

Abstract
Although metagenomic sequencing has revealed high numbers of viruses in mosquitoes sampled globally, our understanding of how their diversity and abundance varies in time and space as well as by host species and gender remains unclear. To address this, we collected 23,109 mosquitoes over the course of 12 months from a bat-dwelling cave and a nearby village in Yunnan province, China. These samples were organized by mosquito species, mosquito gender, and sampling time for meta-transcriptomic sequencing. A total of 162 eukaryotic virus species were identified, of which 101 were novel, including representatives of seventeen RNA virus multi-family supergroups and four species of DNA virus from the families Parvoviridae, Circoviridae, and Nudiviridae. In addition, two known vector-borne viruses—Japanese encephalitis virus and Banna virus—were found. Analyses of the entire virome revealed strikingly different viral compositions and abundance levels in warmer compared to colder months, a strong host structure at the level of mosquito species, and no substantial differences between those viruses harbored by male and female mosquitoes. At the scale of individual viruses, some were found to be ubiquitous throughout the year and across four mosquito species, while most of the other viruses were season and/or host specific. Collectively, this study reveals the diversity, dynamics, and evolution of the mosquito virome at a single location and sheds new lights on the ecology of these important vector animals.
Keywords: mosquito, virome, virome ecology, virus discovery, time-series, meta-transcriptomics
1. Introduction
The development of metagenomic next-generation sequencing (mNGS) has led to explosion of virus identification in arthropods, with more than 2,000 novel virus species documented over the past 5 years (Li et al. 2015a; Shi et al. 2016; Wu et al. 2020). Among these arthropods, mosquitoes (class Insecta, family Culicidae) are subject to intensive virome investigation because they act as the major vectors of human and animal pathogens such as the dengue, yellow fever, and Zika viruses (Favia 2015; Wu et al. 2020). More than fifty-six mosquito-associated viruses have been identified in China alone (Atoni et al. 2020), although those relevant for mammalian/human infection—the so-called arthropod-borne viruses (arboviruses) or vector-borne viruses—comprise only a very small proportion (Li et al. 2015a; Shi et al. 2017). Most of the expansion has occurred in those mosquito viruses that do not infect vertebrates and hence are classed as arthropod-specific (Shi et al. 2016; Xia et al. 2018a).
While the diversity of the mosquito virome continues to expand, how their genetic structure and ecological dynamics change by host species, through time and space, and by mosquito gender remains largely unclear, with detailed epidemiological investigations limited to individual vector-borne viruses (e.g. dengue (Tian et al. 2016), yellow fever (Cunha et al. 2020), and Zika viruses (Wang et al. 2021)) and some arthropod-specific viruses (e.g. Wutai mosquito phasivirus (Ribeiro et al. 2019) and Culex flavivirus (Moraes et al. 2019)). However, we lack a characterization at the scale of entire viromes. Importantly, total RNA sequencing (meta-transcriptomics) enables characterization of the full range of microbes (i.e. the total ‘infectome’) within species and also provides an estimation of their relative abundance, thereby helping to transform studies of viral ecology (Shi, Zhang, and Holmes 2018, Shi et al. 2018). Recent metagenomic studies in Australia (Shi et al. 2017), China (Xia et al. 2018a), Europe (Pettersson et al. 2019), Guadeloupe (Shi et al. 2019), and the USA (Batson et al. 2021) have demonstrated that virome composition and diversity are generally structured by mosquito taxonomy, suggesting that the host species barrier is an important factor in shaping patterns of interspecies virus transmission. To date, however, few studies have investigated how mosquito viromes change through time within a year, even though there have been suggestions that some arthropod-specific viruses may be effectively endemic (Lee et al. 2019). Revealing the dynamics of mosquito virome diversity and abundance in time requires multiple longitudinal samples taken throughout the course of a year, simultaneously controlling for factors such as geographic location, mosquito species, and gender.
Yunnan province is located in southwest China, sharing borders with Myanmar, Vietnam, and Laos (Wu et al. 2017), and is adjacent to Tibet, Sichuan, and Guangxi provinces (Sun et al. 2009). It is characterized by a tropical to subtropical climate, contains a number of biodiversity hotspots (Cao et al. 2011; Feng et al. 2012; Zuo et al. 2014), and is notorious for harboring viruses that have the potential to result in human infection (Hu et al. 2017a; Yang et al. 2019), including mosquito-borne diseases (Zhang et al. 2014; Wu et al. 2017; Mao and Zhou 2020; Zhao et al. 2020). More than ten arboviruses have been isolated in Yunnan province, including Japanese encephalitis virus (JEV) (Liu et al. 2013), dengue virus (Hu et al. 2017a; Hu et al. 2017b), chikungunya virus (Yin et al. 2021), Zika virus (Zhu et al. 2020), and Banna virus (BAV) (Wang et al. 2015), making Yunnan one of the most important places in China for mosquito control (Xia et al. 2018b). In addition, mNGS has identified vector-borne viruses such as Mammalian rubulavirus 5 and Getah virus (Liu et al. 2021). It is therefore of great importance to understand the diversity and ecological dynamics of mosquito-associated viruses in Yunnan.
To understand the factors that shape mosquito viromes, we conducted a time-series meta-transcriptomic analysis of 23,109 mosquitoes collected at a single location in Yunnan province, comparing the diversity and abundance of all the RNA and DNA viruses that could be detected through time.
2. Materials and methods
2.1. Mosquito sampling and sample processing
From January to December in 2018 we performed year-round sampling at two locations in Xiangyun county, Yunnan province, China: (i) Qinghua cave, a bat-dwelling cave, and (ii) Shuizhangdi, a nearby village located 5 km from the cave. Mosquitoes were trapped using an ultraviolet device without carbon dioxide supplementation at each location, and those with obvious signs of blood-feeding were intentionally removed. Precise sampling location and time were recorded. Mosquito gender was assessed by antennae patterns (Sallum et al. 2020). Species identifications were initially carried out based on morphological characteristics by experienced field biologists and further confirmed by analyzing the cytochrome C oxidase subunit I (COI) gene (Delgado-Serra et al. 2021) from the sequencing results. The samples were then transported by liquid nitrogen to the laboratory and transferred to −80°C storage freezer.
2.2. RNA library construction and sequencing
Samples were organized into forty-eight pools based on time, host, and gender and homogenized with a TissueLyser in 600 µl of lysis buffer. Total RNA extraction and purification were performed using the RNeasy Plus Mini Kit (QIAGEN), following the manufacturer’s instructions. An Agilent 2100 Bioanalyzer was used to evaluate the quality of the extracted RNA. Total RNA sequencing libraries were constructed after removing host rRNA using the Ribo-Zero-Gold (Human–Mouse–Rat) Kit (Illumina). Paired-end (100 bp) sequencing of the forty-eight dual-indexed libraries was then performed on an Illumina HiSeq 4000 platform. All library construction and sequencing were performed by Novogene (Beijing, China). All sequencing reads have been deposited in the Sequence Read Archive (SRA) database under the BioProject accession PRJNA778885.
2.3. Virus discovery and characterization
Adaptor sequences and low-quality reads were first removed from the raw sequencing reads. The quality-controlled reads were then assembled de novo into contigs using Megahit version 1.2.8 (Li et al. 2015a; Li et al. 2015b). The assembled contigs were subsequently compared against the nonredundant protein (nr) database downloaded from GenBank using Diamond blastx version 0.9.25 (Buchfink, Xie, and Huson 2015), with an e‐value threshold of 1E − 5 to retain high sensitivity at a low false-positive rate. Viral contigs were first identified based on the taxonomic information of each blast hit, and potential false positives were removed by blasting against the nonredundant nucleotide database (nt). The remaining viral contigs were merged to form longer genomes. To eliminate mis-assembly, reads were mapped back to the virus genome using Bowtie2 version 2.3.5.1 (Deorowicz et al. 2019) and inspected using Geneious Prime version v.9.1.5 (Kearse et al. 2012).
To identify potentially novel viral species, we used a threshold of <90 per cent amino acid identity for the RdRp (RNA-dependent RNA polymerase) protein (RNA viruses), NS1 protein (parvoviruses), replicase protein (circoviruses), and DNA polymerase B protein (nudiviruses), in comparison with the closest related viruses. All viral genome sequences have been deposited in National Center for Biotechnology Information GenBank under accession numbers OL700045–OL700212.
2.4. Virus genome annotation and phylogenetic analysis
For each virus genome, open reading frames (ORFs) were predicted using TransDecoder version 5.5.0 (Onimaru et al. 2018). For annotation, these ORFs were compared against the Conserved Domain Database (Yang et al. 2020) as well as the nr database.
To infer the evolutionary history of the newly identified viruses, we performed interspecies phylogenetic analyses based on alignments of conserved proteins. Specifically, RdRp alignments were used for RNA viruses, while nonstructural protein 1 (NS1), replicase, and DNA polymerase alignments were used for DNA viruses. Reference viral genomes representative of background phylogenetic diversity were downloaded from the GenBank and aligned with viral sequences obtained from this study using MAFFT version 7 (Katoh and Standley 2013), with ambiguously aligned regions removed using TrimAL version 1.2rev59 (Capella-Gutiérrez, Silla-Martínez, and Gabaldón 2009). Phylogenetic trees were estimated using the maximum likelihood approach implemented in PhyML version 3.0 (Guindon et al. 2010), utilizing the LG + Γ model of amino acid substitution and the Subtree Pruning and Regrafting branch-swapping algorithm. The approximate likelihood ratio test with the Shimodaira–Hasegawa-like procedure was used to assess nodal support. In the case of JEV and BAV, intraspecific phylogenies were also estimated, based on the nucleotide genome sequences identified in this study as well as those from GenBank. Similar parameters were used as the phylogenetic analysis described above, with the exception that the General Time Reversible model was employed as the DNA substitution model.
2.5. Characterization of viral abundance and diversity
To determine virus abundance, we first removed reads associated with rRNA by mapping against the total rRNA database obtained from the SILVA website (https://www.arb-silva.de/). The remaining reads were mapped against the viral genome templates using Bowtie2 version 2.3.5.1 (Langmead and Salzberg 2012). The abundance for each virus species was subsequently estimated as the number of reads per million (RPM) using the formula ‘total mapped viral reads / total non-rRNA reads * 1,000,000’. To prevent false positives due to index hopping, a threshold of 0.1 per cent was applied for each virus species present in the same sequencing lane: that is, the libraries containing < 0.1 per cent of the most abundant library were treated as ‘negative’. For each library, alpha diversity, which reflects the observed richness (number of taxa) or evenness (the relative abundance of those taxa) of an average sample, was estimated using the Shannon diversity index (Lagkouvardos et al. 2017) at the virus species level using the vegan package (Komesu et al. 2020). The overall significance levels for each factor—i.e. time, host species, and gender—were evaluated using analysis of variance (ANOVA) and t-tests. Beta diversity (Loraine et al. 2015) (i.e. virus diversity between different libraries) was calculated using the Euclidean distance matrix and virome structure and plotted as a function of multidimensional scaling ordination (Alencar et al. 2018). A generalized linear model was used to investigate the effect of time, species, and gender on richness, abundance, and alpha diversity. All these statistical analyses were performed using R v 4.0.1 implemented in RStudio version 1.3.959, and graphs were plotted using the pheatmap (Ni et al. 2019) and ggplot2 (Ito and Murphy 2013) packages implemented in R. The datasets and R code for the statistical tests and generalized linear model analysis are available at the figshare website under the link: https://doi.org/10.6084/m9.figshare.19064360.v1.
3. Results
3.1. Study design and sampling
In 2018, we performed year-round sampling of mosquitoes in Xiangyun county, Yunnan province, China (Fig. 1A). A total of 23,109 mosquitoes were sampled, which contained four dominant species from two genera (Culex and Anopheles), among which Culex pipiens was mainly identified in Qinghua cave, while Culex theileri, Culex tritaeniorhynchus, and Anopheles sinensis were mainly identified in Shuizhangdi village (Fig. 1B). A representative proportion of the mosquitoes were subsequently divided into forty-eight pools, each comprising approximately fifty individuals, for meta-transcriptomic sequencing. These pools were further organized into sixteen groups based on mosquito species (n = 4), gender (n = 2, i.e. male versus female), and sampling time (n = 7, i.e. January–April, May, June, July, August, September, and October–December) (Fig. 1C). For most groups at least three pools were assigned as replicates (Fig. 1C). These groups formed the basis for later comparisons of the diversity and abundance of mosquito viromes.
Figure 1.
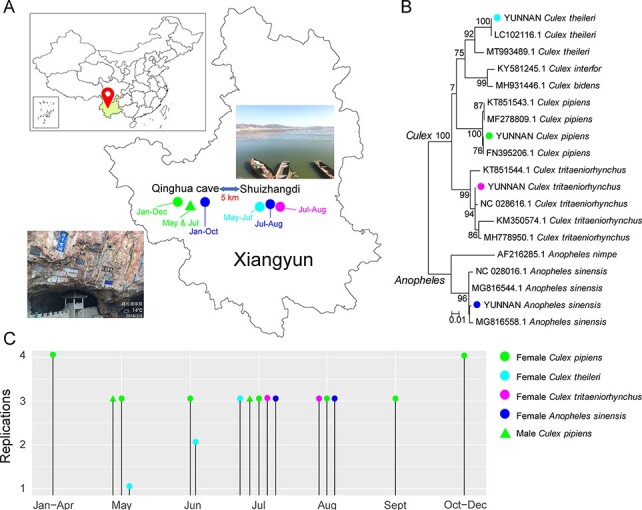
Organization of sampling and sequencing pools. (A) Locations and surrounding environments of Qinghua cave and Shuizhangdi village, Yunnan province, China, where the mosquito sampling was performed. (B) Mosquito species identification based on a maximum likelihood phylogeny of the COI gene revealed by meta-transcriptomic sequencing. (C) The experimental setting for this study: each line represents a group with a unique sampling location, time, and mosquito species, and the height of the line represents replications in that group. Throughout the figure, the color green represents C. pipiens, light blue denotes C. theileri, purple for C. tritaeniorhynchus, and deep blue for A. sinensis. Circles denote female mosquitoes while triangles denote males.
3.2. Overview of the mosquito virome
Meta-transcriptomic sequencing of forty-eight pools resulted in an average of 75,781,758 (range 59,410,848–95,817,340) reads per pool, assembled de novo into 24,164–410,121 contigs for virus discovery and characterization. These data revealed a huge diversity of eukaryotic viruses, comprising 162 species from seventeen major RNA virus supergroups (Shi et al. 2016) and three DNA virus families (Circoviridae, Nudiviridae, and Parvoviridae) (Fig. 2A, Table S1). Importantly, diversity compositions were highly variable across months, gender, and mosquito species. For example, the ‘Bunya-Arena’ clade was highly abundant in Anopheles mosquito pools but not in Culex pools. Similarly, the ‘Nido’ clade only appeared at high abundance in the summer months (Fig. 2B). Conversely, the ‘Narna-levi’ clade was consistent in all seasons and mosquito hosts and was commonplace (>10 per cent of total reads) in most pools (Fig. 2B).
Figure 2.
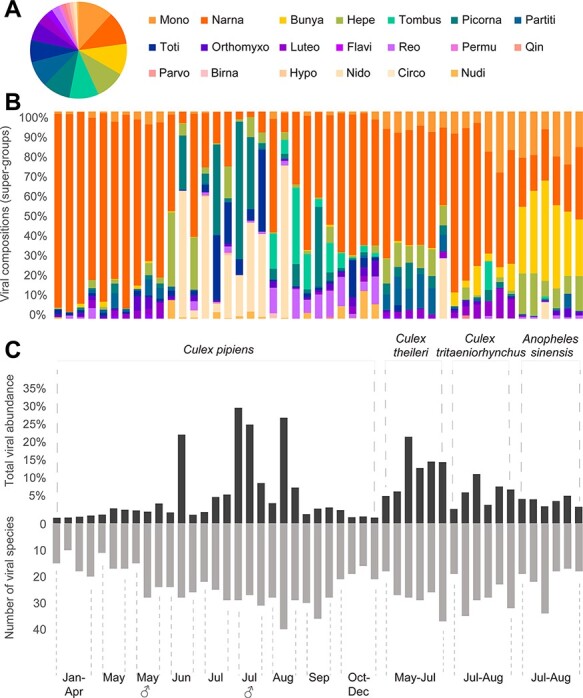
Overview of the mosquito eukaryotic virome determined in this study. (A) Proportion of eukaryotic virus species in each of the clades (RNA viruses) or families (DNA viruses). (B) Viral compositions of each pool. Time, host, and gender information for each pool follows Fig. 2C, and the viral diversity color scheme follows Fig. 2A. (C) Distribution of total viral abundance (top) and the number of viral species in the forty-eight pools. Time information is shown at the bottom of each bar, while host classifications are shown above the graph.
In addition to diversity, we examined the abundance of the eukaryotic viruses discovered, which comprised 1.55–32.01 per cent of total non-rRNA reads in each pool, with an average of 8.2 per cent (Fig. 2C). The lowest abundance (1.55–2.16 per cent) was observed in C. pipiens mosquitoes during the colder months (i.e. January–April and October–December), whereas the highest abundance (>20 per cent of total RNA) was observed in the same mosquito species during summer months (i.e. June–August). There was no apparent correlation between viral diversity (i.e. number of species) and abundance based on these data (Fig. 2C).
3.3. Diversity of mosquito viruses
A total of 162 virus species were identified here, among which 101 were assigned as potentially new species based on an amino acid identity threshold of 90 per cent in the RdRp (RNA viruses) or replication/nonstructural protein (DNA viruses) in comparison to their closest relatives. These viruses belonged to seventeen RNA virus super-clades (Shi et al. 2016) and three DNA virus families (Fig. 3). In the case of RNA viruses, forty-eight species of negative-sense, seventy-eight positive-sense, and thirty-two double-stranded viruses were identified, with the largest diversity was found in the Mono-Chu (n = 19), Narna-Levi (n = 18), and Bunya-Arena (n = 17) clades (Fig. 3, Figs S1–S7).
Figure 3.
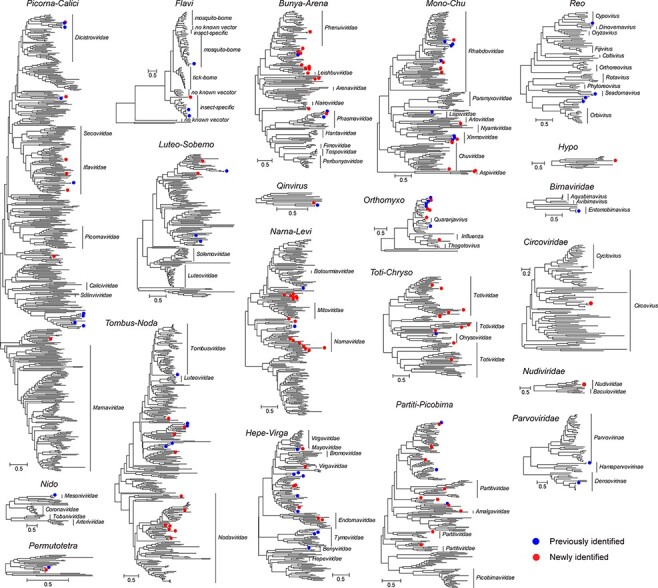
Genetic diversity of the viruses found in this study. A total of twenty phylogenetic trees are shown: seventeen based on RdRp alignments of major RNA virus clades (Shi et al. 2016) and three based on conserved replicase or nonstructural proteins of DNA viruses. Within each phylogeny, existing virus species are denoted by solid blue circles, while new viruses identified here are denoted by solid red circles. The names of the families or genera within each clade are shown to the right of each phylogeny. Each scale bar indicates 0.5 amino acid substitution per site.
Our phylogenetic analysis revealed that the majority (n = 93) of the RNA viruses clustered with virus groups associated with mosquitoes or other insects. For example, we identified four members of the genus Flavivirus, one of which (JEV) belonged to the ‘mosquito-borne’ group, while the other three clustered in the ‘insect-specific’ group. The latter included a new species (XiangYun flavivirus) that was genetically distinct (51.17 per cent amino acid identity at the NS5 region) from existing members of the ‘insect-specific’ group (Fig. S3). Conversely, the DNA viruses identified in this study belonged to the Circoviridae (n = 1), Nudiviridae (n = 1), and Parvoviridae (n = 2, one within the Hamaparvovirinae and the other from the Densovirinae) (Fig. S7). The nudivirus identified here was a new species relatively close to Oryctes rhinoceros nudivirus (39.31 per cent identity) identified from Oryctes rhinoceros. RNA sequencing recovered ∼23.3 per cent of the viral genome, covering a set of well-characterized genes, including the DNA polymerase B protein, pif-1, GrBNV gp23-like protein, GrBNV gp97-like protein, late expression factor 4, DNA helicase, 19 kDa protein, and GrBNV gp13-like protein genes.
3.4. Virome diversity and evolution by host species, time, and gender
We first compared the observed richness, total viral abundance, and alpha diversity across mosquito species (i.e. C. pipiens, C. theileri, C. tritaeniorhynchus, and A. sinensis), different sampling months (January–December), and gender (male versus female). Strikingly, observed richness (operational taxonomic units (OTU), ANOVA test, P = 1e − 04) and alpha diversity (ANOVA test, P = 3e − 04) differed significantly across different times of year based on comparisons of female C. pipiens libraries across seven time periods (i.e. January–April, May, June, July, August, September, and October–December) (Fig. 4, left column). Overall, observed richness and alpha diversity were higher in the warmer months (i.e. from June to September) than the colder months (i.e. January–May or October–December) (Fig. 4, left column). Although virus abundance did not differ across all time points (ANOVA test, P = 0.16), pairwise comparisons between individual time periods revealed significantly higher abundance level in June, July, and August compared to January–April and October–December (t-tests, P values < 0.05).
Figure 4.
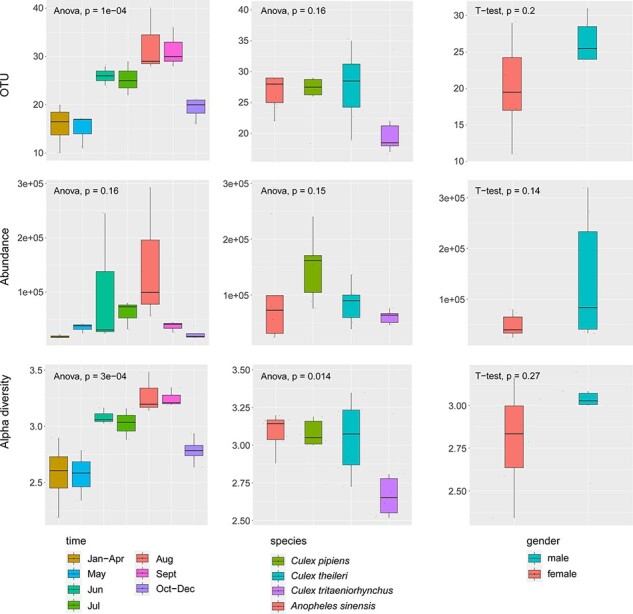
Comparisons of observed richness (OTU, upper row), abundance (RPM, middle row), and alpha diversity (lower row) between different months (left column), mosquito species (middle column), and gender (right column). An ANOVA test was performed for the time and species groups, while a t-test was used to text for associations with gender. P values are shown at the top left of each graph. In the boxplots, bold lines show the median, and upper and lower hinges show the first and third quartiles. Colors correspond to different ecological groups.
With respect to comparisons between mosquito species, which were limited to female mosquitoes collected between May and August, we revealed substantial differences in alpha diversity (ANOVA test, P = 0.014) across the four species (Fig. 4, middle column). Pairwise comparisons indicated that the difference mainly existed between the Culex and Anopheles genera (t-tests, P values < 0.005), with a generally higher alpha diversity in Culex mosquitoes. However, no significant differences were found in observed richness or abundance comparisons across the four mosquito species (OTU, ANOVA test, P = 0.16; abundance, ANOVA, P = 0.15). Similarly, no significant differences were found in observed richness, abundance, or alpha diversity in comparisons of the mosquito (C. pipiens, May–July) virome between genders (OTU, t-test, P = 0.2; abundance, t-test, P = 0.14; alpha diversity, t-test, P = 0.27). Finally, a generalized linear model that included all three factors, namely, time, species, and gender, also revealed that alpha diversity was predicted by time and mosquito species, but not sex (Table S2), which is consistent with the results from separate comparisons.
We next performed multidimensional scaling to examine the effect of time, species, and gender on the mosquito virus community composition. This revealed that viral communities were primarily structured by mosquito species and less so by month/season or gender (Fig. 5A). Nevertheless, some monthly structuring was observed (Fig. 5A, left panel). Specifically, virome compositions in June and July were distinct from the remaining months, most likely due to increasing virus diversity and abundance (Fig. 5B). However, there was also substantial overlap among viromes (Fig. 5B). Of 118 virus species identified, sixty-nine (58.5 per cent) viruses were identified in more than two mosquito species and twenty-two (18.6 per cent) viruses were found in all four species (Fig. 5C). Similarly, thirty-four out of sixty-six virus species were shared between male and female mosquitoes from the same species and regions, suggesting substantial overlap of virome between genders.
Figure 5.
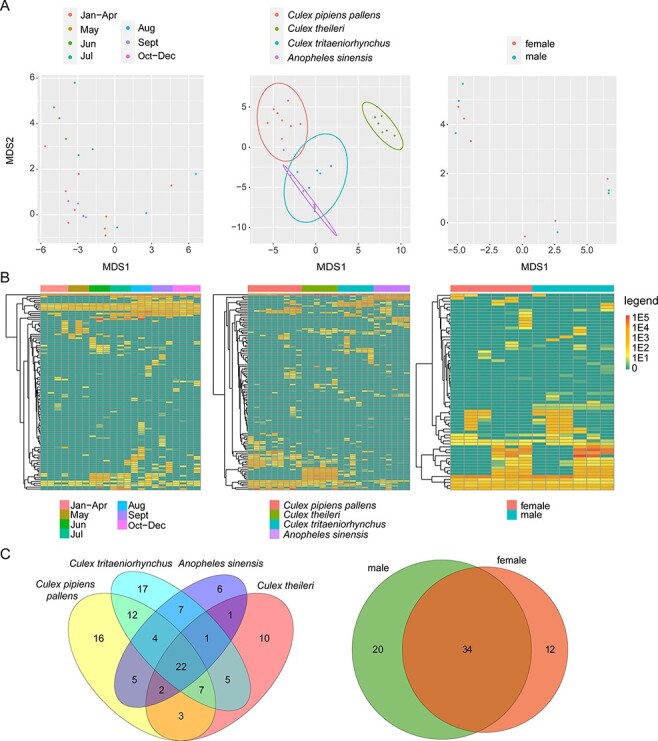
Effect of species, time, and gender on the mosquito virus community composition. (A) Multidimensional scaling plot (applying the Euclidean distance matrix) for viral composition over time, by species and by gender. The circles show the 95 per cent normal probability ellipse for each species group (middle panel). (B) Heatmap showing the normalized abundance of each viral species at different times, species, and gender groups. (C) Venn diagrams of the virus species shared between different hosts and gender. The size of the oval is not indicative of the number of viruses. Colors reflect different ecology groups.
Since these comparisons utilized total viromes, some of the viruses identified here may be associated with diet, parasites, or microbes within the mosquitoes rather than replicating in the mosquitoes themselves. To exclude the impact of these non-mosquito viruses, we identified and selected those that were most likely associated with true mosquito replication using three criteria: (i) they have been shown to infect mosquitoes or mosquito cells in previous studies; (ii) they clustered with other mosquito- or insect-related viruses on phylogenetic trees; and (iii) their abundance measured as RPM was greater than 1,000. Analyses repeated on this mosquito virus data subset generated similar results to those of the initial dataset (Fig. S8), suggesting that these observations are robust.
3.5. High-resolution characterization of individual virus species
For each individual viral species identified here we compared changes in prevalence and abundance over time. Two major temporal patterns were observed. A small number of viruses were characterized by a continuous presence and stable abundance all year round (e.g. Hubei chryso-like virus 1, Culex mononega-like virus 2, and Zhejiang mosquito virus 3). Interestingly, among those at stable abundance, Zhejiang mosquito virus 3 had much higher abundance (13,240–29,570 RPM) than Hubei chryso-like virus 1 and Culex mononega-like virus 2, which were present at 225–402 and 153–260 RPM, respectively (Fig. 6A). However, most viruses were not present all year round and some only appeared sporadically, although they may occasionally reach very high abundance (up to 107,768 RPM in the case of Nam Dinh Virus). Furthermore, seasonal variation is apparent in many virus species, with higher abundance and prevalence in months with higher temperatures (i.e. from May to September) than those with lower temperatures (i.e. January–April and October–December) (Fig. 6A).
Figure 6.
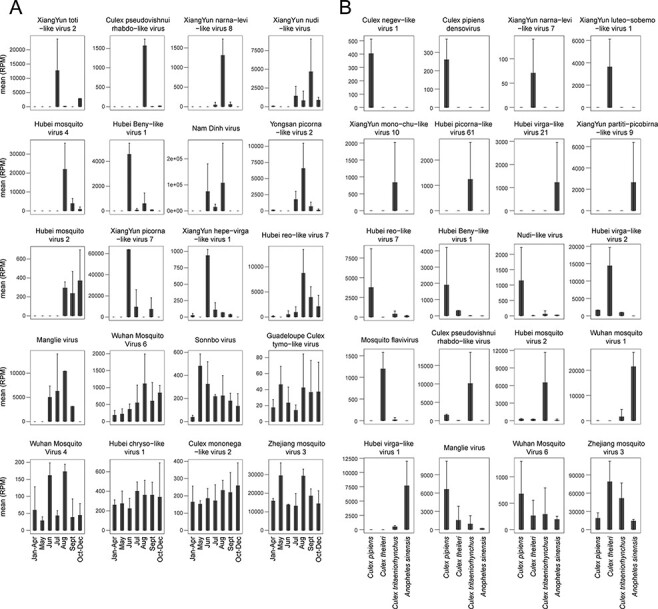
Temporal and host species distribution of selected individual viral species reflected by mean and standard deviation of abundance levels. (A) Temporal distribution of representative individual virus species. The viruses were selected and ordered based on their unique temporal distributions. Date information is provided at the bottom of each bar. The error bar represents standard deviation of mean. (B) Mosquito species distribution of representative individual virus species. The viruses were selected and ordered based on unique host distributions. Host information is shown at the bottom of each bar. The error bar represents standard deviation of mean.
We also investigated the host range for each of the virus species identified. This identified some generalist viruses that were present in all four mosquito species at high abundance (Fig. 6B), including Zhejiang mosquito virus 3 and Wuhan mosquito virus 6 which were also present throughout the year (Fig. 6A). The other virus species revealed strong host specificity or host preference. Specifically, Culex pipiens densovirus, XiangYun luteo-sobemo-like virus 1, and XiangYun partiti-picobirna-like virus 9 were only associated with C. pipiens, C. theileri, and A. sinensis, respectively. Conversely, although Hubei reo-like virus 7, Wuhan mosquito virus 1, and Hubei virga-like virus 1 were detected in multiple hosts, they were consistently at much higher prevalence, suggesting a strong host preference (Fig. 6B).
3.6. Characterization of vector-borne viruses
We also searched for viruses with a potential impact on human health, identifying those that were phylogenetically related to species or genera associated with vector-borne viruses, including flaviviruses, alphaviruses, bunyaviruses, and reoviruses. Two potential vector-borne viruses were identified: JEV and BAV, belonging to the genera Flavivirus and Seadornavirus, respectively (Fig. 7A). Among these, JEV was identified in one C. pipiens (Qinghua cave) and one C. tritaeniorhynchus pool (Shuizhangdi village), both sampled in August. In contrast, BAV was identified in March, April, and August from C. pipiens and in July from A. sinensis (Fig. 7A). Within each library, JEV had 26–155 RPM while BAV was at 1.5–1114 RPM abundance.
Figure 7.
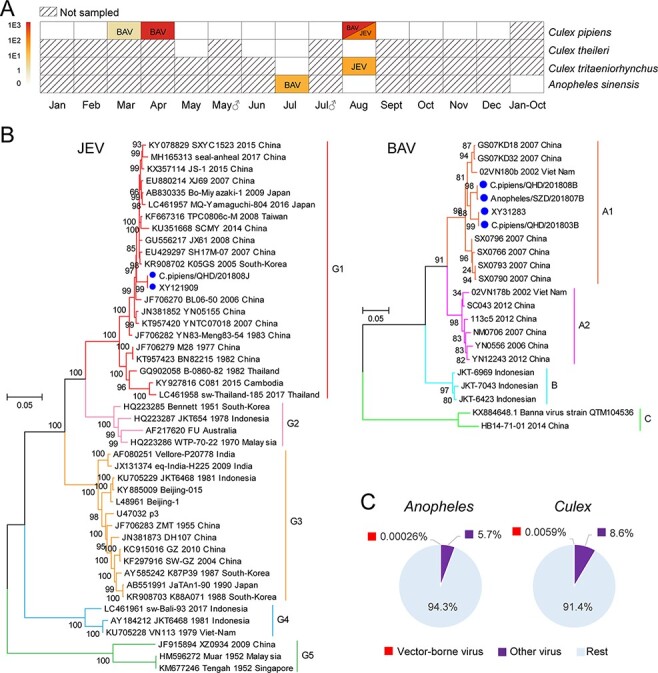
Discovery and characterization of vector-borne viruses. (A) The sampling time, mosquito host, and mosquito gender information of the pools positive for the vector-borne viruses JEV and BAV. Mosquito species information is shown on the right, while month and gender are shown at the bottom. The color of the box indicates both the positivity for vector-borne viruses and their corresponding abundance. The shaded box indicates that no sampling was performed in this category. (B) Genotyping of the JEV and BAV genomes sequenced identified in this study. The blue circle indicates vector-borne viruses identified in this study. Genotype information for JEV and BAV is shown to the right of each phylogeny. (C) Pie chart showing the proportion of total non-rRNA reads associated with vector-borne viruses and the entire virome for the Culex and Anopheles genera, respectively. Vector-borne viruses are marked in red and other viruses are marked in purple.
The JEV sequences identified in this study were named C.pipiens/QHD/201808J and XY121909 and were identical at the nucleotide level. Based on the phylogenetic analyses, they belonged to Genotype I and were closely related to variants from China, Japan, and Korea (Fig. 7B). The BAV sequences identified formed two different phylogenetic groups that shared 96.3 per cent genome sequence identity. Both groups belonged to the A1 genotype and were most closely related to sequences sampled in Vietnam (97.81–98.56 per cent, Fig. 7B).
Overall, our results indicate that vector-borne viruses represent only a tiny proportion of the total virome (Fig. 7C). Indeed, in the case of Anopheles, the proportion of total non-ribosomal RNA reads for vector-borne viruses was only 0.00026 per cent, some four orders of magnitude lower than that of the entire virome (5.7 per cent). With respect to Culex, the proportion was 0.0059 per cent for vector-borne viruses and 8.6 per cent for the entire virome.
4. Discussion
We performed a systematic time-series analysis of the mosquito virome, revealing its diversity and abundance over different mosquito species, sampling seasons, and genders. Although our analyses were limited to one location and four mosquito species, a highly diverse and abundant virome was documented, again demonstrating the capacity of arthropods to tolerate very high virus levels (Bose and Banerjee 2003; Li et al. 2015a; Shi et al. 2016; Wu et al. 2020). Indeed, this study alone identified 162 viral species from twenty families of DNA and RNA viruses. Among these, 101 viral species were novel, including the first mosquito-associated nudivirus (Nudiviridae, Fig. S7) as well as a new member of the genus Flavivirus (Fig. S3). Importantly, most of these newly identified viruses are likely to be mosquito viruses because they were related to those previously identified in mosquitoes, insects from the order Diptera, or other arthropod species and were often at high prevalence and/or abundance in mosquitoes and are hence unlikely to be associated with nonhost organisms. Hence, the presence of such high virus diversity in a small mosquito population again highlights that our understanding of mosquito virus diversity is far from complete (Atoni et al. 2018; Sadeghi et al. 2018; Shi et al. 2019; Du et al. 2020; Ramírez et al. 2020; Nebbak et al. 2021). In addition, the presence of arthropod-specific viruses in mosquitoes might impact the replication and transmission of arboviruses that also infect vertebrates, which has been demonstrated both in vitro (Kenney et al. 2014; Schultz, Frydman, and Connor 2018) and in vivo (Bolling et al. 2012). It will be of considerable interest to determine which, if any, of the arthropod-specific viruses discovered impacts arboviruses in this manner.
Meta-transcriptomics simultaneously reveals the diversity and abundance of the total virome, and it is now frequently used to explore viromes across different host species, geographical distributions, climate conditions, as well as sampling years (Wille et al. 2018, 2019, 2020; Du et al. 2020). Nevertheless, we believe this is the first characterization of a virome over the course of a single year. A key finding was the obvious seasonal pattern: the abundance and diversity of viruses were generally higher in summer than winter. Indeed, many of viruses discovered here, such as XiangYun picorna-like virus 7, Yongsan picorna-like virus 2, Hubei reo-like virus 7, as well as the vector-borne virus JEV, only appeared or had much higher abundance in warmer months. This is expected because seasonal variation in temperature, precipitation, and humidity contributes to the survival, replication, development, and distribution of mosquitoes, in turn impacting their virome structure (Bai, Morton, and Liu 2013; Li et al. 2018, 2019; Mordecai et al. 2020; Caldwell et al. 2021). For example, the reproductive activity of mosquitoes increases with temperature and rainfall, resulting in more efficient virus transmission (Tian et al. 2015). Other factors, such as mosquito population density and mammalian host activities, may also contribute to virome diversity and abundance, although this needs to be examined with more data.
While many viruses identified here experienced seasonal variation within the mosquito population, some were at stable presence throughout the year and characterized by 100 per cent pool prevalence rate and either high (i.e. Zhejiang mosquito virus 3, median 15,982 RPM) or moderate (i.e. Culex mononega-like virus 2, Hubei chryso-like virus 1, and Wuhan mosquito virus 4, median 45–342 RPM) abundance in C. pipiens (Fig. 6A). Among these, Culex mononega-like virus 2, Hubei chryso-like virus 1, and Zhejiang mosquito virus 3 viruses were also constantly detected in pooled Culex australicus and Culex globocoxitus sampled in Australia (Shi et al. 2017). Similarly, for individually sequenced mosquitoes, an almost ubiquitous presence (95.8–100 per cent prevalence) was observed for Wenzhou sobemo-like virus 4 and Wuhan mosquito virus 9 associated with Culex mosquitoes (Shi et al. 2019). The possible reasons why that these viruses are so commonplace include (i) a lack of adaptive immunity that is able to completely clear viral infection (Messaoudi et al. 2013; Gillich et al. 2015; Cheng et al. 2016, 2020; Goic et al. 2016; Filho et al. 2019; Lee et al. 2019; Reyes-Ruiz et al. 2019) and (ii) transovarial transmission such that viromes are passed from infected females to their progeny (Joshi, Mourya, and Sharma 2002; Lequime, Paul, and Lambrechts 2016; da Costa et al. 2017; Li et al. 2017; Yang 2017; Ferreira-de-lima and Lima-Camara 2018; Satoto et al. 2018; Heath et al. 2020). Unfortunately, however, due to limitations in winter sampling the hypothesis of over-wintering of these viruses cannot be addressed with the currently available data. Despite this, it is notable that most of the viruses identified do not persist over the course of the year. One possibility for this variation in prevalence is the fitness cost associated with virus infection. Indeed, it is possible that year-round presence is more often associated with viruses that have no or low fitness cost (Habayeb, Ekengren, and Hultmark 2006; Merkling and van Rij 2015; Wang et al. 2019), although this needs to be examined in the future studies.
Our study compared the viromes of four different mosquito species while controlling for location (i.e. Xiangyun county and Yunnan province) and season (i.e. summer) and revealed that overall viral compositions differ significantly across the four mosquito species—C. pipiens, C. theileri, C. tritaeniorhynchus, and A. sinensis. Previous studies have suggested that Aedes aegypti and Culex quinquefasciatus harbor very distinctive viromes (Marcelino et al. 2019), as do Aedes and Culex genera in general (Shi et al. 2017). Our study is also noteworthy because it identified barriers at the intra-genus level (i.e. between different Culex species). Indeed, the majority of the viruses identified here showed either (i) host species specificity, such that they were only present in one species, or (ii) host species preference, such that they exhibited higher abundance in one particular species, although this is indistinguishable from a generalist virus causing out-of-phase epidemics in different species. Despite this, we did observe some generalist viruses, such as Wuhan mosquito virus 6 and Zhejiang mosquito virus 3 that appeared, at comparable abundance, in all four species (Fig. 6B). These viruses were also reported in Wuhan, China (C. quinquefasciatus) (Li et al. 2015a), and Australia (C. australicus and C. globocoxitus) (Shi et al. 2017), suggesting a much wider geographic distribution and host range for these viruses.
Although most of the viruses identified here belonged to the arthropod-specific category that can be distinguished from the vertebrate-infecting groups based on phylogenetic analyses, we identified two mosquito-borne viruses—JEV and BAV—that have been isolated in diseased humans as well as domestic animals in China (Gao, Nasci, and Liang 2010; Zheng et al. 2012). However, these arboviruses only appeared sporadically, with only a low pool prevalence and low to moderate abundance (Fig. 7). This accords with previous studies in which arboviruses only represent a small proportion of the total mosquito viral population (Li et al. 2015a; Shi et al. 2017; Du et al. 2020). Nevertheless, the presence of JEV in both locations investigated in this study, consistent with a previous report that isolated 17 JEV strains from Shuizhangdi village (Pan et al. 2020), suggests a potential risk of human exposure. Conversely, previous viral surveillance of bat serum collected from Qinghua cave did not find JEV but did identify Yokose virus (YOKV), a flavivirus isolated from a serum sample of Myotis daubentonii (Daubenton’s bat; Feng et al. 2019). However, YOKV was not identified in the mosquito samples here, compatible with its status a member of the ‘no known vector’ (i.e. NKV flavivirus) group (Blitvich and Firth 2017) such that mosquitoes may not be involved in its transmission. Alternatively, the virus may be transmitted by a day-time feeder from the genus Aedes which were not sampled in this study.
Despite its size, this study has several limitations. First, meta-transcriptomics does not provide data on the functional viability of newly discovered viruses such that future isolation of these viruses is needed to confirm their existence as transmissible viral particles. Second, only female C. pipiens were sampled across different months, whereas sampling of other mosquito species and genders were limited to the summer months, limiting our capability to study the interactions among different factors. Third, some potentially important data, such as mosquito population size, temperature, humidity, and anthropogenic activities, were not collected, limiting our capability to reveal all the factors that influence virome structure. Finally, although Qinghua cave and Shuizhang village were only 5 km apart, they have distinct ecological features. While it is difficult to distinguish their effect with current data, future studies are needed to understand the potential impact of geographical/ecological factors on the virome structure.
Collectively, the data generated here revealed strong seasonal variation and strong host structure among the mosquitoes sampled but no substantial differences by gender. Notably, some viruses displayed a ubiquitous presence throughout the year and across four different mosquito species, although the majority was season and host specific. More broadly, our study further highlights how meta-transcriptomics can be used to reveal the diversity of the mosquito virome at both great depth and precision.
Supplementary Material
Contributor Information
Yun Feng, Department of Viral and Rickettsial Disease Control, Yunnan Provincial Key Laboratory for Zoonosis Control and Prevention, Yunnan Institute of Endemic Disease Control and Prevention, No. 5 Wenhua Road, Xiaguan, Dali, Yunnan 671000, China.
Qin-yu Gou, Shenzhen Campus of Sun-Yat Sen University, Sun-Yat Sen University Shenzhen Campus, Guangming New District, Shenzhen, Guangdong 518107, China.
Wei-hong Yang, Department of Viral and Rickettsial Disease Control, Yunnan Provincial Key Laboratory for Zoonosis Control and Prevention, Yunnan Institute of Endemic Disease Control and Prevention, No. 5 Wenhua Road, Xiaguan, Dali, Yunnan 671000, China.
Wei-chen Wu, Shenzhen Campus of Sun-Yat Sen University, Sun-Yat Sen University Shenzhen Campus, Guangming New District, Shenzhen, Guangdong 518107, China.
Juan Wang, Department of Viral and Rickettsial Disease Control, Yunnan Provincial Key Laboratory for Zoonosis Control and Prevention, Yunnan Institute of Endemic Disease Control and Prevention, No. 5 Wenhua Road, Xiaguan, Dali, Yunnan 671000, China.
Edward C Holmes, Sydney Institute for Infectious Diseases, School of Life and Environmental Sciences and School of Medical Sciences, The University of Sydney, Sydney, NSW 2006, Australia.
Guodong Liang, State Key Laboratory of Infectious Disease Prevention and Control, National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, 155 Changbai Road, Changping District, Beijing 102206, China.
Mang Shi, Shenzhen Campus of Sun-Yat Sen University, Sun-Yat Sen University Shenzhen Campus, Guangming New District, Shenzhen, Guangdong 518107, China.
Data availability
All sequencing reads have been deposited in the SRA databases under the project accession PRJNA778885. Relevant virus genome/gene sequences have been deposited in GenBank under the accessions OL700045–OL700212.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
‘Medicine Discipline Leader of Yunnan’ (No. D-2017055) and ‘Middle-aged Academy and Technology Backups of Leaders’ (No. 2019HB052) Fellowships; Shenzhen Science and Technology Program (KQTD20200820145822023) and Guangdong Province ‘Pearl River Talent Plan’ Innovation and Entrepreneurship Team Project (2019ZT08Y464) to M.S.; Australian Research Council Australia Laureate Fellowship (FL170100022) to E.H.
Conflict of interest:
None declared.
References
- Alencar J. et al. (2018) ‘Distribution of Haemagogus and Sabethes Species in Relation to Forest Cover and Climatic Factors in the Chapada Dos Guimarães National Park, State of Mato Grosso, Brazil’, Journal of the American Mosquito Control Association, 34: 85–92. [DOI] [PubMed] [Google Scholar]
- Atoni E. et al. (2018) ‘Metagenomic Virome Analysis of Culex Mosquitoes from Kenya and China’, Viruses, 10: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. (2020) ‘A Dataset of Distribution and Diversity of Mosquito-associated Viruses and Their Mosquito Vectors in China’, Scientific Data, 7: 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L., Morton L. C., and Liu Q. (2013) ‘Climate Change and Mosquito-borne Diseases in China: A Review’, Globalization and Health, 9: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batson J. et al. (2021) ‘Single Mosquito Metatranscriptomics Identifies Vectors, Emerging Pathogens and Reservoirs in One Assay’, Elife, 10: e68353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitvich B. J., and Firth A. E. (2017) ‘A Review of Flaviviruses that Have No Known Arthropod Vector’, Viruses, 9: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolling B. G. et al. (2012) ‘Transmission Dynamics of an Insect-specific Flavivirus in a Naturally Infected Culex Pipiens Laboratory Colony and Effects of Co-infection on Vector Competence for West Nile Virus’, Virology, 427: 90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose S., and Banerjee A. K. (2003) ‘Innate Immune Response against Nonsegmented Negative Strand RNA Viruses’, Journal of Interferon & Cytokine Research, 23: 401–12. [DOI] [PubMed] [Google Scholar]
- Buchfink B., Xie C., and Huson D. H. (2015) ‘Fast and Sensitive Protein Alignment Using DIAMOND’, Nature Methods, 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Caldwell J. M. et al. (2021) ‘Climate Predicts Geographic and Temporal Variation in Mosquito-borne Disease Dynamics on Two Continents’, Nature Communications, 12: 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y. et al. (2011) ‘Distribution of Mosquitoes and Mosquito-Borne Arboviruses in Inner Mongolia, China’, Vector-Borne and Zoonotic Diseases, 11: 1577–81. [DOI] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J. M., and Gabaldón T. (2009) ‘TrimAL: A Tool for Automated Alignment Trimming in Large-scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C. C. et al. (2020) ‘Cell-to-Cell Spread of Dengue Viral RNA in Mosquito Cells’, BioMed Research International, 2020: 2452409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G. et al. (2016) ‘Mosquito Defense Strategies against Viral Infection’, Trends in Parasitology, 32: 177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha M. S. et al. (2020) ‘Genomic Evidence of Yellow Fever Virus in Aedes Scapularis, Southeastern Brazil, 2016’, Acta Tropica, 205: 105390. [DOI] [PubMed] [Google Scholar]
- da Costa C. F. et al. (2017) ‘Transovarial Transmission of DENV in Aedes Aegypti in the Amazon Basin: A Local Model of Xenomonitoring’, Parasites & Vectors, 10: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Serra S. et al. (2021) ‘Molecular Characterization of Mosquito Diversity in the Balearic Islands’, Journal of Medical Entomology, 58: 608–15. [DOI] [PubMed] [Google Scholar]
- Deorowicz S. et al. (2019) ‘Whisper: Read Sorting Allows Robust Mapping of DNA Sequencing Data’, Bioinformatics, 35: 2043–50. [DOI] [PubMed] [Google Scholar]
- Du J. et al. (2020) ‘Characterization of Viromes within Mosquito Species in China’, Science China Life Sciences, 63: 1089–92. [DOI] [PubMed] [Google Scholar]
- Favia G. (2015) ‘Engineered Mosquitoes to Fight Mosquito Borne Diseases: Not a Merely Technical Issue’, Bioengineered, 6: 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y. et al. (2012) ‘Distribution of Mosquitoes and Mosquito-Borne Viruses along the China-Myanmar Border in Yunnan Province’, Japanese Journal of Infectious Diseases, 65: 215–21. [DOI] [PubMed] [Google Scholar]
- —— et al. (2019) ‘Genetic Diversity of the Yokose Virus, XYBX1332, Isolated from Bats (Myotis Daubentonii) in China’, Virology Journal, 16: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira-de-lima V. H., and Lima-Camara T. N. (2018) ‘Natural Vertical Transmission of Dengue Virus in Aedes Aegypti and Aedes Albopictus: A Systematic Review’, Parasites & Vectors, 11: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filho W. L. et al. (2019) ‘Climate Change, Health and Mosquito-Borne Diseases: Trends and Implications to the Pacific Region’, International Journal of Environmental Research and Public Health, 16: 5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Nasci R., and Liang G. (2010) ‘The Neglected Arboviral Infections in Mainland China’, PLoS Neglected Tropical Diseases, 4: e624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillich N. et al. (2015) ‘Persistent Natural Infection of a Culex Tritaeniorhynchus Cell Line with a Novel Culex Tritaeniorhynchus Rhabdovirus Strain’, Microbiology and Immunology, 59: 562–6. [DOI] [PubMed] [Google Scholar]
- Goic B. et al. (2016) ‘Virus-derived DNA Drives Mosquito Vector Tolerance to Arboviral Infection’, Nature Communications, 7: 12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21. [DOI] [PubMed] [Google Scholar]
- Habayeb M. S., Ekengren S. K., and Hultmark D. (2006) ‘Nora Virus, a Persistent Virus in Drosophila, Defines a New Picorna-like Virus Family’, Journal of General Virology, 87: 3045–51. [DOI] [PubMed] [Google Scholar]
- Heath C. J. et al. (2020) ‘Evidence of Transovarial Transmission of Chikungunya and Dengue Viruses in Field-caught Mosquitoes in Kenya’, PLoS Neglected Tropical Diseases, 14: e0008362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B. et al. (2017a) ‘Discovery of a Rich Gene Pool of Bat SARS-related Coronaviruses Provides New Insights into the Origin of SARS Coronavirus’, PLoS Pathogens, 13: e1006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T. S. et al. (2017b) ‘Epidemiological and Molecular Characteristics of Emergent Dengue Virus in Yunnan Province near the China-Myanmar-Laos Border, 2013–2015’, BMC Infectious Diseases, 17: 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., and Murphy D. (2013) ‘Application of Ggplot2 to Pharmacometric Graphics’, CPT: Pharmacometrics and Systems Pharmacology, 2: e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi V., Mourya D. T., and Sharma R. C. (2002) ‘Persistence of Dengue-3 Virus through Transovarial Transmission Passage in Successive Generations of Aedes Aegypti Mosquitoes’, The American Journal of Tropical Medicine and Hygiene, 67: 158–61. [DOI] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M. et al. (2012) ‘Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data’, Bioinformatics, 28: 1647–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney J. L. et al. (2014) ‘Characterization of a Novel Insect-specific Flavivirus from Brazil: Potential for Inhibition of Infection of Arthropod Cells with Medically Important Flaviviruses’, Journal of General Virology, 95: 2796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komesu Y. M. et al. (2020) ‘Defining the Relationship between Vaginal and Urinary Microbiomes’, American Journal of Obstetrics and Gynecology, 222: 154.e1–154.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagkouvardos I. et al. (2017) ‘Rhea: A Transparent and Modular R Pipeline for Microbial Profiling Based on 16S rRNA Gene Amplicons’, PeerJ, 5: e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., and Salzberg S. L. (2012) ‘Fast Gapped-read Alignment with Bowtie 2’, Nature Methods, 9: 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. S. et al. (2019) ‘Mosquito Antiviral Defense Mechanisms: A Delicate Balance between Innate Immunity and Persistent Viral Infection’, Parasites & Vectors, 12: 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lequime S., Paul R. E., and Lambrechts L. (2016) ‘Determinants of Arbovirus Vertical Transmission in Mosquitoes’, PLoS Pathogens, 12: e1005548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. et al. (2018) ‘Climate Change and Dengue Fever Transmission in China: Evidences and Challenges’, Science of the Total Environment, 622–623: 493–501. [DOI] [PubMed] [Google Scholar]
- Li C. X. et al. (2015a) ‘Unprecedented Genomic Diversity of RNA Viruses in Arthropods Reveals the Ancestry of Negative-sense RNA Viruses’, Elife, 4: e05378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. (2017) ‘Vector Competence and Transovarial Transmission of Two Aedes Aegypti Strains to Zika Virus’, Emerging Microbes & Infections, 6: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. et al. (2015b) ‘MEGAHIT: An Ultra-fast Single-node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph’, Bioinformatics, 31: 1674–6. [DOI] [PubMed] [Google Scholar]
- Li R. et al. (2019) ‘Climate-driven Variation in Mosquito Density Predicts the Spatiotemporal Dynamics of Dengue’, Proceedings of the National Academy of Sciences of the United States of America, 116: 3624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. et al. (2013) ‘Japanese Encephalitis Virus in Mosquitoes and Swine in Yunnan Province, China 2009–2010’, Vector-Borne and Zoonotic Diseases, 13: 41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. et al. (2021) ‘Comparative Viromes of Culicoides and Mosquitoes Reveal Their Consistency and Diversity in Viral Profiles’, Briefings in Bioinformatics, 22: bbaa323. [DOI] [PubMed] [Google Scholar]
- Loraine A. E. et al. (2015) ‘Analysis and Visualization of RNA-Seq Expression Data Using RStudio, Bioconductor, and Integrated Genome Browser’, Methods in Molecular Biology, 1284: 481–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X., and Zhou H. (2020) ‘The Spatiotemporal Distribution of Japanese Encephalitis Cases in Yunnan Province, China, from 2007 to 2017’, PLoS One, 15: e0231661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelino V. R. et al. (2019) ‘Meta-transcriptomics Reveals a Diverse Antibiotic Resistance Gene Pool in Avian Microbiomes’, BMC Biology, 17: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkling S. H., and van Rij R. P. (2015) ‘Analysis of Resistance and Tolerance to Virus Infection in Drosophila’, Nature Protocols, 10: 1084–97. [DOI] [PubMed] [Google Scholar]
- Messaoudi I. et al. (2013) ‘Chikungunya Virus Infection Results in Higher and Persistent Viral Replication in Aged Rhesus Macaques Due to Defects in Anti-viral Immunity’, PLoS Neglected Tropical Diseases, 7: e2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes O. S. et al. (2019) ‘Natural Infection by Culex Flavivirus in Culex Quinquefasciatus Mosquitoes Captured in Cuiabá, Mato Grosso Mid‐Western Brazil’, Medical and Veterinary Entomology, 33: 397–406. [DOI] [PubMed] [Google Scholar]
- Mordecai E. A. et al. (2020) ‘Climate Change Could Shift Disease Burden from Malaria to Arboviruses in Africa’, The Lancet Planetary Health, 4: e416–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebbak A. et al. (2021) ‘Virome Diversity among Mosquito Populations in a Sub-Urban Region of Marseille, France’, Viruses, 13: 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni W. et al. (2019) ‘Identification of Cancer-related Gene Network in Hepatocellular Carcinoma by Combined Bioinformatic Approach and Experimental Validation’, Pathology, Research and Practice, 215: 152428. [DOI] [PubMed] [Google Scholar]
- Onimaru K. et al. (2018) ‘A de Novo Transcriptome Assembly of the Zebra Bullhead Shark, Heterodontus Zebra’, Scientific Data, 5: 180197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H. et al. (2020) ‘Investigation of Mosquito-borne Viruses in Xiangyun County, Yunnan Province’, International Journal of Virology, 27: 484–7. [Google Scholar]
- Pettersson J. H. et al. (2019) ‘Meta-Transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex Pipiens and Culex Torrentium in Northern Europe’, Viruses, 11: 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez A. L. et al. (2020) ‘Metagenomic Analysis of the Virome of Mosquito Excreta’, mSphere, 5: e00587–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Ruiz J. M. et al. (2019) ‘Mosquito Cells Persistently Infected with Dengue Virus Produce Viral Particles with Host-dependent Replication’, Virology, 531: 1–18. [DOI] [PubMed] [Google Scholar]
- Ribeiro M. S. et al. (2019) ‘High Prevalence of a Newly Discovered Wutai Mosquito Phasivirus in Mosquitoes from Rio de Janeiro, Brazil’, Insects, 10: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghi M. et al. (2018) ‘Virome of > 12 Thousand Culex Mosquitoes from Throughout California’, Virology, 523: 74–88. [DOI] [PubMed] [Google Scholar]
- Sallum M. A. M. et al. (2020) ‘Identification Keys to the Anopheles Mosquitoes of South America (Diptera: Culicidae). I. Introduction’, Parasites & Vectors, 13: 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoto T. B. T. et al. (2018) ‘Vertical Transmission of Dengue Virus in the Yogyakarta Airport Area’, Environmental Health and Preventive Medicine, 23: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz M. J., Frydman H. M., and Connor J. H. (2018) ‘Dual Insect Specific Virus Infection Limits Arbovirus Replication in Aedes Mosquito Cells’, Virology, 518: 406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C. et al. (2019) ‘Stable Distinct Core Eukaryotic Viromes in Different Mosquito Species from Guadeloupe, Using Single Mosquito Viral Metagenomics’, Microbiome, 7: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M. et al. (2016) ‘Redefining the Invertebrate RNA Virosphere’, Nature, 540: 539–43. [DOI] [PubMed] [Google Scholar]
- —— et al. (2017) ‘High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia’, Journal of Virology, 91: e00680–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. (2018) ‘No Detectable Effect of Wolbachia wMel on the Prevalence and Abundance of the RNA Virome of Drosophila Melanogaster’, Proceedings of the Royal Society B: Biological Sciences, 285: 20181165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M., Zhang Y. Z., and Holmes E. C. (2018) ‘Meta-transcriptomics and the Evolutionary Biology of RNA Viruses’, Virus Research, 243: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X. et al. (2009) ‘Distribution of Arboviruses and Mosquitoes in Northwestern Yunnan Province, China’, Vector-Borne and Zoonotic Diseases, 9: 623–30. [DOI] [PubMed] [Google Scholar]
- Tian H. et al. (2016) ‘Surface Water Areas Significantly Impacted 2014 Dengue Outbreaks in Guangzhou, China’, Environmental Research, 150: 299–305. [DOI] [PubMed] [Google Scholar]
- Tian H. Y. et al. (2015) ‘How Environmental Conditions Impact Mosquito Ecology and Japanese Encephalitis: An Eco-epidemiological Approach’, Environment International, 79: 17–24. [DOI] [PubMed] [Google Scholar]
- Wang J. et al. (2015) ‘Isolation and Genetic Characterization of Mangshi Virus: A Newly Discovered Seadornavirus of the Reoviridae Family Found in Yunnan Province, China’, PLoS One, 10: e0143601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. (2021) ‘Emergence of Zika Virus in Culex Tritaeniorhynchus and Anopheles Sinensis Mosquitoes in China’, Virologica Sinica, 36: 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. et al. (2019) ‘Short-term Persistence Precedes Pathogenic Infection: Infection Kinetics of Cricket Paralysis Virus in Silkworm-derived Bm5 Cells’, Journal of Insect Physiology, 115: 1–11. [DOI] [PubMed] [Google Scholar]
- Wille M. et al. (2018) ‘Virus-virus Interactions and Host Ecology are Associated with RNA Virome Structure in Wild Birds’, Molecular Ecology, 27: 5263–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. (2019) ‘Virome Heterogeneity and Connectivity in Waterfowl and Shorebird Communities’, The ISME Journal, 13: 2603–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. (2020) ‘Sustained RNA Virome Diversity in Antarctic Penguins and Their Ticks’, The ISME Journal, 14: 1768–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. et al. (2017) ‘Behaviors Related to Mosquito-Borne Diseases among Different Ethnic Minority Groups along the China-Laos Border Areas’, International Journal of Environmental Research and Public Health, 14: 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H. et al. (2020) ‘Abundant and Diverse RNA Viruses in Insects Revealed by RNA-Seq Analysis: Ecological and Evolutionary Implications’, mSystems, 5: e00039–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H. et al. (2018a) ‘Comparative Metagenomic Profiling of Viromes Associated with Four Common Mosquito Species in China’, Virologica Sinica, 33: 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. (2018b) ‘Mosquito-Associated Viruses in China’, Virologica Sinica, 33: 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. M. (2017) ‘The Transovarial Transmission in the Dynamics of Dengue Infection: Epidemiological Implications and Thresholds’, Mathematical Biosciences, 286: 1–15. [DOI] [PubMed] [Google Scholar]
- Yang M. et al. (2020) ‘NCBI’s Conserved Domain Database and Tools for Protein Domain Analysis’’, Current Protocols in Bioinformatics, 69: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. L. et al. (2019) ‘Characterization of a Filovirus (M ěnglà Virus) from Rousettus Bats in China’, Nature Microbiology, 4: 390–5. [DOI] [PubMed] [Google Scholar]
- Yin X. et al. (2021) ‘Emergent Chikungunya Fever and Vertical Transmission in Yunnan Province, China, 2019’, Archives of Virology, 166: 1455–62. [DOI] [PubMed] [Google Scholar]
- Zhang F. C. et al. (2014) ‘Severe Dengue Outbreak in Yunnan, China, 2013’, International Journal of Infectious Diseases, 27: 4–6. [DOI] [PubMed] [Google Scholar]
- Zhao X. et al. (2020) ‘Spatiotemporal Trends of Malaria in Relation to Economic Development and Cross-Border Movement along the China–Myanmar Border in Yunnan Province’, The Korean Journal of Parasitology, 58: 267–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. et al. (2012) ‘Japanese Encephalitis and Japanese Encephalitis Virus in Mainland China’, Reviews in Medical Virology, 22: 301–22. [DOI] [PubMed] [Google Scholar]
- Zhu C. et al. (2020) ‘Vertical Transmission of Zika Virus by Jiegao and Mengding Aedes Aegypti (Diptera: Culicidae) Strains in Yunnan Province in China’, Vector-Borne and Zoonotic Diseases, 20: 664–9. [DOI] [PubMed] [Google Scholar]
- Zuo S. et al. (2014) ‘Detection of Quang Binh Virus from Mosquitoes in China’, Virus Research, 180: 31–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing reads have been deposited in the SRA databases under the project accession PRJNA778885. Relevant virus genome/gene sequences have been deposited in GenBank under the accessions OL700045–OL700212.