

Abstract
Over the last 20 years, both severe acute respiratory syndrome coronavirus-1 (SARS-CoV-1) and SARS-CoV-2 was transmitted from animal hosts to humans causing zoonotic outbreaks of severe disease. Both viruses originate from a group of betacoronaviruses known as subgroup 2b. The emergence of two dangerous human pathogens from this group along with previous studies illustrating the potential of other subgroup 2b members to transmit to humans has underscored the need for antiviral development against them. Coronaviruses modify the host innate immune response in part through the reversal of ubiquitination and ISGylation with their papain-like protease (PLpro). To identify unique or overarching subgroup 2b structural features or enzymatic biases the PLpro from a subgroup 2b bat coronavirus, BtSCoV-Rf1.2004, was biochemically and structurally evaluated. This evaluation revealed that PLpros from subgroup 2b coronaviruses have narrow substrate specificity for K48 polyubiquitin and ISG15 originating from certain species. The PLpro of BtSCoV-Rf1.2004 was used as a tool alongside PLpro of CoV-1 and CoV-2 to design 30 novel non-covalent drug-like pan subgroup 2b PLpro inhibitors that included determining the effects of using previously unexplored core linkers within these compounds. Two crystal structures of BtSCoV-Rf1.2004 PLpro bound to these inhibitors aided in compound design as well as shared structural features among subgroup 2b proteases. Screening of these three subgroups 2b PLpros against this novel set of inhibitors along with cytotoxicity studies can provide new directions for pan-coronavirus subgroup 2b antiviral development of PLpro inhibitors.
Keywords: severe acute respiratory syndrome 2, coronavirus, COVID-19, PLpro, ubiquitin, ISG5
Graphical Abstract
Coronaviruses from the 2b group have significant potential to cross over to humans with deleterious consequences. SARS-CoV2 being the last one to make this transition. Here, the three-representative papain-like proteases from the 2b group CoVs are depicted, which were used to investigate substrate preferences as well as explore the potential for drug-like pan inhibitors of these proteases. A sampling of their substrates and pan-inhibitor scaffolds investigated are shown at the bottom.
INTRODUCTION
Over the past two decades, betacoronaviruses have spilled over from animals to humans on with deleterious outcomes1–3. Coronaviruses (CoVs) can be divided into four genera: alpha, beta, gamma, and delta coronaviruses. Betacoronaviruses can be further broken down into four subgroups: 2a, 2b, 2c, and 2d. In 2015, a study highlighted that subgroup 2b contained not only the 2003 pandemic-causing CoV-1 but a cluster of 15 SARS-like coronaviruses4. Many SARS-like CoVs are prevalent among Chinese horseshoe bats but they also have been shown to be able to infect a range of hosts2. These viruses are similar enough that after replacing the spike protein of CoV-1 with that of another subgroup 2b virus from bats they remain capable of productively infecting primary human airway cells. For example, these 2b SARS-like CoVs were shown to reach in vitro viral titers equivalent to CoV-14. The 2b SARS-like CoVs have been found to include over 30 human and zoonotic viruses. This list now includes CoV-2 that caused a worldwide pandemic with over 122 million cases and 2.7 million deaths5. As was foreshadowed, the emergence of CoV-2 emphasizes that CoV-2 and CoV-1 are not likely to be the last CoVs to cross over to humans4. The 2003 and 2019 epidemic and pandemic highlight the urgent need to develop effective coronavirus therapeutics that can thwart current and future coronavirus subgroup 2b health threats.
As shown by the COVID-19 pandemic, the development and large-scale distribution of an effective mRNA spike vaccine is achievable. Unfortunately, the two-shot regimen takes around a month to generate robust immunity, and cannot be utilized by individuals with certain immune conditions reducing their ability to respond to outbreaks6. Additionally, some of the most vulnerable populations such as the elderly, newborns, and the immunocompromised are typically unable to receive vaccines7. The spike protein has shown a propensity for mutation, emphasizing the need for alternative disease intervention strategies8, thus, the development of antivirals for CoV is vital.
As with other ssRNA(+) coronaviruses, betacoronaviruses translate two polypeptides pp1a and pp1ab, which are cleaved into 16 nonstructural proteins (Nsp1 to Nsp16). Nsp1-Nsp16 form the viral membrane-bound replicase complex, which is necessary to transcribe the viral RNA genome before translation9. CoVs encode two proteases that process the CoV polyproteins into functional units10. The 3C-like protease, also known as the main protease, cleaves Nsp4-Nsp16. The papain-like protease (PLpro) cleaves Nsp1-Nsp311, 12. The genomes of coronaviruses can contain up to two PLPs. However, the genomes of subgroup 2b viruses mirror that of Middle East respiratory syndrome (MERS) CoV, a subgroup 2c virus, by encoding only one PLpro10.
In addition to serving a critical function in cleaving the viral polypeptide, PLpros as well as papain-like protease 2 (PLP2) from CoVs have immunosuppressive effects on their host by reversing post-translational modification by ubiquitin (Ub) and interferon-stimulated gene product-15 (ISG15)12, 13. Several forms of ubiquitination and ISGylation have been shown to regulate various aspects of the innate immune system14–16. Post-translational modification with Ub and ISG15 has a profound impact on host type I IFN and NFκB inflammatory responses, as well as upregulating the production of cytokines, chemokines, and other ISGs17, 18. Additionally, some Ub-like protein (Ubl) modifiers have been shown to facilitate inhibition, sequestration, or proteasomal degradation of marked proteins19–22. By reversing these modifications, viral encoded PLpros allow critical viral proteins to remain intact, active, and suppress an anti-viral immune state until the virus can replicate. To determine if there are identifiable trends in enzymatic activity within subgroup 2b PLpros, we examined the PLpro from BtSCoV-Rf1.2004. BtSCoV-Rf1.2004 is a subgroup 2b virus isolated from greater horseshoe bats, and of the currently identified 2b viruses it is the most distantly related to CoV-1 other than CoV-2 (Figure S1)4.
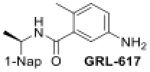
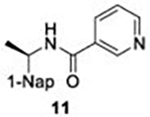
The dual activities of PLpro, i.e, aiding viral replication and suppressing the innate immune system, makes it a highly sought drug target23–26. Its conserved nature among subgroup 2b coronaviruses also presents an opportunity to develop inhibitors that have broad specificity for future subgroup 2b viral threats. Several approaches have been taken to inhibit CoV-1 PLpro, including covalent modification of the catalytic cysteine by electrophilic warheads, as well as competitive inhibition by several non-covalently binding compounds27. This includes two sets of non-covalent drug-like naphthalene-containing compounds that bind to a pocket adjacent to the active site28. Excitingly, these naphthalene-based scaffolds, originally identified to inhibit CoV-1 PLpro, were discovered to possess similar efficacy against CoV-229. This subsequently has drawn significant attention to the potential of these scaffolds to serve as a basis for therapeutic development for CoV-224, 30, 31. These inhibitors typically consist of a naphthyl group and an arene group with various backbones linking them. Thus far the effective inhibitors can be categorized into two sets: series I compounds such as GRL0617 (Figure 1A), which typically utilize an amide backbone, and series II compounds such as 6577871, which commonly have a piperidine scaffold (Figure 1B)27, 29, 32. Both categories have proven effective in inhibiting PLpros from CoV-1 and CoV-2 but neither are fully-optimized in backbone structure or arene ring decoration. Recently many series I analogs have been synthesized based on GRL0617. Some new series I and series II compounds inhibit CoV-2 PLpro with low nanomolar potency but are comparable to existing series I compounds when tested for antiviral activity and cytotoxicity in Vero E6 cells23, 30. Additionally, it remains unknown whether these naphthalene-based inhibitors would be effective against a wider array of coronavirus subgroup 2b PLpros.
Figure 1. PLpro interactions with series I and II naphthalene-based inhibitors.
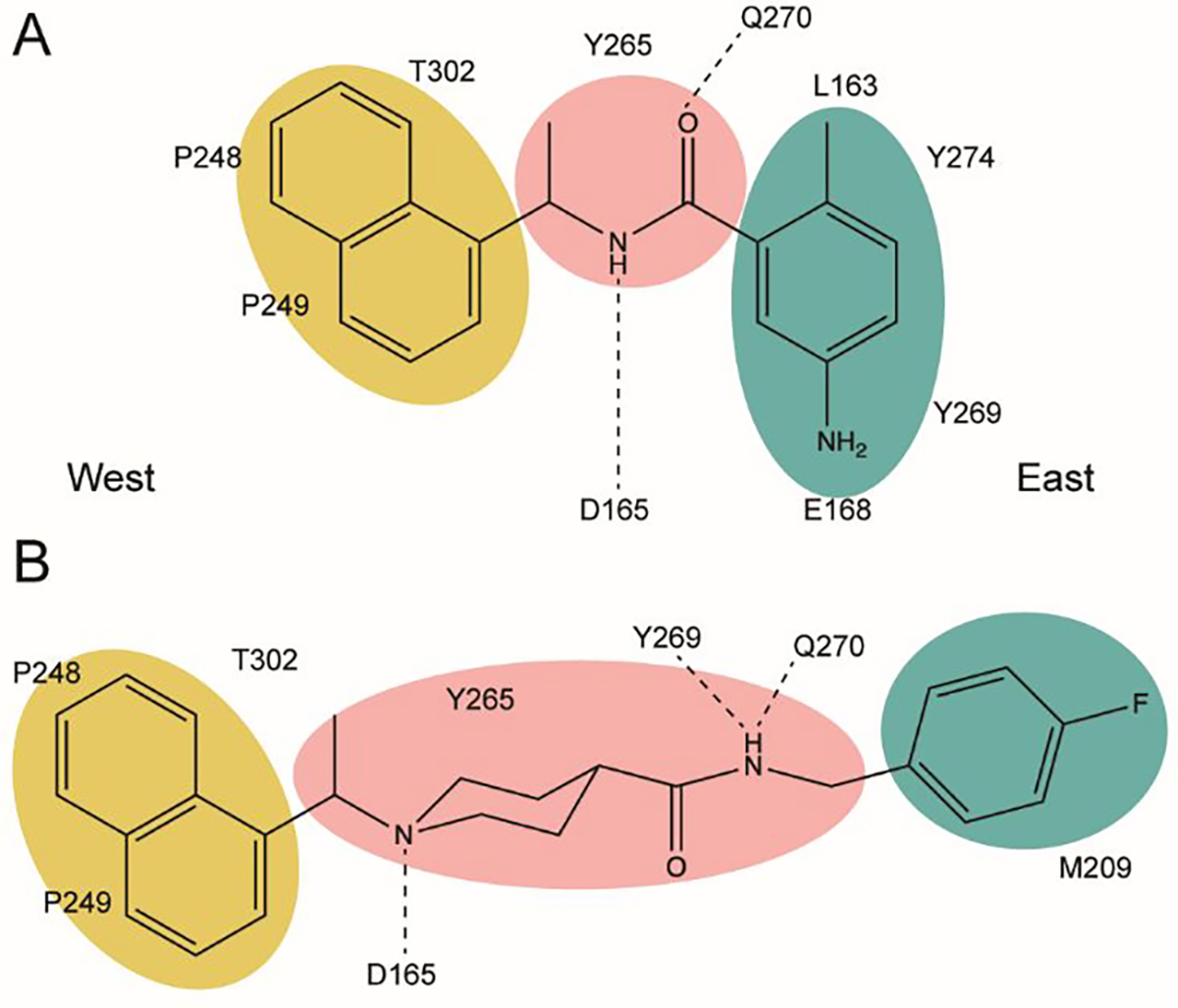
Western naphthyl groups are highlighted in yellow, easter arene groups are highlighted in teal, and core linker groups are highlighted in pink. A. Key hydrophobic and hydrogen bonding interactions of series I inhibitor GRL0617; B. Key hydrophobic and hydrogen bonding interactions of the series II initial hit 6577871.
Here, we evaluate the enzymatic activity of the PLpro of BtSCoV-Rf1.2004 and reveal shared substrate preferences among subgroup 2b coronavirus PLpros. Two structures of BtSCoV-Rf12004 PLpro were solved in complex with non-covalent drug-like naphthalene inhibitors. This new information revealed signature structural motifs that separate subgroup 2b PLpros from those of other coronaviruses. Additionally, these structures in combination with structural activity relationship data obtained with a library composed of compounds with unexplored chemical cores illustrate the potential of developing a novel inhibitor with pan subgroup 2b potency. Evaluation of the cytotoxicity of these compounds against multiple cell lines further emphasizes the potential of non-covalent drug-like naphthalene scaffolds for future therapeutic development against subgroup 2b members.
RESULTS
PLpros are Highly Conserved Proteins Among SARS-like Coronaviruses
Before the emergence of CoV-2, subgroup 2b contained fifteen known SARS-like viruses originating from several species of old-world bats, primarily Horseshoe bats2, 4. Sequence analysis of these fifteen viruses along with that of CoV-2 revealed that each encoded one of six unique PLpro sequences (Figure S1). Point mutations within CoV-2 variant strains have increased the number of unique PLpros with the United Kingdom and Delta variants having A146D mutations, and the Brazilian strain having a K233Q mutation, while the new variant Omicron does not contain any mutations to its PLpro33, 34. Both the genomes and PLpros of subgroup 2b viruses share sequence identity greater than 80 percent. However, PLpros and PLP2 vary in terms of substrate preference and activity. To gain a better understanding of subgroup 2b substrate preferences, a third subgroup 2b PLpro originating from BtSCoV-Rf1.2004 was selected to identify subgroup 2b DUB and deISGylase activity, as well as to assist in subgroup 2b broad-spectrum inhibitor development. BtSCoV-Rf1.2004 was isolated from the greater horseshoe bat (R. ferrumequinum) in Yichang, China, just 320 km from the location of the initial outbreak of the CoV-2 pandemic2. BtSCoV-Rf1.2004 was chosen because at 93.9% conservation it is the most distantly related subgroup 2b virus from CoV-1 other than CoV-2. Highlighting PLpro conservation is among subgroup 2b viruses, the PLpro of BtSCoV-Rf1.2004 retains 97.8% and 82.2% sequence homology, respectively. Initial examination of key functional areas, particularly the active site, naphthalene inhibitor binding pocket, and the Ub interacting motif (UIM) reveal that these areas are highly conserved among subgroup 2b PLpros (Figure S1). Despite three major emergent strains which are now observed only two mutations within CoV-2 PLpro have been detected. The Brazilian strain, K233Q, is located in a functionally relevant location; however, this mutation is conserved converting a lysine residue to the subgroup 2b consensus glutamine residue.
Deubiquitinase and DeISGylase Activity of BtSCoV-Rf1.2004 PLpro
To identify differences in enzyme kinetics between BtSCoV-Rf1.2004 PLpro and those of other subgroup 2b viruses, the kinetic parameters of BtSCoV-Rf1.2004 PLpro for various substrates were determined. The kinetic values for PLpro substrates Ub-AMC and human ISG15-AMC, as well as the last five consensus amino acids between them (RLRGG; peptide-AMC), were determined by monitoring the release of the fluorogenic reporter group 7-amino-4-methylcoumarin (AMC) moiety (Table 1). The catalytic efficiency of BtSCoV-Rf1.2004 PLpro for peptide-AMC is 0.100±0.007 μM−1min−1, which is within the range of other subgroup 2b PLpros. The efficiency of BtSCoV-Rf1.2004 PLpro for Ub-AMC is 1.0±0.1 μM−1min−1 with KM and kcat values of 14.3±0.9 and 13.9±0.4, respectively. Similar to other subgroup 2b viruses, BtSCoV-Rf1.2004 PLpro had a clear preference for ISG15 over Ub. BtSCoV-Rf1.2004 PLpro was not able to be saturated with ISG15-AMC at the concentrations used, however using first-order enzyme kinetics the catalytic efficiency was determined to be 56.6±5.3μM−1min−1. The difficulty in saturating the PLpro appears to be due to its robust deISGylase activity which is twice as efficient as any other SARS-CoV PLpro. Despite not being able to calculate a maximum turnover number, a rate of 595 min−1 was observed. The calculated maximum turnover for ISG15-AMC by CoV-1 is 436min−1, making BtSCoV-Rf1.2004 PLpro the most robust deISGylase among coronavirus PLpros evaluated so far29, 35, 36.
Table 1.
Kinetic analysis of BtSCoV-Rf1.2004 PLpro with PEP-AMC, Ub-AMC, and ISG15-AMC
| Substrate | |||
|---|---|---|---|
|
| |||
| RLRGG-AMC | Ub-AMC | ISG15-AMC | |
|
| |||
| BtSCoV-Rfl.2004 PLpro | |||
| Kcat/Km (μM−1min−1) | 0.100 ± 0.007a | 1.0 ± 0.1 | 56.6 ± 5.3a |
| Kcat (min−1) | 13.9 ± 0.4 | ||
| Km (μM−1) | 14.3 ± 0.9 | ||
|
| |||
| SARS-2 PLprob | |||
| Kcat/Km (μM−1min−1) | 0.0051a | 1.3 ± 0.1 | 10.3 ± 0.5 |
| Kcat (min−1) | 10.0 ± 0.8 | 40.0 ± 1.8 | |
| Km (μM−1) | 7.9 ± 1.4 | 3.9 ± 0.5 | |
|
| |||
| SARS PLproc | |||
| Kcat/Km (μM−1min−1) | 0.3a | 1.5 ± 0.3 | 28.9 ± 5.3 |
| Kcat (min−1) | 75.9 ± 8.1 | 436 ± 40 | |
| Km (μM−1) | 50.6 ± 7.4 | 15.1 ± 2.4 | |
|
| |||
| MERS PLproc | |||
| Kcat/Km (μM−1min−1) | 0.003a | 1.3 ± 0.2 | 9.9 ± 1,6 |
| Kcat (min−1) | 18.8 ± 1.2 | 32.6 ± 1.8 | |
| Km (μM−1) | 14.3 ± 2.0 | 3.3 ± 0.5 | |
|
| |||
| MHV PLP2+d | |||
| Kcat/Km (μM−1min−1) | 0.0016a | 38.3 ± 6.3 | 2.3 ± 0.1a |
| Kcat (min−1) | 49.8 ± 2.9 | ||
| Km (μM−1) | 1.3 ± 0.2 | ||
For nonsaturing substrates kapp is calculated to approximate kcat/Km
The kinetic parameters of SARS-2 PLpro and MERS-CoV is based on Freitas et al. (37)
The kinetic parameters of SARS PLpro and MERS Plpro are from Baez-Santos et al. (35)
The kinetic parameters of MHV PLP2 are from Chen et al. (36)
Poly-Ub Linkage Preferences for BtSCoV-Rf1.2004
PLpros have often shown a greater ability to process poly-Ub chains than those of mono-Ub conjugates24. To determine if this is the case with BtSCoV-Rf1.2004, PLpro cleavage activity was tested against the eight different linkage types of di-Ub M1, K6, K11, K27, K29, K33, K48, and K63. Utilizing similar experimental conditions to those used to evaluate other PLpros29, 38, 10 μM of each di-Ub was incubated with 20 nM PLpro from BtSCoV-Rf1.2004. Divergent from PLpros, and related PLP2s originating out of subgroup 2a and 2c, BtSCoV-Rf1.2004 had modest ability to cleave di-Ub and only a small amount of K48 was cleaved after 60 minutes by BtSCoV-Rf1.2004 PLpro (Figure 2A). This mirrors previous observations seen in assays using other SARS-CoV PLpros which showed little to no activity toward di-Ub29, 38. However, when tested against tetra-Ub chains BtSCoV-Rf1.2004 PLpro demonstrated significantly higher DUB activity (Figure 2B). The K48 linked tetra-Ub was entirely converted to di-Ub in <5 minutes and well-defined bands of tri and di-Ub appeared in the K63 tetra-Ub cleavage experiment after 40 minutes.
Figure 2. Deubiquitination and deISGylation Activity of BtSCoV-Rf1.2004 PLpro.
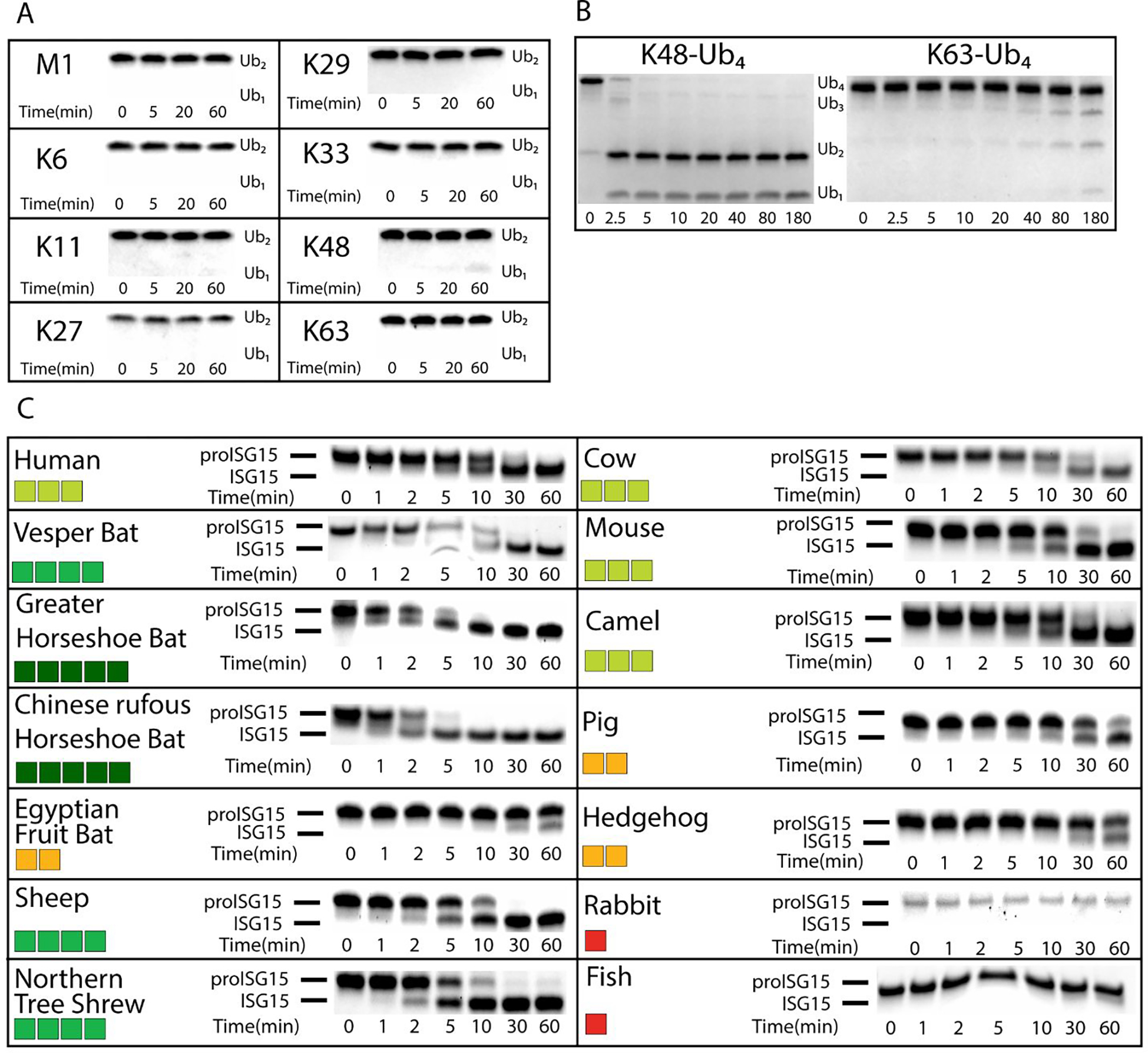
. Different poly-Ub linkages were tested against BtSCoV-Rf1.2004 A. At 37°C 10 μM each of M1, K6, K11, K27, K29, K33, K48, and K63 linked di-Ub were incubated with 20nM BtSCoV-Rf1.2004 PLpro. Samples were taken from the reaction tube at indicated time points. B. Under similar reaction conditions 13.65μM each of K48 and K63 linked tetra-Ub was incubated with 23nM PLpro for 3 hours with samples taken at given time points. C. Activity of BtSCoV-Rf1.2004 PLpro for proISG15 from multiple species. BtSCoV-Rf1.2004 PLpro was evaluated for the cleavage of proISG15s from the following species: human (Homo sapiens; AAH09507.1), cow (Bos taurus; NP_776791.1), vesper bat (Myotis davidii; ELK23605.1), Egyptian fruit bat (Rousettus aegyptiacus; XP_015999857.1), pig (Sus scrofa; ACB87600.1), hedgehog (Erinaceus europaeus; XP_007525810.2), mouse (Mus musculus; AAB02697.1), dromedary camel (Camelus dromedarius; XP_010997700.1), sheep (Ovis aries; AF152103.1), northern tree shrew (Tupaia belangeri; AFH66859.1), greater horseshoe bat (Rhinolophus ferrumequinum; XP_032969719.1), Chinese rufous horseshoe bat (Rhinolophus sinicus; XP_019567580.1), rabbit (Oryctolagus cuniculus; XP_017195918), and jackknife fish (Oplegnathus fasciatus; BAJ16365.1). At 37°C, 10μM of each ISG15 was incubated with 20nM of SARS-CoV-2 PLpro for at least 1hr with samples taken at the time points indicated. The summary of the proISG15 cleavage assays for BtSCoV-Rf1.2004 PLpro is presented as a heat map. Colors range from dark red (no cleavage) to green (relatively robust cleavage)
Species Specific DeISGylase Activity of BtSCoV-Rf1.2004 PLpro
DeISGylases are selective for ISG15s from species which they have been found to productively infect10, 29, 39–41. Unlike Ub, which is almost completely conserved between species, ISG15 can vary with sequence similarity as low as 60% within the Mammalia class39. In vivo, ISG15 is translated as a pro-form that consists of the mature ISG15 with several amino acids following the LRLRGG cleavage site (proISG15). Given the similarity of BtSCoV-Rf1.2004 PLpro with other subgroup 2b PLpros in substrate preference for ISG15 over mono-Ub, molecular weight shift experiments using proISG15s from various species were used to assess ISG15 species preference among a collection of fourteen species10, 29, 39. The experimental parameters that were employed were consistent with previous studies focusing on other PLpros, incubating 20 nM PLpro with 10 μM proISG15 originating from the various species (Figure 2C)10, 29.
In line with previous findings of other PLpros preferring ISG15s from species that serve as reservoirs for their parent virus, BtSCoV-Rf1.2004 PLpro distinctly possesses the highest activity towards ISG15s encoded by the greater horseshoe bat and the Chinese horseshoe bat. Horseshoe bats are known reservoirs for subgroup 2b viruses and are present throughout much of southeast Asia, sub-Saharan Africa, and southern Europe2. BtSCoV-Rf1.2004 PLpro also has varying levels of proficiency for cleaving vesper bat, sheep, and northern tree shrew proISG15s with greater activity toward human, cow, camel, and mouse. Relatively weak activity towards pig, hedgehog, and Egyptian fruit bat was observed with no detectable activity against rabbit or fish ISG15. The cleavage rate of several important ISG15s such as human, mouse, and vesper bat by BtSCoV-Rf1.2004 PLpro closely matched what was observed with CoV-1 and CoV-210, 29. While some species ISG15s are cleaved more slowly than long-chain K48-Ub, those of BtSCoV-Rf1.2004’s natural hosts R. ferrumequinum and R. sinicus appear to be the preferred substrates.
X-ray crystal structure of betacoronavirus subgroup 2b BtSCoV-Rf1.2004 PLpro
To evaluate any differences between subgroup 2b PLpros, an X-ray crystal structure of the BtSCoV-Rf1.2004 PLpro was obtained for comparison to structures of CoV-1 and CoV-2 PLpros. The structure was determined to a resolution of 3.16Å in space group P21212 (Table S1). A homology model of BtSCoV-Rf1.2004 PLpro based on a CoV-1 PLpro (PDB 3E9S) catalytic core was used as a search model. Upon finding two monomers of the catalytic core in the asymmetric unit, the Ubl domain for the monomers was subsequently located using the Ubl of 3E9S as a search model.
Globally, the BtSCoV-Rf1.2004 PLpro resembles other subgroup 2b PLpros in secondary and tertiary structure (Figure 3A/B). It consists of a catalytic core made up of a palm, thumb, and zinc finger domain. The core was found in a holo, open conformation32. Additionally, the PLpros contain an N-terminal Ubl domain. The Ubl domain of BtSCoV-Rf1.2004 is shifted approximately 90 degrees from previously seen elongated forms10 and is tucked against the catalytic domain when compared to the typical extended conformation (Figure S1). Until now this tucked conformation had only been seen in CoV-1 PLpro in complex with mouse ISG15 (PDB 5TL7). When in this conformation a seam of electrostatic interactions is formed between residues on the Ubl domain and α-helix 3 of the thumb domain. These interactions appear to stabilize the association of the two domains.
Figure 3. Tertiary structure of subgroup 2b PLpros compared to groups 2a and 2c.
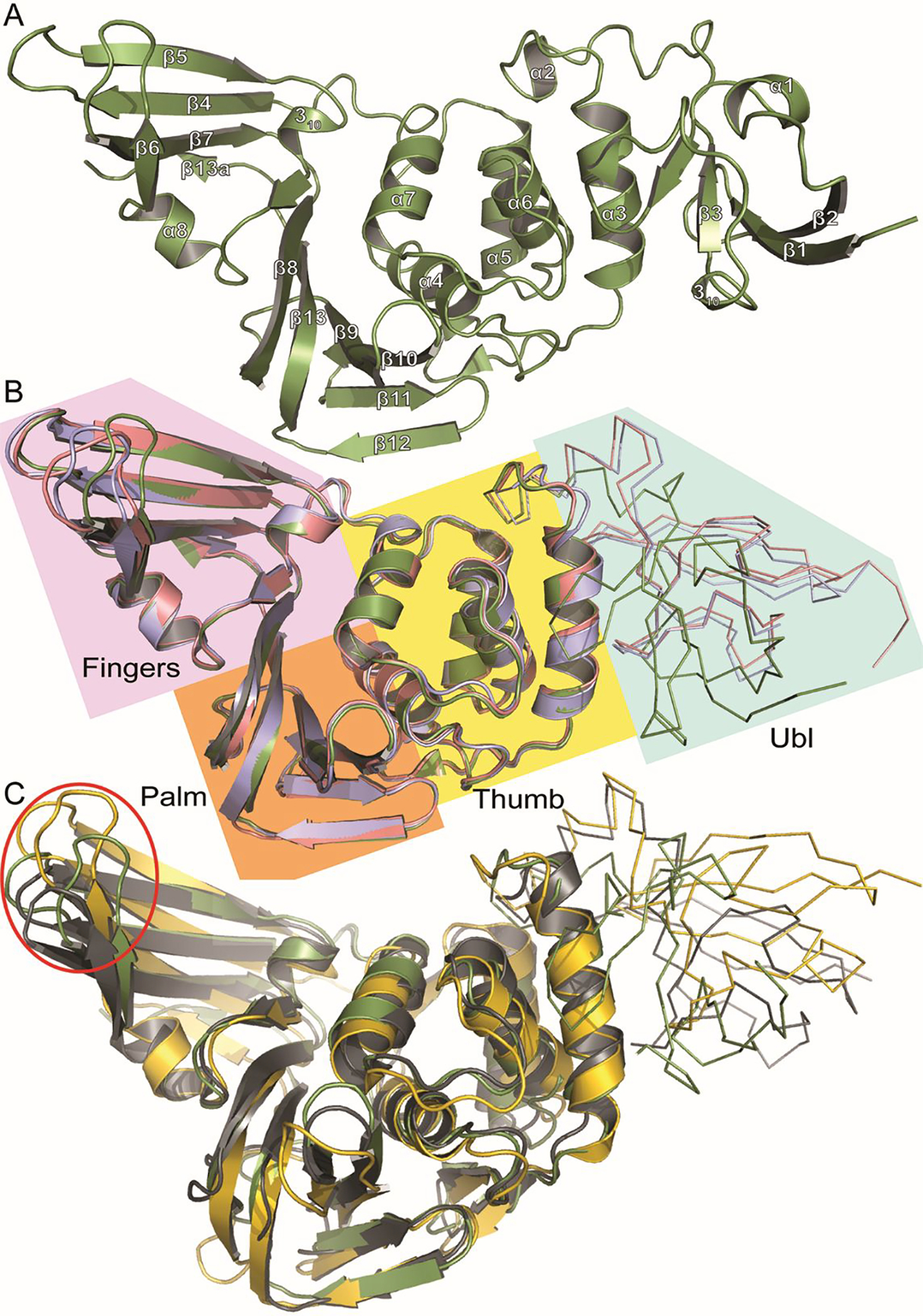
A. Cartoon representation of BtSCoV-Rf1.2004 PLpro (Green) secondary structure with helix and sheet labels corresponding to Figure S1 dssp calculations. B. Overlaid cartoon representations of BtSCoV-Rf1.2004 PLpro (Green), SARS-CoV-1 PLpro (PDB 3E9S) (Pink), and SARS-CoV-2 PLpro (PDB 7JIR) (Blue). The four PLpro domains are labeled and color coded: Fingers (Purple), Palm (Orange), Thumb (Yellow), Ubl (Cyan). C. Overlaid cartoon representations of BtSCoV-Rf1.2004 PLpro (Green), MERS-CoV PLpro (PDB 5W8T)(Yellow), and MHV PLP (5WFI)(Grey) with their Ubl domains represented by ribbons. The Red Circle denotes the Finger domain
When compared with the PLP of the group 2a MHV (PDB 5WFI) and the PLpro of group 2c MERS-CoV (PDB 5W8T), it appears that most secondary structures are conserved between these proteases, however, there is variation in the overall tertiary structure between the PLpros of coronavirus subgroups (Figure 3C). The finger domains vary the most between subgroups, with not only loops shifting, but β-sheets orienting differently. This is most pronounced in the MERS PLpro, where the finger domain is shifted approximately 6 Å away from the P3/P4 pocket relative to MHV and BtSCoV-Rf1.2004. This variance seems to go beyond the open and closed conformations of the finger domain induced by substrate binding previously observed42, 43.
Interactions between GRL0617 and the P3-P4 pocket of BtSCoV-Rf1.2004 PLpro
When examining the BL2 loop Fo-Fc density was readily observed for GRL0617 in both monomers (Figure 4A). Within the pocket GRL0617 is oriented similarly in all subgroup 2b PLpros, forming hydrogen bonds with D165 and Q270, as well as hydrophobic interactions with P248 and P249 (Figure 4C). However, between the monomers of BtSCoV-Rf1.2004, there are differences in the positions of the Q270 side chain, the eastern arene ring, and most notably the western naphthyl group (Figure 4C). The differences appear to be due to a crystal contact present on the B chain that is absent on the A chain in which V226 of the B chain zinc finger forms a hydrophobic interaction with the western naphthyl group of B chain GRL0617 (Figure 5).
Figure 4. Inhibitor binding pocket of three subgroup 2b viruses.
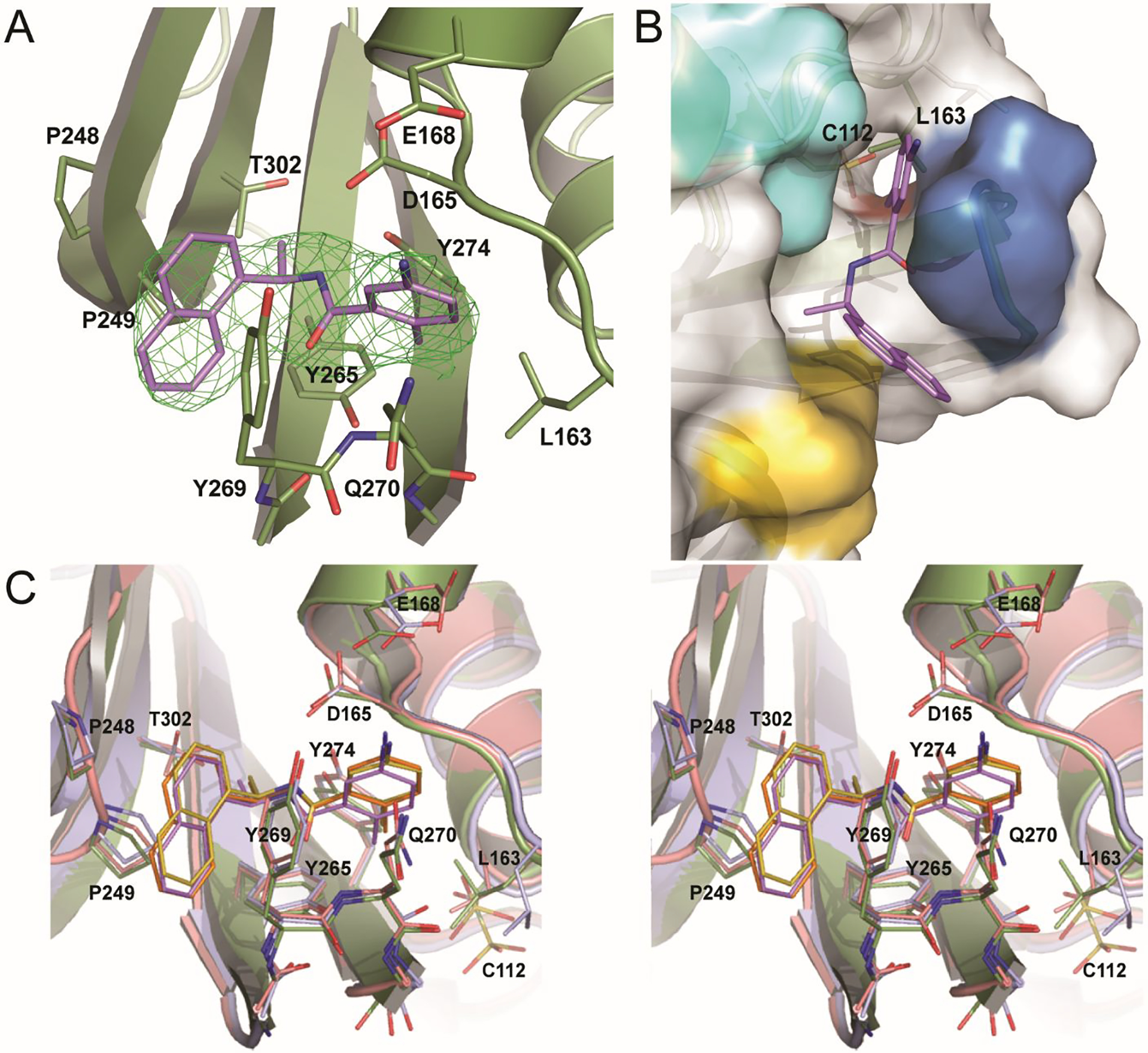
A. A Fo-Fc electron density composite omit map is shown contoured at 3σ (green mesh). With GRL0617 shown in purple and BtSCoV-Rf1 PLpro shown in green. B. GRL0617 (purple) bound to BtSCoV-Rf1 PLpro (green cartoon) overlaid with SARS-CoV-2 (white surface and cartoon) showing a possible path to active site for future inhibitors. C. Stereoview overlay of GRL0617 bound to three different different SARS-CoV PLpros: BtSCoV-Rf1 (green and purple) (PDB 7SKQ), SARS-CoV-1 (pink and orange) (PDB 3E9S), and SARS-CoV-2 (blue and yellow) (PDB 7JIR).
Figure 5. Crystal contacts affecting PLpro conformation (PDB 7SKQ).
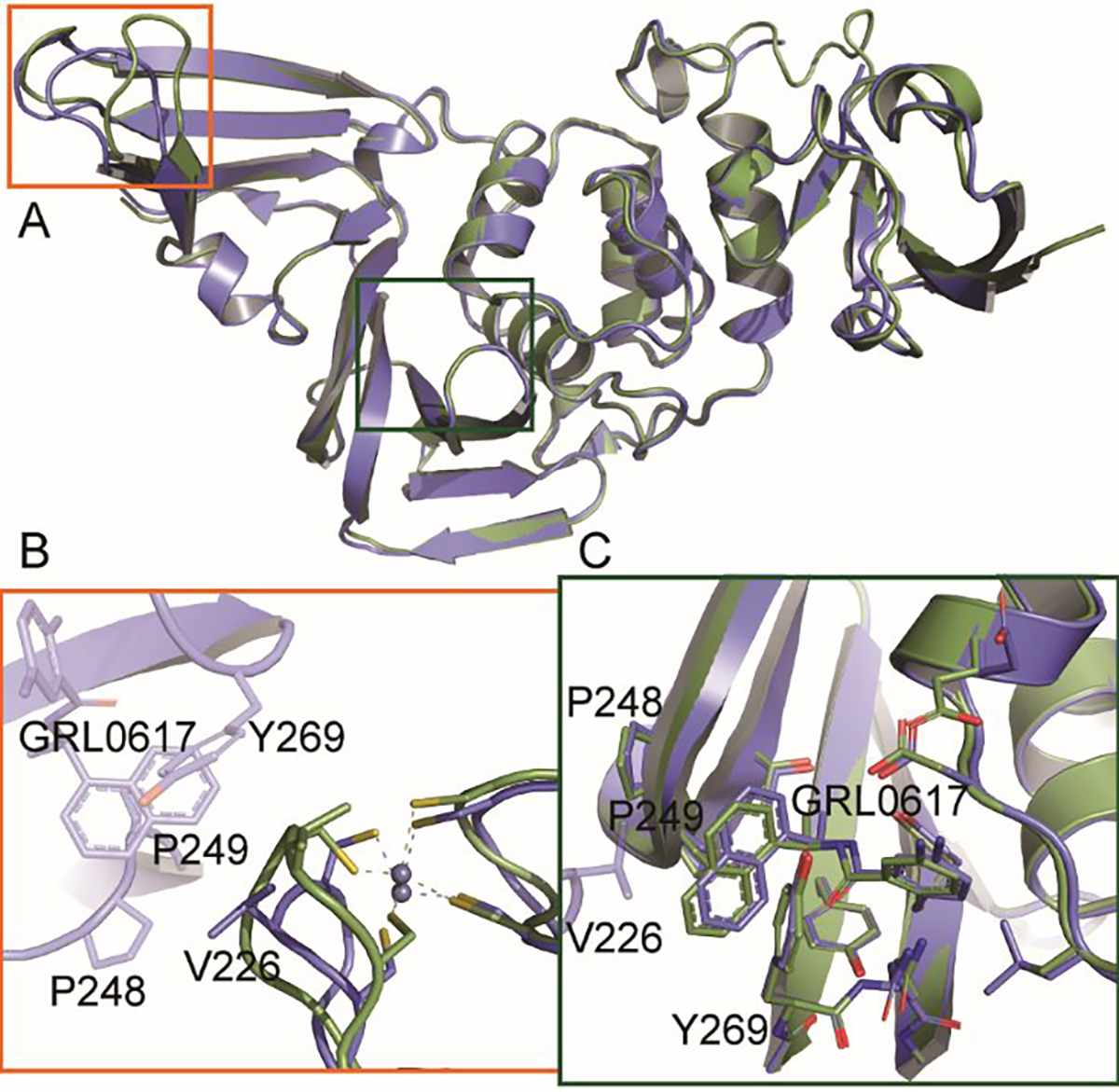
A. Overlaid cartoon representations of BtSCoV-Rf1.2004 PLpro chain A (green) and chain B (blue). B. Zinc finger loops of the two domains differ due to a crystal contact being made by chain B. C. GRL0617 of chain B is shifted in the binding pocket due to a crystal contact with chain B V226.
Crystal contacts on the BL2 loops of CoV-1 and CoV-2 PLpros influence the positioning of the loops and GRL0617 in these structures as well. The absence of a BL2 loop contact on the A chain of BtSCoV-Rf1.2004 PLpro likely places GRL0617 in its most natural orientation within the pocket. The chain B V226 contact is also responsible for a shift in the zinc finger loop (Figure 5). The zinc finger loop appears to be the most variable region within the catalytic core of the PLpro (Figure 3B). CoV-1 and CoV-2 PLpro structures form different crystal contacts at similar sites to those of BtSCoV-Rf1.2004 chain B that cause shifts in the zinc finger and BL2 loops. This suggests the influence of crystal contacts should be considered when using these structures in a structure-based drug design approach.
Beyond the crystal lattice induced changes to the BL2 loop among the subgroup 2b PLpro structures, the only other notable difference is a rotamer in residue L163, which in CoV-1 and BtSCoV-Rf1.2004 is angled towards the 2-methyl group of GRL0617, but in CoV-2 angled away from the pocket (Figure 4B). This altered conformation is seen in structures of CoV-1 PLpro bound to human and mouse ISG15, along with a shifted BL2 loop to accommodate the C-terminal GG cleavage motif of Ubl substrates (PDB 5TL6 and 5TL7). These changes provide insight into the flexibility of the BL2 loop when accommodating BL2 inhibitors.
Inhibition of SARS-CoV Family PLpros with Novel Inhibitors
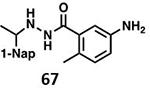
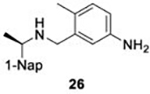
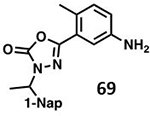
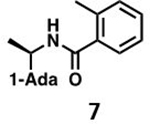
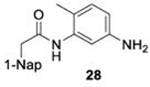
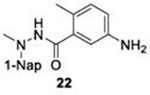
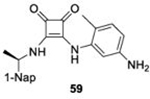
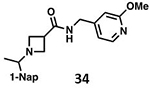
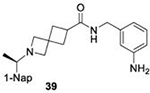
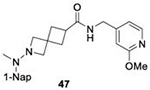
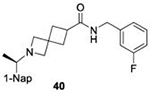
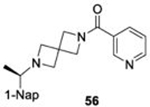
With GRL0617 being readily accommodated within the BtSCoV-Rf1.2004 PLpro P3/P4 site, the practicality of targeting PLpro broadly appears feasible. As a result, 30 compounds were synthesized based on series I and series II compounds designed for the inhibition of CoV-1 PLpro. All compounds were assayed for their ability to inhibit CoV-2, CoV-1, and BtSCoV-Rf1.2004 PLpros, with an initial survey determining percent inhibition at a fixed concentration of 50μM (Table 2).
Table 2. Naphthalene-based inhibitor efficacy against subgroup 2b PLpros.
Percent inhibition of CoV-2 (blue), CoV-1 (red), and BtSCoV-Rf1.2004 (green) PLpros ability to cleave peptide-AMC when incubated with series I (A) and II (B) compounds at 50μM.
| A | ||||||
|---|---|---|---|---|---|---|
| Compound | Percent Inhibition | Compound | Percent Inhibition | Compound | Percent Inhibition | |
|
97.2 ± 0.1 |
|
22.5 ± 7.4 |
|
13.8 ± 1.4 | |
| 98.3 ± 0.0 | 28.4 ± 3.4 | NC | ||||
| 97.7 ± 0.2 | 17.6 ± 3.7 | NC | ||||
|
* |
|
8.9 ± 2.5 |
|
NC | |
| 29.1 ± 5.3 | 20.0 ± 7.7 | |||||
| 19.3 ± 1.9 | NC | |||||
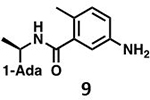
|
9.3 ± 5.4 |
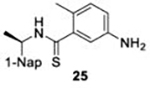
|
38.0 ± 3.8 |
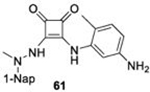
|
14.3 ± 3.8 | |
| 12.4 ± 6.2 | 45.6 ± 2.9 | 29.0 ± 1.9 | ||||
| 18.3 ± 2.6 | 18.3 ± 3.4 | 16.9 ± 5.1 | ||||
|
73.0 ± 0.8 |
|
53.0 ± 2.7 |
|
11.4 ± 0.8 | |
| 73.7 ± 1.0 | 56.6 ± 3.2 | NC | ||||
| 66.4 ± 1.1 | 66.4 ± 1.1 | NC | ||||
|
12.0 ± 1.4 |
|
10.0 ± 1.1 |
|
44.6 ± 2.5 | |
| 22.9 ± 3.4 | 29.2 ± 1.6 | 51.6 ± 1.3 | ||||
| 21.8 ± 6.6 | NC | 17.0 ± 5.2 | ||||
|
39.4 ± 3.0 |
|
18.0 ± 3.1 | |||
| 44.4 ± 3.9 | 18.1 ± 5.4 | |||||
| 12.9 ± 7.0 | 19.3 ± 1.4 | |||||
| B | ||||||
| Compound | Percent Inhibition | Compound | Percent Inhibition | Compound | Percent Inhibition | |
|
77.6 ± 0.5 |
|
63.8 ± 0.6 |
|
98.4 ± 0.1 | |
| 83.2 ± 0.8 | 68.5 ± 2.7 | 94.6 ± 0.2 | ||||
| 76.3 ± 0.3 | 59.6 ± 2.8 | 89.1 ± 0.4 | ||||
|
82.1 ± 1.2 |
|
67.6 ± 2.2 |
|
97.2 ± 0.1 | |
| 92.5 ± 0.2 | 72.0 ± 1.2 | 98.0 ± 0.1 | ||||
| 87.2 ± 0.2 | 64.7 ± 0.5 | 97.1 ± 0.2 | ||||
|
78.3 ± 0.3 |
|
21.9 ± 2.2 |
|
NC | |
| 80.0 ± 0.3 | 19.7 ± 3.5 | 19.5 ± 3.9 | ||||
| 69.1± 0.2 | 11.1 ± 1.3 | 17.0 ± 5.2 | ||||
|
49.3 ± 5.0 |
|
9.6 ± 1.7 | SARS-CoV-2 PLpro SARS-CoV-1 PLpro BtSCoV-Rf1.2004 PLpro | ||
| 66.1 ± 0.8 | NC | |||||
| 24.8 ± 2.5 | 8.0 ± 5.1 | |||||
|
66.0 ± 4.9 |
|
NC | |||
| 77.8 ± 0.4 | NC | |||||
| 41.6 ± 4.1 | 5.5 ± 1.6 | |||||
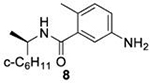
Due to solubility limitations the inhibition of subgroup 2b PLpros by 50μM 8 could not be accurately assessed.
While variation in PLpro sequences exists among subgroup 2b viruses, the residues lining the active site and the adjacent P3 and P4 sites are fully conserved (Figure S1). These residues bind RLRGG residues of Ubl substrates to facilitate cleavage13. They have also been shown to be critical in binding naphthalene-based compounds that successfully inhibit PLpro cleavage activity in CoV-1 and CoV-232. Preliminary studies show that inhibitors designed to bind at this site in CoV-1 exhibit similar IC50 and EC50 values when tested against CoV-229 (Table 3).
Table 3.
IC50 values of series I and II compounds when tested against PLpros of CoV-2 (Blue), CoV-1(Red), and BtSCoV-Rf1.2004 (green) PLpros.
| Compound | IC50 (μM) | Compound | IC50 (μM) | Compound | IC50 (μM) |
|---|---|---|---|---|---|
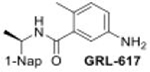
|
22.4 ± 0.2 |
|
27.3 ± 3.6 |
|
1.1 ± 0.1 |
| 1.4 ± 0.0 | 11.4 ± 0.5 | 1.0 ± 0.0 | |||
| 1.2 ± 0.0 | 15.8 ± 1.7 | 8.4 ± 0.8 | |||
|
23.5 ± 1.7 |
|
5.4 ± 0.2 |
|
1.8 ± 0.1 |
| 14.5 ± 0.6 | 10.7 ± 0.7 | 1.6 ± 0.0 | |||
| 26.2 ± 1.4 | 7.5 ± 0.5 | 2.4 ± 0.1 | |||
|
41.1 ± 3.2 |
|
19.6 ± 3.0 |
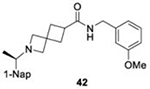
|
28.6 ± 2.0 |
| 36.8 ± 2.3 | 15.2 ± 1.3 | 9.1 ± 1.1 | |||
| 26.2 ± 1.4 | 21.0 ± 3.1 | 22.5 ± 1.7 |
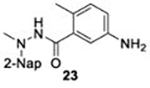
The naphthalene-based inhibitors can be categorized into two groups by their backbone structure. The series I and II hits share a common binding site for their western hydrophobic moieties. The two series differ in the eastern arene moieties, which occupy proximal but different binding pockets, and by the central core – a simple amide unit in series I and a piperidine ring in series II (Figure 1). This suggests that there exists considerable capacity for innovation in the central core, in terms of both the spacing it provides between the eastern and western arenes and the basic structure employed. With this in mind, we set out to explore alternative central cores based on piperidine analogs such as azetidines, and spiroazetidines44, and other bicyclic systems that would allow for variation of the separation between the eastern and western arene while retaining the basic geometry of a 4-substituted piperidine ring.
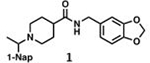
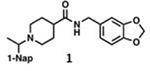
The X-ray crystallographically-derived essential interactions of series I compound GRL0617 in complex with the CoV-1 PLpro (PDB 3E9S) are summarized in Figure 1A. Early X-ray structures of a series II inhibitor 1 with CoV-1 PLpro (PDB 3MJ5) revealed the occupancy of the same hydrophobic pocket by the naphthalene ring but showed the piperidine ring in an unlikely high-energy twist-boat conformation with no interaction of either the basic ring nitrogen or the adjacent amide NH with D16545. A subsequent reevaluation of this X-ray structure, however, represented the piperidine ring in a more standard chair conformation with an H-bond from the basic piperidine nitrogen to D16546. Adopting the latter structure as the more likely, the strong parallels between the interactions of the western halves of the series I and II compounds with the protein are evident and form a solid base on which to engineer next-generation compounds for the inhibition of COV-2 PLpro. The aromatic rings of the eastern halves of the series I and II compounds occupy different sites because of the dissimilar dimensions of the central portions. In series I the backbone amide of Q270 H-bonds to the amide carbonyl of the inhibitor, whereas in series II the hydrocarbon part of the Q270 side chain forms one face of hydrophobic pocket encapsulating the eastern arene, which is completed by the side chain of M209 and other residues. Y269 interacts with the amino group in the eastern arene of the series I compound, but with the amide carbonyl of the series II compound. Chemical Synthesis of all compounds is described in full in the SI.
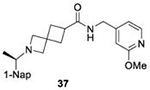
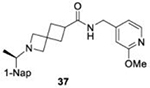
Structure of BtSCoV-Rf1.2004 PLpro – 37 Complex
There is a paucity of data on how series II compounds interact with subgroup 2b PLpros, in addition to no structures of compounds with spiroazetidine backbones. SAR data indicate that the use of either piperidine or spiroazetidine backbones will affect the potency of series II inhibitors. Perhaps more importantly, it shows that the optimal decoration of the eastern arene ring differs depending on which backbone is used. To explore these differences, we crystallized BtSCoV-Rf1.2004 PLpro in complex with 37. Utilizing X-ray diffraction its structure was determined to a resolution of 2.89Å in space group P6422 (Table S1). The BtSCoV-Rf1.2004 PLpro-GRL0617 complex structure was used as a search model for phasing the catalytic core. Upon finding a single monomer in the asymmetric unit, the Ubl domain was added. As with the GRL0617 co-crystal structure, the Ubl domain of this structure was in a tucked conformation (Figure S1).
Within the active site, Fo-Fc density fitting 37 is readily observed within the P3/P4 binding pocket (Figure 6A). In this pocket 37 forms five hydrogen bonds with the protein. The amide scaffold forms hydrogen bonds with the main chains of L163, Y269, and Q270, while spiroazetidine nitrogen forms bonds with the side chains of D165 and Y274. Additionally, strong hydrophobic interactions are formed between the western naphthyl group and P248 and P249 as is typical of naphthalene-based PLpro inhibitors. However, when compared with a structure of CoV-1 PLpro in complex with 1 (PDB 3MJ5) we see that the naphthyl group of 37 is situated approximately 0.9Å further away from the two prolines (Figure 6B). The difference in naphthyl group location could be due to a shift of the BL2 loop caused by the bulky 1,3-dioxolane ring. The eastern arene groups of both 37 and 1 cause a shift in the BL2 loop that is similar to that observed in order to accommodate Ubl substrates. However, this shift is more pronounced in the 1 co-crystal structure, even causing Q270 to flip away from the arene rings to avoid a steric clash.
Figure 6. BtSCoV-Rf1 PLpro in complex with 37 (PDB 7SKR).
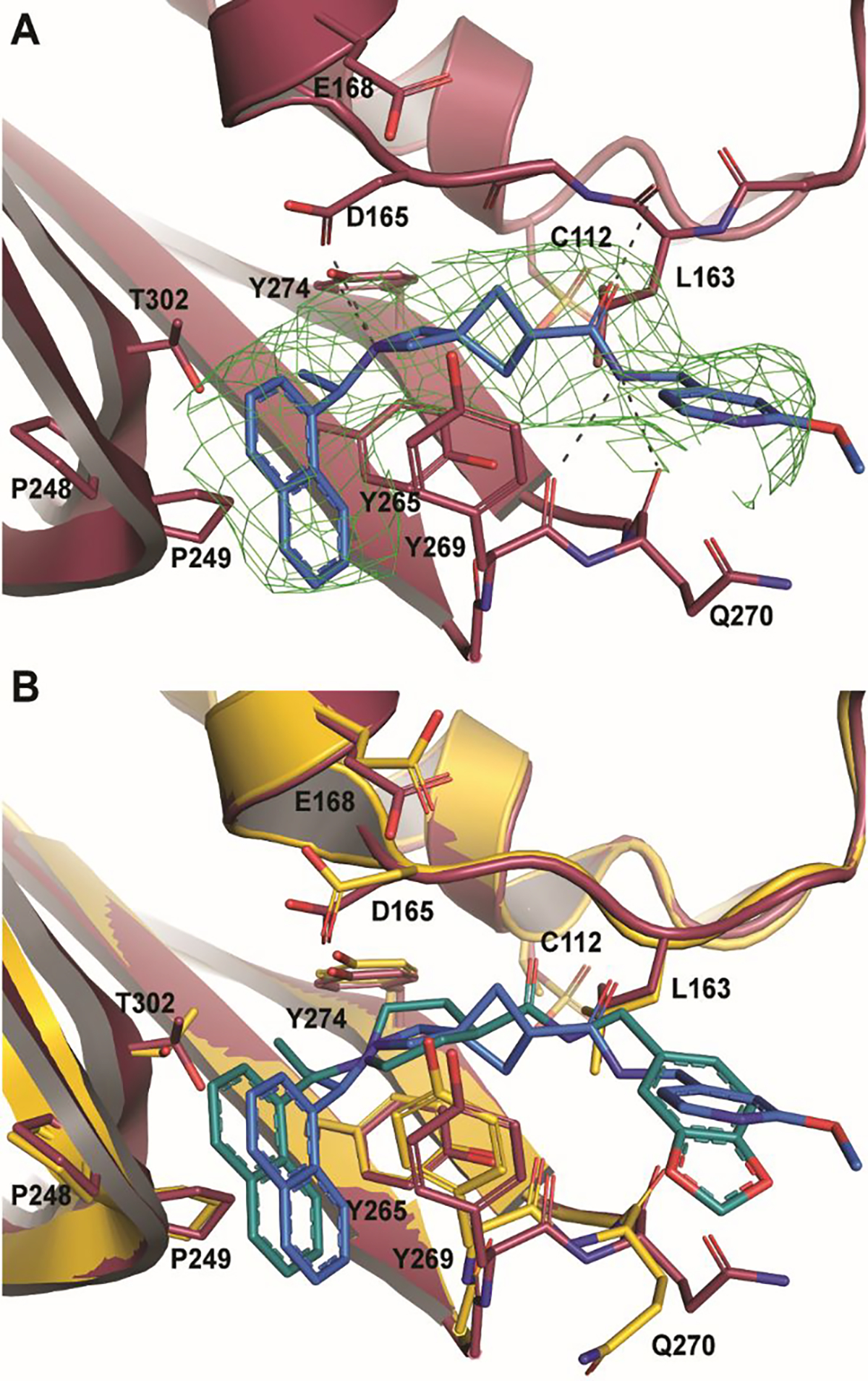
A. An Fo-Fc electron density composite omit map is shown contoured at 3σ (green mesh). With 37 shown in blue and BtSCoV-Rf1 PLpro shown in raspberry. B. Overlay of 1 (teal) bound to SARS-CoV-2 PLpro (yellow).
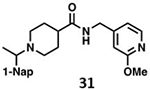
In vitro cytotoxicity, serum binding, and stability of protease inhibitors
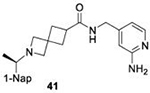
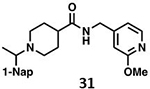
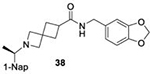
We determined the toxicity of these inhibitors by assessing their ability to decrease the reductive capability of numerous cells using the MTT assay (Figure 7A) and determining CC50 values. We compared these values to those for GRL0617, 1, and 31, all of which have been previously shown to have CC50s greater than 100 μM in Vero E6 cells with the latter two tested in HEK293 cells as well27, 32. To confirm this trend in immunologically relevant human cell lines all three compounds, along with 37 and 38 were assessed for CC50 in multiple cells, including human renal proximal tubule cells (RPTEC), Beas-2B, A549, and Sh-SH5Y cells. None of the other compounds were able to decrease MTT staining below 50% in A549 of Sh-SY5Y cells at concentrations as high as 100 μM. Focusing on RPTEC and BEAS-2b cells, 37 displayed the highest CC50 values in comparison to 1, 38, and 41. The CC50 reported in these cells is folds higher than the IC50 reported for inhibition of PLpro degradation, as determined above. In general, among the newly prepared compounds, alteration of the east moiety appeared to alter the CC50, as compared to the west moiety or the central rings. The addition of the central ring decreased the CC50 as compared to GRL0617, as shown by comparison to other of the new series of compounds. The effect of these compounds on MTT staining was validated using cell morphology, which demonstrated morphological characteristics of cell death in combination with reduced cell number (Supplemental Information). When tested for in vitro efficacy against Vero E6 cells infected with CoV-2, treatment with 37 resulted in significant cytotoxicity in concentrations >25 μM and some viral plaque reduction observed between 25 μM and 12 μM (Figure 7B). Plaque reduction in cells treated with 6 μM, 3 μM, or 1 μM, were not significantly different when compared to the 0 μM control treatment suggesting limited efficacy of 37 as a CoV-2 PLpro inhibitor.
Figure 7. Cytotoxicity Studies of Inhibitors.
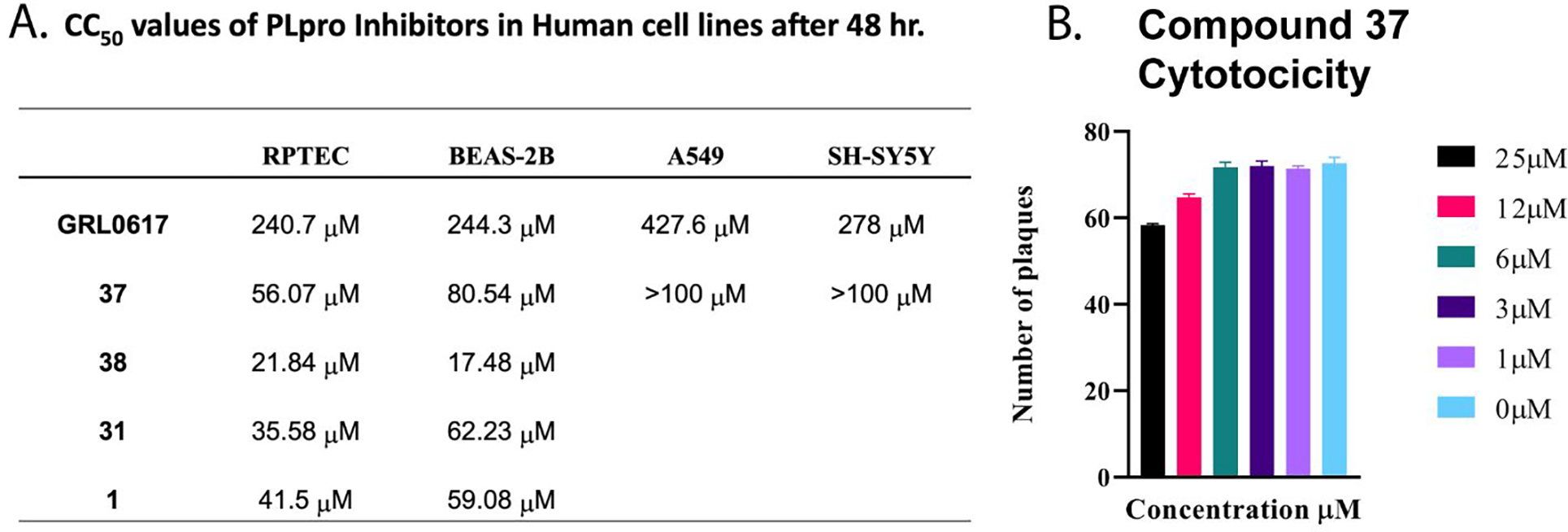
A. CC50 values of PLpro inhibitors in human cell lines after 48 hrs. B. Testing of Compound 37 for Cytotoxicity Vero E6 cells were plated at 6 × 105 cells/well and incubated overnight at 37°C. Subsequently, the cells were washed 1x with PBS. Compound 37 drug dilutions were prepared to 100 uM, 50 uM, 25 uM, 12 uM, 6 uM, 3 uM and 1 uM in overlay media (DMEM supplemented with 1% serum). Culture media was decanted, and 1 mL of virus diluted in infection media to a MOI=0.1 was added to the cells and incubated for 1 hour at 37°C. Following incubation, the inoculum was removed, and 3mL/well of Compound 37 dilutions were added and incubated for 4 days at 37°C and 5% CO2. Control wells included a virus only, uninfected, or DMSO well. Following the 4-day incubation, cells were fixed with methanol: acetone (80:20) for 20 min at room temperature then stained with 0.2% crystal violet. Plaques were counted and analyzed by Prism8 by GraphPad.
DISCUSSION
Effects of Variation in Betacoronavirus PLpros on DUB activity
The PLpros of subgroup 2b viruses are highly conserved in primary, secondary, and tertiary structures, particularly at important function sites. The residues lining the active site, BL2 loop, P3 site, and P4 site are identical among all seven subgroup 2b PLpros but vary considerably in other betacoronaviruses. This results in both series I and II naphthalene-based compounds having similar inhibitory properties in all subgroup 2b PLpros but having negligible inhibition towards other betacoronaviruses24, 47. Furthermore, the UIM, is known to accommodate both Ub and ISG15 binding, is fully conserved between BtSCoV-Rf1.2004 and CoV-1. The UIM of CoV-2 PLpro differs from those of CoV-1 and BtSCoV-Rf1.2004 at six positions, T171(S), H172(Y), K196(Q), L217(F), V226(T), and Q233(K). Of the six points of difference in CoV-2, five are identical across the other subgroup 2b PLpros. The high conservation of Q233 across subgroup 2b is noteworthy because experiments have shown inserting a Q233(E) mutation in CoV-1 PLpro notably diminished DUB activity but increased deISGylase activity. Additionally, CoV-2 has a lysine residue at site 233. and has reduced DUB activity, compared to CoV-1 and BtSCoV-Rf1.2004.
The only difference within the UIM of BtSCoV-Rf1.2004 is L188, which is a valine in CoV-1 (Figure S5). This change may result in the BtSCoV-Rf1.2004 finger domain having stronger interactions with the hydrophobic pocket of Ub, particularly F45 when compared to SARS. Additionally, the increased proximity of L188 to I223 and V226 on the Zn finger loop seems to cause the loop to adopt a slightly more closed conformation around the Ub, increasing the proximity of the hydrophobic PLpro surface. A stronger interaction at the proximal Ub binding site would explain the lower Km BtSCoV-Rf1.2004 PLpro has toward mono-Ub. When BtSCoV-Rf1.2004 PLpro DUB activity is examined through cleavage of various poly-Ub substrates it is apparent that its activity towards long-chain poly-Ub is most similar to CoV-1. Like other subgroup 2b viruses, it follows a distinct di-distributive cleavage pattern, in that it struggles to process mono-Ub-AMC and any linkage type of di-Ub, but readily cleaves K48 linked poly-Ub chains of three or more (Figure 2).
Conversely, MHV PLP and MERS PLpro share secondary structure topology with subgroup 2b PLpros but vary widely in sequence homology. These residue differences, particularly in areas such as the UIM, are responsible for differences in kinetics and substrate preferences. Both have mono-distributive DUB activity, in that they can cleave any length poly-Ub chain, and do not have as strong a preference for K48 linked poly-Ub as subgroup 2b PLpros. The trend in conservation extends to the residues stabilizing the interaction between the Ubl and thumb domain as well and may provide insight into the nature of mono vs di-distributive DUB activity. These residues are highly conserved among subgroup 2b PLpros but are not found in other betacoronaviruses, which points to the possibility that the tucked conformation is a distinct feature of subgroup 2b PLpros. It is unclear if this affects PLpro activity, but the correlation between di-distributive DUB activity and the ability to adopt tucked Ubl conformations warrants further study.
A significant trait that BtSCoV-Rf1.2004 PLpro differs from CoV-1 on is a reduced KM to mono-Ub. Like MERS and CoV-2 PLpros, BtSCoV-Rf1.2004 PLpro is readily saturated with mono-Ub at concentrations equivalent to those found in mammalian cells which range from 10 to 23 μM. The ability to be saturated at cellular Ub concentrations and relative inability to process mono-Ub make BtSCoV-Rf1.2004 susceptible to product inhibition similar to CoV-2 and MERS. Meanwhile, at physiological concentrations, CoV-1 PLpro would not even be at its KM.
Trends in Subgroup 2b PLpro DeISGylating Activities.
Despite Ubls being inherently similar substrates viral proteases, including coronavirus PLpros, display a high degree of substrate specificity. Differences in poly-Ub chain linkage can affect how they fit within the UIM, and ISG15 mutations between species can affect interactions at critical specificity sites. Typically, viral proteases will be adapted to rapidly process substrates involved in pathways detrimental to their survival and may retain off-target activity towards highly similar substrates. While all betacoronaviruses prefer the ISG15 of their natural host over Ub, host species, and therefore substrate preferences vary widely between viruses. Similar to what has been observed for CoV-1 and CoV-2, the deISGylase activity of BtSCoV-Rf1.2004 PLpro exceeds its DUB activity, except in the case of long-chain K48-Ub. The consistently robust deISGylase activity displayed by subgroup 2b PLpros relative to their modest DUB activity suggests that ISG15 regulated immune pathways may be more detrimental to viral replication than pathways regulated by Ub. Despite this, the specific utility deISGylase activity that serves coronaviruses is unknown. Because deISGylase activity appears critical to coronavirus replication, species-specific deISGylase activity can be a factor in determining what species a virus can productively infect. Interspecies variation in ISG15 has been shown to limit the zoonotic range of influenza B viruses48, 49. The similarity in species specificity between subgroup 2b viruses indicates that these viruses probably infect many of the same host species. Humans, palm civets, pangolins, minks, and several bats have all been identified as host species for subgroup 2b viruses, and some have been shown to host multiple 2b viruses2, 4, 5, 50. The largely conserved species preferences of subgroup 2b viruses may also enable them to productively infect new species that already serve as reservoirs for other 2b viruses with relatively few mutations.
BtSCoV-Rf1.2004 PLpro may be able to serve as a useful tool in determining residues that affect interspecies differences between CoV-1 and CoV-2. The deISGylase activities of these PLpros differ against ISG15s from sheep, camel, northern tree shrew, and jackknife fish. The PLpro of BtSCoV-Rf1.2004 matches the activity of CoV-2 against sheep, camel, and jackknife fish, but matches that of CoV-1 against northern tree shrew. While mammalian respiratory viruses would not naturally infect fish, CoV-1, MERS-CoV, and MHV PLpros all demonstrate some off-target activity towards jackknife fish ISG15. CoV-2 was the first betacoronavirus to show no appreciable activity towards fish ISG15, but the lack of activity demonstrated by BtSCoV-Rf1.2004 PLpro, which is more closely related to CoV-1, could help discern which differences are responsible.
The ability of BtSCoV-Rf1.2004 PLpro to quickly process horseshoe bat and vesper bat ISG15s, while having little activity towards Egyptian fruit bats is not unusual. This was observed in CoV-2 as well and is likely due to the diversity within order Chiroptera. Bats make up over 20% of all mammal species and ISG15 can share as little as 60% sequence identity between bats from different families, which is no greater than the similarity expected between any two mammalian ISG15s.
Naphthalene-based inhibitors as lead compounds for coronavirus subgroup 2b therapeutic development
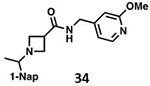
Initial evaluation of each novel compound at 50 μM revealed that most compounds have equivalent levels of inhibition against all three subgroup 2b PLpros regardless of inhibitor efficacy. This trend was further supported by IC50 testing of top compounds, which determined that IC50 values for each inhibitor for all three PLpros were usually within one to two-fold, and always within one order of magnitude. In addition to previously examined compounds, several of the newly designed compounds demonstrated low μM IC50 values and show promise as potential lead compounds. Of the compounds tested in this study GRL0617, 1, 31, and 37 were the most potent inhibitors. In all four cases IC50 values are below 10 μM and CC50 values are more than five times the IC50 in RPTEC and BEAS-2B cells and greater than 100 μM in A549 and SH-SY5Y cells. Within series II the eastern arene ring was the most critical factor affecting CC50 compared to the western naphthyl group or backbone ring system. Alteration of the 1,3-dioxolane ring of 1 and 38 to a 2-methoxy-4-pyridine ring yielded larger therapeutic windows, but only when paired with a spiroazetidine backbone to orient the group properly within the pocket. These two modifications result in 37 having the highest CC50 when compared to other series II compounds tested.
Investigation of 37 in plaque reduction assays against CoV-2 in Vero E6 cells resulted in limited plaque reduction between 25 μM and 12 μM with cytotoxicity limiting observations to concentrations ≤25 μM. However, this observed cytotoxicity in 37 is not particularly surprising considering previous studies synthesized naphthalene-based inhibitor, recently referred to as rac5c, which is number as 31 in our studies, also displays some levels of toxicity in Vero E6 cells30. As described above, when the toxicity of 37 was investigated more closely and compared to compound 31 CC50 values for the former were 56.07 μM and 80.54 μM in RPTEC andBEAS-2B cells respectively with greater than 100 μM CC50in A549 and SH-SY5Y cells. In contrast, 31, showed increased toxicity in RPTEC and BEAS-2B with 35.58 μM and 62.23 μM in these cells respectively. Taken together, these data suggest GRL0617, 1, 31, and 37 are potent CoV-2 PLpro inhibitors and have antiviral activity, with 37 outperforming 31 from a toxicity standpoint among series II leads but with still too narrow of a therapeutic window for viable use. This suggests further optimization of the series II scaffold is necessary to match the more promising series I lead like GRL0617 in terms of toxicity as a pan subgroup 2b virus therapeutic. Overall, the results illustrate that compounds based on these lead scaffolds would likely have the potential to inhibit a broad swath of subgroup 2b viruses.
CONCLUSIONS
The ability of all three 2b PLpros to productively cleave ISG15 from humans as well as several important livestock species further highlights the need to monitor these viruses and develop effective therapeutics to mitigate events linked to future spillover. Most naphthalene-based compounds inhibit all three subgroup 2b PLpros at similar levels, indicating that P3-P6 binding drugs will be useful for the treatment of future subgroup 2b coronavirus outbreaks as well as the current COVID-19 pandemic. Structural data indicate that these compounds could be optimized to bind these pockets, enhancing potency and specificity. Current naphthalene-based PLpro inhibitors are effective at treating both CoV-1 and CoV-2 in vitro with variable levels of cytotoxicity. While further compound optimization and significant in vivo testing is required before these compounds could be used therapeutically, they are promising lead compounds for treating immunocompromised patients that cannot receive vaccines or 2b viruses that induce short-lived immunity.
Methods
Chemicals and Reagents.
5-Amino-2-methyl-N-[(R)-1-(1-naphthyl)ethyl]benzamide (GRL0617) was purchased from Raystar, CN; 5-(acetylamino)-2-methyl-N-[(1R)-1-(1-napthanlenyl)ethyl]-benzamide (compound 6) was purchased from MedChem Express; Z-RLRGG-7-amino-4-methyl-courmarin (peptide-AMC) was purchased from Bachem; Ubiquitin–7-amino-4-methylcourmarin (Ub-AMC) was purchased from Boston Biochem; human ISG15-7-amino-4-methylcourmarin (ISG15-AMC) was purchased from Boston Biochem; Lys6, Lys11, Lys29, Lys33, Lys48, Lys63, and linear linked di-Ub were obtained from Boston Biochem; DL-dithiothroitol (DTT) was purchased from GoldBio; Isopropyl β-D-thiogalactopyranoside (IPTG) was purchased from GoldBio; 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) was purchased from Fisher BioReagents; Imidazole was purchased from Acros Organics; tris(hydroxymethyl)aminomethane (Tris) was purchased from Fisher Scientific; Sodium chloride (NaCl) was purchased from Fisher Chemical; Bovine serum albumin (BSA) was purchased from Sigma Life Science; Dehydrated Luria-Bertani (LB) broth was purchased from Fisher Scientific; Ampicillin was purchased from GoldBio.
Construction, Expression, and Purification of Viral Deubiquitinases.
The ubiquitin-like domain (Ubl) and the catalytic core of BtSCoV-Rf1.2004 PLpro (pp1ab 1536–1850; 2–316) were cloned into pET-15b by Genscript and transformed into T7 express E. coli. Cells were cultured in 4.5L of LB broth containing 100μg/mL ampicillin at 37 °C until the OD600 reached 0.6. Once reached, the expression was induced by the addition of 0.5mM isopropyl β-D-thiogalactopyranoside (IPTG), and the culture was incubated at 18 °C overnight. The culture was centrifuged at 12,000g for 10min, and the pellet was collected and stored in a −80°C freezer. The cell pellet was dissolved into lysis buffer (500mM NaCl and 50mM Tris-HCl [pH = 7.5]) and then sonicated in Fisher Scientific series 150 on ice at 50% power with 5s pulses for 6min. The lysate was centrifuged at 30,000g for 30min to remove all insoluble products. The supernatant was then filtered and placed onto Ni-nitrilotriacetic agarose resin (Qiagen). The resin was washed using five column volumes of lysis buffer containing 10mM imidazole. The protein was eluted using 5 column volumes of lysis buffer containing 250mM imidazole. Thrombin was added to the elution to remove the 6X His-tag, and the combined solution was dialyzed in size exclusion buffer (100mM NaCl, 5mM HEPES, and 2mM dithiothreitol (DTT) [pH = 7.4]) and run over a Size Exclusion Superdex 200 column (GE Healthcare, Pittsburgh PA). Purity was confirmed by gel electrophoresis.
Co-crystallization of BtSCoV-Rf1.2004 PLpro with GRL0617
Size-exclusion chromatography fractions containing BtSCoV-Rf1.2004 PLpro were pooled based on the chromatogram, confirmed by SDS–PAGE and concentrated to 10.5 mg/mL and 10.4 mg/mL for co-crystallization with GRL0617 and 37 respectively. GRL0617 in 100% DMSO was added to the protein sample at a 5:1 molar ratio with a final DMSO concentration of 3%. The sample was screened in 96-well hanging-drop plates against a suite of 1728 commercially available conditions (Qiagen and Hampton) using a Mosquito robot (TTP Labtech). Plate-like crystals in different conditions formed in a time ranging from one to three weeks. Hits from the screen were scaled up to hanging-drop 24-well plates containing a 500 μL reservoir of the crystallization solution and were optimized using varying salt, precipitant, pH, additive and protein concentration gradients. The final crystallization condition for the optimized BtSCoV-Rf1.2004 PLpro-GRL0617 co-crystal was 0.2M Ammonium Acetate, 20% PEG 1000. 37 in 100% DMSO was added to the protein sample at a 5:1 molar ratio and a final DMSO concentration of 0.5%. A crystal screen was set up for the sample against a suite of 768 commercially available conditions (Qiagen) in 96-well hanging-drop plates using a Mosquito robot (TTP Labtech). The final crystallization condition for the BtSCoV-Rf1.2004 PLpro-37 co-crystals was 0.1 M Tris-HCl pH 8.5, 0.6M sodium fluoride, resulting in bipyramidal crystals in 1–3 weeks.
Data Collection and Processing
All crystals were flash-cooled in liquid nitrogen. BtSCoV-Rf1.2004 PLpro-GRL0617 complex structural data were collected under a dry N2 stream on the 17-ID (AMX) beamline at Brookhaven National Laboratory using an Eiger-9M detector. BtSCoV-Rf1.2004 PLpro-37 complex structural data were collected on the 22-ID beamline using an Eiger-16M detector at Argonne National Laboratory under a dry N2 stream. Collection wavelengths for both data sets was 1 Å. The data were indexed, integrated and scaled in HKL-200051. Data-collection statistics are included in Table S1.
Structure Solution and Refinement
Phases for the BtSCoV-Rf1.2004 PLpro–GRL0617 and 37 co-crystal structures were solved by molecular replacement in Phaser52–54. A homology model of BtSCoV-Rf1.2004 PLpro based on a CoV-1 PLpro–GRL0617 co-crystal structure (PDB 3E9S) was used as a reference for the GRL0617 complex, while the BtSCoV-Rf1.2004 PLpro–GRL0617 structure was used as the model for the 37 complex. In both cases the finger, thumb, and palm domains of the PLpro were used as a search model, placing 2 copies in the asymmetric unit of the GRL0617 complex and 1 in the asymmetric unit of the 37 complex. Afterward the Ubl domains were built in manually. The phased models were modified through alternating rounds of manual building and refinement in Coot and Phenix55. The final models were validated in MolProbity56, and the associated refinement statistics are included in Table S1. The coordinates and structure factors have been deposited and will be released upon publication with PDB accession codes 7SKQ (GRL0617 complex) and 7SKR (Compound 37 complex).
BtSCoV-Rf1.2004 PLpro Deubiquitinase and deISGylating Assays.
All assays were run using Corning Costar half-volume 96-well plates containing AMC buffer (100mM NaCl, 50mM HEPES [pH = 7.5], 0.01 mg/mL bovine serum albumin (BSA), and 5mM DTT) to a final volume of 50 μL and performed in triplicate. The CLAIROstar plate reader (BMG Lab Tech, Inc.) was used to measure the fluorescence of the AMC cleavage, and the data was analyzed using MARS (BMG Lab Tech, Inc.). The AMC fluorescence was observed from the cleavage of Ub-AMC and ISG15-AMC obtained from Boston Biochem, MA. ISG15-AMC concentrations of substrate ranged from 625 nM to 20 μM, and Ub-AMC ranged from 235 nM to 30 μM. Protease concentrations used for the Ub-AMC and ISG15-AMC assays were 5 and 1 nM, respectively. To calculate KM and Vmax values, the initial rates were fitted to the Michalis-Menten equation, υ = Vmax/(1 + (KM/[S])), using the Enzyme Kinetics (v. 1.3) module of SigmaPlot (v. 10.0, SPSS Inc.). Vmax was translated into kcat using kcat = Vmax/[E].
BtSCoV-Rf1.2004 PLpro Poly-Ub Cleavage Assays.
Lys6, Lys11, Lys29, Lys33, Lys48, Lys63, and linear linked di-Ub obtained from Boston Biochem were incubated at 10 μM with 20 nM BtSCoV-Rf1.2004 PLpro. Reactions were performed in AMC buffer at a volume of 45 μL and a temperature of 37°C. 10 μL samples were taken at the indicated time points and heat-shocked at 98°C for 5 min. Lys48 and Lys63 linked tetra-Ub obtained from Boston Biochem were incubated at 13.67 μM with 23 nM BtSCoV-Rf1.2004 PLpro. Reactions were performed in AMC buffer at a volume of 70 μL and a temperature of 37°C. 8 μL samples were taken at the indicated time points and heat-shocked at 98°C for 5 min. SDS-PAGE analysis was performed using Mini-PROTEAN TGX and Coomassie blue.
Protease Activity Assay with proISG15 Substrates.
At 37°C, 20nM BtSCoV-Rf1.2004 PLpro was run against 10 μM of each ISG15. Reaction mixtures were 100 μL in AMC buffer. 10μL samples were taken at the indicated time points, and the reaction was quenched in 2×Laemmli sample buffer followed by boiling at 98°C for 5min. SDS-PAGE analysis was performed using Mini-PROTEAN TGX Stain-Free.
BtSCoV-Rf1.2004 PLpro Inhibition IC50 Value Determination.
IC50 assays were performed using similar methods to peptide-AMC, Ub-AMC, and ISG15-AMC cleavage experiments and those described previously. BtSCoV-Rf1.2004 PLpro was run at 1 μM against 50 μM peptide-AMC in 98% AMC buffer/2% DMSO. Reactions were performed in triplicate with inhibitor concentrations ranging from 195nM to 100 μM, depending on compound tested. IC50 calculations were performed using Prism8 from GraphPad.
Compound Synthesis
The synthesis of series I and series II compounds is described in the supplemental information section.
Cells and Culture Conditions
Non-targeted toxicity was assessed in numerous diverse cell lines, including Renal Proximal Tubular Epithelial Cells (RPTECs, ATCC CRL-4031), BEAS-2B bronchial epithelial cells (ATCC CRL-9609), A549 human alveolar epithelial cells (ATCC, CCL-185), and SH-SY5Y neuroblastoma cells (ATCC, CRL-2266). RPTECs were grown in DMEM/F12 (ATCC, 30-2006) supplemented with hTERT Immortalized RPTEC Growth Kit (ATCC, ACS-4007); A549 and SH-SY5Y cells were grown in DMEM (ATCC, 30-2002) supplemented with 10% Fetal Bovine Serum (VWR, 97068-085) and 1% penicillin-streptomycin solution (ATCC, 30-2300). BEAS-2B cells were grown using the BEGM BulletKit (Lonza, CC-3170) and flasks were coated with 0.03 mg/mL bovine collagen (Fisher, CB-40231) and 0.01 mg/mL human fibronectin (Fisher, CB-40008A). All cells were maintained at 37°C in a 5% CO2 incubator.
Cytotoxicity was assessed using MTT staining and cell morphology. Cells were seeded in 48-well tissue culture plates at densities between 25 – 50,000 (A549, BEAS-2B), 100,000 (RPTEC), and 50 – 150,000 (SH-SY5Y) cells per well depending on growth rate and experimental conditions. Cells were maintained at 37°C in a 5% CO2 incubator for a minimum of 24 hr and were at least 80% confluent before dosing. Inhibitors were dissolved in DMSO and diluted in different culture media to their final concentrations. Cells were treated with DMSO alone (vehicle control, no greater than 0.1% (v/v)) or 1, 5, 10, 50, 75, or 100 μM of each inhibitor; except for GRL0617, where cells were also treated with 175, 250, 500, 750, or 1000 μM concentrations. For serum binding assays, inhibitors (and DMSO controls) were pre-incubated in DMEM containing 10% (v/v) Fetal Bovine Serum and 1% penicillin-streptomycin for 5 minutes prior before exposure. Stability was assessed by incubation of inhibitors for 24 hr at 37°C in media. Alterations in cytotoxicity as compared to un-incubated controls were indicative of serum binding or inhibitor instability. Regardless of the experiment, MTT (Sigma, M2128-1G) was added after 48 hr treatment at a final concentration of 0.1 mg/mL, and plates were incubated for 2 hr at 37°C. After media aspiration, the remaining MTT formazan crystals were dissolved in DMSO, and absorbance was determined for each well at 490 nm using a BMG CLARIOstar plate reader. Experiments were performed in triplicate per passage in at least 3 distinct passages of cells. The concentration of protease inhibitor that resulted in 50% growth inhibition (CC50) as compared to DMSO control was estimated from a non-linear regression curve as calculated in GraphPad Prism 7. When GraphPad could not automatically determine accurate CC50 values, the concentration on the linear regression curve at which MTT staining was half of the control was utilized. Changes in MTT staining were compared to changes in cell morphology at either 24 or 48 hr after exposure to inhibitors using a Nikon Eclipse Ti. Figures were assembled in Photoshop.
In vitro efficacy of 37 against SASR-CoV-2 was performed using the CoV-2 isolate nCoV-WA1-2020 (MN985325.1). This isolate was received from BEI resources under contract by American Type Culture Collection (ATCC) and propagated in Vero E6 cells. Propagation was performed at a multiplicity of infection (MOI) of 0.1 in serum-free DMEM for 1 hour after which the inoculum was removed and replaced with DMEM supplemented with 1% heat-inactivated fetal bovine serum57. The virus was harvested 72 hours post-inoculation. Viral titers were determined by plaque assay using Vero E6 cells58. Cells were seeded in a 6-well plate at 6 × 105 cells/well and incubated overnight at 37°C. 100 μM, 50 μM, 25 μM, 12 μM, 6 μM, 1 μM, and 0 μM dilutions of compounds were prepared the following day in DMEM supplemented with 1% fetal bovine serum. The cells were washed once with PBS 1X then inoculated at an MOI of 0.1 for 1 hour, after which the inoculum was removed, and compound dilutions were added in triplicate wells and incubated for 96 hours at 37°C and 5% CO2. The cells were fixed, then stained with crystal violet to determine plaque numbers. Analysis was performed using Prism8 from GraphPad.
Supplementary Material
Acknowledgments
This research used resources (beamline 17-ID-1) of the National Synchrotron Light Source II, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Center for Biomolecular Structure (CBMS) is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893) and by the DOE Office of Biological and Environmental Research (KP1605010). X-ray data were collected on the EIGER 16M detector at Brookhaven National Laboratory. Additional data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. SER-CAT is supported by its member institutions, and equipment grants (S10_RR25528, S10_RR028976 and S10_OD027000) from the National Institutes of Health. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Funding Information
The work was supported partly by National Institute of Allergy and Infectious Diseases grant No. R01AI151006 (SDP) and Sunshine biopharma AWD00012621 (SDP, RT, DC)
Abbreviations Used:
- SARS-CoV-2
Severe acute respiratory syndrome coronavirus 2
- PLpro
papain-like protease
- NSP
nonstructural proteins
- CoV
coronaviruses
- PLPs
papain-like proteases
- MHV
mouse hepatitis virus
- MERS-CoV
Middle East respiratory syndrome CoV
- Ub
ubiquitin
- ISG15
interferon-stimulated gene product 15
- poly-ub
polyubiquitin
- UIM
ubiquitin interacting motif
- AMC
7-amino-4-methyl coumarin
- proISG15
precursor ISG15
- GRL-0617
5-Amino-2-methyl-N-benzamide
- compound 6
5-(acetylamino)-2-methyl-N-[(1R)-1-(1-napthanlenyl)ethyl]-benzamide
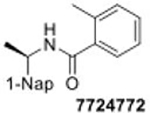
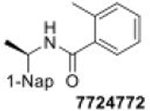
- 7724772
2-methyl-N-[1-(2-naphthyl)ethyl]benzamide
- 6577871
N-(4-methoxybenzyl)-1-(1-naphthylmethyl)-4-piperidinecarboxamide oxalate
- 9247873
1-benzyl-N-(3,4,5-trimethoxybenzyl)-4-piperidinecarboxamide
- DTT
DL-dithiothreitol
- IPTG
Isopropyl β-D-thiogalactopyranoside
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- Tris
tris(hydroxymethyl)aminomethane
- BSA
Bovine Serum Albumin
- DMSO
dimethyl sulfoxide
- UIM
Ub interacting motif
Footnotes
Conflict of Interest
BTF, SDP, RAT, and RJH have submitted a provisional application U.S.S.N. 62/992,895 pertaining to the work enclosed in the manuscript. Also, U.S.S.N 63/086,137 has been submitted by BTF, DAA, RSB, IAD, JH, EO, MP, YPS, RAT, BSC, DC, SDP pertaining to work enclosed in the manuscript.
Supporting Information Available: Includes x-ray crystallographic statistics table, additional structural figures, toxicity studies, compound reaction schemes, and compound synthesis/characterization. The Supporting Information is available free of charge on the ACS Publications Website at http://pubs.acs.org.
References
- (1).Wang LF; Shi Z; Zhang S; Field H; Daszak P; Eaton BT, Review of bats and SARS. Emerg Infect Dis 2006, 12 (12), 1834–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Li W; Shi Z; Yu M; Ren W; Smith C; Epstein JH; Wang H; Crameri G; Hu Z; Zhang H; Zhang J; McEachern J; Field H; Daszak P; Eaton BT; Zhang S; Wang LF, Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310 (5748), 676–9. [DOI] [PubMed] [Google Scholar]
- (3).Ji W; Wang W; Zhao X; Zai J; Li X, Cross-species transmission of the newly identified coronavirus 2019-nCoV. J Med Virol 2020, 92 (4), 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Menachery VD; Yount BL Jr.; Debbink K; Agnihothram S; Gralinski LE; Plante JA; Graham RL; Scobey T; Ge XY; Donaldson EF; Randell SH; Lanzavecchia A; Marasco WA; Shi ZL; Baric RS, A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med 2015, 21 (12), 1508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lam TT; Jia N; Zhang YW; Shum MH; Jiang JF; Zhu HC; Tong YG; Shi YX; Ni XB; Liao YS; Li WJ; Jiang BG; Wei W; Yuan TT; Zheng K; Cui XM; Li J; Pei GQ; Qiang X; Cheung WY; Li LF; Sun FF; Qin S; Huang JC; Leung GM; Holmes EC; Hu YL; Guan Y; Cao WC, Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583 (7815), 282–285. [DOI] [PubMed] [Google Scholar]
- (6).Dudley MZ; Halsey NA; Omer SB; Orenstein WA; O’Leary ST; Limaye RJ; Salmon DA, The state of vaccine safety science: systematic reviews of the evidence. Lancet Infect Dis 2020, 20 (5), e80–e89. [DOI] [PubMed] [Google Scholar]
- (7).Chiu M; Bao C; Sadarangani M, Dilemmas With Rotavirus Vaccine: The Neonate and Immunocompromised. Pediatr Infect Dis J 2019, 38 (6S Suppl 1), S43–S46. [DOI] [PubMed] [Google Scholar]
- (8).Dilucca M; Forcelloni S; Georgakilas AG; Giansanti A; Pavlopoulou A, Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes. Viruses 2020, 12 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Thiel V; Ivanov KA; Putics A; Hertzig T; Schelle B; Bayer S; Weissbrich B; Snijder EJ; Rabenau H; Doerr HW; Gorbalenya AE; Ziebuhr J, Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol 2003, 84 (Pt 9), 2305–2315. [DOI] [PubMed] [Google Scholar]
- (10).Daczkowski CM; Dzimianski JV; Clasman JR; Goodwin O; Mesecar AD; Pegan SD, Structural Insights into the Interaction of Coronavirus Papain-Like Proteases and Interferon-Stimulated Gene Product 15 from Different Species. J Mol Biol 2017, 429 (11), 1661–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Barretto N; Jukneliene D; Ratia K; Chen Z; Mesecar AD; Baker SC, The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol 2005, 79 (24), 15189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mielech AM; Deng X; Chen Y; Kindler E; Wheeler DL; Mesecar AD; Thiel V; Perlman S; Baker SC, Murine coronavirus ubiquitin-like domain is important for papain-like protease stability and viral pathogenesis. J Virol 2015, 89 (9), 4907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ratia K; Saikatendu KS; Santarsiero BD; Barretto N; Baker SC; Stevens RC; Mesecar AD, Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci U S A 2006, 103 (15), 5717–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhao C; Denison C; Huibregtse JM; Gygi S; Krug RM, Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci U S A 2005, 102 (29), 10200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Davis ME; Gack MU, Ubiquitination in the antiviral immune response. Virology 2015, 479–480, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sadler AJ; Williams BR, Interferon-inducible antiviral effectors. Nat Rev Immunol 2008, 8 (7), 559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Frieman M; Ratia K; Johnston RE; Mesecar AD; Baric RS, Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J Virol 2009, 83 (13), 6689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Devaraj SG; Wang N; Chen Z; Chen Z; Tseng M; Barretto N; Lin R; Peters CJ; Tseng CT; Baker SC; Li K, Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem 2007, 282 (44), 32208–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Scholte FEM; Zivcec M; Dzimianski JV; Deaton MK; Spengler JR; Welch SR; Nichol ST; Pegan SD; Spiropoulou CF; Bergeron E, Crimean-Congo Hemorrhagic Fever Virus Suppresses Innate Immune Responses via a Ubiquitin and ISG15 Specific Protease. Cell Rep 2017, 20 (10), 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhao C; Sridharan H; Chen R; Baker DP; Wang S; Krug RM, Influenza B virus non-structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nat Commun 2016, 7, 12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dzimianski JV; Scholte FEM; Bergeron E; Pegan SD, ISG15: It’s Complicated. J Mol Biol 2019, 431 (21), 4203–4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Freitas BT; Scholte FEM; Bergeron E; Pegan SD, How ISG15 combats viral infection. Virus Research 2020, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shen Z; Ratia K; Cooper L; Kong D; Lee H; Kwon Y; Li Y; Alqarni S; Huang F; Dubrovskyi O; Rong L; Thatcher GR; Xiong R, Potent, Novel SARS-CoV-2 PLpro Inhibitors Block Viral Replication in Monkey and Human Cell Cultures. bioRxiv 2021. [Google Scholar]
- (24).Shin D; Mukherjee R; Grewe D; Bojkova D; Baek K; Bhattacharya A; Schulz L; Widera M; Mehdipour AR; Tascher G; Geurink PP; Wilhelm A; van der Heden van Noort GJ; Ovaa H; Muller S; Knobeloch KP; Rajalingam K; Schulman BA; Cinatl J; Hummer G; Ciesek S; Dikic I, Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587 (7835), 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rut W; Lv Z; Zmudzinski M; Patchett S; Nayak D; Snipas SJ; El Oualid F; Huang TT; Bekes M; Drag M; Olsen SK, Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chen X; Chou CY; Chang GG, Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir Chem Chemother 2009, 19 (4), 151–6. [DOI] [PubMed] [Google Scholar]
- (27).Baez-Santos YM; Barraza SJ; Wilson MW; Agius MP; Mielech AM; Davis NM; Baker SC; Larsen SD; Mesecar AD, X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J Med Chem 2014, 57 (6), 2393–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Baez-Santos YM; St John SE; Mesecar AD, The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 2015, 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Freitas BT; Durie IA; Murray J; Longo JE; Miller HC; Crich D; Hogan RJ; Tripp RA; Pegan SD, Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect Dis 2020, 6 (8), 2099–2109. [DOI] [PubMed] [Google Scholar]
- (30).Klemm T; Ebert G; Calleja DJ; Allison CC; Richardson LW; Bernardini JP; Lu BG; Kuchel NW; Grohmann C; Shibata Y; Gan ZY; Cooney JP; Doerflinger M; Au AE; Blackmore TR; van der Heden van Noort GJ; Geurink PP; Ovaa H; Newman J; Riboldi-Tunnicliffe A; Czabotar PE; Mitchell JP; Feltham R; Lechtenberg BC; Lowes KN; Dewson G; Pellegrini M; Lessene G; Komander D, Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J 2020, 39 (18), e106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pitsillou E; Liang J; Ververis K; Lim KW; Hung A; Karagiannis TC, Identification of Small Molecule Inhibitors of the Deubiquitinating Activity of the SARS-CoV-2 Papain-Like Protease: in silico Molecular Docking Studies and in vitro Enzymatic Activity Assay. Front Chem 2020, 8, 623971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ratia K; Pegan S; Takayama J; Sleeman K; Coughlin M; Baliji S; Chaudhuri R; Fu W; Prabhakar BS; Johnson ME; Baker SC; Ghosh AK; Mesecar AD, A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci U S A 2008, 105 (42), 16119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sander AL; Yadouleton A; Moreira-Soto A; Tchibozo C; Hounkanrin G; Badou Y; Fischer C; Krause N; Akogbeto P; E F. d. O. F.; Dossou A; Brunink S; Drosten C; Aissi MAJ; Harouna Djingarey M; Hounkpatin B; Nagel M; Drexler JF, An Observational Laboratory-Based Assessment of SARS-CoV-2 Molecular Diagnostics in Benin, Western Africa. mSphere 2021, 6 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mlcochova P; Kemp SA; Dhar MS; Papa G; Meng B; Ferreira IATM; Datir R; Collier DA; Albecka A; Singh S; Pandey R; Brown J; Zhou J; Goonawardane N; Mishra S; Whittaker C; Mellan T; Marwal R; Datta M; Sengupta S; Ponnusamy K; Radhakrishnan VS; Abdullahi A; Charles O; Chattopadhyay P; Devi P; Caputo D; Peacock T; Wattal C; Goel N; Satwik A; Vaishya R; Agarwal M; Chauhan H; Dikid T; Gogia H; Lall H; Verma K; Dhar MS; Singh MK; Soni N; Meena N; Madan P; Singh P; Sharma R; Sharma R; Kabra S; Kumar S; Kumari S; Sharma U; Chaudhary U; Sivasubbu S; Scaria V; Oberoi JK; Raveendran R; Datta S; Das S; Maitra A; Chinnaswamy S; Biswas NK; Parida A; Raghav SK; Prasad P; Sarin A; Mayor S; Ramakrishnan U; Palakodeti D; Seshasayee ASN; Thangaraj K; Bashyam MD; Dalal A; Bhat M; Shouche Y; Pillai A; Abraham P; Potdar VA; Cherian SS; Desai AS; Pattabiraman C; Manjunatha MV; Mani RS; Udupi GA; Nandicoori V; Tallapaka KB; Sowpati DT; Kawabata R; Morizako N; Sadamasu K; Asakura H; Nagashima M; Yoshimura K; Ito J; Kimura I; Uriu K; Kosugi Y; Suganami M; Oide A; Yokoyama M; Chiba M; Saito A; Butlertanaka EP; Tanaka YL; Ikeda T; Motozono C; Nasser H; Shimizu R; Yuan Y; Kitazato K; Hasebe H; Nakagawa S; Wu J; Takahashi M; Fukuhara T; Shimizu K; Tsushima K; Kubo H; Shirakawa K; Kazuma Y; Nomura R; Horisawa Y; Takaori-Kondo A; Tokunaga K; Ozono S; Baker S; Dougan G; Hess C; Kingston N; Lehner PJ; Lyons PA; Matheson NJ; Owehand WH; Saunders C; Summers C; Thaventhiran JED; Toshner M; Weekes MP; Maxwell P; Shaw A; Bucke A; Calder J; Canna L; Domingo J; Elmer A; Fuller S; Harris J; Hewitt S; Kennet J; Jose S; Kourampa J; Meadows A; O’Brien C; Price J; Publico C; Rastall R; Ribeiro C; Rowlands J; Ruffolo V; Tordesillas H; Bullman B; Dunmore BJ; Fawke S; Gräf S; Hodgson J; Huang C; Hunter K; Jones E; Legchenko E; Matara C; Martin J; Mescia F; O’Donnell C; Pointon L; Pond N; Shih J; Sutcliffe R; Tilly T; Treacy C; Tong Z; Wood J; Wylot M; Bergamaschi L; Betancourt A; Bower G; Cossetti C; De Sa A; Epping M; Fawke S; Gleadall N; Grenfell R; Hinch A; Huhn O; Jackson S; Jarvis I; Krishna B; Lewis D; Marsden J; Nice F; Okecha G; Omarjee O; Perera M; Potts M; Richoz N; Romashova V; Yarkoni NS; Sharma R; Stefanucci L; Stephens J; Strezlecki M; Turner L; De Bie EMDD; Bunclark K; Josipovic M; Mackay M; Rossi S; Selvan M; Spencer S; Yong C; Allison J; Butcher H; Caputo D; Clapham-Riley D; Dewhurst E; Furlong A; Graves B; Gray J; Ivers T; Kasanicki M; Le Gresley E; Linger R; Meloy S; Muldoon F; Ovington N; Papadia S; Phelan I; Stark H; Stirrups KE; Townsend P; Walker N; Webster J; Scholtes I; Hein S; King R; Mavousian A; Lee JH; Bassi J; Silacci-Fegni C; Saliba C; Pinto D; Irie T; Yoshida I; Hamilton WL; Sato K; Bhatt S; Flaxman S; James LC; Corti D; Piccoli L; Barclay WS; Rakshit P; Agrawal A; Gupta RK; The Indian S-C-GC; The Genotype to Phenotype Japan C; The C-NBC-C, SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Baez-Santos YM; Mielech AM; Deng X; Baker S; Mesecar AD, Catalytic function and substrate specificity of the papain-like protease domain of nsp3 from the Middle East respiratory syndrome coronavirus. J Virol 2014, 88 (21), 12511–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen Y; Savinov SN; Mielech AM; Cao T; Baker SC; Mesecar AD, X-ray Structural and Functional Studies of the Three Tandemly Linked Domains of Non-structural Protein 3 (nsp3) from Murine Hepatitis Virus Reveal Conserved Functions. J Biol Chem 2015, 290 (42), 25293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Freitas BT; Durie IA; Crabtree J; Longo JE; Miller HC; Crich D; Hogan RJ; Tripp RA; Pegan SD, Characterization and Noncovalent Inhibition of the Deubiquitinase and DeISGylase Activity of SARS-COV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [DOI] [PubMed] [Google Scholar]
- (38).Bekes M; Rut W; Kasperkiewicz P; Mulder MP; Ovaa H; Drag M; Lima CD; Huang TT, SARS hCoV papain-like protease is a unique Lys48 linkage-specific di-distributive deubiquitinating enzyme. Biochem J 2015, 468 (2), 215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Langley C; Goodwin O; Dzimianski JV; Daczkowski CM; Pegan SD, Structure of interferon-stimulated gene product 15 (ISG15) from the bat species Myotis davidii and the impact of interdomain ISG15 interactions on viral protein engagement. Acta Crystallogr D Struct Biol 2019, 75 (Pt 1), 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Dzimianski JV; Scholte FEM; Williams IL; Langley C; Freitas BT; Spengler JR; Bergeron E; Pegan SD, Determining the molecular drivers of species-specific interferon-stimulated gene product 15 interactions with nairovirus ovarian tumor domain proteases. PLoS One 2019, 14 (12), e0226415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Deaton MK; Dzimianski JV; Daczkowski CM; Whitney GK; Mank NJ; Parham MM; Bergeron E; Pegan SD, Biochemical and Structural Insights into the Preference of Nairoviral DeISGylases for Interferon-Stimulated Gene Product 15 Originating from Certain Species. J Virol 2016, 90 (18), 8314–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bekes M; van der Heden van Noort GJ; Ekkebus R; Ovaa H; Huang TT; Lima CD, Recognition of Lys48-Linked Di-ubiquitin and Deubiquitinating Activities of the SARS Coronavirus Papain-like Protease. Mol Cell 2016, 62 (4), 572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Daczkowski CM; Goodwin OY; Dzimianski JV; Farhat JJ; Pegan SD, Structurally Guided Removal of DeISGylase Biochemical Activity from Papain-Like Protease Originating from Middle East Respiratory Syndrome Coronavirus. J Virol 2017, 91 (23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Rogers-Evans M; Knust H; Plancher JM; Carreira EM; Wuitschik G; Burkhard J; Li DB; Guerot C, Adventures in drug-like chemistry space: from oxetanes to spiroazetidines and beyond! Chimia (Aarau) 2014, 68 (7–8), 492–9. [DOI] [PubMed] [Google Scholar]
- (45).Ghosh AK; Takayama J; Rao KV; Ratia K; Chaudhuri R; Mulhearn DC; Lee H; Nichols DB; Baliji S; Baker SC; Johnson ME; Mesecar AD, Severe Acute Respiratory Syndrome-Coronavirus Papain-Like Protease Inhibitors: Design, Synthesis, Protein-Ligand X-ray Structure and Biologial Evaluation J. Med. Chem. 2010, 53, 4968–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Báez-Santos y. M.; St. John SE; Mesecar AD, The SARS-coronavirus Papain-Like Protease: Structure, function and Inhibition by Designed Antiviral Compounds. Antiviral Res. 2015, 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kilianski A; Baker SC, Cell-based antiviral screening against coronaviruses: developing virus-specific and broad-spectrum inhibitors. Antiviral Res 2014, 101, 105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sridharan H; Zhao C; Krug RM, Species specificity of the NS1 protein of influenza B virus: NS1 binds only human and non-human primate ubiquitin-like ISG15 proteins. J Biol Chem 2010, 285 (11), 7852–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Versteeg GA; Hale BG; van Boheemen S; Wolff T; Lenschow DJ; Garcia-Sastre A, Species-specific antagonism of host ISGylation by the influenza B virus NS1 protein. J Virol 2010, 84 (10), 5423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Oreshkova N; Molenaar RJ; Vreman S; Harders F; Oude Munnink BB; Hakze-van der Honing RW; Gerhards N; Tolsma P; Bouwstra R; Sikkema RS; Tacken MG; de Rooij MM; Weesendorp E; Engelsma MY; Bruschke CJ; Smit LA; Koopmans M; van der Poel WH; Stegeman A, SARS-CoV-2 infection in farmed minks, the Netherlands, April and May 2020. Euro Surveill 2020, 25 (23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Otwinowski Z; Minor W, Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–26. [DOI] [PubMed] [Google Scholar]
- (52).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011, 67 (Pt 4), 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J Appl Crystallogr 2007, 40 (Pt 4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Harcourt J; Tamin A; Lu X; Kamili S; Sakthivel SK; Murray J; Queen K; Tao Y; Paden CR; Zhang J; Li Y; Uehara A; Wang H; Goldsmith C; Bullock HA; Wang L; Whitaker B; Lynch B; Gautam R; Schindewolf C; Lokugamage KG; Scharton D; Plante JA; Mirchandani D; Widen SG; Narayanan K; Makino S; Ksiazek TG; Plante KS; Weaver SC; Lindstrom S; Tong S; Menachery VD; Thornburg NJ, Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerg Infect Dis 2020, 26 (6), 1266–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zhang Y; Wei Y; Li J; Li J, Development and optimization of a direct plaque assay for human and avian metapneumoviruses. J Virol Methods 2012, 185 (1), 61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.