

Abstract
Extracellular vesicles (EVs) have been proved a promising small interfering RNA (siRNA) delivery vehicle to mediate gene‐silencing. Tumour‐derived extracellular vesicles (TDEVs) as genetic exchange vectors in the tumour microenvironment, enable intercellular communication for a wide range of endogenous cargo molecules, such as RNAs and proteins. However, the oncogenic cargo of TDEVs limits their application in siRNA delivery for cancer therapy. Herein, we isolated TDEVs from hepatocellular carcinoma (HCC) cells and derived TDEV membranes by abandoning their content. Innovative TDEV membrane hybrid lipid nanovesicles (LEVs) were then fabricated by fusion of TDEV membranes and phospholipids to realize precise delivery to tumours and highly efficient transfection of siRNA. The TDEV membranes endow LEVs with ‘homing’ targeting ability, facilitating specific internalisation into parent HCC cells primarily through heparan sulfate proteoglycan‐mediated pathways. Unlike conventional lipid‐based nanovesicles, LEVs can bypass the endosomal degradation pathway, boost the delivery of siRNA through the Golgi and endoplasmic reticulum (ER) intracellular ‘freeway’ transportation, achieving a 1.7‐fold improvement in siRNA transfection efficiency compared with liposomes. Additionally, siRNA loaded LEVs were demonstrated to enhance the antitumour efficacy in HCC bearing mice through effective gene silencing in the tumour sites. Our results highlight the potential application of the TDEV membrane‐derived nanovesicles as an advanced siRNA delivery strategy for cancer therapy.
Keywords: tumour‐derived extracellular vesicles, hybrid lipid nanovesicles, tumour homing, siRNA delivery, hepatocellular carcinoma
1. INTRODUCTION
Small interfering RNA (siRNA)‐based gene therapy for specific gene silencing, holds great promise in the treatment of various diseases including cancer, genetic disorders and viral infections (Alterman et al., 2019; Chen, Mangala, et al., 2018; Zou et al., 2020) As free siRNA is unstable in the bloodstream and does not readily cross membranes to enter cells, effective delivery systems for siRNA, including polymer‐ or lipid‐based carriers, are used for the application of siRNA therapeutics (Dong et al., 2019). In recent years, extracellular vesicles (EVs) have attracted extensive interest in therapeutic siRNA delivery, which is further enhanced by their potential clinical utility. The endogenous nature, intrinsic physiological activity and cell transfection properties endow EVs with long circulation ability for siRNA delivery, as well as little immunogenicity (Barile & Vassalli, 2017; Kalluri & LeBleu, 2020). Guo et al. reported that EVs derived from mesenchymal stem cells could deliver siRNA specifically and safely after systemic administration (Guo et al., 2019). Kamerkar et al. described that human foreskin fibroblast‐derived EVs with siRNA loading facilitate delivery to oncogenic KARS in pancreatic cancer by evading phagocytosis of circulating monocytes, thus exerting superior siRNA transfection efficacy compared with liposomes (Kamerkar et al., 2017). Didiot et al. showed that stem cells‐derived EVs could deliver siRNA to brain and mediate effective internalisation in primary cortical neurons, which is expected to advance the development of therapies for the treatment of neurodegenerative disorders (Didiot et al., 2016).
The interaction of EVs with recipient cells relies on the specific proteins, lipids and glycans on their surface. It is currently difficult to control the interaction and uptake of EVs depending on their subtype (O'Brien et al., 2020). Recently, engineered EVs through exogenous peptide, protein or lipid modification, have been reported to facilitate EV targeting to specific recipient cells (Belhadj et al., 2020). For example, RGD and RVG modified EVs could target cancer cells and brain regions for chronic neurodegenerative disorders, respectively (Alvarez‐Erviti et al., 2011; Cao et al., 2019). However, the engineering approach of affinity‐based peptide coating is transient and unstable, while chemical conjugation may cause undesirable loss of EV protein function (Gao et al., 2018; Kooijmans et al., 2018; Pham et al., 2021;). In addition, the transfection efficiency of siRNA is another factor which affects the efficacy of gene therapy. Because a prominent role of intracellular pathway is to shuttle cargo to the lysosome for degradation, endosomal escape of EVs is critical to siRNA function within recipient cells. It is possible that EVs might naturally incorporate some mechanisms for endosomal escape, which so far remains unknown. In general, limited targeting ability as well as unclear intracellular transportation mechanism, exert an unpredictable impact on EVs internalisation and exogenous nucleic acid transfection efficiency for cancer therapy.
Recently, tumour‐derived extracellular vesicles (TDEVs) have aroused the interest of researchers. TDEVs are emerging as key players in conveying molecular and genetic messages from tumour cells to tissues residing at close or distant sites through transfer of information via their cargo, which includes messenger RNAs, DNAs and proteins, thus promoting tumour proliferation and metastasis (Mashouri et al., 2019). Although TDEVs were reported to home to specific recipient cells depending on the protein expression on their surface, the cargos carried by TDEVs are critical components of oncogenic transformation (Sun et al., 2018), leading to less application of TDEVs in drug delivery. To address this problem, we suppose the emerging cell membrane extraction technology may expand the application of TDEVs. The cell membrane‐camouflaged technology has been widely used in various cell‐bionic nanocarriers for disease treatment, such as cancer cells, macrophages and red blood cells (Zhen et al., 2019). Briefly, the cell membrane was collected by removing the contents of the cells, with proteins on the cell membrane inherited, and then combined with flexible substrate materials to prepare cell‐membrane coating nanoparticles.
Herein, inspired by the fact that biomimetic nanoparticles engineered by coating cell membrane could be endowed with surface antigenic diversity and biological benefits of natural cells (Thanuja et al., 2018), we proposed innovative nanovesicles (LEVs) fabricated by hybridisation of TDEV membranes and phospholipids for siRNA delivery. LEVs containing hepatocellular carcinoma (HCC) Sk‐hep1 cell‐derived EV membranes are expected to inherit the intercellular communication ability of TDEVs for the enhanced delivery of siRNA to parent tumour cells. Lipid, hybrid with TDEV membranes, is expected to improve the drug loading capacity and stability of nanovesicles and to be widely used in the treatment of various diseases, just like liposomes. Moreover, cyclin‐dependent kinase 1 (CDK1)‐siRNA (siCDK1), which inhibits the CDK1 gene to kill c‐Myc‐overexpressing HCC cells effectively by synthetic lethality (Goga et al., 2007), was used as a model siRNA to be encapsulated into LEVs to obtain additional benefits for systemic delivery of siRNA. We further explored the endocytic and intracellular pathways of siCDK1‐loaded LEVs (siCDK1@LEVs) both in vitro and in vivo and evaluated siRNA transfection efficiency.
2. MATERIALS AND METHODS
2.1. Cell culture and TDEVs isolation
Human HCC cell line Sk‐hep1 and normal liver cell line LO2 were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin‐streptomycin. Human HCC cell line HepG2 and human umbilical vein endothelial cell line HUVEC were cultured in DMEM medium supplemented with 10% FBS and 1% penicillin‐streptomycin. The cells were cultured at 37°C in a 5% CO2 atmosphere.
To isolate TDEVs from Sk‐hep1 cells, cells were cultured in serum‐free medium for 48 h to eliminate the interference of plasma EVs. Then, the culture medium was collected for TDEVs isolation by differential centrifugation as reported previously (Zhang et al., 2015). Briefly, the culture medium was centrifuged at 300 × g, 2000 × g and 10,000 × g in turn, to remove dead cells and apoptotic bodies. TDEVs were collected via centrifugation at 100,000 × g for 1 h at 4°C (Beckman Coulter Optima XPN, SW45 rotor, USA). Membranes of TDEVs were derived by suspending the TDEVs in water, followed by centrifugation at 25,000 × g for 15 min at 4°C to remove the content. The TDEV membrane (TDEV Mem) was quantified by the BCA protein assay kit and stored at ‐80°C. EVs derived from LO2 cells were isolated using the same procedure as the TDEVs.
2.2. Preparation of LEVs
LEVs composed of TDEV membranes and 1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphocholine (DPPC, A.V.T. Pharmaceutical Co., Ltd, China) were prepared by thin film hydration. Briefly, DPPC film evaporated at 40°C was hydrated in PBS containing TDEV membranes (1 mg/ml). Then the mixture was extruded through a microextruder device (Hamilton Co., Reno, Nevada) to obtain LEVs.
To optimise the hybrid ratio of DPPC and TDEV membranes in LEVs, DPPC films and TDEV membranes were separately labelled with DiO and DiI (Meilunbio, China). LEVs were prepared using different ratios of DPPC and TDEV membranes, and then detected by a microplate reader (Synergy H1, Biotek, USA) for fluorescence resonance energy transfer (FRET) study. The fluorescence emission spectrum of dual fluorescence‐labelled LEVs from 470 to 650 nm was recorded with an excitation wavelength of 440 nm.
siCDK1 (5’‐GGAACUUCGUCAUCCAAAUTT‐3’, Shanghai GenePharma Co. Ltd, China) was loaded into LEVs through electroporation using a Gene Pulser Xcell (BIO‐RAD, USA). LEVs and siCDK1 were gently mixed at a 1:1 (v/v) ratio in the prepared electroporation solution and then rapidly electroporated in a 2 mm cuvette at 400 mV and 125 μF capacitance to obtain siCDK1‐loaded LEVs (siCDK1@LEVs). The free siCDK1 was removed by Sephadex G‐50 gel chromatography. Conventional liposomes (Lips) without TDEV membranes were prepared by DPPC film hydrated in PBS, with a lipid concentration of 2 mg/ml. The siCDK1‐loaded Lips (siCDK1@Lips) were obtained using the same procedure as the siCDK1@LEVs. For fluorescence labelling, DiI (or DiO) was added to DPPC and then hydrated with or without TDEV membranes in PBS to obtain DiI‐labelled LEVs and Lips (or DiO‐labelled LEVs and Lips).
2.3. Characterisation of LEVs
To confirm the successful hybridisation of DPPC films and TEDV membranes, LEVs composed of DiO‐labelled DPPC films and DiI‐labelled TDEV membranes were imaged using an Olympus FV1000 confocal laser scanning microscope (Olympus, Tokyo, Japan) and Stimulated Emission Depletion microscope (Leica TCS SP8 STED, Leica, Germany). The morphology of Lips and LEVs was observed by cryo‐TEM (200 kV, FEI Tecnai G2 F20, USA). The hydrodynamic size and zeta potential of Lips and LEVs were measured by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern Instruments). To investigate the storage stability of LEVs and Lips, the nanovesicles were kept in pH 7.4 PBS containing 10% FBS at 4°C and measured by DLS over 5 days.
2.4. Characterisation of TDEV membrane in LEVs
SDS‐PAGE was employed to characterise the TDEV membrane proteins. The TDEVs and TDEV‐derived samples (TDEV membranes and LEVs) which were obtained from equivalent TDEVs were lysed and boiled in SDS‐PAGE sample loading buffer. Samples were loaded on a 10% SDS‐PAGE gel, and electrophoresis was performed at 120 V using a Mini‐PROTEIN Tetra System (BIO‐RAD, USA). The resultant gel was stained in Coomassie Blue and washed overnight for subsequent imaging with a GBOX gel documentation system (Syngene, UK). For western blotting, TDEVs, TDEV membranes and LEVs lysed in RIPA buffer were separated on SDS‐PAGE gels and transferred onto a PVDF membrane (Beyotime, China). The blots were incubated with primary antibodies against CD9 (1:1000, Abcam, UK), CD63 (1:5000, Abcam, UK), Alix (1:5000, Abcam, UK), albumin (1:2000, Abcam, UK), CD44 (1:1000, Bioss, China), or CD47 (1:5000, Abcam, UK) overnight at 4°C. Horseradish peroxidase (HRP) conjugated secondary antibodies (1:2000, Beyotime, China) were incubated with the PVDF membranes at 37°C for 1 h. Signals were visualised by a Clinx‐6100 chemiluminescence imaging system (Clinx Science Instruments Co. Ltd., China).
Dot blotting was conducted to verify the direction of the TDEV membrane. The dot blot standards, LEVs and TDEV membranes were blotted onto a PVDF membrane. After blotting, the samples were air‐dried for complete adsorption and immobilisation of proteins onto the PVDF membrane. After blocking with 5% skim milk in 0.05% PBST solution, the membranes were incubated with extracellular CD47 antibody (Aviva Systems Biology, China) for primary antibody binding and exposed to chemiluminescence for detection of extracellular CD47 antibody binding after incubation with HRP‐conjugated secondary antibody. Image J was used to count the greyscale value of each dot, the sidedness of each sample was then calculated by comparing its greyscale to the greyscales of the standards.
To investigate the biosafety of the formulations, Sk‐hep1 cells (5 × 104 cells/ml) were seeded into a 96‐well plate and incubated with TDEVs, LEVs and Lips at different concentrations for 48 h. LO2 and HUVEC cells were incubated with LEVs at different concentrations for 48 h. A MTT kit was used to assess cell viability using a microplate reader.
2.5. siCDK1 loading capacity and siCDK1 release from LEVs
The siCDK1@LEVs and siCDK1@Lips after electroporation were centrifuged at 100,000 × g for 1 h. The unencapsulated siCDK1 in the supernatant was detected at 260 nm using a microplate reader, to calculate the siRNA loading of nanovesicles through the weight ratio of encapsulated siCDK1 to siCDK1‐loaded nanovesicles.
To further evaluate the protection of LEVs on siRNA, the stability of siCDK1 loaded in nanovesicles against serum degradation was evaluated by gel electrophoresis. Free siCDK1, siCDK1@LEVs and siCDK1@Lips were incubated with 10% FBS at 37°C, and aliquots were collected at different time points. Triton X‐100 was then added to dissociate the siCDK1, and the mixture was loaded on a 4% agarose gel. RNA fractions were visualised by staining with ethidium bromide.
To study the release of siCDK1 from Lips and LEVs, Cy3‐ siCDK1 (RiboBio Co., Ltd., China) loaded nanovesicles were added to dialysis bags (MWCO 100 kDa, Millipore, Germany), which were immersed in a simulated physical environment (pH 7.4 PBS) or endosomal environment (pH 5.0 PBS) at 37°C. At predetermined time intervals, samples were withdrawn from the dissolution media followed by replacement of fresh buffer solution. The released Cy3‐siCDK1 was quantified using a microplate reader.
2.6. Cellular uptake and tumour cell selectivity study of LEVs
To evaluate the cellular uptake of nanovesicles, Sk‐hep1 or LO2 cells were seeded on coverslips in 12‐well plates and incubated with DiI‐labelled nanovesicles (10 μg/ml) for 4 h. The cells were then fixed with 4% paraformaldehyde and stained with DAPI, followed by imaging using confocal laser scanning microscope (CLSM). For flow cytometric analysis, Sk‐hep1 and LO2 cells were separately seeded in 12‐well plates and incubated with DiI‐labelled nanovesicles (10 μg/ml) for 4 h. Thereafter, the cells were washed and detached for analysis using a BD FACS flow cytometer.
To explore the mechanism of selective internalisation of LEVs by tumour cells, heparin (10 μg/ml), arginine‐glycine‐aspartate (RGD, 1 mM) and tyrosine‐isoleucine‐glycine‐serine‐arginine (YIGSR, 1 mM) were preincubated with Sk‐hep1 or LO2 cells for 1 h. Then the cells were incubated with DiI‐labelled nanovesicles (10 μg/ml) for 4 h and detached for analysis using a BD FACS flow cytometer. For confocal imaging, Sk‐hep1 or LO2 cells were fixed and stained with DAPI after incubation with DiI‐labelled nanovesicles (10 μg/ml). The expression of HSPGs and integrins was evaluated by western blot. The blots were probed overnight with primary antibodies against HSPG or integrin β1 (1:250, Abcam, UK) at 4°C, and incubated with HRP conjugated secondary antibodies (1:2000, Beyotime, China).
To further observe the effect of HSPGs or integrins on the specific internalisation of LEVs in HCC cells, immunofluorescence staining was used to label HSPGs or integrins in Sk‐hep1 and LO2 cells. Briefly, DiI‐labelled LEVs (10 μg/mL) were incubated with Sk‐hep1 or LO2 cells seeded on coverslips for 1 h. After fixation and blockage, the cells were incubated overnight at 4°C with anti‐HSPG (1:250, Abcam, UK) and anti‐integrin β1 (1:150, Abcam, UK), respectively. Subsequently, a fluorescence‐labelled secondary antibody was added and incubated with the cells for 1 h, and the nuclei were stained with DAPI followed by CLSM imaging. In addition, the cellular uptake of LEVs in Sk‐hep1 cells preincubated with anti‐HSPG or anti‐integrin β1 was detected by flow cytometry. Sk‐hep1 cells were incubated with anti‐HSPG or anti‐integrin β1 for 1 h, followed by incubation with DiI‐labelled LEVs (10 μg/ml) for 4 h. The cells were then detached for analysis using a BD FACS flow cytometer.
2.7. Endocytosis and intracellular trafficking of LEVs
To study the endocytosis mechanism of LEVs, Sk‐hep1 cells were separately pretreated with amiloride (2.5 mM), chlorpromazine (CPZ, 30 μM) and filipin (5 μg/ml) at 37°C for 1 h, then incubated with DiI‐labelled nanovesicles (10 μg/ml) for 4 h. Cells were harvested for flow cytometry analysis.
For investigation of the intracellular trafficking of nanovesicles, DiI‐labelled LEVs and Lips (10 μg/ml) were incubated with Sk‐hep1 cells seeded on coverslips for 4 h. After fixation with paraformaldehyde, the cells were blocked with 1% bovine serum albumin containing Triton X‐100 (0.3%, v/v) in PBS for 1 h, and then incubated overnight at 4°C with anti‐LAMP1 (1:200, Cell Signaling Technology, USA), anti‐Golgin‐97 (1:100, Cell Signaling Technology, USA) and anti‐KDEL (1:100, Abcam, UK) to label lysosomes, Golgi and endoplasmic reticulum (ER), respectively. Subsequently, a fluorescence‐labelled secondary antibody was added and incubated with cells for 1 h, and the nuclei were stained with DAPI followed by CLSM imaging. Pearson's coefficient was calculated using Coloc2 analysis in Image J software. To study the localisation of nanovesicles in subcellular organelles by FRET, Sk‐hep1 cells were seeded into plates and incubated with DiI‐labelled nanovesicles (10 μg/mL) for 4 h. After fixation and labeling with primary antibody and fluorescence‐labelled secondary antibody, the cells were detached for analysis by a microplate reader. The fluorescence emission spectrum of the samples from 470 to 700 nm was recorded at selected time points with an excitation wavelength of 440 nm.
2.8. Transfection efficiency and antitumour activity of siCDK1@LEVs
Sk‐hep1 cells were seeded in 6‐well plates for 24 h. Free siCDK1, free scramble siRNA, siCDK1‐loaded nanovesicles with a siRNA concentration of 100 nM and LEVs (1 μg/ml) were then added to each well. After incubating for 36 h, total RNA was extracted from cell pellets using TRIzol reagent (Invitrogen, USA) according to the manufacturer's instructions. Reverse transcription was performed using the PrimeScript RT Reagent Kit (Takara, Japan), and mRNA expression was quantified with the Applied Biosystems 7500 Fast Real‐Time PCR System (ABI, USA) using SYBR Premix Ex Taq II (Takara, Japan). The crossing threshold value for each gene was noted for the transcripts. Target gene expression was normalised to GAPDH expression. The sequences of the primers were as follows: CDK1, forward: 5’‐CAG GAT GTG CTT ATG CAG GA‐3’, reverse: 5’‐CCA CAA AAT GCA GGG ACT TC‐3’, GAPDH, forward: 5’‐CAT GGC CTT CCG TGT TCC TA‐3’, reverse: 5’‐CCT GCT TCA CCA CCT TCT TGA T‐3’. The protein expression of CDK1 was measured by western blot.
To study the antitumour activity of siCDK1 formulations, Sk‐hep1 cells were seeded in 96‐well plates (5 × 103 cells/well) and treated with LEVs, free siCDK1, scramble siRNA, siCDK1@Lips and siCDK1@LEVs at different concentrations for 36 h. Then, the MTT assay was performed. For apoptosis analysis, Sk‐hep1 cells were exposed to different formulations with a siRNA concentration of 250 nM for 36 h. Then, the cells were trypsinized, harvested, and stained using an Annexin V/PI Apoptosis Detection Kit. Apoptosis was measured with a BD Bioscience FACSCalibur flow cytometer.
2.9. Biodistribution and pharmacokinetic study
BALB/c nude mice, C57/BL6 and ICR mice were obtained from Shanghai Lab. Animal Research Center. All animal experiments were performed according to the Institutional Animal Care and Use Committee at Shanghai Institute of Materia Medica (IACUC code: 2020‐05‐GY‐58). SK‐hep1 cell suspension (1 × 107 cells) was subcutaneously injected into the axilla of BALB/c nude mice. When tumour volumes reached approximately 200 mm3 after 2 weeks of tumour implantation, DiR‐labelled Lips and LEVs which were prepared using the same procedure as the DiI‐labelled nanovesicles, were intravenously injected into tumour‐bearing mice. At 2, 4 and 8 h after injection, the mice were imaged by the IVIS Spectrum System (Caliper Cor., Waltham, Massachusetts, USA), and major tissues were harvested at 8 h and tumours were collected at 2, 4 and 8 h for ex vivo imaging, followed by quantification using a region of interest tool. To investigate the distribution of nanovesicles in tumour tissues, tumours were isolated from mice at 4 h after injection of DiI‐labelled Lips and LEVs. The tissue samples were sectioned into 10 μm thick slices (Leica 2800 Frigocut‐E slicer, Germany), followed by fixation and DAPI staining. The fluorescent signals were observed using confocal microscopy (Olympus FV 1000, Japan). For analysis of the intracellular trafficking pathway in vivo, the tumour sections were stained with anti‐LAMP1, anti‐Golgin‐97 and anti‐KDEL, followed by fluorescence‐labelled secondary antibody staining and confocal imaging.
To study the pharmacokinetics of nanovesicles, DiO‐labelled Lips and LEVs were intravenously injected into C57/BL6 mice (n = 3 for each group) at a dosage of 0.8 mg/kg. At predetermined time intervals, blood samples were collected and centrifuged at 3000 × g for 10 min. The plasma was mixed with acetonitrile and centrifuged again. The supernatant was collected to measure the fluorescence intensity of the nanovesicles using a microplate reader. The pharmacokinetic profiles were analysed by DAS software.
2.10. In vivo antitumour efficacy
Subcutaneous xenografts were established by implanting Sk‐hep1 cell suspension (1 × 107 cells) into the axilla of male BALB/c nude mice. When tumour volumes reached 50–100 mm3, the mice were randomly divided into five groups for the following treatments: 1) saline, 2) free siCDK1, 3) scramble siRNA, 4) siCDK1@Lips and 5) siCDK1@LEVs. Each group was intravenously injected with different formulations at siRNA dosage of 1.0 mg/kg every other day. The tumour volume and body weight of the mice (n = 6 for each group) were monitored over time, and survival was defined as natural death. The tumour volume was calculated as follows: volume = (length × width2)/2.
To further evaluate the transfection efficiency and assess safety, the same tumour model and treatment schedule as above was followed (n = 3 for each group). After treatment for 21 days, mice were sacrificed. Tumours collected from mice were homogenised to analyse CDK1 mRNA and protein levels by RT‐qPCR and western blot. The major organs (heart, liver, spleen, lung and kidney) and the tumour were dissected and sectioned for H&E staining. All histological analysis were carried out by the Center for Drug Safety Evaluation and Research (CDSER, SIMM CAS). Whole blood (n = 5 for each group) was collected from Sk‐hep1 tumour‐bearing mice of each group on day 21, and plasma was isolated to measure representative blood parameters including alaninetransaminase (AST) and alanine aminotransferase (ALT) by AST and ALT activity kits.
To investigate the immunogenicity of siCDK1‐loaded nanovesicles, ICR mice were intravenously injected with saline, siCDK1@Lips and siCDK1@LEV (n = 3 for each group) at a siCDK1 dosage of 1.0 mg/kg. At 24 h after injection, plasma was collected to measure TNF‐α and IL‐6 levels using an enzyme‐linked immunosorbent assay (ELISA) kit.
2.11. Statistical analysis
Significant differences were determined using unpaired Student's t test or Wilcoxon rank test for two group comparisons and ANOVA for comparisons of multiple treatment groups within individual experiments. p < 0.05 was considered significant. All values were presented as mean ± standard deviation (SD).
3. RESULTS AND DISCUSSION
3.1. Preparation of LEVs
The LEVs were fabricated by TDEV membranes and phospholipids through a thin‐film hydration and extrusion procedure, as illustrated in Figure 1a. To obtain LEVs with perfect hybridisation of TDEV membranes and phospholipids, FRET was performed with DiI‐labelled TDEV membranes and DiO‐labelled DPPC film to screen the ratio of these components (Figure 1b). A 1:1 weight ratio of TDEV membrane to DPPC film with the highest FRET efficiency, which represents the best hybridisation of both components and obvious colocalisation of red and green fluorescence signals, was chosen as the optimised LEVs for subsequent studies (Figure 1c). The LEVs displayed a nanovesicle structure with an average size of 117.2 nm, similar to that of DPPC liposomes (Lips) (Figure 1d, e and S1). The zeta potential of LEVs (‐25.9 ± 0.6 mV) was lower than that of Lips (‐14.9 ± 0.9 mV), further indicating the successful insertion of TDEV membrane debris into lipid vesicles (Figure 1e). To determine the stability of the developed LEVs under physiological conditions, samples were dispersed in pH 7.4 PBS containing 10% fetal bovine serum (FBS), and size was measured over time (Figure 1f). LEVs and Lips exhibited little difference in size over 5 days, indicating their good stability in biologically relevant media. In general, we successfully prepared TDEV membrane‐hybridised LEVs.
FIGURE 1.

Preparation and characterisation of LEVs. (a) Schematic illustration describing the design of LEVs that are expected to display ‘homing’ targeting to the parent tumour cells and intracellular ‘freeway’ transportation through the Golgi‐ER pathway for enhanced siRNA delivery efficiency. (b) The optimal ratio of DPPC and TDEV membranes in LEVs analysed by FRET. DPPC was labelled with DiO and TDEV membranes were labelled with DiI. (c) Confocal microscopy images and super‐resolution microscopy images of LEVs composed of TDEV membranes (red) and DPPC (green). Scale bar = 2 μm. (d) Representative TEM images of Lips and LEVs. Scale bar = 100 nm. (e) Average size and zeta potential of Lips and LEVs. (f) Stability of Lips and LEVs in pH 7.4 PBS containing 10% FBS. Data are displayed as the mean ± SD (n = 3)
3.2. Characterisation of LEVs and siCDK1 loading
The endogenous molecules on the TDEV membrane surface are expected to enhance the delivery of LEVs to tumour cells. To confirm the membrane protein retention on LEVs, the protein profiles of the TDEVs, TDEV membranes and LEVs were determined via SDS‐PAGE. As shown in Figure 2a, TDEV cargo was removed during the TDEV membrane extraction, and the associated membrane proteins were transferred to the LEVs. The retention of the specific biomarkers was further verified by western blotting (Figure 2b). Albumin, which is rich in the serum, was negligible in the TDEVs and TDEV membranes, indicating the high purity of TDEVs isolated from the cell culture medium. EV membrane markers CD9 and CD63 were rich in TDEV membrane and LEVs, while cytosolic protein Alix highly expressed in TDEVs showed the decreased expression level in TDEV membranes and LEVs, suggesting that LEVs inherited TDEV membranes proteins and lost the TDEV cargos. CD44, which is highly expressed in tumour cells and TDEVs (Chen, Zhao, et al., 2018), was also transferred to the TDEV membranes and LEVs (Figure 2b and c). Additionally, CD47, a widely reported long‐term circulation transmembrane protein (Belhadj et al., 2020; Tang et al., 2019), was also present on the developed LEVs. To characterise the right‐side‐out coating of the TDEV membranes, which is important for retention of the membrane's biological functions, non‐denaturing immunoblotting was explored to determine the extracellular domain content of the transmembrane protein CD47 in LEVs (Figure 2d). The extracellular domain of CD47 on the LEVs was nearly 100% of that in the TDEV Mem, suggesting the right‐side‐out orientation of TDEV membranes in the LEVs (Figure 2e).
FIGURE 2.

Surface protein characterisation and siRNA loading capacity of LEVs. (a) SDS‐PAGE protein analysis of TDEVs, TDEV membranes, and LEVs for the characteristic TDEV membrane proteins. (b) Western blot analysis of protein markers albumin, CD9, CD63, Alix, CD44 and CD47. (c) Western blot and quantitative analysis of CD44 expression on EVs derived from Sk‐hep1 and LO2 cells. (d) Dot blot images and (e) quantitative intensities of TDEV Mem and LEVs probed with antibodies against the extracellular domains of CD47. (f) Cell viability of Sk‐hep1 cells incubated with TDEVs, Lips, and LEVs at various concentrations. (g) Cell viability of HUVEC and LO2 cells incubated with LEVs at various concentrations. (h) siRNA loading of Lips and LEVs determined by a microplate reader. (i) Electrophoresis strips of free siCDK1, siCDK1@Lips and siCDK1@LEVs after incubation with fetal bovine serum. (j) Release profiles of siCDK1 from Lips and LEVs in pH 7.4 and pH 5.0 PBS. Data are displayed as the mean ± SD (n = 3). n.s. p > 0.05; ***p < 0.001; ****p < 0.0001.
It has been reported that TDEVs promote tumour proliferation through tumourigenic contents (Azmi et al., 2013). To investigate the safety of the developed LEVs, the viability of Sk‐hep1 cells, which are the origin of TDEVs, was chosen as an evaluation index. As shown in Figure 2f, the viability of Sk‐hep1 cells treated with TDEVs increased to 229.8%, indicating the tumour growth‐promoting effect of TDEVs. In contrast, LEVs exhibited no effect on Sk‐hep1 viability, indicating the safety of LEVs without tumourigenicity, similar to conventional Lips. Moreover, LEVs showed good biological safety in normal cell lines LO2 and HUVEC (Figure 2g). These results further demonstrated the desirable safety of LEVs for siRNA delivery.
To explore the potential of LEVs in siRNA delivery, we encapsulated siCDK1 in LEVs and examined the drug loading and stability of siRNA. As shown in Figure 2h, siRNA was incorporated into LEVs with a high loading efficiency up to 56%. Then, the degradation protection of nanovesicles for siCDK1 was studied by testing the siCDK1 contents in different formulations incubated with FBS. As shown in Figure 2i, electrophoresis strips of siCDK1 in Lips and LEVs were still detectable at 12 h, while the strip of free siCDK1 solution nearly disappeared at 8 h, indicating that encapsulation of siCDK1 molecules into LEVs could protect siCDK1 from degradation by serum nuclease. Furthermore, the in vitro release profile showed that siRNA loaded Lips exhibited increased release of siRNA in pH 5.0 PBS, while the LEVs showed sustained release characteristics both in pH 7.4 and 5.0 PBS (Figure 2j). This result suggested that the LEVs could reduce siRNA leakage in the physiological environment and protect siRNA from degradation in the intracellular acid environment.
3.3. The cellular uptake selectivity of LEVs
EVs are extracellular nano‐shuttles that mediate intercellular communication between cells and organs. To investigate the cellular internalisation of LEVs, we explored the cellular uptake of LEVs in the human HCC cell line Sk‐hep1, a parental cell line of TDEVs, and in the human normal liver cell line LO2 in vitro (Figure 3a and b). Compared to Lips, LEVs showed 2.0‐fold higher fluorescence intensity in Sk‐hep1 cells, suggesting the enhanced uptake of LEVs in tumour cells (Figure 3c). Additionally, the fluorescence intensity of LEVs in Sk‐hep1 cells was 3.7‐fold higher than that in LO2 cells. These results demonstrated that LEVs could be specifically internalised in the parent tumour cells, which could be called ‘homing’ targeting.
FIGURE 3.

The cellular uptake selectivity of LEVs. (a) Representative CLSM images of Sk‐hep1 and LO2 cells incubated with DiI‐labelled Lips and LEVs. Scale bar = 50 μm. (b) Representative flow cytometry histograms of Sk‐hep1 cells and LO2 cells treated with DiI‐labelled Lips and LEVs. (c) Mean fluorescence intensities from (b). (d) Representative CLSM images of DiI‐labelled Lips and LEVs within Sk‐hep1 and LO2 cells pretreated with heparin, RGD and YIGSR. Scale bar = 50 μm. (e) Representative flow cytometry histograms and (f) mean fluorescence intensity of Sk‐hep1 cells incubated with DiI‐labelled LEVs in the presence of heparin, RGD and YIGSR. (g) Inhibitory rate of DiI‐labelled LEVs internalised into Sk‐hep1 and LO2 cells by heparin, RGD and YIGSR. (h) Expression level of HSPG and integrin in Sk‐hep1 and LO2 cells determined by western blot. (i) Representative immunofluorescence staining images of DiI‐labelled LEVs in Sk‐hep1 or LO2 cells. Nuclei were stained with DAPI (blue). HSPG and integrin were labelled with anti‐HSPG and anti‐integrin (green), respectively. Scale bar = 20 μm. (j) Fluorescence intensity of DiI‐labelled LEVs in Sk‐hep1 cells incubated with anti‐HSPG or anti‐integrin. (k) Inhibitory rate of DiI‐labelled LEVs internalisation into Sk‐hep1 cells with anti‐HSPG or anti‐integrin pretreatment. (l) Representative flow cytometry histograms of Sk‐hep1 cells and HepG2 cells treated with DiI‐labelled Lips and LEVs. (m) Mean fluorescence intensities from (l). Data are displayed as the mean ± SD (n = 3). **p < 0.01; ****p < 0.0001.
To further investigate the homing targeting mechanism of LEVs, we explored the specific uptake of LEVs into Sk‐hep1 cells. Heparan sulfate proteoglycans (HSPGs), a family of membrane proteins highly expressed in most HCCs, have been reported to act as internalizing receptors of TDEVs and virus particles (Christianson et al., 2013). Integrins are transmembrane receptors that have been reported to relate to the cellular uptake of EVs (Nolte et al., 2021). We speculated that integrins and HSPGs on Sk‐hep1 cell membranes may play important roles in the ‘homing’ targeting of LEVs. Then, the role of the two proteins in ‘homing’ targeting was investigated through heparin, RGD and YIGSR pretreatment, which are inhibitors of HSPGs, integrin subunits αV and β1, respectively. As shown in Figure 3d–f and S2, LEVs showed decreased fluorescence signals in Sk‐hep1 cells after shielding HSPGs and integrins, and the inhibitory rate of LEV cellular uptake in Sk‐hep1 cells was up to 3.5 times higher than that in LO2 cells (Figure 3g). Meanwhile, we found that Sk‐hep1 cells possessed 4.3‐fold higher HSPGs expression and 2.7‐fold higher integrin β1 expression than LO2 cells (Figure 3h). The results suggested that the ‘homing’ targeting capacity of LEVs may be correlated with HSPG‐ and integrin‐dependent pathways.
To further explore the mechanism of specific internalisation of LEVs in Sk‐hep1 cells, cells were pretreated with anti‐HSPG and anti‐integrin, and then incubated with LEVs. As shown in Figure 3i a large amount of LEVs distributed around the signal of HSPGs in Sk‐hep1 cells, which may be attributed to the EV‐cell interaction mediated by the linkage of fibronectin between LEV‐HSPGs and cell membrane‐HSPGs, consistent with the literature reported (Cerezo‐Magaña et al., 2020; Purushothaman et al., 2016). After specific blockage of HSPGs through antibodies, LEVs showed an approximately 40% decrease of internalisation in Sk‐hep1 cells (Figure 3j, k and S3). In contrast, a small fraction of LEVs was colocalised with integrins, and only 12.8% of decrease was observed in LEV uptake within Sk‐hep1 cells after blockage of integrins. These results demonstrated that the HSPG‐mediated pathway plays a major role in the ‘homing’ targeting capacity of LEVs.
It is worth noting that LEVs also exhibited selective cellular uptake in HepG2 cells, similar to that in Sk‐hep1 cells. The fluorescence intensity of LEVs in HepG2 cells was 3.2 times higher than that of Lips (Figure 3l and m). This may be attributed to the fact that HSPGs overexpressed in HepG2 cells as hepatic carcinoma cells, promoting LEVs internalisation through a HSPG‐dependent pathway. These results demonstrated that LEVs could specifically home to HCC cells.
3.4. Intracellular freeway transportation and gene silencing efficiency of LEVs
The intracellular transportation of LEVs plays an important role in the transfection efficiency of siRNA. Hence, we investigated the intracellular fate of LEVs in Sk‐hep1 cells. The endocytic pathway was studied by using different inhibitors, including chlorpromazine (CPZ, inhibitor of clathrin‐mediated endocytosis), filipin (inhibitor of caveolae‐dependent endocytosis) and amiloride (inhibitor of Na+/H+ pump related macropinocytosis). As shown in Figure 4a and S4, Lips in cells treated with filipin showed a significant decrease in cellular uptake, indicating that Lips mainly transported through caveolae‐mediated endocytosis. Compared to Lips, LEVs showed decreased internalisation in Sk‐hep1 cells separately incubated with CPZ and amiloride, indicating that clathrin‐dependent endocytosis and macropinocytosis were partially involved. These results suggested that the endocytic pathway of LEVs was different from that of Lips, which may lead to different intracellular fates for Lips and LEVs. Moreover, there was no difference in the uptake of traditional Lips and LEVs by Sk‐hep1 cells after using prazosin hydrochloride to inhibit the endosomal recycling pathway (Figure S5), indicating that LEVs can avoid the recycling pathway.
FIGURE 4.

Intracellular pathway study and gene silencing efficacy of siCDK1@LEVs. (a) Endocytosis pathway of Lips and LEVs probed by different inhibitors. (b) Biodistribution of DiI‐labelled nanovesicles (red) in Sk‐hep1 cells. Nuclei were stained with DAPI (blue), and subcellular organelles were labelled with anti‐LAMP1, anti‐Golgin‐97 and anti‐KDEL (green), respectively. Scale bar = 50 μm. (c) Pearson's R value for the colocalisation of nanovesicles and subcellular organelles from (b). (d) Localisation of nanovesicles (red) in subcellular organelles (green) detected by FRET. (e) Schematic illustration describing the endocytic pathway and intracellular transport of LEVs. (f) mRNA and protein levels of CDK1 in Sk‐hep1 cells with different treatments determined by RT‐qPCR and western blotting, respectively. (g) Cell viability of Sk‐hep1 cells after incubation with different formulations for 36 h. (h) IC50 values of siCDK1@Lips and siCDK1@LEVs. (i) Flow cytometric examination and (j) quantitative analysis of Sk‐hep1 cell apoptosis after different treatments. Data are displayed as the mean ± SD (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
The different endocytic pathway of LEVs may further affect the intracellular transport pathway for siRNA delivery. We then evaluated the colocalisation of LEVs with LAMP1 (a marker of lysosome), Golgin‐97 (a marker of Golgi) and KDEL (a marker of ER). As shown in Figure 4b and c, Lips showed 37.3% colocalisation with lysosome, which suggested that traditional liposomes transported mainly through the lysosome pathway, leading to the loaded siRNA prone to be degraded by lysosome. In contrast, LEVs obviously enhanced colocalisation with the Golgi (∼40%) and ER (∼60%), which is 6.7‐ and 2.2‐fold higher than that of the Lips group, indicating that the TDEV membranes incorporated in LEVs contributed to avoiding degradation by the lysosome pathway. To precisely evaluate the distribution of LEVs in the lysosomes, Golgi apparatus and ER, FRET was performed by labeling the subcellular organelles of Sk‐hep1 cells after incubation with different nanovesicles. Lips showed little increase of fluorescence intensity at 570 nm in the Golgi apparatus and ER at 4 h of incubation, while LEVs showed notably increased fluorescence intensity (Figure 4d), suggesting high colocalisation ratios between LEVs and the Golgi apparatus or ER. LEVs were demonstrated to deliver siRNA from Golgi to ER in the same way as endogenous protein sorting, which has been proven to be a rapid and efficient transport pathway (Gomez‐Navarro & Miller, 2016), thus achieving efficient siRNA transfection, similar to the electronic toll collection (ETC) channel on the freeway. Collectively, the uptake of LEVs depends on multiple endocytic pathways, including clathrin‐ and caveolae‐dependent endocytosis and macropinocytosis. In addition, through the Golgi‐ER intracellular freeway, LEVs could avoid the inefficient delivery of siRNA to enzyme‐filled lysosomes, achieving desirable gene delivery efficiency (Figure 4e).
To investigate gene transfection in vitro, the expression of the target gene CDK1 was quantitated at the mRNA and protein levels through quantitative reverse transcription polymerase chain reaction (RT‐qPCR) and western blotting assays, respectively. As shown in Figure 4f, free siRNA and LEVs displayed no effect on CDK1 expression in Sk‐hep1 cells. In contrast, siCDK1@LEV showed a remarkable inhibitory effect on the mRNA expression of CDK1 with a knockdown efficiency up to 86.7%, which was 1.7‐fold higher than that of siCDK1@Lips. A similar result was also found in the western blotting analysis, indicating enhanced gene silencing through siCDK1@LEVs treatment. The results can be attributed to the efficient delivery and excellent uptake (Figure 3c) of LEVs by Sk‐hep1 cells.
As reported in our previous work (He et al., 2018), c‐Myc‐overexpressing HCC cells were synthetically lethal with CDK1 inhibition, leading to specific apoptosis of HCC cells for increased safety and efficiency. We then tested the capability of siRNA‐loaded nanovesicles to inhibit cancer cell growth. As expected, inhibition of CDK1 expression by siCDK1@LEVs dramatically reduced the viability of Sk‐hep1 cells (IC50 = 141.0 ± 9.9 nM), which was 4.2‐fold lower than that of siCDK1@Lips (Figure 4g and h). Furthermore, a flow cytometric assay was used to investigate the effect on apoptosis of Sk‐hep1 cells. For the cells treated with the siCDK1@LEVs, the apoptotic efficiency reached 80.5%, which was significantly higher than that of the siCDK1@Lips (40.9%), and there was negligible inhibition in cancer cell growth after incubation with scramble siRNA, free siCDK1 or LEVs (Figure 4i and j). In general, this desirable siRNA transfection efficacy and antitumour activity were attributable to the ‘homing’ targeting proteins from TDEV membranes and the intracellular transport pathway different from the liposomes, permitting further application in HCC treatment.
3.5. In vivo ‘homing’ targeting and efficient intracellular transport
To investigate the ‘homing’ targeting ability of LEVs in vivo, we examined the biodistribution of DiR‐labelled Lips and LEVs in Sk‐hep1 tumour‐bearing BALB/c nude mice. As shown in Figure 5a, a near‐infrared signal was distributed in various organs and tumours over time. Ex vivo tissue fluorescence imaging showed that the LEVs exhibited reduced nonspecific distribution in the main organs and enhanced tumour accumulation, with 2.1‐fold higher fluorescence intensity than Lips in tumours at 8 h (Figure 5b–d). The biodistribution of nanovesicles in tumour sections was further observed at high magnification using CLSM. LEVs showed stronger red fluorescence in the tumour sections than Lips (Figure 5e), due to the enhanced intracellular uptake produced by the surface proteins on TDEV membranes (Figure 3). We then evaluated the pharmacokinetics of nanovesicles after intravenous injection. As shown in Figure 5f and g, LEVs showed an extended elimination half‐life of 44.3 h, indicating their prolonged blood circulation compared with Lips. This may be attributed to the expression of CD47, a ‘don't eat me’ signal on the surface of LEVs as illustrated in Figure 2, thus evading clearance by the reticular endothelial system. The immune escape effect of LEVs was verified by the low internalisation in J774 macrophages (Figure S6). This may promote the homing targeting and selective uptake of LEVs in hepatic carcinoma cells. All these results suggested that the LEVs with membrane proteins inherited from TDEV membranes, which bound to HSPGs and integrins of Sk‐hep1, possessed ‘homing’ targeting capacity, resulting in superior accumulation in tumours.
FIGURE 5.
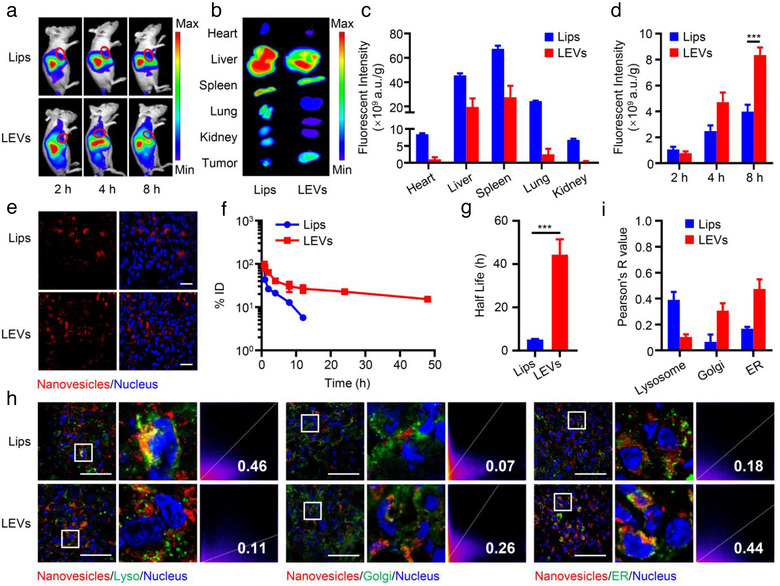
In vivo ‘homing’ targeting and intracellular tracking. (a) In vivo NIR fluorescent images of tumour‐bearing mice after injection of DiR‐labelled Lips and LEVs. (b) Ex vivo NIR fluorescent images of the main organs and tumours at 8 h post‐injection. (c) Quantitative biodistribution of Lips and LEVs in the main organs. (d) NIR fluorescence intensities of DiR‐labelled nanovesicles in tumours at the indicated time points. (e) Biodistribution of DiI‐labelled nanovesicles in tumour sections. Nuclei were stained with DAPI. Scale bar = 20 μm. (f) Pharmacokinetic curves and (g) related elimination half‐life of DiO‐labelled Lips and LEVs after intravenous injection. (h) Fluorescence colocalisation of nanovesicles and subcellular organelles in tumour sections. DiI‐labelled nanovesicles are represented in red, and subcellular organelles separately labelled with anti‐LAMP1, anti‐Golgin‐97 and anti‐KDEL are displayed in green. Scale bar = 50 μm. (i) Pearson's R value for each formulation in (h). Data are displayed as the mean ± SD (n = 3). ***p < 0.001
To investigate the intracellular transportation pathway of LEVs in vivo, fluorescence images of the tumour sections were collected after intravenous administration. As expected, LEVs showed higher colocalisation ratios with the Golgi and ER (Figure 5h and i), while little colocalisation with lysosomes, contrary to Lips. Collectively, the in vivo data exhibited agreement with the in vitro results, suggesting that LEVs delivered siRNA through the endosome‐Golgi‐ER freeway transportation, evading the degradation of siRNA and improving the delivery efficiency of siRNA.
3.6. Gene silencing and antitumour efficacy study in vivo
Encouraged by the superior performance of LEVs, we further evaluated the antitumour activity of siCDK1@LEVs against Sk‐hep1 tumour models. CDK1 expression in tumours after 21 days of treatment was evaluated by RT‐qPCR and western blotting. As shown in Figure 6a, the CDK1 mRNA expression of the siCDK1@LEVs group downregulated to 21%, which was 3‐fold lower than that of the siCDK1@Lips group (62%), due to the synthetic lethal effect of CDK1 gene in HCC cells. The tumour volume in siCDK1@LEVs groups was obviously smaller than that in other groups (Figure 6b and S7), indicating that tumour progression was significantly depressed with siCDK1@LEV treatment, as expected. Notably, 33% of the mice receiving siCDK1@LEVs had complete regression and remained tumour‐free. Moreover, siCDK1@LEVs prolonged mice survival to 80 days, compared with that of 59 days for the Lips group (Figure 6c). The siCDK1@LEVs increased the specific accumulation in the tumours by ‘homing’ targeting effect and the transfection of siCDK1 in the SK‐hep1 cells through the freeway of endosome‐Golgi‐ER, downregulating the expression of CDK1 mRNA.
FIGURE 6.

In vivo gene silencing and antitumour activity of siCDK1@LEVs. (a) CDK1 mRNA and protein expression in tumours treated with different formulations for 21 days (n = 3; mean ± SD). (b) Tumour growth curves of tumour‐bearing mice treated with different formulations (n = 6; mean ± SD). ****p < 0.001 compared to free siCDK1. (c) Survival rate of tumour‐bearing mice in various groups. (d) Bodyweight of treated tumour‐bearing mice over time (n = 6; mean ± SD). (e) Representative H&E staining images of tumours and livers collected from mice after 21 days of treatment. Scale bar = 100 μm. (f) AST and ALT levels in mice blood at day 21 of treatment (n = 5; mean ± SD). (g) Histological assessment of the heart, spleen, lung and kidney via H&E staining. Scale bar = 100 μm. (h) TNF‐α and IL‐6 levels in ICR mice blood after 24 h of systematic administration tested using ELISA kits (n = 3; mean ± SD). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
After the treatment, no obvious abnormality was found in the body weight when the mice were treated with siCDK1@LEVs (Figure 6d). Through histological analysis using hematoxylin and eosin (H&E) staining of tissue slices, a more marked reduction in tumour cells in siCDK1@LEVs treated mice was detected than in other groups, with undetectable metastasis (Figure 6e), and significantly downregulated the AST and ALT levels (Figure 6f), indicating the effective treatment of HCC, while free siCDK1 group showed tumour metastasis (Figure S8). As shown in Figure 6g, the H&E staining assay showed undetectable systematic toxicity of siCDK1@LEVs, due to reduced nonspecific tissue accumulation and synthetic lethal effects. Furthermore, compared with other treatments, the levels of AST and ALT in siCDK1@LEVs group were similar to the healthy group, indicating good in vivo safety of siCDK1@LEVs (Figure 6f). The LEVs group showed considerable liver‐friendly properties, suggesting superior inhibition of tumour metastasis and biosafety, which was suitable for long‐term treatment. These results proved the safety of LEVs delivery and the specificity of siCDK1 to Sk‐hep1 cells rather than other healthy liver cells.
The current siRNA delivery systems, regardless of laboratory or clinical research, were almost cleared by the immune system before they reached the tumour site and started work. Moreover, it has been reported that the proteins expressed on the membrane of TDEVs could depress the immune system (Abusamra et al., 2005; Diao et al., 2015; Zhang and Grizzle, 2011). We found a decrease in TNF‐α and IL‐6 levels after administration of siCDK1@LEVs in immune‐responsive ICR mice (Figure 6h), suggesting that the LEVs had low immunogenicity by inheriting the TDEV membranes. Taken together, our results indicated that siCDK1@LEVs controlled tumour growth more effectively than siCDK1@Lips and free siCDK1 while also displaying reduced toxicity and low immunogenicity.
4. DISCUSSION
Gene therapy has shown considerable potential in the treatment of diverse diseases as it enables more precise modulation of gene expression in specific cells. However, given the biological barriers including instability, potential degradation and off‐target gene silencing effects, the delivery of nucleic acids, such as siRNA, mRNA, microRNA (miRNA) still needs to surmount various obstacles. In particular, the extracellular barriers and the inefficient intracellular delivery of siRNA during administration make siRNA treatment difficult. The non‐precise targeted delivery and low transfection efficiency of the existing vectors make the design and development of siRNA carriers particularly important. EVs, as natural carriers of functional small RNAs, transfer their cargo to target cells for cell‐cell interactions. Considering the advantages of EVs over other vectors, including their long circulation and intrinsic cell targeting properties, we developed a siRNA delivery system composed of TDEV membranes and phospholipids for precise delivery and efficient transfection of siRNA. LEVs inherited the membrane proteins of TDEVs, which possessed the ability of cellular communication provided by TEDV membrane proteins and stable siRNA encapsulation by phospholipid hybridisation, efficiently delivering siRNA to target sites and achieving efficient intracellular transportation.
LEVs could be specifically internalised into Sk‐hep1 cells, the parent cells of TDEVs and improve the gene silencing efficiency of siRNA through a unique intracellular transportation pathway. LEVs exhibited enhanced cellular uptake in Sk‐hep1 cells mainly mediated by HSPGs which are overexpressed on HCC. After targeting Sk‐hep1 cells, LEVs were internalised into cells through clathrin‐ and caveolae‐dependent endocytosis and macropinocytosis, followed by the intercellular trafficking through the Golgi‐ER pathway. The ‘homing’ targeting and intracellular ‘freeway’ transportation promoted the tumour accumulation and distribution of LEVs in HCC xenograft model. Benefiting from the superior accumulation and internalisation in tumours, siCDK1@LEVs could significantly inhibit tumour growth and prolong the survival of tumour‐bearing mice, compared to siCDK1@Lips. TDEV membranes hybrid with lipid vesicles are especially capable of delivering siRNA specifically and efficiently after systemic administration, thus offering a promising novel approach for precise gene therapy in clinical cancer treatment.
TDEVs from different cell lines can be constructed into LEVs using the proposed protocol and subsequently loaded with various therapeutic siRNAs for ‘homing’ targeting to parent cells. Moreover, considering the similarities of nucleic acid drugs in structure, size and charge (Yin et al., 2014), our strategy can be further adapted to the in vivo delivery of other small RNAs, expected to achieve more precise and efficient nucleic acid‐based drug delivery. Therefore, the strategy may serve as a promising platform for next‐generation gene therapy, including the development in material sciences and clinical applications.
AUTHOR CONTRIBUTIONS
Xinxin Zhang and Yong Gan conceived the study, designed and supervised the overall project. Xin Zhou, Yunqiu Miao, Ying Wang, Shufang He performed most of the experiments and analysed the data. Junsong Mao and Yuting Yang contributed to the preparation and characterisation of nanovesicles. Xin Zhou, Yunqiu Miao, Ying Wang, Linmiao Guo, Mingshu Chen discussed the results and wrote the manuscript. All authors conducted the quality assurance of the paper and reviewed the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The study was supported by the National Natural Science Foundation of China (81973250 and 82025032), Natural Science Foundation of Shanghai (21ZR1475800), State Key Laboratory of Drug Research (SIMM2103ZZ‐01) and the Major International Joint Research Project of Chinese Academy of Sciences (grant number: 153631KYSB20190020). We also thank the Electron Microscopy System at the National Facility for Protein Science in Shanghai, Zhangjiang Laboratory (NFPS, ZJLab), China, for cryo‐TEM and flow cytometry.
Zhou, X. , Miao, Y. , Wang, Y. , He, S. , Guo, L. , Mao, J. , Chen, M. , Yang, Y. , Zhang, X. , & Gan, Y. (2022). Tumour‐derived extracellular vesicle membrane hybrid lipid nanovesicles enhance siRNA delivery by tumour‐homing and intracellular freeway transportation. Journal of Extracellular Vesicles, 11, e12198. 10.1002/jev2.12198
Xin Zhou, Yunqiu Miao and Ying Wang contributed equally to this work.
Contributor Information
Xinxin Zhang, Email: xinxinzhang@simm.ac.cn.
Yong Gan, Email: ygan@simm.ac.cn.
REFERENCES
- Abusamra, A. J. , Zhong, Z. , Zheng, X. , Li, Mu , Ichim, T. E. , Chin, J. L. , Min, W.‐P. (2005). Tumor exosomes expressing Fas ligand mediate CD8+ T‐cell apoptosis. Blood Cells, Molecules, and Diseases, 35, 169–173. [DOI] [PubMed] [Google Scholar]
- Alterman, J. F. , Godinho, B. M. D. C. , Hassler, M. R. , Ferguson, C. M. , Echeverria, D. , Sapp, E. , Haraszti, R. A. , Coles, A. H. , Conroy, F. , Miller, R. , Roux, L. , Yan, P. , Knox, E. G. , Turanov, A. A. , King, R. M. , Gernoux, G. , Mueller, C. , Gray‐Edwards, H. L. , Moser, R. P. , … Khvorova, A. (2019). A divalent siRNA chemical scaffold for potent and sustained modulation of gene expression throughout the central nervous system. Nature Biotechnology, 37, 884–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Erviti, L. , Seow, Y. , Yin, H. , Betts, C. , Lakhal, S. , Wood, M. J. A. (2011). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature Biotechnology, 29, 341–345. [DOI] [PubMed] [Google Scholar]
- Azmi, A. S. , Bao, B. , Sarkar, F. H. (2013). Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer and Metastasis Reviews, 32, 623–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barile, L. , & Vassalli, G. (2017). Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacology & Therapeutics, 174, 63–78. [DOI] [PubMed] [Google Scholar]
- Belhadj, Z. , He, B. , Deng, H. , Song, S. , Zhang, H. , Wang, X. , Dai, W. , Zhang, Q. (2020). A combined “eat me/don't eat me” strategy based on extracellular vesicles for anticancer nanomedicine. Journal of Extracellular Vesicles, 9, 1806444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, Yu , Wu, T. , Zhang, K. , Meng, X. , Dai, W. , Wang, D. , Dong, H. , & Zhang, X. (2019). Engineered exosome‐mediated near‐infrared‐II region V2C quantum dot delivery for nucleus‐target low‐temperature photothermal therapy. ACS Nano, 13, 1499–1510. [DOI] [PubMed] [Google Scholar]
- Cerezo‐Magaña, M. , Bång‐Rudenstam, A. , Belting, M. (2020). The pleiotropic role of proteoglycans in extracellular vesicle mediated communication in the tumor microenvironment. Seminars in Cancer Biology, 62, 99–107. [DOI] [PubMed] [Google Scholar]
- Chen, C. , Zhao, S. , Karnad, A. , & Freeman, J. W. (2018). The biology and role of CD44 in cancer progression: therapeutic implications. Journal of Hematology & Oncology, 11, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Mangala, L. S. , Rodriguez‐Aguayo, C. , Kong, X. , Lopez‐Berestein, G. , Sood, A. K. (2018). RNA interference‐based therapy and its delivery systems. Cancer and Metastasis Reviews, 37, 107–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson, H. C. , Svensson, K. J. , Van Kuppevelt, T. H. , Li, J.‐P. , Belting, M. (2013). Cancer cell exosomes depend on cell‐surface heparan sulfate proteoglycans for their internalization and functional activity. Proceedings of the National Academy of Sciences of the United States of America, 110, 17380–17385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao, J. , Yang, X. , Song, X. , Chen, S. , He, Y. , Wang, Q. , Chen, G. , Luo, C. , Wu, X. , Zhang, Y. (2015). Exosomal Hsp70 mediates immunosuppressive activity of the myeloid‐derived suppressor cells via phosphorylation of Stat3. Medical Oncology, 32, 453. [DOI] [PubMed] [Google Scholar]
- Didiot, M.‐C. , Hall, L. M. , Coles, A. H. , Haraszti, R. A. , Godinho, B. M. , Chase, K. , Sapp, E. , Ly, S. , Alterman, J. F. , Hassler, M. R. , Echeverria, D. , Raj, L. , Morrissey, D. V. , Difiglia, M. , Aronin, N. , Khvorova, A. (2016). Exosome‐mediated delivery of hydrophobically modified siRNA for Huntingtin mRNA silencing. Molecular Therapy: The Journal of The American Society of Gene Therapy, 24, 1836–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, Y. , Siegwart, D. J. , Anderson, D. G. (2019). Strategies, design, and chemistry in siRNA delivery systems. Advanced Drug Delivery Reviews, 144, 133–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X. , Ran, N. , Dong, X. , Zuo, B. , Yang, R. , Zhou, Q. , Moulton, H. M. , Seow, Y. , Yin, H. (2018). Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Science Translational Medicine, 10, eaat0195. [DOI] [PubMed] [Google Scholar]
- Goga, A. , Yang, D. , Tward, A. D. , Morgan, D. O. , Bishop, J. M. (2007). Inhibition of CDK1 as a potential therapy for tumors over‐expressing MYC. Nature Medicine, 13, 820–827. [DOI] [PubMed] [Google Scholar]
- Gomez‐Navarro, N. , Miller, E. (2016). Protein sorting at the ER‐Golgi interface. Journal of Cell Biology, 215, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, S. , Perets, N. , Betzer, O. , Ben‐Shaul, S. , Sheinin, A. , Michaelevski, I. , Popovtzer, R. , Offen, D. , Levenberg, S. (2019). Intranasal delivery of mesenchymal stem cell derived exosomes loaded with phosphatase and tensin homolog siRNA repairs complete spinal cord injury. ACS Nano, 13, 10015–10028. [DOI] [PubMed] [Google Scholar]
- He, S. , Fan, W. , Wu, Na , Zhu, J. , Miao, Y. , Miao, X. , Li, F. , Zhang, X. , Gan, Y. (2018). Lipid‐based liquid crystalline nanoparticles facilitate cytosolic delivery of siRNA via structural transformation. Nano Letters, 18, 2411–2419. [DOI] [PubMed] [Google Scholar]
- Kalluri, R. , Lebleu, V. S. (2020). The biology, function, and biomedical applications of exosomes. Science, 367, eaau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerkar, S. , Lebleu, V. S. , Sugimoto, H. , Yang, S. , Ruivo, C. F. , Melo, S. A. , Lee, J. J , Kalluri, R. (2017). Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature, 546, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijmans, S. A. A. , Gitz‐Francois, J. J. J. M. , Schiffelers, R. M. , Vader, P. (2018). Recombinant phosphatidylserine‐binding nanobodies for targeting of extracellular vesicles to tumor cells: A plug‐and‐play approach. Nanoscale, 10, 2413–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashouri, L. , Yousefi, H. , Aref, A. R. , Ahadi, A. M. , Molaei, F. , Alahari, S. K. (2019). Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Molecular Cancer, 18, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte, M. A. , Nolte‐'T Hoen, E. N. M. , Margadant, C. (2021). Integrins control vesicular trafficking; New tricks for old dogs. Trends in Biochemical Sciences, 46, 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'brien, K. , Breyne, K. , Ughetto, S. , Laurent, L. C. , Breakefield, X. O. (2020). RNA delivery by extracellular vesicles in mammalian cells and its applications. Nature Reviews Molecular Cell Biology, 21, 585–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham, T. C. , Jayasinghe, M. K. , Pham, T. T. , Yang, Y. , Wei, L. , Usman, W. M. , Chen, H. , Pirisinu, M. , Gong, J. , Kim, S. , Peng, B. , Wang, W. , Chan, C. , Ma, V. , Nguyen, N. T. H. , Kappei, D. , Nguyen, X.‐H. , Cho, W. C. , Shi, J. , Le, M. T. N. (2021). Covalent conjugation of extracellular vesicles with peptides and nanobodies for targeted therapeutic delivery. Journal of Extracellular Vesicles, 10, e12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman, A. , Bandari, S. K. , Liu, J. , Mobley, J. A. , Brown, E. E. , Sanderson, R. D. (2016). Fibronectin on the surface of myeloma cell‐derived exosomes mediates exosome‐cell interactions. The Journal of Biological Chemistry, 291, 1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Z. , Shi, Ke , Yang, S. , Liu, J. , Zhou, Q. , Wang, G. , Song, J. , Li, Z. , Zhang, Z. , Yuan, W. (2018). Effect of exosomal miRNA on cancer biology and clinical applications. Molecular Cancer, 17, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Y. , Wang, X. , Li, J. , Nie, Yu , Liao, G. , Yu, Y. , Li, C. (2019). Overcoming the reticuloendothelial system barrier to drug delivery with a “Don't‐Eat‐Us” Strategy. ACS nano, 13, 13015–13026. [DOI] [PubMed] [Google Scholar]
- Thanuja M. Y., Anupama C., Ranganath, S. H. (2018). Bioengineered cellular and cell membrane‐derived vehicles for actively targeted drug delivery: So near and yet so far. Advanced Drug Delivery Reviews, 132, 57–80. [DOI] [PubMed] [Google Scholar]
- Yin, H. , Kanasty, R. L. , Eltoukhy, A. A. , Vegas, A. J. , Dorkin, J. R , Anderson, D. G. (2014). Non‐viral vectors for gene‐based therapy. Nature Reviews Genetics, 15, 541–555. [DOI] [PubMed] [Google Scholar]
- Zhang, H‐Ge , Grizzle, W. E. (2011). Exosomes and cancer: a newly described pathway of immune suppression. Clinical Cancer Research, 17, 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Zhang, S. , Yao, J. , Lowery, F. J. , Zhang, Q. , Huang, W.‐C. , Li, P. , Li, M. , Wang, X. , Zhang, C. , Wang, H. , Ellis, K. , Cheerathodi, M. , Mccarty, J. H. , Palmieri, D. , Saunus, J. , Lakhani, S. , Huang, S. , Sahin, A. A. , Aldape, K. D. , Steeg, P. S. , Yu, D. (2015). Microenvironment‐induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature, 527, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen, Xu , Cheng, P. , Pu, K. (2019). Recent advances in cell membrane – Camouflaged nanoparticles for cancer phototherapy. Small, 15, 1804105. [DOI] [PubMed] [Google Scholar]
- Zou, Y. , Sun, X. , Wang, Y. , Yan, C. , Liu, Y. , Li, J. , Zhang, D. , Zheng, M. , Chung, R. S. , Shi, B. (2020). Single siRNA nanocapsules for effective siRNA brain delivery and glioblastoma treatment. Advanced Materials (Deerfield Beach, Fla.), 32, 2000416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material