

Abstract
The development of long-lived immune memory cells against pathogens is critical for the success of vaccines to establish protection against future infections. However, the mechanisms governing the long-term survival of immune memory cells remain to be elucidated. Here we show that the maintenance mitochondrial homeostasis by autophagy is critical for restricting metabolic functions to protect IgG memory B cell survival. Knockout of mitochondrial autophagy genes, Nix and Bnip3, leads to mitochondrial accumulation, and increases in oxidative phosphorylation and fatty acid synthesis, resulting in the loss of IgG+ memory B cells in mice. Inhibiting fatty acid synthesis or silencing necroptosis gene Ripk3 rescued Nix–/–Bnip3–/– IgG memory B cells, indicating that mitochondrial autophagy is important for limiting metabolic functions to prevent cell death. Our results suggest a critical role for mitochondrial autophagy in the maintenance of immunological memory by protecting the metabolic quiescence and longevity of memory B cells.
Introduction
The immune system can develop long-term memory after encountering pathogens from infections or vaccination (1). Stimulation by antigens from pathogens induces the activation and significant expansion of lymphocytes. Most of these activated lymphocytes are cleared by programmed cell death during and after the clearance of pathogens (2). However, a small number of these cells can develop into long-lived memory cells, including memory B cells (3–5) and memory T cells (6, 7). Memory B cells can arise from both germinal center (GCs) dependent and independent pathways, and comprise of a heterogeneous population of metabolically quiescent, antigen-experienced, long-lived B cells that mediate humoral immunological memory (4, 8, 9). The T cell-dependent memory B cells arise from the GCs through interactions between antigen-activated B cells and T cells. In response to previously encountered pathogens, memory B cells rapidly proliferate and either re-seed GCs or directly differentiate into antibody-producing cells.
The long-term survival of immune memory cells is critical for the maintenance of immunological memory against infections. In comparison with activated GC B cells that are short-lived (10, 11), memory B cells are quiescent and can survive for prolonged durations in the absence of their cognate antigens (12). Memory B cells express high levels of anti-apoptotic Bcl-2, which protects them from caspase-dependent apoptosis (11, 13, 14). Autophagy, a cellular process for the sequestration of cytoplasmic components into double-membraned autophagosomes for degradation (15), plays an important role in the protection of lymphocyte survival (16–19). It has been shown that autophagy is essential for the maintenance of memory B cells, memory T cells, and long-lived plasma cells (11, 20–24). Autophagy also protects the survival of autoantigen-specific memory B cells for persistent autoantibody production (25). Blocking Atg7-dependent autophagy in memory B cells results in mitochondrial dysfunction, oxidative stress and lipid peroxidation-induced cell death (11). This suggests that autophagy protects memory B cells in part by clearing damaged mitochondria to prevent oxidative stress.
It has been shown that memory T cells undergo a metabolic switch to support their development that involves the regulation of mitochondrial homeostasis (26, 27). Memory T cells can oxidize fatty acids in the mitochondria to fulfill their energy need, while promoting fatty acid oxidation further enhances T cell memory (28). Whether metabolic programming is involved in the development and maintenance of memory B cells remains to be elucidated. Interestingly, it has been shown that AMPK, an important regulator for cellular metabolic functions and autophagy, is required for the development and maintenance of memory B cells (29). Whether the homeostasis and functions of mitochondria are critical for the regulation of metabolic function in memory B cells remains to be investigated.
Two BH3-only proteins of the Bcl-2 family located on the outer mitochondrial membrane, Nix and Bnip3, are functionally overlapping in promoting mitochondrial autophagy (30–32). Nix- and Bnip3-mediated mitochondrial autophagy are required for the protection of precursors of memory NK cells (30). Nix is also required for the protection of precursors of CD8+ effector memory T cells (33, 34). The mitochondrial kinase Pink1 and the ubiquitin ligase Parkin regulate the targeting of damaged mitochondria to autophagosomes for degradation (35). Nix and Bnip3 can mediate mitochondrial autophagy independently of Pink1/Parkin (36–39). In this study, we show that Nix- and Bnip3-mediated mitochondrial autophagy is important for the maintenance of mitochondrial homeostasis and restriction of metabolic functions, thereby protecting the quiescence and longevity of memory B cells.
Materials and Methods
Mice
Aicda-Cre (AID-Cre) mice (The Jackson Laboratory) were crossed with Nixflox mice and Bnip3−/− mice (from Gerald Dorn, Washington University at St. Louis, crossed to C57BL/6 background for more than 10 generations) to generate AID-Cre+Nixf/fBnip3−/− mice with double knockouts of Nix and Bnip3 in B cells (DKO). For experiments involving Nix−∕− or DKO mice, AID-Cre+/− age-matched mice as appropriate were used as wild-type controls. CD45.1 mice (The Jackson Laboratory) were used as recipients for adoptive transfer experiments. All mice were maintained on a C57BL/6J background. Mice of both genders, ranging from 6 weeks to 3 months old, were used in this study, but were sex and age-matched for all experiments. Mice were immunized with 100 μg NP-KLH (Biosearch Technologies) precipitated in an equal volume of Imject alum adjuvant (Thermo Scientific), or 100 μl of 10% sheep red blood cell suspension (MP Biomedicals) intraperitoneally (i.p.). Experiments were performed according to federal and institutional guidelines, and with the approval of the Institutional Animal Care and Use Committee of the Houston Methodist Research Institute and Baylor College of Medicine.
Flow cytometry and cell sorting
For flow cytometry, single cell suspensions were prepared from spleens of mice and subjected to ACK lysis to remove red blood cells. Cells were then incubated with Fc blocker (anti-CD16/CD32) for 10 min on ice to reduce background staining. Subsequently, cells were stained with appropriate fluorophore-conjugated antibodies on ice, protected from light, for 15 min. After staining, DAPI was added where appropriate to mark dead cells. Splenocytes were stained with antibodies against IgM, IgD, CD138, CD11b, CD11c, Gr-1, CD4, and CD8a (DUMP markers), B220, IgG, GL-7, and CD38. DUMP−B220+IgG+GL-7+CD38– GC B cells or DUMP−B220+IgG+GL-7−CD38+ memory B cells were analyzed using an LSR-II or sorted using a FACSAria (BD Biosciences). Data were analyzed on FlowJo (TreeStar).
The following antibodies or conjugated molecules were used for staining of cell surface markers: biotin-anti-IgD (405734), biotin-anti-IgM (406504), biotin-anti-CD138 (142512), biotin-anti-CD11b (101204), biotin-anti-CD11c (117304), biotin-anti-Gr-1 (108404), biotin-anti-CD4 (100404), biotin-anti-CD8a (100704), PE-anti-IgD (405706), PE-anti-IgM (406508), PE-anti-CD138 (142504), PE-anti-CD11b (101208), PE-anti-CD11c (117308), PE-anti-Gr-1 (108408), PE-anti-CD4 (100408), PE-anti-CD8a (100708), PE-anti-CD38 (102708), PE-anti-CD62L (104408), BV421-anti-IgM (406518), BV421-anti-CD138 (142508), Pacific Blue-anti-IgD (405712), Pacific Blue-anti-CD11b (101224), Pacific Blue-anti-CD11c (117322), Pacific Blue-anti-Gr-1 (108430), Pacific Blue-anti-CD4 (100428), Pacific Blue-anti-CD8a (100725), Pacific Blue-anti-CD38 (102720), APC/Fire-750-anti-B220 (103260), PerCP/Cy5.5-anti-B220 (103236), PerCP/Cy5.5-anti-CD38 (102722), PerCP/Cy5.5-anti-GL-7 (144610), PE/Cy7-anti-B220 (103222), PE/Cy7-anti-CD38 (102718), PE/Cy7-anti-CD4 (100422), anti-CD16/CD32 (101302), Alexa Fluor 647-anti-GL-7 (144606), Alexa Fluor 647-anti-IgG1 (406618), Alexa Fluor 647-anti-IgG2a (407116), APC-anti-IgG2b (406712), APC-anti-IgG (405308), APC-anti-CD44 (103012), APC-anti-CD21 (123412), Alexa Fluor 488-anti-IgG1 (406626), Alexa Fluor 488-anti-GL-7 (144612) and FITC-anti-IgM (406506) were obtained from BioLegend. FITC-anti-TCRβ (553171) from BD Bioscience and APC-anti-IgG3 (1100–31) from SouthernBiotech were also used. NP-PE (N-5070–1) was obtained from Biosearch.
For intracellular staining, cells were fixed with 1% methanol-free formaldehyde for 15 min on ice and permeabilized using Perm/Wash Buffer from Cytofix/Cytoperm Kit (BD Biosciences). Cells were labeled with unconjugated primary antibodies against the desired intracellular markers and then stained with fluorophore-conjugated secondary antibodies. Cells were analyzed by flow cytometry. The following antibodies were used for intracellular flow cytometry: phospho-Ripk3 (Thr231/Ser232) (91702); phospho-Ripk3 (Thr231/Ser232) (57220), anti-active caspase-9 (cat. no. 9509); anti-Gapdh (2118) were from Cell Signaling Technology; Alexa Fluor 647-anti-TOM20 was from Abcam; anti-Nix (cat. no. AF4030) were obtained from R&D Biosystem; anti-Pink1 (7859) was from ProSci; anti-Parkin (PA-13399), anti-TFAM (PA5–80107), FITC-mouse anti-rabbit IgG (31584), Alexa Fluor 647-donkey anti-goat IgG (A-21447) were from ThermoFisher Scientific.
For sorting of memory B cells, splenocytes were harvested from pooled spleens of wild type and DKO mice 8 weeks after NP-KLH immunization. Cells were subjected to negative selection prior to fluorescent antibody staining, to remove unwanted cells. Briefly, cells were labeled with biotinylated DUMP antibodies listed above, incubated with BioMag Streptavidin beads (QIAGEN), and then placed in a magnet stand. The negative fraction was collected and stained with fluorophore-conjugated antibodies as indicated above. DUMP−NP+B220+IgG1+GL-7−CD38+ memory B cells and DUMP−NP+B220+IgG1+GL-7+CD38– GC B cells were sorted using a FACSAria (BD Biosciences). In some experiments, B220+CD23+CD21+IgD+IgM+ naïve follicular B cells were also sorted for experiments.
Immunocytochemistry and deconvolution microscopy
Cells were attached to poly-L-lysine-coated slides by cytospin and fixed with 4% formaldehyde for 15 min. The cells were then permeabilized using 0.1% Triton-X in 0.1% Tween-20 for 10 min and blocked using Image-iT FX Signal Enhancer (Invitrogen) for 30 min. Cells were labeled with unconjugated primary antibodies (1:100 dilution) at 4°C overnight in a humidified chamber and then stained with Alexa Fluor-conjugated secondary antibodies (1:250 dilution) for 2 h at room temperature. Nuclei were counterstained with DAPI. 0.1% Tween-20 was used for all washing steps. Slides were mounted with ProLong Gold (Life Technologies) and allowed to cure at room temperature. Images were acquired using a 100x oil objective lens on a DeltaVision Elite deconvolution microscope (GE Healthcare Life Sciences) and analyzed using the ImageJ software. Pearson’s correlation coefficient (40) for LC3 and COXIV co-localization was quantified using the SoftWoRx software (GE Healthcare Life Sciences).
Detection of lipid droplets
For flow cytometric analysis of lipid droplets, cells were surface stained and incubated with Live/Dead Fixable Violet Dead Cell Stain. Cells were incubated with HCS LipidTOX Red or Deep Red Neutral Lipid Stain (Invitrogen) for 30 min, at room temperature and analyzed by flow cytometry. For deconvolution microscopy analysis of lipid droplets, sorted cells were attached to poly-L-lysine-coated slides by cytospin, fixed using 4% formaldehyde for 15 min, and incubated with HCS LipidTOX Deep Red Neutral Lipid Stain for 30 min, at room temperature. Nuclei were counterstained with DAPI.
Measurement of mitochondrial membrane potential, mitochondrial ROS and BODIPY FL C16 uptake
WT and DKO memory B cells were incubated in RPMI 1640 complete medium containing 25 nM TMRM (Invitrogen) or 5 μM MitoSOX Red (Invitrogen) for 30 min at 37°C and 5% CO2. Cells were analyzed by deconvolution microscopy or flow cytometry. To measure fatty uptake, memory B cells were incubated in RPMI 1640 complete medium containing 1 μM BODIPY FL C16 (ThermoFisher) for 30 min at 37°C and 5% CO2. Cells were then surface stained and analyzed by flow cytometry.
Transmission electron microscopy
Sorted WT and DKO memory B cells were fixed and embedded in Spurr’s low viscosity resin, and sections were cut using a U6 ultramicrotome (Leica), stained with saturated uranyl acetate, and counterstained with Reynold’s lead citrate (31). Images (5,000X or 12,000X magnification) were acquired using a H-7500 transmission electron microscope (Hitachi).
Adoptive transfer of NP-specific memory B cells
NP-specific memory B cells were sorted from pooled spleens of WT and DKO mice on a CD45.2 background, 6 weeks after immunization with NP-KLH. WT or DKO NP-specific memory B cells (5×104) were mixed with congenic CD45.1+ Naïve B cells (1:10) as filler cells to minimize cell loss. The cells were injected into each CD45.1 recipient mouse retro-orbitally. The spleen cells of recipients was analyzed by flow cytometry 4 weeks later. B220+DUMP– cells were gated and transferred CD45.2+CD45.1–NP+IgG+CD38+ memory B cells were quantified. In parallel experiments, mice with or without adoptive transfer of memory B cells (5×104 memory B cells/recipient) were challenged with soluble NP-KLH without adjuvants the day after adoptive transfer. Sera were then collected and anti-NP IgG levels were measured by ELISA 5 days after transfer. Antibody titers were quantified by ELISA similar to described procedures (11, 41).
Real-time quantitative RT-PCR (qRT-PCR)
To measure gene expression, RNA was prepared from sorted cells using TRIzol LS (ThermoFisher Scientific) or a MagMAX 96 Blood RNA Isolation Kit (Applied Biosystems). RNA was reverse transcribed to cDNA using a SuperScript VILO cDNA Synthesis Kit (Invitrogen). A master mix consisting of iTaq Universal SYBR Green Supermix (Bio-Rad) and pre-designed primers (0.9 μM) against target genes (Sigma-Aldrich or Harvard Primer Bank) was prepared. Each reaction consisted of 5.5 μl of master mix and 4.5 μl of cDNA diluted 9-fold in nuclease-free water. Real-time PCR was performed in 384-well PCR plates (Applied Biosystems) on a QuantStudio 5 Real-Time PCR System (Applied Biosystems). Cycling conditions were as follows: 50°C for 2 min, 95°C for 10 min, then 40 cycles of 95°C for 15 s and 60°C for 1 min, and a melt curve stage consisting of 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. Actb was used as an internal control. The following primers were used: Actb forward (GATGTATGAAGGCTTTGGTC) and reverse (TGTGCACTTTTATTGGTCTC); Tfam forward (ATTCCGAAGTGTTTTTCCAGCA) and reverse (TCTGAAAGTTTTGCATCTGGGT); Bnip3 forward (ACCACAAGATACCAACAGAG) and reverse (AATCTTCCTCAGACAGAGTG); Nix forward (GAAATGGAACTCTTTGGGTG) and reverse (CAGAAGAAGAAGTTGTAGAAGG); Sdhc forward (CTTTGTATCAGAAATGCTGC) and reverse (AGATAGTCAAATGGGGAGAC); Sdhaf2 forward (CATTCTCCTTAGCCTGTTTG) and reverse (GTTAATCAGGCGATCATAGAG); Srebf1 forward (GCAGCCACCATCTAGCCTG) and reverse (CAGCAGTGAGTCTGCCTTGAT); Slc25a1 forward (GAGGCATCGAAATCTGCATCA) and reverse (GGATGGAGCCGTAGAGCAA); Acly forward (CAGCCAAGGCAATTTCAGAGC) and reverse (CTCGACGTTTGATTAACTGGTCT); Acaca forward (CTGTATGAGAAAGGCTATGTG) and reverse (AACCTGTCTGAAGAGGTTAG); Fasn forward (GATTCAGGGAGTGGATATTG) and reverse (CATTCAGAATCGTGGCATAG); Bcl2 forward (ATGCCTTTGTGGAACTATATGGC) and reverse (GGTATGCACCCAGAGTGATGC). Real-time PCR for autophagy gene was performed using Taqman Universal PCR Master Mix with specific primers from the TaqMan Gene Expression Assay Kit (ThermoFisher Scientific) as described (11). The assay IDs for the primers of the analyzed genes are as follows: Mm01265461_m1 (Becn1), Mm00504340_m1 (Atg5) and Mm00512209_m1 (Atg7).
To measure mtDNA content, sorted WT and DKO memory B cells were lysed in TRIzol LS (ThermoFisher). Chloroform was added at a 1:1 v/v ratio to partition all nucleic acids into the aqueous layer. Nucleic acids were isolated from the aqueous layer according to the manufacturer’s protocol. Real-time PCR was carried out as above, but with primers specific to mtDNA or genomic Actb DNA. Data was normalized to Actb DNA. The following primers were used: mtDNA forward (ACCATTTGCAGACGCCATAA) and reverse (TGAAATTGTTTGGGCTACGG); Actb DNA forward (TGTTCCCTTCCACAGGGTGT) and reverse (TCCCAGTTGGTAACAATGCCA).
In vivo drug treatment
WT and DKO mice were immunized with NP-KLH and drug treatments were started at 2 weeks post-immunization. For the NAC treatment experiment, mice were given i.p. injection of NAC (A7250, Sigma-Aldrich; 100 mg/kg) or DPBS vehicle control once every 2 d for 4 weeks. For the CTPI-2 treatment experiment, mice were given i.p. injection of CTPI-2 (S2968, Selleck Chemicals; 50 mg/kg) or DMSO vehicle control daily for 4 weeks.
In vitro analysis of apoptosis and necroptosis signaling
For analysis of apoptosis signaling, WT or DKO splenocytes were incubated for 6 h in RPMI 1640 complete medium with 0.1% FBS at 37°C and 5% CO2. Cells were then subjected to surface staining for GC and memory B cell markers, followed by intracellular staining for cleaved caspase-9 (9509) or caspase-3 (9661) (Cell Signaling Technology). The cells were then incubated with secondary antibodies and analyzed by flow cytometry. For analysis of necroptosis signaling, WT or DKO splenocytes were first pre-incubated with 20 µM z-VAD-FMK (ALX-260–020-M001, ENZO Life Sciences) for 1 h in RPMI 1640 medium containing 10% FBS at 37°C and 5% CO2. 1 SM-164 (1 µM; S7089, Selleckchem) was then added to the cells, and incubation was continued for 10 h at 37°C and 5% CO2. Cells were then subjected to surface staining for GC and memory B cell markers, followed by intracellular staining for phospho-Ripk3 (Thr231/Ser232) (Cell Signaling Technology) and analyzed by flow cytometry.
siRNA transfection and adoptive transfer of memory B cells
IgG+CD38+ memory B cells were sorted from spleens of 20 WT or B/Nix–/–Bnip3–/– mice 2 weeks after immunization with SRBCs and mixed with naïve B cells (1:10) as carriers. The cells were transfected with non-targeting control siRNA (ThermoFisher Scientific) or Ripk3 siRNA (Santa Cruz Biotechnology) using the Neon Transfection System (ThermoFisher Scientific) and adoptively transferred into CD45.1 recipient mice intravenously (5×104/mouse). Seven days later, recipient spleen cells were analyzed by flow cytometry. DUMP– cells were gated and transferred CD45.2+CD45.1– memory B cells were quantified. For Western blot analysis, the transfected B cells were cultured for 48 h in the presence of 5 μg/mL anti-CD40 (102811), 5 μg/mL anti-CD180 (11709) and 20 ng/mL IL-4 (all from BioLegend). Cells were then lysed in lysis buffer consisting of 1% Triton X-100, 50 mM Tris pH 8.0, 150 mM sodium chloride, 10 mM sodium fluoride, 1 mM sodium orthovanadate, and 1x protease inhibitor mixture (Roche Applied Science). Lysates were used for Western blot by probing with rabbit anti-mouse Ripk3 (US Biologicals cat. no. 351904), followed with HRP-conjugated goat anti-rabbit IgG (Abcam). The blots were also probed with HRP-conjugated anti-mouse Gapdh (Cell Signaling Technology cat. no. 3683). The blots were developed with Clarity Western ECL Substrate (Bio-Rad), followed by chemiluminescence image acquisition on a Bio-Rad ChemiDoc Touch Imaging System.
Extracellular flux analysis
An XFe96 plate (Seahorse Bioscience) was coated with Cell-Tak Cell and Tissue Adhesive (5 μg/ml, Agilent). Sorted WT and DKO memory B cells were seeded on the plate at 3×105 cells/well, in XF Base Medium (Seahorse Bioscience) supplemented with sodium pyruvate (1 mM) and L-glutamine (2 mM). The plate was centrifuged and cells were allowed to attach to the wells in a non-CO2 incubator at 37°C for 30 min. The OCR and ECAR were measured on an XFe96 Analyzer (Seahorse Bioscience) at baseline and upon sequential addition of 0.2 μM oligomycin, 0.1 μM FCCP, 50 μM etomoxir, and antimycin A+rotenone (0.5 μM each).
RNA-sequencing
QIAseq FX Single Cell RNA Library Kit (QIAGEN) was used to extract RNA from ~1000 sorted WT and DKO memory B cells and prepare cDNA libraries enriched for polyadenylated RNAs, according to manufacturer’s instructions. Library cleanup was performed using GeneRead Size Selection Kit (QIAGEN). Libraries were sent to Novogene Corporation (Sacramento, California), where sequencing was performed on a HiSeq 4000 instrument (Illumina) with paired-end 150 bp reads. Raw sequencing data were processed by Novogene and converted into read counts per gene. R software (version 3.6.0) was used for subsequent analysis of the data. The edgeR package in R was used to identify differentially expressed genes (false discovery rate (FDR)-adjusted P value ≤ 0.05). Expression values for selected differentially expressed genes (DEGs) were log2-transformed (after addition of a pseudocount of 1) and plotted on a heatmap generated using the R package pheatmap. DEGs are shown in Supplemental Table 1. Functional classification of differentially expressed genes was performed using PANTHER GO-slim.
Statistics
Data are presented as mean ± s.e.m and statistical analysis was performed using Prism 8.1.2 (GraphPad) as follows: Populations were compared using unpaired two-tailed Student’s or Welch’s t-tests. P values of < 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, ns = not statistically significant.
Results
Maintenance of mitochondrial homeostasis in IgG+ memory B cells by mitochondrial autophagy.
The regulation of mitochondrial homeostasis is critical for metabolic rewiring and the development of memory T cells (26, 27). To determine whether mitochondrial changes play a functional role in memory B cell homeostasis, we examined mitochondrial content in memory B cells. We generated class-switched germinal center (GC) and memory B cells from the mice immunized with a T cell-dependent antigen, 4-hydroxy-3-nitrophenylacetyl (NP) conjugated to keyhole limpet hemocyanin (KLH) (11, 42) (Fig. 1a and Supplemental Fig. 1). Compared to GC B cells, memory B cells had less mitochondrial content as shown by intracellular staining of mitochondrial import receptor subunit Tom20 (Fig. 1A), or by Mitotracker green (43) staining (Fig. 1B). These data indicate that memory B cells have reduced mitochondrial content in comparison with GC B cells.
FIGURE 1.
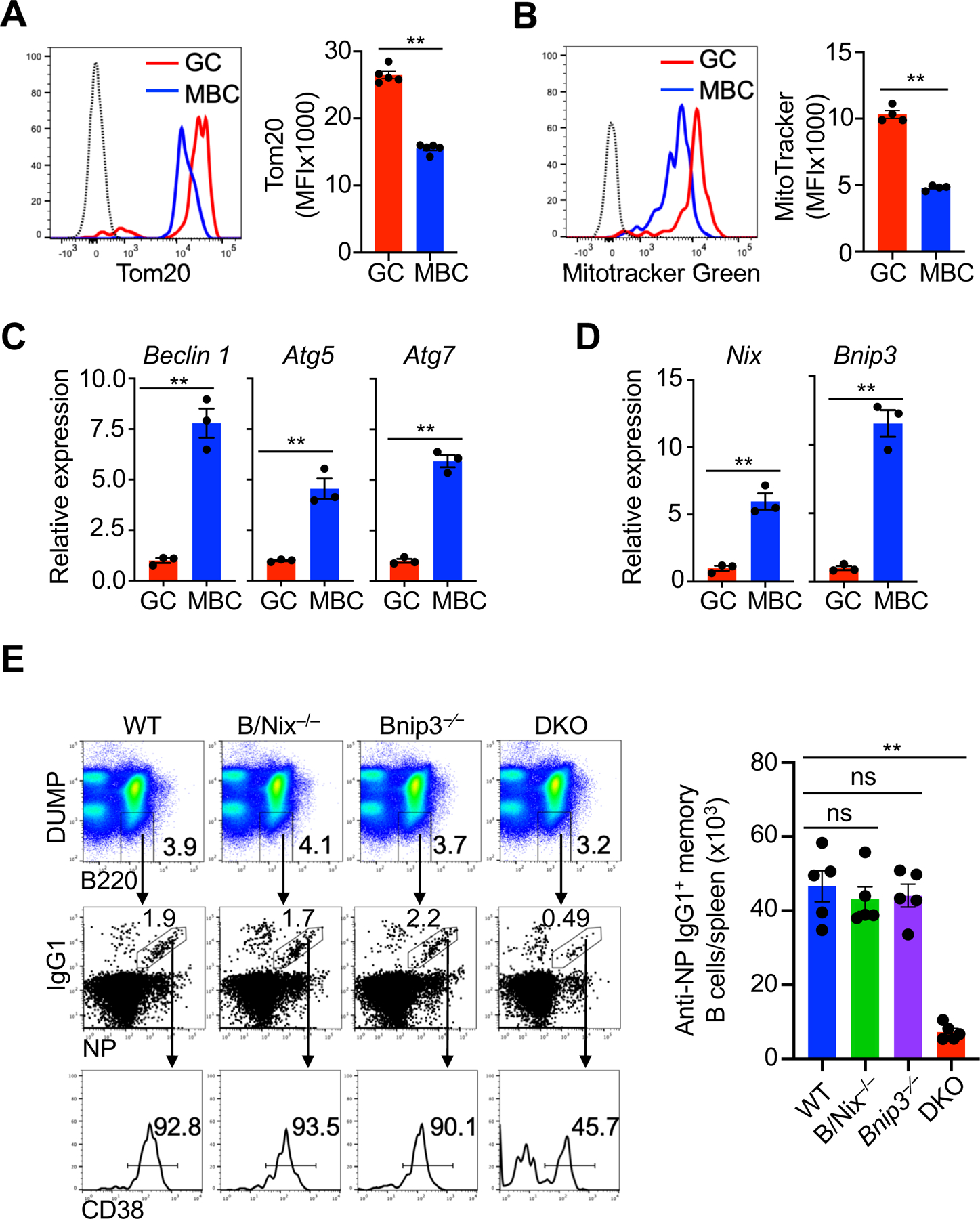
Memory B cells have reduced mitochondrial content and require Bnip3 and Nix for mitochondrial homeostasis. (A) Intracellular staining of Tom20 in GC and memory B cells. Dashed line: isotype control (n=5 mice per group). Data are representative of three independent experiments and are presented as mean ± s.e.m. **P < 0.01. (B) Flow cytometry analysis of Mitotracker Green staining in germinal center (GC) and memory B cells (MBC). Dashed line: isotype control (n=4 mice per group). Data are representative of three independent experiments and are presented as mean ± s.e.m. **P < 0.01. (C, D) Real-time RT-PCR for Beclin 1, Atg5, Atg7 (C), Nix and Bnip3 (D) in GC and memory B ( n=3). Data are representative of two independent experiments and are presented as mean ± s.e.m. **P < 0.01; ns, not significant. (E) Analyses of NP-specific memory B cells 8 weeks after immunization in the spleens of wild type (WT), B/Nix–/–, Bnip3−∕−, B/Nix–/–Bnip3−∕− (DKO) mice (n=5 mice per group) by flow cytometry. Data are representative of three independent experiments and are presented as mean ± s.e.m. **P < 0.01; ns, not significantly different.
Autophagy plays an important role in the regulation of mitochondrial homeostasis (44). We found that memory B cells had increased expression of autophagy genes Beclin 1, Atg5 and Atg7 compared to GC B cells (Fig. 1C). In addition, Nix and Bnip3, two functionally overlapping molecules for mitochondrial autophagy (30–33, 45), were increasingly expressed in memory B cells compared to GC B cells (Fig. 1D). In contrast, the levels of Pink1 and Parkin that regulate mitochondrial autophagy independent of Nix and Bnip3 (38, 46), were comparable between memory and GC B cells (Supplemental Fig. 2A). We therefore determined whether Nix and Bnip3 might be important for the regulation of mitochondrial autophagy during memory B cell development.
We crossed Nixflox mice with AID-Cre mice with the expression of Cre controlled by the promoter/enhancer of activation-induced cytidine deaminase, a gene expressed in GC B cells (47). Such expression of AID-Cre allows the deletion of the floxed genes at GC B cell stage and in cell types subsequently differentiated from GC B cells, including GC-derived memory B cells and plasma cells. AID-Cre/Nixflox mice were then crossed to Bnip3−/− mice to generate mice with double knockout of Nix and Bnip3 (DKO) starting from GC B cell stage. We observed deletion of Nix in memory B cells but not memory T cells or dendritic cells from AID-Cre/Nixflox mice (Supplemental Fig. 2B). We observed that NP-specific memory B cell numbers 8 weeks after immunization were comparable between wild-type (WT), B/Nix–/– and Bnip3−⁄− mice (Fig. 1E). However, NP-specific memory B cells were significantly reduced in DKO mice, suggesting that Nix and Bnip3 are required for memory B cell generation or survival (Fig. 1E). Total T cells, total B cells and the percentages of naïve follicular and marginal zone (MZ) B cell subsets (48) were normal in B/Nix–/–Bnip3−⁄− mice (Supplemental Fig. 3). These results suggests that Nix/Bnip3-dependent mitochondrial autophagy specifically affects the IgG+ memory B cell compartment but not naïve or GC B cells.
Normal formation but impaired persistence of Nix−/−Bnip3−/− memory B cells
Next, we examined whether formation or maintenance was defective in Nix−/−Bnip3−/− memory B cells. At 2 weeks after immunization with NP-KLH, NP-specific GC and memory B cells were comparable between DKO mice and wild type controls (Fig. 2A). These data suggest that primary antibody responses and memory B cell formation were normal in DKO mice. By 6 weeks post immunization, however, NP-specific memory B cells in DKO mice were substantially reduced (Fig. 2B), indicating that memory B cells cannot persist in the absence of Nix and Bnip3.
FIGURE 2.
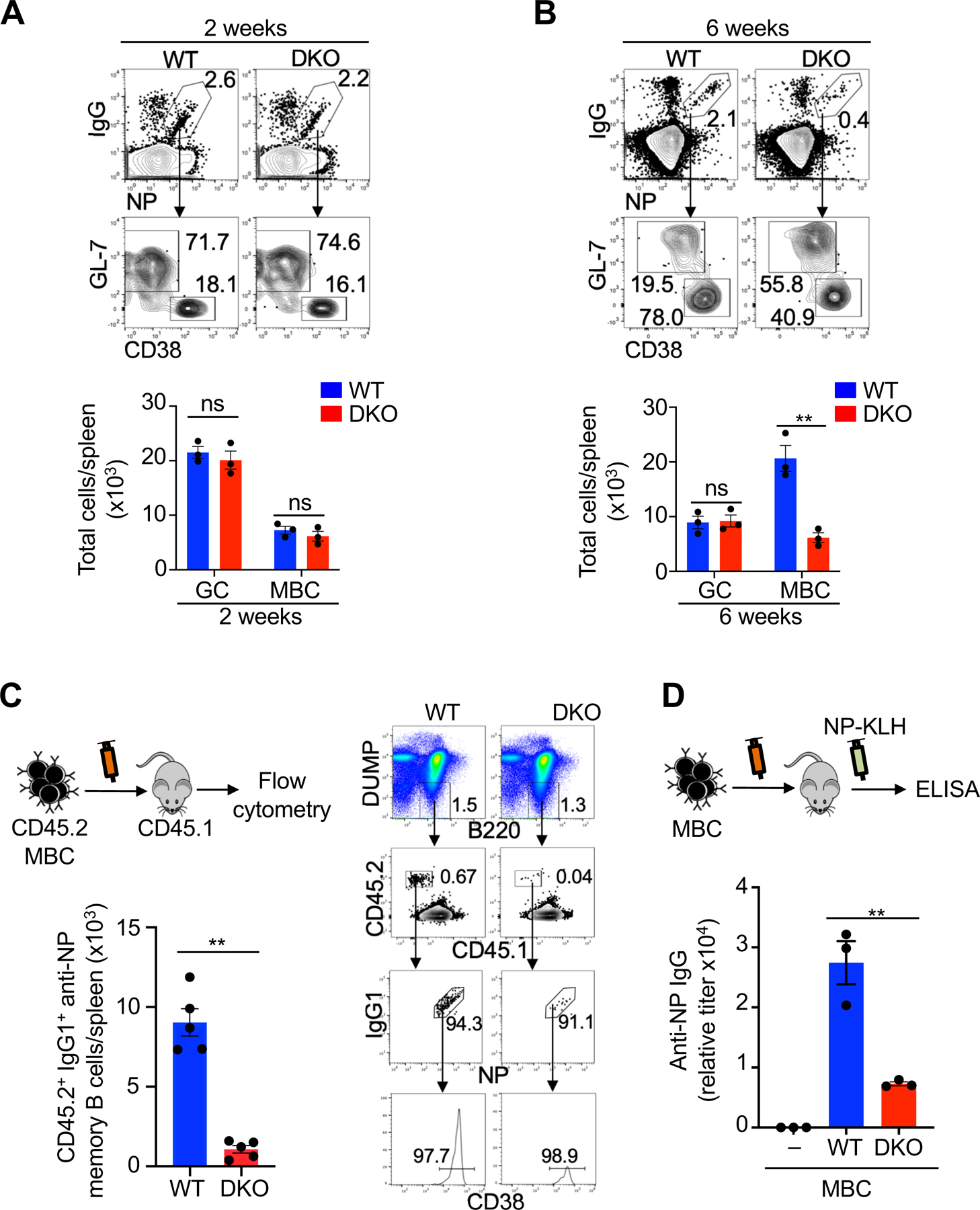
Nix−∕−Bnip3−∕− memory B cells form normally but have impaired maintenance and mediate defective secondary antibody responses. (A, B) Flow cytometry analyses of NP-specific GC and memory B cells in the spleens of DKO and wild type control mice at 2 weeks (A) or 6 weeks (B) after immunization with NP-KLH (n=3 mice per group). Data are representative of five independent experiments and are presented as mean ± s.e.m. **P < 0.01; ns, not significantly different. (C) NP-specific memory B cells (DUMP–B220+NP+IgG+GL-7–CD38+) were sorted from CD45.2 WT or DKO mice and mixed with congenic CD45.1 naïve B cells (1:10) as carriers, followed by adoptive transfer into CD45.1 recipients (5×104 memory B cells/mouse) retroorbitally (n=5 mice per group). Four weeks later, spleen cells from recipient mice were analyzed by flow cytometry The DUMP–CD45.2+CD45.1–NP+IgG+CD38+ transferred memory B cells were quantified. Data are representative of three independent experiments and are presented as mean ± s.e.m. **P <0.01. (D) In parallel experiments, the recipient mice as in (C) were injected with soluble NP-KLH. Anti-NP IgG in serum was measured by ELISA. Data are representative of two independent experiments and are presented as mean ± s.e.m. **P <0.01.
It has been shown that limiting nutrients or cytokines may cause stress and trigger autophagy in lymphocytes to sustain cell survival (16, 49). Adoptive transfer of memory B cells to unimmunized mice would put memory B cells under stress in such a local microenvironment. To test whether the failure of memory B cells deficient in Nix and Bnip3 depended on extrinsic factors elicited by prior immunization, we performed adoptive transfer of sorted memory B cells into unimmunized congenic recipients according to the established procedures (50, 51). We could detect transferred wild type CD45.2+CD45.1– memory B cells in the recipient mice (Fig. 2C). However, the number of transferred Nix–/–Bnip3–/– memory B cells were significantly lower in the recipients (Fig. 2C). These data suggest that Nix–/–Bnip3–/– IgG1+ memory B cells are defective in long-term survival.
We next examined whether Nix–/–Bnip3–/– memory B cells with decreased survival potential were also defective in mounting antibody recall responses. Although antigens alone without adjuvants cannot induce immune response efficiently, it has been shown that NP-KLH without adjuvants can stimulate adoptively transferred memory B cells to become antibody-producing cells (8). To measure antibody recall responses, equal numbers of wild type and Nix–/–Bnip3–/– memory B cells were adoptive transferred into recipient mice, followed by challenge of recipient mice with soluble NP-KLH. We detected the production of IgG anti-NP in mice transferred with wild type memory B cells, but not in mice that did not receive memory B cells after challenge with soluble NP-KLH (Fig. 2D). Interestingly, mice transferred with Nix–/–Bnip3−⁄− memory B cells showed much lower secondary antibody responses (Fig. 2D). These data suggest that NIX−/−BNIP3−/− memory B cells with decreased survival potential are defective in mounting antibody recall responses.
Nix−/−Bnip3−/− memory B cells have impaired mitochondrial autophagy and accumulate mitochondria
Since Nix and Bnip3 mediate mitochondrial autophagy (31–33, 52), we next examined whether they affect the mitochondrial compartment in memory B cells by transmission electron microscopy (TEM) studies. Because of significantly reduced number of NP+IgG1+ memory B cells in DKO mice (Fig. 1E), we sorted DUMP−B220+IgG+GL-7−CD38+ memory B cells, which contain both NP-specific and non-specific memory B cells, to obtain sufficient number of cells for TEM studies. TEM showed the accumulation of mitochondria in Nix–/–Bnip3−⁄− IgG+ memory B cells (Fig. 3A, yellow arrows). Autophagosomes could be detected in both wild type and Nix–/–Bnip3−⁄− memory B cells (Fig. 3A, red arrows). However, degrading mitochondria can only be found in autophagosomes in wild type but not Nix–/–Bnip3−⁄− IgG+ memory B cells (Fig. 3A). Immunocytochemistry analyses showed more staining for mitochondrial protein CoxIV in NP-specific Nix–/–Bnip3−⁄− memory B cells (Fig. 3B). Mitochondrial accumulation can induce mitochondrial fusion (53). Interestingly, many accumulated mitochondria were potentially linked or fused together in the TEM sections of Nix–/–Bnip3−⁄− memory B cells (Fig. 3A). Moreover, increased mitochondrial DNA (mtDNA) was detected in Nix–/–Bnip3−⁄− memory B cells (Fig. 3C). These data suggest that excessive mitochondria are accumulated in Nix–/–Bnip3−⁄− memory B cells. We also found that Nix–/–Bnip3−⁄− memory B cells and wild type controls had similar expression of the mitochondrial biogenesis gene TFAM (Fig. 3D), indicating that the accumulation of mitochondria in Nix–/–Bnip3−⁄− IgG+ memory B cells is not due to elevated mitochondrial biogenesis.
FIGURE 3.
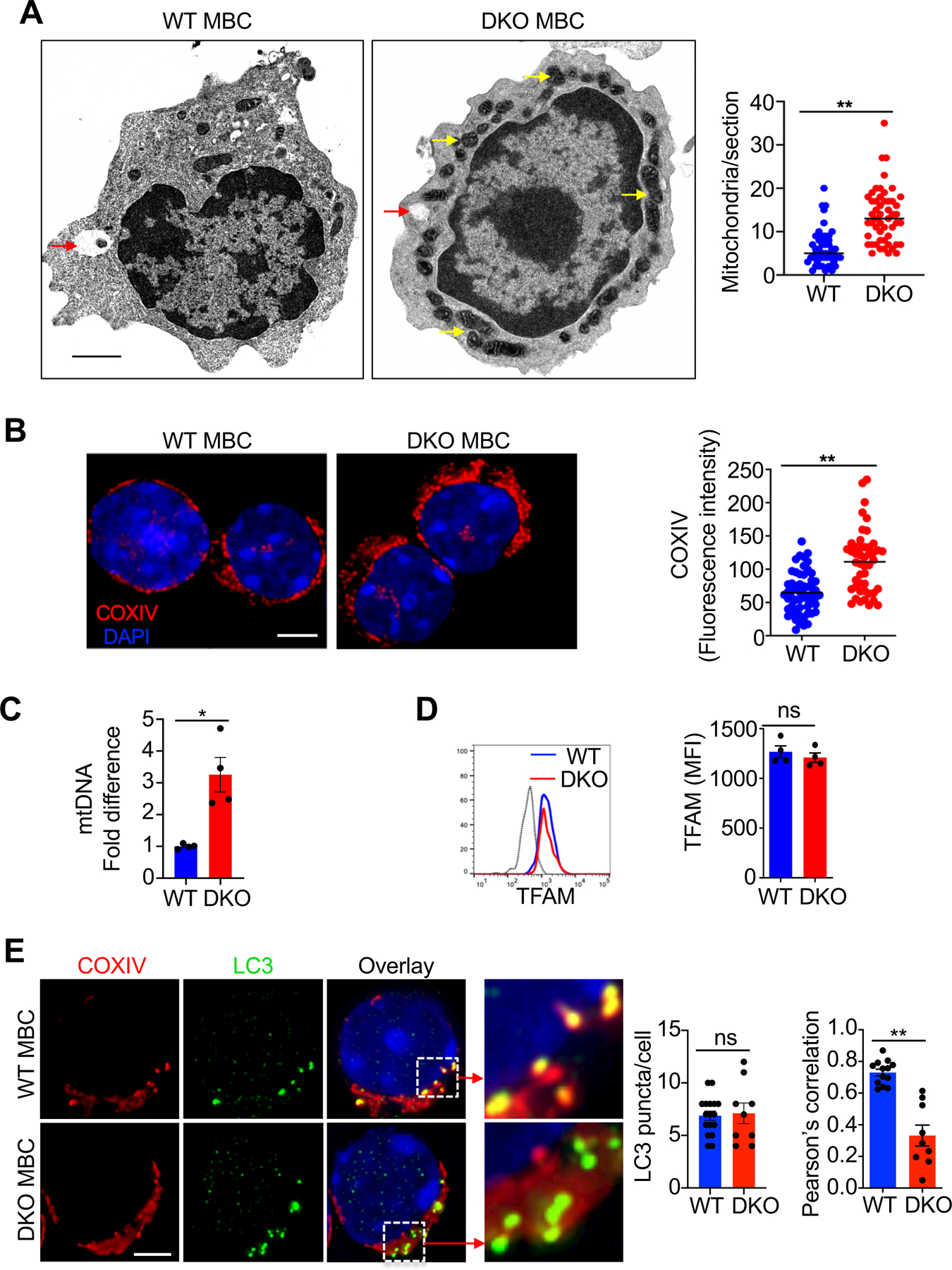
Nix−∕−Bnip3−∕− memory B cells exhibit impaired mitochondrial autophagy and accumulate mitochondria. (A) Transmission electron microscopy images (12,000X) of DUMP−B220+IgG+GL-7−CD38+ memory B cells sorted from WT or DKO mice (8 weeks post-immunization with NP-KLH). Red arrows indicate autophagosomes. Yellow arrows indicate enlarged mitochondria in DKO memory B cells. Scale bar, 500 nm. Data are representative of analyses of 21 (WT) or 30 (DKO) sections of different memory B cells. **P < 0.01. (B) Deconvolution microscopy analyses of WT and DKO NP-specific memory B cells sorted as DUMP−B220+NP+IgG1+GL-7−CD38+ (WT, n=47 cells; DKO, n=55 cells) stained for COXIV. Fluorescence level of COXIV staining was determined using ImageJ. Nuclei were counterstained with DAPI. Scale bar, 3 µm. Data are representative of three independent experiments and are presented as mean ± s.e.m. **P < 0.01. (C) Real-time PCR for mtDNA in WT or DKO NP-specific memory B cells (n=4 mice per group). *P< 0.05. (D) Intracellular staining for TFAM in WT and DKO NP-specific memory B cells (n=4 mice per group) was analyzed by flow cytometry. Dashed line: isotype control. ns = not statistically significant. (E) WT and DKO NP-specific memory D cells sorted as above were stained for LC3 and COXIV and analyzed by deconvolution microscopy. Nuclei were counterstained with DAPI. Scale bar, 3 µm. LC3 puncta per cell were counted. Pearson’s correlation coefficient for the colocalization between LC3 puncta and COXIV (WT, 0.73; DKO, 0.33. P = 0.002). A Pearson’s correlation coefficient value greater than 0.5 is considered to represent colocalization between the two signals. Data are representative of two independent experiments and are presented as mean ± s.e.m. **P < 0.01; ns, not statistically significant.
To determine whether mitochondrial autophagy is defective in Nix–/–Bnip3−⁄− memory B cells, we examined the staining of LC3 puncta that are characteristic of the formation of autophagosome during active autophagy (54, 55), and mitochondrial protein, CoxIV. We found that LC3 puncta were comparable in wild type and Nix–/–Bnip3−⁄− memory B cells (Fig. 3E), suggesting that autophagosomal formation is normal in Nix–/–Bnip3−⁄− memory B cells. In areas where LC3 were present, the colocalization between LC3 puncta and CoxIV was significantly reduced in Nix–/–Bnip3−⁄− memory B cells, as measured by decreased Pearson’s correlation coefficient (40) for LC3 and CoxIV staining (Fig. 3E). These data suggest that the targeting of mitochondria to autophagosomes was defective in Nix−⁄−Bnip3−⁄− IgG+ memory B cells, resulting in the failure of maintaining mitochondrial homeostasis.
Nix−/−Bnip3−/− memory B cells contain more active mitochondria
Increased mitochondrial content can lead to greater production of mitochondrial reactive oxygen species (ROS) (56). Staining with MitoSOX Red (57) indicated elevated levels of mitochondrial ROS in Nix–/–Bnip3−⁄− memory B cells (Fig. 4A). The increased ROS in Nix–/–Bnip3−⁄− memory B cells could be caused by increased total mitochondrial content or accumulation of dysfunctional mitochondria. To determine whether ROS could account for the loss of Nix–/–Bnip3−⁄− memory B cells, we treated WT and AID/Nix–/–Bnip3−⁄− mice with N-acetylcysteine (NAC), a ROS scavenger (11, 30, 33). We started NAC treatment at 2 weeks post-immunization, when normal formation of memory B cells was observed (Fig. 2A). However, we did not observe the rescue of Nix–/–Bnip3−⁄− memory B cells by NAC treatment (Fig. 4B). These data suggest that inhibition of oxidative stress alone is not sufficient to protect Nix–/–Bnip3−⁄− IgG+ memory B cells. Additional mechanisms likely exist to account for the loss of these cells.
FIGURE 4.
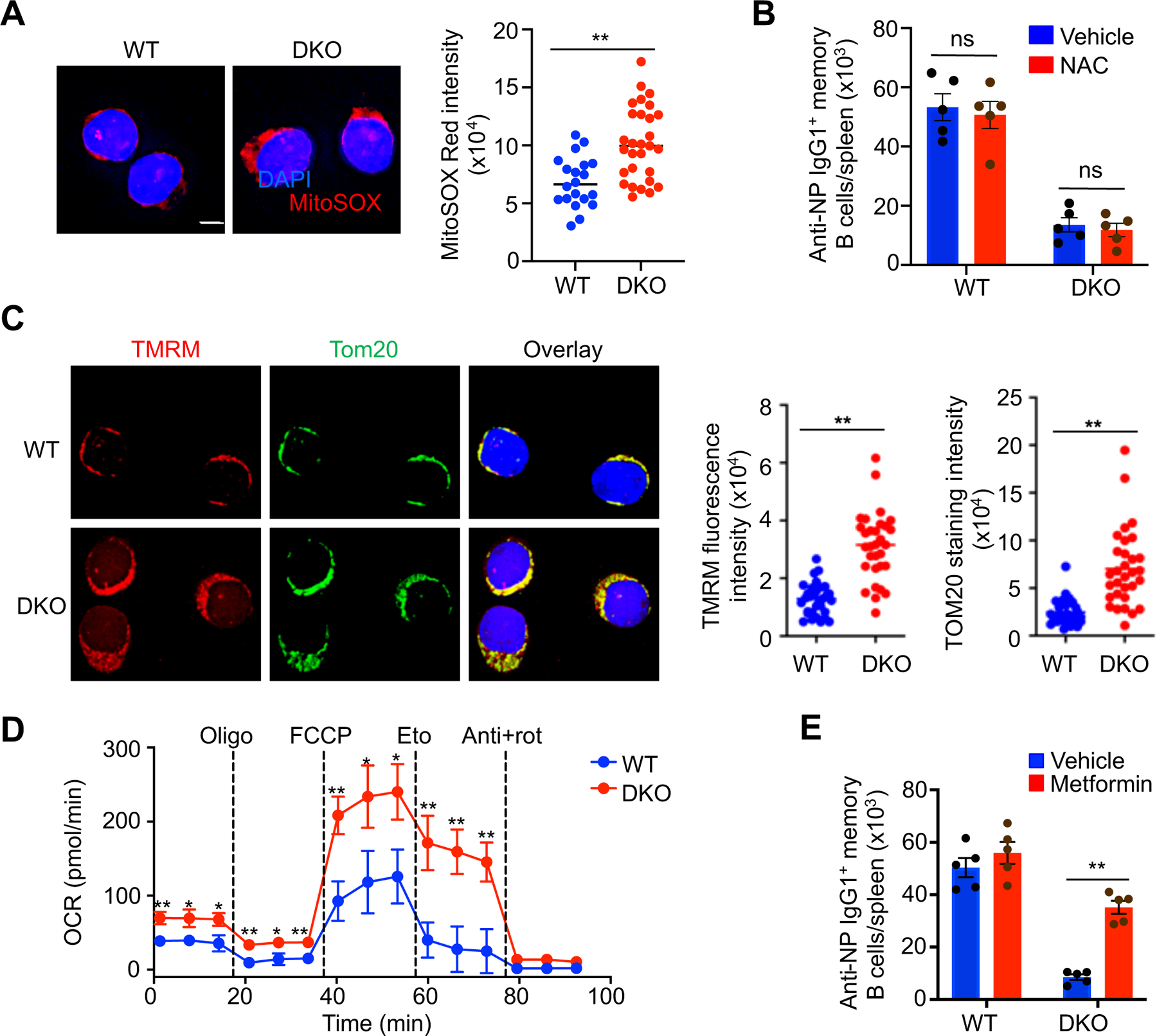
Nix−∕−Bnip3−∕− memory B cells have increased active, functional mitochondria. (A) Deconvolution microscopy analysis of MitoSOX Red staining in memory B cells from WT or DKO mice. Nuclei were counterstained with DAPI. Scale bar, 3 µm. Fluorescence level of MitoSOX Red staining was determined using ImageJ. Data are representative of two independent experiments and are presented as mean ± s.e.m. **P < 0.01. (B) WT and DKO mice were immunized with NP-KLH and treated with vehicle or NAC once every two days from 2 to 6 weeks post-immunization. Splenic NP-specific memory B cells were quantified by flow cytometry (n=5 mice/group). Data are representative of two independent experiments and are presented as mean ± s.e.m. ns, not statistically significant. (C) Deconvolution microscopy analysis of staining with TMRM and anti-TOM20 in NP-specific memory B cells from WT or DKO mice sorted as above. Nuclei were counterstained with DAPI. Scale bar, 3 µm. The fluorescence intensity of TMRM and TOM20 staining per cell was determined using ImageJ. Data are representative of three independent experiments and are presented as mean ± s.e.m. **P < 0.01. (D) Seahorse extracellular flux analysis of (left) ECAR and (right) OCR of WT and DKO NP-specific memory B cells sorted as above. The cells were sequentially treated with the drugs oligomycin (oligo), FCCP, etomoxir (eto), antimycin+rotenone (anti+rot) at the indicated time points. Data are representative of two independent experiments and are presented as mean ± s.e.m. *P < 0.05; **P < 0.01.
Mitochondrial retention due to defective mitochondrial autophagy may lead to accumulation of damaged mitochondria with reduced membrane potential (30, 33, 58). We therefore stained Nix–/–Bnip3−⁄− memory B cells with tetramethyl rhodamine methyl ester (TMRM) to measure mitochondrial membrane potential (59). In addition to increased mitochondrial content by TOM20 staining, Nix–/–Bnip3−⁄− memory B cells exhibited increased TMRM staining compared to wild type controls (Fig. 4C). These results indicate that Nix–/–Bnip3−⁄− IgG+ memory B cells contained more functional mitochondria.
We next examined whether the accumulation of active mitochondria could lead to increased metabolic functions in Nix–/–Bnip3−⁄− memory B cells. We measured oxygen consumption rate (OCR), a readout of oxidative phosphorylation, in Nix–/–Bnip3−⁄− memory B cells and wild type controls at baseline and after sequential addition of oligomycin (inhibits ATP synthase), FCCP (uncouples ATP synthesis from the electron transport chain), etomoxir (inhibits mitochondrial fatty acid oxidation), and rotenone plus antimycin (inhibit complex I and III, respectively) (60, 61). Nix–/–Bnip3−⁄− memory B cells showed higher OCR at the basal level (Fig. 4D). OCR was also significantly higher in Nix–/–Bnip3−⁄− memory B cells after the addition of FCCP (Fig. 4D), suggesting the increase in the maximum potential of oxidative phosphorylation. Inhibition of oxidation of endogenous fatty acids with etomoxir (62) showed suppression of OCR in both Nix–/–Bnip3−⁄− IgG+ memory B cells and wild type controls (Fig. 4D). Together, these data suggest that Nix–/–Bnip3−⁄− IgG+ memory B cells containing more active mitochondria have increases in oxidative phosphorylation.
We next examined whether altered metabolism in Nix–/–Bnip3−⁄− memory B cells might contribute cell loss. It has been shown that AMPK, a key regulator for cellular metabolic functions and autophagy, is important for the development and maintenance of memory B cells (29). Loss of AMPKa1 resulted in decreased mitochondrial respiratory activity, decreased mitophagy, and increased lipid peroxidation. Because metformin can activate AMPK in vivo (63), we next examined whether AMPK activated mitophagy pathway depends on Nix and Bnip3 in memory B cells by treating mice with metformin. Interestingly, we found that treatments with metformin partially rescued Nix–/–Bnip3−⁄− IgG+ memory B cells in vivo (Fig. 4E). These results are consistent with the possibility that dysregulated metabolic functions in Nix–/–Bnip3−⁄− IgG+ memory B cells contribute to the reduction in memory B cells.
Elevated expression of de novo fatty acid synthesis genes in Nix−/−Bnip3−/− memory B cells
To determine potential transcriptional changes due to the loss of Nix and Bnip3, we performed RNA-sequencing (RNA-seq) analyses of IgG+ WT and Nix–/–Bnip3−⁄− memory B cells. We found 204 differentially expressed genes (DEGs) in IgG+ Nix−/−Bnip3−/− memory B cells (Supplemental Table 1). Functional classification using PANTHER GO-slim annotations indicated that most of the DEGs were related to cellular and metabolic processes (Fig. 5A). The oxidative phosphorylation genes Sdhc and Sdhaf2 (succinate dehydrogenase complex) (64) were among the DEGs elevated in Nix–/–Bnip3−⁄− IgG+ memory B cells (Fig. 5, A and B), indicating that Nix–/–Bnip3−⁄− memory B cells may have increased oxidative phosphorylation capacity.
FIGURE 5.
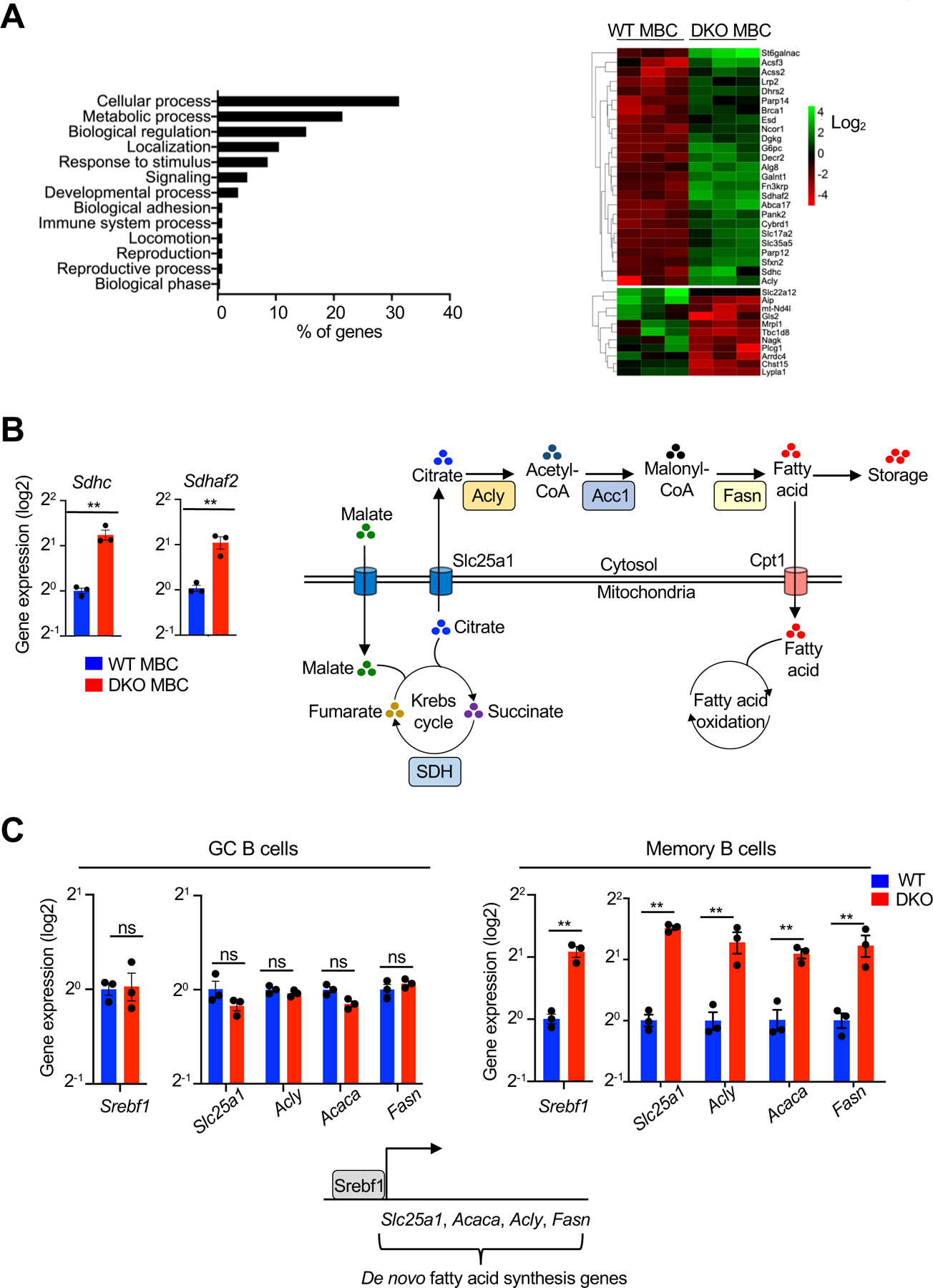
Nix−∕−Bnip3−∕− memory B cells upregulate de novo fatty acid synthesis-related genes. (A) RNA-seq analysis of WT and DKO memory B cells. Left panel: functional classification of differentially expressed genes (DEGs) on the basis of biological process using PANTHER GO-slim. Right panel: Heatmap of metabolism-related DEG expression in WT and DKO MBCs. Data represent the analyses of three independent samples in each group. (B) Real-time RT-PCR analysis for Sdhc and Sdhaf2 in memory B cells sorted from WT or DKO mice as above. Relative gene expression was normalized to actin. Data are representative of two independent experiments and are presented as mean ± s.e.m. **P < 0.01. A schematic diagram shows that the Krebs cycle in the mitochondria is linked to de novo fatty acid synthesis in the cytosol. In Nix–/–Bnip3−⁄− memory B cells, excessive de novo fatty acid synthesis van lead to more lipid storage in the cells or changes in cellular biosynthesis. (C) qRT-PCR analyses showing the fold of changes for Srebf1, Slc25a1, Acly, Acaca, and Fasn in GC or memory B cells sorted from WT or DKO mice as in Fig. 1. Schematic diagram shows the role for Srebf1 in the induction of genes involved in de novo fatty acid synthesis. Data are representative of four independent experiments and are presented as mean ± s.e.m. **P < 0.01; ns, not statistically significant.
Interestingly, de novo fatty acid synthesis genes, including Acly, Acss2, and Acsf3 (32, 65, 66), were also among the DEGs enriched in Nix–/–Bnip3−⁄− memory B cells (Fig. 5A). it has been shown that active mitochondria can stimulate de novo fatty acid synthesis (64, 67). In exchange for cytosolic malate, the mitochondrial citrate transporter (Slc25a1) diverts citrate from the Krebs cycle to the cytosol where it can be used as a substrate for de novo fatty acid synthesis (64, 68) (Fig. 5B). De novo fatty acid synthesis proceeds in a series of reactions mediated sequentially by ATP citrate lyase (Acly), acetyl-coA carboxylase 1 (Acaca/Acc1), and fatty acid synthase (Fasn) (64, 68). The transcription factor Srebf1 is a master regulator of de novo fatty acid synthesis genes (32, 69). Real-time RT-PCR confirmed the upregulation of Srebf1 and its targets Slc25a1, Acly, Acaca, and Fasn (32, 69) in memory but not GC B cells in the absence of Nix and Bnip3 (Fig. 5C). Since Nix–/–Bnip3−⁄− IgG+ memory B cells accumulate active mitochondria, enhanced Slc25a1-dependent mitochondrial citrate export may lead to increases in de novo fatty acid synthesis.
Nix−/−Bnip3−/− IgG memory B cells are rescued in vivo by inhibiting de novo fatty acid synthesis
Elevated de novo fatty acid synthesis can lead to increased storage of fatty acids in lipid droplets if there is no decrease in exogenous fatty acid uptake or increase in fatty acid oxidation (70). Staining with LipidTOX (71) revealed that Nix–/–Bnip3−⁄− memory but not GC B cells accumulated lipid droplets by microscopy (Fig. 6A) or flow cytometry analyses (Fig. 6B). We also observed that Nix–/–Bnip3−⁄− memory B cells and wild type controls acquired similar amounts of exogenous fatty acids as measured by the uptake of a fluorescently-labeled form of fatty acid, BODIPY FL C16 (72) (Fig. 6C), indicating that increased lipid droplets in Nix–/–Bnip3−⁄− memory B cells is not due to elevated fatty acid uptake.
FIGURE 6.
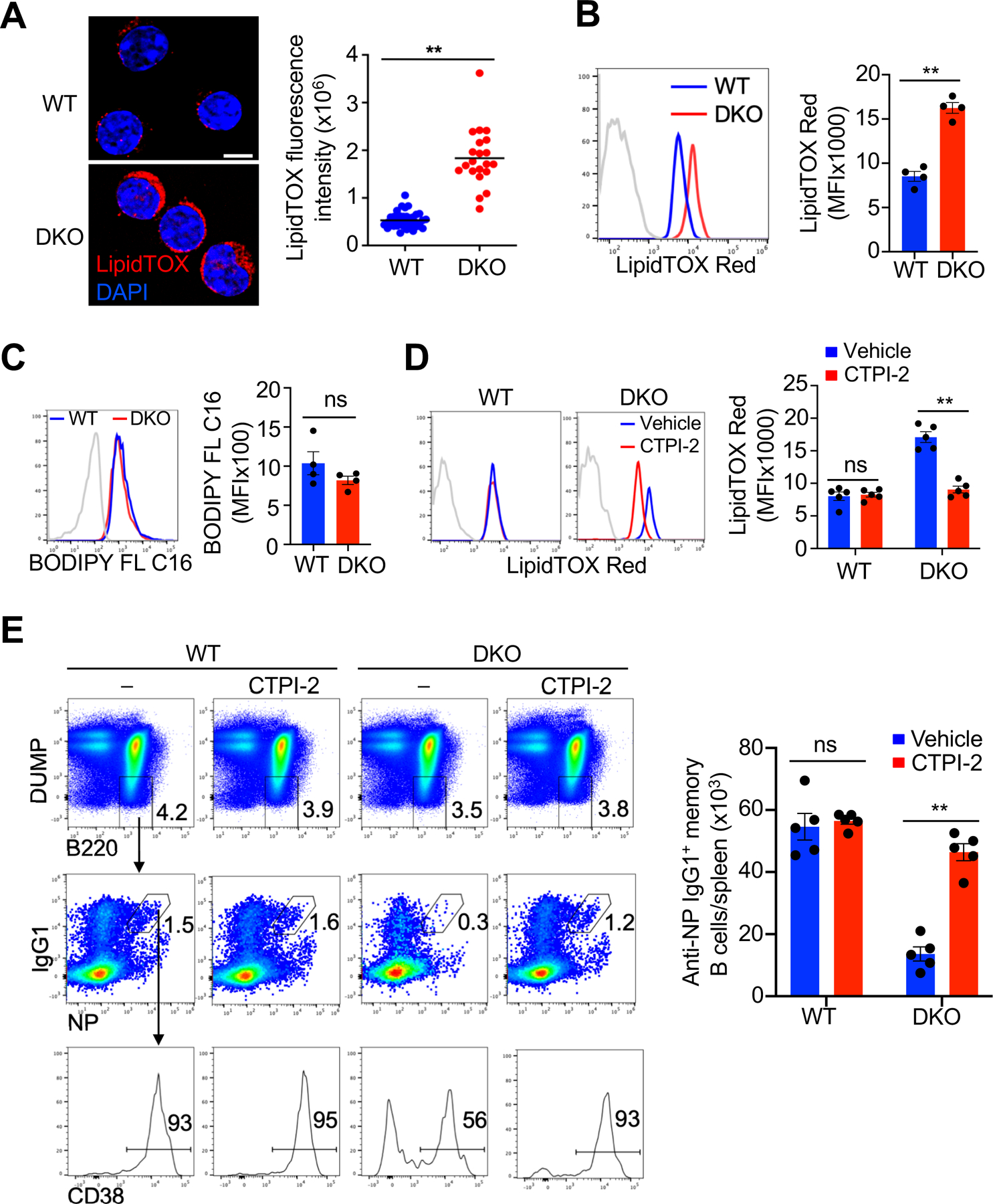
Accumulation of lipid droplets in Nix−∕−Bnip3−∕− memory B cells. (A) Determination of LipidTOX Deep Red staining in WT and DKO memory B cells by deconvolution microscopy (WT, n=21 cells; DKO, n=40 cells). Nuclei were counterstained with DAPI. Scale bar, 5 µm. Data are representative of two independent experiments and are presented as mean ± s.e.m. **P < 0.01. (B) Flow cytometry analyses of LipidTOX Red staining in WT and DKO GC and memory B cells (n=4 mice per group). Dashed line: isotype control. Data are representative of five independent experiments. **P < 0.01. (C) Uptake of BODIPY FL C16 by WT and DKO memory B cells was analyzed by flow cytometry (n=4 mice per group). Data are representative of two independent experiments. ns, not statistically significant. (D, E) WT and DKO mice were immunized with NP-KLH and treated with vehicle or CTPI-2 every day starting from day 14 post-immunization until day 28 post-immunization. LipidTOX Red staining (D) and quantification of NP-specific memory B cells (E) were determined by flow cytometry (n=5 mice per group). Data are representative of two independent experiments and are presented as mean ± s.e.m. *P < 0.05; **P < 0.01; ns, not statistically significant.
It has been shown that increases in mitochondrial content can stimulate de novo fatty acid synthesis (64, 67). We observed that the loss of Nix and Bnip3 resulted in elevated expression of Slc25a1 (Fig. 5C). Mitochondrial accumulation in Nix–/–Bnip3−⁄− memory B cells may lead to increased export of citrate via Slc25a1 to fuel excessive de novo fatty acid synthesis. Therefore, we next examined whether blocking mitochondrial citrate export with a highly specific Slc25a1 inhibitor, CTPI-2 (68), could rescue Nix–/–Bnip3−⁄− memory B cells. We found that administration of CTPI-2 reduced lipid droplets in Nix–/–Bnip3−⁄− memory B cells, and rescued their survival in vivo (Fig. 6, D and E). These data are consistent with the possibility that accumulation of active mitochondria leads to abnormal de novo fatty acid synthesis and lipid accumulation, resulting in the loss of Nix–/–Bnip3−⁄− memory B cells.
Increased necroptosis signaling in Nix−/−Bnip3−/− IgG memory B cells
We next investigated mechanisms for the loss of Nix–/–Bnip3−⁄− memory B cells. In contrast to adipocytes, non-adipose cells typically have a limited capacity to store excess fatty acid. Fatty acid overload may result in cellular dysfunction or cell death, a phenomenon known as lipotoxicity (73, 74). Therefore, we next examined whether Nix–/–Bnip3−⁄− memory B cells underwent accelerated apoptosis. We have previously shown that in vitro culture with reduced serum induces caspase activation and apoptosis in GC but not memory B cells (11, 75). We first examined caspase-9, the initiator caspase in the mitochondrion-dependent apoptosis pathway (76) in Nix–/–Bnip3−⁄− GC and memory B cells. While caspase-9 could be activated in Nix–/–Bnip3−⁄− GC B cells and wild type controls, we did not find significant caspase-9 activation in wild type or Nix–/–Bnip3−⁄− memory B cells by intracellular staining of the active cleaved form of caspase-9 (Fig. 7A, upper panels). Similarly, a downstream effector, caspase-3, could be activated in GC B cells but not memory B cells (Fig. 7A, lower panels). Moreover, Nix/Bnip3-deficiency did not cause increased caspase-3 activation in Nix–/–Bnip3−⁄− memory B cells, as shown by intracellular staining of the active cleaved form of caspase-3 (Fig. 7A, lower panels). In addition, anti-apoptotic Bcl-2 that is highly expressed in memory B cells (11, 77), was also comparable between WT and Nix–/–Bnip3−⁄− memory B cells (Fig. 7B). These data suggest that Nix–/–Bnip3−⁄− memory B cells do not have elevated apoptosis signaling.
FIGURE 7.
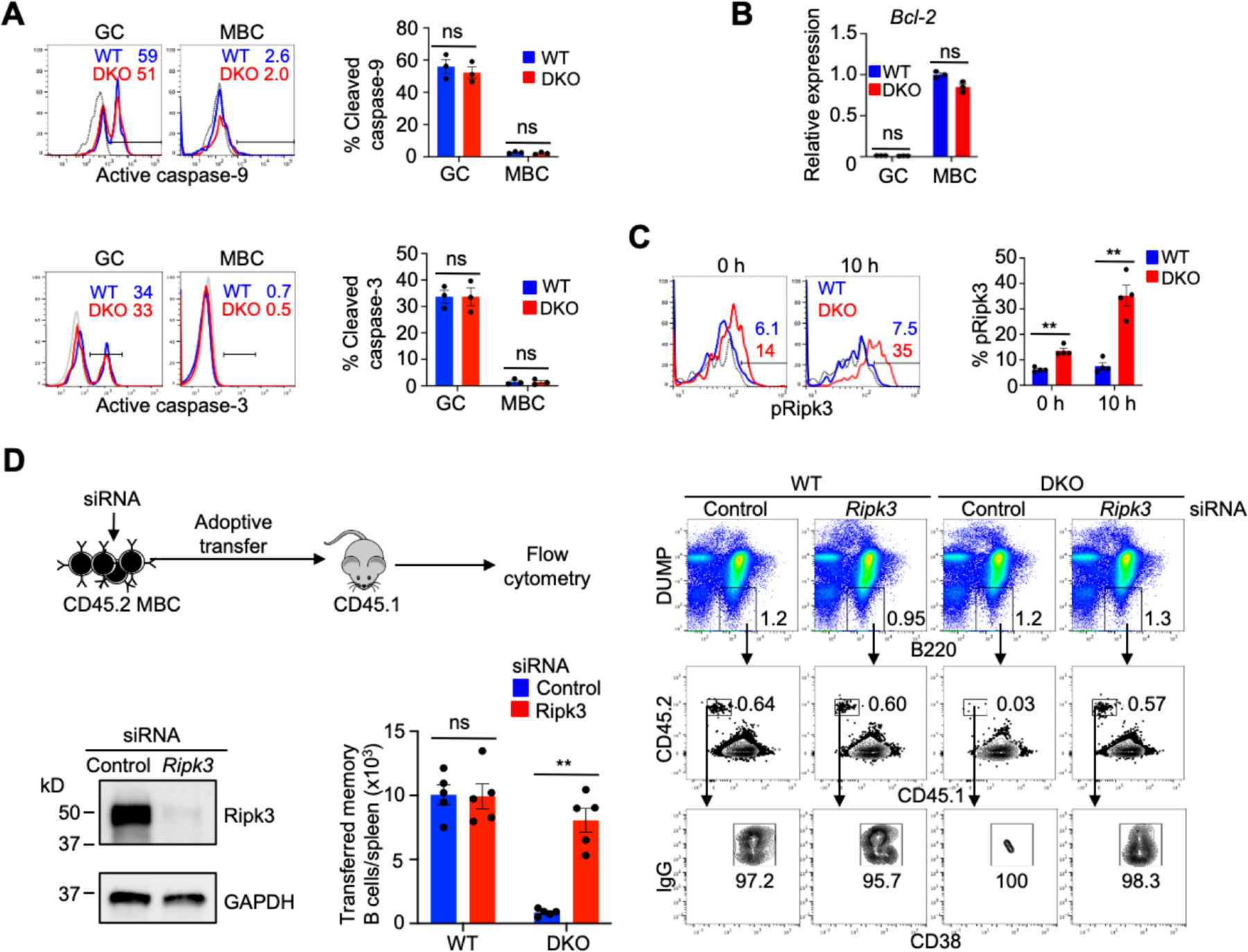
Nix−∕−Bnip3−∕− memory B cells show increased necroptosis but not apoptosis signaling. (A) Flow cytometry analysis of active cleaved forms of caspase-9 (upper panels) and caspase-3 (lower panels) in GC and memory B cells from spleens of WT or DKO mice after 6 h of in vitro culture under a low serum (1% fetal bovine serum) condition (n=3 mice per group). Dashed line: isotype control. Data are representative of two independent experiments. ns, not statistically significant. (B) qRT-PCR analysis for Bcl2 in GC and memory B cells sorted from pooled spleens of WT or DKO mice (n=3 mice/group). Data are representative of two independent experiments and are presented as mean ± s.e.m. ns, not statistically significant. (C) Flow cytometry analysis of intracellular staining for phospho-Ripk3 (Thr231/Ser232) in memory B cells from spleens of WT and DKO mice (n=4 mice per group) after 0 or 10 h of in vitro culture in the presence of zVAD-FMK. Dashed line: isotype control. Data are representative of two independent experiments and are presented as mean ± s.e.m. **P < 0.01. (D) DUMP-IgG+CD38+ Memory B cells from WT or DKO mice immunized with SRBCs were sorted and mixed with CD45.1 naïve B cells (1:10) as carriers. After transfected with siRNA targeting Ripk3 or control siRNA, the cells were adoptively transferred into recipient CD45.1 mice (5×104 memory B cells/mouse). DUMP− cells were gated to analyze CD45.2+CD45.1–IgG+CD38+ transferred memory B cells in the spleens of recipient mice by flow cytometry (n=5 mice/group). Representative flow cytometry analyses are show in the right panel. The transfected cells were also stimulated in vitro with anti-CD40, anti-CD180 and IL-4 for 2 days and used for Western blot analyses. Data are representative of two independent experiments and are presented as mean ± s.e.m. **P < 0.01; ns, not statistically significant.
We have previously observed that caspase-9-deficient GC B cells spontaneously undergo necroptotic cell death, but can be rescued by deletion of Ripk3 (75). It has been reported that accumulation of fatty acids from de novo fatty acid synthesis induces lipotoxicity through necroptosis (78, 79). Therefore, we tested whether Nix–/–Bnip3−⁄− memory B cells that displayed increases in de novo fatty acid synthesis also underwent necroptosis. It has been shown that activation of Ripk3 by phosphorylation is required for necroptotic signaling (80). Inhibition of caspases can typically increase necroptosis signaling (81, 82). We therefore cultured wild type and memory B cells in the presence of a pan-caspase inhibitor, zVAD-FMK. We found that Nix–/–Bnip3−⁄− memory B cells displayed increased phosphorylation of Ripk3, which was enhanced after culture with zVAD (Fig. 7C), suggesting that Nix–/–Bnip3−⁄− memory B cells display active necroptosis signaling.
We next determined whether the loss of Nix–/–Bnip3−⁄− memory B cells is due to necroptosis. It has been shown that memory B cell formation during GC responses peaks at days 6–8 after immunization (42). We have observed that the formation of memory B cells at 2 weeks are not decreased in the absence of Nix/Bnip3 (Fig. 2A). To determine whether these cells could be rescued by inhibiting cell death, we used memory B cells at 2 weeks after immunization before significant cell loss is expected to take place. We purified memory B cells from DKO mice and wild type controls after immunization with sheep red blood cells (SRBCs), which have been shown to induce robust GC and memory B cell generation (83). Sorted memory B cells were mixed with naïve B cells as carriers and transfected with siRNAs targeting Ripk3, and then transferred into congenic CD45.1+ recipient mice. Seven days later, we examined the CD45.2+CD45.1– transferred Nix–/–Bnip3−⁄− memory B cells in recipient mice (Fig. 7D). Consistent with the observation of decreased survival of transferred NP-specific Nix–/–Bnip3−⁄− memory B cells (Fig. 2C), SRBC-induced Nix–/–Bnip3−⁄− memory B cells transfected with non-targeting siRNA also showed reduced survival in vivo compared to wild type controls (Fig. 7D). Interestingly, transfection with Ripk3 siRNA rescued Nix–/–Bnip3−⁄− memory B cells (Fig. 7D). In comparison, Ripk3 siRNA did not have any effect on wild type memory B cells (Fig. 7D). These data indicate that the loss of Nix–/–Bnip3−⁄− memory B cells involves Ripk3-dependent necroptosis.
Discussion
Here we demonstrate that Nix- and Bnip3-mediated mitochondrial autophagy (also called mitophagy) plays an important role in the protection of IgG+ memory B cells by maintaining mitochondrial homeostasis. Deficiency in Nix- and Bnip3-dependent mitochondrial autophagy leads to mitochondria accumulation and increases in oxidative phosphorylation and fatty acid synthesis in memory B cells, resulting in increased lipid droplets in the cytoplasm and cell loss. Silencing of the necroptosis mediator Ripk3 protected Nix–/–Bnip3−⁄− memory B cells, indicating that the loss of these cells is due to necroptosis. These results suggest that Nix- and Bnip3-mediated mitochondrial autophagy is required for maintaining mitochondrial homeostasis, thereby limiting excessive glucose and fatty acid metabolism to prevent lipotoxicity and cell death. Our study suggests that restriction of cellular metabolism by mitochondrial autophagy is critical for the protection of quiescence and longevity of memory B cells.
Our results indicate that Nix and Bnip3 are upregulated in IgG+ memory B cells and may therefore be uniquely important for the maintenance of these cells. In CD8+ T cells, Nix is upregulated after antigen stimulation, while Bnip3 is downregulated (33). Nix but not Bnip3 plays an important role in the protection of CD8+ effector memory T cell precursors from cell death by clearing damaged mitochondria (33). In NK cells, Nix and Bnip3 are both upregulated during the effector to memory transition (30). Loss of either Nix or Bnip3 leads to the loss of memory NK cell precursors (30). Nix and Bnip3 can mediate mitochondrial autophagy independently of Pink1 and Parkin (36–39). Nix/Bnip3 but not Pink1/Parkin are upregulated in memory B cells, suggesting a unique role for Nix and Bnip3-mediated mitochondrial autophagy in memory B cells. Our results indicate that Nix and Bnip3 are functionally overlapping in the regulation of mitochondrial homeostasis in memory B cells.
Results from adoptive transfer experiments showed that Nix–/–Bnip3–/– IgG+ memory B cells had a poor survival potential after adoptive transfer. We have previously shown that Atg7-dependent canonical autophagy removes damaged mitochondria to protect memory B cells from ROS-induced cell death (11, 24). Although the loss of Nix and Bnip3 causes increased mitochondrial ROS in memory B cells, inhibition of ROS did not rescue these cells. This indicates that Nix/Bnip3 and Atg7 regulate mitophagy with partially overlapping and partially distinct mechanisms, and additional mechanisms other than increased mitochondrial ROS are likely involved in the loss of Nix–/–Bnip3–/– memory B cells. It has been reported that Atg5/Atg7-independent noncanonical autophagy could be induced in mammalian cells in response to some stress stimuli (84, 85). While Atg5 and Atg7 are not essential for mitophagy in red blood cells (84, 86, 87), Nix is required for this process (31, 84). Therefore, Nix likely interacts with both canonical and non-canonical autophagy pathways to regulate mitophagy in memory B cells. Since Nix–/–Bnip3–/– memory B cells have increased mitochondrial membrane potential and mitochondrial mass, Nix- and Bnip3-mediated mitochondrial autophagy may play an especially important role in maintaining mitochondrial homeostasis by preventing the accumulation of actively respiring mitochondria, thereby suppressing lipid accumulation and preventing lipotoxicity in memory B cells.
It has been well established that metabolic switches are important for supporting the development of memory T cells (26). In contrast, studies of metabolic programming in memory B cells are still scarce. Interestingly, it has been shown that AMPKa1 that plays diverse roles in the regulation of metabolism, autophagy and cell growth differentiation, is important for protecting the longevity of NP+IgD–GL7–CD38+ memory B cells, which include unswitched IgM+ and switched IgG+ memory B cells (29, 88). Loss of AMPKa1 resulted in decreased mitochondrial respiratory activity, decreased mitophagy, and increased lipid peroxidation (29, 88). These important studies of signaling nodes in metabolism pathways open a very promising avenue of investigation regarding metabolic regulation of mitophagy and memory B cells. Our research comes from a different angle to study how mitophagy signaling molecules regulate metabolic functions to support the persistence of IgG+ memory B cells. Our results support and extend from these previous studies and implicate a possible crosstalk between AMPKa1 and Nix/Bnip3 mediated mitochondrial autophagy. Nix/Bnip3 have been shown to mediate mitophagy via Atg5/7-dependent canonical autophagy, as well as Atg5/7-independent and Ulk1-dependent noncanonical autophagy in several tissues. Future studies will be required to determine whether Nix/Bnip3 mediates mitochondrial autophagy via noncanonical autophagy in memory B cells, and if so, whether this noncanonical mitophagy pathway could be regulated by AMPK in memory B cells. Studies from different angles will enable the elucidation of the role of metabolic programming in the development and long-term maintenance of memory B cells.
While memory B cells exhibited increased lipid accumulation and impaired survival in the absence of Nix and Bnip3, we did not observe these effects in Nix–/–Bnip3−⁄− GC B cells. This may be explained by metabolic and lifespan differences between GC and memory B cells. GC B cells have a high rate of fatty acid oxidation (89), suggesting that they are resistant to lipid accumulation. Since GC B cells are short-lived, they may not persist long enough to experience deleterious consequences from the absence of Nix and Bnip3. We observed that memory B cells contained more lipid droplets than GC B cells despite smaller size, suggesting a tendency to store, rather than immediately oxidize fatty acids. In addition to the maintenance of protective memory B cells against infections (11), autophagy is also required for the persistence of autoantigen-specific memory B cells and autoantibody responses (25). Interestingly, autophagy has also been shown to be important for the protection of long-lived plasma cells and autoantibody responses (23, 90). It will be interesting to determine whether Nix and Bnip3-dependent mitochondrial autophagy regulates mitochondrial homeostasis, fatty acid synthesis or other metabolic functions in long-lived and autoimmune memory B cells and plasma cells, and could be targeted to regulate autoimmune responses.
De novo fatty acid synthesis takes place in the cytosol and requires the conversion of citrate to acetyl-CoA by Acly (64, 68). Citrate is produced by active mitochondria as part of the Krebs cycle, and can be redirected to the cytosol via the mitochondrial citrate transporter, Slc25a1 (68). Thus, increased numbers of viable mitochondria could represent a greater source of citrate to drive de novo fatty acid synthesis. Indeed, isolated mitochondria can be used to replace citrate in soluble enzyme systems in order to stimulate de novo fatty acid synthesis (67). In support of this possibility, we found that a specific inhibitor for Slc25a1, CTPI-2 (68), prevented lipid accumulation and rescued Nix−/−Bnip3−∕− memory B cells in vivo. Our data suggest that elevated de novo fatty acid synthesis leads to lipotoxicity-induced cell death in IgG+ memory B cells. Interestingly, an integrative transcriptome and chromatin study reveals that the development of memory B cells is accompanies by the changes in the landscape of gene expression, including FASN, ACACA and ACLY in the metabolic pathways (91). It is possible that these metabolism-related genes are important for the maintenance of memory B cells, but a delicate balance in their expression is critical for the control of metabolic quiescence in these cells.
Lipotoxicity has been associated with apoptosis in multiple cell types (73). However, we have observed that memory B cells are resistant to caspase-dependent apoptosis (11). We did not detect increases in caspase-9 activation in Nix−/−Bnip3−∕− memory B cells, suggesting that alternative cell death mechanisms account for their loss. We have previously found that caspase-9 deficiency leads to increased necroptosis in GC B cells (75). It has been shown that inhibition of de novo fatty acid synthesis can protect cells from necroptosis (79). Interestingly, we found that Nix−/−Bnip3−∕− memory B cells displayed increases in necroptosis signaling. Silencing of Ripk3 rescued Nix−/−Bnip3−∕− memory B cells in vivo. Fatty acid accumulation due to de novo fatty acid synthesis has been reported to induce necroptosis (78, 79). Our results indicate that lipotoxicity due to increased de novo fatty acid synthesis induces necroptosis in Nix−/−Bnip3−∕− IgG+ memory B cells.
Memory B cells comprise a heterogeneous population of metabolically quiescent long-lived B cells, which include not only switched IgG memory B cells, but also unswitched IgM memory B cells memory (4, 8, 9). IgM memory B cells play a particular important role in recall responses against future similar pathogens (92). It will be interesting to determine whether the survival and maintenance of unswitched IgM memory B cells may be regulated by different mechanisms from their switched counterparts. Whether Nix and Bnip3 regulates the maintenance of unswitched IgM memory B cells through affecting mitochondrial homeostasis and lipid metabolism will be important to study in the future.
Our work reveals a mechanism for the protection of quiescence and longevity of IgG+ memory B cells by mitochondrial autophagy. Accumulation of mitochondria due to defective mitochondrial autophagy leads to increased oxidative phosphorylation to fuel excessive de novo synthesis of fatty acids, leading to memory B cell death through necroptosis. These findings indicate that mitochondrial autophagy is crucial for the metabolic reprogramming required to protect the long-term survival of memory B cells. Our results support the role for mitochondrial autophagy in the protection of immunological memory by the maintenance of mitochondrial homeostasis and restriction of metabolic functions in memory B cells.
Supplementary Material
Key points:
Nix and Bnip3 regulate mitochondrial homeostasis by autophagy in memory B cells.
Mitochondrial autophagy is required for metabolic quiescence in memory B cells.
Uncontrolled metabolic function increases cell death in memory B cells.
Acknowledgments
We thank Gerald Dorn for Nixf/f and Bnip3–/– mice.
This study was supported by funding from the NIH (R01AI116644 and R01AI123221 to J.W), the Cancer Prevention and Research Institute of Texas (RP160384 to J.W. and M.C.).
Abbreviations used in this article:
- DKO
double knockout
- GC
germinal center
- MBC
memory B cells
- NP-KLH
NP–keyhole limpet hemocyanin
- SRBC
sheep red blood cells
- WT
wild-type
References
- 1.Pulendran B, Ahmed R. 2011. Immunological mechanisms of vaccination. Nat Immunol 12: 509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. 1999. Mature T lymphocyte apoptosis--immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol 17: 221–253. [DOI] [PubMed] [Google Scholar]
- 3.McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. 2011. Molecular programming of B cell memory. Nat Rev Immunol 12: 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurosaki T, Kometani K, Ise W. 2015. Memory B cells. Nat Rev Immunol 15: 149–159. [DOI] [PubMed] [Google Scholar]
- 5.Shlomchik MJ, Weisel F. 2012. Germinal center selection and the development of memory B and plasma cells. Immunol Rev 247: 52–63. [DOI] [PubMed] [Google Scholar]
- 6.Laidlaw BJ, Craft JE, Kaech SM. 2016. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol 16: 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Omilusik KD, Goldrath AW. 2019. Remembering to remember: T cell memory maintenance and plasticity. Curr Opin Immunol 58: 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McHeyzer-Williams LJ, Milpied PJ, Okitsu SL, McHeyzer-Williams MG. 2015. Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat Immunol 16: 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. 2011. Different B cell populations mediate early and late memory during an endogenous immune response. Science 331: 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, Pai J, Oliveira TY, Wang Q, Escolano A, Medina-Ramirez M, Sanders RW, Nussenzweig MC. 2017. The microanatomic segregation of selection by apoptosis in the germinal center. Science 358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, Corry DB, Kheradmand F, Wang J. 2014. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med 20: 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maruyama M, Lam KP, Rajewsky K. 2000. Memory B-cell persistence is independent of persisting immunizing antigen. Nature 407: 636–642. [DOI] [PubMed] [Google Scholar]
- 13.Clybouw C, Fischer S, Auffredou MT, Hugues P, Alexia C, Bouillet P, Raphael M, Leca G, Strasser A, Tarlinton DM, Vazquez A. 2011. Regulation of memory B-cell survival by the BH3-only protein Puma. Blood 118: 4120–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer SF, Bouillet P, O’Donnell K, Light A, Tarlinton DM, Strasser A. 2007. Proapoptotic BH3-only protein Bim is essential for developmentally programmed death of germinal center-derived memory B cells and antibody-forming cells. Blood 110: 3978–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morishita H, Mizushima N. 2019. Diverse Cellular Roles of Autophagy. Annu Rev Cell Dev Biol 35: 453–475. [DOI] [PubMed] [Google Scholar]
- 16.McLeod IX, Jia W, He YW. 2012. The contribution of autophagy to lymphocyte survival and homeostasis. Immunol Rev 249: 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munz C. 2020. Autophagy in immunity. Prog Mol Biol Transl Sci 172: 67–85. [DOI] [PubMed] [Google Scholar]
- 18.Clarke AJ, Simon AK. 2019. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol 19: 170–183. [DOI] [PubMed] [Google Scholar]
- 19.Sandoval H, Kodali S, Wang J. 2018. Regulation of B cell fate, survival, and function by mitochondria and autophagy. Mitochondrion 41: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murera D, Arbogast F, Arnold J, Bouis D, Muller S, Gros F. 2018. CD4 T cell autophagy is integral to memory maintenance. Sci Rep 8: 5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, Watson AS, Cerundolo V, Townsend AR, Klenerman P, Simon AK. 2014. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife 3: e03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, Green DR, Jones DP, Virgin HW, Ahmed R. 2014. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol 15: 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, Fagioli C, Merlini A, Mariani E, Pasqualetto E, Orfanelli U, Ponzoni M, Sitia R, Casola S, Cenci S. 2013. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol 14: 298–305. [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Kodali S, Jang A, Kuai L, Wang J. 2015. Requirement for autophagy in the long-term persistence but not initial formation of memory B cells. J Immunol 194: 2607–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang A, Sharp R, Wang JM, Feng Y, Wang J, Chen M. 2021. Dependence on autophagy for autoreactive memory B cells in the development of pristane-induced lupus. Front. Immunol 12: 701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. 2012. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36: 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, Chen Q, Huang SC, O’Neill CM, Edelson BT, Pearce EJ, Sesaki H, Huber TB, Rambold AS, Pearce EL. 2016. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 166: 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. 2009. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460: 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brookens SK, Cho SH, Basso PJ, Boothby MR. 2020. AMPKalpha1 in B Cells Dampens Primary Antibody Responses yet Promotes Mitochondrial Homeostasis and Persistence of B Cell Memory. J Immunol 205: 3011–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Sullivan TE, Johnson LR, Kang HH, Sun JC. 2015. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 43: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. 2008. Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J, Dorn GW 2nd, Brady MJ, Macleod KF. 2012. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol 32: 2570–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta SS, Sharp R, Hofferek C, Kuai L, Dorn GW 2nd, Wang J, Chen M. 2019. NIX-Mediated Mitophagy Promotes Effector Memory Formation in Antigen-Specific CD8(+) T Cells. Cell Rep 29: 1862–1877 e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta SS, Wang J, Chen M. 2020. Metabolic Reprogramming in CD8(+) T Cells During Acute Viral Infections. Front Immunol 11: 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narendra D, Tanaka A, Suen DF, Youle RJ. 2008. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Ney PA. 2009. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 16: 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novak I, Dikic I. 2011. Autophagy receptors in developmental clearance of mitochondria. Autophagy 7: 301–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pickles S, Vigie P, Youle RJ. 2018. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol 28: R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macleod KF. 2020. Mitophagy and Mitochondrial Dysfunction in Cancer. Annual Review of Cancer Biology 4: 41–60. [Google Scholar]
- 40.Dunn KW, Kamocka MM, McDonald JH. 2011. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300: C723–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jash A, Wang Y, Weisel FJ, Scharer CD, Boss JM, Shlomchik MJ, Bhattacharya D. 2016. ZBTB32 Restricts the Duration of Memory B Cell Recall Responses. J Immunol 197: 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. 2016. A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity 44: 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Brito Monteiro L, Davanzo GG, de Aguiar CF, Correa da Silva F, Andrade JR, Campos Codo A, Silva Pereira JAD, Freitas LP, Moraes-Vieira PM. 2020. M-CSF- and L929-derived macrophages present distinct metabolic profiles with similar inflammatory outcomes. Immunobiology 225: 151935. [DOI] [PubMed] [Google Scholar]
- 44.Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q. 2020. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front Cell Dev Biol 8: 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, Ney PA. 2012. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy 8: 1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teresak P, Lapao A, Subic N, Boya P, Elazar Z, Simonsen A. 2021. Regulation of PRKN-independent mitophagy. Autophagy 10.1080/15548627.2021.1888244: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montano C, Feigenbaum L, Wilson P, Janz S, Papavasiliou FN, Casellas R. 2007. Regulation of AID expression in the immune response. J Exp Med 204: 1145–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cariappa A, Boboila C, Moran ST, Liu H, Shi HN, Pillai S. 2007. The recirculating B cell pool contains two functionally distinct, long-lived, posttransitional, follicular B cell populations. J Immunol 179: 2270–2281. [DOI] [PubMed] [Google Scholar]
- 49.Wei J, Raynor J, Nguyen TL, Chi H. 2017. Nutrient and Metabolic Sensing in T Cell Responses. Front Immunol 8: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zuccarino-Catania G, Shlomchik M. 2015. Adoptive Transfer of Memory B Cells. Bio Protoc 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McHeyzer-Williams LJ, Cool M, McHeyzer-Williams MG. 2000. Antigen-specific B cell memory: expression and replenishment of a novel b220(−) memory b cell compartment. J Exp Med 191: 1149–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding WX, Yin XM. 2012. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 393: 547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. 2004. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117: 2805–2812. [DOI] [PubMed] [Google Scholar]
- 55.Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wellen KE, Thompson CB. 2010. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 40: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jang KJ, Mano H, Aoki K, Hayashi T, Muto A, Nambu Y, Takahashi K, Itoh K, Taketani S, Nutt SL, Igarashi K, Shimizu A, Sugai M. 2015. Mitochondrial function provides instructive signals for activation-induced B-cell fates. Nat Commun 6: 6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, Dufour E, Spelbrink JN, Weinberg SE, Zhao Y, DeBerardinis RJ, Chandel NS. 2016. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol Cell 61: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akkaya M, Traba J, Roesler AS, Miozzo P, Akkaya B, Theall BP, Sohn H, Pena M, Smelkinson M, Kabat J, Dahlstrom E, Dorward DW, Skinner J, Sack MN, Pierce SK. 2018. Second signals rescue B cells from activation-induced mitochondrial dysfunction and death. Nat Immunol 19: 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Costa R, Peruzzo R, Bachmann M, Monta GD, Vicario M, Santinon G, Mattarei A, Moro E, Quintana-Cabrera R, Scorrano L, Zeviani M, Vallese F, Zoratti M, Paradisi C, Argenton F, Brini M, Cali T, Dupont S, Szabo I, Leanza L. 2019. Impaired Mitochondrial ATP Production Downregulates Wnt Signaling via ER Stress Induction. Cell Rep 28: 1949–1960 e1946. [DOI] [PubMed] [Google Scholar]
- 61.van der Windt GJ, Chang CH, Pearce EL. 2016. Measuring Bioenergetics in T Cells Using a Seahorse Extracellular Flux Analyzer. Curr Protoc Immunol 113: 3 16B 11–13 16B 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr., Alt FW, Kahn CR, Verdin E. 2010. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464: 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. 2002. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 51: 2420–2425. [DOI] [PubMed] [Google Scholar]
- 64.Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, Kluger Y, Maher LJ 3rd, Rabinowitz J, Craft J, Flavell RA. 2019. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571: 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Witkowski A, Thweatt J, Smith S. 2011. Mammalian ACSF3 protein is a malonyl-CoA synthetase that supplies the chain extender units for mitochondrial fatty acid synthesis. J Biol Chem 286: 33729–33736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo YM, Frey AJ, Meurs N, Viola JM, Blair IA, Weljie AM, Metallo CM, Snyder NW, Andrews AJ, Wellen KE. 2016. ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep 17: 1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bhaduri AM, Srere PA. 1964. Mitochondrial Stimulation of Fatty Acid Biosynthesis. J Biol Chem 239: 1357–1363. [PubMed] [Google Scholar]
- 68.Tan M, Mosaoa R, Graham GT, Kasprzyk-Pawelec A, Gadre S, Parasido E, Catalina-Rodriguez O, Foley P, Giaccone G, Cheema A, Kallakury B, Albanese C, Yi C, Avantaggiati ML. 2020. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ 27: 2143–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rudolph MC, Monks J, Burns V, Phistry M, Marians R, Foote MR, Bauman DE, Anderson SM, Neville MC. 2010. Sterol regulatory element binding protein and dietary lipid regulation of fatty acid synthesis in the mammary epithelium. Am J Physiol Endocrinol Metab 299: E918–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rosas-Ballina M, Guan XL, Schmidt A, Bumann D. 2020. Classical Activation of Macrophages Leads to Lipid Droplet Formation Without de novo Fatty Acid Synthesis. Front Immunol 11: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chhabra R, Nanjundan M. 2020. Lysophosphatidic acid reverses Temsirolimus-induced changes in lipid droplets and mitochondrial networks in renal cancer cells. PLoS One 15: e0233887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karsenty J, Helal O, de la Porte PL, Beauclair-Deprez P, Martin-Elyazidi C, Planells R, Storch J, Gastaldi M. 2009. I-FABP expression alters the intracellular distribution of the BODIPY C16 fatty acid analog. Mol Cell Biochem 326: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., Ory DS, Schaffer JE. 2003. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 100: 3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jimenez-Preitner M, Berney X, Uldry M, Vitali A, Cinti S, Ledford JG, Thorens B. 2011. Plac8 is an inducer of C/EBPbeta required for brown fat differentiation, thermoregulation, and control of body weight. Cell Metab 14: 658–670. [DOI] [PubMed] [Google Scholar]
- 75.Zhang J, Kodali S, Chen M, Wang J. 2020. Maintenance of Germinal Center B Cells by Caspase-9 through Promotion of Apoptosis and Inhibition of Necroptosis. J Immunol 205: 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Green DR. 2005. Apoptotic pathways: ten minutes to dead. Cell 121: 671–674. [DOI] [PubMed] [Google Scholar]
- 77.Resch U, Schichl YM, Winsauer G, Gudi R, Prasad K, de Martin R. 2009. Siva1 is a XIAP-interacting protein that balances NFkappaB and JNK signalling to promote apoptosis. J Cell Sci 122: 2651–2661. [DOI] [PubMed] [Google Scholar]
- 78.Khan MJ, Rizwan Alam M, Waldeck-Weiermair M, Karsten F, Groschner L, Riederer M, Hallstrom S, Rockenfeller P, Konya V, Heinemann A, Madeo F, Graier WF, Malli R. 2012. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J Biol Chem 287: 21110–21120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parisi LR, Li N, Atilla-Gokcumen GE. 2017. Very Long Chain Fatty Acids Are Functionally Involved in Necroptosis. Cell Chem Biol 24: 1445–1454 e1448. [DOI] [PubMed] [Google Scholar]
- 80.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. 2009. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Preyat N, Rossi M, Kers J, Chen L, Bertin J, Gough PJ, Le Moine A, Rongvaux A, Van Gool F, Leo O. 2016. Intracellular nicotinamide adenine dinucleotide promotes TNF-induced necroptosis in a sirtuin-dependent manner. Cell Death Differ 23: 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sawai H. 2014. Characterization of TNF-induced caspase-independent necroptosis. Leuk Res 38: 706–713. [DOI] [PubMed] [Google Scholar]
- 83.McAllister EJ, Apgar JR, Leung CR, Rickert RC, Jellusova J. 2017. New Methods To Analyze B Cell Immune Responses to Thymus-Dependent Antigen Sheep Red Blood Cells. J Immunol 199: 2998–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. 2009. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461: 654–658. [DOI] [PubMed] [Google Scholar]
- 85.Codogno P, Mehrpour M, Proikas-Cezanne T. 2011. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol 13: 7–12. [DOI] [PubMed] [Google Scholar]
- 86.Matsui M, Yamamoto A, Kuma A, Ohsumi Y, Mizushima N. 2006. Organelle degradation during the lens and erythroid differentiation is independent of autophagy. Biochem Biophys Res Commun 339: 485–489. [DOI] [PubMed] [Google Scholar]
- 87.Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, Shimizu S. 2014. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun 5: 4004. [DOI] [PubMed] [Google Scholar]
- 88.Brookens SK, Boothby MR. 2021. AMPK Metabolism in the B Lineage Modulates Humoral Responses. Immunometabolism 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weisel FJ, Mullett SJ, Elsner RA, Menk AV, Trivedi N, Luo W, Wikenheiser D, Hawse WF, Chikina M, Smita S, Conter LJ, Joachim SM, Wendell SG, Jurczak MJ, Winkler TH, Delgoffe GM, Shlomchik MJ. 2020. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat Immunol 21: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arnold J, Murera D, Arbogast F, Fauny JD, Muller S, Gros F. 2016. Autophagy is dispensable for B-cell development but essential for humoral autoimmune responses. Cell Death Differ 23: 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moroney JB, Vasudev A, Pertsemlidis A, Zan H, Casali P. 2020. Integrative transcriptome and chromatin landscape analysis reveals distinct epigenetic regulations in human memory B cells. Nat Commun 11: 5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weill JC, Reynaud CA. 2020. IgM memory B cells: specific effectors of innate-like and adaptive responses. Curr Opin Immunol 63: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.