

Abstract
Canine bufavirus (CBuV), a novel protoparvovirus of dogs that is associated with enteric and respiratory symptoms, has been reported only in Italy and China. The enteric prevalence of CBuV in India was investigated, and the nearly complete genome sequence (4292 bp) was amplified and reconstructed for one strain. A nucleotide sequence alignment indicated 93.42–98.81% identity to the other available CBuV sequences and 70.88–73.39% and 54.4–54.8% identity to human bufavirus and canine parvovirus 2 (CPV-2), respectively. The current strain is most closely related to Chinese CBuV strains, which together form an Asian lineage. This first report of the prevalence of CBuV in India emphasizes the need for further epidemiological surveillance.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00705-022-05398-7.
Keywords: Canine bufavirus, Protoparvovirus, Phylogenetics
Bufaviruses (BuVs) are small, non-enveloped, single-stranded DNA viruses that belong to the family Parvoviridae and genus Protoparvovirus [1, 2]. The genome ranges in size from 4.3 to 4.8 kb, with complex hairpin structures at the 5′ and 3′ ends. It has two open reading frames (ORFs): ORF1 and ORF2. ORF1 encodes a non-structural protein, and ORF2 encodes a capsid protein [3]. BuVs were first reported in 2012 in faecal samples from a child with acute gastroenteritis [4]. Since then, BuVs have been reported in several other humans, as well as in wild and domestic animals [1, 5, 6–15]. Canine bufavirus (CBuV) was first reported in Italy in 2016 in a litter of three 5-month-old mixed-breed puppies with canine infectious respiratory disease (CIRD) [11] and, more recently, in China [16, 17]. Although the mechanism is still unknown, CBuVs are known to be associated with canine enteritis [11, 16, 17]. Information on the epidemiology and genetic features of CBuVs is rather limited; the only reports of its existence have been from Italy and China [11, 16–18]. To this end, in the current study, we investigated the prevalence of CBuV in India. For this, fecal samples from dogs presenting with enteritis at veterinary hospitals in Hyderabad, Telangana State, India, were collected. Also, the nearly full-length genome sequence of one strain was determined and characterized, providing a significant reference for further studies on the evolution and epidemiology of CBuV.
A total of 186 fecal samples were collected from dogs (1 month to 10 years old) presenting with clinical gastroenteritis at veterinary hospitals in Hyderabad, Telangana State, India, during 2019–2020. Of these, 138 were from puppies (< 1 year old), and 48 were from adult dogs (> 1 year old) (Supplementary Table S1). The fecal samples were collected in sterile centrifuge tubes using rectal swabs and stored at -20ºC until used. The rectal swabs were homogenized in 3 ml of 0.1 M PBS (pH 7.4) containing antibiotics (100 IU of benzyl penicillin and 100 µg of streptomycin sulphate per ml) and centrifuged at 6000 rpm for 10 min at 4ºC, and the supernatant was filtered through a 0.22-µm syringe filter. Viral DNA was extracted using phenol chloroform and isoamyl alcohol as described by Sambrook and Russel [19]. The extracted DNA was stored at -20ºC until use. For detection of CBuV, real-time PCR was performed as described previously [11]. The CBuV-positive samples were screened for coinfections with other canine enteric viruses, namely, CPV-2 [20], canine adenovirus 2 (CAdV-2) [21], canine distemper virus (CDV) [22], canine coronavirus (CCoV) [23], canine astrovirus (CAstV) [24], and canine rotavirus (CRV) [25]. Of the 186 samples screened by real-time PCR, eight were found to be positive for CBuV, resulting in a prevalence rate of 4.3%. Of these, six samples were from diarrheic puppies (< 1 year old), and two samples were from diarrheic adult dogs (> 1 year old). Seven of the eight CBuV-positive samples had coinfections with other enteric viruses, including canine parvovirus CPV-2, CAdV-2, and CAdV, and they were negative for CCoV, CRV, and CDV (Table 1). One of the PCR-positive samples was selected for amplification of the complete CBuV genome using five different primer pairs as described previously [26], with slight modifications in the reaction conditions (Supplementary Table S2). The amplified PCR products (Supplementary Fig. S1) were purified using a DNA purification kit (QIAGEN, USA), following the manufacturer’s instructions, and were sequenced at the sequencing facility of Bioserve, Pvt. Ltd, Hyderabad, India. The PCR products were sequenced twice. The nearly complete genome sequence of CBuV strain 407/PVNRTVU/2020 was assembled using SeqMan software (DNASTAR). ORFs were identified using the NCBI ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/), and the sequences were uploaded to the GenBank database (accession number: MZ574435). The sequenced region of the genome was 4292 nt in length, and ORF-1 and ORF-2 were found to be 1917 nt and 2178 nt in length, respectively (Fig. 1). The length of NS1, VP1, and VP2 was 1917 nt (638 amino acids), 2133 nt (710 amino acids), and 1707 nt (568 amino acids), respectively (Fig. 1).
Table 1.
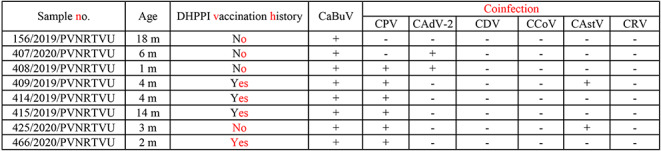
Detection of CBuV in fecal samples and coinfection with CPV, CAdV-2, CDV, CCoV, and CAstV
Figure 1.
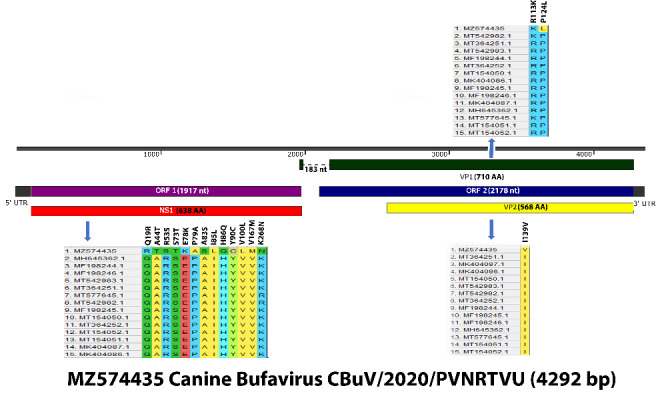
Graphical representation of the whole genome sequence of CBuV (MZ574435). Non-synonymous substitutions in NS1, VP1, and VP2 genes are indicated in a format of the actual amino acid followed by the substitution position and the substituted amino acid.
Nucleotide and amino acid sequences were compared by multiple sequence BLAST analysis. A multiple sequence alignment of the whole genome was performed using the CLUSTALW algorithm in MEGA X software, and a phylogenetic tree was generated by the maximum-likelihood method with the Tamura 3-parameter model [27]. The reliability of the phylogenetic tree was tested by applying a bootstrap test with 1000 replicates. When compared with the other CBuV reference strains, the nucleotide sequence identity of the complete genome was 93.42–98.81% (Supplementary Table S3a). The nucleotide sequence identity in the NS1, VP1, and VP2 coding regions was 96.48–98.13%, 89.91–99.48%, and 88.19–99.47%, respectively (Supplementary Table S3b), and the amino acid sequence identity in the NS1, VP1, and VP2 proteins was 94.98–97.81%, 94.79–99.72%, and 94.19–99.82%, respectively (Supplementary Table S3b). Various novel mutations were observed in the NS1, VP1, and VP2 coding regions, with 23, eight, and seven synonymous mutations and 13, two, and one non-synonymous substitutions, respectively (Fig. 1). Compared to the other members of the family Parvoviridae including other CBuVs, the nucleotide sequence identity of the complete genome sequence obtained in the current study was 39.5–98.81% (Supplementary Table S3a). Phylogenetic analysis performed with the nearly complete genome sequence from this study, other publicly available CBuV sequences, and other protoparvovirus sequences available in the GenBank database showed that the CBuV isolate from India was most closely related to Chinese strains, and together they formed an Asian lineage (Fig. 2a). Furthermore, the CBuVs showed the closest relationship to bat and sea otter parvoviruses and all other bufaviruses (human, porcine, rat, and megabat), whereas the canine and avian parvoviruses together formed a separate branch (Fig. 2b).
Figure 2.
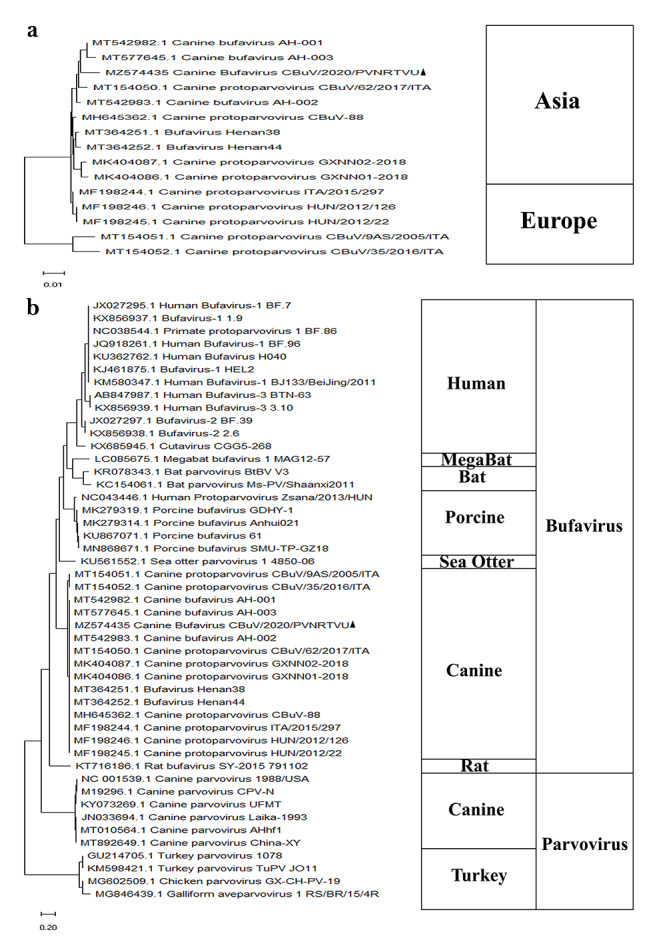
Evolutionary analysis of CBuV (MZ574435). (a) Phylogenetic tree based on whole genome sequences of CBuVs. A total of 15 whole-genome sequences of CBuV strains published earlier in GenBank were used as references to reconstruct the phylogenetic tree by the maximum-likelihood method with 1000 bootstrap replicates and the Tamura 3-parameter model. The strain from this study is indicated by a filled triangle. (b) Phylogenetic tree of members of the family Parvoviridae based on whole genome sequence. A total of 46 whole-genome sequences of strains infecting different species obtained from the GenBank database were used as references to reconstruct the phylogenetic tree by the maximum-likelihood method with 1000 bootstrap replicates and the Tamura 3-parameter model. The strain from this study is indicated by a filled triangle.
The recombination analysis tool (RAT) and recombination detection program (RDP) package Beta 4.101 were used for identification of recombinant sequences in default mode. A recombination event identified with a significance of p < 0.01 by at least four of the nine selected algorithms: RDP, GENECONV, Boot-Scan, Maxchi, Chimaera, SiScan, 3Seq, LARD, and PhylPro, was considered reliable [28]. RAT is a distance-based method of recombination detection [29]. To investigate whether recombination has played a role in CBuV evolution, CPVs, CBuVs, HuBuVs, PoBuVs, megabat BuV, rat BuV, bat BuV, avian BuV, human cutavirus, and sea otter parvovirus were analyzed using multiple algorithms in RAT and RDP software. Although no recombination events could be found in the CBuVs, it should be noted that major recombination events possibly drove evolution of other BuVs in the genus Protoparvovirus (Supplementary Fig. S2).
CBuV is a novel protoparvovirus that was first identified during an outbreak of CIRD [11]. Since then, only a handful of studies from a few countries (Italy, Hungary, and China) have reported its prevalence [11, 16–18]. Here, we report the prevalence of CBuV in India for the first time. Varying prevalence rates have been reported for this virus so far, ranging from 1.74–42.15% [11, 16–18, 30]. In our study, we found the prevalence rate to be 4.3%. It has been suggested that geographical and temporal factors as well as the target canine population included in the studies can affect the prevalence rates [18]. A statistically non-significant correlation of CBuV with enteritis has been reported before; although non-significant, the authors observed higher loads of CBuV in diarrheic samples than in controls [11, 18]. Moreover, our study, along with another study in which the prevalence of CBuV was studied extensively in diarrheic dogs, stresses the role of CBuV in causing enteritis [16]. We found that seven out of eight CBuV-positive samples had coinfections with other enteric viruses, with CPV-2 being the predominant coinfecting virus (6/8), followed by CAstV (2/8) and CAdV-2 (2/8). Similar to our findings, a few other studies also showed CPV-2 to be the major coinfecting agent with CBuV, with the prevalence of CPV-2 ranging from 87.5–100% [17, 18]. CPV-2 is a well-known viral agent that is associated with severe gastroenteritis in dogs, especially pups [31]. Further pathogenicity studies will help to clarify the role of coinfections on the severity of gastroenteritis in dogs. We have determined the nearly complete genome sequence of one strain of CBuV detected in an Indian dog population. Comparison with other publicly available CBuV sequences revealed NS1 to be the most conserved gene and VP2 to be the most variable, both at the level of nucleotides (NS1: 96.48%-98.13%; VP2: 88.19%-99.47%) and amino acids (NS1: 94.98%-97.81%; VP2: 94.19%-99.82%). Phylogenetic analysis based on whole genome sequences showed that the current CBuV strain clustered with CBuV strains identified in China. Our analysis differentiates all of the available CBuV sequences into two groups: Asian and European. Like other DNA viruses, parvoviruses have a low mutation rate [17]. The genetic grouping observed in the current study indicates unique evolutionary forces driving evolution of CBuVs. Furthermore, CBuV 407/2020/PVNRTVU showed the highest nucleotide sequence identity (77.89%) to a megabat bufavirus strain, followed by bat parvovirus (70.34–79.5%), porcine bufavirus (73.16–73.4%), human bufavirus (70.92–73.39%), sea otter parvovirus (72.7%), rat bufavirus (63%), canine parvovirus (54.4–54.8%), and avian parvovirus (39.5–40%). Individual pairwise comparisons of the NS1, VP1, and VP2 coding regions of 407/2020/PVNRTVU revealed the presence of 13, two, and one unique non-synonymous substitutions when compared to other available CBuV sequences. Future experimental studies investigating the biological significance of these mutations can elucidate their role (if any) in driving the evolution of this virus and adaptation to local host populations.
In summary, this is the first report of the prevalence and complete genome sequence of a CBuV strain from India. The Indian strain was found to be related to CBuVs strains from China. The current study forms a foundation for further studies on the epidemiology and genetic diversity of CBuV in India.
Electronic Supplementary Material
Below is the link to the electronic supplementary material
Acknowledgements
We wish to express our gratitude to the animal owners who participated in the collection of rectal swab specimens.
Authors’ contributions
KP conceived and designed the study. VG and BBL performed the experiments. KP, VG, and YNR analyzed the data and wrote the manuscript. The authors read and approved the final manuscript.
Funding
The authors acknowledge the Department of Biotechnology (No. BT/ADV/Canine Health/TANUVAS), India, for the financial support.
Declaration
Ethical approval
This research did not involve any human participants or experiments on live animals. All of the work was performed in accordance with standard operating procedures adopted by the Department of Veterinary Biotechnology, PVNRTVU, Hyderabad, India. Before sample collection, the dogs' owners were informed of the purpose of this study, and consent was obtained.
Conflict of interest
The authors declare no competing interests with respect to the research, authorship, and/or publication of this article.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hargitai R, Pankovics P, Kertész AM, et al. Detection and genetic characterization of a novel parvovirus distantly related to human bufavirus in domestic pigs. Arch Virol. 2016;161:1033–1037. doi: 10.1007/s00705-015-2732-4. [DOI] [PubMed] [Google Scholar]
- 2.Huang H, Li Y, Wang W et al (2020) Detection and molecular characterization of novel porcine bufaviruses in Guangxi province. Infect Genet Evol 82:104286. 10.1016/j.meegid.2020.104286 [DOI] [PubMed]
- 3.Vaïsänen E, Paloniemi M, Kuisma I, et al. Epidemiology of two human protoparvoviruses, bufavirus and tusavirus. Sci Rep. 2016;6:1–8. doi: 10.1038/srep39267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phan TG, Vo NP, Bonkoungou IJO, et al. Acute Diarrhea in West African Children: Diverse Enteric Viruses and a Novel Parvovirus Genus. J Virol. 2012;86:11024–11030. doi: 10.1128/jvi.01427-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altay A, Yahiro T, Bozdayi G et al (2015) Bufavirus genotype 3 in Turkish children with severe diarrhoea. Clin Microbiol Infect 21:965.e1–e4. 10.1016/j.cmi.2015.06.006 [DOI] [PubMed]
- 6.Huang DD, Wang W, Lu Q, Bin et al (2015) Identification of Bufavirus-1 and Bufavirus-3 in Feces of Patients with Acute Diarrhea, China. Sci Rep 5:13272. 10.1038/srep13272 [DOI] [PMC free article] [PubMed]
- 7.Smits SL, Schapendonk CME, van Beek J, et al. New viruses in idiopathic human diarrhea cases, the Netherlands. Emerg Infect Dis. 2014;20:1218–1222. doi: 10.3201/eid2007.140190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yahiro T, Wangchuk S, Tshering K, et al. Novel human bufavirus genotype 3 in children with severe diarrhea, Bhutan. Emerg Infect Dis. 2014;20:1037–1039. doi: 10.3201/eid2006.131430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diakoudi G, Lanave G, Capozza P, et al. Identification of a novel parvovirus in domestic cats. Vet Microbiol. 2019;228:246–251. doi: 10.1016/j.vetmic.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Handley SA, Thackray LB, Zhao G, et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell. 2012;151:253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martella V, Lanave G, Mihalov-Kovács E, et al. Novel Parvovirus Related to Primate Bufaviruses in Dogs. Emerg Infect Dis. 2018;24:1061. doi: 10.3201/EID2406.171965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melegari I, Di Profio F, Palombieri A, et al. Molecular detection of canine bufaviruses in wild canids. Arch Virol. 2019;164:2315–2320. doi: 10.1007/s00705-019-04304-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki M, Orba Y, Anindita PD, et al. Distinct lineages of bufavirus in wild shrews and nonhuman primates. Emerg Infect Dis. 2015;21:1230–1233. doi: 10.3201/eid2107.141969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasaki M, Gonzalez G, Wada Y, et al. Divergent bufavirus harboured in megabats represents a new lineage of parvoviruses. Sci Rep 2016. 2016;61 6:1–8. doi: 10.1038/srep24257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang S, Liu D, Wang Y, et al. Bufavirus Protoparvovirus in feces of wild rats in China. Virus Genes. 2016;52:130–133. doi: 10.1007/s11262-015-1262-1. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Cui L, Deng X, et al. Canine bufavirus in faeces and plasma of dogs with diarrhoea, China. Emerg Microbes Infect. 2019;8:245–247. doi: 10.1080/22221751.2018.1563457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun W, Zhang S, Huang H, et al. First identification of a novel parvovirus distantly related to human bufavirus from diarrheal dogs in China. Virus Res. 2019;265:127–131. doi: 10.1016/j.virusres.2019.03.020. [DOI] [PubMed] [Google Scholar]
- 18.Di Martino B, Sarchese V, Di Profio F, et al. Genetic heterogeneity of canine bufaviruses. Transbound Emerg Dis. 2021;68:802–812. doi: 10.1111/tbed.13746. [DOI] [PubMed] [Google Scholar]
- 19.Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press
- 20.Decaro N, Elia G, Martella V, et al. A real-time PCR assay for rapid detection and quantitation of canine parvovirus type 2 in the feces of dogs. Vet Microbiol. 2005;105:19–28. doi: 10.1016/j.vetmic.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 21.Balboni A, Dondi F, Prosperi S, Battilani M. Development of a SYBR Green real-time PCR assay with melting curve analysis for simultaneous detection and differentiation of canine adenovirus type 1 and type 2. J Virol Methods. 2015;222:34–40. doi: 10.1016/j.jviromet.2015.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Elia G, Decaro N, Martella V, et al. Detection of canine distemper virus in dogs by real-time RT-PCR. J Virol Methods. 2006;136:171–176. doi: 10.1016/j.jviromet.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Decaro N, Pratelli A, Campolo M, et al. Quantitation of canine coronavirus RNA in the faeces of dogs by TaqMan RT-PCR. J Virol Methods. 2004;119:145–150. doi: 10.1016/j.jviromet.2004.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martella V, Moschidou P, Lorusso E, et al. Detection and characterization of canine astroviruses. J Gen Virol. 2011;92:1880–1887. doi: 10.1099/vir.0.029025-0. [DOI] [PubMed] [Google Scholar]
- 25.Logan C, O’Leary JJ, O’Sullivan N. Real-time reverse transcription-PCR for detection of rotavirus and adenovirus as causative agents of acute viral gastroenteritis in children. J Clin Microbiol. 2006;44:3189–3195. doi: 10.1128/JCM.00915-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Guo X, Zhang D, et al. Genetic and phylogenetic analysis of canine bufavirus from Anhui Province, Eastern China. Infect Genet Evol. 2020;86:104600. doi: 10.1016/J.MEEGID.2020.104600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar S, Stecher G, Li M, et al. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin DP, Murrell B, Golden M et al (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1(1):vev003.10.1093/VE/VEV003 [DOI] [PMC free article] [PubMed]
- 29.Etherington GJ, Dicks J, Roberts IN. Recombination Analysis Tool (RAT): a program for the high-throughput detection of recombination. Bioinformatics. 2005;21:278–281. doi: 10.1093/BIOINFORMATICS/BTH500. [DOI] [PubMed] [Google Scholar]
- 30.Shao R, Zheng F, Cai S, et al. Genomic sequencing and characterization of a novel group of canine bufaviruses from Henan province, China. Arch Virol. 2020;165:2699–2702. doi: 10.1007/s00705-020-04785-2. [DOI] [PubMed] [Google Scholar]
- 31.Nandi S, Kumar M. Canine parvovirus: Current perspective. Indian J Virol. 2010;21:31–44. doi: 10.1007/s13337-010-0007-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.