

Abstract
Targeting autophagy might be a promising anticancer strategy; however, the dual roles of autophagy in cancer development and malignancy remain unclear. NSCLC (non-small cell lung cancer) cells harbour high levels of SQSTM1 (sequestosome 1), the autophagy receptor that is critical for the dual roles of autophagy. Therefore, mechanistic insights into SQSTM1 modulation may point towards better approaches to treat NSCLC. Herein, we used multiple autophagy flux models and autophagy readouts to show that aldo-keto reductase family 1 member C1 (AKR1C1), which is highly expressed in NSCLC, promotes autophagy by directly binding to SQSTM1 in a catalytic-independent manner. This interaction may be strengthened by reactive oxygen species (ROS), important autophagy inducers. Further mechanistic research demonstrated that AKR1C1 interacts with SQSTM1 to augment SQSTM1 oligomerization, contributing to the SQSTM1 affinity for binding cargo. Collectively, our data reveal a catalytic-independent role of AKR1C1 for interacting with SQSTM1 and promoting autophagy. All these findings not only reveal a novel functional role of AKR1C1 in the autophagy process but also indicate that modulation of the AKR1C1-SQSTM1 interaction may be a new strategy for targeting autophagy.
Keywords: AKR1C1, SQSTM1, autophagy, NSCLC
Introduction
Aldo-keto reductase family 1 member C1 (AKR1C1) mainly catalyses the reduction of progesterone by utilising NADP+ (oxidised form of nicotinamide adenine dinucleotide phosphate) as a cofactor [1]. Sharing high homology with AKR1C2/3, AKR1C1 displays overlapping substrate specificity with two members in its same cluster [2]. Higher AKR1C1 levels are observed in many types of cancers, which predicts the overall survival of patients [3–5]. AKR1C1 causes resistance to cisplatin and doxorubicin, participates in bladder cancer metastasis and contributes to malignant transformation of fibroblasts [6–9]. Notably, these studies focused on the catalytic functions of AKR1C1, the blockade of which causes failure in efficient prevention of cancer progression [8]. AKR1C1 overexpression is also extensively observed in nonhormone-related cancers, especially lung cancer [5, 10]. This evidence indicates a catalytically independent role of AKR1C1 in cancer development and/or progression. In our previous work, AKR1C1 was shown to promote the metastasis of NSCLC (non-small cell lung cancer) cells in a catalytic-independent manner by directly interacting with STAT3 (signal transducer and activator of transcription 3), thus reinforcing its transcriptional activity [4, 11]. As a potent prometastatic factor [12], STAT3 may dictate the roles of AKR1C1 in NSCLC metastasis. Therefore, we speculated that the proteins interacting with AKR1C1 are critical for their catalytic-independent functions in cancer.
Autophagy is a catabolic process that maintains cellular homoeostasis by recycling cellular metabolites, proteins or organelles via lysosomes [13]. Upon exposure stressful stimuli (drug treatment, toxin exposure, metabolic disorders and microbe invasion), a double membrane forms at a site in the endoplasmic reticulum to create a phagophore, which engulfs cellular cargo and closes, forming an autophagosome that subsequently fuses with a lysosome to produce an autolysosome, in which degradation occurs [14]. Autophagy signalling cascades ultimately converge on autophagosomal proteins, including LC3B (microtubule-associated protein 1 light chain 3 beta), which are routinely used as indicators of autophagic flux [15]. Another essential component of autophagy is a receptor protein. Similar to other autophagy receptors, SQSTM1 (sequestosome 1) binds to cargo as well as autophagosomal proteins [16]. Although autophagy exhibits pleiotropic functions during the whole process of cancer development (transformation, initiation, metastasis and drug resistance), strategies for targeting autophagy are limited [17, 18]. This dilemma is attributed to the context-dependent functions of autophagy receptors, making autophagy a ‘Janus-faced’ player [19, 20].
As the best known autophagy receptor, SQSTM1 is commonly dysregulated in cancers. Increasing evidence has revealed that SQSTM1 is upregulated in more than 37% of lung cancer tissues and that its aggregates accumulate in ~50% of hepatocellular carcinomas [21–24]. On the one hand, SQSTM1 promotes tumour metastasis and proliferation [25, 26]. On the other hand, SQSTM1 hinders tumour inflammation and chemically induced malignancy [27]. Collectively, mounting evidence has highlighted the dual roles of SQSTM1 in cancers, which inspires extensive exploration of the underlying mechanisms. SQSTM1 is sensitive to ubiquitination, phosphorylation and oxidation, which are essential for its autophagy receptor function [28–31]. Recent studies underscored the importance of SQSTM1-binding proteins in the SQSTM1 signalling hub [16, 19, 32, 33]. Therefore, the identification of SQSTM1-interacting partners may shed light on the details of SQSTM1 signalling.
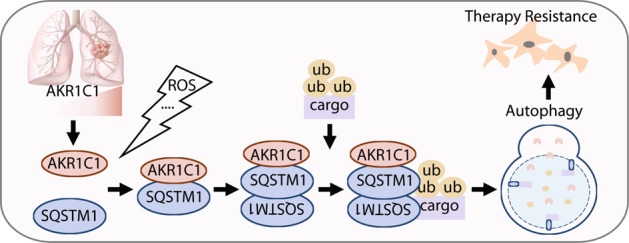
In the current study, we demonstrated for the first time, that AKR1C1 binds SQSTM1 in a catalytic-independent manner. Multiple autophagy flux models and autophagy readouts indicate that AKR1C1 promotes autophagy through the activation and maintenance of the receptor functions of SQSTM1. Our study provides the rationale for interrupting the AKR1C1-SQSTM1 interaction and subsequent autophagy as therapeutic strategies for NSCLC patients presenting with high levels of AKR1C1.
Materials and methods
Chemicals
Enzymatic inhibitor of AKR1C1 (3-bromo-5-phenylsalicylic acid, 5BPSA) was purchased from Cayman Chemical Company (13574, Ann Arbor, MI, USA) [34]. Hydrogen peroxide (H2O2) was obtained from Sinopharm (10011208, Shanghai, China). Antioxidants were all purchased from Sigma-Aldrich (St. Louis, MO, USA), including N-acetyl-L-cysteine (NAC, A9165), glutathione (GSH, G4251) and vitamin C/ascorbic acid (VC, A5960). Autophagy inhibitor, chloroquine (CQ, S4157), was obtained from Selleckchem (Houston, TX, USA).
Cell lines
293FT, 293T and HeLa cells were cultured in DMEM (Gibco, Grand Island, NY, USA). NCI-H1299, NCI-H292 and NCI-H460 lung cancer cells were maintained in RPMI-1640 (Gibco, Grand Island, NY, USA). All cell culture mediums were supplemented with 10% FBS (Gibco, Grand Island, NY, USA). Cells were all cultured in a 5% CO2 incubator at 37 °C.
Plasmids
pCMV6-AKR1C1-Myc-Flag, pCMV6-AKR1C3-Myc-Flag and pCMV6- SQSTM1-Myc-Flag was available from OriGene (Rockville, MD, USA). pENTER-AKR1C2-Flag-His was available from Vigene Bioscience (Rockville, MD, USA). pBABE-mCherry-EGFP-LC3B was available from Addgene (Cambridge, MA, USA). The other plasmids were constructed from the aforementioned plasmids, as shown in Supplementary Table S1. The hairpin sequence was 5′-AAGCTTTAGAGGCCACCAAATCTCGAGATTTGGTGGCCTCTAAAGCTT-3′ for pLKO.1-shAKR1C1.
Mass spectrometry
First of all, stable overexpression of AKR1C1 and AKR1C1-Flag in NCI-H1299 were achieved by lentivirus technology. Secondly, stable overexpression cell lines were cultured in routine culture medium for 5 passages. And then equal number of NCI-H1299 cells from both AKR1C1- and AKR1C1-Flag-expressing groups were mixed to produce two biological replicates (batch 1 and batch 2). Finally, the two samples were subjected to Flag immunoprecipitation and assessed by LC-MS/MS (Jingjie PTM BioLab, Hangzhou, China).
Immunoprecipitation
Cells were seeded into six-well plates at 40% confluence overnight. Plasmids were then introduced into cells using X-tremeGENE™ HP DNA Transfection Reagent (06366236001, Roche, Mannheim, Germany). At 48 h posttransfection, cells were harvested and lysed in ice-cold Nonidet-P40 lysis buffer (1% Nonidet-P40, 10% Glycerol, 25 mM Tris-Base, 150 mM NaCl, 5 μg/mL leupeptin, 1 μL/mL Na3VO4·12H2O, pH 8.0). Measured by Bradford reagent (B6916, Sigma-Aldrich, St. Louis, MO, USA), 500 μg/group protein was applied to immunoprecipitation. Anti-Myc magnetic beads were available from Biotool (B26302, Bimake, Houston, TX, USA) and anti-Flag agarose resin were from GenScript (L00425, Nanjing, China). Precipitates were then heated in 1.25× Loading buffer (5×Loading: 62.5 mM Tris-HCl, 50% glycerol, 10% SDS, 5% mercaptoethanol, 0.1% bromophenol blue, pH 6.8) for 10 min at 95 °C and assessed by Western blotting. Antibodies were obtained as shown in Supplementary Table S2.
GST pull-down assay
GST and GST-AKR1C1 were purified as previously described [35]. NCI-H1299 cells were lysed in ice-cold Nonidet-P40 lysis buffer (1% Nonidet-P40, 10% glycerol, 25 mM Tris-Base, 150 mM NaCl, 5 μg/mL leupeptin, 1 μL/mL Na3VO4·12H2O, pH 8.0) and measured by Bradford reagent. Purified GST-AKR1C1 or GST of 1 μg was preincubated with or without 5BPSA for 30 min in 37 °C to investigate the effects of 5BPSA on GST-AKR1C1 and SQSTM1 interaction. GST beads of 10 μL (C600031, Sangon Biotech, Shanghai, China) were added into the mixtures of 300 μg NCI-H1299 cell lysates and 1 μg purified GST-AKR1C1/GST with or without 5BPSA. Mixtures were rotated for 1 h in 4 °C. The immunoprecipitates and lysates were probed with antibodies against SQSTM1, AKR1C1 and GST (Supplementary Table S2).
Detection of LC3B puncta formation
Stable overexpression of EGFP-LC3B in HeLa cells was achieved by lentivirus technology and cultured in routine medium for five passages. Then these HeLa cells were seeded into Lab-Tek® II Chamber Slide™ (154534, Thermo Fisher Scientific, Rochester, NY, USA) at 30% confluency. After 24 h, these cells were transiently transfected with AKR1C1 plasmids using X-tremeGENE™ HP DNA Transfection Reagent (06366236001, Roche, Mannheim, Germany). At 24 h posttransfection, these cells were treated with CQ (10 μM, 15 h) or HBSS (2 h). Finally, these HeLa cells were subjected to immunofluorescence and photographed. The numbers of LC3B puncta were quantified by Image J software. DAPI was from DOJINDO (D523, Kumamoto, Japan).
Evaluation of SQSTM1 punta formation
Stable AKR1C1 silencing in NCI-H460 was achieved by lentivirus-delivered RNAi silencing technology. After multipled three passages, NCI-H460 cells were seeded into Lab-Tek® II Chamber Slide™ (154534, Thermo Fisher Scientific, Rochester, NY, USA) at 50% confluency. Thirty-six hours later, cells were stimulated with 1 mM H2O2 for 1 h and treated by 4% paraformaldehyde immediately. Then these NCI-H460 cells were subjected to immunofluorescence and photographed. The numbers of SQSTM1 punta were quantified by Image J software. Antibodies were shown in Supplementary Table S2.
SQSTM1 disulphide-linked conjugate (DLC) status
AKR1C1 silencing in NCI-H292 was achieved by lentivirus-delivered RNA silencing technology. At 5 days postinfection, cells were stimulated with 1 mM H2O2 for 2 h and harvested on ice. Then cells were lysed in RIPA lysis buffer (1% Nonidet-P40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-Base, 150 mM NaCl, 5 μg/mL leupeptin, 1 μL/mL Na3VO4·12H2O, pH 7.4) plus 50 mM N-ethylmaleimide. Next, the supernatant from each group was divided into two tubes and boiled in Laemmli buffer (non-reduced samples)/Loading buffer (reduced samples) separately at 100 °C for 5 min. Western blotting was next implemented to explore DLC status of SQSTM1. 5× loading: 62.5 mM Tris-HCl, 50% glycerol, 10% SDS, 5% mercaptoethanol, 0.1% bromophenol blue, pH 6.8. Laemmli buffer: 62.5 mM Tris-HCl, 25% glycerol, 2% SDS, 0.1% bromophenol blue, pH 6.8. Antibodies were shown in Supplementary Table S2.
Statistical analysis
Significance was determined by two-tailed Student’s t-test, n = 3. *P < 0.05; **P < 0.01; ***P < 0.001.
Results
AKR1C1 interacts with SQSTM1 in a catalytic-independent manner
Our previous work showed that AKR1C1 binds to STAT3 in a catalytic-independent manner and results in the activation of the STAT3 pathway and the subsequent metastasis of NSCLC cells [4, 11]. Therefore, we reasoned that interacting proteins, not catalytic activity, may be essential for the tumour-promoting functions of AKR1C1. To systematically address this point, we generated exogenous AKR1C1- and AKR1C1-Flag-expressing cells and performed analysis with mass spectrometry. To obtain an unbiased profile of protein interactions, we transfected NCI-H1299 cells with AKR1C1 to generate a control group and with AKR1C1-Flag to generate an experimental group. Analysing Venn diagrams generated on the basis of two independent experiments, we identified seven proteins according to the average number of unique peptides (≥2) (Fig. 1a, left). Notably, among the seven potential interacting proteins, SQSTM1 was identified with the top average scores for AKR1C1 binding (Fig. 1a, right).
Fig. 1. SQSTM1 interacts with AKR1C1.

a NCI-H1299 cells were transfected with AKR1C1-Flag or AKR1C1. Cells were subjected to Flag IP and assessed by LC-MS/MS. Venn diagrams were used to visualize the potential AKR1C1-interacting proteins (left). Vertical bars represent proteins (unique peptides ≥ 2) from both batch 1 and batch 2 according to the rank of protein scores (right). b GST pulldown assay was utilized to evaluate the interaction of recombinant GST-AKR1C1 with SQSTM1 from whole cell lysates of NCI-H1299. c AKR1C1-HA or SQSTM1-Myc or their combination were expressed in 293T cells for 36 h. Myc IP and IB were applied to the indicated cells. d Vector or SQSTM1-Myc plasmids were introduced into NCI-H460 for 36 h. Cells were subjected to Myc IP and IB.
We confirmed that SQSTM1 is a positive candidate for AKR1C1 interaction through a GST pull-down assay with NCI-H1299 cell lysates (Fig. 1b). Then, we further utilized coimmunoprecipitation and confirmed the AKR1C1-SQSTM1 interaction by immunoprecipitating exogeneous AKR1C1 and SQSTM1 (Fig. 1c, d). Collectively, these data indicated that AKR1C1 binds SQSTM1 both in vivo and in vitro.
As mentioned above, although AKR1C1 is an aldo-keto reductase family member, its tumour-promoting function may depend on catalytic activity. We were thus interested in exploring whether catalytic activity is required for the AKR1C1-SQSTM1 interaction. AKR1C2 and AKR1C3, the other two family members possessing similar enzymatic activities, were introduced to cells, and the coimmunoprecipitation assay showed that SQSTM1 preferentially bound to AKR1C1 compared to the other family members (Fig. 2a). To verify the contribution of catalytic activity, we employed AKR1C1 enzymatic mutants, namely, Y55F, E127D and H222I. A proton donor, Y55 is considered a catalytically dead mutant [36]. All of these mutants led to a ≥24-fold decrease in progesterone reduction [34, 36, 37]. As depicted in Fig. 2b, all the catalytic mutants showed similar interactions with SQSTM1.
Fig. 2. The enzymatic activity of AKR1C1 is not involved in its binding with SQSTM1.

a AKR1C1/2/3-HA with or without SQSTM1-Myc-Flag was introduced into 293T cells and assessed by Myc IP and IB. b 293T cells were transfected with AKR1C1-HA enzymatic mutants or SQSTM1-Myc-Flag or their combinations and assessed by Myc IP and IB. c AKR1C1 and AKR1C2 share approximately 97% sequence similarity. Y55, E127, H222 and R304 account for AKR1C1 catalytic activity. AKR1C1 differs from AKR1C2 in only seven amino acid residues. d AKR1C2-HA mimic mutants were constructed using AKR1C1-HA as the template: including T38V, R47H, L54V, C87S, V151M, R170H, Q172L, R47H & L54V and R170H & Q172L. These mimic mutants were transfected with or without SQSTM1-Myc into 293T cells and assessed by Myc IP and IB.
Taken together, these data indicate that SQSTM1 is an AKR1C1-interacting protein, and this interaction is not dependent on the catalytic activity of AKR1C1.
The R47H & L54V and R170H & Q172L mutants of AKR1C1 do not interact with SQSTM1
Next, we sought to map the region(s) of AKR1C1 accounting for the interaction with SQSTM1. Although studies revealed ~97% sequence similarity between AKR1C1 and AKR1C2, SQSTM1 binds only to AKR1C1, not to AKR1C2 (Fig. 2a). Therefore, we asked whether the amino acid residues that are different between AKR1C1 and AKR1C2 are critical for the interaction. As mapped in Fig. 2c, AKR1C1 differs from AKR1C2 in only seven amino acid residues: T38, R47, L54, C87, V151, R170 and Q172. Considering these seven amino acid residues, we generated AKR1C2-HA mimic mutants using AKR1C1-HA as the template. Notably, only the mutants of R47H & L54V and R170H & Q172L failed to interact with SQSTM1 (Fig. 2d). This outcome may be attributed to dual mutations (R47H & L54V and 170H & Q172L) having lower affinity for SQSTM1 compared to WT AKR1C1, which is worthy of an in-depth future investigation. R47, L54, R170 and Q172 in AKR1C1 were not essential for catalytic activity. In conclusion, R47H & L54V and R170H & Q172L play vital roles in the AKR1C1–SQSTM1 interaction.
ROS augments the AKR1C1–SQSTM1 interaction
The abovementioned data showed that AKR1C1 bound to SQSTM1 in a catalytic-independent manner. In this context, 3-bromo-5-phenylsalicylic acid (5BPSA), one of the most potent AKR1C1 enzymatic inhibitors (Ki = 4 nM) [37], was expected to impose no effect on the interaction between AKR1C1 and SQSTM1. This hypothesis was verified with a GST pull-down assay (Fig. 3a), but interestingly, a paradoxical observation was observed in the cellular co-IP system. As shown in Fig. 3b, after 5BPSA cell treatment, the interaction of AKR1C1 and SQSTM1 was greatly impeded, but this effect was significantly attenuated by FBS (Fig. 3b). Accumulating evidence implies that FBS includes a variety of antioxidants [38–40], which inspired us to investigate the roles of reactive oxygen species (ROS) in the AKR1C1–SQSTM1 interaction. We introduced hydrogen peroxide (H2O2), one of the most important ROS, and found that H2O2 exposure remarkably promoted the AKR1C1–SQSTM1 interaction (Fig. 3c), an effect that was attenuated by antioxidants, including NAC, GSH and VC (Fig. 3d).
Fig. 3. ROS (reactive oxygen species) generation plays a vital role in AKR1C1-SQSTM1 interaction.

a Purified GST-AKR1C1 was incubated with or without 5BPSA for 30 min in 37 °C. Then NCI-H1299 cell lysates and GST beads were added into the mixtures of different groups and assessed by GST pulldown assay. b–e Cells were transfected with AKR1C1-HA or SQSTM1-Myc or their combinations and assessed by Myc IP and IB. b Cells were treated with or without 5BPSA for 10 h plus FBS starvation or not. c ROS was mimicked by 1 mM H2O2 in fresh DMEM medium for 2 h. d Antioxidants treatment for 1 h, including 1 mM NAC, GSH and VC were used for the rescue experiment. e Cells were treated with fresh medium (10% FBS and 90% DMEM) or with HBSS (hank’s balanced salt solution) for the indicated time.
ROS participate in multiple signaling pathways and biological functions in tumorigenesis and progression [41, 42]. Recent studies showed that by managing autophagic clearance, the autophagy receptor SQSTM1 is susceptible to ROS, a condition that is essential in the autophagy process [29, 43, 44]. Therefore, Hank’s balanced salt solution (HBSS), the canonical autophagy induction buffer, was introduced to establish a ROS induction model [44–46]. Interestingly, the AKR1C1-SQSTM1 complex accumulated gradually during the autophagy process upon HBSS treatment (Fig. 3e). As shown by immunoprecipitation, upon exposure to HBSS, the interaction between AKR1C1 and SQSTM1 was enhanced in a time-dependent manner (Fig. 3e). This observation indicates that AKR1C1 may play a role in autophagy.
These observations collectively identify ROS as important physiological stimuli controlling the interaction between AKR1C1 and SQSTM1 and implicate the potential roles of AKR1C1 in the autophagy process.
AKR1C1 promotes autophagic flux
Autophagy is a homoeostatic cellular process that clears damaged or unnecessary organelles and proteins [13]. Cumulative evidence underpins the importance of autophagy in tumour progression, including drug resistance, metastasis, inflammation and immunity [17, 47–49]. At the core of autophagy are autophagy-related gene 8 (Atg8) family proteins, including LC3, which are routinely used as indicators of autophagic flux [15].
The abovementioned data showed the increased AKR1C1-SQSTM1 interaction along with HBSS-induced autophagy; thus, we were encouraged to explore the role of AKR1C1 in autophagy. First, exogenous AKR1C1 was introduced into cells. As shown in Fig. 4a, b, LC3B-II protein levels were increased (4.94 ± 1.02)-fold in HeLa cells (P = 0.0028, vs. the control) and (2.70 ± 0.39)-fold for NCI-H1299 (P = 0.0066, vs. the control), respectively. These results suggested that AKR1C1 overexpression led to enhanced LC3 conjugation to phosphatidylethanolamine, creating LC3B-II, a commonly used marker used to monitor the number of autophagosomes formed in a cell [50].
Fig. 4. AKR1C1 accelerates autophagy flux.

HeLa (a and c) or NCI-H1299 cells (b and d) were transfected with AKR1C1 or vector plasmid and assessed by Western blotting. Band intensities of LC3B-II were measured and normalised to β-Actin. c HeLa cells were exposed to CQ (10 μM) for 15 h at 40% confluency. d NCI-H1299 cells were cultured in HBSS for 2 h. e–h LC3B-EGFP stably-expressed HeLa cells were constructed and cultured for 5 generations. These cells were transfected with AKR1C1 and treated by CQ (10 μM, 15 h) or HBSS (2 h). Then cells were subjected to IF. LC3B puncta were quantified by Image J. Green: LC3B-EGFP. Red: AKR1C1. Blue: cell nuclei. Scale bar: 20 μm. Large: size ≥ 5.0 pixel units. Total: size ≥ 2.5 pixel units.
However, increased amounts of LC3B-II can correlate with either an induction of autophagy or a block at the late steps of this pathway [51]. To distinguish between these two possibilities for AKR1C1-primed LC3B-II accumulation, we next assessed the effects of AKR1C1 on autophagic flux. A lysosome inhibitor of autophagy flux, chloroquine (CQ), was introduced [52]. If the LC3B-II induced by AKR1C1 was caused by autophagic flux inhibition, then cotreatment with CQ would not have an additive effect. As shown in Fig. 4c, e, f, CQ + AKR1C1 overexpression led to higher numbers of LC3B-II and LC3B puncta than CQ treatment alone, indicating that autophagy is induced, rather than inhibited, in the AKR1C1-transfected cells. Moreover, AKR1C1 overexpression further amplified the increase in LC3B-II and LC3B puncta caused by HBSS (Fig. 4d, g, h). Taken together, these data demonstrate that AKR1C1 is an autophagy inducer.
AKR1C1 interacts with SQSTM1 to modulate its autophagy receptor function
We next asked how AKR1C1 promoted autophagy, and we particularly focused on the AKR1C1 interacting protein SQSTM1, as this was the first identified autophagy receptor. Mounting evidence has revealed that ubiquitination and oligomerization are essential for the autophagy receptor function of SQSTM1 [28, 30, 53]. Therefore, we evaluated the oligomerized and ubiquitinated states of SQSTM1 in the AKR1C1–SQSTM1 complex. Ubiquitination of SQSTM1 in this complex reflects the affinity of SQSTM1 for autophagy substrates. We found that AKR1C1 overexpression led to more ubiquitin conjugated to the SQSTM1 in the AKR1C1-SQSTM1 complex, whereas SQSTM1 imposed no effects on the ubiquitination of AKR1C1 (Fig. 5a, b). These data suggest that AKR1C1 may not be the autophagy substrate for SQSTM1; however, it may increase the substrate binding affinity for SQSTM1, as indicated by the long polyubiquitin chain on SQSTM1.
Fig. 5. AKR1C1 promotes SQSTM1 oligomerization.

a, b AKR1C1-HA or SQSTM1-tags or their combinations were transfected for 36 h. Ubiquitination of complex was then assessed by Myc IP or HA IP. c Plamids were introduced into cells for 36 h and assessed by Myc IP and IB. d, e NCI-H460 (4 × 105 in each well) cells were seeded onto eight-well chamber slides. After cultured for 36 h, cells were stimulated with or without 1 mM H2O2 for 1 h and assessed by IF. SQSTM1 bodies were quantified by Image J. Green: SQSTM1. Red: AKR1C1. Blue: cell nuclei. Scale bar: 20 μm. f NCI-H292 cells were infected with lentivirus-shAKR1C1 for 5 days. Then cells were stimulated with or without 1 mM H2O2 for 2 h and assessed by SQSTM1 DLC detection.
Several lines of evidence have shown that SQSTM1 oligomerization is required for its ubiquitin- and ubiquitinated substrate-binding ability [28]. In this context, we asked whether AKR1C1 promotes the oligomerization of SQSTM1. As shown in Fig. 5c, the oligomerization of exogenous SQSTM1 was markedly augmented in the presence of AKR1C1. To visualize SQSTM1 oligomerization, we employed immunofluorescence to assess SQSTM1 puncta formation. In response to H2O2, SQSTM1 puncta levels increased from (1.08 ± 0.68)-fold to (14.80 ± 2.67)-fold (P = 0.00099, H2O2 vs. the control) (Fig. 5d). These results suggest that in NCI-H460 cells harbouring high AKR1C1 protein levels, SQSTM1 puncta formation was greatly induced by H2O2, which has been well documented [29]. More interestingly, AKR1C1 shRNA obviously impeded the formation of SQSTM1 puncta induced by H2O2 (Fig. 5e). In addition, we also implemented nonreduced Western blotting to assess endogenous SQSTM1 disulfide-linked conjugate (DLC) status, which can promote SQSTM1 oligomerization [29]. Upon H2O2 stimulation, SQSTM1 formed DLCs, including dimers and polymers, and this increase was blocked by shAKR1C1 (Fig. 5f).
Collectively, these data show that AKR1C1 plays a vital role in promoting the function of SQSTM1 as an autophagy receptor.
Discussion
AKR1C1 is more highly expressed in NSCLC than it is in other cancer types, predicting poor prognosis for NSCLC patients. However, catalytic inhibitors of AKR1C1 fail to exhibit potent anticancer efficiency [8], emphasising the importance of the catalytic-independent roles of AKR1C1 in tumour progression. In this study, we performed an LC/MS/MS assay to explore the catalytic-independent roles of AKR1C1 and identified the autophagy receptor SQSTM1 as a newly discovered AKR1C1 interacting partner, which subsequently promotes autophagy by activating the receptor functions of SQSTM1.
Reductases endow AKR1C1 with resistance-inducing effects towards cisplatin, methotrexate, daunorubicin and progesterone [7–9, 54]. However, the catalytic-independent roles of AKR1C1 remain enigmatic in therapy resistance. In the present work, we demonstrate that AKR1C1 is a key regulator of autophagy. As shown in HeLa and NCI-H1299 cells, the presence of AKR1C1 leads to greater LC3B-II accumulation and LC3B-EGFP puncta than autophagy induction/inhibition treatments administered in the absence of AKR1C1 (Fig. 4), which hints at the possibility that AKR1C1 promotes autophagy. Increasing literature has underscored the role of cytoprotective autophagy in therapy resistance, which ultimately results in treatment failure [18, 55]. We thus speculate that lung cancer patients presenting high levels of AKR1C1 may have previously undergone autophagy, leading to therapy resistance.
Although autophagy inhibition has been a focus of recent studies to overcome therapy resistance, the outcomes remain limited [18]. Preliminary results indicate that EGFR TKIs induce autophagy, partly contributing to chemoresistance; thus, NIH sponsored clinical trials to evaluate the regimen of autophagy inhibitors (e.g. chloroquine and hydroxychloroquine) and EGFR inhibitors in the treatment of NSCLC [56, 57]. Previous work also suggested that different BRAF-mutant cancers resist targeted therapy through autophagy, leading to the launch of clinical trials combining dabrafenib, trametinib, and HCQ [18, 57]. To date, although some HCQ and HCQ combinations have exhibited some activity in clinical trials, these treatments are not efficient or sufficiently safe for further study, compared with contemporary therapies [18]. A deepening understanding of autophagy modulation will contribute to new therapeutic strategies to target autophagy. Here, we identified AKR1C1 as a new binding partner of the autophagy receptor SQSTM1, contributing to autophagy (Figs. 1, 4 and 5). In light of this finding, it seems possible to overcome autophagy-addicted therapy resistance by interrupting the AKR1C1-SQSTM1 complex, which deserves further research.
Many groups have underscored the essential roles of ROS in autophagy [29, 43, 44]. One group reported that the Cys81 residue is a target for the redox regulation of autophagy-related 4A cysteine peptidase, which degrades and recycles Atg8 [44]. Similarly, another group demonstrated that in response to age-associated oxidative stress, Cys105/113 residues together with Lys102 locate to a charged bridge and promote SQSTM1 oligomerization, which is required for its autophagy receptor functions [29]. In line with these findings, our data reveal that ROS plays vital role in the AKR1C1-SQSTM1 interaction (Fig. 3). Upon H2O2 exposure, more AKR1C1–SQSTM1 complex accumulated, which was abolished by antioxidants (Fig. 3c, d). Interestingly, SQSTM1-mediated Keap1 degradation restores Nrf2, which can target AKR1C1 for transcription [58, 59]. This finding prompted us to assume that a signalling loop is established, namely, a AKR1C1–SQSTM1–Nrf2 loop, in tumour cells in response to oxidative stress, which may accelerate the protective function of antioxidants in tumours.
According to reported studies as well as our previous studies, AKR1C1 actually exerts catalytic-dependent and catalytic-independent activity in cancer cells [4, 8, 9, 11]. Although most studies treat AKR1C1 as a reductase in cancers, AKR1C1 is upregulated in nonhormone-related cancer as well, particularly lung cancer, although progesterone is a major substrate for AKR1C1 [4]. In addition, AKR1C1 and AKR1C2 both function as targets of Nrf2 to detoxify other toxic by-products, whereas only AKR1C1 predicts overall survival for lung cancer patients [4, 60]. Moreover, our immunoprecipitation results indicated that, among three similar reductase family members, AKR1C1 is a novel SQSTM1 binding partner (Fig. 2a). All this evidence is corroborated by our previous work, in which AKR1C1 promotes the metastasis of NSCLC cells in a catalytic-independent manner [4, 11]. In our present work, we also show that AKR1C1 is endowed with the ability to bind SQSTM1 despite losing its catalytic ability, as induced by either treatment with 5BPSA or the introduction of AKR1C1 mutants (Figs. 2b and 3a). These data explained the mechanisms underlying AKR1C1-promoted autophagy in which AKR1C1 directly binds the autophagy receptor SQSTM1 independent of AKR1C1 catalytic activity.
In summary, our present work revealed a catalytic-independent role for AKR1C1 in promoting autophagy. In response to oxidative stress, AKR1C1 directly interacts with the autophagy receptor SQSTM1 to facilitate SQSTM1 oligomerization required for its increased affinity for autophagy substrates. With these newly acquired insights, this work not only increases our understanding of signalling cascades for autophagy machinery in cancer cells but also provides a potential avenue to disrupt the AKR1C1-SQSTM1 interaction to target autophagy, thus shedding light upon the design of anti-resistance therapies against lung cancer.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81673458 and 81803556), the Natural Science Foundation of Zhejiang Province (Grant no. LR19H310002), Jiangsu Young Medical Talents (QNRC2016180), Six Talent Peaks Project of Jiangsu Province (WSN-173), the Science and Technology Research Project of Henan Province (No. 212102311032).
Author contributions
HZ and LLC conceived and designed the study. LLC, YKL, CXZ, CMZ, FJG and JMD performed the experiments. LLC, WZZ, PHL, QJH and BY performed the data analysis. HZ and LLC contributed to writing the manuscript. ZH and BY contributed to the materials. All the authors read and approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Hong Zhu, Email: hongzhu@zju.edu.cn.
Bo Yang, Email: yang924@zju.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00673-w.
References
- 1.Dhagat U, Endo S, Sumii R, Hara A, El-Kabbani O. Selectivity determinants of inhibitor binding to human 20alpha-hydroxysteroid dehydrogenase: crystal structure of the enzyme in ternary complex with coenzyme and the potent inhibitor 3,5-dichlorosalicylic acid. J Med Chem. 2008;51:4844–8. doi: 10.1021/jm8003575. [DOI] [PubMed] [Google Scholar]
- 2.Ohta T, Ishikura S, Shintani S, Usami N, Hara A. Kinetic alteration of a human dihydrodiol/3alpha-hydroxysteroid dehydrogenase isoenzyme, AKR1C4, by replacement of histidine-216 with tyrosine or phenylalanine. Biochem J. 2000;352:685–91. [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou C, Shen GW, Yang F, Duan JL, Wu Z, Yang MQ, et al. Loss of AKR1C1 is a good prognostic factor in advanced NPC cases and increases chemosensitivity to cisplatin in NPC cells. J Cell Mol Med. 2020;24:6438–47. doi: 10.1111/jcmm.15291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu H, Chang LL, Yan FJ, Hu Y, Zeng CM, Zhou TY, et al. AKR1C1 activates STAT3 to promote the metastasis of non-small cell lung cancer. Theranostics. 2018;8:676–92. doi: 10.7150/thno.21463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian H, Li X, Jiang WL, Lv CT, Sun WZ, Huang CG, et al. High expression of AKR1C1 is associated with proliferation and migration of small-cell lung cancer cells. Lung Cancer. 2016;7:53–61. doi: 10.2147/LCTT.S90694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chien CW, Ho IC, Lee TC. Induction of neoplastic transformation by ectopic expression of human aldo-keto reductase 1C isoforms in NIH3T3 cells. Carcinogenesis. 2009;30:1813–20. doi: 10.1093/carcin/bgp195. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto R, Tsuda M, Yoshida K, Tanino M, Kimura T, Nishihara H, et al. Aldo-keto reductase 1C1 induced by interleukin-1beta mediates the invasive potential and drug resistance of metastatic bladder cancer cells. Sci Rep. 2016;6:34625. doi: 10.1038/srep34625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsunaga T, Yamaguchi A, Morikawa Y, Kezuka C, Takazawa H, Endo S, et al. Induction of aldo-keto reductases (AKR1C1 and AKR1C3) abolishes the efficacy of daunorubicin chemotherapy for leukemic U937 cells. Anticancer Drugs. 2014;25:868–77. doi: 10.1097/CAD.0000000000000112. [DOI] [PubMed] [Google Scholar]
- 9.Matsunaga T, Hojo A, Yamane Y, Endo S, El-Kabbani O, Hara A. Pathophysiological roles of aldo-keto reductases (AKR1C1 and AKR1C3) in development of cisplatin resistance in human colon cancers. Chem Biol Interact. 2013;202:234–42. doi: 10.1016/j.cbi.2012.09.024. [DOI] [PubMed] [Google Scholar]
- 10.Gene: AKR1C1; Analysis type: cancer vs. normal analysis; data type: mRNA; sample type: clinical specimen. https://www.oncomine.org/resource/login.html.
- 11.Zhu H, Hu Y, Zeng CM, Chang LL, Ge FJ, Wang WH, et al. The SIRT2-mediated deacetylation of AKR1C1 is required for suppressing its pro-metastasis function in non-small cell lung cancer. Theranostics. 2020;10:2188–200. doi: 10.7150/thno.39151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–48. doi: 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–72. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 14.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 15.Jatana N, Ascher DB, Pires DEV, Gokhale RS, Thukral L. Human LC3 and GABARAP subfamily members achieve functional specificity via specific structural modulations. Autophagy. 2020;16:239–55. doi: 10.1080/15548627.2019.1606636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem Sci. 2012;37:230–6. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L, Yan L, Liao N, Wu WQ, Shi JL. A review of ULK1-mediated autophagy in drug resistance of cancer. Cancers. 2020;12:352. doi: 10.3390/cancers12020352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9:1167–81. doi: 10.1158/2159-8290.CD-19-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282:4672–8. doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- 20.Liang C, Xu J, Meng QC, Zhang B, Liu J, Hua J, et al. TGFB1-induced autophagy affects the pattern of pancreatic cancer progression in distinct ways depending on SMAD4 status. Autophagy. 2020;16:486–500. doi: 10.1080/15548627.2019.1628540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103:760–6. doi: 10.1111/j.1349-7006.2012.02216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Denk H, Stumptner C, Fuchsbichler A, Muller T, Farr G, Muller W, et al. Are the Mallory bodies and intracellular hyaline bodies in neoplastic and non-neoplastic hepatocytes related? J Pathol. 2006;208:653–61. doi: 10.1002/path.1946. [DOI] [PubMed] [Google Scholar]
- 24.Nuttli T, McGoey RR. Pathology case of the month. Altered mental status, alcohol abuse, and hyperammonemia. DIAGNOSIS: Mallory-Denk bodies (a.k.a. Mallory’s hyaline)-seen most commonly in alcoholic liver disease. J La State Med Soc. 2014;166:46–8. [PubMed] [Google Scholar]
- 25.Ling JH, Kang YA, Zhao RY, Xia QH, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–20. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karras P, Riveiro-Falkenbach E, Canon E, Tejedo C, Calvo TG, Martinez-Herranz R, et al. p62/SQSTM1 fuels melanoma progression by opposing mRNA decay of a selective set of pro-metastatic factors. Cancer Cell. 2019;35:46–63. doi: 10.1016/j.ccell.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Duran A, Hernandez ED, Reina-Campos M, Castilla EA, Subramaniam S, Raghunandan S, et al. p62/SQSTM1 by binding to Vitamin D receptor inhibits hepatic stellate cell activity, fibrosis, and liver cancer. Cancer Cell. 2016;30:595–609. doi: 10.1016/j.ccell.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Mun SR, Linares JF, Towers CG, Thorburn A, Diaz-Meco MT, et al. Mechanistic insight into the regulation of SQSTM1/p62. Autophagy. 2019;15:735–7. doi: 10.1080/15548627.2019.1569935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carroll B, Otten EG, Manni D, Stefanatos R, Menzies FM, Smith GR, et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat Commun. 2018;9:256. doi: 10.1038/s41467-017-02746-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng H, Yang J, Li GY, You Q, Han W, Li TR, et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res. 2017;27:657–4. doi: 10.1038/cr.2017.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–31. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Puissant A, Fenouille N, Auberger P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res. 2012;2:397–413. [PMC free article] [PubMed] [Google Scholar]
- 33.Moscat J, Karin M, Diaz-Meco MT. p62 in cancer: signaling adaptor beyond autophagy. Cell. 2016;167:606–9. doi: 10.1016/j.cell.2016.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El-Kabbani O, Scammells PJ, Day T, Dhagat U, Endo S, Matsunaga T, et al. Structure-based optimization and biological evaluation of human 20alpha-hydroxysteroid dehydrogenase (AKR1C1) salicylic acid-based inhibitors. Eur J Med Chem. 2010;45:5309–17. doi: 10.1016/j.ejmech.2010.08.052. [DOI] [PubMed] [Google Scholar]
- 35.Zeng CM, Zhu DF, You J, Dong XW, Yang B, Zhu H, et al. Liquiritin, as a natural inhibitor of AKR1C1, could interfere with the progesterone metabolism. Front Physiol. 2019;10:833. doi: 10.3389/fphys.2019.00833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Couture JF, Legrand P, Cantin L, Luu-The V, Labrie F, Breton R. Human 20alpha-hydroxysteroid dehydrogenase: crystallographic and site-directed mutagenesis studies lead to the identification of an alternative binding site for C21-steroids. J Mol Biol. 2003;331:593–604. doi: 10.1016/s0022-2836(03)00762-9. [DOI] [PubMed] [Google Scholar]
- 37.El-Kabbani O, Scammells PJ, Gosling J, Dhagat U, Endo S, Matsunaga T, et al. Structure-guided design, synthesis, and evaluation of salicylic acid-based inhibitors targeting a selectivity pocket in the active site of human 20alpha-hydroxysteroid dehydrogenase (AKR1C1) J Med Chem. 2009;52:3259–64. doi: 10.1021/jm9001633. [DOI] [PubMed] [Google Scholar]
- 38.Faure P, Tamisier R, Baguet JP, Favier A, Halimi S, Levy P, et al. Impairment of serum albumin antioxidant properties in obstructive sleep apnoea syndrome. Eur Respir J. 2008;31:1046–53. doi: 10.1183/09031936.00062707. [DOI] [PubMed] [Google Scholar]
- 39.Song JH, Harris MS, Shin SH. Effects of fetal bovine serum on ferrous ion-induced oxidative stress in pheochromocytoma (PC12) cells. Neurochem Res. 2001;26:407–14. doi: 10.1023/a:1010907316475. [DOI] [PubMed] [Google Scholar]
- 40.Zheng X, Baker H, Hancock WS, Fawaz F, McCaman M, Pungor E., Jr Proteomic analysis for the assessment of different lots of fetal bovine serum as a raw material for cell culture. Part IV. Application of proteomics to the manufacture of biological drugs. Biotechnol Prog. 2006;22:1294–300. doi: 10.1021/bp060121o. [DOI] [PubMed] [Google Scholar]
- 41.Harris IS, DeNicola GM. The complex interplay between antioxidants and ROS in cancer. Trends Cell Biol. 2020;30:440–51. doi: 10.1016/j.tcb.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 42.Chio IIC, Tuveson DA. ROS in cancer: the burning question. Trends Mol Med. 2017;23:411–29. doi: 10.1016/j.molmed.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang JY, Ou-Yang F, Hou MF, Huang HW, Wang HR, Li KT, et al. Oxidative stress-modulating drugs have preferential anticancer effects - involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin Cancer Biol. 2019;58:109–17. doi: 10.1016/j.semcancer.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 44.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang YG, Zhu XR, Lu R, Messer JS, Xia YL, Chang EB, et al. Intestinal epithelial HMGB1 inhibits bacterial infection via STAT3 regulation of autophagy. Autophagy. 2019;15:1935–53. doi: 10.1080/15548627.2019.1596485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soutar MPM, Kempthorne L, Annuario E, Luft C, Wray S, Ketteler R, et al. FBS/BSA media concentration determines CCCP’s ability to depolarize mitochondria and activate PINK1-PRKN mitophagy. Autophagy. 2019;15:2002–11. doi: 10.1080/15548627.2019.1603549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung C, Seo W, Silwal P, Jo EK. Crosstalks between inflammasome and autophagy in cancer. J Hematol Oncol. 2020;13:100. doi: 10.1186/s13045-020-00936-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El Hout M, Cosialls E, Mehrpour M, Hamai A. Crosstalk between autophagy and metabolic regulation of cancer stem cells. Mol Cancer. 2020;19:27. doi: 10.1186/s12943-019-1126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14:243–51. doi: 10.1080/15548627.2017.1402992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. 2017;86:193–224. doi: 10.1146/annurev-biochem-061516-044908. [DOI] [PubMed] [Google Scholar]
- 51.Huang HS, Zhu JL, Li Y, Zhang LP, Gu JY, Xie QP, et al. Upregulation of SQSTM1/p62 contributes to nickel-induced malignant transformation of human bronchial epithelial cells. Autophagy. 2016;12:1687–703. doi: 10.1080/15548627.2016.1196313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schrezenmeier E, Dorner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol. 2020;16:155–66. doi: 10.1038/s41584-020-0372-x. [DOI] [PubMed] [Google Scholar]
- 53.Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, et al. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife. 2015;4:e08941. doi: 10.7554/eLife.08941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Selga E, Noe V, Ciudad CJ. Transcriptional regulation of aldo-keto reductase 1C1 in HT29 human colon cancer cells resistant to methotrexate: role in the cell cycle and apoptosis. Biochem Pharmacol. 2008;75:414–26. doi: 10.1016/j.bcp.2007.08.034. [DOI] [PubMed] [Google Scholar]
- 55.Smith AG, Macleod KF. Autophagy, cancer stem cells and drug resistance. J Pathol. 2019;247:708–18. doi: 10.1002/path.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei YJ, Zou ZJ, Becker N, Anderson M, Sumpter R, Xiao GH, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154:1269–84. doi: 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2016;35:1–11. doi: 10.1038/onc.2015.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee DH, Park JS, Lee YS, Han J, Lee DK, Kwon SW, et al. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy. 2020;16:1949–73. doi: 10.1080/15548627.2020.1712108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi YL, Fan SQ, Wu MG, Zuo ZX, Li XY, Jiang LP, et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. 2019;10:4892. doi: 10.1038/s41467-019-12801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murray JR, de la Vega L, Hayes JD, Duan L, Penning TM. Induction of the antioxidant response by the transcription factor NRF2 increases bioactivation of the mutagenic air pollutant 3-nitrobenzanthrone in human lung cells. Chem Res Toxicol. 2019;32:2538–51. doi: 10.1021/acs.chemrestox.9b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.