

Abstract
Neurodegenerative diseases (NDs) are characteristic with progression of neuron degeneration, resulting in dysfunction of cognition and mobility. Many neurodegenerative diseases are because of proteinopathies that results from unusual protein accumulations and aggregations. The aggregation of misfolded proteins like β-amyloid, α-synuclein, tau, and polyglutamates are hallmarked in Alzheimer’s disease (AD), which are undruggable targets, and usually do not respond to conventional small-molecule agents. Therefore, developing novel technology and strategy for reducing the levels of protein aggregates would be critical for treatment of AD. Recently, the emerging proteolysis targeting chimeras (PRPTACs) technology has been significantly considered for artificial and selective degradation of aberrant target proteins. These engineered bifunctional molecules engage target proteins to be degraded by either the cellular degradation machinery in the ubiquitin-proteasome system (UPS) or via the autophagy-lysosome degradation pathway. Although the application of PROTACs technology is preferable than oligonucleotide and antibodies for treatment of NDs, many limitations such as their pharmacokinetic properties, tissue distribution and cell permeabilities, still need to be corrected. Herein, we review the recent advances in PROTACs technology with their limitation for pharmaceutical targeting of aberrant proteins involved in Alzheimer’s diseases. We also review therapeutic potential of dysregulated signaling such as PI3K/AKT/mTOR axis for the management of AD.
Keywords: PROTAC, ubiquitin-proteasome system, protein degradation, Autophagy, Alzheimer’s disease
1. introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that accountable for the leading case of dementia in elder people and ranks the sixth causes of death [1]. In 2020, it is estimated that 6.2 million American ages 65 and older are living with and suffering from Alzheimer’s disease [1]. Early-onset AD has also been reported for patients under age of 65, which is approximately occurring in 200,000 Americans because of the inherited genetic mutations, such as APP, APOE4, PS1, etc [2]. The most early symptom is trouble in memorizing new events, but in advanced level the problems include with linguistic, mood swing, confusion, losing motivation, self neglects, and behavioral issues [3]. The pathogenesis of Alzheimer’s disease is still not fully understood to date, and importantly its pathogenesis is irreversible at late stage, such as brain atrophy, thereby it requires unmet need for early diagnosis and treatment.
There are two pathological and aberrant structures in damaged neuronal cells that have been identified as main pathological hallmarks in AD, including senile plaques and neurofibrillary tangles (NFT) [4]. Usually senile plaques are caused by the aberrant deposition of aggregated protein fragments that is named beta-amyloid (Aβ42 and Aβ40) and constructed among nerve cells [5]. This event is called “amyloidosis of brain”, where Aβ peptides are cleaved from the amyloid precursor protein (APP) and aggregated as oligomeric Aβ. These pathogenic proteins aggregate into soluble toxic oligomers for the creation of hydrophobic surfaces before formation of insoluble fibrils and disruption of the phospholipid bilayer [6], which are considered as the main reason of AD [7]. On the other hand, the neurofibrillary tangles are constructed from the abnormal fibers of the hyper-phosphorylated tau protein inside neuronal cells [8]. Notably, hyper-phosphorylation of tau constructs tangles that eventually damage the structure and function of neuronal cells, which is considered another main pathology in AD [9]. In fact, the mechanistic principal of both senile plaques and neurofibrillary tangles have been well established and regarded as key hallmarks of Alzheimer disease [10, 11] (Figure 1).
Figure 1. Therapeutic targets in Alzheimer’s disease signaling.
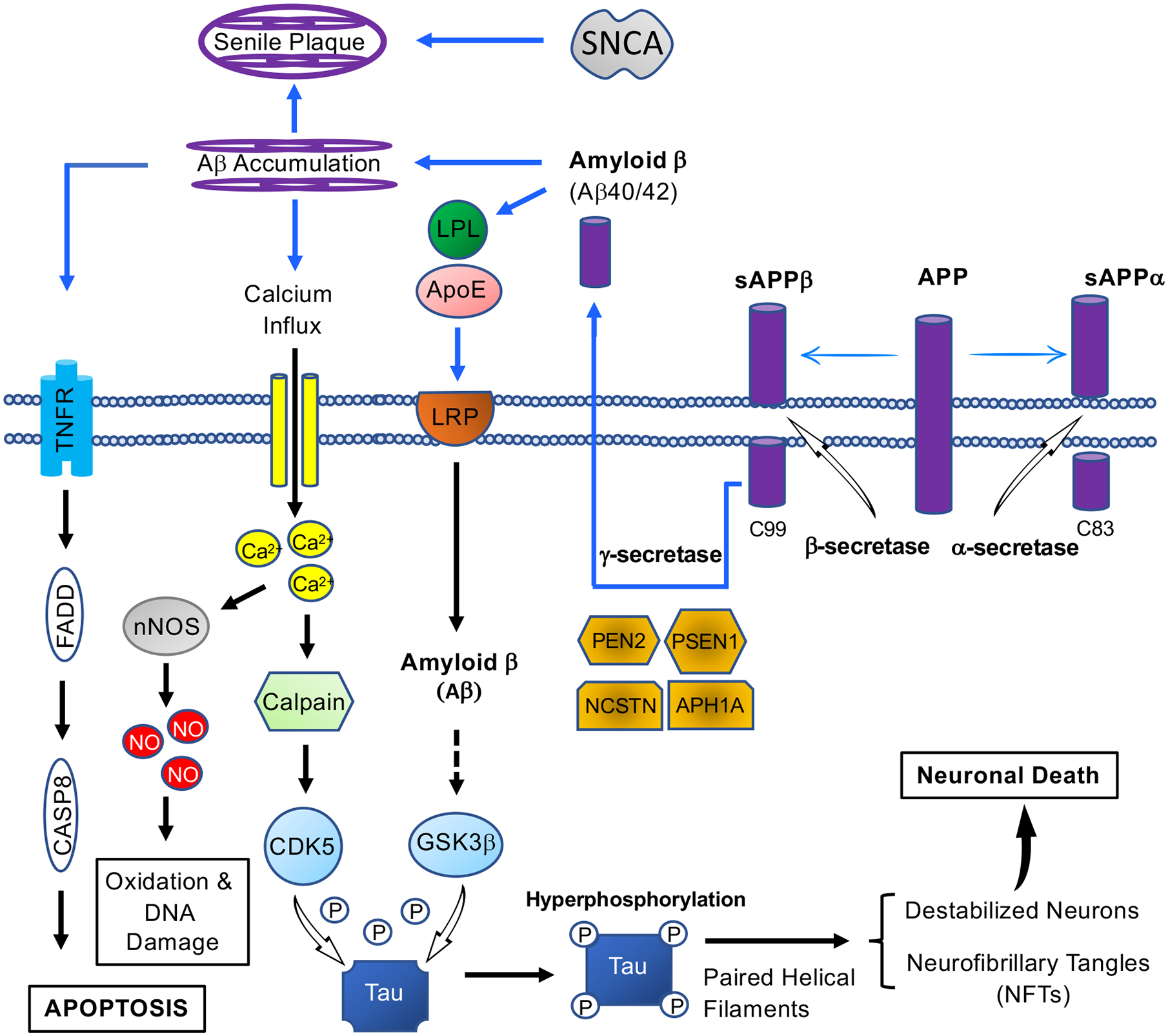
The formation of NFTs and senile plaques in AD signaling introduces several therapeutic targets. Four genes are pathogenetically committed to AD: amyloid precursor protein (APP), apolipoprotein E (ApoE), presenilin 1 (PSEN1) and presenilin2 (PSEN2). An increased level of amyloid-beta peptides is caused by mutations in APP and PSEN, leading to the formation of amyloid-beta 42, the main component of senile plaques. Cleavage of APP either by alpha-secretase or beta-secretase initiates extracellular release of soluble APP peptides, sAPPα and sAPPβ, and retains the corresponding membrane-anchored C-terminal fragments, C83 and C99. Alternatively, PSEN1/Nicastrin (NCSTN)-mediated gamma-secretase processing of C99 releases the amyloid-beta proteins, Amyloid β (Aβ40/42). In neuronal cell bodies, neurite outgrowth is stimulated by ApoE-containing lipoprotein lipase (LPL), via interacting with and letting LRP to bind APP for production of proteolytic fragment (Aβ) [161]. The accumulation Amyloid β results in blocked ion channels, mitochondrial oxidative stress, and activation of TNFR-regulated Caspase 8, and ultimately neuronal cell death. On the other hand, GSK-3 phosphorylates Tau at several sites for partial inhibition of tau biological activity in AD [162]. Upon abnormal Ca2+ homeostasis disturbance, the stimulation of calpain mediates the cleavage of p35 to p25 for the activation of CDK5, leading to hyperphosphorylation of Tau, and also promotion of APP truncation [163]. Finally, the elevated Ca2+ stimulates neuronal NO synthase, leading to production of nitrogen species and reactive oxygen [164]. SNCA: Alpha synuclein; PEN2: Presenilin Enhancer (Gamma-Secretase Subunit); APH1A: Aph-1 Homolog A (Gamma-Secretase Subunit); TNFR: Tumor necrosis factor receptors; FADD: Fas-associated protein with death domain; CASP8: Caspase 8; nNOS: Neuronal nitric oxides synthase
2. Protein Degradation Machinery in Alzheimer’s disease (AD)
Normal cells are gained self-controlling quality process to achieve homeostasis through prevention of prolonged damage in response to environmental conditions. Several neurodegenerative diseases are caused by misfolded proteins that aggregated into a β-sheet structure [6]. Usually cells are equipped with a defense machinery against misfolded and aggregated proteins to preserve homeostasis through two main strategies: (a) they refold misfolded proteins using a plethora of molecular chaperones, and (b) if cannot refold the misfolded proteins, cells will eliminate the forms of aggregated proteins for relieving neurodegenerative diseases. Outcome of losing these defensive machinery systems increase the deposition of protein aggregates, subsequently develop neurodegenerative diseases. Below we introduce the ubiquitin proteasome system (UPS) and autophagy pathways, which are main defensive machinery systems for protein quality control in neuronal cells.
2.1. The Ubiquitin Proteasome system
The ubiquitin proteasome system (UPS) is essential for protein homeostasis for quality control in cells [12]. Ubiquitination activities degradation of “unwanted” proteins via the 26S proteasome in which ubiquitin, a 76-amino-acid protein, is critical for composition of monoubiquitylation and/or polyubiquitination structure through covalent attachment to a target protein. This product is organized by multistep and reversible enzymatic cascade reactions including E1, E2, and E3 enzymes [13]. The eight amino group of N-terminus or Lysine residues (K6, K11, K27, K29, K33, K48, K63 and M1) in ubiquitin protein provide different probabilities for producing the “ubiquitination” signals [14] with diverse functional consequences (Figure 2).
Figure 2. A schematic representation of the E3 ligase biology.
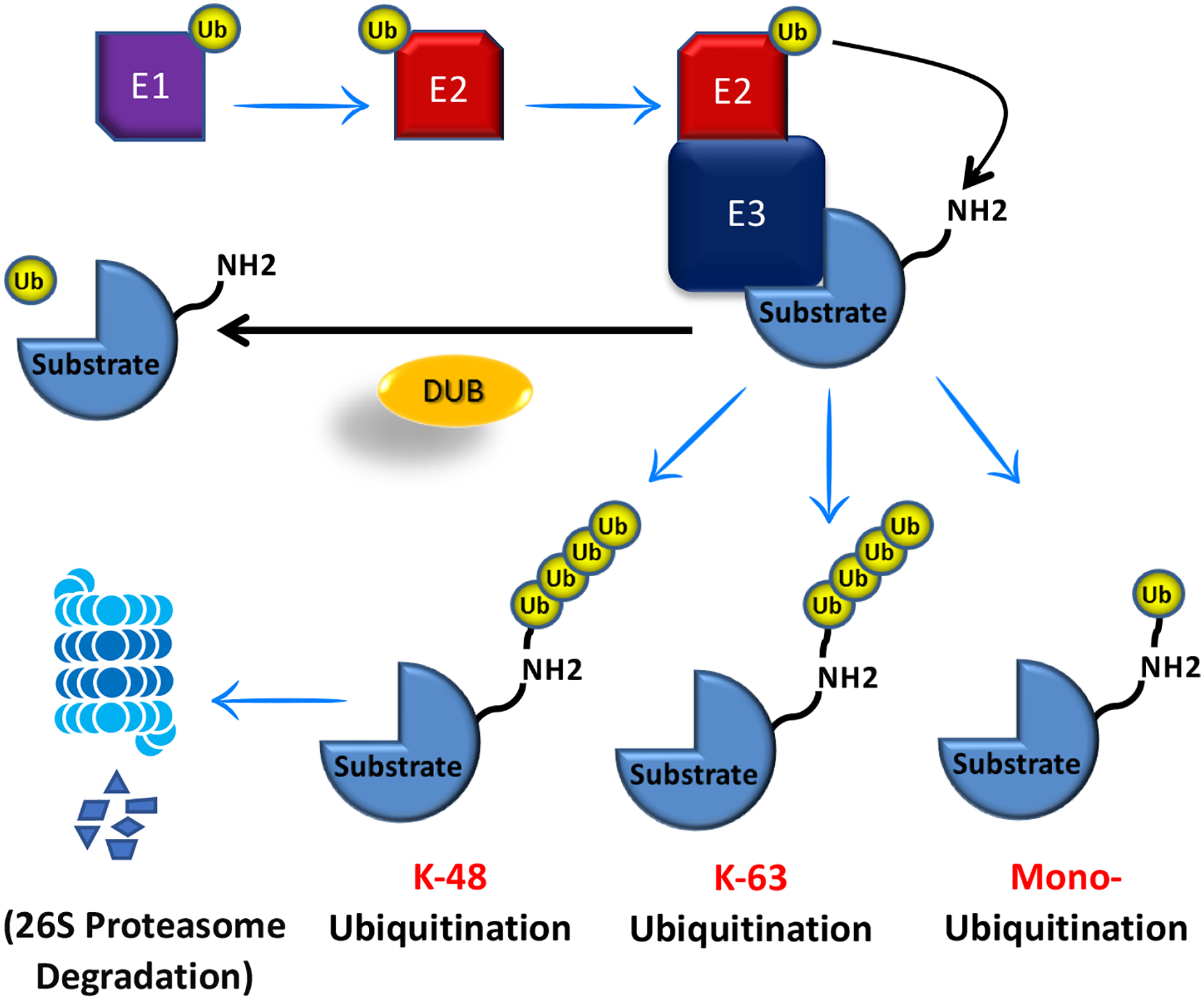
The ubiquitination of cellular proteins is triggered by the E1 enzyme that utilizes the formation of Ub-AMP. Following this catalytic reaction, ubiquitin is subsequently transferred to an E2 enzyme for activation of the thioester-linked E2-Ub complex. Finally, an E3 ligase enzyme transfers the ubiquitin protein from the E2 enzyme to a target protein. This ubiquitination process can be reversed by deubiquitinating enzymes (DUBs), which catalyze the cleavage of ubiquitin from target protein or substrate.
E1, Ubiquitin-activating enzymes; E2, ubiquitin-conjugating enzymes; and E3, ubiquitin ligases and Ub, Ubiquitin.
Protein turnover is essential for the synaptic plasticity and memory in the nervous system, which should be considered in the regulation of protein stability and functions in neuronal cells [15]. To this end, the UPS system controls most of the protein functions in the postsynaptic response in neuronal cells. Meanwhile, it is demonstrated that protein aggregate is largely the outcome of decreasing degradation process, but not from increasing the synthesis [16]. This indicates that neurons are failed in clearing of abnormal proteins in the process of neurodegenerative proteinopathies. Thus, understanding the process of UPS system in each neuron is needed for developing novel therapeutic approaches by enhancing proteasomal degradation for removing pathogenic aggregates in neuronal cells.
2.2. Autophagy
Autophagy is another important system and conserved degradation process for supporting protein homeostasis in eukaryotic cells. Autophagy mainly processes larger cytosolic structure such as cellular protein aggregates and organelles within the lysosome [17]. The best well-known form of autophagy is macro-autophagy, in which the protein targets are sequestered in the phagophore as a cytosolic membrane compartment [18]. Mechanistically, cellular targets are swallowed by autophagosomes, which is then transported and fused with lysosomes through the cytoskeleton. This mechanism expedites the degradation of cytoplasmic substrates such as misfolding proteins (aggrephagy), high loaded peroxisomes (perophagy), abnormal mitochondria (mitophagy) and pathogenic organisms (xenophagy) [19]. In fact, several intracellular proteins are known to govern the autophagy processes, which are known as autophagy-related proteins (ATG) [20] (Figure 3).
Figure 3. Protein degradation pathways in the neuronal cells along with targeted protein degradation strategies.
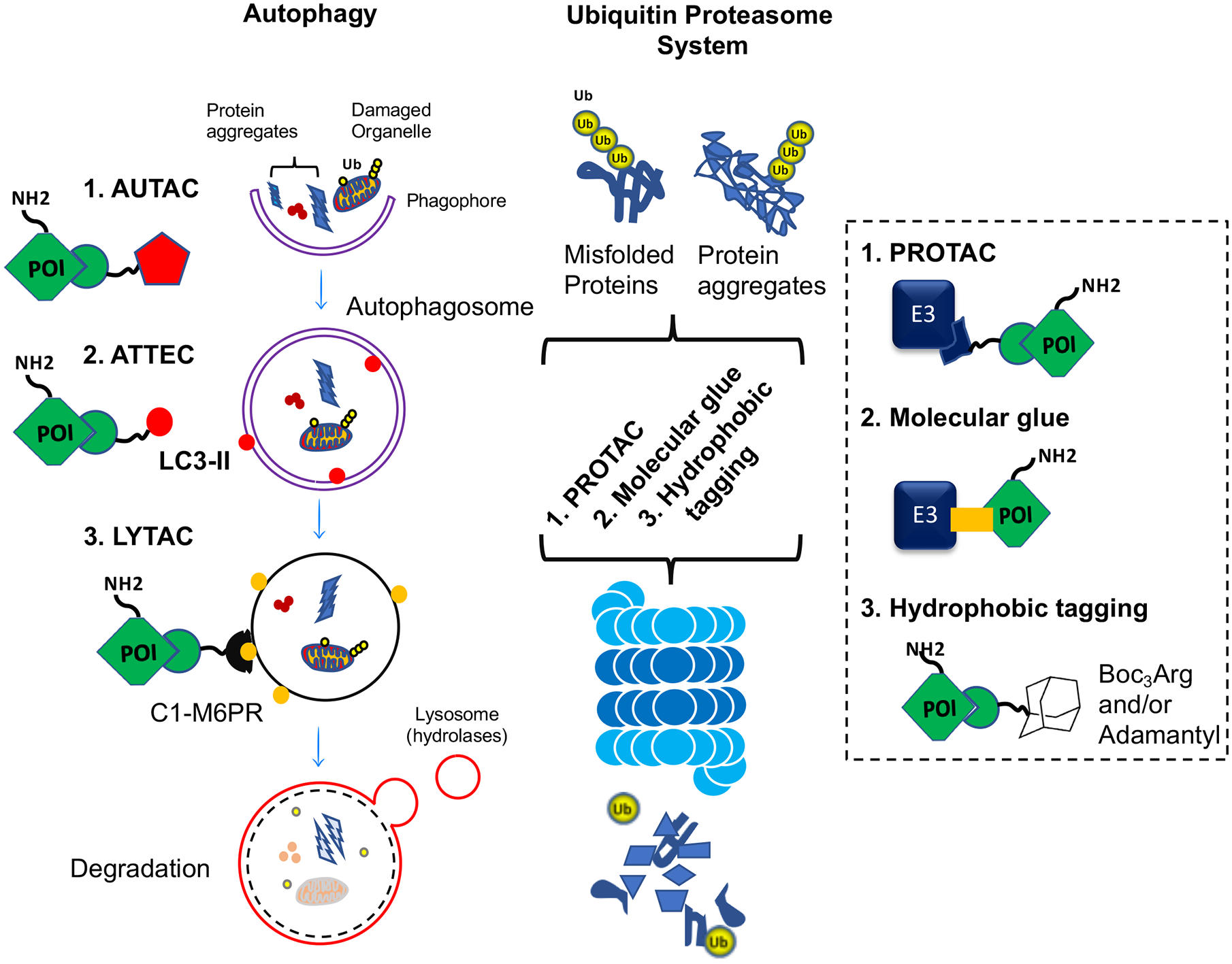
PROTAC, proteolysis-targeting chimera; AUTAC, autophagy-targeting chimeras; ATTEC, autophagosome-tethering compound; LYTAC, lysosome-targeting chimeras; C1M6PR, cation-independent mannose 6-phosphate receptor; LC3, Microtubule-associated proteins 1A/1B light chain 3B Ub, ubiquitin; E3, ubiquitin ligase; POI, protein of interest.
In regard to the protective function of autophagy in the physiology of neurons, the accumulation of abnormal tau proteins might be due to impaired autophagy within neurons [21]. It was shown that elevated activation of the autophagy increases degradation of the tau protein along with reduction in intracellular tau aggregation [22]. It was also discovered that increased activity of autophagy effectively reduces Aβ content, particularly in the early stage of Aβ accumulation [23, 24]. This is in the line with the report showing that Aβ can be resulted from amyloid precursor protein within the autophagosome, which indicates a unique link between the autophagy pathway and the formation of Aβ plaques [25].
3. Chemical-mediated targeted protein degradation
The strategies of targeted protein degradation (TPD) have recently emerged as modalities in drug discovery where the bifunctional small molecules hijack the cellular degradation machinery and direct protein targets for ubiquitin-mediated destruction. In 2001, the Crews group developed the first, peptide-based, bifunctional molecules including the ligands of target protein and a ubiquitin E3, for construction of proteolysis-targeting chimera (PROTAC). Thereafter this technology has become promising for the regulation of key target proteins that are otherwise untargetable. There are several methods have been established based on chemical-mediated targeted protein for degradation, which include Hydrophobic Tagging; HyT, Molecular Glues, Autophagy-Targeting Chimeras; ATTAC, Autophagosome-Tethering compound; ATTEC, Lysosome-Targeting Chimeras; LYTAC, and PROTACs (Figure 3).
In HyT experimental system, the addition of hydrophobic tags (e.g. Boc3Arg or Adamantyl) to ligands induce structural changes along with hydrophobic patches that initiates unfolding of protein targets for subsequent degradation via the protein homeostasis machinery systems [26]. On the other hand, in the molecular glue strategy, degraders such as lenalidomide interact the target protein with a specific, hijacked ubiquitin ligase complex for initiation of the ubiquitination process for the target protein to trigger its subsequent degradation by the 26S proteasome [27].
Moreover, Takahashi et al. have recently developed a novel targeted-clearance strategy termed as autophagy-targeting chimeras AUTACs, which are bifunctional molecules conjugated by small molecule that induce autophagy [28]. In brief, they added S-guanylation (guanine derivatives) as a tag to the chimeric molecules (including the guanine component and a specific ligand to a “target of interest” protein) to selectively direct protein substrates into the autophagy system for programmed destruction. They showed that AUTAC activated the removal of fragmented mitochondria and also the biogenesis of normal mitochondria. Thus, AUTAC could possibly provide a modality for developing autophagy-based drugs to its target specificity, and as an arsenal to combat Alzheimer’s disease. In addition, other strategies have been reported using autophagy including autophagosome-tethering compound, ATTEC, and also lysosome-targeting chimers, LYTAC, for targeted protein degradation [29]. In brief, molecules of ATTEC bind to the target protein along with LC3, an autophagosome protein, that requites target proteins to the autophagosome for subsequent induction of autophagy. Meanwhile, LYTAC which is a heterobifunctional molecule, targets extracellular and also membrane-associated proteins by the conjugation that engage the endosome and lysosome by recruitment of protein targets to the lysosome-shuttling receptor in cell-surface [29].
4. PROTACs
The proteolysis targeting chimeras (PROTACs) are heterobifunctional molecules that degrade a target protein by hijacking the powerful cellular degradation systems [30–32]. PROTACs recruit target protein to the E3 ubiquitin ligase (usually MDM2, VHL, IAPs, and CRBN ligases) with an optimal linker (Figure 4). This formation of complex between the protein of interest (POI) and the E3 ligases triggers POI ubiquitination for subsequent degradation by the 26S proteasome in eukaryotic cells [33–38] (Figure 5). The first PROTAC degrader was developed to target the androgen receptor in metastatic castration-resistant prostate cancer for treatment [39, 40]. After validation of the proof of concept, there is an intensive increase in the usage and development of the PROTAC technology in industry and academy. This is mainly because of the following reasons associated with PROTACs: 1) capable of reducing both the enzymatic and non-enzymatic functions of proteins, 2) able to destroy the “undruggable” protein of interest, 3) potent in improving specificity and selectivity, 4) elimination of protein targets rapidly and reversibly, and 5) with promise in overcoming drug resistance [41]. Drug resistance is a serious problem in anticancer therapy, thereby PROTACs technology is going to generate more effective drugs to overcome drug resistance. The usage of PROTACs should be also a great beneficial because of its novel mechanism of action for reducing the level of the target protein using less amount of drug compared to conventional ones. The first oral PROTAC that has been assessed in clinical trials is ARV-110 for prostate cancer and also ARV-471 for breast cancer both from Arvinas [42]. In brief, PROTAC molecules can be used to cure autoimmune and inflammatory and treat diseases such allergies, asthma, cardiovascular and Alzheimer [43].
Figure 4. E3 ligase ligands that are commonly used for PROTACs.
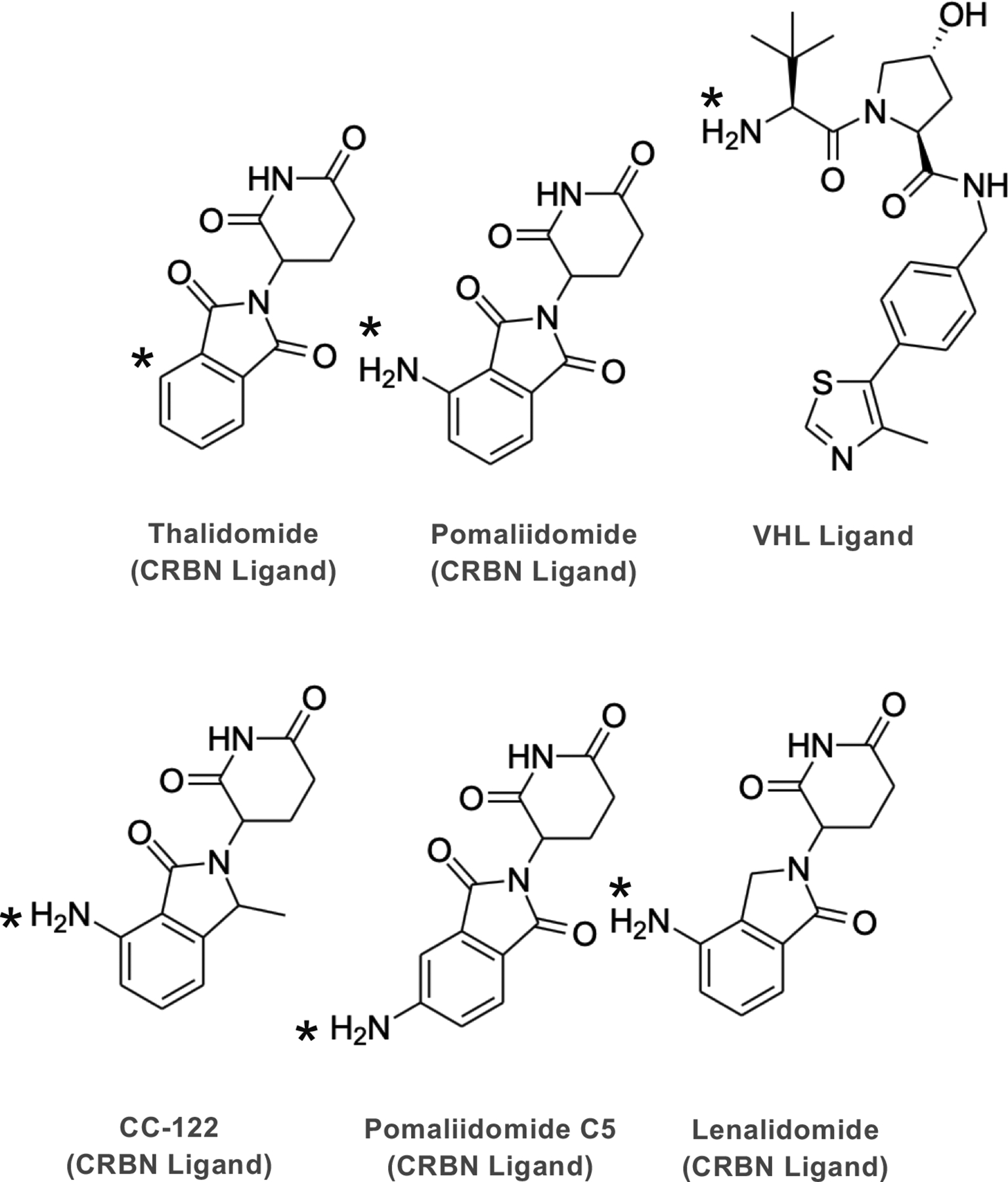
Derivatives of Thalidomide targeting Cereblon (CRBN) and the ligand of Von Hippel Lindau (VHL). The asterisk shows the point of attachment to the linker.
Figure 5. A schematic illustration of PROTAC mechanism(s) of actions.
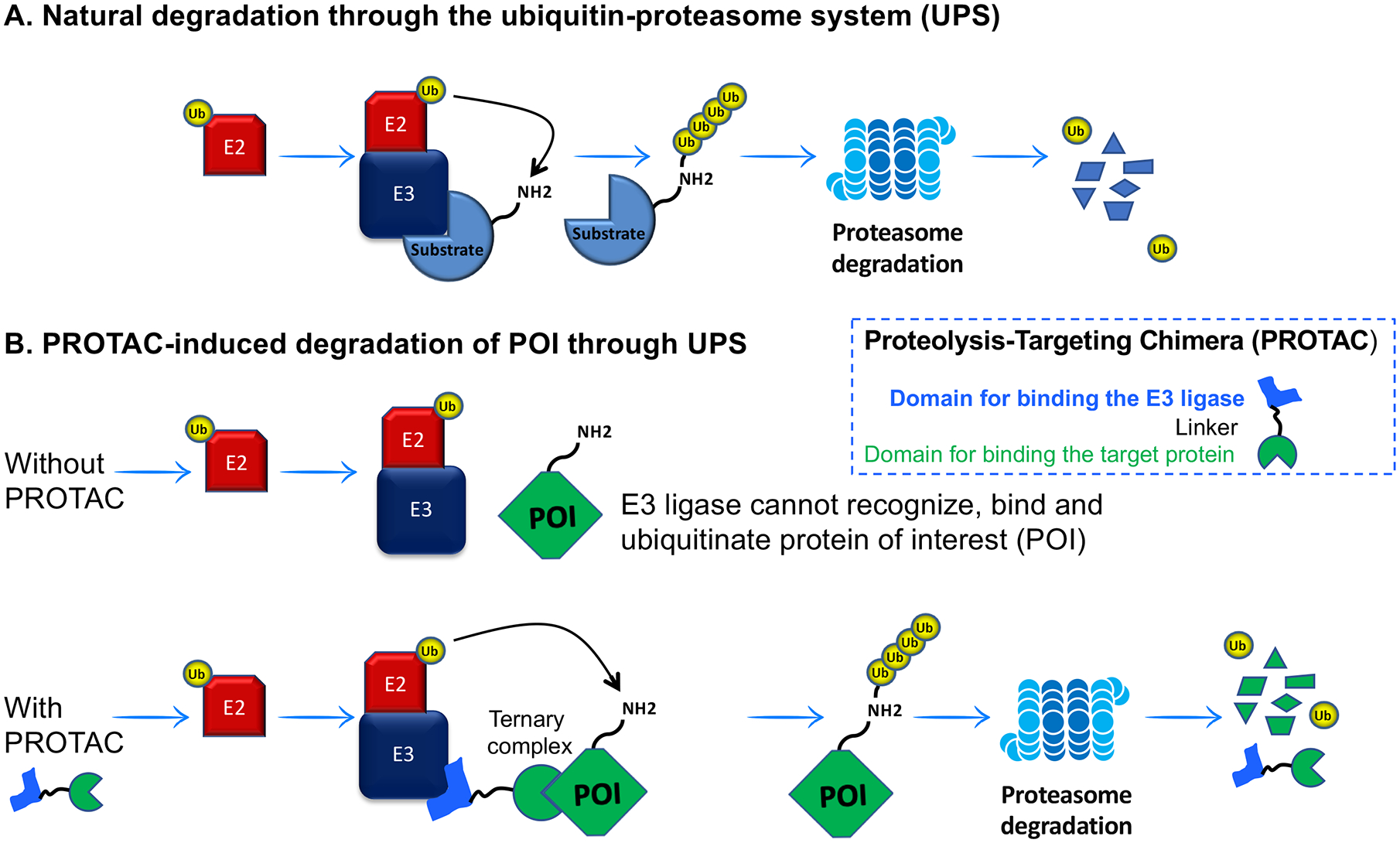
The E1 Ubiquitin-activating enzyme initiates transferring of Ubiquitin (Ub) to a target protein through the E1-E2-E3 enzymatic cascade. Then, the ubiquitinated target protein is degraded by the 26S proteasome. Without PROTAC, target protein is not recruited for the process of ubiquitination. The molecule of PROTAC attaches protein of interest (POI) with the E3 ubiquitin ligase for ubiquitination and subsequent proteasomal degradation.
5. Proteins targeting PROTACs in the management of Alzheimer’s disease
Here we introduce several PROTACs that have been developed for elimination of misfolded proteins, which is a major cause of neurodegenerative diseases. This provides an incredible therapeutic benefit to target a pathogenic protein for degradation. We will introduce the major dysregulated proteins including tau and α-synuclein that cause neurodegenerative diseases including AD. We will also discuss the main aberrant protein/signaling pathways including Hypoxia/HIF1α signaling, PI3K/mTOR/AKT, GSK-3β and BET that are characterized to contribute to Alzheimer disease.
5.1. The main pathological targets involved in Alzheimer’s disease
5.1.1. Tau
The tau proteins are abundant in neuronal cells, which functions to stabilize microtubules in axons, thereby is known as microtubule-associated proteins (MAPs) for axonal transport [44–48]. The increased aggregation of the tau protein is correlated with synaptic dysfunction, leading to abnormal localization of tau from axons to the somatodendritic region. Neurofibrillary tangles is the aggregation of tau protein inside neuronal cells of Alzheimer’s disease patients [44]. Herein, hyperphosphorylation of tau proteins develop aggregation into neurofibrillary tangles. Tauopathies which is specified by development of neurofibrils from hyperphosphorylation of tau, can arise in atypical Parkinson syndromes [49]. Dysregulation of tau is also an important issue in frontotemporal dementia (FTA), and in the toxicity of amyloid-β (Aβ). Thereby, tau is suggested as an attractive target for potential treatment of Alzheimer’s disease.
In 2016 and also 2018, Chu and Lu et al. reported that using peptide form of PROTACs could degrade tau proteins [50, 51]. Interestingly, peptidic PROTAC named as TH006 could degrade tau in CA3 region of hippocampus in vivo (Table 1). Later in 2019, Silva et al. constructed a set of unique PROTACs using a tau PET tracer as a warhead [52] for targeting tau in human differentiated frontotemporal dementia (FTD) neurons. They could sufficiently degrade both wildtype (WT) and tau variants, A152T and P301L, in neurons using PROTAC T807 with the Kd value of 1.8, 2.1 and 1.7 μM, respectively. They also established second PROTAC QC-01-175 that could specially degrade tau variants in neurons with FTD compared to healthy ones (Figure 6) (Table 1). In addition, they reported that QC-01-175 could rescue stress vulnerability in FTD patient-derived neuronal cell models.
Table 1.
A description of the properties for peptide- or chemical mediated targeting PROTACs.
| PROTAC | Entityc | Recognize Tau | Linker | Recruit E3 Ligasea | Cell-Penetrating Peptide |
IC50 (μM) | K d value (μM) | In Vivo study | PK study | PMID |
|---|---|---|---|---|---|---|---|---|---|---|
| Peptid-mediated targeting | TH006 | YQQYQDATADEQG | GSGS | ALAPYIP (VHL) | RRRRRRRRa | None | 0.3944 ±0.1589 | Tau degradation in CA3 region of hippocampus | Not reported | 27105281 |
| Peptide 1 | YQQYQDATADEQG | GSGS | LDPETGEYL(Keap1) | RRRRRRRR | None | Not reported | Not reported | Not reported | 29407955 | |
| Chemical-mediated targeting | QC-01–175 | WTb | CRL4CRBN | 8.559 | 1.2 ± 0.44 | Not reported | Not reported | 30907729 | ||
| A152Tb | 1.7 ± 0.54 | |||||||||
| P301Lb | 2.5 ± 1.31 | |||||||||
| T807 | WT | CRL4CRBN | 0.144 | 1.8 ± 0.99 | Not reported | Not reported | 30907729 | |||
| A152T | 2.1 ± 0.50 | |||||||||
| P301L | 1.7 ± 0.77 | |||||||||
| C004019 | Total, pS214 and pS404 tau | VHL | 0.00785 | Not reported | Robust tau clearance in hippocampus and cortex; Improvement of synaptic and cognitive functions | Tmax (h)=0167; Cmax (ng/ml)=10.8; AUClast (h*ng/ml)=8.42 | 33859747 |
Represents poly-D-arginine
Represents human recombinant biotinylated-tau WT, A152T and P301L, respectively
Tmax: the time point at which the drug concentration was the highest; Cmax: the maximum drug concentration; AUClast: area under the curve (the integral from the beginning to the last point in time).
Figure 6.
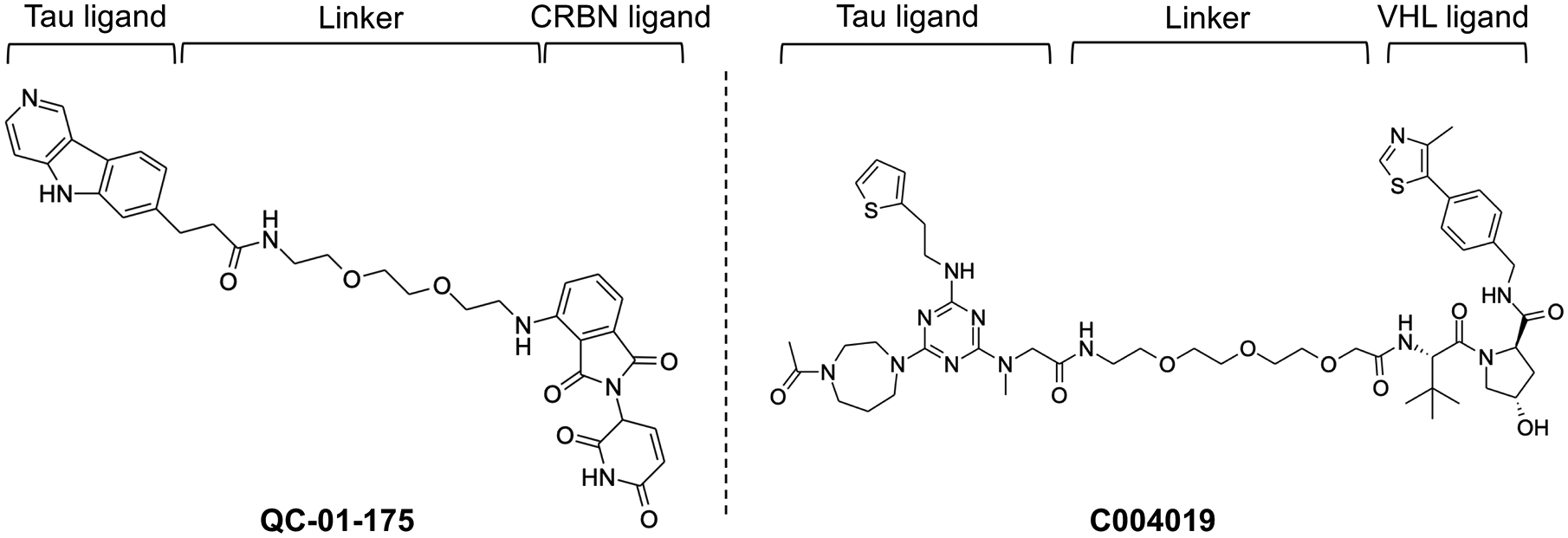
Chemical structure of Tau targeting PROTACs, QC-01-175 and C004019.
In 2021, Wang et al. developed a PROTAC using a linker that could connect tau to a VHL ligand [53] (Figure 6). Through in vitro and in vivo experiments, they found that their PROTAC, C004019, could clear tau protein in both physiological and pathological conditions. Interestingly, single dose or infrequent administration of the PROTAC (once per 6 days) could sustain tau reduction and alleviate Aβ-induced neurotoxicity in the brain of 3xTg-AD mouse model without showing obvious abnormalities. They could also determine the robust tau clearance in hippocampus and cortex of mice along with improvement of synaptic and cognitive functions. This indicates that their PROTAC was efficient drug candidate for tauopathies and treatment of AD (Table 1).
5.1.2. α-Synuclein
The amino acid sequence of α-synuclein includes three distinct domains: N-terminus, C-terminus and a central hydrophobic region which assembles α-synuclein into amyloid fibrils [54]. The misfolded and aggregation of α-synuclein are expanded in a prion-like fashion from cell to cell, resulting the amplification and progression of the fibrils into synucleopathies [55]. Since deposition of α-synuclein initiates in the enteric nervous system, the pathogenesis of synucleopathies is assumed to begin in enteric nerves prior to deposition in the brain. In fact, patients with Parkinson’s disease (PD) have gastrointestinal problems before displaying motor deficits [56]. The symptoms of PD and AD diseases are moderately similar, but pathologically AD affects the cerebral cortex and hippocampus, while PD mainly occurs in the substantia nigra [57]. Even though patients with AD show PD symptom and vice versa, PD patients compared to AD reveal more obvious cognitive dysfunction, suggesting that the event of pathological synergy between Aβ and αSyn. More studies revealed that αSyn is highly expressed in the brain region with abundant AD lesions, and the enrichment of αSyn in cortical region is correlated with existence of Aβ [58].
Recently, Kargbo et al developed bifunctional (PROTAC) compound that targets αSyn protein [59]. Their compound was developed using a Von Hippel Lindau (VHL) ends to moiety which could target protein of interest. The linker could place VHL E3 ubiquitin ligase in proximity to target protein for degradation in the UPS system. Subsequently, the representative PROTAC could prevent the accumulation and aberrant aggregation of αSyn protein in HEK293 cells stably expressing TREX αSyn A53T [59].
5.2. Aberrant target proteins dysregulated in AD microenvironment
5.2.1. Hypoxia/HIF1α signaling
Insufficient amount of oxygen known as hypoxia effects on pathological and physiological function of cells [60]. The prolyl hydroxylases (PHD) is recognized as a main molecular sensor of oxygen, which is disabled upon hypoxia for prolyl hydroxylation of hypoxia-induced factor alpha (HIF1α) and also AKT [61, 62]. The hydroxylation of HIF1α is recognized and polyubiquitinated by an E3 ligase, Von Hippel Lindau (VHL), leading to HIF1α for further degradation by the UPS system [63, 64]. Hypoxia-driven and/or HIF1α accumulation in VHL-deficient cells (ccRCCs), recruits HIF1β for transcriptional regulation of numerous genes [64]. It was also shown that phosphorylation of histone H2AX plays an important role upon hypoxia for the maintenance and inhibition of HIF1α degradation [65, 66].
The reduction of oxygen in the brain also associates with neurodegenerative disease such as Alzheimer’s. In fact, central nervous system is highly sensitive to oxygen supply which should be due to aging that weakens delivery of oxygen via cardiovascular system [67]. It was also shown that hypoxia supports the formation of plaque, leading to memory deficit in the mouse model for AD [68]. In the brain of hypoxia-driven AD, several molecular signaling is also activated to support oxidative stress [69], AKT/mTOR activation [70], angiogenesis [71], and metabolic activation. [72]. In the short period of time, hypoxia induces the expression of Aβ, cyclooxygenase-2 (COX-2) and presenilin 1 (PS1) for leading neuroinflammation in the brain with AD [73], but in the extended hypoxic condition, Ca2+ channels are upregulated for production of Aβ [74]. Importantly, HIF1α triggers transcriptional expression of VEGF [75] and BACE1 (main biomarker for AD) [76], thereby increasing Aβ deposition and also neurotic plaque production in transgenic mouse model [76]. This indicates that HIF1α induces tau phosphorylation in part through regulation of several signaling pathway including GSK3β, CDK5 and mTOR [77, 78]. Moreover, Glut1 and Glut3, other HIF1α-target genes and the main brain glucose transporters, are expressed less in AD [79].
In early 2004, Schneekloth JS Jr et al. reported peptide-based PROTACs could recognize the transcription HIF1α and had also the potentiality to bind to VHL [80]. They constructed a poly-D-arginine tag on the peptide sequence which could assist in cell penetration. Interestingly, the discovery of small-molecule that could inhibit the interaction between HIF1α and VHL, initiated the establishment of several PROTACs, which link VHL to other target protein of interest with high specificity and high-affinity [81, 82]. Taken together, aging could induce hypoxia/HIF1α for dysregulation of several molecular signaling in neurons for AD activation, which should be considered for careful and selective degradation using novel PROTACs technology.
5.2.2. PI3K/mTOR signaling
Phosphatidylinositol 3-kinases (PI3K) is a member of the PI3K/AKT/mTOR signaling pathway functions as phosphatidylinositol kinases. It mainly regulates apoptosis, proliferation and differentiation of cell and its overexpression drives the main feature of tumorigeneses. Moreover, the mammalian target of rapamycin (mTOR) responds to wide range of extracellular stimuli for the regulation of cell growth and metabolic homeostasis [83]. mTOR pathway also regulates several diseases including cancer [84], diabetes [85] and neurodegenerative pathological condition [86]. In fact, inhibiting mTOR expands life span of several organism such as C. elegans [87] and mice [88]. Constantly, Rapamycin as a mTOR specific inhibitor extends their lifespan [89–93]. Importantly, mTOR kinase promotes the phosphorylation of tau by regulating multiple kinases including glycogen synthase kinase 3 (GSK3), protein kinase A (PKA), and cyclin-dependent kinase 5 (CDK5) [94]. Moreover, mTOR directly phosphorylates and inhibits protein phosphatase 2A (PP2A), which is a main phosphatase downregulated in the brain with AD [95], for increasing tau phosphorylation [96]. Furthermore, downstream targets of mTOR such as S6K and eukaryotic translation factor 4E (eIF4E), elevate mRNA translation level of Tau, indicates that overactivation of mTOR could accumulate tau protein [97]. Surprisingly, the administration of Aβ in the hippocampus of normal mice could activate the mTOR pathway, representing the role of the amyloid precursor in the activity of mTOR signaling [98]. Furthermore, rapamycin could reduce cognitive deficits in the tau pathology through amelioration of Aβ in the AD brains of mice [99]. Taken together, mTOR imbalances the homeostasis of tau, resulting in the aggregation of tau and formation of neurofibrillary tangles for AD generation. This suggests potential strategy for treatment of AD through targeting mTOR.
Recently several PI3K inhibitors have been stablished in which most of them have limitation because of less selectivity and also side effects [100, 101]. It means that improvement of unique PI3K-targeting PROTACs has been recognized as potent strategy. In 2018, Jiang et al. has established a set of prospective PI3K-degreders using lenalidomide links to the ZSTK474 as a PI3K inhibitor [102]. Although this PROTAC revealed less enzymatic activity compared to ZSTK474, they could successfully degrade PI3K at 10 μM along with decreasing the phosphorylation of GSK-3β, S6K and AKT in the PI3K/AKT/mTOR signaling pathway.
5.2.3. AKT
AKT is a central member of the PI3K/AKT/mTOR signaling pathways and known as a serine/threonine kinase which regulates several cellular processes such as survival, proliferation and metabolism. Some mutations with gain of function and/or activation of oncogenes such as PI3K and receptor tyrosine kinases and/or loss of tumor suppressor function of PTEN, lead to hyperactivation of AKT for cancer progression [103]. It was also shown that GSK3β-mediated phosphorylation of tau is critical for initiation of AKT-sulfhydration [104]. Therefore, AKT is considered in the central of the PI3K/AKT/mTOR signaling which should be an interesting therapeutic target to combat AD.
In 2019, Toker et al. developed a specific small molecule degrader using the conjugation of CRBN ligand along with GDC-0068 as an AKT inhibitor [105]. This engineered PROTAC could accordingly inhibit AKT1, AKT2, and AKT3 with IC50 values at 2.0 nM, 6.8 nM, and 3.5 nM, whereas IC50 values of GDC-0068 was measured at 5 nM, 18 nM, and 8 nM, respectively. In addition, this PROTAC could destabilize all three isoforms followed by reduction in AKT-downstream signaling. They also found that the engineered PROTAC could downregulate cell proliferation much efficient that its parental inhibitor, suggesting its beneficial for targeted degradation of AKT.
5.2.4. GSK-3β
Glycogen synthase kinase 3; GSK-3, is known as serine/threonine protein kinase and a member of phosphotransferase family [106]. Pathologically, GSK-3β regulates several process through phosphorylation of tau and also produce amyloid beta peptide which cause neurofibrillary tangles and amyloid in Alzheimer’s disease [104]. Moreover, proinflammatory function of GSK-3β causes the loss of neuron [107–109] and develop neurodegenerative disease [110], thereby should be considered as therapeutic target to fight against AD [111–113]. In 2021, Jiang et al. reported a first set of GSK-3β targeting PROTACs which was developed using CRBN as an E3 ubiquitin ligase [114]. Their PROTAC could efficiently induce degradation of GSK-3β protein (44%) in less IC50 value at 2.8 μM, which should be considered as effective GSK-3β degraders in the PROTAC system.
5.2.5. BET
Dysregulation of inflammation is critical process in the pathology of many CNS diseases, which occurs through several pathways including NF-kB and Nrf2 signaling involved in inflammation. Extra-terminal domain (BET) proteins include four members (BRD2, BRD3, BRD4 and BRDT) that play critical roles for transcriptional regulation of inflammatory response [115, 116]. For example, BETs assemble the complex of histone acetylation-dependent chromatin for the expression of inflammatory genes. Early studies have also shown that inhibition of BET activates anti-inflammation, suggesting that BRD2, BRD3 and BRD4 proteins could play critical roles in AD and other neuroinflammation disorders. [116–120]. Targeting BET proteins using small-molecule inhibitor (JQ1) downregulates several pro-inflammatory regulators such as IL-1β and TNF-α followed by the phosphorylation of tau at Ser396 in the frontal cortex and the hippocampus of the 3xTg mouse model for AD [121]. However, mice did not show deficits in memory and the ameliorating in learning. In the AD mouse model of the APP/PS1, JQ1 could enhance long-term potentiation and cognitive function [122]. Further study showed that JQ1 could activate the gene expression of hippocampal genes, which were responsible to the activation of ion channel and DNA repair [122]. The chemical probes such as pan-BETi(s) serve as an ideal PROTAC for targeting BET proteins due to their potentiality for identification and recruitment of BET.
The first PROTACs targeted BET proteins (dBET1) were identified in 2015 and contained by an E3 ligase and a BRD4 BD binding moiety (JQ1 or OTX015) [123]. The BET PROTACs could identify and recruit the CRBN E3 ubiquitin ligase for efficient and selective degradation of BRD4 protein in vitro and in vivo [123–127]. Moreover, dBET1 could eliminate BET proteins 8–10-fold along with several other BET-target proteins revealed by the proteomic study [123].
6. The Strengths and weaknesses of PROTACs Technology
6.1. Advantages
The PROTACs technology has several advantages over traditional approaches that make them exclusively suitable for the treatment of CNS diseases [128]. The main advantage is the capability of PROTACs for utilizing targeted degradation of undruggable proteins in the CNS. The specificity of PROTACs is high in comparison to other methods, in which they can selectively degrade different isotypes of proteins which are expressed by the same gene. Furthermore, PROTACs bypass the potential toxic effects that can be developed by pharmacological approaches. In fact, they do not need to directly attach target protein for long period of time for inactivation of the target protein function. Moreover, development of PROTACs is designed based on catalytic reaction, which can be reused for many cycles until the target proteins in cells are eliminated. Furthermore, PROTACs have sub-stoichiometric catalytic activity [129], letting the administration of very low concentrations to be sufficient enough to degrade a target protein. Therefore, there is strong feasibility to develop an active drug using PROTACs, in which the inhibition of the target protein is not required molar mass of drug [130, 131]. Overall, these notions suggest that PROTACs could be a very effective method to target and destroy undruggable proteins that is depictive in the pathophysiology of many CNS disorders including AD.
6.2. Disadvantages
There are a number of limitations that could prevent development of PROTAC drug for the clinical applications in the future. Because PROTACs technology employ drugs for dual targets, thereby the constructed compounds would have a high molecular weight for being easily synthesized [30, 131]. Therefore, they cannot be simply dissolved for oral absorption because of transmural issue, which reveal a serious barrier in pharmacokinetics. For example, one main issue is the blood-brain barrier permeability, which is a limiting factor for many pharmacological approaches. As we reviewed the efficacy study of PROTAC named as C004019 (section 5.1.1) [53], it is noteworthy to suggest that they might also effectively penetrate the blood-brain barrier [30].
Another possible limitation is localization of the PROTAC to the specific brain regions, which are aimed to combat neurodegenerative diseases such as Alzheimer’s disease. For successful use of PROTAC, the E3 ligase must be expressed in the target region(s). However, some E3 ligases (e.g. CRBN) [132] are differentially expressed across brain areas. This differential expression pattern can complicate treating diseases. For example, tau accumulation occurs in a progressive manner in the AD brain regions that is started in locus coeruleus and also entorhinal cortex and ended in primary visual cortex [133]. This suggests that depending on disease progression, PROTACs would need to target tau precisely in some specific regions of the brain. Therefore, it is challenging to manage drug for colocalization with and/or without expression of E3 ligase in the affected brain regions, and it needs establishment of new technology to solve this emerging concern [134]. Furthermore, it was shown that many CNS disorders are associated with expansive reductions in proteasome catalytic activity [135]. This indicates that even if a PROTAC can ubiquitinate the protein of interest, it still may not possibly degrade it. Thus, reduced proteasome function remains a barrier for PROTAC-mediated degradation of target proteins for treatment of AD, which are divergently expressed in the brain.
7. Discussion
PROTACs is a powerful and attractive strategy studied and developed in both academy and industry. Recently, they have been widely investigated for treatment of several diseases including cancer, neurodegeneration, immune disorders, cardiovascular dysfunction, fatty liver disease (FLD), and for controlling viral infections. Here we discuss the AD-specific PROTACs, and also several others that have been used for targeting aberrant proteins/signaling which are dysregulated in tumor microenvironment. However, due to their similar structure and mechanism, we introduce them as potential PROTACs to fight against AD. We reviewed two peptidic PROTACs that have been developed for targeting tau in treatment of AD [50, 51]. Although they could degrade tau in the center of the aberrant signaling in AD, their application has been limited in part due to their intrinsic weakness against protease degradation and their poor membrane permeability in vivo. The stability of those peptides might be improved by substitution of unnatural amino acid, backbone modification, and cyclization [136] and their cell membrane permeability could be modified by increasing lipophilicity and decreasing hydrogen bonding [137]. Silva et al. have also established another type of PROTACs using a tau PET tracer as a warhead, however, their application was not validated in vivo [52]). In fact, the construction of PROTAC such as warhead with high affinity to target protein might be not enough for the generation of an active PROTAC [138]. Therefore, the induction of steric structure by the formation of target protein linked to hijacked E3 ligase could facilitate the transfer of ubiquitin from E2 to target protein and potentiate PROTAC activity. This ternary complex structure enables PROTAC to degrade large molecules such as protein aggregates, which are normally not allowed in the proteasome’s barrel-like structure for degradation [15]. Through this approach, Wang et al. established PROTAC, named as C004019, that could efficiently induce clearance of tau in the brain of hTau and 3xTg-AD transgenic mice. This indicates that C004019 could potentially pass through the proteasome’s barrel-like structure and also the brain-blood barrier, in which most of large drugs unable to do so. Although C004019 could remarkably improve synaptic and cognitive functions in the mice, its selectivity was revealed poor for recognition of tau species (wild type and phosphorylated tau). This might be in part due to the structure of tau which is natively unfolded protein, thereby opening new challenge for the improvement of their PROTAC capability.
Although aging is a main cause of cancer and AD [139], there are some studies revealing pathogenesis of AD protects patients against cancer and vice versa, but the underlying mechanisms remain elusive to date [140, 141]. Notably, cancer and AD pathologically share common feature, for example, a high level of cell cycle re-entry, which is required for cancer pathogenesis, is also revealed in the AD patients [142, 143], but instead of cell division, neuronal cell cycle is aborted going to death [144, 145]. This includes a significant elevation in the activation of CDK2, CDK4, CDK5 and caspases during cell cycling for APP phosphorylation, APP proteolysis, and production of Aβ [146–148]. The role of mTOR activation for the initiation of proliferation suggests its function in the neuronal cell cycle re-entry and pathogenesis of AD [149]. The activation of mTOR is also believed for driving the neurodegeneration through activation of Tau [150], leading to the accumulation of NFTs. Moreover, inactivation of mTOR pathway results in the activation of autophagy for cell clearance from Aβ [151]. In the brain tumor microenvironment, the adenosine monophosphate protein kinase protein (AMPK) might be activated upon energy stress for positively activation of FOXO3 [152] to protect neuronal cells. This energy stress is due to high metabolomic rate of cancer cell proliferation that hijacking energy in the brain microenvironment. Meanwhile, the mTOR signaling and the consequent activation of cell cycle re-entry is inhibited by AMPK [153], in which FOXO increases the expression of antioxidant enzymes to diminish the damage of cell [154, 155] and inhibit neurodegeneration. Taken together, mTOR-mediated cell growth and proliferation not only drives cancer cell progression, but also causes neuronal cell arrest that is a readout of disabled cell division in neuronal cells. Therefore, targeting the PI3K/mTOR/AKT axis introduces a great therapeutic option for the management of AD and tumor microenvironments (Figure 7).
Figure 7. Therapeutic potential of the PI3K/AKT/mTOR axis for the management of AD and cancer.
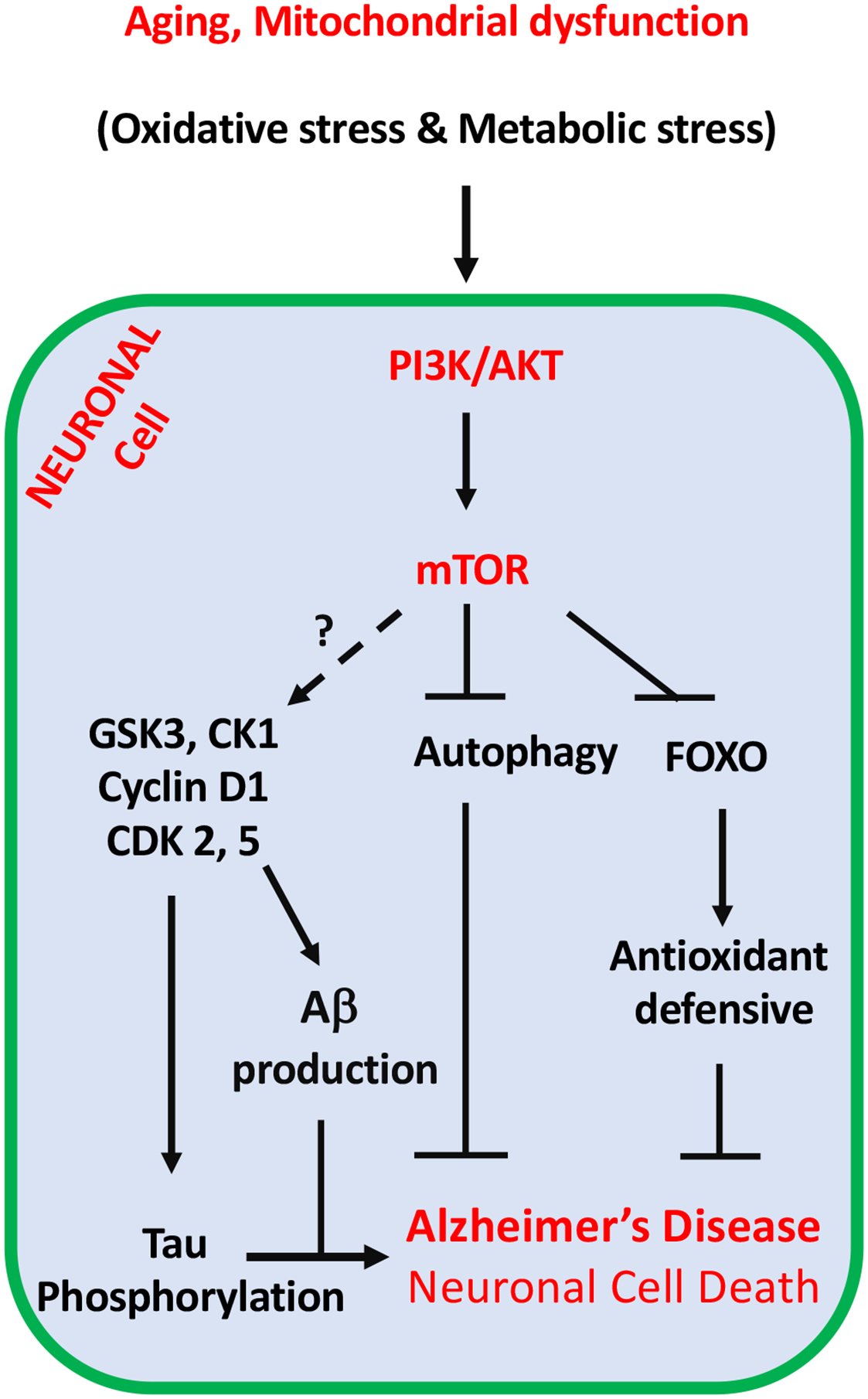
A long-term function of physiological stimulators such as aging followed by mitochondrial dysfunction might accumulate oxidative stress and metabolic stress, which both trigger the activation of the PI3K/AKT/mTOR signaling for highly activation of cell cycle re-entry in neuronal cells. In contrast with cancer, neuronal cell cycle is aborted and going to death instead of cell division. Moreover, a significant elevation of CDK2, CDK4, CDK5 and activation of caspases during cell cycle re-entry leading to APP phosphorylation, APP proteolysis, and production of Aβ. Furthermore, the activation of mTOR pathway results in the inactivation of autophagy, which is required for the clearance of Aβ in the neuronal cells. Upon cancer cell proliferation in the brain and hijacking energy from neurons, AMPK is stimulated upon this energy stress and positively activate FOXO3 to protect neuronal cells in the brain tumor microenvironment. FOXO increases the expression of antioxidant enzymes to diminish the damage of neuronal cell. Dashed arrow required additional investigation for that molecular network in the certain condition.
In section 5.2, we briefly introduced several PROTACs that have been established to target HIF1α, PI3K, AKT, and BET in tumor microenvironment with aberrant signaling. Most of these PROTACs could efficiently degrade their own target proteins, such as BET, with high sensitivity and selectivity. Therefore, the application of these PROTACs shows great potential to target AD microenvironment. Although cancer and AD are pathologically share common feature, the disease-causing mechanisms differ dramatically. Given that most established PROTACs were mainly studied in the cancer disease setting, their formulation and mechanism of actions should be carefully designed to combat AD. Currently, two PROTACs, ARV-110 and ARV-47, have been used in clinical trials for treatment of prostate and breast cancer, respectively by targeting of androgen receptor and estrogen receptor (from Arvinas). They both show an acceptable safety trial in which ARV-471 is well-tolerated at all tested dose levels without serious side effects. Interestingly, ARV-471 could produce a synergistic effect on the inhibition of tumor in the combination treatment using kinase inhibitors such as CDK4/6 inhibitors. This indicates that combination of PROTAC with chemotherapy, antibody (in immunotherapy), and small molecule inhibitors might be an alternative approach for treatment of disease, including cancer and AD.
8. Conclusion and Perspectives
The PROTACs technology enables us to discover new therapeutic agents with unique capability for degradation of “undruggable” proteins instead of inhibition. They are capable of answering some concerns related to the use of traditional small molecules which have poor selectivity, side effects along with drug resistance. However, the toxicity of PROTACs can limit future drug development in part due to the effects of off-target degradation. The PROTACs degrade proteins completely in the ubiquitin proteasome system, while parental small molecules or compounds are just required to inhibit protein functions. Therefore, the combination of small molecules with a tissue specific E3 ligase provided great advantages for selective degradation of target proteins. Moreover, the structure of PROTACs is generally complex and their syntheses are complicated because of high molecular weight (800–1000 kDa), which should be recognized for further modification and screening for the improvement of the brain membrane permeability. This needs further optimization in the structure and synthesis of PROTACs, which is essential in the stable platform for drug discovery. To this end, the application of crystallography can help us to understand their structural mechanism. For example, structural analyses of VHL and/or Cereblon E3 ligase proteins demonstrate the shape and the position of the Cullin-RING E3 ubiquitin ligase along with E2 and target proteins in details that enable us to target ubiquitin transfer [156].
As we mentioned, it is impossible to confirm that all proteins of interest will be degraded by the PROTAC in the absence of the ligands. Therefore, selection of new ligands and validation of targets will be another focus in the PROTAC development. There are approximately over six hundreds of E3 ligases existing in the human genome, but only a few of them have been used in PROTACs technology. This shows the possibility for expanding effort to screen and find suitable E3 ligases for this issue. Furthermore, more E3 ligases need to be investigated for avoiding off-target in timely response to brain diseases especially in Alzheimer’s disease. Finally, recent advancements in the CRISPR-dCas9 technology could be helpful for stimulation of proteasome function in the brain disorders with expansive reductions in proteasome catalytic activity [157]. Interestingly, it was found that IU1, as a compound for inhibition of ubiquitin-specific protease 14 (USP14), could improve protein degradation in vitro [158] and in mouse brain tissue [159], although proteasome function was not broadly enhanced in the brain [160]. This suggests a viable avenue in careful designing of combination therapy using PROTACs, monoclonal antibodies, RNA interferes, and/or small-molecule inhibitors to combat Alzheimer’s disease.
9. Data Retrieval
This review was accomplished using electronic databases, such as PubMed, Scopus, Web of Medline for finding articles linked to Alzheimer’s disease, aberrant signaling pathways, as well as the application of PROTACs. We used the keywords such as (“Alzheimer’s disease” OR “PROTAC” OR “Ubiquitin Proteasome system”, OR “Therapeutic target”, OR “aberrant signaling”) (title/abstract/keywords). In general, the entitled principles were PROTACs affecting aberrant signaling pathway in Alzheimer’s disease. Data were gathered without date limitations until November 2021. The list of reference in the articles were also reviewed by the hand search for aberrant signaling pathways, which was recognized as a crucial therapeutic target in AD.
Acknowledgments
We would like to sincerely thank our colleagues for critical reading of the manuscript. We are also very sorry that due to space limitation, we could not include all of the studies related to Alzheimer’s disease and PROTACs in this review. This study was partially supported by US National Institutes of Health (NIH) grants to W.W. (R35CA253027).
Footnotes
Declaration of Competing Interest
The authors declare no conflicts of interest associated with this manuscript.
References
- 1.2021 Alzheimer’s disease facts and figures. Alzheimers Dement, 2021. 17(3): p. 327–406. [DOI] [PubMed] [Google Scholar]
- 2.Alzheimer’s A, 2009 Alzheimer’s disease facts and figures. Alzheimers Dement, 2009. 5(3): p. 234–70. [DOI] [PubMed] [Google Scholar]
- 3.Burns A and Iliffe S, Alzheimer’s disease. BMJ, 2009. 338: p. b158. [DOI] [PubMed] [Google Scholar]
- 4.Zhu X, et al. , Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta, 2007. 1772(4): p. 494–502. [DOI] [PubMed] [Google Scholar]
- 5.Murphy MP and LeVine H 3rd, Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis, 2010. 19(1): p. 311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartl FU, Protein Misfolding Diseases. Annu Rev Biochem, 2017. 86: p. 21–26. [DOI] [PubMed] [Google Scholar]
- 7.Hardy J and Selkoe DJ, The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science, 2002. 297(5580): p. 353–6. [DOI] [PubMed] [Google Scholar]
- 8.O’Brien RJ and Wong PC, Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci, 2011. 34: p. 185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alonso AC, Grundke-Iqbal I, and Iqbal K, Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med, 1996. 2(7): p. 783–7. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong RA, The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol, 2009. 47(4): p. 289–99. [PubMed] [Google Scholar]
- 11.Serrano-Pozo A, et al. , Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med, 2011. 1(1): p. a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pohl C and Dikic I, Cellular quality control by the ubiquitin-proteasome system and autophagy. Science, 2019. 366(6467): p. 818–822. [DOI] [PubMed] [Google Scholar]
- 13.Deol KK, Lorenz S, and Strieter ER, Enzymatic Logic of Ubiquitin Chain Assembly. Front Physiol, 2019. 10: p. 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heride C, Urbe S, and Clague MJ, Ubiquitin code assembly and disassembly. Curr Biol, 2014. 24(6): p. R215–20. [DOI] [PubMed] [Google Scholar]
- 15.Tai HC and Schuman EM, Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci, 2008. 9(11): p. 826–38. [DOI] [PubMed] [Google Scholar]
- 16.Ding Q, Cecarini V, and Keller JN, Interplay between protein synthesis and degradation in the CNS: physiological and pathological implications. Trends Neurosci, 2007. 30(1): p. 31–6. [DOI] [PubMed] [Google Scholar]
- 17.Kerr JS, et al. , Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci, 2017. 40(3): p. 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Y, et al. , The machinery of macroautophagy. Cell Res, 2014. 24(1): p. 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menzies FM, et al. , Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron, 2017. 93(5): p. 1015–1034. [DOI] [PubMed] [Google Scholar]
- 20.Cheng J, et al. , Autophagy regulates MAVS signaling activation in a phosphorylation-dependent manner in microglia. Cell Death Differ, 2017. 24(2): p. 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q, Liu Y, and Sun M, Autophagy and Alzheimer’s Disease. Cell Mol Neurobiol, 2017. 37(3): p. 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zare-Shahabadi A, et al. , Autophagy in Alzheimer’s disease. Rev Neurosci, 2015. 26(4): p. 385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian Y, et al. , A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J, 2011. 25(6): p. 1934–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vingtdeux V, et al. , Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J, 2011. 25(1): p. 219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boland B, et al. , Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci, 2008. 28(27): p. 6926–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cromm PM and Crews CM, Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem Biol, 2017. 24(9): p. 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schreiber SL, The Rise of Molecular Glues. Cell, 2021. 184(1): p. 3–9. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi D, et al. , AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol Cell, 2019. 76(5): p. 797–810 e10. [DOI] [PubMed] [Google Scholar]
- 29.Banik SM, et al. , Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature, 2020. 584(7820): p. 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu S, et al. , PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. Bioessays, 2018. 40(4): p. e1700247. [DOI] [PubMed] [Google Scholar]
- 31.Sakamoto KM, et al. , Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A, 2001. 98(15): p. 8554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, et al. , Simple Structural Modifications Converting a Bona fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders. J Med Chem, 2019. 62(21): p. 9471–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.An S and Fu L, Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine, 2018. 36: p. 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farnaby W, et al. , BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol, 2019. 15(7): p. 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gadd MS, et al. , Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol, 2017. 13(5): p. 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowak RP, et al. , Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol, 2018. 14(7): p. 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riching KM, et al. , Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem Biol, 2018. 13(9): p. 2758–2770. [DOI] [PubMed] [Google Scholar]
- 38.Smith BE, et al. , Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun, 2019. 10(1): p. 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raina K, et al. , PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A, 2016. 113(26): p. 7124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salami J, et al. , Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol, 2018. 1: p. 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, et al. , PROTACs: A novel strategy for cancer therapy. Semin Cancer Biol, 2020. 67(Pt 2): p. 171–179. [DOI] [PubMed] [Google Scholar]
- 42.Qi SM, et al. , PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front Pharmacol, 2021. 12: p. 692574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kargbo RB, Treatment of Cancer and Alzheimer’s Disease by PROTAC Degradation of EGFR. ACS Med Chem Lett, 2019. 10(8): p. 1098–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ballatore C, Lee VM, and Trojanowski JQ, Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci, 2007. 8(9): p. 663–72. [DOI] [PubMed] [Google Scholar]
- 45.Hof PR and Morrison JH, The aging brain: morphomolecular senescence of cortical circuits. Trends Neurosci, 2004. 27(10): p. 607–13. [DOI] [PubMed] [Google Scholar]
- 46.Jellinger KA, Neuropathological aspects of Alzheimer disease, Parkinson disease and frontotemporal dementia. Neurodegener Dis, 2008. 5(3–4): p. 118–21. [DOI] [PubMed] [Google Scholar]
- 47.Sergeant N, et al. , Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev Proteomics, 2008. 5(2): p. 207–24. [DOI] [PubMed] [Google Scholar]
- 48.Tracy TE and Gan L, Tau-mediated synaptic and neuronal dysfunction in neurodegenerative disease. Curr Opin Neurobiol, 2018. 51: p. 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang X, et al. , Tau Pathology in Parkinson’s Disease. Front Neurol, 2018. 9: p. 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chu TT, et al. , Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem Biol, 2016. 23(4): p. 453–61. [DOI] [PubMed] [Google Scholar]
- 51.Lu M, et al. , Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem, 2018. 146: p. 251–259. [DOI] [PubMed] [Google Scholar]
- 52.Silva MC, et al. , Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife, 2019. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang W, et al. , A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models. Theranostics, 2021. 11(11): p. 5279–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ueda K, Saitoh T, and Mori H, Tissue-dependent alternative splicing of mRNA for NACP, the precursor of non-A beta component of Alzheimer’s disease amyloid. Biochem Biophys Res Commun, 1994. 205(2): p. 1366–72. [DOI] [PubMed] [Google Scholar]
- 55.Burre J, Sharma M, and Sudhof TC, Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb Perspect Med, 2018. 8(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colosimo C, et al. , Non-motor symptoms in atypical and secondary parkinsonism: the PRIAMO study. J Neurol, 2010. 257(1): p. 5–14. [DOI] [PubMed] [Google Scholar]
- 57.Filippini A, Gennarelli M, and Russo I, alpha-Synuclein and Glia in Parkinson’s Disease: A Beneficial or a Detrimental Duet for the Endo-Lysosomal System? Cell Mol Neurobiol, 2019. 39(2): p. 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yokota O, et al. , NACP/alpha-synuclein, NAC, and beta-amyloid pathology of familial Alzheimer’s disease with the E184D presenilin-1 mutation: a clinicopathological study of two autopsy cases. Acta Neuropathol, 2002. 104(6): p. 637–48. [DOI] [PubMed] [Google Scholar]
- 59.Kargbo RB, PROTAC Compounds Targeting alpha-Synuclein Protein for Treating Neurogenerative Disorders: Alzheimer’s and Parkinson’s Diseases. ACS Med Chem Lett, 2020. 11(6): p. 1086–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harris AL, Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer, 2002. 2(1): p. 38–47. [DOI] [PubMed] [Google Scholar]
- 61.Guo J, et al. , pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science, 2016. 353(6302): p. 929–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Safran M and Kaelin WG Jr., HIF hydroxylation and the mammalian oxygen-sensing pathway. J Clin Invest, 2003. 111(6): p. 779–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Epstein AC, et al. , C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell, 2001. 107(1): p. 43–54. [DOI] [PubMed] [Google Scholar]
- 64.Ivan M, et al. , HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science, 2001. 292(5516): p. 464–8. [DOI] [PubMed] [Google Scholar]
- 65.Rezaeian AH, et al. , A hypoxia-responsive TRAF6-ATM-H2AX signalling axis promotes HIF1alpha activation, tumorigenesis and metastasis. Nat Cell Biol, 2017. 19(1): p. 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rezaeian AH, Wang YH, and Lin HK, DNA damage players are linked to HIF-1alpha/hypoxia signaling. Cell Cycle, 2017. 16(8): p. 725–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peers C, et al. , Hypoxia and neurodegeneration. Ann N Y Acad Sci, 2009. 1177: p. 169–77. [DOI] [PubMed] [Google Scholar]
- 68.Zhang CE, et al. , Hypoxia-induced tau phosphorylation and memory deficit in rats. Neurodegener Dis, 2014. 14(3): p. 107–16. [DOI] [PubMed] [Google Scholar]
- 69.Guglielmotto M, Tamagno E, and Danni O, Oxidative stress and hypoxia contribute to Alzheimer’s disease pathogenesis: two sides of the same coin. ScientificWorldJournal, 2009. 9: p. 781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wouters BG and Koritzinsky M, Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer, 2008. 8(11): p. 851–64. [DOI] [PubMed] [Google Scholar]
- 71.Grammas P, et al. , Brain microvasculature and hypoxia-related proteins in Alzheimer’s disease. Int J Clin Exp Pathol, 2011. 4(6): p. 616–27. [PMC free article] [PubMed] [Google Scholar]
- 72.Masson N and Ratcliffe PJ, Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab, 2014. 2(1): p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bazan NG, Palacios-Pelaez R, and Lukiw WJ, Hypoxia signaling to genes: significance in Alzheimer’s disease. Mol Neurobiol, 2002. 26(2–3): p. 283–98. [DOI] [PubMed] [Google Scholar]
- 74.Green KN, Boyle JP, and Peers C, Hypoxia potentiates exocytosis and Ca2+ channels in PC12 cells via increased amyloid beta peptide formation and reactive oxygen species generation. J Physiol, 2002. 541(Pt 3): p. 1013–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pugh CW and Ratcliffe PJ, Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med, 2003. 9(6): p. 677–84. [DOI] [PubMed] [Google Scholar]
- 76.Sun X, et al. , Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A, 2006. 103(49): p. 18727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fang H, et al. , Acute hypoxia promote the phosphorylation of tau via ERK pathway. Neurosci Lett, 2010. 474(3): p. 173–177. [DOI] [PubMed] [Google Scholar]
- 78.Liu Y, et al. , Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett, 2008. 582(2): p. 359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mobasheri A, et al. , Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol Histopathol, 2005. 20(4): p. 1327–38. [DOI] [PubMed] [Google Scholar]
- 80.Schneekloth JS Jr., et al. , Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc, 2004. 126(12): p. 3748–54. [DOI] [PubMed] [Google Scholar]
- 81.Buckley DL, et al. , Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew Chem Int Ed Engl, 2012. 51(46): p. 11463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Buckley DL, et al. , Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc, 2012. 134(10): p. 4465–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saxton RA and Sabatini DM, mTOR Signaling in Growth, Metabolism, and Disease. Cell, 2017. 169(2): p. 361–371. [DOI] [PubMed] [Google Scholar]
- 84.Cornu M, Albert V, and Hall MN, mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev, 2013. 23(1): p. 53–62. [DOI] [PubMed] [Google Scholar]
- 85.Chong ZZ and Maiese K, Mammalian target of rapamycin signaling in diabetic cardiovascular disease. Cardiovasc Diabetol, 2012. 11: p. 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sarkar S and Rubinsztein DC, Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst, 2008. 4(9): p. 895–901. [DOI] [PubMed] [Google Scholar]
- 87.Vellai T, et al. , Genetics: influence of TOR kinase on lifespan in C. elegans. Nature, 2003. 426(6967): p. 620. [DOI] [PubMed] [Google Scholar]
- 88.Selman C, et al. , Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science, 2009. 326(5949): p. 140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anisimov VN, et al. , Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle, 2011. 10(24): p. 4230–6. [DOI] [PubMed] [Google Scholar]
- 90.Bjedov I, et al. , Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab, 2010. 11(1): p. 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harrison DE, et al. , Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature, 2009. 460(7253): p. 392–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Powers RW 3rd, et al. , Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev, 2006. 20(2): p. 174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Robida-Stubbs S, et al. , TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab, 2012. 15(5): p. 713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Caccamo A, et al. , mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell, 2013. 12(3): p. 370–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sontag JM and Sontag E, Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci, 2014. 7: p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kickstein E, et al. , Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A, 2010. 107(50): p. 21830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pei JJ, et al. , P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett, 2006. 580(1): p. 107–14. [DOI] [PubMed] [Google Scholar]
- 98.Lafay-Chebassier C, et al. , mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J Neurochem, 2005. 94(1): p. 215–25. [DOI] [PubMed] [Google Scholar]
- 99.Majumder S, et al. , Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One, 2011. 6(9): p. e25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Burke JE and Williams RL, Synergy in activating class I PI3Ks. Trends Biochem Sci, 2015. 40(2): p. 88–100. [DOI] [PubMed] [Google Scholar]
- 101.Thorpe LM, Yuzugullu H, and Zhao JJ, PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer, 2015. 15(1): p. 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li W, et al. , Phthalimide conjugations for the degradation of oncogenic PI3K. Eur J Med Chem, 2018. 151: p. 237–247. [DOI] [PubMed] [Google Scholar]
- 103.Fruman DA, et al. , The PI3K Pathway in Human Disease. Cell, 2017. 170(4): p. 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Correction for Sen et al. , Sulfhydration of AKT triggers Tau-phosphorylation by activating glycogen synthase kinase 3beta in Alzheimer’s disease. Proc Natl Acad Sci U S A, 2021. 118(42). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.You I, et al. , Discovery of an AKT Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem Biol, 2020. 27(1): p. 66–73 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Embi N, Rylatt DB, and Cohen P, Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem, 1980. 107(2): p. 519–27. [PubMed] [Google Scholar]
- 107.L’Episcopo F, et al. , GSK-3beta-induced Tau pathology drives hippocampal neuronal cell death in Huntington’s disease: involvement of astrocyte-neuron interactions. Cell Death Dis, 2016. 7: p. e2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Phiel CJ, et al. , GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature, 2003. 423(6938): p. 435–9. [DOI] [PubMed] [Google Scholar]
- 109.Sirerol-Piquer M, et al. , GSK3beta overexpression induces neuronal death and a depletion of the neurogenic niches in the dentate gyrus. Hippocampus, 2011. 21(8): p. 910–22. [DOI] [PubMed] [Google Scholar]
- 110.Maqbool M, Mobashir M, and Hoda N, Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur J Med Chem, 2016. 107: p. 63–81. [DOI] [PubMed] [Google Scholar]
- 111.Beurel E, Grieco SF, and Jope RS, Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther, 2015. 148: p. 114–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eldar-Finkelman H, Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med, 2002. 8(3): p. 126–32. [DOI] [PubMed] [Google Scholar]
- 113.Jope RS, Yuskaitis CJ, and Beurel E, Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res, 2007. 32(4–5): p. 577–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jiang X, et al. , PROTACs suppression of GSK-3beta, a crucial kinase in neurodegenerative diseases. Eur J Med Chem, 2021. 210: p. 112949. [DOI] [PubMed] [Google Scholar]
- 115.Cochran AG, Conery AR, and Sims RJ 3rd, Bromodomains: a new target class for drug development. Nat Rev Drug Discov, 2019. 18(8): p. 609–628. [DOI] [PubMed] [Google Scholar]
- 116.Hargreaves DC, Horng T, and Medzhitov R, Control of inducible gene expression by signal-dependent transcriptional elongation. Cell, 2009. 138(1): p. 129–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bao Y, et al. , Brd4 modulates the innate immune response through Mnk2-eIF4E pathway-dependent translational control of IkappaBalpha. Proc Natl Acad Sci U S A, 2017. 114(20): p. E3993–E4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Belkina AC, Nikolajczyk BS, and Denis GV, BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol, 2013. 190(7): p. 3670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nicodeme E, et al. , Suppression of inflammation by a synthetic histone mimic. Nature, 2010. 468(7327): p. 1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu Y and Vakoc CR, Brd4 is on the move during inflammation. Trends Cell Biol, 2014. 24(11): p. 615–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Magistri M, et al. , The BET-Bromodomain Inhibitor JQ1 Reduces Inflammation and Tau Phosphorylation at Ser396 in the Brain of the 3xTg Model of Alzheimer’s Disease. Curr Alzheimer Res, 2016. 13(9): p. 985–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Benito E, et al. , The BET/BRD inhibitor JQ1 improves brain plasticity in WT and APP mice. Transl Psychiatry, 2017. 7(9): p. e1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Winter GE, et al. , DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science, 2015. 348(6241): p. 1376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.DeMars KM, Yang C, and Candelario-Jalil E, Neuroprotective effects of targeting BET proteins for degradation with dBET1 in aged mice subjected to ischemic stroke. Neurochem Int, 2019. 127: p. 94–102. [DOI] [PubMed] [Google Scholar]
- 125.DeMars KM, et al. , Selective degradation of BET proteins with dBET1, a proteolysis-targeting chimera, potently reduces pro-inflammatory responses in lipopolysaccharide-activated microglia. Biochem Biophys Res Commun, 2018. 497(1): p. 410–415. [DOI] [PubMed] [Google Scholar]
- 126.Lu J, et al. , Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol, 2015. 22(6): p. 755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zengerle M, Chan KH, and Ciulli A, Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol, 2015. 10(8): p. 1770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang Y, et al. , Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B, 2020. 10(2): p. 207–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bondeson DP, et al. , Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol, 2015. 11(8): p. 611–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Churcher I, Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? J Med Chem, 2018. 61(2): p. 444–452. [DOI] [PubMed] [Google Scholar]
- 131.Crews CM, Inducing Protein Degradation as a Therapeutic Strategy. J Med Chem, 2018. 61(2): p. 403–404. [DOI] [PubMed] [Google Scholar]
- 132.Higgins JJ, et al. , Temporal and spatial mouse brain expression of cereblon, an ionic channel regulator involved in human intelligence. J Neurogenet, 2010. 24(1): p. 18–26. [DOI] [PubMed] [Google Scholar]
- 133.Franzmeier N, et al. , Functional brain architecture is associated with the rate of tau accumulation in Alzheimer’s disease. Nat Commun, 2020. 11(1): p. 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rich MC, et al. , Focused ultrasound blood brain barrier opening mediated delivery of MRI-visible albumin nanoclusters to the rat brain for localized drug delivery with temporal control. J Control Release, 2020. 324: p. 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Thibaudeau TA, Anderson RT, and Smith DM, A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun, 2018. 9(1): p. 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yao JF, et al. , Metabolism of Peptide Drugs and Strategies to Improve their Metabolic Stability. Curr Drug Metab, 2018. 19(11): p. 892–901. [DOI] [PubMed] [Google Scholar]
- 137.Di L, Strategic approaches to optimizing peptide ADME properties. AAPS J, 2015. 17(1): p. 134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Konstantinidou M, et al. , PROTACs- a game-changing technology. Expert Opin Drug Discov, 2019. 14(12): p. 1255–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guerreiro R and Bras J, The age factor in Alzheimer’s disease. Genome Med, 2015. 7: p. 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Musicco M, et al. , Inverse occurrence of cancer and Alzheimer disease: a population-based incidence study. Neurology, 2013. 81(4): p. 322–8. [DOI] [PubMed] [Google Scholar]
- 141.Romero JP, et al. , Alzheimer’s disease is associated with decreased risk of cancer-specific mortality: a prospective study (NEDICES). J Alzheimers Dis, 2014. 40(2): p. 465–73. [DOI] [PubMed] [Google Scholar]
- 142.Klein JA and Ackerman SL, Oxidative stress, cell cycle, and neurodegeneration. J Clin Invest, 2003. 111(6): p. 785–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kruman II, Why do neurons enter the cell cycle? Cell Cycle, 2004. 3(6): p. 769–73. [PubMed] [Google Scholar]
- 144.McShea A, et al. , Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim Biophys Acta, 2007. 1772(4): p. 467–72. [DOI] [PubMed] [Google Scholar]
- 145.Yang Y, Mufson EJ, and Herrup K, Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci, 2003. 23(7): p. 2557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Judge M, et al. , Mitosis-specific phosphorylation of amyloid precursor protein at threonine 668 leads to its altered processing and association with centrosomes. Mol Neurodegener, 2011. 6: p. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liu F, et al. , Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett, 2003. 547(1–3): p. 193–6. [DOI] [PubMed] [Google Scholar]
- 148.Folch J, et al. , Role of cell cycle re-entry in neurons: a common apoptotic mechanism of neuronal cell death. Neurotox Res, 2012. 22(3): p. 195–207. [DOI] [PubMed] [Google Scholar]
- 149.Norambuena A, et al. , mTOR and neuronal cell cycle reentry: How impaired brain insulin signaling promotes Alzheimer’s disease. Alzheimers Dement, 2017. 13(2): p. 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rogaeva E, Kawarai T, and George-Hyslop PS, Genetic complexity of Alzheimer’s disease: successes and challenges. J Alzheimers Dis, 2006. 9(3 Suppl): p. 381–7. [DOI] [PubMed] [Google Scholar]
- 151.Spilman P, et al. , Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One, 2010. 5(4): p. e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Greer EL, et al. , The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem, 2007. 282(41): p. 30107–19. [DOI] [PubMed] [Google Scholar]
- 153.Xu J, Ji J, and Yan XH, Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr, 2012. 52(5): p. 373–81. [DOI] [PubMed] [Google Scholar]
- 154.Gomez-Crisostomo NP, Rodriguez Martinez E, and Rivas-Arancibia S, Oxidative stress activates the transcription factors FoxO 1a and FoxO 3a in the hippocampus of rats exposed to low doses of ozone. Oxid Med Cell Longev, 2014. 2014: p. 805764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hay N, Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta, 2011. 1813(11): p. 1965–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schapira M, et al. , Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov, 2019. 18(12): p. 949–963. [DOI] [PubMed] [Google Scholar]
- 157.Devulapalli R, et al. , Males and females differ in the regulation and engagement of, but not requirement for, protein degradation in the amygdala during fear memory formation. Neurobiol Learn Mem, 2021. 180: p. 107404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lee BH, et al. , Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature, 2010. 467(7312): p. 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Min JW, et al. , USP14 inhibitor attenuates cerebral ischemia/reperfusion-induced neuronal injury in mice. J Neurochem, 2017. 140(5): p. 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jarome TJ, et al. , The ubiquitin-specific protease 14 (USP14) is a critical regulator of long-term memory formation. Learn Mem, 2013. 21(1): p. 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zerbinatti CV and Bu G, LRP and Alzheimer’s disease. Rev Neurosci, 2005. 16(2): p. 123–35. [DOI] [PubMed] [Google Scholar]
- 162.Wang JZ, et al. , Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett, 1998. 436(1): p. 28–34. [DOI] [PubMed] [Google Scholar]
- 163.Mahaman YAR, et al. , Involvement of calpain in the neuropathogenesis of Alzheimer’s disease. Med Res Rev, 2019. 39(2): p. 608–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zundorf G and Reiser G, Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid Redox Signal, 2011. 14(7): p. 1275–88. [DOI] [PMC free article] [PubMed] [Google Scholar]