

Abstract
Intermittent hypoxia (IH) is a hallmark manifestation of obstructive sleep apnea (OSA). Rodents treated with IH exhibit hypertension. Hypoxia-inducible factor (HIF)-1-dependent transcriptional activation of NADPH oxidases (Nox) and the resulting increase in reactive oxygen species (ROS) levels is a major molecular mechanism underlying IH/OSA-induced hypertension. Jumanji C (JmjC)-containing histone lysine demethylases (JmjC-KDMs) are coactivators of HIF-1-dependent transcriptional activation. In the present study, we tested the hypothesis that JmjC-KDMs are required for IH-evoked HIF-1 transcriptional activation of Nox4 and the ensuing hypertension. Studies were performed on pheochromocytoma (PC)12 cells and rats. IH increased KDM6B protein and enzyme activity in PC12 cells in an HIF-1-independent manner as evidenced by unaltered KDM6B activation by IH in HIF-1α shRNA-treated cells. Cells treated with IH showed increased HIF-1-dependent Nox4 transcription as indicated by increased HIF-1α binding to hypoxia-responsive element (HRE) sequence of the Nox4 gene promoter demonstrated by chromatin immunoprecipitation (ChiP) assay. Pharmacological blockade of KDM6B with GSKJ4, a specific KDM6 inhibitor, or genetic silencing of KDM6B with shRNA abolished IH-induced Nox4 transcriptional activation by blocking HIF-1α binding to the promoter of the Nox4 gene. Treating IH-exposed rats with GSKJ4 showed: 1) absence of KDM6B activation and HIF-1-dependent Nox4 transcription in the adrenal medullae, and 2) absence of elevated plasma catecholamines and hypertension. Collectively, these findings indicate that KDM6B functions as a coactivator of HIF-1-mediated Nox4 transactivation and demonstrates a hitherto uncharacterized role for KDMs in IH-induced hypertension by HIF-1.
Keywords: hypoxia-inducible factor, intermittent hypoxia, lysine demethylases, NADPH oxidase, sleep apnea
INTRODUCTION
Obstructive sleep apnea (OSA) is a widespread disorder of breathing affecting nearly 30% of adult males and 15% of adult females (1, 2). OSA is characterized by periodic interruption of airflow during sleep due to partial or complete collapse of the upper airway. Patients with OSA exhibit several comorbidities including hypertension (3–5), type 2 diabetes (T2D) (6–8), and cognitive decline (9) to name a few. Repeated disruption of air flow leads to intermittent hypoxia (IH), mild hypercapnia, and arousals from sleep. However, rodents treated with IH alone patterned after blood O2 profiles during OSA exhibit many of the OSA comorbidities including hypertension (10).
Rodent and cell culture models provided insights into the molecular mechanisms underlying IH-induced hypertension. IH is a potent activator of the hypoxia-inducible factor (HIF)-1 (11–13). Cell cultures and rodents treated with IH show HIF-1-dependent transcriptional activation of NADPH oxidases (Nox) (14–16), resulting in increased levels of reactive oxygen species (ROS) in the central and peripheral nervous system associated with regulation of blood pressure (17, 18). Increased ROS generation by IH contributes to hypertension through activation of the sympathetic nervous system by the carotid body chemoreflex (19). Mice partially deficient in HIF-1α, the O2-regulated subunit of the HIF-1 complex, exhibit remarkable absence of increased ROS levels, sympathetic activation, and hypertension (12) However, little information is available on the mechanism(s) underlying HIF-1-dependent Nox transcriptional activation by IH.
Lysine demethylases (KDMs) catalyze the oxidative demethylation of methylated lysine residues of histones (20) and thereby regulate gene expression (21). Two distinct families of KDMs have been identified: the LSD1 family of flavin-dependent monoamine oxidases (KDM1) and the JmjC domain-containing proteins (JmjC-KDMs). Based on the JmjC domain sequence homology and their demethylase activities (22), the JmjC-KDMs, which are the larger of the two KDM families, have been categorized into seven subfamilies (KDM2–8). Continuous hypoxia activates JmjC-KDMs (KDM3A, KDM4A, KDM4B, KDM4C, KDM5B, KDM5C, KDM6B) in an HIF-1 dependent manner (23, 24). In addition, evidence also suggests that KDMs facilitate HIF-1-dependent transcriptional activation of certain genes by hypoxia (23). Based on this information, the current study tested the hypothesis that IH activates JmjC-KDMs in an HIF-1-dependent manner, which in turn facilitate HIF-1-dependent Nox transcriptional activation and the resulting increase in ROS generation contribute to IH-induced hypertension.
METHODS
Experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Chicago (Protocol No. 71811), and were performed on adult male Sprague-Dawley rats weighing 160–180 g between ages 2 and 3 mo.
Exposure of Rats to Intermittent Hypoxia
The protocols for exposing rats to chronic IH were essentially as described previously (25). Briefly, conscious rats were placed in a specialized chamber and exposed to alternating cycles of hypoxia (15 s of ∼5% O2 followed by 5 min of room air, 8 h/day). Control experiments were simultaneously performed on rats exposed to alternating cycles of room air in an identical chamber. The duration of the gas flow was regulated by timer-controlled solenoid valves. Ambient O2 and CO2 levels in the chamber were continuously monitored, and the CO2 levels were maintained at ∼0.1%.
Measurement of Blood Pressure and Plasma Norepinephrine
Blood pressure (BP) was measured in unsedated rats between 0900 h and 1100 h by the tail cuff method using a noninvasive BP system (IITC Life Science Inc., CA). Blood samples were collected in heparinized vials (30 IU/mL) from rats anesthetized with urethane (1.2 g/kg ip) and plasma was separated by centrifugation. Norepinephrine (NE) was extracted from the plasma with a cis-diol-specific affinity gel, acetylated, and then quantified by a competitive ELISA kit (Labor-Diagnostika, Nord Gmbh & Co. KG, Nordhorn, Germany).
Exposure of PC12 Cells to IH
Pheochromocytoma (PC) 12 cells (original clone from Dr. Green, Columbia University, New York, NY) (26) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS) and 10% horse serum under 10% CO2 and 90% room air (20% O2) at 37°C (27). Experiments were performed on cells starved overnight in serum-free DMEM medium. Cell cultures were exposed to IH (1.5% O2 for 30 s followed by 20% O2 for 5 min at 37°C) as described (28). Ambient O2 levels in the IH chamber were monitored using an O2 analyzer (Alpha Omega Instruments). In experiments involving treatment with drugs, cells were treated with either drug or vehicle for 30 min before and during IH exposure.
Transient Transfection with Lentivirus
Recombinant lentivirus was generated by transfection of HEK293T cells with the third-generation packaging plasmids, pMDLg/pRRE, pRSV-Rev, pMD2-G (No. 12251, No. 12253, No. 12259, respectively, from Addgene) and recombinant pLKO-1 expression vector encoding HIF-1α shRNA ( CCGG CCAGTTACGATTGTGAAGTTAC TCGAGTAAC TTCACAATCGTAACTGGTTTTTG; target sequence in italics: NM-024359.2) provided by Dr. G. L. Semenza (The Johns Hopkins University, Baltimore, MD) or KDM6B shRNA ( CCGG CTCCTACACCCAGCATTTATT CTCGAGAATAAATGCTGGGTGTAGGAGTTTTTG: target sequence in italics: NM-001108829.1) and nontargeting control shRNA (Sigma SHC016) using Trans IT-Virus GEN transfection Reagent (Mirus). Lentiviral supernatants were harvested, passed through a 0.45-μm filter, and aliquots were frozen at −80°C. PC12 cells transfected with lentivirus were cultured in complete medium for 48 h before exposure to IH. Transfection efficiency was determined by counting number of green fluorescent-positive cells treated with green fluorescent protein (GFP) lentiviral vector and was between 80% and 90%.
Immunoassay
Total cell extracts were prepared in lysis buffer (phosphate buffer, pH 7.4 containing 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mM EDTA, and protease inhibitor cocktail). Nuclear extracts of PC12 cells were prepared using nuclear extraction kit (Active Motif, Carlsbad, CA). Briefly, cells (1 × 106) were homogenized in 200 µL of hypotonic buffer provided in the kit and centrifuged at 850 g for 10 min at 4°C. The pellet was resuspended in 200 µL of hypotonic buffer and a small sample was checked under the microscope to verify that cells were efficiently lysed and nuclei were released. The suspension was centrifuged at 14,000 g for 30 s. The nuclear pellet was resuspended in 40 µL of complete lysis buffer provided in the kit. Cell or nuclear lysates were analyzed by the WES System from Protein Simple according to the manufacturer’s instructions. Briefly, SDS/heat-denatured proteins were subjected to automated capillary electrophoresis size-based separation, and proteins were detected directly in the capillary by primary and secondary antibodies. Primary antibodies against the following proteins were used at a dilution of 1/50 for the assays: mouse HIF-1α (No. NB100-123), rabbit NOX4 (No. NB100-58851), and rabbit KDM6B (NBP1-06640) were from Novus Biologicals (Centennial, CO). Rabbit KDM3A (No. ab106456), rabbit KDM4A (No. ab70786), and rabbit KDM4B (No. ab191434) were from Abcam (Cambridge, MA). Mouse KDM4C (No. sc-515767) and rabbit KDM6A (No. CST33510) were from Santa Cruz Biotechnology (Dallas, TX) and Cell Signaling (Danvers, MA), respectively. Sample loading variability was normalized to mouse tubulin (No. T6199, Sigma, St. Louis, MO) or mouse TBP (TATA-binding protein; No. ab51841, Abcam) antibodies. Signals were detected using secondary anti-rabbit and anti-mouse detection modules [module includes luminol-S, peroxide, secondary antibody, and Streptavidin-horseradish peroxidase (HRP); Nos. DM-001 and DM-002, respectively; Protein Simple]. Data were analyzed by the integrated Compass software (Protein Simple). Briefly, relative amount of each protein was calculated by peak area of protein/peak area of tubulin/TBP (loading control) from the chromatogram and normalized to normoxic controls. For analysis of HIF-1 binding to KDM6, cell lysates were immunoprecipitated with rabbit HIF-1α (NB100-479; Novus Biologicals) antibody, and the immunoprecipitates were analyzed as described above.
qRT-PCR
Total RNA was isolated with TRIzol (Invitrogen) and cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). Quantitative reverse transcription-PCR (qRT-PCR) was performed using SsoFast EvaGreen (Bio-Rad) as a fluorogenic binding dye; housekeeping genes, 18S RNA and β-actin, were included as quantitation controls. Changes in target mRNA expression were calculated based on the Δ(ΔCt) method as described previously (29). Primer sequences used for real-time PCR amplification were NOX4 (NM_053524.1): forward; CGG GTG GCT TGT TGA AGT AT; reverse; TGG AAC TTG GGT TCT TCC AG. 18S (NR_003278): forward; CGCCGCTAGAGGTGAAATTC; reverse; CGAACCTCCGACTTTCGTTCT. β-actin (NM_031144.3): forward; GTA CCC CAT TGA ACA CGG CA; reverse; GTA CCC CAT TGA ACA CGG CA.
ChIP Assays
Cells (15–20 million) exposed either to normoxia or IH were cross linked with 1% formaldehyde for 5 min at 37°C and quenched with 125 mM glycine for 5 min at room temperature. Cells were washed with PBS and centrifuged at 800 g for 5 min. The pellet was flash-frozen in liquid nitrogen and stored at −80°C. Pellet was resuspended in nuclear lysis buffer with protease inhibitors and sonicated using Branson Digital Sonifier. Sonication efficiency was checked on an agarose gel so that the final cross-linked, sheared chromatin DNA has an average size between 200 kb and 1,000 kb. An aliquot was taken to be used for input control and DNA was immunoprecipitated from sonicated lysates with chip grade HIF-1α antibody (NB100–479, Novus Biological), after preclearing with salmon sperm DNA and Protein G Magnet Beads (Millipore). Rabbit IgG was used as a negative control. Chipped DNA and input DNA were purified using PCR clean-up kits (Qiagen) and the products were analyzed by qPCR. Fold enrichment was calculated based on the cycle threshold (Ct) as 2 − Δ(ΔCt), where ΔCt = Ct immunoprecipitated (IP) − Ct Input and Δ(ΔCt) = ΔCt antibody − ΔCt IgG. Primer sequences used for qRT-PCR are HRE site 1: forward; CACCCGGGACATCCTGAAC; reverse: ATCTCCTGCCTGGCCTCTAG and HRE site 2: forward; ATGTGGGCCTGGCGGTCTGTCT; reverse; ACAGATTGCTGATGAGCGGG.
Measurement of KDM and NADPH Oxidase Activity
KDM3, KDM4, and KDM6 activities were measured in nuclear lysates by EpiQuik Histone Demethylase (H3K4 Specific) Activity/Inhibition Fast Assay Kit, JMJD2 Demethylase Activity/Inhibition Assay Kit, and Epigenase JMJD3/UTX demethylase activity/inhibition assay kit (Epigentik, Farmington, NY), respectively. Briefly, 5 μg of nuclear extract was added to a plate previously coated with specific methylated H3 substrate. Active KDM binds and demethylates H3, which is recognized with a specific antibody and measured by reading the absorbance at 450 nm. The activity is directly proportional to the intensity of absorbance and expressed as ng/min/mg protein. NADPH oxidase activity in the membrane-enriched protein fractions (100 μg) was measured by superoxide dismutase-inhibitable rate of cytochrome c reduction, and reading the absorbance at 550 nm. NADPH oxidase activity was calculated based on the extinction coefficient (21 mmol/L) per cm, and is expressed as nmol/min/mg protein (30).
Statistical Analysis
Data are expressed as means ± SE from three to five independent cell culture experiments or from six to eight rats per group per experiment. Statistical analysis was performed by analysis of variance (ANOVA). The Wilcoxon–Mann–Whitney test was used for analysis of normalized data. P < 0.05 was considered significant. Power analysis was performed for both animal and cell culture experiments and found to be at a power level of >80%.
RESULTS
Studies on PC12 Cells
The following studies determined whether IH activates JmjC-KDMs in an HIF-1-dependent manner, and if KDMs facilitate HIF-1-dependent Nox transcription. Given that these experiments require genetic silencing approaches and chromatin immunoprecipitation (ChiP) assays, which can be best performed in cell cultures rather than tissues, following experiments were performed on PC12 cells subjected to in vitro IH as described previously (31). We previously reported that IH is a potent activator of the HIF-1 in PC12 cells (31).
IH increases KDM6B protein and enzyme activity.
Continuous hypoxia activates JmjC-KDMs in an HIF-1-dependent manner (23, 24). We determined whether IH also activates one or more of the KDMs in an HIF-1-dependent manner. To this end, PC12 cells were treated with either alternating cycles of room air (controls) or 60 cycles of IH (IH60), each cycle consisting of 30 s of hypoxia and 5 min of room air as described previously (28). Protein abundance of JmjC-KDM isoforms was determined by immunoassay. IH60 increased KDM3A and KDM6B proteins, but not KDM4A, KDM4B, KDM4C, or KDM6A proteins (Fig. 1A). To assess whether the increased protein abundance reflected in enzyme activity, KDM3, KDM4, and KDM6 enzyme activities were determined. KDM6 enzyme activity increased in IH60-treated cells (Fig. 1B), whereas enzyme activities of KDM3 and KDM4 were below detection limits in control and IH-exposed cells (data not shown). These data suggest that KDM6 activity is selectively upregulated, and that KDM6B is the major isoform responsible for increased JmjC-KDM enzyme activity in IH-treated PC12 cells.
Figure 1.
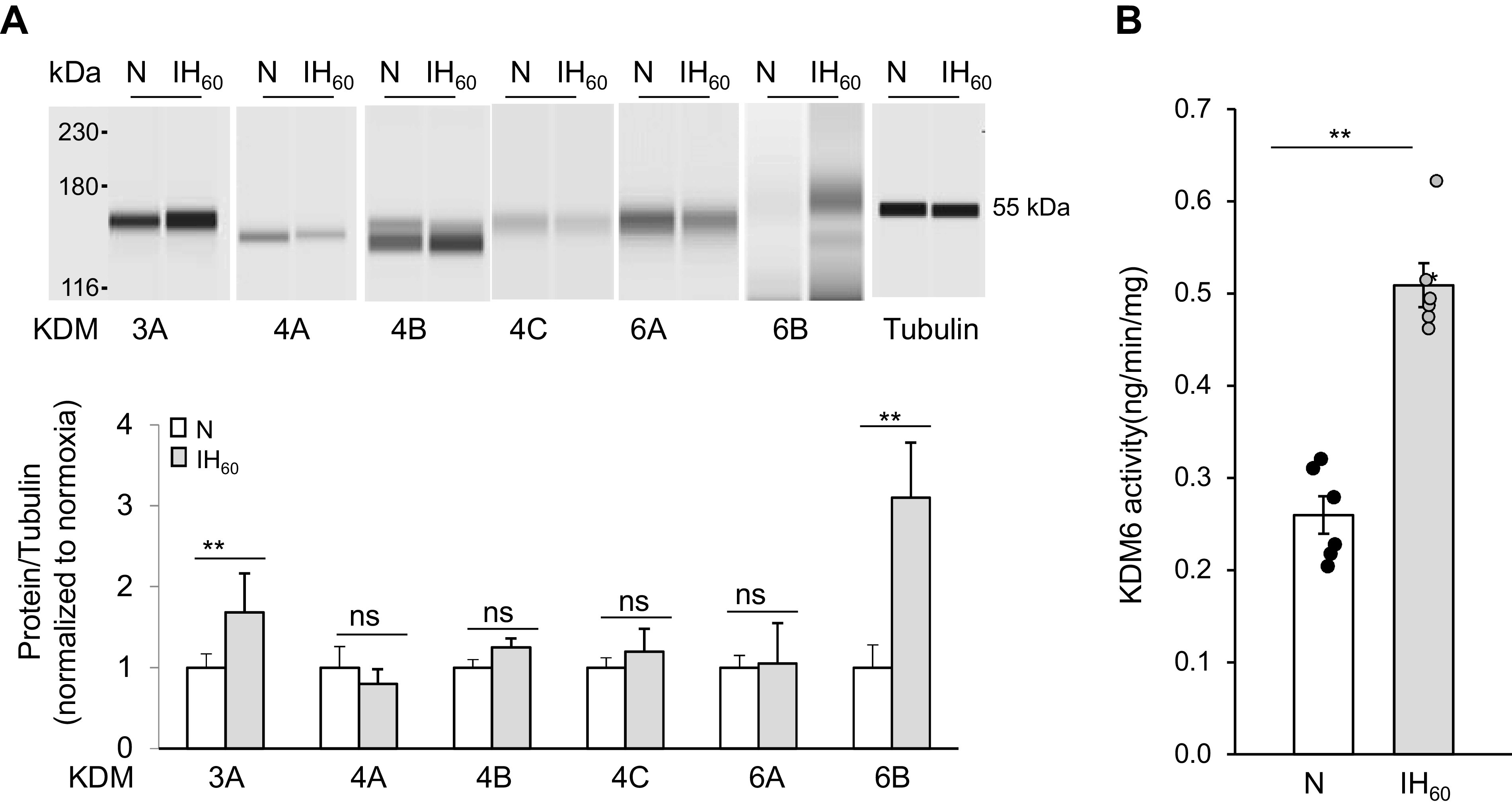
Effect of IH on KDM proteins and activity in PC12 cells exposed to IH60 or normoxia (N). A, top: representative example of KDM and tubulin (loading control) protein expression in cell lysates from control (N) and IH-exposed PC12 cells. Bottom: densitometric analysis of the gels. B: KDM6 activity in nuclear lysates from control (N) and IH-exposed PC12 cells (means ± SE; n = 3 independent experiments run in duplicates). **P ≤ 0.05; ns = not significant (P > 0.05) as determined by one-way ANOVA test. IH, intermittent hypoxia; KDM, lysine demethylases; PC, pheochromocytoma.
HIF-1-independent KDM6 activation by IH.
To test whether HIF-1 contributes to KDM6 activation by IH, cells were treated with scrambled shRNA or shRNA directed to HIF-1α, the O2 regulated subunit of the HIF-1 complex. Cells treated with scrambled shRNA showed elevated HIF-1α and KDM6B proteins, whereas HIF-1α shRNA-treated cells showed an absence of increased HIF-1α protein, but still showed elevated KDM6B protein (Fig. 2, A and B). KDM6B mRNA expression was unaltered in IH60-treated cells, and silencing HIF-1α had no effect (Fig. 2C). These results indicate that the increased KDM6B protein by IH is unlikely due to HIF-1-dependent or independent transcriptional induction of KDM6B.
Figure 2.
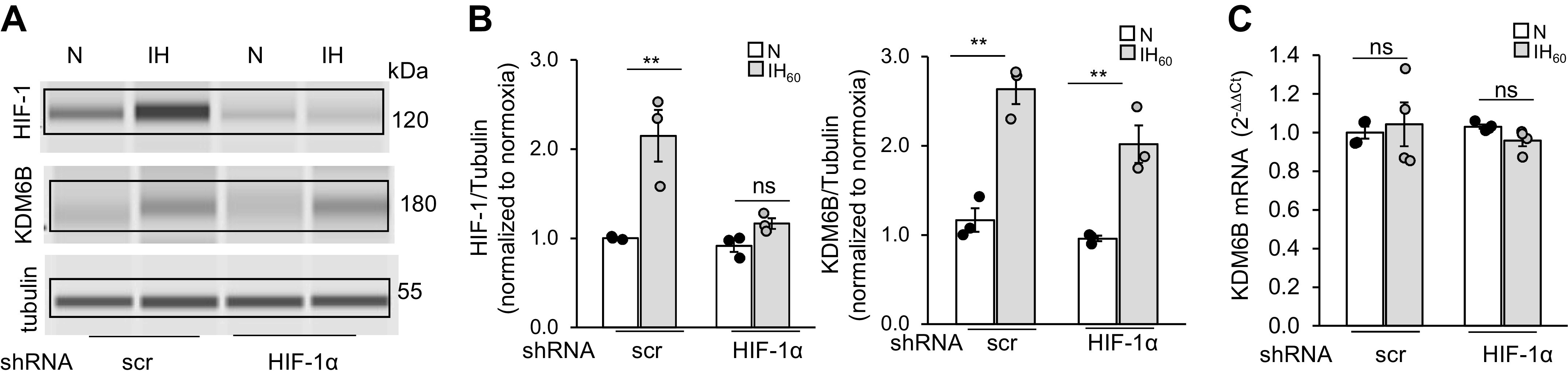
IH-augmented KDM6B protein in PC12 cells is independent of HIF-1α. A: representative example of HIF-1α and KDM6B protein expression with tubulin as loading control in total cell lysates from normoxia (N) and IH60-exposed cells treated with lentiviral scr shRNA or HIF-1α shRNA. B: densitometric analysis of gels (means ± SE; n = 3 independent experiments). C: KDM6B mRNA expression in lysates from PC12 cells treated with lentiviral HIF-1α shRNA or scr shRNA and exposed to 60 cycles of IH (IH60) or normoxia (N) (means ± SE; n = 3 independent experiments with triplicates). **P ≤ 0.05; ns = not significant (P > 0.05) as determined by Mann-Whitney test. HIF, hypoxia-inducible factor; IH, intermittent hypoxia; KDM, lysine demethylases; scr, scramble.
IH activates HIF-1-dependent NOX4 transcription.
We next sought to determine whether JmjC-KDMs facilitate HIF-1-transcriptional activation by IH. HIF-1 is a potent transcriptional activator of Nox4 (32), which is a major pro-oxidant enzyme activated by IH (14–16). We first determined whether IH activates Nox4 through HIF-1. IH60-treated cells showed increased Nox4 mRNA, and this effect was absent in cells treated with HIF-1α shRNA (Fig. 3A).
Figure 3.

IH leads to HIF-1α-dependent increase in NOX4 mRNA expression in PC12 cells. A: Nox4 mRNA expression in lysates from PC12 cells treated with lentiviral HIF-1α shRNA or scr shRNA and exposed to 60 cycles of IH (IH60) or normoxia (N) (means ± SE; n = 4 independent experiments with duplicates). B: two putative HRE HIF-binding sites (red, boxed), one (HRE1) found at base pairs −147 to −151 relative to the transcription start site (shown in blue), and a second (HRE2) found approximately +167 bases downstream from the transcription start site of the Nox4 gene. C: ChIP assay of PC12 cells exposed to IH60 or normoxia (N) using antibodies against HIF-1α. Enrichment of each sequence in the immunoprecipitates relative to the starting lysate was determined by qRT-PCR. Data are presented as means ± SE from two independent experiments with triplicates. **P ≤ 0.05; n.s. = not significant (P > 0.05) as determined by Mann-Whitney test. ChIP, chromatin immunoprecipitation; HIF, hypoxia-inducible factor; HRE, hypoxia-responsive element; IH, intermittent hypoxia; KDM, lysine demethylases; Nox4, NADPH oxidases; PC, pheochromocytoma; qRT-PCR, quantitative reverse transcription-PCR; scr, scramble.
HIF-1 binds to hypoxia-responsive elements (HRE) in the promoter regions of target genes to drive transcription (33, 34). To further establish a role for HIF-1, chromatin immunoprecipitation (ChIP) assay was performed to establish the binding of HIF-1 to the HRE sequence of the Nox4 gene. Analysis of the rat Nox4 gene sequence identified two HRE sites that correspond to HIF-1 consensus binding sequence 5′-RCGTG-3′. A first putative HRE site (HRE1) was found at base pairs −147 to −151 relative to the transcription start site of Nox4 gene, a location similar to several other known HREs in HIF target genes (33). A second putative HRE site (HRE2) was found approximately +167 bases away from the transcription start site (Fig. 3B). ChIP assay showed recruitment of HIF-1α to HRE1 but not to the HRE2 of the Nox promoter in IH-treated cells (Fig. 3C).
KDM6B is required for IH-evoked HIF-1-dependent Nox4 transcription.
Two approaches were employed to assess the role of KDM6B in HIF-1-mediated Nox4 transcriptional activation by IH: 1) pharmacological blockade of KDM6 with GSKJ4 (35, 36) and 2) genetic silencing of KDM6B with shRNA. Cells treated with GSKJ4 blocked the IH60-induced increase in KDM6B enzyme activity, protein, and Nox4 mRNA (Fig. 4, A–C). GSKJ4 can inhibit both KDM6A and KDM6B (35). Therefore, we used a genetic approach to specifically silence KDM6B by treating PC12 cells with KDM6B shRNA or a scrambled shRNA (control), and then exposing them to either room air or IH60. Compared with scrambled shRNA controls, KDM6B shRNA-treated cells exposed to IH60 exhibited absence of KDM6 enzyme activity, upregulation of KDM6B protein, and Nox4 mRNA (Fig. 4, D–F). These findings demonstrate that KDM6B is required for IH-evoked Nox4 mRNA expression in PC12 cells.
Figure 4.
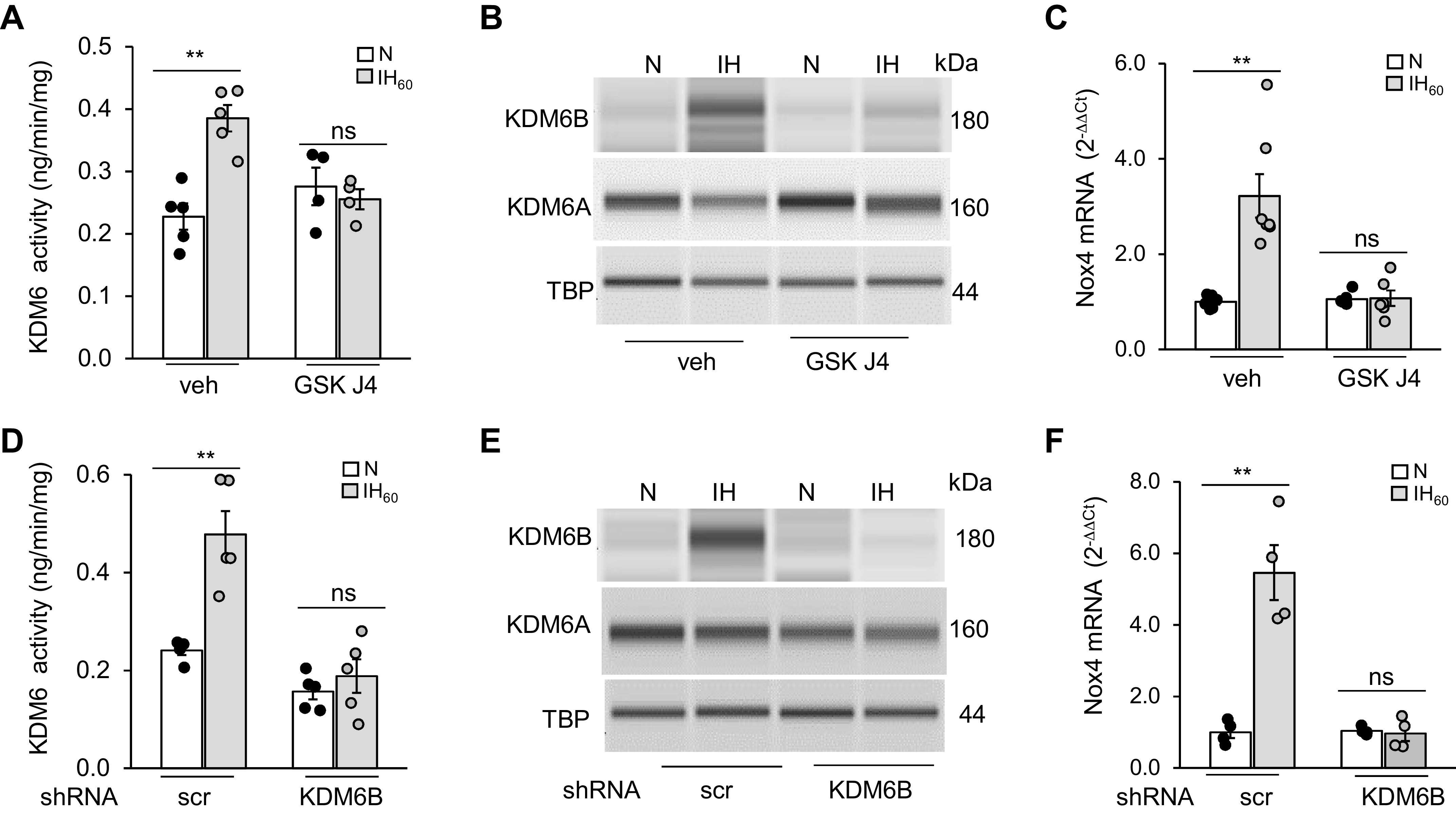
KDM6B is required for IH-induced transcriptional activation of Nox4 in PC12 cells. A: KDM6 activity; B: KDM6A and KDM6B protein expression in nuclear lysates with TBP as loading control; and C: NOX4 mRNA in PC12 cells treated with GSKJ4 (50 µM), a specific KDM6 inhibitor, and exposed to IH60 or normoxia (N). D: KDM6 activity (means ± SE; n = 4 independent experiments run in duplicates); E: KDM6A and 6B protein expression with TBP as loading control (means ± SE; n = 3 independent experiments); and F: Nox4 mRNA in PC12 cells treated with lentiviral KDM6B siRNA or scr siRNA, and exposed to IH60 or normoxia (N) (means ± SE; n = 5). **P ≤ 0.05 as determined by one-way ANOVA. ns = not significant (P > 0.05). IH, intermittent hypoxia; KDM, lysine demethylases; Nox4, NADPH oxidases; PC, pheochromocytoma; scr, scramble; TBP, TATA-binding protein; veh, vehicle.
KDM6B binds to HIF-1α.
We then sought to determine the mechanism(s) underlying the participation of KDM6B in HIF-1-dependent Nox4 transcriptional activation. JmjC-KDMs bind to sequence-specific transcription factors and associate with RNA- and DNA-binding factors in the chromatin (37–39). We analyzed whether KDM6B physically interacts with HIF-1α. To this end, coimmunoprecipitation assays were performed on nuclear lysates of PC12 cells treated with either IH60 or room air (controls). Lysates were immunoprecipitated with anti-HIF-1α antibody, and the immunoprecipitates were probed with anti-HIF-1α and KDM6B antibodies. HIF-1α protein abundance increased in immunoprecipitates of IH60-treated nuclear lysates compared with room air-exposed control cells (Fig. 5A). KDM6B co-immunoprecipitated with HIF-1α in nuclear lysates from IH but not in room air-treated cell lysates (Fig. 5A). These findings suggest that KDM6B physically interacts with HIF-1α.
Figure 5.

KDM6B stimulates HIF-1α-mediated Nox4 transcription in PC12 cells. A: PC12 cells were exposed to IH60 or normoxia (N) and then immunoprecipitated with anti-HIF1α antibody. Precipitates were separated on WES protein simple system and probed with anti-HIF-1α or anti-KDM6B. HIF-1α and KDM6B protein expression shown in black boxes. B: Nox4 enrichment determined by ChIP assay in the immunoprecipitates using antibodies against HIF-1α from PC12 cells treated with lentiviral KDM6B siRNA or scr siRNA, and exposed to IH60 or normoxia (N). Data are presented as means ± SE from two independent experiments. **P ≤ 0.05; ns: not significant (P > 0.05) as determined by Mann-Whitney test. ChIP, chromatin immunoprecipitation; HIF, hypoxia-inducible factor; IH, intermittent hypoxia; KDM, lysine demethylases; Nox4, NADPH oxidases; PC, pheochromocytoma; scr, scramble.
We next assessed whether KDM6B interaction with HIF-1α is required for binding of HIF-1 to the HRE1 site of the Nox4 gene. ChIP assay revealed absence of HIF-1α binding to the HRE1 site in KDM6B shRNA-treated cells exposed to IH60 compared with control cells treated with scrambled shRNA (Fig. 5B). These data demonstrate that physical interaction between KDM6B and HIF-1α is necessary for binding of HIF-1 to HRE1 site of the Nox4 gene.
Studies On Rats
Thus far, studies on PC12 cell cultures suggest a role for KDM6 in HIF-1-dependent Nox4 transcriptional activation by IH. We next sought to establish the effect of IH on KDM6B in rats and determine the potential contribution of KDM6B in IH-evoked sympathetic activation and hypertension. To this end, experiments were performed with 2- to 3-mo-old male rats to exclude potential confounding factors such as menstrual changes in females.
IH activates Nox4 transcription through HIF-1interaction with KDM6B.
Rats were treated with IH for 10 days and KDM6 activity and protein expressions were analyzed in adrenal medullae (AM). We selected AM tissue because: 1) PC12 cells studied in the cell culture experiments are of adrenal medullary chromaffin cell origin (26), and 2) AM plays a critical role in IH-induced hypertension (40). AM of IH-treated rats showed increased KDM6 enzyme activity as well as increased KDM6B protein, and these effects were absent in IH-exposed rats treated with GSKJ4 (10 mg/kg ip, every day for 10 days), an inhibitor of KDM6 (Fig. 6, A and B).
Figure 6.
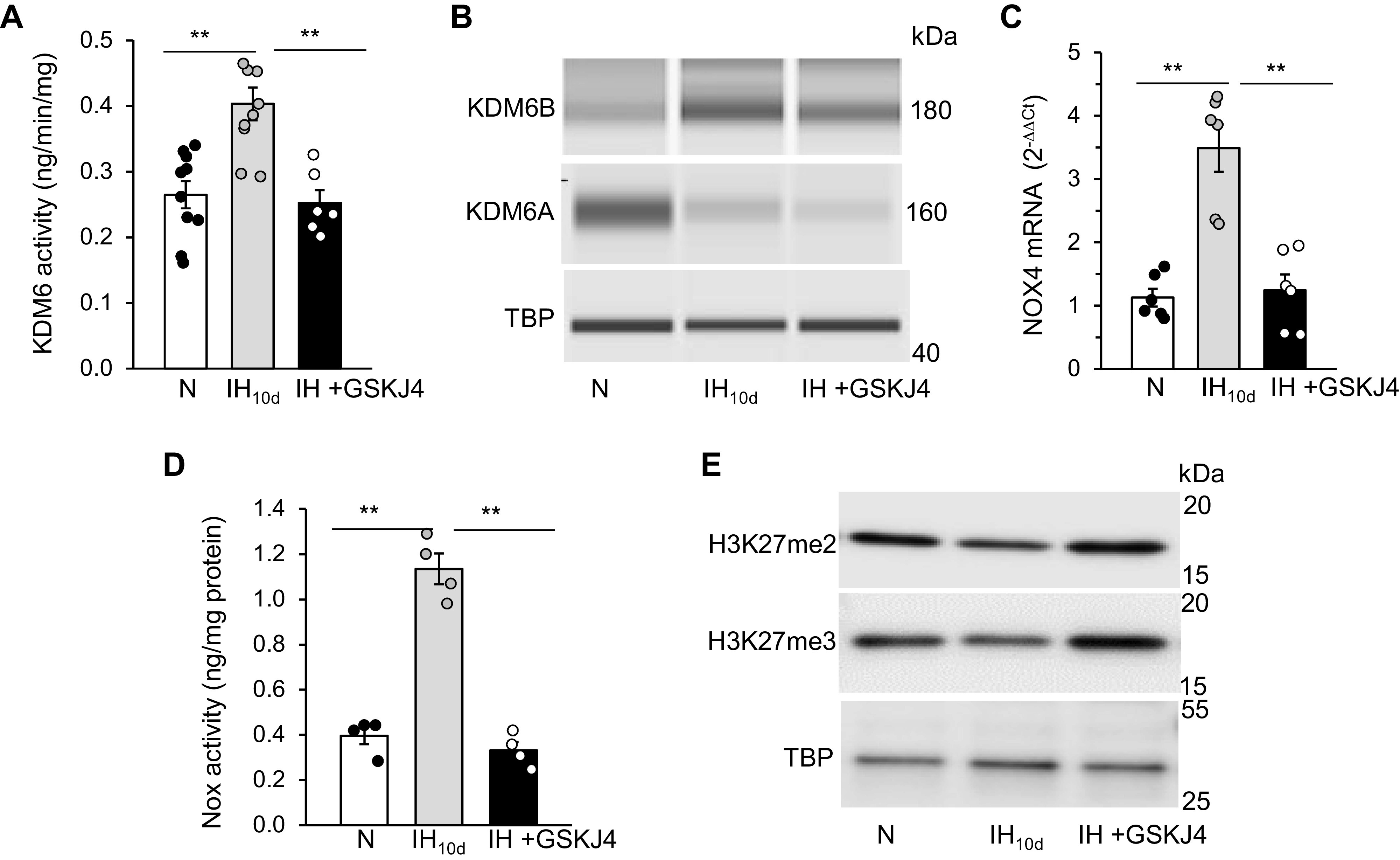
IH activates Nox4 through HIF-1/KDM6B in rat adrenal medulla (AM). Adult male rats were exposed to 10 days of IH along with daily treatment with either vehicle (IH10d) or GSKJ4 (10 mg/kg/day ip). Effect of GSKJ4 treatment on A: KDM6 activity; B: KDM6A and KDM6B protein expression; C: Nox4 mRNA; D: Nox activity; and E: H3K27me2 and me3 methylation status in cell lysates from AM. Data are means ± SE from 8 rats per group. **P ≤ 0.05; ns: not significant (P > 0.05). HIF, hypoxia-inducible factor; IH, intermittent hypoxia; KDM, lysine demethylases; N, normoxia; Nox4, NADPH oxidases; TBP, TATA-binding protein.
To assess whether activation of KDM6B contributes to Nox4 transcription by IH, Nox4 mRNA and Nox enzyme activities were determined in AM of rats treated with GSKJ4 (10 mg/kg ip). AM of rats treated with IH showed elevated Nox4 mRNA and Nox enzyme activity (Fig. 6, C and D), and these responses were blocked with GSKJ4 treatment (Fig. 6, C and D).
Methylation Status of H3K27 in AM from IH-Exposed Rats.
Removal of H3K27-methylated chromatin marks is a key mechanism by which KDM6B represses gene expression (21). H3K27 can be mono-, di-, or tri-methylated. KDM6 demethylates H3K27me2 and me3. We, analyzed the methylation status of H3K27 in AM of IH-exposed rats treated with either vehicle or GSKJ4. IH decreased methylation of H3K27me2 and me3, albeit to different extents (me2 > me3); and GSKJ4 treatment restored the methylation status of H3K27 to control levels (Fig. 6E).
Pharmacological blockade of KDM6 prevents IH-evoked activation of sympathetic nervous system and hypertension.
HIF-1-dependent transcriptional activation of Nox contribute to sympathetic nerve activation and hypertension in IH-treated wild-type mice, and these effects are absent in HIF-1α+/− mice (12). We hypothesized that if KDM6B plays a central role in HIF-1-dependent Nox4 transcription by IH, then blockade of KDM6 activity by GSKJ4 should prevent IH-induced sympathetic nerve activation and hypertension. These possibilities were tested by measuring plasma norepinephrine (NE) levels as an index of activation of the sympathetic nervous system as well as by monitoring BP in unsedated rats exposed to IH10d with and without GSKJ4 (10 mg/kg/day for 10 days) treatment. Systolic, diastolic, and mean BP as well as plasma NE were elevated in IH10d-treated rats compared with control rats treated with room air, and these effects were either absent or markedly attenuated in IH10d rats treated with GSKJ4 (Fig. 7, A and B).
Figure 7.

Effect of KDM6 inhibitor (GSKJ4) on IH-evoked sympathetic nerve activation and elevated BP in rats. A: systolic (SBP), diastolic (DBP), and mean (MBP) blood pressure expressed as difference between post- and pre-IH and B: Plasma norepinephrine levels in male rats exposed to IH10d treatment with either vehicle (IH) or GSKJ4 (10 mg/kg/day ip). Data are means ± SE from 6 rats per group. **P ≤ 0.05; ns: not significant (P > 0.05) as determined by one-way ANOVA. BP, blood pressure; IH, intermittent hypoxia; KDM, lysine demethylases; N, normoxia; TBP, TATA-binding protein; veh, vehicle.
DISCUSSION
The present study examined the role of lysine demethylases (KDMs) in transcriptional responses to IH patterned after blood O2 profiles during obstructive sleep apnea (OSA) in cell cultures (in vitro) and intact rats (in vivo). The major findings from cell culture studies were: 1) IH selectively increased KDM6B protein and enzyme activity and this effect was independent of HIF-1; 2) IH-induced HIF-1 transcriptional activity was abolished either by pharmacological or genetic blockade of KDM6B; and 3) KDM6B physically interacts with HIF-1α protein in IH-treated cells. Studies on rats showed that: 1) IH increased KDM6B protein and enzyme activity in the AM, and this effect was associated with transcriptional activation of Nox4 by HIF-1; and 2) pharmacological blockade of KDM6B prevented IH-induced HIF-1-dependent Nox4 transcription, sympathetic nerve activation, and hypertension. These findings suggest a hitherto uncharacterized role for KDM6B as an important molecular mechanism underlying IH-induced Nox4 activation by HIF-1 and ensuing hypertension.
IH increased KDM6 enzyme activity both in PC12 cell cultures treated with IH in vitro, and rat AM treated with IH in vivo. The following findings demonstrate that KDM6B is the major isoform responsible for the increased KDM6 enzyme activity by IH: 1) KDM6B but not KDM6A protein expression increased in IH-treated PC12 cells and adrenal chromaffin cells of rats treated with IH; and 2) genetic silencing of KDM6B blocked IH-evoked increase in KDM6 enzyme activity. Although IH increased KDM3A and KDM6B protein abundances in PC12 cells; KDM3 enzyme activity was below detection limits both under normoxia and IH. These findings suggest variations in basal enzyme activity of the KDMs in PC12 cell cultures. KDM knockout mice exhibit large phenotypic variations ranging from embryonic lethality (KDM5B and KDM8), perinatal lethality (KDM6A and KDM6B), neurological development defects (KDM2B and KDM6A) to no detectable phenotypic changes (KDM4D), suggesting differential expression of KDM members in diverse tissues may account for specific roles in various biological processes in both physiological and pathophysiological states (38).
Continuous hypoxia activates KDM6B in an HIF-1-dependent manner (4, 16). IH being a potent activator of HIF-1 (31, 41), we tested whether IH-evoked KDM6B activation also requires HIF-1. Consistent with earlier reports (31), IH increased HIF-1α protein in PC12 cell cultures (Fig. 2A). Although HIF-1α shRNA prevented IH-evoked upregulation of HIF-1α protein, it had no effect on increased KDM6B protein abundance elicited by IH (Fig. 2, A and B). Moreover, IH had no effect on KDM6B mRNA. Collectively, these findings suggest that increased KDM6B protein by IH is unlikely due to HIF-1-dependent or -independent transcriptional induction of KDM6B. Histone demethylases oxidize the methyl group to formaldehyde, thereby linking intracellular redox state to histone demethylation. Given that IH increases intracellular ROS levels, it remains to be determined whether ROS contribute to increased KDM6B protein by IH through post-translational modifications.
Although HIF-1 plays no role in KDM6B activation by IH, our study demonstrates that KDM6B facilitates transcriptional activation of HIF-1-dependent Nox4 transcription in IH-treated PC12 cells. Evidence includes that either pharmacological blockade of KDM6B by GSK J4 or genetic silencing of KDM6B with shRNA blocked elevated Nox4 mRNA levels in IH-treated cells as well as binding of HIF-1 to HRE1 site on the Nox4 gene promoter.
How might KDM6B facilitate HIF-1 activation by IH? JmjC-KDMs regulate transcription by maintaining or removing histone lysine methylation states such as H3K27me3 or H3K4me3, which are known to repress or activate gene expression, respectively (20, 21). In vitro studies have shown that KDMs have particular selectivity for histone residues. KDM3 members are specific for demethylation of H3K9, whereas KDM6 members are selective to H3K27 (38). Consistent with an earlier study (38), we found decreased H3K27 methylation in response to IH, and this effect was blocked by GSKJ4, a KDM6 inhibitor. These findings suggest that increased KDM6 enzyme activity regulates methylation of H3K27 under the setting of IH. It is likely under IH that KDM6B is recruited to HRE-binding sites through interaction with HIF-1α and by demethylating H3K27, KDM6B regulates HIF-1 transcriptional activity (Fig. 8). Recent studies have shown that HIF-1α transcriptional activity is also regulated by methylation of its lysine residues (42, 43). Additional studies are needed to assess whether KDM6B regulate methylation of lysine residues in HIF-1α under IH condition.
Figure 8.

Schematic representation of molecular mechanisms by which KDM6B regulates IH-induced HIF-1α transcriptional activation of Nox4. IH-induced HIF-1α binds directly to KDM6B, targeting it to HRE sites in promoters of target genes including Nox4. KDM6B mediates H3K27 demethylation, resulting in HIF-1 transcriptional activity from the Nox4 promoter. HIF-1α-dependent increase in Nox4 mRNA leads to augmented ROS abundance, resulting in elevated catecholamines and hypertension in response to IH. HIF, hypoxia-inducible factor; HRE, hypoxia-responsive element; IH, intermittent hypoxia; KDM, lysine demethylases; Nox4, NADPH oxidases; ROS, reactive oxygen species; TSS, transcriptional start site.
In contrast to our findings with IH, Chakraborty et al. (44) have shown that continuous hypoxia inactivates KDM6A but not KDM6B, and promotes methylation of H3K27me3. Unlike continuous hypoxia, IH is characterized with periodic reoxygenation. KDM6 enzyme activity require oxygen, iron, and 2-oxoglutarate as cofactors. It is likely that KDM6 enzyme is activated during the reoxygenation phase of IH, resulting in demethylation of H3K27, which may explain the differences between our study with IH and earlier study by Chakraborty et al. (44) with continuous hypoxia. For these reasons, we believe the KDM6B activation resulting in H3K27 demethylation is selective to IH.
Hypertension is an important comorbidity associated with OSA (3–5). Emerging evidence suggests that IH is a major contributor to hypertension associated with OSA (45). Earlier studies showed that HIF-1-dependent ROS production from Nox family of enzymes is an important cellular mechanism underlying IH-induced sympathetic nerve activation and the ensuing hypertension (45). Systemic administration of GSKJ4, which inhibited KDM6B activation in AM of IH-treated rats prevented HIF-1-dependent Nox4 activation, and this effect was associated with absence of sympathetic nervous system activation as indicated by lack of elevated plasma NE levels and hypertension.
We have previously reported long-term IH initiates epigenetic reprogramming of the redox state involving transcriptional repression of antioxidant enzyme genes through DNA hypermethylation leading to persistent cardiorespiratory abnormalities (46). Unlike DNA methylation, activation of KDM6B is observed with only a few days of IH treatment, suggesting that post-translational modifications involving methylation of histone tails is an early epigenetic mechanism initiated by short-term IH
Limitations
The following are the limitations of the current study. First, our studies were performed on adult male rats only. Additional studies in female rats are necessary to further establish a role for KDM6B in HIF-1-dependent transcriptional responses to IH. Second, we used an IH paradigm consisting of 15 s of hypoxia (average duration of apnea reported in human subjects), followed by 5 min of room air with 9 episodes/h for 8 h/day. This IH paradigm decreases blood O2 saturation from 97% to 80% (47–49). This IH paradigm differs from IH protocols reported by Nichols and Mitchell (50) for evoking respiratory motoneurons plasticity. These investigators subjected rats to four to five episodes of acute IH, with each episode lasting ∼5 min. Although this paradigm evokes robust plasticity of phrenic motoneurons, the duration of hypoxic episodes encountered with OSA lasted on an average no more than tens of seconds. In cell culture studies, we employed IH with 30 s of hypoxic episodes instead of 15 s of hypoxic episodes used in in vivo studies, because a previous study showed that this paradigm of IH produces robust increase in HIF-1α protein in PC12 cell cultures in vitro (27).
Despite the above limitations, the current study demonstrates that pharmacological disruption of HIF-1α activation with KDM6B inhibitor GSKJ4 might offer a novel therapeutic alternative for preventing cardiovascular morbidity caused by IH associated with OSA. Clinically, this is of importance because continuous positive airway pressure (CPAP), the current treatment of choice for OSA is not effective in all patients in reversing hypertension. Abnormal KDM6B activity has been implicated in a variety of pathological conditions including inflammation and atherosclerosis (51–53). GSK-J1/J4, a specific inhibitor, targeting the KDM6 subfamily, has proven to be an effective therapeutic intervention tool for multiple diseases as indicated by earlier study (54), and OSA/IH-induced hypertension as shown by the present study.
GRANTS
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute Grant P01 HL144454.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.N. conceived and designed research; N.W. and B.L.W. performed experiments; J.N., N.W., and B.L.W. analyzed data; J.N. interpreted results of experiments; J.N. prepared figures; J.N. drafted manuscript; J.N., B.L.W., and N.R.P. edited and revised manuscript; J.N. and N.R.P. approved final version of manuscript.
REFERENCES
- 1.Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 177: 1006–1014, 2013. doi: 10.1093/aje/kws342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc 5: 136–143, 2008. doi: 10.1513/pats.200709-155MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 283: 1829–1836, 2000. doi: 10.1001/jama.283.14.1829. [DOI] [PubMed] [Google Scholar]
- 4.Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 342: 1378–1384, 2000. doi: 10.1056/NEJM200005113421901. [DOI] [PubMed] [Google Scholar]
- 5.Lavie P, Herer P, Hoffstein V. Obstructive sleep apnoea syndrome as a risk factor for hypertension: population study. BMJ 320: 479–482, 2000. doi: 10.1136/bmj.320.7233.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Idris I, Hall AP, O'Reilly J, Barnett A, Allen M, Andrews R, Grunstein P, Lewis K, Goenka N, Wilding JP. Obstructive sleep apnoea in patients with type 2 diabetes: aetiology and implications for clinical care. Diabetes Obes Metab 11: 733–741, 2009. doi: 10.1111/j.1463-1326.2009.01045.x. [DOI] [PubMed] [Google Scholar]
- 7.Laaban JP, Daenen S, Léger D, Pascal S, Bayon V, Slama G, Elgrably F. Prevalence and predictive factors of sleep apnoea syndrome in type 2 diabetic patients. Diabetes Metab 35: 372–377, 2009. doi: 10.1016/j.diabet.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Tasali E, Mokhlesi B, Van Cauter E. Obstructive sleep apnea and type 2 diabetes: interacting epidemics. Chest 133: 496–506, 2008. doi: 10.1378/chest.07-0828. [DOI] [PubMed] [Google Scholar]
- 9.Khuu MA, Pagan CM, Nallamothu T, Hevner RF, Hodge RD, Ramirez JM, Garcia AJ. Intermittent hypoxia disrupts adult neurogenesis and synaptic plasticity in the dentate gyrus. J Neurosci 39: 1320–1331, 2019. doi: 10.1523/JNEUROSCI.1359-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fletcher EC. An animal model of the relationship between systemic hypertension and repetitive episodic hypoxia as seen in sleep apnoea. J Sleep Res 4: 71–77, 1995. doi: 10.1111/j.1365-2869.1995.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 11.Nanduri J, Peng YJ, Yuan G, Kumar GK, Prabhakar NR. Hypoxia-inducible factors and hypertension: lessons from sleep apnea syndrome. J Mol Med (Berl) 93: 473–480, 2015. doi: 10.1007/s00109-015-1274-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol 577: 705–716, 2006. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semenza GL, Prabhakar NR. HIF-1-dependent respiratory, cardiovascular, and redox responses to chronic intermittent hypoxia. Antioxid Redox Signal 9: 1391–1396, 2007. doi: 10.1089/ars.2007.1691. [DOI] [PubMed] [Google Scholar]
- 14.Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK, Prabhakar NR. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci 29: 4903–4910, 2009. doi: 10.1523/JNEUROSCI.4768-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Souvannakitti D, Nanduri J, Yuan G, Kumar GK, Fox AP, Prabhakar NR. NADPH oxidase-dependent regulation of T-type Ca2+ channels and ryanodine receptors mediate the augmented exocytosis of catecholamines from intermittent hypoxia-treated neonatal rat chromaffin cells. J Neurosci 30: 10763–10772, 2010. doi: 10.1523/JNEUROSCI.2307-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol 226: 2925–2933, 2011. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prabhakar NR. Novel role for reactive oxygen species as amplifiers of intermittent hypoxia. Focus on “Reactive oxygen species mediate central cardiorespiratory network responses to acute intermittent hypoxia". J Neurophysiol 97: 1877, 2007. doi: 10.1152/jn.01322.2006. [DOI] [PubMed] [Google Scholar]
- 18.Prabhakar NR, Kumar GK, Nanduri J. Intermittent hypoxia augments acute hypoxic sensing via HIF-mediated ROS. Respir Physiol Neurobiol 174: 230–234, 2010. doi: 10.1016/j.resp.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prabhakar NR, Peng YJ, Nanduri J. Recent advances in understanding the physiology of hypoxic sensing by the carotid body. F1000Res 7, 2018. doi: 10.12688/f1000research.16247.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kouzarides T. Chromatin modifications and their function. Cell 128: 693–705, 2007. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 21.Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol 13: 297–311, 2012. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 22.Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell 25: 1–14, 2007. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 23.Hancock RL, Dunne K, Walport LJ, Flashman E, Kawamura A. Epigenetic regulation by histone demethylases in hypoxia. Epigenomics 7: 791–811, 2015. doi: 10.2217/epi.15.24. [DOI] [PubMed] [Google Scholar]
- 24.Lee HY, Choi K, Oh H, Park YK, Park H. HIF-1-dependent induction of Jumonji domain-containing protein (JMJD) 3 under hypoxic conditions. Mol Cells 37: 43–50, 2014. doi: 10.14348/molcells.2014.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol (1985) 96: 1236–1242, 2004. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- 26.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73: 2424–2428, 1976. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan G, Nanduri J, Bhasker CR, Semenza GL, Prabhakar NR. Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J Biol Chem 280: 4321–4328, 2005. doi: 10.1074/jbc.M407706200. [DOI] [PubMed] [Google Scholar]
- 28.Yuan G, Adhikary G, McCormick AA, Holcroft JJ, Kumar GK, Prabhakar NR. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J Physiol 557: 773–783, 2004. doi: 10.1113/jphysiol.2003.058503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci USA 106: 1199–1204, 2009. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khan SA, Nanduri J, Yuan G, Kinsman B, Kumar GK, Joseph J, Kalyanaraman B, Prabhakar NR. NADPH oxidase 2 mediates intermittent hypoxia-induced mitochondrial complex I inhibition: relevance to blood pressure changes in rats. Antioxid Redox Signal 14: 533–542, 2011. doi: 10.1089/ars.2010.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol 217: 674–685, 2008. doi: 10.1002/jcp.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diebold I, Petry A, Hess J, Görlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell 21: 2087–2096, 2010. doi: 10.1091/mbc.e09-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem 268: 21513–21518, 1993. doi: 10.1016/S0021-9258(20)80571-7. [DOI] [PubMed] [Google Scholar]
- 34.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE 2005: re12, 2005. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 35.Heinemann B, Nielsen JM, Hudlebusch HR, Lees MJ, Larsen DV, Boesen T, Labelle M, Gerlach LO, Birk P, Helin K. Inhibition of demethylases by GSK-J1/J4. Nature 514: E1–E2, 2014. doi: 10.1038/nature13688. [DOI] [PubMed] [Google Scholar]
- 36.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488: 404–408, 2012. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci USA 109: E3367–E3376, 2012. doi: 10.1073/pnas.1217394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shmakova A, Batie M, Druker J, Rocha S. Chromatin and oxygen sensing in the context of JmjC histone demethylases. Biochem J 462: 385–395, 2014. doi: 10.1042/BJ20140754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams K, Christensen J, Rappsilber J, Nielsen AL, Johansen JV, Helin K. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS One 9: e96545, 2014. doi: 10.1371/journal.pone.0096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prabhakar NR, Kumar GK, Peng YJ. Sympatho-adrenal activation by chronic intermittent hypoxia. J Appl Physiol (1985) 113: 1304–1310, 2012. doi: 10.1152/japplphysiol.00444.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arias-Cavieres A, Khuu MA, Nwakudu CU, Barnard JE, Dalgin G, Garcia AJ 3rd.. A HIF1a-Dependent Pro-Oxidant State Disrupts Synaptic Plasticity and Impairs Spatial Memory in Response to Intermittent Hypoxia. eNeuro 7: ENEURO.0024-20.2020, 2020. doi: 10.1523/ENEURO.0024-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bao L, Chen Y, Lai HT, Wu SY, Wang JE, Hatanpaa KJ, Raisanen JM, Fontenot M, Lega B, Chiang CM, Semenza GL, Wang Y, Luo W. Methylation of hypoxia-inducible factor (HIF)-1α by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res 46: 6576–6591, 2018. doi: 10.1093/nar/gky449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim Y, Nam HJ, Lee J, Park DY, Kim C, Yu YS, Kim D, Park SW, Bhin J, Hwang D, Lee H, Koh GY, Baek SH. Methylation-dependent regulation of HIF-1α stability restricts retinal and tumour angiogenesis. Nat Commun 7: 10347, 2016. doi: 10.1038/ncomms10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chakraborty AA, Laukka T, Myllykoski M, Ringel AE, Booker MA, Tolstorukov MY, Meng YJ, Meier SR, Jennings RB, Creech AL, Herbert ZT, McBrayer SK, Olenchock BA, Jaffe JD, Haigis MC, Beroukhim R, Signoretti S, Koivunen P, Kaelin WG Jr.. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363: 1217–1222, 2019. doi: 10.1126/science.aaw1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prabhakar NR, Peng YJ, Nanduri J. Hypoxia-inducible factors and obstructive sleep apnea. J Clin Invest 130: 5042–5051, 2020. doi: 10.1172/JCI137560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nanduri J, Peng YJ, Wang N, Khan SA, Semenza GL, Kumar GK, Prabhakar NR. Epigenetic regulation of redox state mediates persistent cardiorespiratory abnormalities after long-term intermittent hypoxia. J Physiol 595: 63–77, 2017. doi: 10.1113/JP272346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nanduri J, Makarenko V, Reddy VD, Yuan G, Pawar A, Wang N, Khan SA, Zhang X, Kinsman B, Peng YJ, Kumar GK, Fox AP, Godley LA, Semenza GL, Prabhakar NR. Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc Natl Acad Sci USA 109: 2515–2520, 2012. doi: 10.1073/pnas.1120600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peng YJ, Nanduri J, Zhang X, Wang N, Raghuraman G, Seagard J, Kumar GK, Prabhakar NR. Endothelin-1 mediates attenuated carotid baroreceptor activity by intermittent hypoxia. J Appl Physiol (1985) 112: 187–196, 2012. [Erratum in J Appl Physiol 112: 1800, 2012]. doi: 10.1152/japplphysiol.00529.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng YJ, Yuan G, Khan S, Nanduri J, Makarenko VV, Reddy VD, Vasavda C, Kumar GK, Semenza GL, Prabhakar NR. Regulation of hypoxia-inducible factor-α isoforms and redox state by carotid body neural activity in rats. J Physiol 592: 3841–3858, 2014. doi: 10.1113/jphysiol.2014.273789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nichols NL, Mitchell GS. Mechanisms of severe acute intermittent hypoxia-induced phrenic long-term facilitation. J Neurophysiol 125: 1146–1156, 2021. doi: 10.1152/jn.00691.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burchfield JS, Li Q, Wang HY, Wang RF. JMJD3 as an epigenetic regulator in development and disease. Int J Biochem Cell Biol 67: 148–157, 2015. doi: 10.1016/j.biocel.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johansson C, Tumber A, Che K, Cain P, Nowak R, Gileadi C, Oppermann U. The roles of Jumonji-type oxygenases in human disease. Epigenomics 6: 89–120, 2014. doi: 10.2217/epi.13.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X, Liu L, Yuan X, Wei Y, Wei X. JMJD3 in the regulation of human diseases. Protein Cell 10: 864–882, 2019. doi: 10.1007/s13238-019-0653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hong BJ, Park WY, Kim HR, Moon JW, Lee HY, Park JH, Kim SK, Oh Y, Roe JS, Kim MY. Oncogenic KRAS sensitizes lung adenocarcinoma to GSK-J4-induced metabolic and oxidative stress. Cancer Res 79: 5849–5859, 2019. doi: 10.1158/0008-5472.CAN-18-3511. [DOI] [PubMed] [Google Scholar]