

Summary
Mutations in leucine-rich repeat kinase 2 (LRRK2) are commonly implicated in the pathogenesis of both familial and sporadic Parkinson’s disease (PD). LRRK2 regulates critical cellular processes at membranous organelles and forms microtubule-based pathogenic filaments, yet the molecular basis underlying these biological roles of LRRK2 remains largely enigmatic. Here we determined high-resolution structures of full-length human LRRK2, revealing its architecture and key interdomain scaffolding elements for rationalizing disease-causing mutations. The kinase domain of LRRK2 is captured in an inactive state, a conformation also adopted by the most common PD-associated mutation, LRRK2G2019S. This conformation serves as a framework for structure-guided design of conformational specific inhibitors. We further determined the structure of COR-mediated LRRK2 dimers and found that single-point mutations at the dimer interface abolished pathogenic filamentation in cells. Overall, our study provides mechanistic insights into physiological and pathological roles of LRRK2 and establishes a structural template for future therapeutic intervention in PD.
Introduction
Since the identification of LRRK2 mutations as a cause of inherited Parkinson’s disease (PD) (Paisan-Ruiz et al., 2004; Zimprich et al., 2004), the leucine-rich repeat kinase 2 (LRRK2) protein has been one of the central molecules in PD studies (Cookson, 2015; Domingos et al., 2019; Lee et al., 2012). Mutations in LRRK2 are the most frequent cause of inherited PD, accounting for at least 5% of familial and 1%-2% of idiopathic PD cases (Di Maio et al., 2018; Gilks et al., 2005; Monfrini and Di Fonzo, 2017). Studies have identified numerous pathogenic mutations in LRRK2. Generic variations on six sites are disease-causing and high-risk in PD: Asn1437, Arg1441, Tyr1699 and Ser1761 in the ROC–COR domains and Gly2019, Ile2020 in the kinase domain (Christensen et al., 2018). LRRK2G2019S, a missense mutation Gly2019Ser, with hyperactive kinase activity, is the most common one (Greggio and Cookson, 2009; Healy et al., 2008). In addition to PD, an LRRK2 mutation (N2081D) has also been identified by genome-wide association studies as a major susceptibility gene for Crohn’s disease (CD) (Hui et al., 2018). Despite the clinical relevance, the structure-function relationship underlying these disease-causing mutations is still elusive (Domingos et al., 2019).
LRRK2 is a 286-kDa multidomain protein that includes seven sequential domains: armadillo repeat motif (ARM), ankyrin repeat (ANK), leucine-rich repeat (LRR), ras-of-complex (ROC), C-terminal of ROC (COR), kinase (KIN) and WD40 domains. Much progress had been made in illustrating the structure of LRRK2 and its bacterial homolog, ROCO proteins (Deng et al., 2008; Deniston et al., 2020; Deyaert et al., 2019; Gilsbach et al., 2012; Gotthardt et al., 2008; Guaitoli et al., 2016; Sejwal et al., 2017; Zhang et al., 2019), including a recent in-situ cryo-electron tomography (cryoET) analysis of LRRK2 filaments in association with microtubules (Watanabe et al., 2020). Yet molecular details underlying the functional assembly of the seven domains and their functional roles remain mostly unknown due to the lack of a high-resolution full-length LRRK2 structure.
LRRK2 forms quaternary structures in vivo and in vitro through homo-oligomerization (Civiero et al., 2017; Sejwal et al., 2017; Watanabe et al., 2020). LRRK2 can dimerize through both ROC–COR and WD40 domains (Civiero et al., 2017; Deniston et al., 2020; Guaitoli et al., 2016; Sejwal et al., 2017; Zhang et al., 2019). Microtubule-dependent filamentous structures of LRRK2 have also been reported under pathogenic conditions (Deniston et al., 2020; Kett et al., 2012). However, not only are atomic details underlying the ROC–COR-mediated dimer yet to be uncovered, but it is also puzzling why LRRK2 exists mostly as monomers and dimers rather than filaments under physiological conditions (Gilsbach et al., 2018).
One key player in the LRRK2 signaling pathway is the Rab GTPase family, which functions as LRRK2 regulators and kinase substrates (Purlyte et al., 2018; Steger et al., 2017; Steger et al., 2016; Taylor and Alessi, 2020). Membrane-anchored Rab proteins regulate intracellular vesicle trafficking (Homma et al., 2020). Defective vesicle trafficking is suggested as a culprit of PD and has been reported in cells harboring PD-causative LRRK2 mutations (Gao et al., 2018). Rab proteins directly interact with LRRK2 via the ARM and/or ANK domains, recruit LRRK2 to the trans-Golgi network or lysosomes and activate its kinase activity (Liu et al., 2018; McGrath et al., 2019; Purlyte et al., 2019). Yet little is known about the biological assembly between LRRK2 and Rab proteins.
Here we present high-resolution structures of full-length human LRRK2 in three physiologically important states: monomer, dimer and LRRK2G2019S, using single-particle cryo-electron microscopy (cryoEM) analysis. In light of the vast functional data on human LRRK2 (Berwick et al., 2019), our cryoEM structures provide a deeper molecular understanding of this vital molecule.
Results
Architecture of the full-length human LRRK2
The full-length human LRRK2 was purified mainly as monomers in solution (Fig. 1A) and used for structural analysis. To test whether the protein was still functional after our expression and purification procedure, we measured its kinase activity by using an ADP-Glo™ kinase assay. As expected, the purified protein catalyzed ATP hydrolysis in the presence of a peptide substrate, LRRKtide (Jaleel et al., 2007). The LRRK2 inhibitor, GSK2578215A (Reith et al., 2012), greatly inhibited the observed catalytic activity (Fig. 1B). Unexpectedly, we captured structures of LRRK2 in both monomeric and dimeric states at overall resolutions of 3.7 Å and 3.5 Å, respectively (Fig. S1A and Table S1). The N terminus of LRRK2 in both monomeric and dimeric states are flexible (Fig. S1B and S1C). To further improve the map quality, we carried out symmetry expansion for the dimer followed by focused 3D classification and refinement—this improved map resolution of the C-terminal part to 3.1 Å (Fig. S1A and Table S1) and allowed de novo model building for the majority of the protein ranging from residue 558 to the end. We performed homology modeling and rigid-body fitting for the illustration of residues 1-557 (Fig. 1 and S2A).
Figure 1.
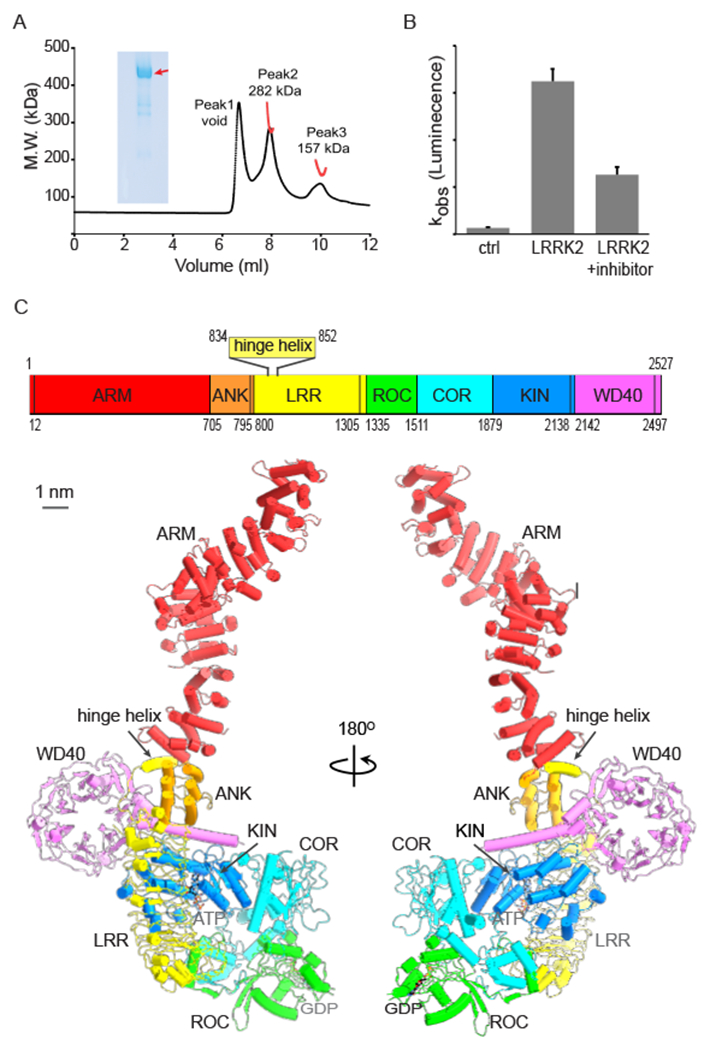
Structure of the LRRK2 monomer
(A) SEC-MALS analysis of LRRK2 protein. The size-exclusion column WTC-030S5 (MW range 5,000–1,250,000 Da) was used to analyze the molecule weight of LRRK2. SEC-MALS data are plotted as a distribution of the weight average molecular weight (red) superimposed on the chromatogram of UV absorbance (black) at 280 nm as a function of elution volume. The final data show three peaks from left to right. Peak 1 is void, peak 2 is LRRK2 and Peak 3 contains contamination and degradation products of LRRK2. The SDS-PAGE of purified LRRK2 is also shown, and the band corresponding to LRRK2 is indicated by a red arrow.
(B) The kinase activity of purified LRRK2. The kinase activity of LRRK2 is measured using an ADP-Glo™ assay. The y-axis is the bio-luminance signal. Data shown are the mean ± SD (n=4)
(C) The overall structure of the LRRK2 monomer. Top: domain scheme of LRRK2. The domain boundary is mapped based on our cryoEM structure. The domain boundary is labeled. Bottom: cylinder model of the full-length LRRK2 at two views. ARM, ANK, LRR, ROC, COR, KIN and WD40 domains are colored in red, orange, yellow, green, cyan, blue and violet, respectively. The linkers connecting adjacent domains are colored using the same color as the previous domain. The same color code is used unless noted.
See also Figures S1 and S2 and Table S1.
The LRRK2 monomer adopts a “J” shape, with the longest dimension of ~225 Å (Fig. 1C). The extended ARM domain (Fig. 1C and S2A) shows flexibility relative to the rest of the protein (Fig. S1B). The LRR domain of LRRK2 differs from the canonical structure and has a long insertion between the first and second repeats (Fig. S2B). The truncated LRRK2RCKW structure, which only contains the ROC, COR, KIN and WD40 domains, was recently reported (Deniston et al., 2020). We compared these four domains from the current structure with LRRK2RCKW, resulting in an RMSD of 5.2 Å. The ROC and COR domains displace significantly relative to the KIN and WD40 domains (Fig. S2C). When aligned with the ROC domain and viewed from that perspective, the COR domain shows a clockwise rotation in the LRRK2RCKW structure (Fig. S2C). Of note, the Thr1343 has a phosphorylation modification in the LRRK2RCKW sample, which may have contributed to the observed conformational differences.
Despite adding GTP before freezing the sample, we likely captured LRRK2 in a GDP-bound form, as suggested by the following two observations. First, the GDP fits better in the cryoEM density (Fig. S2D). Second, the conformation of the ROC “switch 1” motif, which deviates from the GTP-bound state (Fig. S2D), favors a GDP molecule. In general, “switch 1” motifs are structurally similar in GTP-bound ROC domains but show variations in the GDP-bound state (Fig. S2D) (Eathiraj et al., 2005; Guo et al., 2013; McGrath et al., 2019; Vetter and Wittinghofer, 2001).
Interdomain interactions, disease hotspots and scaffolding helices
The full-length structure of LRRK2 monomers revealed its overall architecture and enabled us to dissect the interdomain interactions at atomic details (Fig. 2, 3C–3D, S3A and S3C), which otherwise was impossible with low-resolution or truncated structures (Deniston et al., 2020; Watanabe et al., 2020). Two of the interdomain interfaces show high disease relevance (Fig. 2A–2B). First, the COR–ROC interface that buries a surface area of ~2600 Å2 is a hotspot for PD disease mutations. All four high-risk mutation sites in ROC and COR domains (Asn1437, Arg1441, Tyr1699 and Ser1761) are found at or near the interface (Fig. 2A), indicating the importance of the coupling between the ROC and COR domains in PD pathogenesis. Second, the ANK and LRR domains interact with the KIN domain through three sites: two on the C-lobe (Fig. 2B and 3D) and one on the N-lobe of the KIN domain (Fig. 3D and S3C). One of the C-lobe interacting sites involves Asn2081 (Fig. 2B), whose mutation to aspartic acid (N2081D) confers risk for Cohn’s diseases (Hui et al., 2018). Asn2081 interacts with the LRR domain through hydrophilic interactions. Mutation of Asn2081Asp, which shows increased kinase activity, is expected to destabilize such interactions. Intriguingly, the autophosphorylation site, Ser1292 (Gloeckner et al., 2010), is also localized in the close vicinity (Fig. 2B). It is plausible that Asn2018Asp and Ser1292 phosphorylation lead to similar structural changes by introducing negative charges at the interface.
Figure 2.
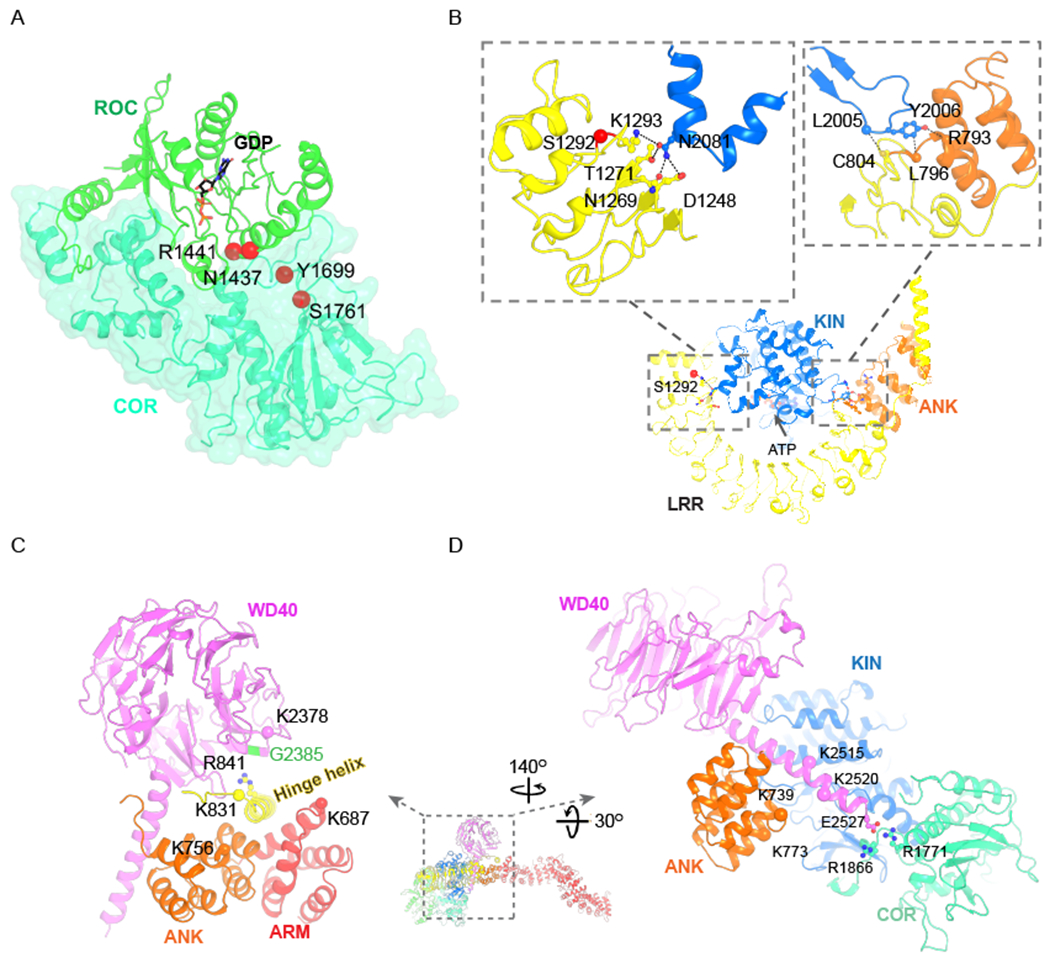
Inter-domain interaction and two scaffolding elements
(A) Interdomain interaction between ROC and COR domains. PD disease mutation sites are shown as red spheres and labeled.
(B) Interdomain interaction between KIN and ANK-LRR. The autophosphorylation site S1292 is shown as a red sphere. Sidechains of interacting residues between KIN and ANK-LRR are shown as ball-and-stick models, and mainchains are represented using spheres. Possible interactions are indicated by dashed lines.
(C) The hinge helix connects the WD40, ANK and ARM domains. Residues that can be crosslinked are shown as spheres. K831-K756 is crosslinked in both our and previous studies (Guaitoli et al., 2016). The K831-K687 is a confident crosslinking pair in our study, and the K831-K2378 pair was reported with medium confidence. The disease mutation, G2385, facing R841 (shown as sticks and balls) of the hinge helix is colored in green.
(D) The C-terminal helix interacts with KIN, ANK and COR domains. The side chains of E2527, R1866 and R1771 are shown as sticks and balls. Residues reported to be crosslinked with high confidence (K739-K2515 and K773-K2520) are shown as spheres.
Figure 3.
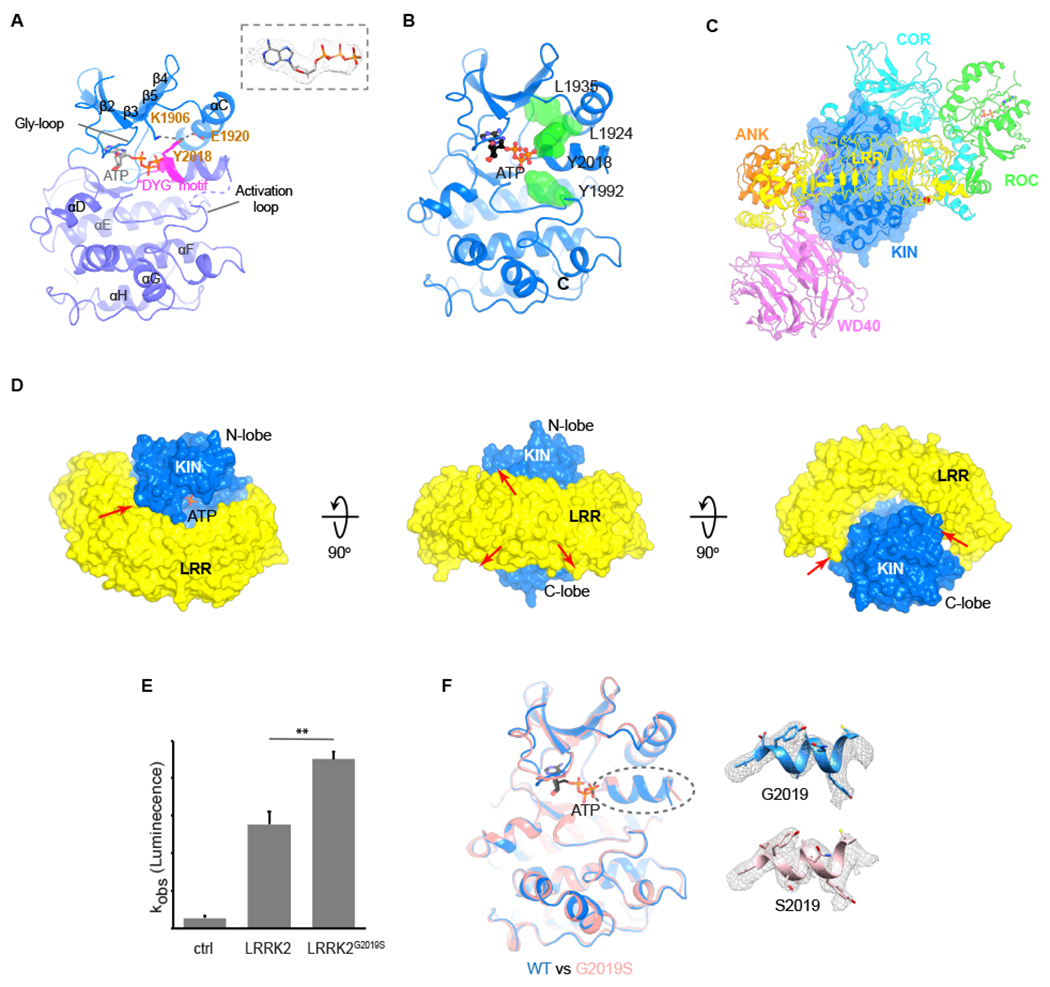
The inactive kinase domain of LRRK2 and LRRK2G2019S.
(A) Structural elements of the LRRK2 kinase domain. N-lobe and C-lobe are colored in blue and purple-blue and the “DYG” motif in magenta. The cryoEM density of the ATP molecule is shown. Interactions between Y2018 and K1906, Y2018 and E1920 are indicated by dashes.
(B) The broken R-spine of the kinase domain. The four residues forming the R-spine (L1935, L1924, Y2018 and Y1992) are shown as green surfaces.
(C) The position of KIN domain relative to ANK, LRR, ROC, COR and WD40 domains.
(D) Interaction between KIN and LRR domains.
(E) The kinase activity of LRRK2 and LRRK2G2019S. LRRK2G2019S shows ~1.4-fold increased kinase activity, compared to that of the wild-type LRRK2 (p<0.01, N=4). Data shown are the mean ± SD.
(F) Comparison of kinase domains between the full-length LRRK2 (blue) and LRRK2G2019S (pink). ATP is shown as sticks. The densities of helices where the mutation is located are shown as grey meshes.
See also Figure S3.
The full-length structure revealed two essential scaffolding helices that mediate multidomain associations (Fig. 2C–D). First, the helix located between the first and second leucine-rich repeats bridges ARM, ANK and WD40 domains (Fig. 2C). Such an interactive mode is validated by our (Table S2) and previously published crosslinking mass-spectrometry (MS) data (Guaitoli et al., 2016). Specifically, Lys831 at the edge of this helix crosslinked with Lys687 of the ARM domain, Lys756 of the ANK domain and Lys2378 of the WD40 domain (Fig. 2C). Because it functions as a hinge for the dynamic motions of the ARM domain, we named it the “hinge helix” (Fig. S1B). Intriguingly, the hinge helix bridges the interaction between ARM and WD40 domains, which prevents homodimerization of WD40 domains (Fig. S2E), thereby providing a possible explanation for why LRRK2 doesn’t form filaments under normal physiological conditions. Second, the “C-terminal helix” following the WD40 domain that leans on the back of the KIN domain interacts with the ANK and COR domains (Fig. 2D). We also found two crosslinking pairs between the ANK domain and the C-terminal helix: Lys739–Lys2515 and Lys773–Lys2520 (Fig. 2D). The last residue of this helix, Glu2527, is in close proximity to Arg1771 and Arg1866 of the COR domain (Fig. 2D). These data support the idea that the C-terminal helix functions as a scaffolding element that connects the ANK, KIN, WD40 and COR domains.
An inactive state of the LRRK2 kinase domain
The KIN domain of LRRK2 displays key features of an inactive kinase with “DYG” motif and “αC helix” flipping out (Fig. 3A) (Ung et al., 2018). The regulatory spine (R-spine) (Kornev and Taylor, 2010), formed by Leu1935, Leu1924, Tyr2018 and Tyr1992 (Schmidt et al., 2019), is broken (Fig. 3B). LRRK2 is a rather unusual kinase, as the phenylalanine residue in the highly conserved “DFG” motif is replaced by Tyr2018. In the current structure, Tyr2018 interacts with and sits between Lys1906 and Glu1920 (Fig. 3A), two of the three hydrophilic regulatory triad residues (Schmidt et al., 2019) and prevents Lys1906–Glu1920 salt bridge formation. As such, Tyr2018 spatially hinders the positioning of Glu1920 to Lys1906 and ATP, thereby stabilizing the inactive state with “αC helix” flipping out (Xie et al., 2020). This observation is consistent with the hypothesis that the hydroxyl moiety of Tyr2018 serves as a “brake” for kinase activation (Schmidt et al., 2019).
The LRR domain seems to modulate and stabilize the inactive state of the KIN domain by shielding substrate entry and by maintaining an open ATP-binding cleft (Fig. 3D). The KIN domain of LRRK2 is surrounded by ANK, LRR, ROC, COR and WD40 domains (Fig. 3C). Spatially, the LRR domain wraps around and shields the ATP-binding cleft (Fig. 3D). As a result, the ATP is not accessible for globular substrates, such as Rab proteins. In addition, LRR contacts the KIN domain through three sites as mentioned (Fig. 2B, 3D and S3C). By interacting with both the N-lobe and C-lobe of the KIN, the LRR functions as if it is a strut to hold the ATP-binding cleft open, which stabilizes the KIN domain in an inactive state.
We compared the KIN domain of LRRK2 with that of truncated LRRK2RCKW (Deniston et al., 2020). The two structures showed three major differences. First, our structure was determined in the presence of ATP and Mg2+, and a strong ATP density was visible in the cryoEM map (Fig. 3A). The presence of unhydrolyzed ATP is consistent with an inactive state of the kinase. Second, part of the activation loop (residues 2017–2027) in our structure, which hosts the “DYG” motif, forms an alpha helix (Fig. 3A and S3B), which may be due to the presence of ATP. Lastly, the N-lobe of the KIN domain in the current study interacts with the LRR domain through the residues preceding the Gly-rich loop (Fig. S3B and S3C). As a result, we observed that the β1 strand of the KIN domain becomes a loop and the N-lobe moves towards the LRR domain (Fig. S3B and S3C).
Because mutation of Gly2019Ser in the “DYG” motif is the most prevalent mutation of LRRK2 in PD, we ask how the Gly2019Ser mutation affects the KIN domain. LRRK2G2019S increases the kinase activity of LRRK2 (~1.4-fold, Fig. 3E) (Greggio and Cookson, 2009), making LRRK2-specific inhibitors of pharmacological interest. To better characterize this mutation, we determined the cryoEM structure of LRRK2G2019S at a 3.8-Å resolution (Table S1). The overall structure of LRRK2G2019S is almost identical to the wild-type LRRK2 (Fig. S3D). The kinase domains from the two structures have an RMSD of 0.4 Å, with minor differences in flexible loop regions (Fig. 3F). The structural similarity between LRRK2 wild type and LRRK2G2019S leads us to hypothesize that Gly2019Ser mutation does not alter the conformation of LRRK2 significantly but instead affects the kinase kinetics, which is supported by the molecular dynamics analysis (Schmidt, 2020).
The architecture of the full-length LRRK2 dimer and its biological significance
We further presented the structure of LRRK2 dimers at an overall resolution of 3.5 Å (Fig. 4A, S1A and Table S1). Each protomer in the LRRK2 dimer is similar to the LRRK2 monomer with an RMSD of 0.6 Å and is also captured in an ATP-bound kinase-inactive state (Fig. S4D). The dimeric interface is mainly mediated by the COR domain—mostly by the COR-B subdomain, forming two partially overlapped β sheets (Fig. 4B). Each β sheet has a “7+1” structure with seven strands from one subunit and one strand from the other subunit. The detailed interactions are shown in Fig. 4B. Since numerous biochemical and structural studies had shown that LRRK2 forms functionally important homodimers through its ROC–COR domains (Berger et al., 2010; Civiero et al., 2017; Rosenbusch and Kortholt, 2016), it leads us to believe that the LRRK2 dimer here is biologically relevant.
Figure 4.
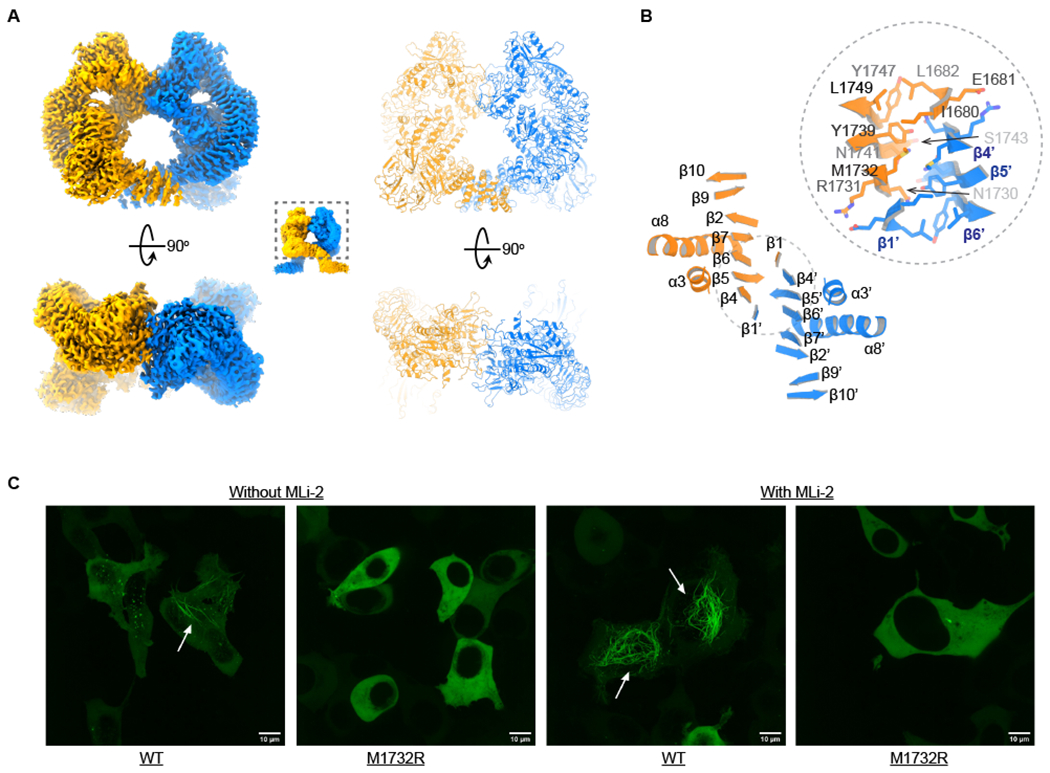
Structure of the LRRK2 dimer
(A) Structure of LRRK2 dimers. The density and model of the well-resolved part of LRRK2 dimers are shown at two views. The two protomers are colored in orange and blue, respectively.
(B) The dimer interface. The secondary structures of the COR-B subdomain are shown and numbered. Side chains of the interface residues are shown.
(C) Imaging results of GFP-LRRK2RCKW wild type and Met1732Arg mutation in the presence and absence of MLi-2. The filaments are indicated by white arrows. The white scale bar is 10 µm in length.
The observed LRRK2 dimer interface should be pathologically significant, as suggested by our following structure-function analyses. We docked the ROC–COR dimer into the in-situ cryoET map of the microtubule-associated LRRK2 filament (Watanabe et al., 2020). The ROC–COR dimer model fitted well in the EM density (Fig. S4A), indicating that the observed dimer is vital for pathogenic LRRK2 filamentation. This observation is further supported by our mutagenesis and live-cell confocal imaging assays. By introducing a point mutation, Met1732Arg, in the dimer interface, we completely abolished the LRRK2 filament formation in HEK293 cells both in the presence and absence of MLi-2 (Fell et al., 2015), a kinase inhibitor that facilitates LRRK2 filamentation (Watanabe et al., 2020). We used a construct expressing GFP-LRRK2RCKW for the imaging analysis, as it shows a higher percentage of filament formation than the full-length LRRK2 (Schmidt, 2020).
We further proposed that the observed LRRK2 dimer is physiologically important through structurally modeling the Rab-LRRK2 complex (Fig. S4B and S4C). Rab proteins are kinase substrates and membrane anchors of LRRK2 and thus are key players in LRRK2 signaling (Liu et al., 2018; Purlyte et al., 2019). Previous studies had shown two interacting modes between Rab proteins and LRRK2: one through the ANK domain and the other through the ARM domain of LRRK2 (McGrath et al., 2019; Purlyte et al., 2019). As the possible Rab-interacting residues of the ANK domain are not accessible in current structures, we modeled the interaction between the ARM domain and Rab32-subfamily proteins (Rab29, Rab32 and Rab38) to understand the Rab-dependent membrane recruitment mechanism of LRRK2 (Fig. S4B and S4C). The potential Rab-interacting regions of the ARM domain (residues 386-392) are located on the same side of LRRK2 dimers. Docking Rab proteins on this region is compatible with membrane geometry without generating any obvious crashing. We think such a complex assembly could represent the membrane-associated inactive state of LRRK2 (GDP-bound) in the “Rab-dependent LRRK2 activation model” proposed by Harvey and colleagues (Berwick et al., 2019).
Discussion
In this study, we reported the cryoEM structures of full-length LRRK2 of different states. The structures enabled us to dissect the domain arrangement of this large multidomain protein, analyze disease mutations and study the biological significance of the dimer arrangement, and thus opened an anotherchapter for the structure-function analysis of LRRK2.
The structures provide a unique opportunity to rationalize the functional consequences of disease mutations guided by structural information. In addition to the six disease-causing PD mutations and one CD mutation discussed above, LRRK2 also hosts many PD-association variants, including the Gly2385Arg mutation (LRRK2G2385R) in the WD40 domain that is common in Chinese population (Mata et al., 2005). LRRK2G2385R was shown to disrupt WD40 dimers and the formation of pathogenic filaments in cells (Watanabe et al., 2020; Zhang et al., 2019). However, this doesn’t explain how LRRK2G2385R is associated with PD. Here we show that Gly2385 is located at the interface between WD40 and the hinge helix (Fig. 2C). Arg841 of the hinge helix is pointing towards Gly2385. It is possible that introducing a positively charged arginine at the position of Gly2385 could disrupt the interaction between the WD40 domain and the hinge helix, destabilize the observed inactive state of LRRK2 and thus cause PD. Consistently, it had been shown that LRRK2G2385R moderately enhanced Rab10 phosphorylation by about twofold (Zhang et al., 2019).
The inactive-state structure of LRRK2G2019S serves as a structural framework for designing conformation-specific LRRK2 inhibitors for treating PD. LRRK2G2019S showed little structural difference from wild type, suggesting the LRRK2G2019S may not cause a significant change in conformation, at least under certain experimental conditions. However, the inactive-state structure of LRRK2G2019S does provide the opportunity to design conformation-specific inhibitors for PD treatment. On the one hand, structural insights into inactive states of kinases are usually leveraged to design selective inhibitors (Noble et al., 2004; Xie et al., 2020). On the other hand, LRRK2G2019S is the most common PD-causing mutation in LRRK2. Conformation-specific small molecules targeting this mutation could be of great pharmacological significance.
Lastly, our structural and functional data have important implications for other possible conformations of LRRK2. LRRK2 is believed to adopt states different from the current structures. Our data suggest that conformational changes between different states of LRRK2 could be large. First, the autophosphorylation site Ser1292 (Sheng et al., 2012) is about 25 Å away from ATP (Fig. 2B); the distance between those two sites in neighboring subunit is about 99 Å. The two sites need to be in close vicinity to carry out autophosphorylation. Second, the dimerization of WD40 for the filament formation requires rearrangement of the hinge helix and ARM domain (Fig. S2E). Third, crosslinking data suggest that the hinge helix can move out from the interface between WD40 and ANK–ARM domains. For example, Lys831 of the hinge helix crosslinks with Lys1963 of the KIN domain in both our (Table S2) and previous published crosslinking results with high confidence (Guaitoli et al., 2016). Although the structural illustration of the above conformation(s) requires further investigation, our current study paves the way for obtaining a complete conformational landscape of LRRK2, dissecting its physiological and pathological roles and developing inhibitors for therapeutic purposes.
Limitations of the Study
The proposed structural model for LRRK2 recruitment by Rab proteins in the context of the membrane bilayer (Fig. S4B) needs to be experimentally verified.
STAR METHODS
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ji Sun (ji.sun@stjude.org).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Data Resources
The three-dimensional cryoEM density maps for human LRRK2 monomer, dimer, symmetry expanded monomer and LRRK2G2019S monomer have been deposited in the EM Database under the accession code EMDB: EMD-23352, EMD-23350, EMD-23360 and EMD-23359, and the coordinates for the structure have been deposited in Protein Data Bank under accession code PDB 7LHW, 7LHT, 7LI4 and 7LI3.
Experimental Model and Subject Details
Cell lines
Adherent HEK293T cells were maintained in DMEM medium (GIBCO) supplemented with 10% fetal bovine serum (FBS) at 37°C. Sf9 cells were cultured in Sf-900 III SFM medium (GIBCO) at 27°C. HEK293F cells were cultured in Freestyle 293 medium (GIBCO) supplemented with 2% FBS and 1% Pen Strep at 37°C.
Method Details
Cloning, expression and purification of human LRRK2
The cDNAs encoding human LRRK2 were purchased from Horizon Discovery and contains three natural variants (R50H, S1647T and M2397T) from the canonical LRRK2 sequence (NP_940980.4). A GFP tag followed by a preScission protease cleavage site was engineered at the N terminus of LRRK2, which was cloned into the BacMam expression vector (Goehring et al., 2014).
Recombinant baculoviruses of LRRK2 were generated using the Bac-to-Bac system according to manufacturer’s instructions (Invitrogen). Then P3 virus of LRRK2 was used for transfection of HEK293F cells for protein expression. Briefly, for 1L cultures of HEK293F cells (~2-3×106 cells/mL) in Freestyle 293 media (Gibco) supplemented with 2% FBS (Gibco), about 100 ml P3 virus was used. Infected cells were incubated at 37oC overnight, and protein expression was induced by adding 10 mM sodium butyrate. Cells were cultured at 30oC for another 48-60 hrs before harvest.
Cell pellet from 600 mL culture was resuspended in 30 ml lysis buffer (20 mM Tris pH 8.0, 200 mM NaCl, 10% glycerol, 2 mM DTT and protease inhibitors), and then cells were lysed by brief sonication. LRRK2 was separated from the insoluble fraction by high-speed centrifugation (38,000g for 1 hr), and incubated with 1 ml CNBr-activated sepharose beads (GE Healthcare) coupled with 1 mg high-affinity GFP nanobodies (GFP-NB) (Kirchhofer et al., 2010). The GFP tag was cleaved by preScission protease at 4oC, and LRRK2 was further purified by size-exclusion chromatography with a Superose 6 increased 10/300 GL column (GE Healthcare) equilibrated with 20 mM Tris pH 8.0, 200 mM NaCl and 2 mM DTT. Fractions of the peak around 14 ml were pooled, and then ATP and Mg2+ were added to a final concentration of 2 mM and 1 mM, respectively. The protein sample was concentrated to 6-8 mg/ml (OD280) using a 100-kDa MWCO centrifugal device (Ambion) and immediately used for cryoEM grid preparation. 0.5 mM GTP and 2 mM Fluorinated Fos-choline-8 (FFC-8) were added 15–30 min before freezing.
Kinase assay
The kinase activity of LRRK2 and LRRK2G2019Swas measured using a commercially available ADP-Glo™ assay. Briefly, 2 µl purified LRRK2 (0.5 µM), 2 µl LRRKtide (0.5 mM) and 1 µl ATP (0.5 mM) were mixed and incubated at room temperature for 100 min. 5 µl ADP-Glo™ reagent was added, followed by another 40 min incubation. 10 µl kinase detection reagent was then added. Luminescence was taken after a 30-min incubation at an integration time of 1 sec. The GSK2578215A was added to the protein sample for the inhibition assay at a final concentration of 1 µM 2 min before the assay.
SEC-MALS assay
The size-exclusion chromatography multi-angle light scattering (SEC-MALS) (Kendrick et al., 2001) experiments were carried out using WTC-030S5 (MW range 5,000–1,250,000 Da) size-exclusion column (Wyatt Technologies, Santa Barbara, CA, USA) with three detectors connected in series: an Agilent 1200 ultraviolet (UV) detector (Agilent Technologies, Santa Clara, CA), a Wyatt DAWN-HELEOS-II multi-angle light scattering (MALS) and a Wyatt Optilab T-rEX differential refractive index (RI) detector (Wyatt Technologies, Santa Barbara, CA, USA). The column was equilibrated with 20 mM Tris pH 8.0, 200 mM NaCl and 2 mM DTT. The data was collected at 25°C. A 100 µL sample in volume was injected into the column using an auto-sample injection method with a flow rate of 0.5 ml/min. Protein in the eluent was detected via UV absorbance at 280 nm, light scattering, and refractive index detectors. The data were recorded and analyzed with the Wyatt Astra software (version 8.0). The refractive index increment, dn/dc, was assumed to be 0.185 ml/g for measuring the concentration of the protein samples. EASI Graphs (Astra software) were exported and plotted as a molar mass distribution superimposed on a light scattering and UV traces versus elution volume.
Crosslinking and mass-spectrometry analysis
LRRK2 (30 µg) was crosslinked with DSSO (Kao et al., 2011) using a 1:900 protein-to-crosslinker molar ratio. The reaction was carried out at room temperature (RT) for 1 hr and then quenched with excess ammonium bicarbonate (ABC) for 30 min at RT. Denaturation buffer was added to reach 8 M urea concentration, followed by reduction with dithiothreitol and alkylation with iodoacetamide. Then the sample was diluted with 50 mM ABC to the final concentration of 2 M urea. Trypsin was added to a protease-to-substrate ratio of 1:10 (w/w), and the sample was incubated at 37°C for 3 h. Asp-N was added to the trypsin digested sample with a protease-to-substrate ratio of 1:10 (w/w) at 37°C overnight. Digestion reaction was stopped with 10% trifluoroacetic acid. A negative control sample with the addition of the same level of DMSO instead of DSSO was also analyzed.
The digested peptides were analyzed by customized two-dimensional liquid chromatography and tandem mass spectrometry (LC/LC-MS/MS) (Liu et al., 2020). Pre-fractionation was performed by basic pH RPLC using a microscale HPLC system (Agilent 1120) coupled with a 3-way flow splitter (IDEX Health & Science). The digested sample was speed vacuumed to almost dry to avoid sample loss and resuspended with mobile phase A (10 mM ammonium formate, pH 8.0), centrifuged at 21,000 × g for 5 min. With the flow splitter closed, the supernatant was loaded onto an XBridge C18 column (1 mm × 50 mm, 3.5 μm beads, Waters) at a flow rate of 50 µl/min by 95% of mobile phase A. After loading, the flow splitter was opened, and the peptides were eluted with a 45 min gradient from 5–90% of mobile phase B (90% acetonitrile, 10 mM ammonium formate, pH 8.0). With the flow splitter, the HPLC was operated at 800 µl/min, but the column was eluted at 30 µl/min. Six fractions were manually collected.
Further LC-MS/MS analysis was performed using a Waters nanoAcquity UPLC coupled with a Thermo Scientific Orbitrap Q Exactive HF. Mobile phase A consisted of 0.2% formic acid (FA) in water and mobile phase B consisted of 0.2% FA in acetonitrile. Peptides were loaded into a self-packed C4 column (75 µm × 20 cm, Halo C4, 3.4 μm particles, 400 Å pore size) at a flow rate of 300 nl/min with the following gradient: 10% to 30% mobile phase B over 40 min, 30% to 50% mobile phase B in 5 min, 50% to 80% mobile phase B in 5 min, static 80% mobile phase B for 3 min, and equilibration at 3% mobile phase B for 15 min (Wang et al., 2015). MS1 scans were detected in the Orbitrap at 120K resolution in the m/z range 400–2000 and AGC target of 1 × 106 with a maximum injection time of 50 ms. Ions with charge states from 3+ to 8+ were selected for fragmentation by stepped high energy collision dissociation (HCD) at 27%, 30%, 33% with AGC of 1 × 105, maximum injection time of 100 ms, and detected in the Orbitrap at 60K resolution.
Raw data were converted to .mzXML format using MSconvert and searched with MeroX2.0 (Iacobucci et al., 2018) for crosslinked peptides and JUMP (Wang et al., 2014) for regular peptides. Settings for crosslinking search were as follows: maximum total missed cleavage, 3; peptide length, 5–30; static modification, carbamidomethylation of cysteine; dynamic modification, oxidation of methionine; cross linker, DSSO, assumed reaction site at lysine, serine, threonine, and tyrosine; modification of fragments, with 54.0105 Da and 85.9826 Da mass modification; MS1 precision, 10 ppm; MS2 precision, 10 ppm; RISEUP Mode with 2 maximum missing ions; Prescore, 10% intensity; FDR cut off, 5%; Score cut off, −1; Include cRAP database; Decoy database generation, shuffle sequence but keep protease sites. Results from the MeroX search were manually validated using the following 5 criteria after the calibration of mass accuracy: (i) 5 ppm mass tolerance of precursor ions; (ii) validation of at least two signature product ions within 5 ppm mass tolerance; (iii) continuity of b and y ions within 5 ppm mass tolerance; (iv) presence of branched product ions within 5 ppm mass tolerance; and (v) absence in the negative control run.
CryoEM analysis
CryoEM grids were prepared with a Vitrobot Mark IV (FEI). Quantifoil R1.2/1.3 holey carbon gold grids were glow-discharged for 15 s. Then 3.5 μL of 6-8 mg/ml protein sample was pipetted onto the grids, which were blotted for 3 s under blot force −3 at 100% humidity and frozen in liquid nitrogen-cooled liquid ethane.
CryoEM data were collected using Titan Krios (Thermo Fisher) transmission electron microscope, equipped with a K3 direct electron detector and post column GIF (energy filter). K3 gain references were collected just before data collection. Data collection was performed in SerialEM software (Mastronarde, 2005) with image shift protocol (9 images were collected with one defocus measurements). Movies were recorded at defocus values from −0.8 to −1.7 μm at a magnification of 105kx, which corresponds to the pixel size of 0.826 Å at the specimen level (super-resolution pixel size is 0.413). During 2.8-second exposure, 70 frames (0.04 s per frame and the dose of 1.154 e/frame/Å2) were collected with a total dose of ~81 e. In total, 12,576 images were collected. Motion correction was performed on raw super-resolution movie stacks and binned by 2 using MotionCor2 software (Zheng et al., 2017). CTF parameters were determined using CTFind and refined later in Relion 3.0 (Zivanov et al., 2018), CisTEM (Grant et al., 2018) and cryoSPARC (Punjani et al., 2017). Prior particles picking micrographs were analyzed for good power spectrum, and the bad ones were discarded (with 9,804 good images remained). Particles were picked within cisTEM and later coordinates were transferred to Relion 3.0 (in total 1,214,310 particles).
After extraction binned by 8 particles in Relion3.0, the particle stack was transferred to cryoSPARC. Several rounds of the 2D classification using 8x binned particles (pixel size = 6.608 Å/pixel) were performed to eliminate ice, carbon edges, and false-positive particles containing noise resulted in approximately 479,170 particles for further analysis. During 2D classification, two groups of classes were observed. The first group was corresponding to the monomer and the other to the dimer. Both groups were selected, and ab initio reconstruction was performed. In order to further separate dimers from monomers, we perform Heterogeneous Refinement in cryoSPARC with particles bin by 2 (pix size 1.652 Å/pixel). As a result, 231,875 particles were assigned to the dimer class and 129,744 particles to the monomer. Both 3D classes were refined in parallel using cryoSPARC and Relion3.1 software after extraction of corresponding particles without binning (pix size 1.652 Å/pixel). For monomer, we performed a focused 3D refinement in addition to a standard refinement. As to the dimer, we have implemented C2 and C1 symmetry refinement. We also perform a symmetry expansion procedure in order to get a better resolution for the C-terminal part of the protein. Particle transfer between Relion and cryoSPARC were done by using script from Daniel Asarnow (UCSF, laboratory of Prof. Yifan Cheng). At the final step for both the monomer and dimer, CTF refinement and high order aberration correction were performed. The density maps sharpened in cryoSPARC or Relion3.1 were used to produce figures.
Model building and refinement
Models were built in Coot (Emsley et al., 2010). First, homology models of LRRK2 individual domains except the WD40 domain were generated using the I-TASSER server (Yang et al., 2015) and docked into the cryoEM map. The WD40 domain from a crystal structure was docked into the cryoEM density. From this starting point, manual rebuilding was carried out for the symmetry expansion map (3.1 Å). The structural model was refined using the phenix.real_space_refine (Afonine et al., 2013) with secondary structure restraints and Coot iteratively. Protein structure quality was monitored using the Molprobity server (Chen et al., 2010). We then modeled the monomer and dimer using the above structure as a starting point, following a similar procedure. Figures were prepared using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.) and UCSF Chimera (Pettersen et al., 2004).
Model the interaction between LRRK2-ARM domain and Rab proteins
To model the membrane recruitment of LRRK2 by Rab32-subfamily members (Rab29, Rab32 and Rab38), we used the Rab38 structure (PDB 6HDU) and the homology model of the LRRK2 ARM domain. Here, Rab38 structure is used for the following reasons. First, it shares high sequence identity (52%) to Rab29, which is the most studied Rab protein that recuits LRRK2 to membrane surface. Second, a GTP-bound structure of Rab38 is available, as LRRK2-Rab interaction is GTP-dependent. Third, Rab38 also interacts with LRRK2 through ARM domain and could potentially recruit LRRK2 to membrane surface. Of note, Rab38, unlike Rab29, is not a substrate as it lacks the conserved Threonine/Serine residue within the Switch 2 motif. The modeling was performed using the PatchDock server(Schneidman-Duhovny et al., 2005). The interacting residues were given based on the mutagenesis data from the previous study (McGrath et al., 2019). The model with the largest interacting interface was used here in Fig. S4B.
Live-Cell Confocal Microscopy
The day before transfection, HEK293T cells incubated in DMEM medium were plated on 30 mm glass bottom dishes pretreated with Poly-D-lysine. The LRRK2RCKW was subcloned from the pDEST53-LRRK2-WT vector (Addgene: 25044). Cells were transfected with 1 µg plasmid expressing GFP-LRRK2RCKW or GFP-LRRK2RCKW(M1732R) using Lipofectamine 3000 reagent (Thermo Fisher). After 48 hrs, DMEM medium was replaced with phenol red free medium. Cells were treated with either DMSO or MLi-2 drugs for 2 hrs before live cell imaging experiments.
All confocal microscopy was performed at 37°C with 5% CO2. Images were acquired on a 3i Marianas system (Denver, CO USA) configured with a Yokogawa CSU-W spinning disk utilizing a 100x (1.46 NA) Zeiss objective. Z-stack images were acquired with a step size of 0.2 microns onto a Prime95B sCMOS camera (Teledyne Photometrics, Tucson AZ, USA). Images were processed using the SlideBook reader (intelligent-imaging.com/slidebook) and the image J software (rsb.info.nih.gov/ij)
Quantification and Statistical Analysis
The kinase activity assay in Fig. 1B and 3C were visualized using Microsoft Excel and the values were averages of the four (N=4) independent determinations with standard deviations. The quantification and statistical analyses for model refinement and validation were generated using MolProbity (Chen et al., 2010).
Supplementary Material
Figure S1. Structural determination of LRRK2, Related to Figure 1
(A) A brief flow chart of single-particle cryoEM data collection and process.
(B) The flexibility of ANK and ARM domains in the context of LRRK2 monomers.
(C) The flexibility of ANK and ARM domains in the context of LRRK2 dimers.
Figure S2. Structural features of LRRK2, Related to Figure 1
(A) Docking of the ARM domain in cryoEM densities. The cryoEM density of the ARM domain is from local refinement (Fig. S1).
(B) The structural model of the LRR domain with an insertion between the first and second repeats. N-terminal and C-terminal ends of the LRR domain are shown as spheres. Secondary structures are colored differently with alpha-helix in red, beta-strand in yellow and loop in green.
(C) Comparison between LRRK2 and LRRK2RCKW. Left: displacement of ROC–COR domains relative to KIN-WD40 domains. ROC, COR, KIN and WD40 domains in full-length LRRK2 are colored in green, cyan, blue and violet, respectively, and LRRK2RCKW is in grey. Right: rotational movement of the COR domain. The blue arrow indicates the phosphorylated T1343.
(D) Comparison of ROC domain with different Rab proteins. “Switch 1” motifs are indicated by black lines. Structural models and the PDB codes are in same color codes, and LRRK2 ROC domain is colored in green. The cryoEM density of the potential GDP molecule is shown. Residues in the GDP-binding site are shown as sticks.
(E) Superimposition of the WD40 dimer (PDB 6DLO) and the full-length LRRK2 structures. Steric clashes between the WD40 dimer and hinge helix and ARM domain of the full-length LRRK2 are indicated by blue arrows. The WD40 dimer is colored in dark grey.
Figure S3. Interdomain interactions and the kinase domain of LRRK2, Related to Figures 2 and 3
(A) Interdomain interactions at secondary structure level. The linker between LRR and ROC domains is colored in grey.
(B) Comparison of kinase domains between the full-length LRRK2 (blue) and truncated LRRK2RCKW (wheat). The displacement of the N-lobe of the kinase domain is indicated by a dashed arrow. The red arrow points to the “DYG” motif with different secondary structures. Gly-loop following the distorted β1 strand of the kinase domain is indicated by a black line.
(C) The interaction between the LRR domain and the N-lobe of the KIN domain. Sidechains of interface residues are shown as sticks.
(D) Comparison of the cryoEM maps and structural models between LRRK2 wild type and LRRK2G2019S monomers. Wild-type LRRK2 and LRRK2G2019S are colored in blue and grey, respectively.
Figure S4. Structural analysis of LRRK2 dimers, Related to Figure 4
(A) Docking of the ROC–COR dimer into the cryo-ET map. One copy of LRRK2 ROC-COR domains is colored in blue, the other in orange.
(B) Modeling of the LRRK2-Rab complex in the context of the membrane bilayer. Rab is colored in magenta. The C-terminal of Rab that is linked to lipid modification is shown as green spheres. LRRK2 dimers are shown in surface representation.
(C) The interaction between ARM domain and Rab38 at the secondary structure level. On the ARM domain side, it involves three regions: res. 386-392, res. 430-437 and res. 472-478. On the Rab protein side, switch 1 motif, α1, β2 and α2 are on the interface.
(D) Structural comparison between the LRRK2 monomer and dimer.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM medium | GIBCO | 11965-092 |
| SF-900 III SFM medium | GIBCO | 10902-088 |
| L-Glutamine (100x) | GIBCO | 25030-081 |
| Pen Strep | GIBCO | 15140-122 |
| Freestyle 293 medium | GIBCO | 12338-018 |
| Fetal bovine serum | GIBCO | 16000-044 |
| DMEM medium without phenol red | Thermo Fisher | 21063029 |
| Lipofectamine 3000 transfection reagent | Invitrogen | 11668019 |
| Cellfectin II reagent | Invitrogen | 10362100 |
| Lauryl Maltose Neopentyl Glycol (LMNG) | Anatrace | NG310 |
| Cholesteryl hemisuccinate | Anatrace | CH210 |
| GDN | Anatrace | GDN-101 |
| Digitonin | Millipore Sigma | 300410 |
| Critical Commercial Assays | ||
| CNBR-activated sepharose beads | GE Healthcare | 17-0430-01 |
| Superose 6, 10/300 GL | GE Healthcare | 17-5172-01 |
| Deposited Data | ||
| LRRK2 monomer | This study | EMD-23352 |
| LRRK2 monomer atomic model | This study | 7LHW |
| LRRK2 dimer | This study | EMD-23350 |
| LRRK2 dimer atomic model | This study | 7LHT |
| LRRK2 dimer symmetry expansion (masked) | This study | EMD-23360 |
| LRRK2 dimer symmetry expansion (masked) model | This study | 7LI4 |
| LRRK2 G2019S | This study | EMD-23359 |
| LRRK2 G2019S atomic model | This study | 7LI3 |
| Experimental Models: Cell Lines | ||
| Sf9 | Thermo Fisher | 12659017 |
| FreeStyle 293-F cells | Thermo Fisher | R79007 |
| HEK293T | Sigma | 12022001 |
| Recombinant DNA | ||
| Human LRRK2 cDNA gene | Horizon Discovery | MHS6278-211691095 |
| pDEST53-LRRK2-WT vector | Addgene | 25044 |
| Software and Algorithms | ||
| SerialEM | Mastronarde, 2005 | http://bio3d.colorado.edu/SerialEM |
| RELION | Scheres, 2012 | http://www2.mrc-lmb.cam.ac.uk/relion |
| CryoSPARC | Punjani et al. 2017 | https://cryosparc.com/ |
| EMAN2 | Tang et al., 2007 | http://blake.bcm.edu/emanwiki/EMAN2 |
| UCSF Chimera | Pettersen et al., 2004 | http://www.cgl.ucsf.edu/chimera |
| PHENIX | Afonine et al., 2013 | https://www.phenix-online.org |
| COOT | Emsley et al., 2010 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu |
| CisTEM | Grant et al., 2018 | http://cistem.org |
| PyMOL | Molecular Graphics System, Version 1.8 Schrodinger, LLC | http://www.pymol.org |
| Other | ||
| Quantifoil R1.2/1.3 400 mesh gold holey carbon grids | SPI supplies | 4240G-XA |
Acknowledgments
We thank the members of the Cryo-electron Microscopy and Tomography Center of St Jude Children’s Research Hospital for help with cryoEM data collection; S. Vaithiyalingam and R. Kalathur of the Protein Technology Center for help with mammalian cell culture and SEC-MALS analysis; V. Centonze and G. Campbell of the Cell & Tissue imaging center for help with the design of imaging assays; members of Halic and Lee labs for helpful discussions; J. Payandeh and C. Kalodimos for critical reading of the manuscript;and I. Chen and Z. Luo for suggestions and help with bioillustration during manuscript revision. This work was partially funded by the NIH grants: HL143037(JS), AG053987 (JP) and American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Declaration of Interests
The authors declare no conflicts of interest.
References
- Afonine PV, Grosse-Kunstleve RW, Adams PD, and Urzhumtsev A (2013). Bulk-solvent and overall scaling revisited: faster calculations, improved results. Acta Crystallogr. D Biol. Crystallogr 69, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z, Smith KA, and Lavoie MJ (2010). Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49, 5511–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berwick DC, Heaton GR, Azeggagh S, and Harvey K (2019). LRRK2 biology from structure to dysfunction: research progresses, but the themes remain the same. Mol. Neurodegener 14, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen KV, Hentzer M, Oppermann FS, Elschenbroich S, Dossang P, Thirstrup K, Egebjerg J, Williamson DS, and Smith GP (2018). LRRK2 exonic variants associated with Parkinson’s disease augment phosphorylation levels for LRRK2-Ser1292 and Rab10-Thr73. BioRxiv. [Google Scholar]
- Civiero L, Russo I, Bubacco L, and Greggio E (2017). Molecular insights and functional implication of LRRK2 dimerization. Adv. Neurobiol 14, 107–121. [DOI] [PubMed] [Google Scholar]
- Cookson MR (2015). LRRK2 pathways leading to neurodegeneration. Curr. Neurol. Neurosci. Rep 15, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Lewis PA, Greggio E, Sluch E, Beilina A, and Cookson MR (2008). Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. USA 105, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniston CK, Salogiannis J, Mathea S, Snead DM, Lahiri I, Matyszewski M, Donosa O, Watanabe R, Bohning J, Shiau AK, et al. (2020). Structure of LRRK2 in Parkinson’s disease and model for microtubule interaction. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyaert E, Leemans M, Singh RK, Gallardo R, Steyaert J, Kortholt A, Lauer J, and Versees W (2019). Structure and nucleotide-induced conformational dynamics of the Chlorobium tepidum Roco protein. Biochem. J 476, 51–66. [DOI] [PubMed] [Google Scholar]
- Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, Zharikov A, Van Laar A, Stepan AF, Lanz TA, et al. (2018). LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingos S, Duarte T, Saraiva L, Guedes RC, and Moreira R (2019). Targeting leucine-rich repeat kinase 2 (LRRK2) for the treatment of Parkinson’s disease. Future Med. Chem 11, 1953–1977. [DOI] [PubMed] [Google Scholar]
- Eathiraj S, Pan X, Ritacco C, and Lambright DG (2005). Structural basis of family-wide Rab GTPase recognition by rabenosyn-5. Nature 436, 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell MJ, Mirescu C, Basu K, Cheewatrakoolpong B, DeMong DE, Ellis JM, Hyde LA, Lin Y, Markgraf CG, Mei H, et al. (2015). MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J. Pharmacol. Exp. Ther 355, 397–409. [DOI] [PubMed] [Google Scholar]
- Gao Y, Wilson GR, Stephenson SEM, Bozaoglu K, Farrer MJ, and Lockhart PJ (2018). The emerging role of Rab GTPases in the pathogenesis of Parkinson’s disease. Mov. Disord 33, 196–207. [DOI] [PubMed] [Google Scholar]
- Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, et al. (2005). A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 365, 415–416. [DOI] [PubMed] [Google Scholar]
- Gilsbach BK, Eckert M, and Gloeckner CJ (2018). Regulation of LRRK2: insights from structural and biochemical analysis. Biol. Chem 399, 637–642. [DOI] [PubMed] [Google Scholar]
- Gilsbach BK, Ho FY, Vetter IR, van Haastert PJ, Wittinghofer A, and Kortholt A (2012). Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc. Natl. Acad. Sci. USA 109, 10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloeckner CJ, Boldt K, von Zweydorf F, Helm S, Wiesent L, Sarioglu H, and Ueffing M (2010). Phosphopeptide analysis reveals two discrete clusters of phosphorylation in the N-terminus and the Roc domain of the Parkinson-disease associated protein kinase LRRK2. J. Proteome. Res 9, 1738–1745. [DOI] [PubMed] [Google Scholar]
- Goehring A, Lee CH, Wang KH, Michel JC, Claxton DP, Baconguis I, Althoff T, Fischer S, Garcia KC, and Gouaux E (2014). Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc 9, 2574–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotthardt K, Weyand M, Kortholt A, Van Haastert PJ, and Wittinghofer A (2008). Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. EMBO J. 27, 2239–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, and Cookson MR (2009). Leucine-rich repeat kinase 2 mutations and Parkinson’s disease: three questions. ASN Neuro. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guaitoli G, Raimondi F, Gilsbach BK, Gomez-Llorente Y, Deyaert E, Renzi F, Li X, Schaffner A, Jagtap PK, Boldt K, et al. (2016). Structural model of the dimeric Parkinson’s protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc. Natl. Acad. Sci. USA 113, E4357–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Hou X, Goody RS, and Itzen A (2013). Intermediates in the guanine nucleotide exchange reaction of Rab8 protein catalyzed by guanine nucleotide exchange factors Rabin8 and GRAB. J. Biol. Chem 288, 32466–32474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, et al. (2008). Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma Y, Hiragi S, and Fukuda M (2020). Rab family of small GTPases: an updated view on their regulation and functions. FEBS J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui KY, Fernandez-Hernandez H, Hu J, Schaffner A, Pankratz N, Hsu NY, Chuang LS, Carmi S, Villaverde N, Li X, et al. (2018). Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobucci C, Gotze M, Ihling CH, Piotrowski C, Arlt C, Schafer M, Hage C, Schmidt R, and Sinz A (2018). A cross-linking/mass spectrometry workflow based on MS-cleavable cross-linkers and the MeroX software for studying protein structures and protein-protein interactions. Nat. Protoc 13, 2864–2889. [DOI] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, and Alessi DR (2007). LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J 405, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao AH, Chiu CL, Vellucci D, Yang YY, Patel VR, Guan SH, Randall A, Baldi P, Rychnovsky SD, and Huang L (2011). Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell Proteomics 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick BS, Kerwin BA, Chang BS, and Philo JS (2001). Online size-exclusion high-performance liquid chromatography light scattering and differential refractometry methods to determine degree of polymer conjugation to proteins and protein-protein or protein-ligand association states. Anal. Biochem 299, 136–146. [DOI] [PubMed] [Google Scholar]
- Kett LR, Boassa D, Ho CC, Rideout HJ, Hu J, Terada M, Ellisman M, and Dauer WT (2012). LRRK2 Parkinson disease mutations enhance its microtubule association. Hum. Mol. Genet 21, 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, Karcher A, Pellis M, Muyldermans S, Casas-Delucchi CS, Cardoso MC, et al. (2010). Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol 17, 133–U162. [DOI] [PubMed] [Google Scholar]
- Kornev AP, and Taylor SS (2010). Defining the conserved internal architecture of a protein kinase. Biochim. Biophys. Acta 1804, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BD, Dawson VL, and Dawson TM (2012). Leucine-rich repeat kinase 2 (LRRK2) as a potential therapeutic target in Parkinson’s disease. Trends Pharmacol. Sci 33, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Yang S, Kavdia K, Sifford JM, Wu Z, Xie B, Wang Z, Pagala VR, Wang H, Yu K, et al. (2020). Deep profiling of microgram-scale proteome by tandem mass tag mass spectrometry. J. Proteome Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Bryant N, Kumaran R, Beilina A, Abeliovich A, Cookson MR, and West AB (2018). LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum. Mol. Genet 27, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T, Hulihan MM, Cobb SA, Wu RM, Lu CS, et al. (2005). LRRK2 pathogenic substitutions in Parkinson’s disease. Neurogenetics 6, 171–177. [DOI] [PubMed] [Google Scholar]
- McGrath E, Waschbusch D, Baker BM, and Khan AR (2019). LRRK2 binds to the Rab32 subfamily in a GTP-dependent manner via its armadillo domain. Small GTPases, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monfrini E, and Di Fonzo A (2017). Leucine-Rich Repeat Kinase (LRRK2) Genetics and Parkinson’s Disease. Adv. Neurobiol 14, 3–30. [DOI] [PubMed] [Google Scholar]
- Noble MEM, Endicott JA, and Johnson LN (2004). Protein kinase inhibitors: Insights into drug design from structure. Science 303, 1800–1805. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, Martinez TN, Tonelli F, Pfeffer SR, and Alessi DR (2018). Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J. 37, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, Martinez TN, Tonelli F, Pfeffer SR, and Alessi DR (2019). Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith AD, Bamborough P, Jandu K, Andreotti D, Mensah L, Dossang P, Choi HG, Deng X, Zhang J, Alessi DR, et al. (2012). GSK2578215A; a potent and highly selective 2-arylmethyloxy-5-substitutent-N-arylbenzamide LRRK2 kinase inhibitor. Bioorg. Med. Chem. Lett 22, 5625–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbusch KE, and Kortholt A (2016). Activation mechanism of LRRK2 and its cellular functions in Parkinson’s disease. Parkinsons Dis. 2016, 7351985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SH, Knape MJ, Boassa D, Mumdey N, Kornev AP, Ellisman MH, Taylor SS, and Herberg FW (2019). The dynamic switch mechanism that leads to activation of LRRK2 is embedded in the DFGpsi motif in the kinase domain. Proc. Natl. Acad. Sci. USA 116, 14979–14988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SH, Weng J, Aoto PC, Boassa D, Mathea S, Silletti S, Hu J, Wallbott M, Komives EA, Knapp S, Herberg FW, and Taylor SS (2020). Conformation and dynamics of the kinase domain drive subcellular location and activation of LRRK2. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, and Wolfson HJ (2005). PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, W363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejwal K, Chami M, Remigy H, Vancraenenbroeck R, Sibran W, Sutterlin R, Baumgartner P, McLeod R, Chartier-Harlin MC, Baekelandt V, et al. (2017). Cryo-EM analysis of homodimeric full-length LRRK2 and LRRK1 protein complexes. Sci. Rep 7, 8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZJ, Zhang SO, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, Solanoy HO, Drummond J, Zhang XL, Ding X, et al. (2012). Ser(1292) autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of pd mutations. Science Translational Medicine 4. [DOI] [PubMed] [Google Scholar]
- Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, Tonelli F, Martinez TN, Lorentzen E, Pfeffer SR, et al. (2017). Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M, and Alessi DR (2020). Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell Biol 63, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ung PM, Rahman R, and Schlessinger A (2018). Redefining the protein kinase conformational space with machine learning. Cell Chem. Biol 25, 916–924 e912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter IR, and Wittinghofer A (2001). The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang YL, Li YX, Bai B, Wang XS, Tan HY, Liu T, Beach TG, Peng JM, and Wu ZP (2015). Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J. Proteome Res 14, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Wu Z, Wang H, Tan H, and Peng J (2014). JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell Proteomics 13, 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Buschauer R, Bohning J, Audagnotto M, Lasker K, Lu TW, Boassa D, Taylor S, and Villa E (2020). The In Situ structure of Parkinson’s disease-linked LRRK2. Cell 182, 1508–1518 e1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Saleh T, Rossi P, and Kalodimos CG (2020). Conformational states dynamically populated by a kinase determine its function. Science 370, 189-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Fan Y, Ru H, Wang L, Magupalli VG, Taylor SS, Alessi DR, and Wu H (2019). Crystal structure of the WD40 domain dimer of LRRK2. Proc. Natl. Acad. Sci. USA 116, 1579–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Structural determination of LRRK2, Related to Figure 1
(A) A brief flow chart of single-particle cryoEM data collection and process.
(B) The flexibility of ANK and ARM domains in the context of LRRK2 monomers.
(C) The flexibility of ANK and ARM domains in the context of LRRK2 dimers.
Figure S2. Structural features of LRRK2, Related to Figure 1
(A) Docking of the ARM domain in cryoEM densities. The cryoEM density of the ARM domain is from local refinement (Fig. S1).
(B) The structural model of the LRR domain with an insertion between the first and second repeats. N-terminal and C-terminal ends of the LRR domain are shown as spheres. Secondary structures are colored differently with alpha-helix in red, beta-strand in yellow and loop in green.
(C) Comparison between LRRK2 and LRRK2RCKW. Left: displacement of ROC–COR domains relative to KIN-WD40 domains. ROC, COR, KIN and WD40 domains in full-length LRRK2 are colored in green, cyan, blue and violet, respectively, and LRRK2RCKW is in grey. Right: rotational movement of the COR domain. The blue arrow indicates the phosphorylated T1343.
(D) Comparison of ROC domain with different Rab proteins. “Switch 1” motifs are indicated by black lines. Structural models and the PDB codes are in same color codes, and LRRK2 ROC domain is colored in green. The cryoEM density of the potential GDP molecule is shown. Residues in the GDP-binding site are shown as sticks.
(E) Superimposition of the WD40 dimer (PDB 6DLO) and the full-length LRRK2 structures. Steric clashes between the WD40 dimer and hinge helix and ARM domain of the full-length LRRK2 are indicated by blue arrows. The WD40 dimer is colored in dark grey.
Figure S3. Interdomain interactions and the kinase domain of LRRK2, Related to Figures 2 and 3
(A) Interdomain interactions at secondary structure level. The linker between LRR and ROC domains is colored in grey.
(B) Comparison of kinase domains between the full-length LRRK2 (blue) and truncated LRRK2RCKW (wheat). The displacement of the N-lobe of the kinase domain is indicated by a dashed arrow. The red arrow points to the “DYG” motif with different secondary structures. Gly-loop following the distorted β1 strand of the kinase domain is indicated by a black line.
(C) The interaction between the LRR domain and the N-lobe of the KIN domain. Sidechains of interface residues are shown as sticks.
(D) Comparison of the cryoEM maps and structural models between LRRK2 wild type and LRRK2G2019S monomers. Wild-type LRRK2 and LRRK2G2019S are colored in blue and grey, respectively.
Figure S4. Structural analysis of LRRK2 dimers, Related to Figure 4
(A) Docking of the ROC–COR dimer into the cryo-ET map. One copy of LRRK2 ROC-COR domains is colored in blue, the other in orange.
(B) Modeling of the LRRK2-Rab complex in the context of the membrane bilayer. Rab is colored in magenta. The C-terminal of Rab that is linked to lipid modification is shown as green spheres. LRRK2 dimers are shown in surface representation.
(C) The interaction between ARM domain and Rab38 at the secondary structure level. On the ARM domain side, it involves three regions: res. 386-392, res. 430-437 and res. 472-478. On the Rab protein side, switch 1 motif, α1, β2 and α2 are on the interface.
(D) Structural comparison between the LRRK2 monomer and dimer.
Data Availability Statement
Data Resources
The three-dimensional cryoEM density maps for human LRRK2 monomer, dimer, symmetry expanded monomer and LRRK2G2019S monomer have been deposited in the EM Database under the accession code EMDB: EMD-23352, EMD-23350, EMD-23360 and EMD-23359, and the coordinates for the structure have been deposited in Protein Data Bank under accession code PDB 7LHW, 7LHT, 7LI4 and 7LI3.