Electrophoretic mobility shift assay (EMSA) confirmed that a 100bp probe spanning the OB11 region was able to bind FLAG-tagged ThPOK, and underwent “supershifting” upon addition of anti-FLAG antibody (Fig. 2b, lanes 3 and 5). Mutation of 2 predicted ThPOK binding sites within the OB11 region (sites 1 and 3) abolished ThPOK binding by Y1H, demonstrating DNA sequence specificity (Fig. 2a). Similarly, mutation of sites 1 and 3 abrogated ThPOK binding in EMSA (Fig. 2b, lane 6). Individually mutating site 3, but not site 1, partially impaired ThPOK binding in EMSA, suggesting greater importance of site 3 relative to site 1, but also partial functional redundancy between this and other sites (Suppl. Fig. 2a). To evaluate contribution of individual binding sites in vivo, we generated 3 additional mutant lines with smaller deletions or mutations within the OB11 region (ThPOKΔRA42,ΔRN93 and ΔQN33) (Suppl. Fig. 2b,c,d). Mutants ThPOKΔRA42 (sites 3 and 4 ablated) and ThPOKΔQN33 (sites 1, 3 and 4 ablated) show slight shift to CD4lo phenotype, indicating that site 2 is individually important for ThPOK expression, but partially redundant with other sites. Finally ThPOK,ΔRN93/ΔRN93, which preserves sites 2, 3 and 4, exhibits no phenotype, indicating functional redundancy between site 1 and other sites. Furthermore, sites 2 and 3 are perfectly conserved between human and mouse, consistent with important functional role of these 2 sites (Suppl. Fig. 2d).
Fig. 2. Regulation of anti-silencer by ThPOK binding.
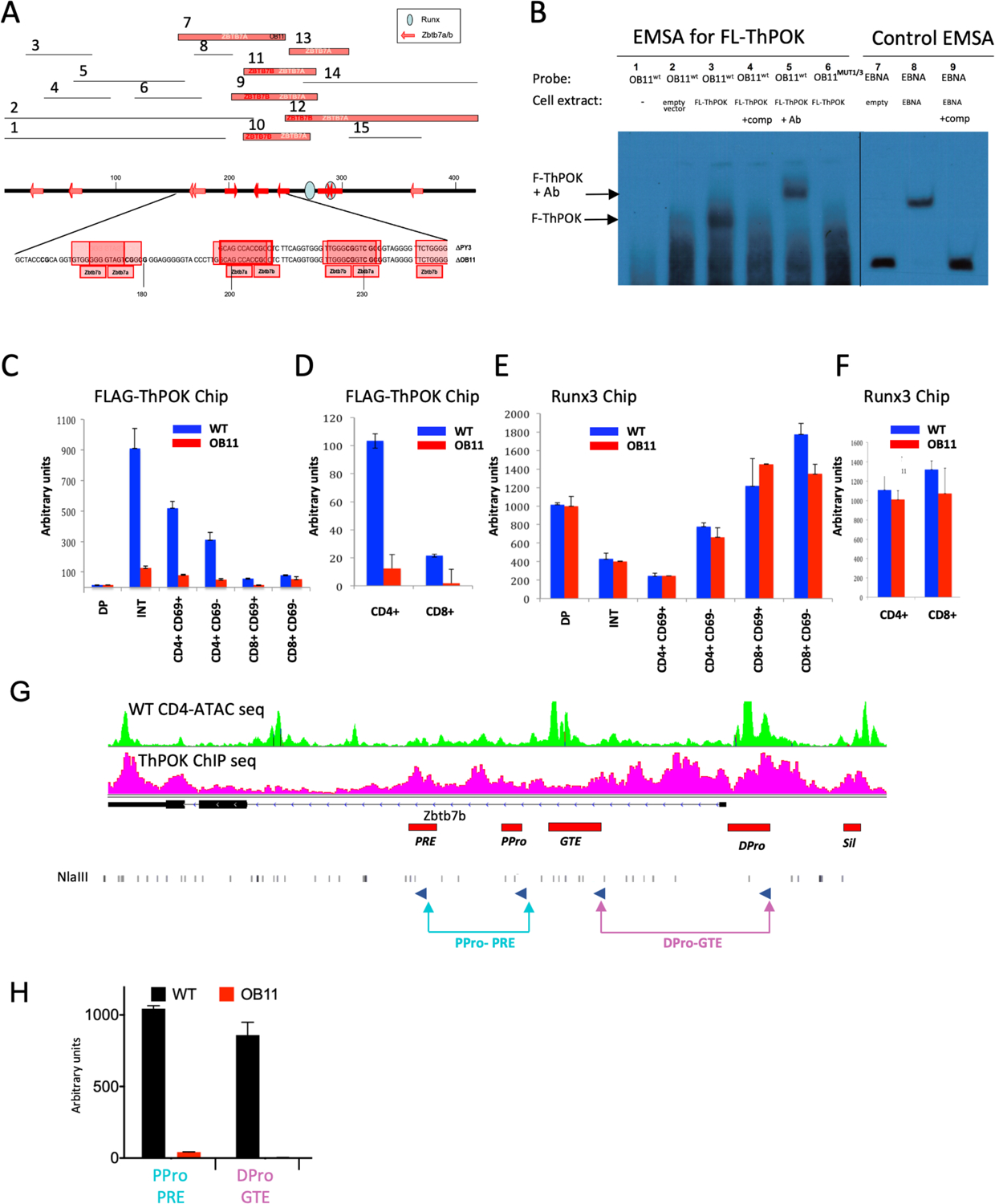
a) Numbered bars (top) represent 15 different mouse ThPOK silencer fragments that were tested as bait in Y1H analyses against ThPOK as prey (Zbtb7b). Fragments that bind ThPOK, and/or the related LRF (Zbtb7a) factor are indicated by thick red bars, whereas those that do not show binding are marked as thin black lines. Thick black line (middle) represents the full-length silencer, with positions of consensus Zbtb7b/a binding sites (as predicted by JASPAR algorithm) marked by red arrows, and conserved Runx binding sites by blue ovals. Sequence of regions deleted in OB11 and PY3 mutants, with positions of predicted Zbtb7b/a binding sites marked in red. b) EMSA analysis, using the 100bp OB11 region (lanes 1–6) or EBNA control DNA (lanes 7–9) as probes. Biotinylated probes were incubated with cell extracts from NIH 3T3 cells transfected with empty vector, Fl-ThPOK or EBNA expression constructs, as indicated. In some lanes unlabeled competitor DNA (lanes 4, 9), anti-Fl-ThPOK antibody (lane 5), or mutant OB11 probe in which the consensus ThPOK binding sites are destroyed (lane 6), were added. c-f) ChIP analysis with antibodies against indicated FL-ThPOK (c, d) or Runx3 (e, f) for indicated sorted thymocyte (c, e) of LN T cell (d, f) subsets. Red and blue bars indicate WT and ThPOKOB11/OB11 cells, respectively. Error bars represent standard deviations of 3 biological replicates. G) 3C assay-coupled with sorted peripheral CD4T cells from WT or ThPOKOB11/OB11 mice. qPCR was performed to reveal interactions between indicated elements (lower panel). Primer positions relative to ThPOK enhancers and promoters are marked (blue arrows, Top panel).
To confirm ThPOK binding to the SilΔOB11 silencer in vivo, we crossed ThPOKOB11 mice with Fh-ThPOK mice, encoding FLAG-tagged ThPOK, to generate compound heterozygous Fh-ThPOK/ThPOKΔOB11 mice. We employed anti-Flag ChIP in combination with allele-specific primer pairs to assess ThPOK binding to the SilΔOB11 and SilWT elements. This demonstrated ThPOK binding specifically to the SilWT but not the SilΔOB11 allele in all thymic (Fig. 2c) and peripheral (Fig. 2d) T cells. Of note, binding of Runx3 to Runx consensus motifs adjacent to the OB11 region was unaffected in the SilΔOB11 allele, demonstrating that the OB11 deletion does not broadly affect TF binding to the SilThPOK (Fig. 2e, f). Taken together these data reveal that the 100bp OB11 region is exclusively responsible for ThPOK binding to the SilThPOK.
Ablation of OB11 region disrupts physical interaction between key cis elements at the ThPOK locus.
Enhancer-promoter dynamics are critical for the spatiotemporal control of gene expression. Several enhancers and promoters have been identified at the ThPOK locus using transgenic and knockin reporter models5, 6, 12 (Fig. 2g). Deletion of the proximal enhancer (PRE) during thymic development leads to redirection of MHC class II-selected cells toward the cytotoxic lineage12, but its continued role in maintaining ThPOK expression in peripheral CD4 T cells remains unclear, as does that of the other enhancers. Therefore we used the 3C approach to test whether ThPOK binding to the OB11 region affects promoter and enhancer interactions in mature naïve CD4 T cells. Indeed, we observed clear interaction between ThPOK cis elements in WT CD4 T cells under homeostatic conditions, between both enhancers and promoters, which disappeared in ThPOK ΔOB11/ΔOB11 T cells (Fig. 2h).
ThPOK binding to its silencer drives a positive autoregulatory loop, that sustains ThPOK expression and CD4 T cell lineage stability.
To assess effect of the OB11 mutation on ThPOK expression at the single-cell level, we generated a SilΔOB11.GFP reporter allele in which GFP expression is regulated by the mutant SilΔOB11 element (note that GFP acts as a “gene trap” precluding ThPOK expression from this allele) (Fig. 3a). As expected, sorted CD4 thymocytes from heterozygous ThPOKΔOB11.GFP/+ mice show high GFP expression, while mature CD4 T cells mostly lack GFP mRNA and protein expression by qPCR and FACS (although a fraction show low GFP protein expression) (Fig. 3 b,d). We conclude that the OB11 mutation selectively prevents transcription from the ΔOB11.GFP reporter allele in peripheral CD4 T cells versus thymocytes. Importantly, reduced transcription of GFP in SilΔOB11.GFP mice cannot be restored by ThPOK expressed from the wild type ThPOK allele in SilΔOB11.GFP/+ mice, confirming that the SilΔOB11.GFP allele is insensitive to positive regulation by ThPOK (Fig. 3c).
Fig. 3. Loss of anti-silencer destabilizes CD4 T cell phenotype.
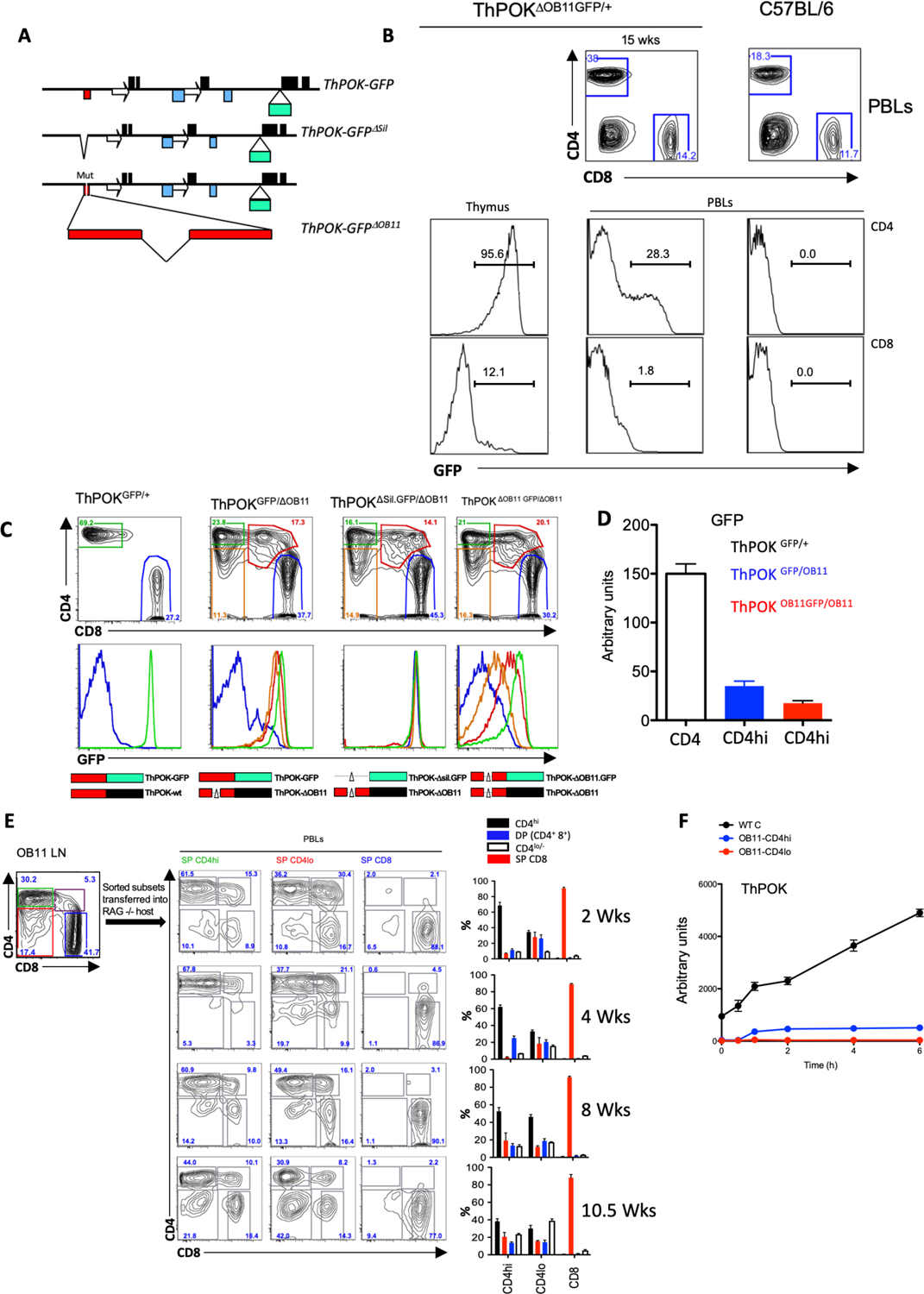
a) Structure of ThPOKGFP, ThPOKΔSil.GFP, and ThPOKDOB11.GFP reporter alleles, showing insertion site of GFP-encoding exon (labeling of elements as in Fig. 1a). b) FACS analysis of CD4, and CD8a expression in total PBLs of WT mice and heterozygous ThPOKΔOB11.GFP/+ mice (top), and GFP expression of gated SP CD4 and CD8 population from PBLs of same mice (bottom rows). c) FACS analysis of CD4, and CD8a expression in gated TCRβ+ PBLs (top), or GFP expression of gated CD4hi (green), CD4lo/− (orange), CD4+ CD8+ (red), or CD8 PBLs (middle), of WT mice and indicated compound heterozygous reporter lines. Bottom row illustrates ThPOK alleles of indicated mouse. Red box represents silencer (thin black line shows extent of deletion); green and black boxes indicate whether allele expresses GFP or ThPOK mRNA. d) RNA was collected from freshly sorted CD4 T cells from ThPOKGFP/+ (white), ThPOKΔOB11.GFP/ΔOB11(red), or ThPOKGFP/ΔOB11 (blue) mice and probed for GFP mRNA expression by qPCR. e) Reconstitution of RAG−/− hosts with CD4hi, CD4lo and SP CD8 cells from ThPOKΔOB11.GFP/ΔOB11 donor mice. FACS analysis showing sort gates for isolation of donor cells (upper left panel). FACS analysis of CD4 and CD8 expression by PBLs from reconstituted hosts at indicated time after transfer (next 3 columns). Plots at right indicate % of CD4hi, CD4lo/−, CD4+ 8+ (DP) and CD8 cells within gated TCRβ+ PBLs of mice reconstituted with indicated ThPOKΔOB11/ΔOB11 donor cells, at each time point. Error bars represent standard deviations of 4 biological replicates. f) Sorted T cell population from indicated mice were stimulated with anti-TCRβ. RNA was prepared at different hour of probed for ThPOK mRNA expression by qPCR.
To test the requirement for ThPOK protein in control of ThPOK transcription in peripheral CD4 T cells, we crossed the ThPOKΔOB11 strain to a second reporter line in which GFP is knocked into the WT ThPOK locus (ThPOKGFP mice), to create compound heterozygous ThPOKGFP/ΔOB11 mice (Fig. 3a, c). In ThPOKGFP mice GFP acts as a “gene trap”, precluding expression of ThPOK, so that the ThPOKΔOB11 allele provides the only potential source of ThPOK in heterozygous ThPOKGFP/ΔOB11 mice. We observe significantly reduced GFP protein levels in ThPOKGFP/ΔOB11compared to ThPOKGFP/+ mice (Fig. 3c, d), implying that continued ThPOK expression is necessary to prevent reactivation of the SilThPOK in peripheral CD4 T cells. To directly confirm that reduced reporter expression in ThPOKGFP/ΔOB11 CD4 T cells is silencer-dependent, we crossed ThPOKΔOB11 mice to a third reporter line in which GFP is knocked into a ThPOK locus in which the SilThPOK is deleted (ThPOKΔSil.GFP mice) (Fig. 3a). Importantly, GFP expression is fully restored in CD4 T cells of compound heterozygous ThPOKΔSil.GFP/ΔOB11 mice, showing that reduced reporter expression by ThPOKGFP/ΔOB11 CD4 T cells is silencer-dependent (Fig. 3c, 3rd column). Overall, these results show that ThPOK binding to SilThPOK mediates an autoregulatory loop that is essential to block activity of the SilThPOK in peripheral CD4 T cells to allow persistent ThPOK expression.
As noted above, 35% of mature CD4 T cells from ThPOKΔOB11.GFP/+ mice express GFP protein by FACS (Fig. 3 b). Given that the OB11 mutation blocks ThPOK transcription in peripheral CD4 T cells, these GFP+ cells could represent recent thymic immigrants that still express long-lived GFP protein produced in the thymus, as is the case for RAG-GFP mice20, and/or peripheral T cells that have undergone transient reactivation of their ThPOK locus post-thymically. To evaluate these possibilities, we adoptively transferred sorted GFP- T cell subsets from ThPOKΔOB11/ΔOB11.GFP and ThPOKGFP/+ mice into RAG−/− hosts (Fig. 3e; data not shown). qPCR analysis of ThPOK and GFP mRNA expression by sorted T cell subsets 4 weeks after adoptive transfer indicated that the transcription from the ThPOK locus remained very low (at the cell population level) (data not shown), but that 20% of transferred CD4hi T cells now expressed GFP protein (at the single cell level) (Suppl. Fig. 3b). Since GFP protein has a half-life of ~26hrs21, newly synthesized GFP protein should degrade to background levels within 5–7 days, implying that GFP+ CD4hi T cells have undergone reactivation of their OB11-GFP reporter allele within the preceding week. To assess whether this transient ThPOK transcription might depend on TCR-mediated induction, we subjected CD4 T cells from SilΔOB11/ΔOB11 versus WT mice to antibody-mediated TCR stimulation (anti-TCRβ). While this increased ThPOK mRNA expression by WT CD4 T cells, neither CD4hi or CD4lo T cells from ThPOKΔOB11/ΔOB11 mice showed significant ThPOK induction (Fig. 3f). Surface expression of TCR, as measured by staining with anti-TCRβ and CD3ε, was unaffected (Suppl. Fig. 3c). We conclude that the OB11 allele in contrast to the wt allele is not subject to TCR-mediated induction.
Finally, we found that both CD4hi cells and CD4lo cells, from ThPOKΔOB11/ΔOB11.GFP mice can give rise to all other subsets upon transfer into RAG−/− mice, i.e. CD4hi, CD4lo, DP and CD8, upon adoptive transfer (Fig. 3e), indicating that the CD4hi and CD4lo phenotypes of ThPOKΔOB11/ΔOB11.GFP cells are metastable and can readily interconvert in vivo. In contrast, transfer of CD8 T cells from ThPOKΔOB11/ ΔOB11.GFP mice gave rise almost exclusively to CD8 T cells, indicating that the SP CD8 phenotype is stable/terminal (Fig. 3e).
In summary, these results indicate that the ThPOK autoregulatory loop is essential for sustaining ThPOK expression and CD4 T cell lineage stability of mature MHC class II-restricted T cells.
Genome-wide changes in gene expression in ThPOKΔOB11/ΔOB11 T cells.
To assess how the anti-silencer regulates ThPOK-dependent gene expression in peripheral CD4 T cells to maintain stable Th lineage, we carried out RNAseq analysis of sorted naïve CD4hi, CD4lo and DP T cells from ThPOKΔOB11/ΔOB11 mice, versus WT CD4 and CD8 T cells. This revealed that 1724, 1862, and 1570 genes were differentially expressed (at least 2-fold) between WT CD4 T cells and either CD4hi, DP, or CD4lo ThPOKΔOB11/ΔOB11 subsets, respectively (Fig. 4 a,b; Suppl. Fig. 4b). 789 (28%) of these differentially expressed genes (DEGs) show similar trend in expression between different ThPOKΔOB11/ΔOB11 T cell subsets, while the remainder show divergent expression between subsets, indicating that the phenotypically distinct ThPOKΔOB11/ΔOB11 T cell subsets are also distinct at the level of gene expression (Fig. 4 a,c). For instance, CD4lo cells show upregulation of 283 unique genes compared to other OB11 subsets.
Fig. 4. Ablation of anti-silencer leads to deregulation of CD4 T cell gene expression program.
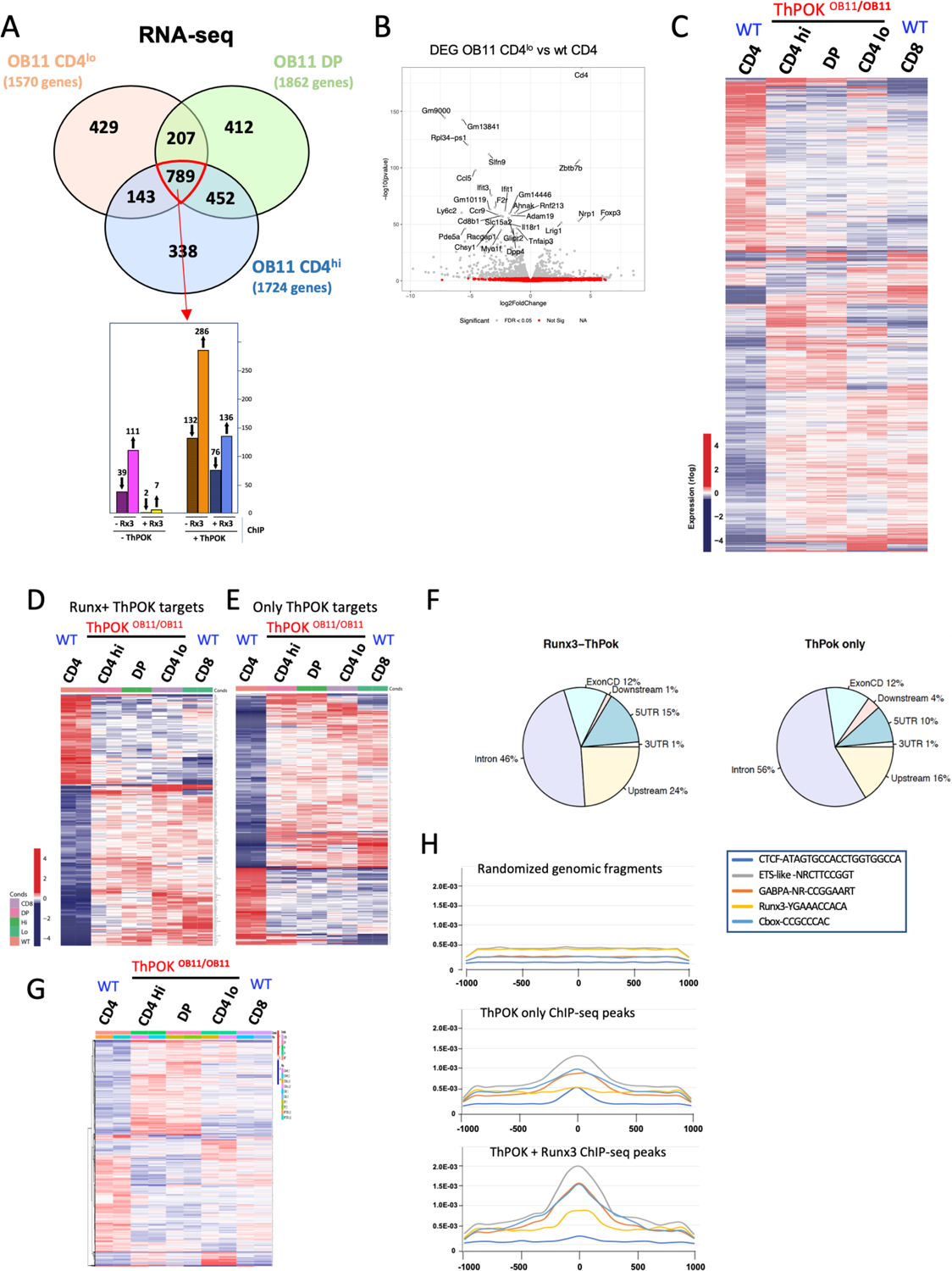
a) Venn diagram illustrating intersection between gene subsets that are differentially expressed (according to RNA-seq analysis) between indicated T cell subsets from ThPOKΔOB11/ΔOB11 mice and WT CD4 T cells. Number of genes that are differentially expressed for each subset is shown in brackets below subset name. Bar graph at bottom indicates number of 789 commonly misregulated (upward and downward arrow indicate up regulated and down regulated genes respectively) that are direct target of ThPOK and Runx3 or both ThPOK and Runx3, as indicated. b) Volcano plot illustrating gene expression differences between OB11 CD4lo and WT CD4 T cells. The grey dots represent genes differentially expressed (adjusted P<0.05) between samples. Genes with the largest negative or positive standardized mean difference are marked. c) Heat map displaying hierarchical clustering of DEGs for indicated OB11 and WT T cell subsets. Analysis is restricted to the union of all 2769 genes differing in expression between any OB11 T cell subset and WT CD4 T cells (FDR 5%). Red indicates increased gene expression levels; blue indicates decreased levels. d-e) Heat maps displaying hierarchical clustering of DEGs for indicated OB11 and WT T cell subsets, that are commonly misregulated between any OB11 T cell subset and WT CD4 T cells (789 genes), and are targets of ThPOK alone (318 genes) (d) or both ThPOK and Runx3 (212 genes) (e) (FDR 5%). Red indicates increased gene expression levels; blue indicates decreased levels. f) Pie charts illustrating distribution of ThPOK binding sites within target DEGs that are direct target of ThPOK (right) or ThPOK+ Runx3 (left) g) Heat map displaying hierarchical clustering of DEGs for indicated ThPOKΔOB11/ΔOB11 and WT T cell subsets. Analysis is restricted to the subset of 1042 DEGs whose expression differs between any ThPOKΔOB11/ΔOB11 subset versus WT CD4 T cells, but NOT between WT CD4 and WT CD8 T cells, i.e. genes that are not CD8 like (FDR 5%). Red indicates increased gene expression levels; blue indicates decreased levels. h) Plots illustrating codistribution of ThPOK ChIP-seq peaks with other TF binding sites (defined by ChIP-seq). Same analysis was carried out for ThPOK peaks associated with Runx consensus motifs (bottom), not associated with Runx motifs (middle), or random genomic fragments, i.e. NOT bound by ThPOK (top).
Importantly, we find that 760 (27%) of the 2770 DEGs are, “CD8 like”, consistent with the idea that lack of ThPOK allows the CD8 transcriptional program to take over in CD4 T cells (Fig. 4c). On the other hand, 1042 DEGs are NOT CD8 like (FDR 5%), suggesting potential roles of ThPOK outside of CD4-CD8 differentiation (Fig. 4g). Intriguingly, some of these genes are characteristically expressed at the ETP to DN2 stage transition in early thymopoiesis (including Id2, Kit, Sca1, Pgk1, Meis1, Cd82, and Sox13) (Suppl. Fig. 4a), or granulocyte differentiation (including Mpo, Tyrobp, Fcerg1, and Lyz, a marker of DN thymocytes destined to differentiate to the granulocytic lineage25.
Next, to identify direct ThPOK target among DEGs, we intersected our RNA-seq data with α-ThPOK ChIP-seq data from WT CD4 T cells (using public anti-ThPOK ChIP-seq data;26, GSE116506). Given the known functional antagonism between ThPOK and Runx3, we further intersected our RNA-seq data with α-Runx3 ChIP-seq data from WT CD8 T cells (using Runx3 Chip Seq data;27 GSE124912). This revealed genome wide 4574 sites associated with 3200 genes where ThPOK and Runx3 bind in close proximity (within 400 bp), representing DUAL targets of ThPOK and Runx3 (Fig. 4 d, f; Suppl. Fig. 4c, d). Among common 789 DEGs in all ThPOKΔOB11/ΔOB11 T cell subsets, 212 (24%) are targets of both ThPOK and Runx3 (Fig.4D) but 630 are direct targets of only ThPOK (80%) (Fig. 4a, bottom panel, Fig. 4e). Interestingly, genes bound by both ThPOK and Runx3 include several critical transcriptional regulators of CD8 differentiation, e.g. Eomes, RORα, and T-bet (Suppl. Table 1a,b). Based on pathway (DAVID) and literature analysis, we find that 15 out of 76 ThPOK and Runx3 dual target genes that are down-modulated in all ThPOKΔOB11/ΔOB11 subsets (Suppl. Table 1a) are implicated in regulation of TCR signaling (as indicated by pink boxes in Suppl. Table 1), especially dampening of TCR signaling (including Camk2d, Kidins20, Pag1, Prkd2, etc.), with most showing decreased expression in ThPOKΔOB11/ΔOB11 T cells, suggesting that ThPOKΔOB11/ΔOB11 prone to TCR stimulation (Suppl. Table 2a,b). Other pathways enriched in this gene subset are those involved in translation (Rpl19, Rpl36, Eif4b etc.), and cytokine signaling (Stat1, Stat4 etc), consistent with widespread and profound effects of Runx3 and ThPOK on control of CD4 T cell biology and function (Suppl. Table 2a,b). We also noted presence of Ets factor binding sites and CTCF binding sites in very close proximity to ThPOK binding sites, as evidenced by genomewide intersection of ChIP-seq data of respective transcription factors (Fig. 4h). CTCF sites were predominantly associated with ThPOK sites not linked with Runx3 sites, suggesting that persistent ThPOK expression acts in concert with CTCF for maintenance of chromosomal territory in CD4 T cells.
Altered chromatin accessibility of super enhancers in ThPOKΔOB11/ΔOB11 T cells.
Our RNA-seq data in concert with our T cell transfer experiments (Fig. 3e) indicate that distinct ThPOKΔOB11/ΔOB11 T cell subsets are metastable at both the phenotypic and transcriptional levels, so that disruption of the ThPOK autoregulatory loop hinders imposition of a stable transcriptional state, while permitting interconversion between different metastable states. Metastable states both in effector and memory precursor progenitors, and hematopoietic thymic progenitors have been shown to be associated with epigenomic plasticity28, 29 metastable chromatin accessibility and multilineage transcriptional signatures30, 31. Therefore, to better understand chromatin organization of peripheral T cells from ThPOKΔOB11/ΔOB1 mice, we carried out ATAC-seq on naïve ThPOKΔOB11/ΔOB11 CD4lo and WT CD4 T cells. This revealed strikingly altered chromatin accessibility in ThPOKΔOB11/ΔOB11 T cells, such that regions normally open in WT CD4 T cells are diminished in height in a genome-wide manner in ThPOKΔOB11/ΔOB11 T cells (Fig. 5a; Suppl. Fig 5a). We identified 15,608 and 2,767 Differentially Accessible Chromatin Regions (DACRs) that were selectively open in WT CD4 and ThPOKΔOB11/ΔOB11 CD4lo T cells, respectively. To ascertain the extent to which altered chromatin accessibility directly depends on ThPOK binding, we intersected ATAC-seq and ThPOK ChIP-seq data. This revealed that ThPOK binding is associated with 566 and 661 chromatin regions that are selectively open in ThPOKΔOB11/ΔOB11 CD4lo and WT CD4 T cells, respectively (Fig. 5b). Regions selectively open in ThPOKΔOB11/ΔOB11 CD4lo T cells showed enrichment of Runx binding motifs, suggesting a major role of ThPOK in masking Runx binding sites in CD4 T cells (Fig 5c; Suppl. Fig. 5b), while regions selectively open in WT CD4 T cells showed enrichment of chromatin territory-defining regulatory factors CTCF and CTCFL (Boris), suggesting a possible role of ThPOK in maintenance of accessible chromatin territory in the CD4 lineage (Fig. 5c).
Fig. 5. Loss of anti-silencer function causes genome-wide change in chromatin accessibility.
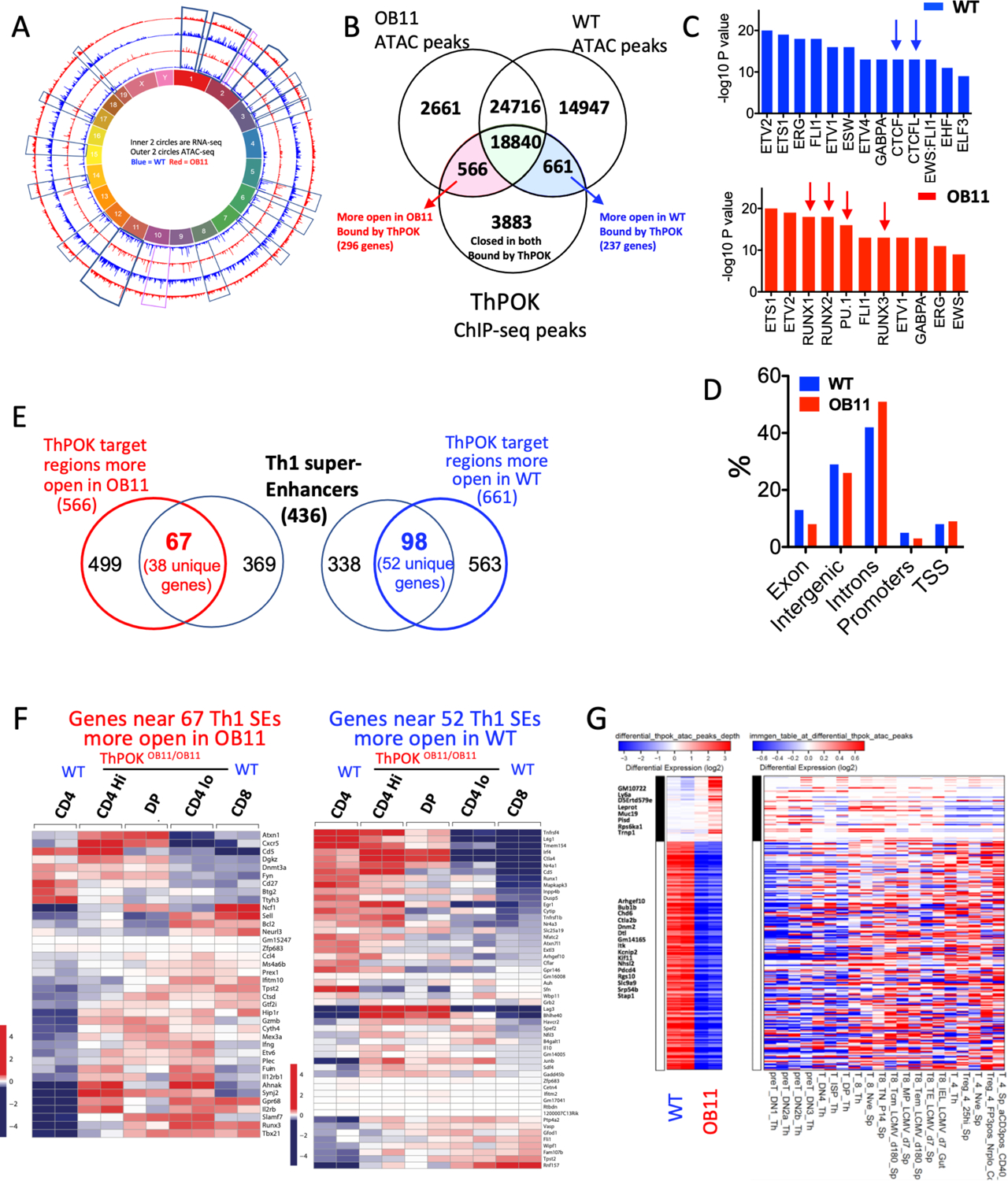
a) Circular plot of mouse chromosomes. Outer 2 rings represent ATAC-seq peaks. Inner 2 rings represent RNA-seq peaks. Blue and red rings represent WT CD4 and OB11 CD4lo T cells, respectively. b) Venn diagram indicating intersection between ATAC-seq peaks that are selectively open in WT CD4 and OB11 CD4lo T cells and α-ThPOK ChIP-seq peaks, as indicated. c) Relative enrichment of TF binding sites associated with open chromatin region in WT CD4 T cells (top) or ThPOKΔOB11/ΔOB11 CD4lo T cells (bottom). d) Relative distribution of DACRs in ThPOKΔOB11/ΔOB1 CD4lo (red) versus WT CD4 T cells (blue) with respect to gene organization (TSS = transcriptional start site). e) Venn diagram indicating intersection between DACRs that are selectively open in WT CD4 and ThPOKΔOB11/ΔOB11 CD4lo T cells, and Th cell associated super enhancers, as indicated. f) Heat maps showing relative expression of genes associated with 67 and 98 SEs that are selectively open in ThPOKΔOB11/ΔOB11 CD4lo versus WT CD4 T cell subsets, respectively (from panel e). g) Heat map showing top 250 regions with highest differential accessibility between WT CD4 and ThPOKΔOB11/ΔOB11 CD4lo T cells, filtered for genes that are also differentially expressed between these cell types (but NOT differentially expressed between WT CD4 and WT CD8 T cells) (left panel). Comparison of chromatin accessibility of these 250 regions with a T cell developmental accessibility panorama (Immgen) (right panel).
To further understand how changes in gene transcription correlate with changes in chromatin accessibility, we mapped DACRs that correlated with ThPOK binding peaks to closely linked genes (i.e. from 5kb upstream of TSS to 1kb downstream of TSS). This identified 296 and 237 ThPOK target genes that are more accessible in ThPOKΔOB11/ΔOB11 and WT CD4 T cells, respectively (Fig. 5b). 190 (80%) genes that are selectively open in ThPOKΔOB11/ΔOB11 T cells are relatively upmodulated in ThPOKΔOB11/ΔOB11 vs WT CD4 T cells, indicating strong correlation between open chromatin and transcription. DACRs which were more open in ThPOKΔOB11/ΔOB1 versus WT CD4 T cells were relatively enriched for introns and depleted of promoters (Fig. 5d).
T cell “identity” relates largely to the precise regulation of key lineage master regulators, cytokines, chemokines and their receptors, and these genes are frequently associated with Super Enhancers (SE) in T cells32–33. Given that many of these key genes are misregulated in ThPOKΔOB11/ΔOB11 T cells, we considered whether ThPOK might be involved in regulation of accessibility of their associated SEs. To test this possibility we intersected 566 and 661 ThPOK-bound DACRs that are more open in ThPOKΔOB11/ΔOB11 and WT CD4 T cells, respectively, with a dataset of 436 Th cell-specific SEs (dbSUPER) (https://asntech.org/dbsuper)34. Strikingly, this revealed that 67 (11%) and 98 (14%) of selectively open DACRs in ThPOKΔOB11/ΔOB11 T cells and WT CD4 T cells, respectively, mapped to Th cell-specific SEs (Fig. 5e), but not to SEs associated with thymic development or early B cell development (Suppl. Fig. 5c). These Th SEs are closely linked (within 20kb) with key genes encoding the Th master regulator T-bet, as well critical cytokines (Ifng, Il10), chemokines (Ccl4), and cytokine receptors (il12rb, Tnfrsf4), Runx factors (Runx1/3), cytotoxic molecules (Gzmb), TCR signal pathways (Egr1, Nfatc2, Grb2), and epigenetic regulators (Dnmt3a). Importantly, most of these genes are disregulated in ThPOKΔOB11/ΔOB11 versus WT CD4 T cells, indicating a key role of ThPOK in regulation of Th SE opening s well as their activity (Fig. 5f).
Finally, since we showed that many DEGs from ThPOKΔOB11/ΔOB11 T cells show similarity with early T cell progenitor gene expression pattern (Suppl. Fig. 4a), we compared chromatin accessibility of 250 chromatin regions located near (within 10kb of) these DEGs against chromatin data for sorted thymic developmental stages from WT mice (Immgen). Indeed, we find that regions which are less accessible in ThPOKΔOB11/ΔOB11 CD4lo T cells are also less accessible in early T cell progenitors (DN1- DP) and vice versa (Fig. 5g).
In summary, ThPOKΔOB11/ΔOB11 T cells display profound changes in gene expression and chromatin accessibility, including Super Enhancer accessibility, resulting in a metastable state and lineage infidelity. Thus ThPOK binding to the anti-silencer promotes persistent ThPOK expression required to maintain the CD4 lineage-specific chromatin conformation and gene expression programs to prevent trans-differentiation to CD8 lineage and and/or de-differentiation to early T cell progenitor stage.
Enhancement of TCR sensitivity, proliferation and cytokine expression in OB11 T cells.
Our transcriptomic and genome-wide accessibility studies indicate that the ThPOK autoregulatory loop plays an important role in regulation of genes involved in TCR signal strength. To functionally validate this proposition, we compared proliferation and cytokine production of sorted WT and ThPOKΔOB11/ΔOB11 T cells in response to weak (anti-TCRβ) versus strong (anti-CD3/CD28) TCR signals. Strikingly, class II-restricted T cells from OB11 mice exhibit stronger proliferative response compared to WT CD4 T cells in response to weak stimulation (Suppl. Fig. 6a), as well as more sustained ERK phosphorylation (Suppl. Fig. 6b), suggesting that the ThPOK autoregulatory loop regulates susceptibility of CD4 T cells to TCR stimulation by sustaining the TCR signal. Further, we uncovered promiscuous cytokine deregulation (IFNγ, IL-9, and IL-17) and associated transcription factors (T-bet, Rorc, Ahr, Irf4) in the absence of any stimulus, suggesting that the ThPOK autoregulatory feedback loop is required to maintain ‘naiveness’ in CD4 T cells and repress diverse Th effector signatures (Suppl. Fig. 6 c, d). These effects may reflect direct control by ThPOK, as we find ThPOK ChIP-seq peaks within the IFNγ and T-bet super enhancers (Fig. 5f), the Ahr promoter, as well as within genes encoding Rorc and the IL9-inducing factor IRF4 to the CNS25 regulatory region of the IL9 gene (Suppl. Fig. 6e). Since TCR signal strength is also known to control Th differentiation35, we further assessed whether ThPOKΔOB11/ΔOB11 T cells show altered Th differentiation. Indeed, we found that activated ThPOKΔOB11/ΔOB11 T cells display strong skewing towards a Th1/inflammatory response relative to WT CD4 T cells, consistent with previous ThPOK knockdown and knockout studies (Suppl. Fig. 6 d).
Abrogation of ThPOK autoregulatory loop redirects Treg differentiation and enhances Treg-mediated control of colitis.
Although previous studies employing conditional Foxp3Cre-mediated deletion of ThPOK have demonstrated a substantial reduction in mature Tregs in the absence of ThPOK, indicating a major role in Treg development and/or homeostasis, this does not result in any overt autoimmune or inflammatory phenotype14, 36. Similarly, ThPOKΔOB11/ΔOB11 mice exhibit a substantial (2-fold) diminution of Foxp3+ Tregs in LN and spleen versus WT control mice (data not shown), yet show no evidence of autoimmunity at least till 15 months of age. Given the absence of an autoimmune phenotype in ThPOKΔOB11/ΔOB11 mice despite reduced Treg numbers, we reasoned that Treg function is maintained or even enhanced in these mice. To evaluate whether ThPOKΔOB11/ΔOB1 mice exhibit more subtle defects in Treg development and differentiation, we crossed ThPOKΔOB11/ΔOB1 mice with Foxp3-RFP mice and examined development of nTregs. As expected, thymic Treg development was relatively normal (data not shown). Interestingly, among peripheral Tregs there was a significant increase in the anti-colitogenic GITRloPD-1lo CD25lo (Triplelo) subset37 relative to the GITRhiPD-1hiCD25hi (Triplehi) subset in ThPOKΔOB11/ΔOB11 versus ThPOKΔOB11/+ mice (Fig. 6a). Of note, WT Triplelo Tregs express much lower level of ThPOK than Triplehi Tregs, as evidenced by FACS analysis and ThPOK qPCR (Fig. 6b), suggesting that differential ThPOK expression plays a physiological role in promoting alternate differentiation of these subsets in WT mice. Next we tested the effect of autoregulatory loop ablation on iTreg generation. For this purpose, we cultured CD4hi cells from WT and ThPOKΔOB11/ΔOB11 mice under Treg polarizing conditions. Both WT and ThPOKΔOB11/ΔOB11 T cells were able to induce Foxp3 under these conditions, although Foxp3 levels were lower for ThPOKΔOB11/ΔOB11 T cells. Most strikingly, ThPOKΔOB11/ΔOB11 T cells gave rise to large numbers of Triplelo Tregs, which also expressed lower levels of FR4 and CD73 compared to WT T cells (Fig. 6c). Hence, maintenance of ThPOK expression via its autoregulatory loop promotes generation of Triplehi Tregs, while ThPOK down-modulation via ablation of this loop favors generation of Triplelo Tregs.
Fig. 6. Ablation of anti-silencer promotes differentiation of Triplelo Treg subset.
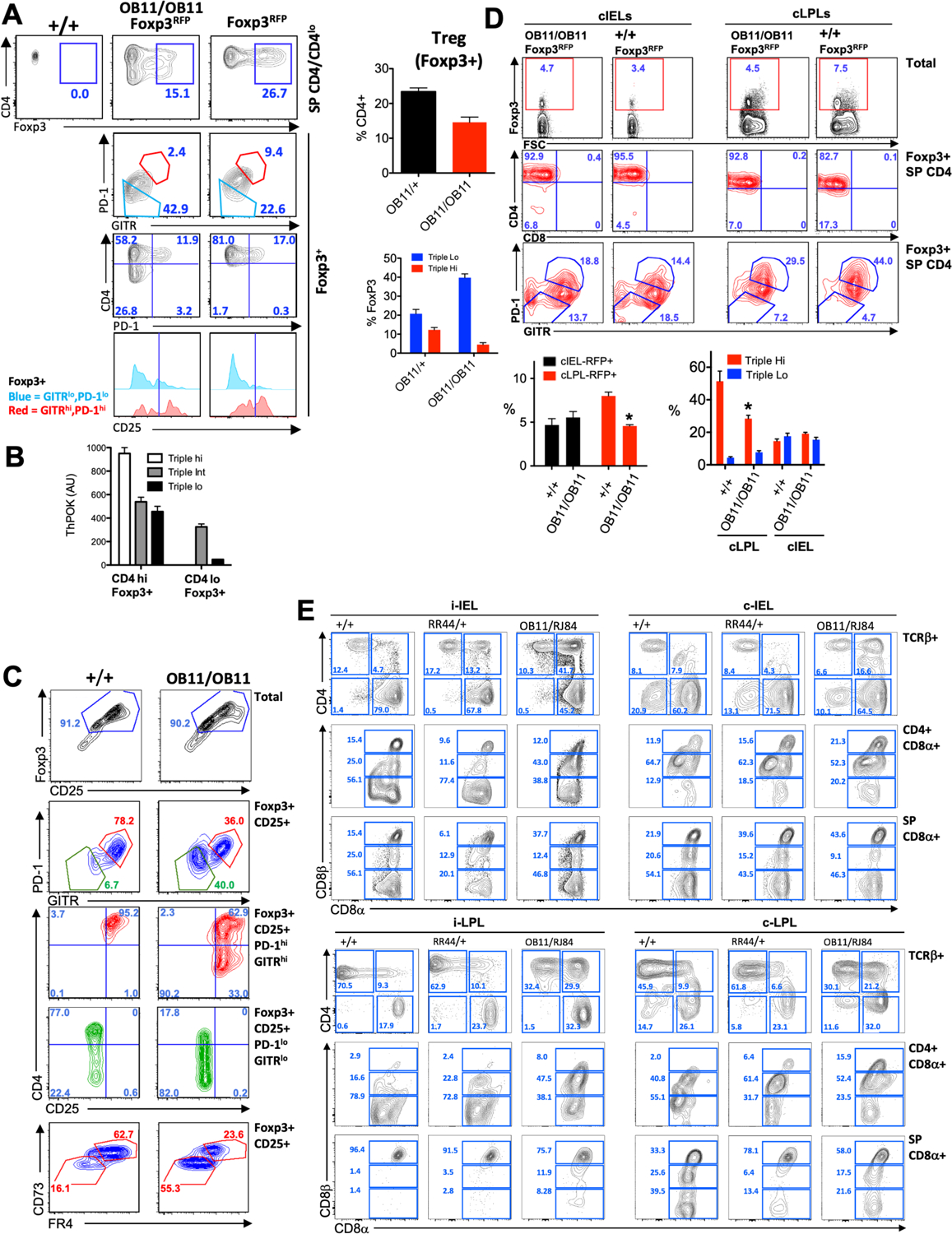
a) FACS analysis of Foxp3 versus CD4 (top row), PD-1 versus GITR (second row), CD4 versus CD25 (third row), or CD25 expression by gated GITRloPD-1lo or GITRhiPD-1hi LN (mesenteric) T cells. Bar graphs at right represent % of Tregs amongst total CD4 lymphocytes (top), or % of Triplelo and Triplehi cells amongst Foxp3+ CD4+ T cells, of ThPOKΔOB11/+ or ThPOKΔOB11/ΔOB11 mice. b) RNA was collected from WT CD4+ Foxp3+T cells subsetted into Triplehi, TripleINT and Triplelo subsets, as indicated, and assayed for ThPOK mRNA expression by qPCR. c) FACS analysis of Foxp3 versus CD25 (top row), PD-1 versus GITR (second row), CD4 versus CD25 (third, fourth rows), and CD73 versus FR4 expression for WT or ThPOKΔOB11/ΔOB11 CD4 LN T cells following in vitro Treg polarization. Plots are for total population or gated subsets, as indicated. d) FACS analysis of Foxp3 versus forward scatter (FSC) (top row), CD4 versus CD8α (second row), and PD-1 versus GITR (bottom row) by total or gated Foxp3+ cLPLs or cIELs T cells, as indicated. Bar graphs at bottom represent % of Tregs amongst total population (left), or % of Triplelo and Triplehi cells amongst gated T cells, of ThPOK +/+ or ThPOKΔOB11/ΔOB11 mice, as indicated. e) FACS analysis of CD4 versus CD8α (top row), or CD8α versus CD8β (second, third rows) expression, for either total TCRβ+ cells or indicated gated subset of freshly isolated iIEL, cIEL, iLPL or cLPL populations, as indicated. i-IEL =intestinal IEL; c-IEL = Colonic IEL; i-LPL = intestinal LPL, c-LPL = colonic -LPL.
Tregs play an important role in suppressing a variety of immune cells and actively preventing inflammatory bowel disease and food allergies. Importantly, intestinal Tregs cannot cross the lamina propria, but can interconvert to Foxp3−CD8αα+CD4+ intraepithelial lymphocytes (CD4+ IELs) by down-modulating ThPOK in a microbiota-dependent fashion and accumulate in the intestinal epithelium14. Since ablation of the ThPOK auto-regulatory loop leads to increase in Triplelo Tregs and Triplelo Tregs have been implicated previously in protection from colitis34, we hypothesized that physiological regulation of ThPOK via its autoregulatory loop might play an important role in maintaining Treg identity, such that disruption of the ThPOK autoregulatory loop would alter CD4 IEL formation from Treg. To test this hypothesis, we first examined intestinal Tregs and CD4 IEL populations in ThPOKΔOB11/ΔOB11 and WT mice. The proportion of Tregs among cIELs was mildly (1.5-fold) increased in ThPOKΔOB11/ΔOB11 versus WT mice, but substantially decreased (2-fold) among cLPLs (Fig. 6d), although in cLPL the representation of Triplelo cells was increased with concomitant decrease in Triplehi. We observed an increase in CD4+CD8α+CD8βlo (CD4IEL precursors) in lamina propria (>4 fold in Intestinal LPL and >3 fold increase in colonic LPL) of ThPOKΔOB11/ΔOB11 mice compared to WT mice (Fig. 6e). Hence, while CD4 IEL are only generated in the intestinal mucosa under physiological conditions, in the absence of the ThPOK autoregulatory loop they are generated before travelling to the intestinal epithelium. We also noted >2 fold increase in CD4-CD8 αα+TCR β+ T cells among IEL populations ThPOKΔOB11/ΔOB11 mice compared to WT.
Recent single cell transcriptomic analysis defined organ-specific gene expression signatures for Tregs transiting from the lymph node to barrier tissues including the colon38. We therefore tested whether ThPOK autoregulatory loop ablation may rewire the CD4 T cell gene expression program to favor conversion of OB11 CD4 T cells into colonic Treg by comparing gene expression profiles of OB11 T cell subsets with the reported gene signature of colonic Tregs. Strikingly, ThPOKΔOB11/ΔOB11 naïve CD4hi and DP T cells showed upmodulation of ~70% of the colon-specific Treg signature genes (Fig. 7a).
Fig. 7. Anti-silencer deficient CD4 T cells display anti-colitogenic activity.
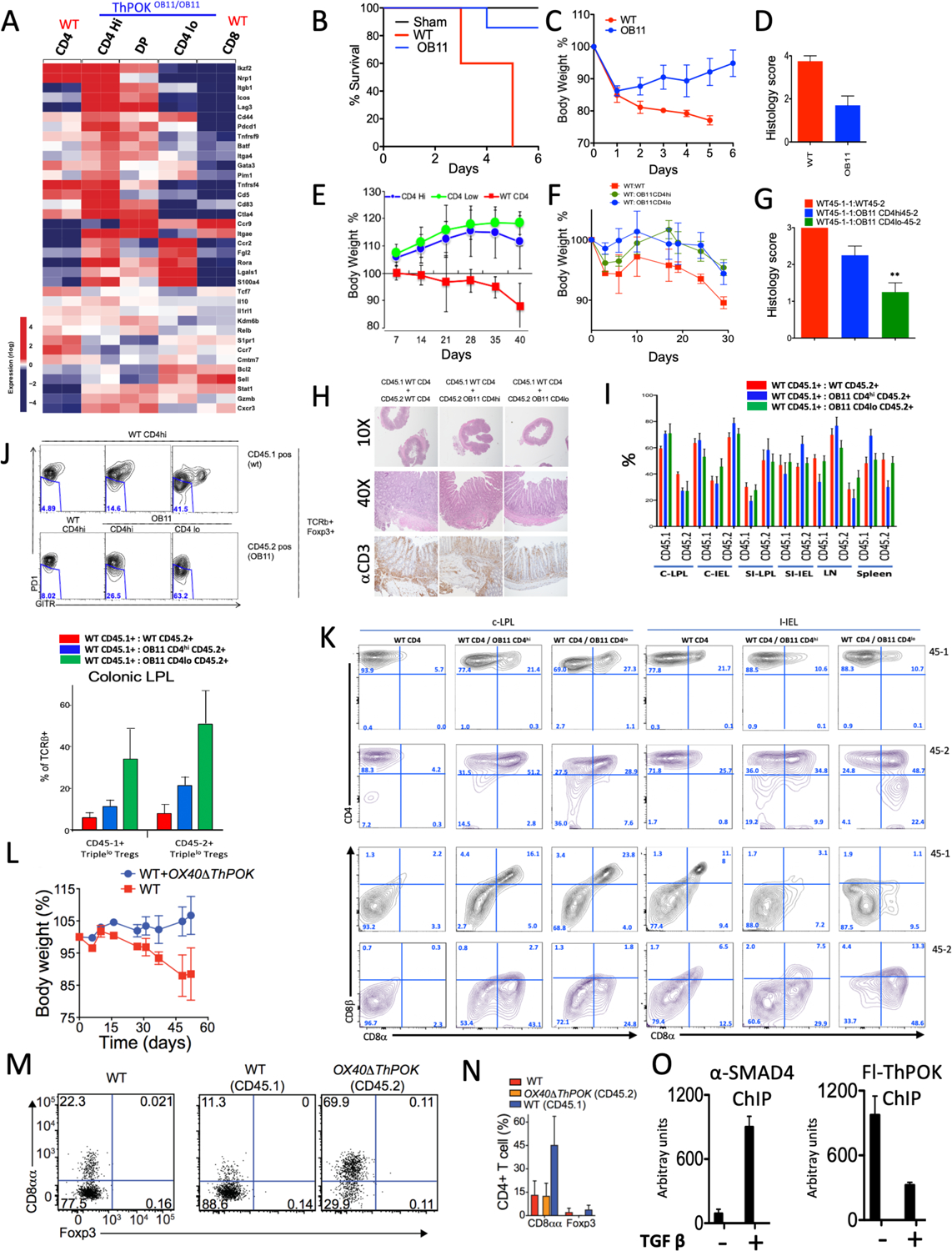
a) Heat map displaying relative expression of colonic Treg signature genes for indicated ThPOKΔOB11/ΔOB11 and WT T cell subsets. Red indicates increased gene expression levels; blue indicates decreased levels. b) Survival plot, c) Weight plot, and d) colitis severity score (based on colonic histology) after colitis induction with TNBS of indicated mouse strains. e) Weight plot of host RAG−/− mice after transfer of sorted naïve ThPOKΔOB11/ΔOB11 or WT T cell subsets, as indicated. f) Similar analysis as in panel e, except all animals received a cotransfer of WT CD4 T cells. g) Bar graph indicating colitis severity score for same mice as in panel f, at 30 day timepoint. h) Histopathological analysis of colon at day 30, from same experiment as panel f. i) Bar graphs depicting proportions of CD45.1+ and CD45.2+ cells for indicated cell subset for each cotransfer at day 30 (same experiment as panel f). j) FACS analysis of PD-1 versus GITR expression, for gated CD45.1+ or CD45.2+ T cell subset, from cotransfer experiment (panel f). Bar graph at bottom indicates proportions of Triplehi and Triplelo cells within gated Foxp3+ Tregs. k) FACS analysis of CD4, CD8α and CD8β expression, for gated TCRβ+ cells, from co-transfer experiment (panel f) from different gut locations. l) Weight plot of host RAG−/− mice after transfer of sorted OX40ΔThPOK or WT CD4 T cells, as indicated. m) FACS analysis of CD8a versus Foxp3 expression, for indicated mice (right panels from cotransfer experiment in panel l). n) Bar graph indicating proportions of CD8aa and Foxp3+ cells among CD4+ IELs (same experiment as panel l). o) SMAD4 and ThPOK binding to the silencer were determined by ChIP assay performed using anti SMAD4 antibody and anti Flag antibody for ThPOK in anti CD3/CD28 stimulated CD4 T cells, cultured (48h) in presence and absence of 2ng/ml of TGFβ.
Since, intestinal Tregs plays important role in prevention of colitis, we directly compared the functional response of ThPOKΔOB11/ΔOB11 mice and WT control mice to TNBS-induced colitis39. Strikingly, we found that while ThPOKΔOB11/ΔOB11 mice and WT control mice both showed similar initial pathology in response to TNBS treatment (weight loss, accompanied by multiple stress indicators such as lethargy and hunched posture), ThPOKΔOB11/ΔOB11 mice exhibited rapid recovery and improved longevity compared to WT controls (Fig. 7 b,c; Suppl. Fig 7e). To directly assess the anti-colitogenic potential of ThPOKΔOB11/ΔOB11 CD4 T cells, we next employed a T cell transfer model of colitis40. In striking contrast to naïve WT CD4 T cell transferred mice, both ThPOKΔOB11/ΔOB11 CD4hi and CD4lo subsets failed to induce colitis, although transferred T cells were readily detected in circulation. To test whether ThPOKΔOB11/ΔOB11 T cells might actually be anti-colitogenic in presence of inflammatory WT CD4, we conducted co-transfers of naïve WT (CD45.1+) and naïve ThPOKΔOB11/ΔOB11 (CD45.2+) T cells into the same hosts (Fig. 7f–h). Strikingly, this revealed that ThPOKΔOB11/ΔOB11 T cells could suppress inflammation in colon by naïve WT CD4 T cells (Fig. 7 f–h). FACS analysis of co-transferred mice reveals that in contrast to WT T cells, ThPOKΔOB11/ΔOB11 T cells readily converted to CD4+ CD8αα T cells and CD8αβ+ T cells (Fig. 7k). The frequency of Tregs was the same among both WT and ThPOKΔOB11/ΔOB11 co-transferred T cells in both intestinal and colonic lamina propria (data not shown). However, the ThPOKΔOB11/ΔOB11 Treg population in colonic lamina propria of co-transferred mice was remarkably enriched for Triplelo Treg cells in contrast to co-transferred WT (Fig. 7j). Of note, WT (CD45.1+) T cells from ThPOKΔOB11/ΔOB11 co-transferred mice were also enriched with Triplelo Treg cells (Fig. 7j).
Finally, to exclude any effect of altered peripheral environment on differentiation/function of ThPOKΔOB11/ΔOB11 T cells, we performed similar co-transfer experiment using naïve CD4 T cells from Ox40-Cre ThPOKFl/Fl mice in which ThPOK is conditionally ablated only in mature peripheral T cells. Consistent with our other results, we found that OX40ΔThPOK CD4 T cells can strongly suppress colitis induction by naïve WT CD4 T cells even at a 5:1 ratio (Fig. 7 l–o) accompanied by striking enhancement of CD4+CD8aa+ IEL generation.
Previously we have shown that TGF-β and RA are required for downmodulation of ThPOK expression and for CD8α expression by intestinal CD4+ T cells in vitro and in vivo13. This may suggest that under normal physiological circumstances TGFβ acts on CD4 T cells arriving in the gut microenvironment to promote SMAD binding to the SilThPOK, which in turn may interrupt the ThPOK autoregulatory loop, and thereby downmodulate ThPOK. Consistent with this possibility, our bioinformatic and Y1H analysis, as well as public Smad4 ChIP-seq data (GSM2706519), indicate presence of SMAD sites within the SilThPOK (data not shown). To directly determine whether TGF-β-mediated SMAD binding can disrupt ThPOK binding to the SilThPOK, we performed ChIP assay on sorted naïve CD4 cells from Fh-ThPOK homozygous mice stimulated for 48h with anti-CD3/CD28 in presence or absence of TGF-β. Indeed, we find that TGF-β treatment induced strong Smad4 binding to the SilThPOK and that this was accompanied by significant diminution of ThPOK binding to the OB11 region (Fig. 7o).
Altogether, our data suggest that under physiological conditions the ThPOK auto-regulatory loop inhibits untimely rewiring of circulating naïve CD4 T cells Tregs towards highly suppressive barrier-specific Treg.
Discussion
In this study we identify a novel regulatory switch, i.e. an anti-silencer located within the SilThPOK, which is necessary for maintenance of helper T cell identity. Genetic disruption of anti-silencer function leads to phenotypic plasticity, drastic change in genome-wide chromosomal accessibility, disregulated gene expression in CD4 T cells, and adoption of a metastable state. Our study provides the first evidence that the SilThPOK silencer encodes distinct regulatory modules that promote or oppose gene silencing. Furthermore, the anti-silencer module we report is conserved in evolution in mammals, implying a conserved function in regulation of peripheral CD4 T lymphocyte identity across mammalian species. Using in vivo colitis and in vitro Treg cell culture models, we further demonstrate a crucial role of this autoregulatory loop for tissue-specific Treg differentiation and maintenance of Treg integrity in the intestinal microenvironment to prevent premature CD4 IEL generation.
Our results suggest that the anti-silencer mediates recruitment of ThPOK, thereby driving a positive feedback loop that stabilizes ThPOK transcription and maintains T helper integrity. Ablation of the anti-silencer disrupts this loop, such that ThPOK expression level is insufficient to maintain T helper integrity. It’s increasingly understood that genes alternate between periods of transcriptional bursts and transcriptional inactivity22, 23, 41, 42. Given that a significant fraction of adoptively transferred OB11 T cells express GFP even many weeks after transfer, we propose that the interval between transcriptional bursts is greatly elongated (compared to the wt ThPOK allele). This could be directly tested in future using live cell and gene imaging approaches.
The metastable state induced by anti-silencer ablation is characterized by 3 distinct cellular phenotypes, CD4hi, CD4lo and DP, each with divergent gene expression profiles. The metastable state is maintained over time, as shown by persistence of all 3 subsets many weeks after adoptive transfer, but is also highly plastic, as evidenced by interconversion of different subsets following adoptive transfer. Finally, permanent escape from the metastable condition to the CD8 state is possible, since CD4hi and CD4lo cells can give rise to CD8 T cells, but not vice versa. It will be interesting to determine whether interconversion between subsets and escape to the CD8 fate are determined stochastically or by external stimuli. The metastable state is apparent in coexistence of multiple distinct lineage- and stage- gene expression signatures in ThPOKΔOB11/ΔOB11 T cell subsets, as exemplified by derepression of ETP-DN2 thymocyte (Kit, Meis1, Pgk1, Flt3, Id2 etc;25), and granulocyte-biased DN thymocyte gene programs (Fcgr1, MPO, Lyz2 etc)44, 45.
The question arises as to how this metastable state is achieved at the level of chromatin organization and transcriptional regulation. First, we find that autoregulatory loop-dependent ThPOK expression targets SE accessibility and activity. Secondly ThPOK controls chromatin accessibility by directly targeting lamina-associated genes (Lmnb1, Lmna, Lbr etc), as well as key mediators of transcriptional silencing (e.g. Satb1, Rreb1, Hdac1/7/10, Tle1, Bcor, Etv3, Dnmt3a, Kdm1 etc.), causing widespread transcriptional derepression. Thirdly, we find genome-wide association of ThPOK binding sites with CTCF consensus motifs, suggesting that ThPOK may functionally interact with CTCF in limiting spread of closed chromatin, consistent with overall reduction in accessible chromatin in ThPOKΔOB11/ΔOB11 T cells revealed by our ATAC-seq analysis.
Fourthly, enrichment of Runx and PU.1 binding motifs in accessible chromatin regions in ThPOKΔOB11/ΔOB11 T cells suggests that absence of ThPOK binding may unmask these sites and promote Runx and PU.1 binding, thereby contributing to multilineage gene derepression. Indeed, the chromatin accessibility pattern of ThPOKΔOB11/ΔOB11 T cells strikingly resembles that of the early T cell progenitor stage (ETP), suggesting that the ThPOK autoregulatory loop plays a key role in preventing de-dedifferentiation to the early T cell progenitor stage.
Survival of naive T cells in the quiescent state requires T cell receptor (TCR) “tickling” by self-MHC46. ThPOK-mediated auto regulatory loop is required to maintain the quiescent state of naïve CD4 T cells as evidenced by derepression of effector cytokine expression, and dispensability of co-stimulation for activation and proliferation of naïve ThPOKΔOB11/ΔOB11 T cells. Active TGF-β signaling has been implicated in maintaining the quiescent state in naive T cells downstream of TCR teasing47. Our RNA seq data suggests that absence of ThPOK auto regulatory loop leads to down-modulation of TGFβR as well as genes involved in restraining TCR signaling. Accordingly, ThPOKΔOB11/ΔOB11 T cells exhibit an activated phenotype, indicating an important role of the ThPOK-mediated auto regulatory loop in maintaining these processes to prevent T cell hyperactivation and autoimmunity. ThPOKΔOB11/ΔOB11 T cell subsets also exhibit upmodulation of genes related to cytotoxic CD4 T cells (ThCTL) obtained from influenza-infected lungs and tumor sites (e.g. Anxa1, Id2, Lat2, Nkg7 and Eomes)48–53, suggesting a key role of anti-silencer dependent ThPOK autoregulatory loop in preventing inappropriate cytotoxic CD4T cell generation.
Tregs and CD4 IEL play major roles in enteric immunity. Upon arrival in the gut Tregs convert to CD4 IELs by down-modulating ThPOK in a microbiota-dependent fashion14, which is a crucial step for maintenance of gut immune-homeostasis. Our study reveals that the ThPOK-mediated autoregulatory loop plays an important role in controlling enteric immune homeostasis. Specifically, we find that abrogation of the ThPOK autoregulatory loop promotes selective differentiation of naive CD4 T cells into anticolitogenic Triplelo Tregs in vitro and in vivo and enrichment of CD4+ IELs in the gut, thereby offering dominant protection from colitis. Indeed, naïve ThPOKΔOB11/ΔOB11 CD4hi and DP cells shows strong bias towards the colonic Treg-specific gene expression signature, while CD4lo cells are more biased towards the CD4 IEL gene signature (Ccl5, Nkg7, Gzm, Cd160, Itgae, Cd244, Cd7). TGF-β has been proposed as a major mediator of ThPOK downmodulation imparting plasticity of the gut homing T cell14. Here we find TGF-β-induced SMAD binding to SilThPOK is able to interrupt the ThPOK autoregulatory loop leading to downmodulation of ThPOK and IEL conversion. Recent findings from our lab suggest that TCR signaling plays a major role in Treg to IEL conversion (in press). Future experiments will address how modulation of ThPOK expression affects TCR signaling to ensure selective Treg repertoire-specific conversion to IEL.
We conclude that perturbation of the ThPOK autoregulatory feedback loop is a physiological mechanism for the post-thymic generation of Treg and CD8aa IEL in the colon and potentially other mucosal barriers. This controls a delicate balance between protective immunity and absence of tissue-destructive autoimmune responses. ThPOK misregulation via interruption of this autoregulatory loop may be responsible for other pathological conditions, e.g. development of auto‐aggressive pathogenic T cells in an IDDM model characterized by an unusual diabetogenic CD4lo T cell subset53–54, and appearance of double-positive CD4+/CD8+ T cells in HIV-infected individuals55.
Methods
Mice:
All experimentation involving animals was approved by Institutional Animal Care and Use Committee (IACUC), Fox Chase Cancer Center. RAG1 −/−, β2m−/−, Foxp3IRES-mRFP, B6.SJL-PtprcaPepcb, Zbtb7bFl have been procured from Jackson Laboratory. All other mouse lines described in this paper have been generated by the FCCC Transgenic Facility. Animal care was in accordance with NIH guidelines.
ZFN-mediated gene targeting in mouse embryos:
Site-specific mutagenesis was carried out as described7. Briefly a pair of ZFN RNAs that recognize a specific target site was designed and generated by Millipore-Sigma (Genome Editing division). mRNAs encoding the 2 site-specific ZFNs (50ng/μl) were introduced into 1-cell mouse oocytes by pronuclear injection, and injected oocytes were transferred to a pseudopregnant surrogate mother. Positive founder pups were identified using mutation-specific primers, and mated to C57BL/6 mice to generate stable heritable knockin lines.
Antibodies:
All fluorescently labeled antibodies used were obtained from commercial sources (eBioscience or Biolegend), including anti-Thy1, TCRβ, γδTCR, CD4, CD8α, CD8β, CD69, HSA, CD62L, CD45.1, CD45.2, Foxp3, CD25, GITR, PD1. Anti SMAD4 procured from ABCAM
EMSA.
Nuclear extracts were prepared from human embryonic kidney 293T cells transfected with Flag tagged murine ThPOK constructs cloned into the pcDNA3 expression vector as described. Negative controls included nuclear extracts from cells transfected with vector alone. ThPOK expression was verified by immunoblot analysis (data not shown) and used as a ThPOK protein sourcefor binding assay. DNA-binding probes were generated by annealing of synthetic double-stranded oligonucleotides corresponding to the OB11 region and end-labeling with polynucleotide kinase and digoxigenin-11-ddUTP using EMSA Kit (Sigma). The anti–Flag Ab (Sigma) was used for ‘supershifting’ of ThPOK protein–DNA complexes.
Quantitative RT-PCR:
qPCR was performed as previously described7.
Th Polarization.
Spleen and lymph node cells were FACS sorted for CD44-CD69-CD62lhi naïve CD4+ T cells. Cells were activated with 5 μg/mL plate-bound anti-CD3/CD28 antibodies (Biolegend, San Diego, CA) with IL-2 (25 IU/ml). For Th0 conditions, anti–IL-4 (11B11; 20 μg/ml) and anti–IFN-γ (20 μg/ml) were added; for Th1 conditions, anti–IL-4 (11B11; 20 μg/ml) and IL-12 (10 ng/ml) were added; for Th2 conditions, anti–IFN-γ (20 μg/ml) and IL-4 (20 ng/ml) were added; for Th9 conditions, anti–IFN-γ (20 μg/ml), IL-4 (20 ng/ml) and TGF-β (2 ng/ml) were added; for Th17 conditions, IL-6 (10 ng/ml), TGF-β (2 ng/ml), anti–IFN-γ (20 μg/ml) and IL-4 (20 ng/ml) were added. For Treg polarization, TGF-β (10 ng/ml), IL-2 (100 U/ml), anti–IFN-γ (20 μg/ml) and IL-4 (20 ng/ml) were added. Mouse IL-12, and IL-4 were from PeproTech, TGF-β and IL-6 were from R&D Systems, human IL-2 was from Roche, and neutralizing antibodies to mouse IFN-γ (XMG1.2) and IL-4 (11B11) were from BD Bioscience. Cells were cultured for 5 days in RPMI medium 1640 containing 10 mM Hepes (pH 7.0), 10% (v/v) FBS, 2 mM L-glutamine, antibiotics (complete medium), and 50 μM β-Mercaptoethanol.
Proliferation assays:
FACS-sorted T cell subsets were incubated with 5-(and 6-) carboxyfluorescein diacetate succinimidyl ester (CFSE; Dojindo; 5 μM) in PBS (37°C; 15 min). CFSE-labeled cells were cultured in plate-bound anti-TCRβ or antiCD3/CD28 antibodies for 1 to 4 days prior to FACS analysis. Generation analysis was performed using FlowJo Software.
Intracellular staining:
Cells were fixed in 100 μl of Cytofix/Cytoperm solution for 30min at 4°C, washed 2X two times in Perm/Wash solution, pelleted by centrifugation and resuspended in 100 μl of Perm/Wash solution with or without (FMO control) fluorochrome-conjugated antibody at room temperature, using the BD Fixation/Permeabilization Solution Kit (Cat. No. 554714), according to manufacturer’s instructions.
Chip.
1×106 thymocyte or naïve (SP CD62lhiCD44lo) peripheral T cell subsets were purified by FACS from compound heterozygous ThPOKFH/ΔOB11 mice. Chromatin crosslinking and immunoprecipitation were performed using the iDeal ChIP-seq kit for Transcription Factors (Diagenode) according to manufacturer’s protocol. The anti-Flag and anti Runx3 (Abcam, Cat#11905) antibody was used for immunoprecipitation and purified DNA sequences were analyzed by qPCR using WT allele (F: GGCGCGCAGTTATAAATAG, R: CCCCTACCGCGACCGCCCAA) and OB11 allele-specific (F:CAGT TATAAATAGAGGCTT, R: CTGCCTCCGCTTCCCTCGAA) primers.
ATAC-seq.
5 × 104 flow sorted naïve CD4 cells from WT or CD4lo cells from OB11 mice were pelleted at 500 g for 5 min at 4 °C, and whole cell pellets resuspended in 50μL ATAC-RSB buffer (10 mM Tris-HCl pH 7.4 10 mM NaCl; 3 mM MgCl2, 0.1% Igepal CA-630), 0.1% Tween 20 (Sigma), and 0.01% Digitonin, and kept on ice for 3 min. 1ml of cold ATAC-RSB + 0.1% Tween20 was added, and samples centrifuged 500g for 10 min at 4°c (fixed angle rotor) to obtain nuclear pellet. Transposase reaction of open chromatin was achieved by resuspending free nuclei in tagmentation mix (22.5 μL Tagment DNA Buffer; 2.5 μL Tagment DNA enzyme; 25μL H2O) (Illumina, FC-121- 1030) and incubating at 37 °C for 30 min. Purification of DNA was performed with Diapure kit (Diagenode), according to the manufacturer’s protocol. Barcoding and amplification was performed using Nextera Index Kit (Illumina, FC-121-1011), as previously described56. Amplified ATAC-seq libraries were purified using Gene-Read Size Selection Kit (Qiagen, 180514) according to the manufacturer’s protocol. Quality and quantity of final ATAC-seq libraries were assessed with the High Sensitivity DNA kit (Agilent, 5067-4626) run on an Agilent 2100 Bioanalyzer. ATAC-seq libraries were sequenced using Illumina 125-bp paired-ends sequencing on a HiSeq2500 platform with, generating between 38 and 43 million reads per condition per biological replicate.
Following quality control by FastQC (URL:https://www.bioinformatics.babraham.ac.uk/projects/:fastqc/), ATAC-Seq reads after were aligned to mouse genome (mm10) using BoWTie2 (PMID: 22388286). Samples were filtered for regions blacklisted by the ENCODE project57 and deduplicated using Picard tools (http://broadinstitute.github.io/picard). Alignment coordinates were converted to BED format using BEDTools v.2.1758 and peak calling was performed using MACS255 with default parameters. To visualize the relative occupancy of ThPOK along with ATAC-Seq peaks and the relative relationship to genes we binned the chromosomes into consequtive 10Kb regions and plotted the read distribution of ATAC-Seq and ThPOK ChIP-Seq data.
ChIP-Seq data analysis.
We obtained ThPOK ChIP-seq data from Vacchio et al.26 from Gene Expression Omnibus (GEO) (GSE116506). ChIP-Seq reads were aligned to mouse genome (mm10) using BWA aligner59. Peak calling was performed using MACS260. Similarly, CD8 Tcell Runx3 ChIP-Seq peaks were obtained from Istaces et al.27 through GEO (GSM3559330). Peak annotations and motif enrichment analyses were performed using HOMER tools61.
Bulk RNA-seq.
For each population, 105 cells were sorted into RLT lysis buffer (Qiagen) containing 1% BME and total RNA purified using the RNA Microprep kit (Zymo Research). All resulting RNA was used as an input for complementary DNA synthesis using the SMART-Seq v.4 kit (Takara Bio) and 10 cycles of PCR amplification. Next, 1 ng cDNA was converted to a sequencing library using the NexteraXT DNA Library Prep Kit and NexteraXT indexing primers (Illumina) with 10 additional cycles of PCR. Final libraries were pooled at equimolar ratios and sequenced on a HiSeq2500 using 100-bp paired-end sequencing. Quality controlled reads after FastQC were aligned to mouse genome (mm10) using Tophat2 (PMID: 23618408). For counting reads from the resulting BAM files for ATAC-Seq, ChIP-Seq and RNA-Seq, HTSeq62 was used with default parameters. To identify differentially expressed genes (DEG), we applied Deseq2 algorithm63. Genes with an FDR < 0.05 and absolute log2(FC) > 1 and < −1 were considered to be significant. Enrichment analyses were done using DAVID bioinformatics resource64.
Colitis models.
Adoptive T cell transfer model: Colitis was induced after transfer of 5 × 105 sorted naive T cells into Rag1−/− mice65, as previously described61. Recipient mice were monitored regularly for signs of disease, including weight loss, hunched appearance, piloerection of the coat, and diarrhea, and analyzed at various times after the initial transfer or when they reached 90% of their initial weight.
TNBS colitis model: Colitis was induced by transfer of intra rectal transfer of TNBS as previously described39. Recipient mice were monitored regularly for signs of disease, including weight loss, hunched appearance, piloerection of the coat, and diarrhea, and analyzed at various times after the initial transfer or when they reached 80% of their initial weight.
3C Assay:
Quantitative analysis of chromosome conformation capture assays has been performed following the proto described earlier66 using 4 BP cutter NlaIII and ThPOK BAC plasmid was used as the positive control.
Supplementary Material
Suppl. Fig. 1. Ablation of anti-silencer does not substantially disrupt development of class II-restricted thymocytes. a) FACS analysis of CD4, and CD8a expression in total thymocytes (top row), or gated mature (TCRβ+ CD69− CD24− CD62L+) thymocytes of WT mice and homozygous ThPOKΔOB11/ΔOB11 mice, as indicated. Plots at right indicate % of SP CD4, CD8, and CD4+ 8+ (DP) thymocytes among total thymocytes (top panel), or gated mature thymocytes (as defined in panel a) (n = 3, for each strain). Error bars represent standard deviations. b) RNA was collected from freshly sorted thymocyte subsets, as indicated, before probing for ThPOK expression by qPCR. c) FACS analysis of TCRβ and CD69 expression in total thymocytes (top row), or gated mature thymocytes of β2m−/− ThPOK+/+ and β2m−/− ThPOKDΔOB11/ΔOB11 mice, as indicated.
Suppl. Fig. 2. Functional analysis of ThPOK binding sites in anti-silencer. a) EMSA analysis, using the 100bp OB11 region (lanes 1–4) or EBNA control DNA (lanes 5,6) as probes. Biotinylated probes were incubated with cell extracts from NIH 3T3 cells transfected with empty vector, Fl-ThPOK or EBNA expression constructs, as indicated. In some lanes mutant OB11 probes in which the consensus ThPOK binding sites 1 (lane 4) or 3 (lane 3) are disrupted were added. b) Organization of mouse ThPOK gene (top), and diagram of silencer deletion mutants (bottom). Black boxes, blue boxes, arrows, and red boxes indicate exons, enhancers, silencers and promoters, respectively. Deletions within the silencer are indicated by thin black lines. “R” indicates positions of conserved Runx binding sites. c) FACS analysis of CD4, and CD8a expression in gated TCRβ+ PBLs of WT mice and homozygous mutant lines, as designated in panel b. d) Sequence of WT murine OB11 region (top row) aligned with corresponding human sequence (second row; mismatches between human and mouse are highlighted in yellow), as well as sequences of indicated mutants used in EMSA or for generation of mutant alleles.
Suppl. Fig. 3. Ablation of anti-silencer leads to transient transcription at ThPOK locus. a) FACS analysis of GFP expression in gated CD4hi (green), CD4lo/− (orange), DP (CD4+ CD8+) (red), or CD8 PBLs (middle) of ThPOKGFP/+ or ThPOKGFP/ΔOB11 mice. Black line indicates MFI of GFP expression by ThPOKGFP/+ CD4 T cells, for comparison (same mice as in Fig. 3c). b) FACS analysis of GFP expression by gated CD4hi, CD4lo/−, CD4+ CD8+ (DP), and CD8 PBLs from RAG−/− hosts reconstituted with indicated sorted T cell subsets from ThPOKGFP/+ or ThPOKΔOB11.GFP/ΔOB11 mice, as indicated (same mice as in Fig. 3e).
Suppl. Fig. 4. Genes misregulated upon anti-silencer ablation are often direct targets of ThPOK and Runx3 binding. a) Heat map displaying relative expression of genes associated with ETP-DN2 transition for indicated ThPOKΔOB11/ΔOB11 and WT T cell subsets. Red indicates increased gene expression levels; blue indicates decreased levels. b) Volcano plots illustrating gene expression differences between individual sorted ThPOKΔOB11/ΔOB11 subsets and WT CD4 T cells. The grey dots represent genes differentially expressed (adjusted P<0.05) between samples. Genes with the largest negative or positive standardized mean difference are marked. Note diminished ThPOK (Zbt7b) expression in all samples. c) ThPOK-ChIP and Runx3-Chip profiles and Refseq gene positions across chromosome regions containing Runx3, CD4 and ThPOK (Zbtb7b) genes, as marked. Input is already subtracted for Runx3 peaks. d) Venn diagram indicating intersection between ThPOK and Runx3 ChIP-seq peaks (from WT CD4 and WT CD8 cells, respectively).
Suppl. Fig. 5. Altered chromatin accessibility in ThPOKΔOB11/ ΔOB11 CD4lo T cells. a) Comparison of distribution of RNA-seq reads (top row), ATAC-seq reads (second row, top), genes (second row, bottom), and ThPOK-ChIP seq reads per 100kb bins across one chromosome (Chr11). b) TF consensus motif enrichment in DACRs selectively open in ThPOKΔOB11/ ΔOB11 CD4lo T cells or WT CD4 T cells, as indicated. c) Venn diagram illustrating intersection between super-enhancers from Th cells, thymocytes and proB cells, as indicated.
Suppl. Fig. 6. Effect Anti-silencer element ablation on CD4 T cell proliferation and cytokine gene expression. a) Bar graph denoting the percentage of cells assigned to each division cycle based on CFSE dilution following stimulation with anti-TCRβ (bottom) or anti-CD3ε/CD28 (top) for 48hrs. b) Time course of phospho-ERK expression in response to anti-TCRβ stimulation of indicated WT or OB11 T cell subsets. c-d) Bar graphs illustrating deregulated expression of indicated mRNAs according to qPCR. Sorted OB11 or WT T cell subsets were cultured with anti-TCRβ or anti-CD3ε/CD28 stimulation, or in the absence of stimulation, as indicated. e) ThPOK-ChIP-seq (for WT CD4 T cells, biological triplicates; pink tracks), and ATAC-seq (for ThPOKΔOB11/ΔOB11 CD4lo T cells or WT CD4 T cells, as indicated, biological duplicates; grey tracks) profiles for gene loci.
Suppl. Fig. 7. Colitis induction in WT and ThPOKΔOB11/ΔOB11 mice. a-d) Bar graphs depicting the percentage of CD45.1+ (a, c) or CD45.2+ (b, d) cells within each indicated intestinal or splenic population. Data is derived from cotransfer experiment depicted in Fig. 7f. Error bars represent standard deviations of at least 3 biological replicates. e) Image of whole colons, or stained cross-sections of the indicated mouse strains after colitis induction with TNBS.
Suppl. Table 1. a) List of genes that are bound by ThPOK and Runx3 (according to ChIP-seq), and are at least 2-fold down- (DN) or up-modulated (UP) in all ThPOKΔOB11/ΔOB11 T cell subsets (see Fig. 4a). b) List of genes that are bound by ThPOK but NOT Runx3 (according to ChIP-seq), and are at least 2-fold down- (DN) or up-modulated (UP) in all ThPOKΔOB11/ΔOB11 T cell subsets (see Fig. 4a). Pink boxes indicate genes implicated in regulation of TCR signaling, especially dampening of TCR signaling.
Suppl. Table 2. a) DAVID gene set analysis showing enriched pathways (KEGG) or functional categories (GO term) for genes that are bound by ThPOK and Runx3 (according to ChIP-seq), and are at least 2-fold down- (DN) or up-modulated (UP) in all ThPOKΔOB11/ΔOB11 T cell subsets (see Fig. 4a). b) DAVID gene set analysis showing enriched pathways (KEGG) or functional categories (GO term) for genes that are are bound by ThPOK but NOT Runx3 (according to ChIP-seq), and are at least 2-fold down- (DN) or up-modulated (UP) in all ThPOKΔOB11/ΔOB11 T cell subsets (see Fig. 4a).
ACKNOWLEDGEMENTS:
This work was supported by NIH grants R01 AI068907 (DJK), R01 GM107179 (DJK), R01 GM082971 (JFB & AJMW), R01CA227629 (SG), R01CA218133 (SG) and P30 CA006927 (FCCC Comprehensive Cancer Center Core Grant). We acknowledge the assistance of the following core facilities of the Fox Chase Cancer Center: Flow Cytometry, Cell Culture, DNA Sequencing, Genomic and Laboratory Animal. We are grateful to D.L. Wiest and G. Rall for critical comments and suggestions on the manuscript.
References
- 1.Etzensperger R et al. Identification of lineage-specifying cytokines that signal all CD8(+)-cytotoxic-lineage-fate ‘decisions’ in the thymus. Nat. Immunol 18, 1218–1227 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dave VP, Allman D, Keefe R, Hardy RR & Kappes DJ HD mice: a novel mouse mutant with a specific defect in the generation of CD4(+) T cells. Proc. Natl. Acad. Sci. U.S.A 95, 8187–8192 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He X et al. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature 433, 826–833 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Keefe R, Dave V, Allman D, Wiest D & Kappes DJ Regulation of lineage commitment distinct from positive selection. Science 286, 1149–1153 (1999). [DOI] [PubMed] [Google Scholar]
- 5.He X et al. CD4-CD8 lineage commitment is regulated by a silencer element at the ThPOK transcription-factor locus. Immunity 28, 346–358 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Setoguchi R et al. Repression of the transcription factor Th-POK by Runx complexes in cytotoxic T cell development. Science 319, 822–825 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Mookerjee-Basu J et al. Functional conservation of a developmental switch in mammals since the Jurassic age. Mol. Biol. Evol 36, 39–53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grueter B et al. Runx3 regulates integrin alpha E/CD103 and CD4 expression during development of CD4−/CD8+ T cells. J. Immunol 175, 1694–1705 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Wang L et al. The zinc finger transcription factor Zbtb7b represses CD8-lineage gene expression in peripheral CD4+ T cells. Immunity 29, 876–887 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vacchio MS et al. A ThPOK-LRF transcriptional node maintains the integrity and effector potential of post-thymic CD4+ T cells. Nat. Immunol 15, 947–956 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciucci T et al. The emergence and functional fitness of memory CD4(+) T cells require the transcription factor Thpok. Immunity 50, 91–105 e104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muroi S et al. Cascading suppression of transcriptional silencers by ThPOK seals helper T cell fate. Nat. Immunol 9, 1113–1121 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Mucida D et al. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat. Immunol 14, 281–289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sujino T et al. Tissue adaptation of regulatory and intraepithelial CD4(+) T cells controls gut inflammation. Science 352, 1581–1586 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka H et al. Epigenetic Thpok silencing limits the time window to choose CD4(+) helper-lineage fate in the thymus. EMBO J. 32, 1183–1194 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I & Mucida D Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nat. Immunol 14, 271–280 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuxman Bass JI et al. Human gene-centered transcription factor networks for enhancers and disease variants. Cell 161, 661–673 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reece-Hoyes JS et al. Yeast one-hybrid assays for gene-centered human gene regulatory network mapping. Nat. Methods 8, 1050–1052 (2011a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reece-Hoyes JS et al. Enhanced yeast one-hybrid assays for high-throughput gene-centered regulatory network mapping. Nat. Methods 8, 1059–1064 (2011b). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boursalian TE, Golob J, Soper DM, Cooper CJ & Fink PJ Continued maturation of thymic emigrants in the periphery. Nat. Immunol 5, 418–425 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Corish & Tyler. Protein Engineering, Design and Selection (1999).
- 22.Larsson AJM et al. Genomic encoding of transcriptional burst kinetics. Nature 565, 251–254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suter DM et al. Mammalian genes are transcribed with widely different bursting kinetics. Science 332, 472–474 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Stefanova I et al. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol 4, 248–254 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Zhou W et al. Single-cell analysis reveals regulatory gene expression dynamics leading to lineage commitment in early T cell development. Cell Syst. 9, 321–337 e329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vacchio MS et al. A Thpok-directed transcriptional circuitry promotes Bcl6 and Maf expression to orchestrate T follicular helper differentiation. Immunity 51, 465–478 e466 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Istaces N et al. EOMES interacts with RUNX3 and BRG1 to promote innate memory cell formation through epigenetic reprogramming. Nat. Commun 10, 3306 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang JA, Mortazavi A, Williams BA, Wold BJ & Rothenberg EV Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell 149, 467–482 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng S, Papalexi E, Butler A, Stephenson W & Satija R Molecular transitions in early progenitors during human cord blood hematopoiesis. Mol. Syst. Biol 14, e8041 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diao & Pipkin. F1000Research (F1000 Faculty Rev) 8, 1278 (2019). [Google Scholar]
- 31.Chen H et al. Single-cell trajectories reconstruction, exploration and mapping of omics data with STREAM. Nat. Commun 10, 1903 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whyte WA. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 53:307 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vahedi G et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature. 520: 558 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan A, Zhang X. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 44: D164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snook JP, Kim C & Williams MA TCR signal strength controls the differentiation of CD4(+) effector and memory T cells. Sci. Immunol 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carpenter AC et al. Control of regulatory T cell differentiation by the transcription factors Thpok and LRF. J. Immunol 199, 1716–1728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyss L et al. Affinity for self antigen selects Treg cells with distinct functional properties. Nat. Immunol 17, 1093–1101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miragaia RJ et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity 50, 493–504 e497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neurath M, Fuss I, Strober W TNBS-colitis Int Rev Immunol. 19: 51 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Powrie F et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562 (1994). [DOI] [PubMed] [Google Scholar]
- 41.Chubb JR, Trcek T, Shenoy SM & Singer RH Transcriptional pulsing of a developmental gene. Curr. Biol 16, 1018–1025 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raj A, Peskin CS, Tranchina D, Vargas DY & Tyagi S Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 4, e309 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balazsi G, van Oudenaarden A & Collins JJ Cellular decision making and biological noise: from microbes to mammals. Cell 144, 910–925 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Obaldia ME, Bell JJ & Bhandoola A Early T-cell progenitors are the major granulocyte precursors in the adult mouse thymus. Blood 121, 64–71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothenberg EV, Hosokawa H & Ungerback J Mechanisms of action of hematopoietic transcription factor PU.1 in initiation of T-cell development. Front Immunol. 10, 228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takada K & Jameson SC Naive T cell homeostasis: from awareness of space to a sense of place. Nat. Rev. Immunol 9, 823–832 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Tu E et al. T cell receptor-regulated TGF-beta Type I receptor expression determines T cell quiescence and activation. Immunity 48, 745–759 e746 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marshall NB et al. NKG2C/E marks the unique cytotoxic CD4 T cell subset, ThCTL, generated by influenza infection. J. Immunol 198, 1142–1155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pearce EL et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302, 1041–1043 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Curran MA et al. Systemic 4–1BB activation induces a novel T cell phenotype driven by high expression of Eomesodermin. J. Exp. Med 210, 743–755 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirschhorn-Cymerman D et al. Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J. Exp. Med 209, 2113–2126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qui HZ et al. CD134 plus CD137 dual costimulation induces Eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J. Immunol 187, 3555–3564 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner DH Jr. et al. Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc. Natl. Acad. Sci. U.S.A 99, 3782–3787 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waid DM, Vaitaitis GM & Wagner DH Jr. Peripheral CD4loCD40+ auto-aggressive T cell expansion during insulin-dependent diabetes mellitus. Eur J Immunol 34, 1488–1497 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Flamand L et al. Activation of CD8+ T lymphocytes through the T cell receptor turns on CD4 gene expression: implications for HIV pathogenesis. Proc. Natl. Acad. Sci. U.S.A 95, 3111–3116 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21 29 21–21 29 29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffman MM et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 41, 827–841 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quinlan AR BEDTools: The swiss-army tool for genome feature analysis. Curr. Protoc. Bioinformatics 47, 11 12 11–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H & Durbin R Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feng J, Liu T, Qin B, Zhang Y & Liu XS Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang da W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Mucida D et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317, 256–260 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Hagège H et al. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat Protoc. 2:1722 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig. 1. Ablation of anti-silencer does not substantially disrupt development of class II-restricted thymocytes. a) FACS analysis of CD4, and CD8a expression in total thymocytes (top row), or gated mature (TCRβ+ CD69− CD24− CD62L+) thymocytes of WT mice and homozygous ThPOKΔOB11/ΔOB11 mice, as indicated. Plots at right indicate % of SP CD4, CD8, and CD4+ 8+ (DP) thymocytes among total thymocytes (top panel), or gated mature thymocytes (as defined in panel a) (n = 3, for each strain). Error bars represent standard deviations. b) RNA was collected from freshly sorted thymocyte subsets, as indicated, before probing for ThPOK expression by qPCR. c) FACS analysis of TCRβ and CD69 expression in total thymocytes (top row), or gated mature thymocytes of β2m−/− ThPOK+/+ and β2m−/− ThPOKDΔOB11/ΔOB11 mice, as indicated.
Suppl. Fig. 2. Functional analysis of ThPOK binding sites in anti-silencer. a) EMSA analysis, using the 100bp OB11 region (lanes 1–4) or EBNA control DNA (lanes 5,6) as probes. Biotinylated probes were incubated with cell extracts from NIH 3T3 cells transfected with empty vector, Fl-ThPOK or EBNA expression constructs, as indicated. In some lanes mutant OB11 probes in which the consensus ThPOK binding sites 1 (lane 4) or 3 (lane 3) are disrupted were added. b) Organization of mouse ThPOK gene (top), and diagram of silencer deletion mutants (bottom). Black boxes, blue boxes, arrows, and red boxes indicate exons, enhancers, silencers and promoters, respectively. Deletions within the silencer are indicated by thin black lines. “R” indicates positions of conserved Runx binding sites. c) FACS analysis of CD4, and CD8a expression in gated TCRβ+ PBLs of WT mice and homozygous mutant lines, as designated in panel b. d) Sequence of WT murine OB11 region (top row) aligned with corresponding human sequence (second row; mismatches between human and mouse are highlighted in yellow), as well as sequences of indicated mutants used in EMSA or for generation of mutant alleles.
Suppl. Fig. 3. Ablation of anti-silencer leads to transient transcription at ThPOK locus. a) FACS analysis of GFP expression in gated CD4hi (green), CD4lo/− (orange), DP (CD4+ CD8+) (red), or CD8 PBLs (middle) of ThPOKGFP/+ or ThPOKGFP/ΔOB11 mice. Black line indicates MFI of GFP expression by ThPOKGFP/+ CD4 T cells, for comparison (same mice as in Fig. 3c). b) FACS analysis of GFP expression by gated CD4hi, CD4lo/−, CD4+ CD8+ (DP), and CD8 PBLs from RAG−/− hosts reconstituted with indicated sorted T cell subsets from ThPOKGFP/+ or ThPOKΔOB11.GFP/ΔOB11 mice, as indicated (same mice as in Fig. 3e).
Suppl. Fig. 4. Genes misregulated upon anti-silencer ablation are often direct targets of ThPOK and Runx3 binding. a) Heat map displaying relative expression of genes associated with ETP-DN2 transition for indicated ThPOKΔOB11/ΔOB11 and WT T cell subsets. Red indicates increased gene expression levels; blue indicates decreased levels. b) Volcano plots illustrating gene expression differences between individual sorted ThPOKΔOB11/ΔOB11 subsets and WT CD4 T cells. The grey dots represent genes differentially expressed (adjusted P<0.05) between samples. Genes with the largest negative or positive standardized mean difference are marked. Note diminished ThPOK (Zbt7b) expression in all samples. c) ThPOK-ChIP and Runx3-Chip profiles and Refseq gene positions across chromosome regions containing Runx3, CD4 and ThPOK (Zbtb7b) genes, as marked. Input is already subtracted for Runx3 peaks. d) Venn diagram indicating intersection between ThPOK and Runx3 ChIP-seq peaks (from WT CD4 and WT CD8 cells, respectively).
Suppl. Fig. 5. Altered chromatin accessibility in ThPOKΔOB11/ ΔOB11 CD4lo T cells. a) Comparison of distribution of RNA-seq reads (top row), ATAC-seq reads (second row, top), genes (second row, bottom), and ThPOK-ChIP seq reads per 100kb bins across one chromosome (Chr11). b) TF consensus motif enrichment in DACRs selectively open in ThPOKΔOB11/ ΔOB11 CD4lo T cells or WT CD4 T cells, as indicated. c) Venn diagram illustrating intersection between super-enhancers from Th cells, thymocytes and proB cells, as indicated.
Suppl. Fig. 6. Effect Anti-silencer element ablation on CD4 T cell proliferation and cytokine gene expression. a) Bar graph denoting the percentage of cells assigned to each division cycle based on CFSE dilution following stimulation with anti-TCRβ (bottom) or anti-CD3ε/CD28 (top) for 48hrs. b) Time course of phospho-ERK expression in response to anti-TCRβ stimulation of indicated WT or OB11 T cell subsets. c-d) Bar graphs illustrating deregulated expression of indicated mRNAs according to qPCR. Sorted OB11 or WT T cell subsets were cultured with anti-TCRβ or anti-CD3ε/CD28 stimulation, or in the absence of stimulation, as indicated. e) ThPOK-ChIP-seq (for WT CD4 T cells, biological triplicates; pink tracks), and ATAC-seq (for ThPOKΔOB11/ΔOB11 CD4lo T cells or WT CD4 T cells, as indicated, biological duplicates; grey tracks) profiles for gene loci.
Suppl. Fig. 7. Colitis induction in WT and ThPOKΔOB11/ΔOB11 mice. a-d) Bar graphs depicting the percentage of CD45.1+ (a, c) or CD45.2+ (b, d) cells within each indicated intestinal or splenic population. Data is derived from cotransfer experiment depicted in Fig. 7f. Error bars represent standard deviations of at least 3 biological replicates. e) Image of whole colons, or stained cross-sections of the indicated mouse strains after colitis induction with TNBS.
Suppl. Table 1. a) List of genes that are bound by ThPOK and Runx3 (according to ChIP-seq), and are at least 2-fold down- (DN) or up-modulated (UP) in all ThPOKΔOB11/ΔOB11 T cell subsets (see Fig. 4a). b) List of genes that are bound by ThPOK but NOT Runx3 (according to ChIP-seq), and are at least 2-fold down- (DN) or up-modulated (UP) in all ThPOKΔOB11/ΔOB11 T cell subsets (see Fig. 4a). Pink boxes indicate genes implicated in regulation of TCR signaling, especially dampening of TCR signaling.
Suppl. Table 2. a) DAVID gene set analysis showing enriched pathways (KEGG) or functional categories (GO term) for genes that are bound by ThPOK and Runx3 (according to ChIP-seq), and are at least 2-fold down- (DN) or up-modulated (UP) in all ThPOKΔOB11/ΔOB11 T cell subsets (see Fig. 4a). b) DAVID gene set analysis showing enriched pathways (KEGG) or functional categories (GO term) for genes that are are bound by ThPOK but NOT Runx3 (according to ChIP-seq), and are at least 2-fold down- (DN) or up-modulated (UP) in all ThPOKΔOB11/ΔOB11 T cell subsets (see Fig. 4a).