

Summary
Smooth muscle guides the morphogenesis of several epithelia during organogenesis, including the mammalian airways. However, it remains unclear how airway smooth muscle differentiation is spatiotemporally patterned and whether it originates from transcriptionally distinct mesenchymal progenitors. Using single-cell RNA-sequencing of embryonic mouse lungs, we show that the pulmonary mesenchyme contains a continuum of cell identities, but no transcriptionally distinct progenitors. Transcriptional variability correlates with spatially distinct sub-epithelial and sub-mesothelial mesenchymal compartments that are regulated by Wnt signaling. Live-imaging and tension-sensors reveal compartment-specific migratory behaviors and cortical forces and show that sub-epithelial mesenchyme contributes to airway smooth muscle. Reconstructing differentiation trajectories reveals early activation of cytoskeletal and Wnt signaling genes. Consistently, Wnt activation induces the earliest stages of smooth muscle differentiation and local accumulation of mesenchymal F-actin, which influences epithelial morphology. Our single-cell approach uncovers the principles of pulmonary mesenchymal patterning and identifies a morphogenetically active mesenchymal layer that sculpts the airway epithelium.
Subject areas: Cell biology, Developmental biology, Transcriptomics
Graphical Abstract
Highlights
-
•
The embryonic lung mesenchyme is organized into spatially distinct compartments
-
•
Migratory behaviors and cortical forces differ between compartments
-
•
Diffusion analysis recapitulates airway smooth muscle differentiation
-
•
The early stages of smooth muscle differentiation influence airway branching
Cell biology; Developmental biology; Transcriptomics
Introduction
Smooth muscle and similar contractile cell types influence the morphogenesis of epithelial tissues (Shyer et al., 2013; Neumann et al., 2018; Jaslove and Nelson, 2018). Branching of the mammalian airway epithelium has long been thought to involve smooth muscle differentiation from the surrounding pulmonary mesenchyme (Roman et al., 1998; Yamada et al., 2002; Leslie et al., 1990; Kim and Vu, 2006; Danopoulos et al., 2018). Accordingly, ex vivo culture revealed that the patterned differentiation of airway smooth muscle specifies cleft sites to promote epithelial bifurcation and shapes the domain branches that establish the underlying architecture of the murine lung (Goodwin et al., 2019; Kim et al., 2015). It is, therefore, surprising that the smooth muscle transcription factor myocardin (Myocd) is dispensable for lung branching morphogenesis (Young et al., 2020) and that mice lacking α-smooth muscle actin (αSMA or Acta2) are viable and only develop deleterious phenotypes in smooth muscle and similar contractile tissues under conditions of high mechanical stress (Schildmeyer et al., 2000; Haaksma et al., 2011). These studies highlight the complexity (and robustness) of smooth muscle differentiation and transcriptional identity during development and emphasize the importance of uncovering the factors that define a smooth muscle cell and control its differentiation.
Lung development is intricately regulated by signaling between the epithelium, mesothelium, and mesenchyme. Shh signaling from the epithelium is required for airway smooth muscle differentiation (Miller et al., 2004), while Fgf9 signaling from the mesothelium prevents the differentiation of this tissue (El Agha et al., 2017). The physical layout of the airway smooth muscle and the differing diffusible signals from the epithelium and mesothelium could potentially establish separate mesenchymal compartments. In particular, it appears that epithelial and mesothelial signals directly influence the behavior of the sub-epithelial and sub-mesothelial mesenchyme, respectively (White et al., 2006; De Langhe et al., 2008).
Cells from different locations within the pulmonary mesenchyme possess different capacities for differentiation: mesenchyme that is transplanted from stalk to tip regions fails to migrate toward the epithelium or differentiate into smooth muscle unless it is exposed to Wnt1 (Kumar et al., 2014). Wnt signaling may, therefore, be involved in a compartment-specific manner in specifying smooth muscle progenitor identity and behavior. Consistently, Wnt2a is expressed in the distal mesenchyme (De Langhe et al., 2005) and the expression of Axin2 is strongest in the mesenchyme around branch tips (Ludtke et al., 2016). Deleting Ctnnb1 (β-catenin) from the mesenchyme delays branching morphogenesis and inhibits mesenchymal growth and survival (Yin et al., 2008; Luo et al., 2015; De Langhe et al., 2008). Expressing stabilized β-catenin leads to disorganized smooth muscle wrapping (Kumar et al., 2014), and Wnt2 promotes the expression of myogenic transcription factors to support robust smooth muscle differentiation (Goss et al., 2011).
Lineage-tracing studies based on single marker genes have not identified a distinct airway smooth muscle progenitor (Mailleux et al., 2005; Moiseenko et al., 2017). Additionally, the signals required for patterned smooth muscle differentiation remain unclear. Several pathways have been implicated but studying each one individually will not reveal how they interact to achieve spatiotemporal control. To circumvent these challenges, we used single-cell RNA-sequencing (scRNA-seq) analysis. As smooth muscle differentiates continuously during lung branching morphogenesis (Jaslove and Nelson, 2018), scRNA-seq analysis at a single snapshot in time can be used to interrogate cell identities ranging from putative undifferentiated progenitor to nascent (immature) smooth muscle to mature smooth muscle (Tritschler et al., 2019; Haghverdi et al., 2015).
scRNA-seq analysis of E11.5 mouse lungs revealed a continuum of mesenchymal and smooth muscle cell identities, but no transcriptionally distinct population of smooth muscle progenitors. Instead, mesenchymal cell heterogeneity reflects spatially distinct, Wnt-dependent sub-epithelial and sub-mesothelial compartments. Live-imaging and tension-sensor experiments revealed differences in motility and cortical tension between compartments, and showed that airway smooth muscle cells arise from the sub-epithelial mesenchyme. Computationally reconstructing this differentiation trajectory showed that cytoskeletal, adhesion, and Wnt signaling genes are upregulated early during differentiation and that the airway smooth muscle gene-expression program is largely Myocd-independent. Our computational analyses uncovered a role for Yap1 in mesenchymal patterning and showed that differentiating cells systematically downregulate genes involved in proliferative metabolism. Finally, we found that manipulating Wnt signaling affects the enrichment of nascent smooth muscle cells and F-actin in the mesenchyme, thus altering mesenchymal constraint on the epithelium and influencing epithelial branching. Overall, we provide the first single-cell view of airway smooth muscle differentiation and demonstrate that the earliest steps of this cascade involve Wnt-dependent mesenchymal stiffening that shapes epithelial branches.
Results
scRNA-seq analysis reveals expected populations of cells in the embryonic murine lung
We carried out scRNA-seq using cells isolated from the left lobes of lungs of CD1 mouse embryos harvested at E11.5 and analyzed the data with Seurat (Butler et al., 2018) (Figures 1A, and S1A–S1J). After removing contaminating cells and correcting for the cell-cycle stage (Figures S1K–S1O), clustering identified six transcriptionally distinct populations, each containing cells from both replicates (Figures 1B and 1C). We identified two mesenchymal clusters expressing homeobox B6 (Hoxb6) and pleiotrophin (Ptn), a smooth muscle cluster expressing Acta2, an epithelial cluster expressing thyroid transcription factor 1 (TTF-1 or Nkx2.1), a vascular-endothelial cluster expressing VE-cadherin (Cdh5), and a mesothelial cluster expressing Wilms tumor 1 (Wt1) (Figures 1B–1E, Table S1).
Figure 1.

Clustering scRNA-seq data from E11.5 mouse lungs reveals expected cell populations
(A) Sample image of a lung used for scRNA-seq experiment. Each lung was micro-dissected to isolate the left lobe (indicated by dotted outlines). Scale bar shows 100 μm.
(B and C) UMAP plots of cell cycle-corrected embryonic lung cells isolated by scRNA-seq of E11.5 CD1 mouse lungs, color-coded either by cluster (B) or by replicate (C).
(D) UMAP plots from (B and C) color-coded to show the expression of population-specific genes.
(E) Top 20 markers for each cluster shown in (B) based on log fold-change (FC) and color-coded by adjusted p value.
We then isolated and reclustered the closely-related mesenchymal and smooth muscle populations, and found that the Hoxb6-, Ptn-, and Acta2-expressing cells still clustered together in a similar pattern (Figures S2A–S2C). Previous studies used lineage-tracing of Axin2-, Fgf10-, Gli1-, or Wt1-expressing cells in an attempt to identify smooth muscle progenitors (Mailleux et al., 2005; Moiseenko et al., 2017). However, none of these markers labeled a distinct population in our dataset (Figures S2D–S2G). Further analysis of reclustered mesenchymal cells provided no additional evidence for a transcriptionally distinct smooth muscle progenitor (Figures S2H–S2Q). Similar to other developing tissues (Giraddi et al., 2018; Brunskill et al., 2014), the embryonic pulmonary mesenchyme does not appear to harbor a transcriptionally-distinct progenitor population – whether there exists an airway smooth muscle progenitor characterized by a distinct epigenetic profile or chromatin organization remains unknown.
Undifferentiated mesenchymal cell clusters represent spatially distinct populations
To gain insight into mesenchymal patterning, we examined the expression of markers that distinguished the two mesenchymal clusters. Fluorescence in situ hybridization showed that cells expressing Hoxb6 (cluster 0) and Ptn (cluster 1) are present in spatially-distinct layers within the mesenchyme of E11.5 lungs, with Hoxb6 adjacent to the epithelium and Ptn adjacent to the mesothelium (Figures 2A and 2B). Similarly, immunofluorescence analysis for Lef1 (cluster 0) showed that Lef1+ cells are present in the sub-epithelial mesenchyme adjacent to the epithelium (Figures 2C and 2D, Table S2), suggesting that these sub-epithelial cells represent cluster 0. Conversely, immunofluorescence analysis for Foxp1 (cluster 1) showed that Foxp1+ cells are mainly in sub-mesothelial regions, suggesting that these cells comprise cluster 1 (Figures 2C and 2D). Quantification of intensity profiles of Lef1 as a function of distance from the epithelium and of Foxp1 as a function of distance from the mesothelium showed that these markers are present in opposing gradients across the mesenchyme (Figure 2E). We found similar patterns of Pitx2, an alternate marker for cluster 1 (Figures S3A–S3C). Apart from their differential expression patterns in the mesenchyme, Lef1 and Foxp1 are detected in smooth muscle (Figures 2C, S3D, and S3E), and Foxp1 is detected in the epithelium and mesothelium (Figures 2C and 2D). These data suggest that the computationally identified mesenchymal clusters represent spatially distinct populations within the embryonic pulmonary mesenchyme. Therefore, we delineated two mesenchymal compartments, hereafter referred to as sub-epithelial (cluster 0) and sub-mesothelial (cluster 1), following previously used nomenclature (White et al., 2006; De Langhe et al., 2008) (Figure 2F). The observed gradients of Lef1+ and Foxp1+ cells, and the fact that a portion of mesenchymal cells expresses both markers (Figure S3F), suggests that mesenchymal cells exist in a continuum between these two compartments rather than as two separate tissue types.
Figure 2.

Computationally identified mesenchymal clusters represent spatially distinct populations
(A) Violin plots showing the expression of Hoxb6 or Ptn in each cluster.
(B) Fluorescence in situ hybridization for Hoxb6 and Ptn in E11.5 lungs. Scale bar shows 25 μm.
(C) Violin plots showing the expression of Lef1 or Foxp1 in each cluster.
(D) E11.5 lungs immunostained for cluster 0 marker Lef1 or for cluster 1 marker Foxp1 and counterstained with Hoechst. Scale bars show 25 μm.
(E) Quantifications of Lef1 and Foxp1 intensity profiles emanating from the epithelium (for Lef1) or from the mesothelium (for Foxp1). Schematics show lines and direction along which intensity profiles were measured. Mean and SD are plotted (n = 4).
(F) Schematic depicting the sub-epithelial and sub-mesothelial compartments of the mesenchyme.
(G) Heatmap showing the expression of genes specific to either mesenchymal compartment. Genes (rows) are clustered based on the dendrogram to the right. Cells (columns) are clustered based on the dendrogram above, and each column is color-coded according to the original cluster identity from Figure 1B.
(H) UMAP of mesenchymal and smooth muscle cells color-coded according to the sum of their expression of either sub-epithelial or sub-mesothelial mesenchyme marker genes. Dotted line indicates the location of smooth muscle cells
To further validate these conclusions, we took advantage of published in situ hybridization results available via the Mouse Genome Informatics (MGI) database. We downloaded a list of genes that are detected in the pulmonary mesenchyme from E11.5 to E12.5 and classified them by expression pattern (Table S3). We then focused on those that were clearly sub-epithelial or sub-mesothelial and that were detected at sufficient levels in our scRNA-seq dataset. Unsupervised clustering of mesenchymal cells based on their expression of region-specific genes showed that sub-epithelial cells group together and have similar expression of sub-epithelial markers and, likewise, that sub-mesothelial cells group together and have similar expression of their corresponding markers (Figure 2G). UMAPs color-coded according to the summed expression of either sub-epithelial or sub-mesothelial genes show that the two sets of markers are expressed by cells in complementary regions of the UMAP (Figure 2H). These findings are consistent with our conclusion that the computationally identified clusters represent spatially distinct mesenchymal compartments.
We next asked whether these mesenchymal compartments persist over the pseudoglandular stage of lung development. At E12.5 and E14.5, we detected Lef1+ cells in the sub-epithelial mesenchyme around branch tips and Foxp1+ cells in the sub-mesothelial mesenchyme (Figures S3F–S3I). Individual Foxp1+ cells are also visible in the mesenchyme in between branches, where they likely represent vascular endothelial cells (Figures 2C, S3G, and S3I). Therefore, the lung maintains a sub-epithelial, Lef1+ mesenchymal population and a sub-mesothelial, Foxp1+ mesenchymal population over the course of branching morphogenesis. The mesenchyme in between branches (with low Lef1 and Foxp1 immunostaining) may take on new characteristics, perhaps to support the formation of vasculature. Consistent with this interpretation, other scRNA-seq studies have revealed that the embryonic pulmonary mesenchyme becomes increasingly complex at later stages of development (Liu et al., 2021).
Wnt signaling regulates cell identity in the embryonic pulmonary mesenchyme
We hypothesized that the mesenchymal populations identified in our scRNA-seq dataset represent cell states with important functions in branching morphogenesis. Indeed, GO enrichment analysis of genes upregulated in each mesenchymal cluster revealed that both clusters are enriched for genes involved in “morphogenesis of a branching epithelium” (Figure 3A, Tables S2 and S4). Additionally, our analysis revealed a significant enrichment of genes with GO terms related to Wnt signaling. We, therefore, examined differentially-expressed Wnt signaling-related genes (Figure 3B). Among the genes upregulated in the sub-epithelial cluster were Lef1, a Wnt signaling effector (Eastman and Grosschedl, 1999), and Wnt2, a Wnt ligand implicated in airway smooth muscle differentiation (Goss et al., 2011). Overall, the sub-epithelial cluster expresses more activators and targets of Wnt signaling, while the sub-mesothelial cluster expresses more inhibitors. Each mesenchymal cluster is, therefore, characterized by a distinct Wnt-signaling signature, suggesting that Wnt signaling is likely operating in different ways in each mesenchymal compartment and, consistent with previous studies (Al Alam et al., 2011; De Langhe et al., 2005; Ludtke et al., 2016), the sub-epithelial cluster may be more Wnt-active.
Figure 3.

Wnt signaling regulates cell identity in the embryonic pulmonary mesenchyme.
(A) Bubble plot showing the enrichment percentage and adjusted p value of relevant GO terms identified based on genes upregulated in each mesenchymal cluster.
(B) Heatmap showing the expression of Wnt-associated genes upregulated in either mesenchymal cluster. Activators and targets are colored in blue, inhibitors are colored in red.
(C) Heatmap showing the expression of Wnt ligands, secreted inhibitors, and receptors detected in either mesenchymal cluster and in clusters containing cells from the mesothelium (meso), vascular endothelium (ve), epithelium (ep), and smooth muscle (sm).
(D–G) Confocal sections and quantification of Lef1 and Foxp1 intensity profiles around branch L.L2 in lungs isolated at E11.5 from CD1 embryos and immunostained for Lef1 or Foxp1 after treatment with either DMSO, LiCl (10 mM), or IWR1 (100 μM) for 24 h (n = 2–6). Yellow dashed lines indicate the border of the epithelium. Schematics show lines and direction along which intensity profiles were measured. Mean and SEM are plotted, and curves were compared using two-way ANOVA.
(H–M) E12.5 control and Tbx4-rtTA;tet-O-Cre;Ctnnb1fl/fl lungs immunostained for Lef1 or Foxp1 and quantification of Lef1 and Foxp1 intensity profiles (n = 3). Low-magnification z-projections (H and I) and high-magnification confocal slices (J and K) are shown. Scale bars show 50 μm. ∗ indicates p<0.05, ∗∗ indicates p<0.001, and ∗∗∗ indicates p<0.0001
Signals from the epithelium and mesothelium have previously been shown to regulate behaviors of the sub-epithelial and sub-mesothelial mesenchyme, respectively (White et al., 2006; De Langhe et al., 2008). We, therefore, hypothesized that Wnt signals from these tissues could establish compartments of differential Wnt signaling across the epithelium-to-mesothelium axis. Tissue-specific analysis using our scRNA-seq dataset confirmed that the epithelium expresses Wnt7b and the sub-epithelial mesenchyme expresses Wnt2 (De Langhe et al., 2005) (Figure 3C). The sub-mesothelial mesenchyme expresses Wnt11, the mesothelium expresses Wnt9a, and both tissues express the inhibitors Sfrp1 and Sfrp2, suggesting that Wnt signaling may be inhibited at the mesothelial end of the axis (Figure 3C). Vascular-endothelial and smooth muscle cells do not show any striking expression patterns of Wnt ligands or secreted inhibitors. Fzd1 and Fzd2 are the only Wnt receptors detected in the mesenchyme, but they are not preferentially enriched in either cluster (Figure 3C), suggesting that differential Wnt signaling across the mesenchyme arises from patterned expression of ligands and inhibitors, rather than from differential expression of receptors. However, it is important to note that any genes absent from Figure 3C were simply not detected by our scRNA-seq analysis, but could be expressed at low levels.
To determine whether Wnt signaling regulates the spatially-distinct compartments of the pulmonary mesenchyme, we manipulated this pathway in lungs explanted from E11.5 CD1 mouse embryos. To activate Wnt signaling we inhibited GSK3 by treating lung explants with LiCl (Ludtke et al., 2016), which maintains nuclear β-catenin levels and increases cytoplasmic Axin2 intensity in the mesenchyme (Figures S4A–S4D). Conversely, to downregulate Wnt signaling we treated explants with IWR1, which stabilizes Axin2 leading to enhanced activity of the β-catenin destruction complex (Ludtke et al., 2016). As expected given its mechanism of action, treatment with IWR1 does not affect Axin2 intensity, but reduces mesenchymal levels of nuclear β-catenin (Figures S4A–S4D).
We then quantified the domains of Lef1+ sub-epithelial cells and Foxp1+ sub-mesothelial cells. We plotted Lef1-intensity profiles as a function of distance from the tip of the epithelial branch L.L2 [the second domain branch of the left lobe, which initiates and extends during the culture period (Goodwin et al., 2019; Metzger et al., 2008)] and Foxp1-intensity profiles as a function of distance from the mesothelium. Activating Wnt by treating with LiCl resulted in an expanded domain of Lef1+ cells around epithelial branch tips and fewer Foxp1+ cells than DMSO-treated controls (Figures 3D–3G). Conversely, inhibiting Wnt by treating with IWR1 resulted in a significant reduction in Lef1 intensity and a random distribution of Foxp1+ cells in the mesenchyme adjacent to the tip of the branch L.L2 (Figures 3D–3G). These changes in expression occurred without significantly affecting the number of mesenchymal cells between the epithelium and mesothelium (Figure S4E). Overall, activating Wnt signaling expands the sub-epithelial mesenchyme at the expense of the sub-mesothelial mesenchyme, and inhibiting Wnt shrinks the sub-epithelial compartment.
To confirm our findings and circumvent possible off-target effects of inhibitors on the epithelium, we took advantage of a genetic approach to delete Ctnnb1 (β-catenin) from the pulmonary mesenchyme using Tbx4-rtTA;tet-O-Cre. We found that inhibiting Wnt signaling genetically leads to a dramatic reduction of the Lef1+ sub-epithelial mesenchymal compartment (Figures 3H, 3J, and 3L), in line with previous work (Yin et al., 2008), and a corresponding expansion of the Foxp1+ sub-mesothelial compartment (Figures 3I, 3K, and 3M). Consistently, others have shown that enhancing Wnt signaling via mesenchymal deletion of the Wnt-inhibitor Apc (Adenomatous polyposis coli) results in increased expression of the sub-epithelial mesenchymal marker Vcan (versican; Table S2) (Luo et al., 2015). Our predictions based on scRNA-seq analyses are, therefore, confirmed by both pharmacological and genetic manipulations.
To determine whether mesenchymal patterning also depends on Shh, we treated explants with the Shh antagonist cyclopamine, which prevents smooth muscle differentiation and decreases sub-epithelial mesenchymal cell proliferation (Goodwin et al., 2019; White et al., 2006). In the presence of cyclopamine, the Lef1+ subepithelial domain is reduced around branch tips, but the Foxp1+ sub-mesothelial domain is slightly expanded (Figures S4F–S4I). We then asked whether Wnt could override the compartment-specific effects of Shh. We combined cyclopamine and LiCl treatments and found that activating Wnt signaling rescues the loss of Lef1+ cells caused by inhibiting Shh (Figures S4J and S4L). However, Foxp1 intensity is much lower than controls and similar to LiCl treatment alone (Figures S4K and S4M), suggesting that the positive effects of Shh inhibition on the sub-mesothelial compartment are indirect and cannot overcome the effects of activating Wnt signaling.
Mesenchymal cell motility and cortical forces are spatially patterned during airway branching morphogenesis
Our observations thus far suggest that the two mesenchymal clusters represent a continuum of cell states that can be shifted by Wnt signaling, instead of two distinct tissues. We, therefore, investigated whether mesenchymal cells transition between compartments and/or into the smooth muscle lineage. We used Dermo1-Cre-driven expression of R26R-Confetti to fluorescently label mesenchymal cells for time-lapse imaging analysis of lung explants isolated at E11.5 (Figure 4A). Dermo1-Cre is active throughout the pulmonary mesenchyme (White et al., 2006), including in Lef1+ sub-epithelial and Foxp1+ sub-mesothelial mesenchymal cells and the smooth muscle layer (Figures S5A–S5D). During time-lapse imaging, Confetti-labeled cells elongate around branching epithelial buds (Figure 4A). Strikingly, Confetti-labeled cells are not clustered together, as has been observed in other tissues labeled with Confetti (Snippert et al., 2010), suggesting that embryonic pulmonary mesenchymal cells rearrange extensively as the airway epithelium undergoes branching morphogenesis (Figure 4A). Indeed, mesenchymal cells are surprisingly motile (Video S1) and exhibit a range of migratory behaviors, from sub-diffusive to directed (Figures 4B and S5E–S5J), as revealed by cell tracking and mean squared displacement (MSD) analyses. Migration speeds are highly variable and are greatest among sub-epithelial mesenchymal cells (Figures 4B and 4C), demonstrating that cell motility differs between mesenchymal compartments.
Figure 4.

Embryonic pulmonary mesenchymal cells exhibit spatially patterned migratory behaviors and cortical tension
(A) Snapshots from time-lapse analysis of Dermo1-Cre;Confettifl/lungs isolated at E11.5 showing a bifurcating epithelial bud surrounded by sparsely fluorescently labeled mesenchymal cells.
(B) Plot of cell trajectories from tracking time-lapse analysis of Dermo1-Cre;Confettifl/lungs isolated at E11.5. Tracks are color-coded based on time-point. Shaded areas show mesenchymal compartments and smooth muscle regions. Inset in top left corner shows a snapshot of the time-lapse from which the cell tracks are taken.
(C) Mean speed of tracked cells of each mesenchymal cell type (n = 6 time-lapses, n = 280 cells total). Error bars show SD
(D–F) Snapshots from time-lapse analyses of Dermo1-Cre;Confettifl/lungs isolated at E11.5 showing a mesenchymal cell migrating from the sub-mesothelial to the sub-epithelial mesenchyme (D) and vice versa (E), showing a sub-epithelial mesenchymal cell elongating and presumably differentiating into smooth muscle (F). Yellow arrowhead indicates tracked cell, dashed line indicates the epithelium, and dotted line indicates the mesothelium.
(G and H) Percentage of tracked cells from the sub-mesothelial (G; n = 51) or the sub-epithelial (H; n = 203) mesenchyme that stay within their original compartment, cross compartments, or elongate, presumably differentiating into smooth muscle.
(I) Percentage of tracked smooth muscle cells that were already smooth muscle or that were observed to differentiate from sub-epithelial mesenchymal cells (n = 26).
(J) Schematic of VinTS. Vh is vinculin head, Vt is vinculin tail.
(K) Fluorescence intensity of VinTS in E12.5 lung and lifetime of TFP donor along mesenchymal cell edges.
(L) Fluorescence lifetime of sub-epithelial and sub-mesothelial mesenchymal cells (n = 6 samples, n = 261–282 cells). Boxes show 25th and 75th percentiles, center lines show median. Scale bars show 50 μm. Groups were compared using two-sided t-test and Bartlett’s test for unequal variances. ∗∗∗ indicates p<0.0001 for t-test and # indicates p<0.05, ## indicates p<0.001, and ### indicates p<0.0001 for Bartlett’s test
We observed that mesenchymal cells can move between compartments over timescales that are short (∼5 h) relative to that of branching in culture ex vivo (∼20 h). The majority of cells remain within their compartments over the timescales analyzed, but a fraction cross over into the opposite compartment (Figures 4D, 4E, 4G, and 4H), possibly contributing to the continuum of transcriptional states observed in the mesenchyme. Most smooth muscle cells that we tracked were already elongated around the epithelium, but some originated from rounder, sub-epithelial cells at branch tips (Figures 4F and 4I). Consistent with previous conclusions from fixed tissues (Kumar et al., 2014), our time-lapse data show that airway smooth muscle cells arise exclusively from the sub-epithelial mesenchyme on the timescales of our live-imaging experiments.
While we did not observe sub-mesothelial cells giving rise to airway smooth muscle, our computational clustering suggests that the sub-mesothelial mesenchyme is also closely related to smooth muscle. We, therefore, hypothesized that the sub-mesothelial mesenchyme might give rise to vascular smooth muscle cells. Given that mature vascular smooth muscle is absent from the lung at E11.5 (Badri et al., 2008), and that we isolated the left lobe (excluding upper airways) for our scRNA-seq experiments, we did not expect to find mature vascular smooth muscle cells in our dataset. There could, however, be cells in the early stages of differentiation down this lineage. To test the plausibility of this hypothesis, we examined the levels of markers enriched in either airway or vascular smooth muscle (Jaslove and Nelson, 2018) (Figure S6A). The expression of airway smooth muscle markers is highest in the smooth muscle cluster (Figures S6B–S6F). Among these, Foxf1, Mylk and Nog are enriched in sub-epithelial compared to sub-mesothelial mesenchyme. Conversely, the vascular smooth muscle markers Heyl and Speg are slightly enriched in the sub-mesothelial mesenchyme (Figures S6G and S6H), suggesting that this compartment might contain nascent vascular smooth muscle cells. Reclustering of smooth muscle cells alone did not reveal a distinct vascular smooth muscle population (Figures S6I–S6P). We, therefore, examined lungs at E14.5, when mature vascular smooth muscle is easily detected around blood vessels, and observed that vascular smooth muscle cells are Lef1- and Foxp1+ (Figures S6Q–S6S), consistent with our hypothesis that Foxp1+ sub-mesothelial mesenchyme differentiates into vascular smooth muscle. If Foxp1+ cells are, indeed, the source of vascular smooth muscle, then an expansion of the Foxp1+ domain could lead to increased differentiation of vascular smooth muscle. Consistently, we observed that deleting Ctnnb1 from the pulmonary mesenchyme, which expands the Foxp1+ domain (Figures 3J and 3K), leads to an increase in vascular smooth muscle (Figures S6T and S6U). Additional experiments, including lineage tracing and scRNA-seq at later time points, would help to confirm whether sub-mesothelial mesenchyme gives rise to vascular smooth muscle.
Cell migration and rearrangements are regulated by cortical tension, which can be estimated using the FRET-based vinculin tension sensor (VinTS) (Grashoff et al., 2010) (Figure 4J). A VinTS knock-in mouse was recently generated and validated to infer cortical forces during mandibular arch morphogenesis (Tao et al., 2019). Live-imaging and fluorescence lifetime imaging microscopy (FLIM) of E12.5 VinTS lungs revealed that the epithelium and sub-epithelial mesenchyme have the longest average fluorescence lifetimes (highest tension), while the vasculature has the lowest (Figures S5K–S5L). Consistent with the observed differences in migratory behavior (Tao et al., 2019), we found that sub-epithelial mesenchymal cells have higher and more variable (indicative of population-level heterogeneity and/or fluctuations within individual cells) fluorescence lifetimes than sub-mesothelial mesenchymal cells (Figures 4K and 4L). These data indicate that sub-epithelial cells experience higher cortical tension. We, therefore, conclude that the two transcriptionally-distinct mesenchymal compartments have different migratory behaviors and mechanical properties.
Diffusion analysis of mesenchymal cells provides insight into differentiation trajectories
Our scRNA-seq dataset is comprised of undifferentiated mesenchymal cells and mature smooth muscle cells, and theoretically should include cells at every stage of differentiation between these two states. Live imaging revealed that airway smooth muscle cells originate from the sub-epithelial mesenchyme (Figure 4). We, therefore, focused on transitions from the sub-epithelial mesenchymal cluster to the smooth muscle cluster. To search for differentiation trajectories, we implemented diffusion analysis using Destiny (Haghverdi et al., 2015). Undifferentiated sub-epithelial mesenchymal cells are separated from smooth muscle cells along diffusion component (DC) 1 (Figure 5A). The expression of smooth muscle-related genes is positively correlated with DC1 (Figure 5B). In contrast, gene number, a strong indicator of cell developmental potential (Gulati et al., 2020), is negatively correlated with DC1 (Figure 5C), suggesting that progression along DC1 represents cells moving away from a stem-cell-like state and differentiating into airway smooth muscle.
Figure 5.

Diffusion analysis reveals differentiation trajectory of mesenchymal cells into airway smooth muscle
(A) Diffusion plots of sub-epithelial mesenchymal and smooth muscle cells.
(B) Scaled expression of known smooth muscle markers versus cell loadings along DC1. Pearson correlation coefficients and significance are indicated on plots and lines represent smoothed data with SE shaded in gray. ∗∗∗ indicates p< 0.0001.
(C) Number of genes detected per cell versus DC1. Pearson correlation coefficients and significance are indicated on plots and lines represent smoothed data with SE shaded in gray. ∗∗∗ indicates p< 0.0001.
(D) Heatmap showing five gene sets identified using hierarchical agglomerative clustering of spline-fitted gene-expression profiles along DC1. Genes of interest are indicated to the right.
(E) E11.5 lungs immunostained for Tbx3 (gene set 4), Ecad, and αSMA. Scale bars show 50 μm.
(F) Bubble plot showing the enrichment percentage and adjusted p value of relevant KEGG pathways for the gene sets defined in (E)
(G) Bubble plot showing the p values of motifs for transcription factor-binding sites in the promoters of and DNase-hypersensitive regions near genes of sets 2–5.
To determine how gene expression evolves over the course of airway smooth muscle differentiation, we fit the expression of each gene along DC1 and applied unbiased clustering to group genes with similar profiles. We focused on four resultant gene sets that increased along DC1 and one that decreased (Figure 5D, Table S6). The upregulated gene sets (one to four, numbered from latest to earliest) included genes related to smooth muscle and to Wnt, Shh, TGFβ, Bmp, and Hippo signaling pathways, and the downregulated gene set (5) included the mesenchymal progenitor marker Tbx2 and the distally-expressed ligand Wnt2, suggesting that our analysis had yielded biologically relevant gene sets. Tbx3 was among the earliest genes activated along the smooth muscle differentiation trajectory, suggesting that it may be a marker for immature smooth muscle cells; consistently, immunofluorescence analysis showed that Tbx3+ cells are located in a narrow domain adjacent to the epithelium (Figure 5E), surrounding areas that are not yet wrapped by mature αSMA+ smooth muscle.
To identify signaling pathways implicated along the smooth muscle differentiation trajectory, we used KEGG pathway enrichment analysis and carried out motif discovery in promoter and DNase-hypersensitive regions (indicative of accessible chromatin) proximal to genes of each set (Figures 5F, 5G, and S7). Consistent with our hypothesis that DC1 recapitulates smooth muscle differentiation, we found that Srf-binding sites (CArG boxes) are highly enriched in the promoters and DNase-hypersensitive regions of genes in sets two and 3 (Figure 5G). Furthermore, we found binding sites for Foxf1, a regulator of airway smooth muscle differentiation (Ustiyan et al., 2018), in DNase-hypersensitive regions near genes of set 3 (Figure 5G). We also identified binding sites for the mesenchymal-compartment markers Lef1 and Foxp1 near genes of sets 2, 3, and 4 (Figure 5G).
All gene sets together and, in particular, those that increase in expression along DC1, are enriched for genes involved in Wnt, TGFβ, Hippo, Hedgehog, and MAPK signaling (Figure 5F). Accordingly, we found motifs for binding sites of Smad-family transcription factors in promoters of genes in set 3, and of Tcf- and Tead-family members in DNase-hypersensitive regions near genes in sets 2–5 (Figure 5G). Nearly all of these pathways have been implicated in airway smooth muscle differentiation, with the exception of Hippo signaling. To test the role of this signaling pathway, we deleted the Hippo effector Yap1 from the embryonic mesenchyme using Dermo1-Cre and isolated lungs at E12.5. We found that markers for the sub-epithelial and sub-mesothelial compartments are greatly reduced in the mesenchymal knockouts, as is smooth muscle wrapping (Figure 6A), demonstrating that Yap1 is required for patterning and differentiation in the embryonic pulmonary mesenchyme. Our computational analyses can, therefore, be used to uncover signaling pathways important for lung development.
Figure 6.

Regulators and features of smooth muscle differentiation
(A) Sections of E12.5 Dermo1-Cre/+; Yapfl/fl; mTmG/+ lungs and littermate controls immunostained for GFP and either Yap1, Lef1, Foxp1, or αSMA. Insets show zoomed-in view of the mesenchyme to highlight the decrease in mesenchymal Yap1 levels in mutants. Yap1+ cells in the mesenchyme of mutants are vascular endothelial cells (ve, indicated by white arrowheads), which are not targeted by Dermo1-Cre. ep is epithelium. Scale bars show 50 μm.
(B) Scaled expression of genes involved in cytoskeleton, cell adhesion, and extracellular matrix versus cell loadings along DC1 compared to the expression profiles of the smooth muscle markers Acta2 and Myocd (dotted lines). Pearson correlation coefficients and significance are indicated and lines represent smoothed data with SE shaded in gray.
(C) Simplified pathway diagram depicting the steps of proliferative metabolism and showing relevant enzymes at each step. Enzymes that are significantly downregulated along DC1 are indicated in bold red font, with a significance of spline fit indicated by asterisks. ∗ indicates p < 0.05, ∗∗ indicates p < 0.001, and ∗∗∗ indicates p < 0.0001
Differentiating smooth muscle cells undergo changes in contractility and adhesion to elongate and wrap around the epithelium (Jaslove and Nelson, 2018). Our analyses revealed that genes related to the cytoskeleton, cell adhesion, and the extracellular matrix are upregulated early during differentiation and that their expression accompanies or precedes that of classical smooth muscle markers (Acta2) and transcription factors (Myocd) (Figures 5F and 6B). These data suggest that gene expression at the earliest stages of smooth muscle differentiation is Myocd-independent. In line with this hypothesis, only a minor fraction (0–6%) of the smooth muscle genes identified using diffusion analysis (Figure 5D) and of smooth muscle cluster markers (11%) (Figure 1D) are differentially expressed in a bulk RNA-seq dataset of E13.5 lungs in which Myocd is deleted from the mesenchyme (Young et al., 2020) (Figure S8). Overall, these data show that the airway smooth muscle gene-expression program is largely Myocd-independent.
Finally, we found that genes that decrease in expression along the smooth muscle differentiation trajectory are associated with metabolic pathways (Figure 5F), and that those involved in proliferative metabolism and biomass production are downregulated along DC1 (Figure 6C). Indeed, Myc- and Mycn-binding sites are enriched in DNase-hypersensitive regions near genes in set 5 (Figure 5G), suggesting that proliferative gene expression is downregulated as smooth muscle cells differentiate. Additionally, binding sites for Foxk1 and Foxk2, regulators of glycolysis (Sukonina et al., 2019), are present in the promoters and DNase-hypersensitive regions in gene sets 3 and 4 (Figure 5G). Overall, our analyses uncover a possible role for metabolic reprogramming during smooth muscle differentiation.
Wnt signaling activates the early stages of smooth muscle differentiation to influence epithelial branching
Our computational analyses suggested that the early stages of airway smooth muscle differentiation involve changes in cytoskeletal components and Wnt signaling. We, therefore, evaluated the effects of manipulating Wnt signaling on F-actin accumulation in the mesenchyme around epithelial branch tips and on the expression patterns of early markers of smooth muscle. To visualize smooth muscle cells in the earliest stages of differentiation, we isolated lungs from embryos expressing RFP under the control of the αSMA promoter (SMA-RFP mice). RFP signal can be used to identify nascent smooth muscle cells that do not have sufficient αSMA protein for detection by immunofluorescence analysis (Kim et al., 2015). As an alternative approach, we used Tbx3, an early marker of smooth muscle differentiation identified in our diffusion analyses (Figure 5).
Strikingly, activating Wnt signaling in SMA-RFP explants by treating with LiCl generated a thick layer of F-actin-rich mesenchyme containing many RFP+ cells surrounding the epithelium (Figures 7A–7C). The domain of Tbx3+ nascent smooth muscle cells also expanded after LiCl treatment (Figures S9A and S9B). Conversely, genetically inhibiting Wnt signaling via mesenchymal deletion of Ctnnb1 led to fewer Tbx3+ cells around the epithelium and reduced F-actin throughout the mesenchyme (Figures 7D, 7E, 7F, S9C, and S9D).
Figure 7.

Wnt signaling regulates the accumulation of F-actin and nascent smooth muscle cells in the sub-epithelial mesenchyme to influence epithelial morphology
(A) Confocal sections of the left lobe of lungs isolated from E11.5 SMA-RFP embryos, immunostained for RFP, and labeled with phalloidin (F-actin) after treatment with DMSO or LiCl for 24 h.
(B) Quantification of F-actin intensity profiles emanating from the epithelium (n = 3–6).
(C) Quantification of RFP intensity profiles emanating from the epithelium of lungs isolated from SMA-RFP embryos (n = 4–5).
(D) E12.5 control and Tbx4-rtTA;tet-O-Cre;Ctnnb1fl/fl lungs immunostained for F-actin.
(E) Quantification of F-actin intensity profiles emanating from the epithelium (n = 5).
(F) Quantification of Tbx3 intensity profiles emanating from the epithelium (n = 3).
(G) Confocal sections of the left lobe of lungs isolated from E11.5 CD1 embryos and immunostained for E-cadherin (Ecad) and αSMA after treatment with DMSO or LiCl for 24 hrs.
(H and I) Width and length of branch L.L2 after 24 h of culture (n = 13–16).
(J) Quantification of smooth muscle coverage of the left lobe after 24 h of culture (n = 6–11).
(K) E12.5 control and Tbx4-rtTA;tet-O-Cre;Ctnnb1fl/fl lungs immunostained for Ecad and αSMA.
(L and M) Width and length of branch L.L2 after 24 h of culture (n = 7–8).
(N) Quantification of smooth muscle coverage of the left lobe in control and Tbx4-rtTA;tet-O-Cre;Ctnnb1fl/fl lungs (n = 3). Data in bar graphs were plotted with error bars representing SD and were compared using t test. Intensity profiles were plotted as mean ± SEM and compared using two-way ANOVA. ∗ indicates p < 0.05, ∗∗ p < 0.001 and ∗∗∗ p < 0.0001.
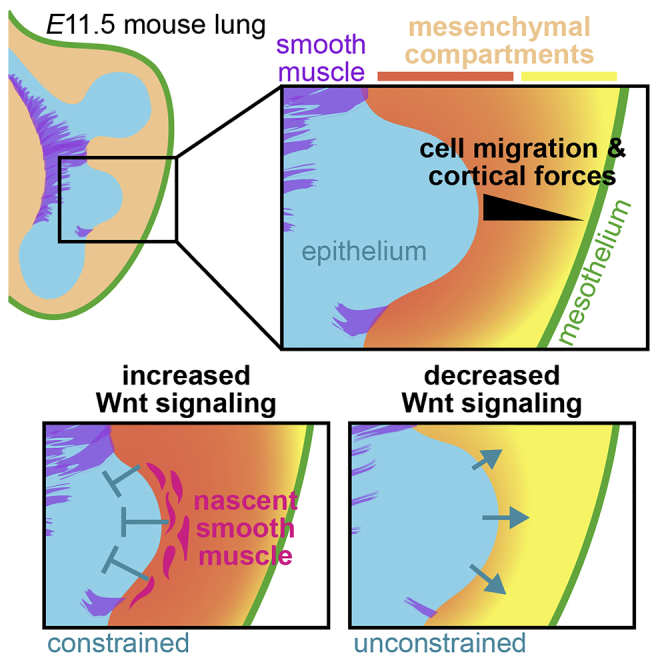
Importantly, these changes in smooth muscle differentiation and mesenchymal F-actin correlated with changes in epithelial morphology and smooth muscle wrapping. LiCl treatment impeded branch extension, suggesting that activating Wnt signaling constrains epithelial branching (Figures 7G–7I). LiCl treatment does not affect mature smooth muscle coverage as assessed by immunofluorescence analysis for αSMA after 24 h of culture (Figures 7G, 7J, and S9D), but the number of nascent smooth muscle cells (which may not yet have robust αSMA protein) is clearly increased (Figures 7A, 7C, and S9A). Conversely, genetically inhibiting Wnt signaling in the pulmonary mesenchyme by deleting Ctnnb1 led to much wider and shorter branches with an irregular shape suggestive of a lack of constraint (Figures 7K–7M). Ctnnb1-mutants also exhibited disorganized αSMA+ smooth muscle wrapping and significantly less smooth muscle coverage (Figures 7K, 7L, 7N, and S9E). These data suggest that Wnt activates the earliest stages of smooth muscle differentiation and local accumulation of F-actin, indicative of mesenchymal stiffening (Solon et al., 2007) (Heer and Martin, 2017), to constrain the branching epithelium (Figure 8).
Figure 8.

Proposed model for the behaviors and roles of mesenchymal compartments in the embryonic mouse lung
The embryonic pulmonary mesenchyme consists of sub-epithelial and sub-mesothelial compartments that are regulated by Wnt signaling and that are characterized by distinct migratory behaviors and cortical forces. Cells can migrate between compartments and sub-epithelial cells give rise to airway smooth muscle. Activating Wnt signaling expands the sub-epithelial compartment, activates the earliest stages of smooth muscle differentiation, and leads to the local accumulation of F-actin in the mesenchyme to constrain the branching epithelium.
Discussion
Here, we provide the first single-cell overview of cell identities and transitions in the embryonic pulmonary mesenchyme during branching morphogenesis of the mouse lung, with a specific focus on airway smooth muscle. scRNA-seq analyses revealed compartment-specific transcriptional heterogeneity across the mesenchyme and uncovered how signaling pathways evolve over the course of airway smooth muscle differentiation. Building on our computational findings, we identify Wnt signaling as a key regulator of mesenchymal patterning in early lung development and show that Wnt-dependent activation of smooth muscle differentiation and mesenchymal stiffening can physically influence airway epithelial branching, even prior to smooth muscle maturation.
Our data show that the pulmonary mesenchyme contains a continuum of cell identities, which may explain why a distinct marker of smooth muscle progenitors has remained elusive (Moiseenko et al., 2017). Unbiased lineage tracing in fixed samples previously showed that new smooth muscle cells at branch tips are members of clones from the adjacent sub-epithelial mesenchyme (Kumar et al., 2014). Our live-imaging data support these findings, and also reveal a surprising level of cell motility within the embryonic pulmonary mesenchyme. Similar dynamic behaviors have been observed in the developing kidney (Combes et al., 2016). The extent and timescales of these rearrangements raise questions about how focal morphogen sources proposed to act as chemoattractants could be maintained as mesenchymal cells migrate and rearrange during branching morphogenesis (Menshykau et al., 2014; Metzger and Krasnow, 1999).
The heterogeneity of mesenchymal cells within developing tissues influences how they respond to morphogenetic signals (Reinhardt et al., 2019) and can define if and when they differentiate. Exposing transplanted stalk mesenchyme to a Wnt signal primes it to migrate into a host tissue and form smooth muscle (Kumar et al., 2014). It is possible that stalk mesenchyme is comprised of sub-mesothelial mesenchymal cells, and that the activation of Wnt signaling converts these cells to a sub-epithelial identity, which we have shown here is associated with greater cytoskeletal tension and motility.
We and others have shown that the embryonic pulmonary mesenchyme is patterned by gradients in Fgf9/Shh (White et al., 2006) and Wnt signaling that spans the epithelium to the mesothelium. Epithelium-derived Wnt signals also radially pattern smooth muscle differentiation in the ureteric mesenchyme (Trowe et al., 2012). In the chick intestine, opposing molecular signals from the epithelium and the mesothelium, as well as mechanical forces generated by growth of the intestinal epithelium and contraction of smooth muscle, are integrated to spatiotemporally pattern the differentiation of multiple smooth muscle layers (Huycke et al., 2019). The role of mechanical forces in regulating pulmonary mesenchymal cell identity has yet to be fully elucidated.
Reconstructing the smooth muscle differentiation trajectory revealed that genes related to the cytoskeleton and cell-matrix adhesion are expressed early during differentiation, prior to and independently of the expression of classical smooth muscle markers. These changes could lead to the stiffening of immature smooth muscle cells to regulate diverse morphogenetic events (Barriga et al., 2018; Heer and Martin, 2017; Zhu et al., 2020). Indeed, we found that Wnt-dependent accumulation of nascent smooth muscle cells and F-actin around emerging branches is associated with the local inhibition of epithelial growth. Similarly, activating Wnt signaling in the ureteric mesenchyme leads to ectopic accumulation of smooth muscle progenitors and a hypoplastic ureter (Trowe et al., 2012). F-actin levels can reflect local tissue stiffness (Solon et al., 2007; Heer and Martin, 2017), and Wnt signaling can activate actomyosin contractility (Lee et al., 2006) and matrix deposition (De Langhe et al., 2005), possibly contributing to tissue stiffening around emerging branches. Our VinTS experiments revealed higher cortical tension in the sub-epithelial mesenchyme, which may allow it to physically sculpt the branching epithelium. These effects could be enhanced by the activation of Wnt signaling to promote sub-epithelial mesenchymal cell states.
Our results raise questions about which stage(s) of smooth muscle differentiation and which aspects of smooth muscle cell identity influence the branching of the airway epithelium. The smooth muscle gene-expression program extends beyond the influence of a single transcription factor (Myocd), suggesting that developing smooth muscle may be robust to the loss or absence of single markers. Indeed, other actin isoforms compensate for the loss of Acta2 (Schildmeyer et al., 2000; Haaksma et al., 2011). In the developing lung, Myocd deletion decreases the expression of only a few smooth muscle genes (Figure S8), and in the developing ureter, expansion of smooth muscle progenitors occurs with minimal increase in the expression of Myocd (Trowe et al., 2012). Consistently, Myocd expression is insufficient to drive the entire smooth muscle gene-expression program in cell culture (Yoshida et al., 2004). Combined with previous findings from Myocd-knockout animals, our data reveal that airway branching is influenced by the initiation of smooth muscle differentiation but does not require mature smooth muscle.
Our scRNA-seq analyses also uncovered regulators of mesenchymal patterning during lung development. We computationally identified and genetically validated a role for Hippo signaling via Yap1 in the embryonic pulmonary mesenchyme. Mesenchymal Yap regulates smooth muscle differentiation around the gastrointestinal epithelium (Cotton et al., 2017), and crosstalk between Wnt and Yap signaling regulates intestinal stem cell homeostasis (Li et al., 2020). Future investigations will elucidate how Yap influences airway smooth muscle differentiation and whether cooperation with Wnt signaling is involved. Our data also reveal that sub-epithelial cells systematically downregulate enzymes required for proliferative metabolism as they differentiate into smooth muscle and that Foxk1/2, regulators of glycolysis (Sukonina et al., 2019), may be involved in differentiation. Metabolic reprogramming is a critical component of stem cell maintenance and differentiation (Intlekofer and Finley, 2019), and enhanced glycolysis acts via Yap to regulate neural crest migration (Bhattacharya et al., 2020). Notably, Foxk1 interacts with Srf to repress the expression of genes including Acta2 (Freddie et al., 2007). Taken together, these findings suggest that metabolic reprogramming is involved in airway smooth muscle differentiation.
In conclusion, our findings reveal the heterogeneity of the embryonic pulmonary mesenchyme and the multitude of signals involved in smooth muscle differentiation. Our computational analyses lay the foundation for future investigations into the mechanisms that regulate cell identity and differentiation in the pulmonary mesenchyme. Furthermore, our approaches could be extended to other organs or organisms to compare tissue patterning and differentiation in diverse developmental contexts.
Limitations of the study
Definitive conclusions about the contributions of each mesenchymal cluster to airway and vascular smooth muscle require lineage tracing in vivo. To our knowledge, there are unfortunately no mice available for lineage tracing these mesenchymal populations, which are distinguished by differences in the relative levels of expression of many marker genes that are also expressed by other cell types.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal αSMA antibody | Sigma | Cat# a5228; RRID:AB_262054 |
| Rabbit polyclonal Axin2 antibody | Abcam | Cat# ab32197; RRID:AB_2290204 |
| Rabbit polyclonal β-catenin antibody | Sigma | Cat# SAB4500541; RRID:AB_10743932 |
| Rabbit monoclonal E-cadherin antibody | Cell Signaling | Cat# 3195; RRID:AB_2291471 |
| Rat monoclonal E-cadherin antibody | Invitrogen | Cat # 13-1900; RRID:AB_86571 |
| Rabbit polyclonal Foxp1 antibody | Cell Signaling | Cat# 2005; RRID:AB_2106979 |
| Rabbit polyclonal GFP antibody | Invitrogen | Cat# A-11122; RRID:AB_221569 |
| Rabbit monoclonal Lef1 antibody | Cell Signaling | Cat# 2230; RRID:AB_823558 |
| Rabbit polyclonal CD31 (PECAM) antibody | Abcam | Cat# ab28364; RRID:AB_726362 |
| Rabbit polyclonal Pitx2 antibody | L’Honoré et al., 2007, gift of Jacques Drouin | N/A |
| Rabbit polyclonal RFP antibody | Abcam | Cat# ab62341; RRID:AB_945213 |
| Rabbit polyclonal Tbx3 antibody | Invitrogen | Cat# 42-4800; RRID:AB_2533526 |
| Goat anti-Mouse IgG1 Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat# A-21240; RRID:AB_141658 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat# A-11006; RRID:AB_141373 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat# A-11007; RRID:AB_141374 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat# A-11012; RRID:AB_141359 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat# A-21244; RRID:AB_2535812 |
| Alexa Fluor 568 Phalloidin | Thermo Fisher Scientific | Cat# A12380 |
| Alexa Fluor 488 Phalloidin | Thermo Fisher Scientific | Cat# A12379 |
| Chemicals, peptides, and recombinant proteins | ||
| LiCl | Sigma | L9650 |
| IWR1 | Sigma | I0161 |
| Cyclopamine | Tocris | 1623 |
| Critical commercial assays | ||
| RNAScope Multiplex Fluorescent V2 Assay | ACD | Cat No. 323100 |
| RNAScope Probe Mm-Hoxb6 | ACD | Cat No. 564171 |
| RNAScope Probe Mm-Ptn-C3 | ACD | Cat No. 486381-C3 |
| Deposited data | ||
| Raw and analyzed scRNA-seq data of E11.5 lungs | This paper | GEO: GSE153069 |
| Raw and analyzed bulk RNA-seq data of E13.5 Myocd mutant lungs | Young et al., 2020 | GEO: GSE143394 |
| ENCODE E14 lung DNase-hypersensitivity dataset | Yue et al., 2014 | http://www.mouseencode.org/ |
| Experimental models: Organisms/strains | ||
| Mouse: CD1 | ||
| Mouse: B6.129-Ctnnb1tm2Kem/KnwJ | The Jackson Laboratory | JAX: 004152 |
| Mouse: B6.129X1-Twist2tm1.1(cre)Dor/J | The Jackson Laboratory | JAX: 008712 |
| Mouse: B6.129P2-Gt(ROSA)26Sortm1(CAG-Brainbow2.1)Cle/J | The Jackson Laboratory | JAX: 017492 |
| Mouse: B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | The Jackson Laboratory | JAX: 007676 |
| Mouse: B6.FVB-Tg(Acta2-DsRed)1Rkl/J | The Jackson Laboratory | JAX: 031159 |
| Mouse: Tbx4-rtTA | Gift from Wei Shi | N/A |
| Mouse: tet-O-Cre | The Jackson Laboratory | JAX: 006234 |
| Mouse: VinTS | Laboratory of Sevan Hopyan, Tao et al., 2019 | N/A |
| Mouse: Yap1tm1.1Dupa/J | The Jackson Laboratory | JAX: 027929 |
| Oligonucleotides | ||
| Primer: Cre, Forward: GCATTACCGGTCGATGCAACGAGTGATGAG | Devenport Lab | N/A |
| Primer: Cre, Reverse: GAGTGAACGAACCTGGTCGAAATCAGTGCG | Devenport Lab | N/A |
| Primer: Tbx4, Forward: CGGCCCCGAATTCACCATGTCTAGA | Zhang et al., 2013 | N/A |
| Primer: Tbx4, Reverse: ACGCGTCGACACTTAGTTACCCGGGGAGCATG | Zhang et al., 2013 | N/A |
| Primer: Ctnnb1-flox, Forward: AAGGTAGAGTGATGAAAGTTGTT | The Jackson Laboratory | Primer no. oIMR1512 |
| Primer: Ctnnb1-flox, Reverse: CACCATGTCCTCTGTCTATTC | The Jackson Laboratory | Primer no. oIMR1513 |
| Primer: Yap1-flox, Forward: AGGACAGCCAGGACTACACAG | The Jackson Laboratory | Primer no. 29878 |
| Primer: Yap1-flox, Reverse: CACCAGCCTTTAAATTGAGAAC | The Jackson Laboratory | Primer no. 29879 |
| Primer: mTmG, Wildtype Forward: AGGGAGCTGCAGTGGAGTAG | The Jackson Laboratory | Primer no. 30298 |
| Primer: mTmG, Mutant Forward: TAGAGCTTGCGGAACCCTTC | The Jackson Laboratory | Primer no. 30297 |
| Primer: mTmG, Common Reverse: CTTTAAGCCTGCCCAGAAGA | The Jackson Laboratory |
Primer no. 12177 |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| MATLAB | MathWorks | N/A |
| CellRanger | 10× Genomics | Version 2.0.1 |
| R | R Foundation for Statistical Computing | N/A |
| Python | N/A | N/A |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| Regulatory Sequence Analysis Tools | Nguyen et al., 2018 | http://rsat.sb-roscoff.fr/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Seurat | Butler et al., 2018 | https://satijalab.org/seurat/ |
| Destiny | Haghverdi et al., 2015 | https://www.bioconductor.org/packages/release/bioc/html/destiny.html |
| Edge | Storey et al., 2019 | https://www.bioconductor.org/packages/release/bioc/html/edge.html |
| clusterProfiler | Yu et al., 2012 | https://www.bioconductor.org/packages//2.10/bioc/html/clusterProfiler.html |
| Other | ||
| Opal 520 Reagent Pack | Akoya Biosciences | FP1487001KT |
| Opal 620 Reagent Pack | Akoya Biosciences | FP1495001KT |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Celeste M. Nelson (celesten@princeton.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
Breeding of CD1, Ctnnb1fl/fl (JAX 004152), Dermo1-Cre (JAX 008712), R26R-Confetti (JAX 017492), mTmG (JAX 007676), SMA-RFP (JAX 031159), Tbx4-rtTA;tet-O-Cre (Zhang et al., 2013) (gift of Wei Shi), and Yapfl/fl (JAX 027929) mice and isolation of embryos were carried out in accordance with institutional guidelines following the NIH Guide for the Care and Use of Laboratory Animals and approved by Princeton’s Institutional Animal Care and Use Committee. Breeding of VinTS mice (Tao et al., 2019) and isolation of E11.5, E12.5, and E14.5 embryos were carried out in accordance with the Animals for Research Act of Ontario and the Guidelines of the Canadian Council on Animal Care, and procedures were approved by the Hospital for Sick Children Animal Care Committee. The sex of the embryos was not determined. Dermo1-Cre;Confettifl/+ embryos were obtained by mating Confetti homozygous females to Dermo1-Cre heterozygous males. Ctnnb1fl/fl or Yapfl/fl and mTmG mice were bred to generate homozygous Ctnnb1fl/fl;mTmG and Yapfl/fl;mTmG mice. Dermo1-Cre males were bred to Yapfl/fl females to generate Dermo1-Cre;Yapfl/+ males, and these were then mated to Yapfl/fl;mTmG females to generate Dermo1-Cre;Yapfl/fl;mTmG embryos. Tbx4-rtTA;tet-O-Cre males were bred to Ctnnb1-flox;mTmG females to generate Tbx4-rtTA;tet-O-Cre;Ctnnb1fl/+;mTmG males, and these were then mated to Ctnnb1fl/fl;mTmG females to generate Tbx4-rtTA;tet-O-Cre;Ctnnb1fl/fl;mTmG embryos. Pregnant dams were administered 0.5 mg/ml doxycycline (Sigma) via drinking water from E6.5 to E12.5, with water replaced every two days. Littermates were used as controls. Pups and embryos were genotyped for Cre, Tbx4-rtTA, mTmG, Ctnnb1fl, and/or Yapfl by isolating DNA from the tail snips (pups) or from the head of each embryo, followed by PCR and gel electrophoresis. The forward primer sequence for Cre was GCATTACCGGTCGATGCAACGAGTGATGAG and the reverse primer sequence was GAGTGAACGAACCTGGTCGAAATCAGTGCG. The primer sequences for Tbx4-rtTA were previously published (Zhang et al., 2013) and the primer sequences for Ctnnb1fl/fl, Yapfl/fl, and mTmG were provided in the Jackson Labs genotyping protocol for each strain. Where possible, mGFP expression from mTmG was used for genotyping.
Method details
scRNA-seq experiments
Lungs were dissected from CD1 mouse embryos collected at E11.5 in cold PBS. Isolated left lobes were grouped by stage, placed in dispase, and mechanically dissociated with tungsten needles. After 10 minutes in dispase at room temperature, DMEM without HEPES and supplemented with 5% fetal bovine serum (FBS, Atlanta Biologicals) was added, and cell suspensions were passed through a filter with 40-μm-diameter pores. The resultant cell suspensions were then processed by the Princeton Genomics Core Facility. The two sample groups were processed separately. Cells were loaded and processed using the Chromium Single Cell 3’ Library and Gel Bead Kit v2 on the Chromium Controller (10× Genomics) following manufacturer protocols. Individual cells were encapsulated in droplets with single gel beads carrying unique barcoded primers and then lysed. Then cDNA fragments were synthesized, barcoded, and amplified by PCR. Illumina sequencing libraries were prepared from the amplified cDNA from each sample group using the Nextra DNA library prep kit (Illumina). The v2 libraries were sequenced on Illumina HiSeq 2500 Rapid flowcells (Illumina) as paired-end 26 + 50 nucleotide reads following manufacturer protocols. Base calling was performed and raw sequencing reads were filtered using the Illumina sequencer control software to keep only pass-filtered reads for downstream analysis. The 10× CellRanger software version 2.0.1 was used to run the count pipeline with default settings on all FASTQ files from each sample to generate gene-barcode matrices using the Mus musculus reference genome mm10-1.2.0.
Organ explant culture and live imaging
Lungs from E11.5 CD1 mice were dissected in cold, sterile PBS supplemented with antibiotics (50 units/mL of penicillin and streptomycin) and then cultured on porous membranes (nucleopore polycarbonate track-etch membrane, 8 μm pore size, 25 mm diameter; Whatman) floating on top of DMEM/F12 medium (without HEPES) supplemented with 5% FBS and antibiotics (50 units/mL of penicillin and streptomycin) for 24 hr. Reagents used to manipulate Wnt signaling included LiCl (10 mM; Sigma) and IWR1 (100 μM; Sigma). To inhibit Shh signaling we used cyclopamine (1 μM; Tocris). For live-imaging analysis, Dermo1-Cre/+;Confetti/+ lungs were cultured on Transwell filters (polyethylene terephthalate membrane, 3 μm pore size, 10.5 mm diameter; Corning) within a stage-top incubator (Pathology Devices). Frames were acquired every 30 or 60 min for up to 48 hr under brightfield (1-2 ms exposure per plane for a total of seven planes per time point) or spinning disk confocal illumination (X-light, 122 ms exposure per plane for 5-7 planes per time point) on an inverted microscope (Nikon Ti).
Vinculin tension sensor (VinTS) experiments
Lungs were isolated from E12.5 VinTS embryos and embedded in 1% low melting point agarose on a coverslip fitted into a custom imaging chamber. After an hour of incubation at 37°C, cell culture medium supplemented with 5% FBS was added to the chamber and samples were imaged by confocal and fluorescence lifetime microscopy (FLIM) (Tao et al., 2019). FLIM was carried out on a Nikon A1R Si laser-scanning confocal microscope with a PicoHarp 300 TCSPC module and a 440 nm pulsed-diode laser (Picoquant). Imaging was performed with a 40×/1.25 NA water immersion objective. For each lung, 2 to 3 z-stacks at different locations along the distal left lobe were acquired. Fluorescence lifetime of the donor fluorophore (mTFP1) was estimated using FLIM Fit 5.1.1 (Warren et al., 2013). Cells of interest were segmented manually in FLIM Fit to obtain an average lifetime per cell. Sub-epithelial mesenchymal cells were at least 2-cell bodies away the from the mesothelium, and sub-mesothelial mesenchymal cells were within 2-cell bodies of the mesothelium.
Immunofluorescence analysis and imaging
Isolated lungs were fixed in 4% paraformaldehyde in PBS for 15 minutes at room temperature. For sectioning, lungs were washed first in 20% sucrose in PBS, then 30% sucrose in PBS, then left overnight in a 1:1 mixture of OCT and 30% sucrose prior to embedding and freezing in OCT. A Leica CM3050S cryostat was then used to create 10-μm-thick sections for staining on slides. For whole-mount staining and for sections on slides, samples were washed with 0.1% Triton X-100 in PBS and then blocked with 5% goat serum and 0.1% BSA. Samples were then incubated with primary antibodies against αSMA (Sigma a5228, 1:400), Axin2 (Abcam ab32197, 1:200), β-catenin (Sigma SAB4500541, 1:200), E-cadherin (Cell Signaling 3195, 1:200 or Invitrogen 13-1900, 1:200), Foxp1 (Cell Signaling 2005, 1:200), GFP (Invitrogen A-11122, 1:500), Lef1 (Cell Signaling 2230, 1:200), PECAM (Abcam ab28364, 1:200), Pitx2 (gift of Jacques Drouin, 1:300), RFP (Abcam ab62341, 1:400), or Tbx3 (Invitrogen 42-4800, 1:200) followed by incubation with Alexa Fluor-conjugated secondary antibodies (1:200), Alexa Fluor-conjugated phalloidin (1:500), and/or Hoechst (1:1000). Sections on slides were then mounted in Fluorosave. Whole lungs were dehydrated in a methanol series (or isopropanol series for phalloidin-stained lungs) and cleared with Murray’s clear (1:2 ratio of benzyl alcohol to benzyl benzoate). To preserve endogenous fluorescence of Confetti and VinTS lungs, samples were not dehydrated or cleared, but instead mounted in Fluorosave. Confocal stacks were collected using a spinning disk confocal (BioVision X-Light V2) on an inverted microscope (Nikon Ti) using either 20× air, 40× oil, or 60× oil objectives (Nikon).
Fluorescence in situ hybridization analysis
Lungs were isolated at E11.5 and immediately fixed in 4% paraformaldehyde in PBS for 24 hr at 4ºC. Lungs were then washed through a sucrose gradient, embedded in OCT (Tissue Tek), and frozen on dry ice. Sectioning was performed on a Leica CM3050S cryostat. Fluorescence in situ hybridization was performed using the standard RNAScope Multiplex Fluorescent V2 Assay (ACD) protocol for fixed-frozen samples. The probes were for Mus musculus Hoxb6 (RNAScope Probe 564171) and Ptn (RNAScope Probe 486381) and the fluorophores were Opal 520 and Opal 620 (Akoya Biosciences FP1487001KT and FP1495001KT). Sections were imaged on a Nikon A1RSi confocal microscope with a 20× objective.
Quantification and statistical analyses
scRNA-seq data analysis
scRNA-seq data exported from Cell Ranger were imported into R and processed using the Seurat package (Butler et al., 2018). Datasets from all groups were normalized and then integrated based on 2000 variable features (genes) identified by the Seurat function FindVariableFeatures. The integrated dataset was then scaled and analyzed to find neighbors and clusters with the Seurat functions FindNeighbors and FindClusters. Finally, the uniform manifold approximation and projection (UMAP) dimensional reduction was performed to visualize clusters. For initial analyses, all cells were used, which revealed large clusters of blood and immune cells (Figures S1K and S1L). Since these were not the cell populations of interest, we removed them from further analysis (Figures S1M and S1N). We then adjusted the dataset to account for cell-to-cell variability based on cell-cycle stage using the Seurat package. The resulting filtered and adjusted dataset was used for all subsequent analyses. Cluster identities were then assigned based on markers enriched in each cluster that are known to be enriched in each lung cell population. To investigate mesenchymal cells specifically, we repeated the Seurat pipeline outlined above including only cells from clusters 0, 1, and 2 (Figure S2). We generated a UMAP, identified markers for each of the 5 new clusters identified, and examined the expression of putative progenitor markers in each cluster (Figure S2).
Using published in situ hybridization results available on MGI, we compiled a list of genes that have been detected in the pulmonary mesenchyme at E11.5-E12.5. Based on MGI entries containing images, we classified the expression pattern of each gene as either sub-epithelial, sub-mesothelial, smooth muscle, everywhere, or weak (Table S3). Genes that did not fit any of these broad categories because of even more limited expression domains were excluded. A subset of the genes classified as sub-epithelial or sub-mesothelial were detected and included as variable features by the Seurat algorithm. Using the heatmap.2 function in R, we clustered sub-epithelial and sub-mesothelial cells based on their expression of this subset of genes (columns in Figure 2E) and clustered the genes from the curated MGI list (rows in Figure 2E).
To search for functional differences between the mesenchymal clusters identified in Figure 1, we identified genes that were differentially expressed in cluster 0 compared to cluster 1 using the FindMarkers function from the Seurat package, and then grouped these based on whether they were upregulated in cluster 0 or in cluster 1. These gene lists were then subjected to GO enrichment analysis using the clusterProfiler package in R (Yu et al., 2012).
Diffusion analysis of mesenchymal cells
To reconstruct smooth muscle differentiation trajectories, we used the cell-by-gene matrix of 2000 variable genes with scaled expression values generated using the Seurat algorithm and applied the Destiny package in R (Haghverdi et al., 2015). Only clusters 0 and 2 were used in this analysis. Three cells from cluster 0 appeared as clear outliers and were excluded from this analysis. To search for groups of genes with similar expression patterns over the course of smooth muscle differentiation, we first used the edgeR package in R to fit the expression of all genes along DC1 using a spline model with three degrees of freedom (Storey et al., 2019). The resulting fits were then filtered by adjusted p value < 0.05 obtained by implementing a generalized likelihood ratio test with the lrt function from the edgeR package (Storey et al., 2019). These fits were then clustered using hierarchical agglomerative clustering with average linkage based on Pearson correlation distance to identify gene sets with similar expression profiles along DC1. Gene sets of interest were then subjected to KEGG pathway enrichment analysis implemented using clusterProfiler (Yu et al., 2012).
Motif discovery
Motif discovery was carried out by examining promoter regions and DNase-hypersensitive regions near genes of interest using a custom Python pipeline and HOMER (Heinz et al., 2010). Genomic regions based on the mouse ENCODE E14 lung DNase-hypersensitivity dataset (Yue et al., 2014) were matched to genes from the gene sets identified in Figure 5D based on proximity to their transcriptional start sites. The sequences of these DNase-hypersensitive regions were then extracted and analyzed using the HOMER findMotifs.pl FASTA motif analysis tool (Heinz et al., 2010) to identify overrepresented motifs compared to a background file of DNase-hypersensitive regions located near the transcriptional start sites of a list of genes randomly generated with the Regulatory Sequence Analysis Tools (RSAT) web interface (Nguyen et al., 2018).
Image analysis and statistics
We developed a simple pipeline in MatLab to quantify fluorescence intensity profiles. First, images of Lef1 or Foxp1 immunostaining were background subtracted and then lines were traced either starting at the edge of branch L.L2 and moving outward into the mesenchyme for Lef1 or starting at the mesothelium near branch L.L2 and moving inward into the mesenchyme for Foxp1. Average fluorescence intensity in a 16 × 16 pixel window (5.7 × 5.7 μm) was measured at each point along the line, and the calculation included only the intensity of pixels within cell nuclei based on a mask generated from Hoechst staining. Intensity profiles were averaged from 5 distinct lines per sample, and the resultant curves were compared using two-way ANOVA in GraphPad Prism 5. A similar pipeline was used to quantify Axin2, β-catenin, and F-actin intensity, with small modifications. Axin2 intensity was only averaged from regions with no Hoechst signal, and β-catenin levels were compared as the ratio of mean nuclear (Hoechst+) to mean cytoplasmic (Hoechst-) pixels. F-actin intensity was measured from all pixels (no mask). To measure PECAM and αSMA immunofluorescence intensity in blood vessels, 10-pixel-thick lines were manually traced across blood vessels in ImageJ and pixel intensity was plotted as a function of distance.
To measure smooth muscle coverage, we first generated masks based on z-projected confocal images of αSMA immunofluorescence. Images were background subtracted and smoothed prior to thresholding. Next, the mask was subject to a “closing” morphological operation in which a binary image is dilated (white regions expand) and then eroded (white regions shrink) in order to close any holes in the initial mask. % coverage was then calculated as the area of the original mask divided by the area of the closed mask (Figures S9D–S9E).
Time-lapse analysis
Cells from time-lapse movies of Dermo1-Cre/+;Confetti/+ embryos were tracked manually in Fiji for as many consecutive time points as possible, and the resultant tracks were analyzed in MatLab. To estimate cell speeds, we first corrected cell tracks for local tissue movement and drift. Since the direction and magnitude of lung growth depends on location within the organ, we corrected cell tracks based on local displacements: for each cell, we computed the mean displacement of all cells within a 150 pixel (111 μm) radius as an estimate of local tissue movement, and then subtracted the cumulative sum of these displacements from the original cell’s track at each time point. This approach allowed us to independently correct for proximal-distal movements near the elongating primary bronchus and lateral movements near expanding branch tips (see tracks for smooth muscle cells vs. sub-epithelial and sub-mesothelial mesenchymal cells in Figure S5F). Corrected tracks had slightly smaller instantaneous speeds and much smaller overall displacements (Figures S5G and S5H), indicating that our approach had removed the effects of drift and tissue growth. MSD curves for each tracked cell were generated based on time intervals of up to 7 hours and fit with a 1st degree polynomial to estimate persistence. Only fits with R2 greater than 0.4 were included in the comparison between samples. To generate the pie charts in Figure 4, cells were assigned to a tissue compartment (sub-epithelial mesenchyme, sub-mesothelial mesenchyme, or smooth muscle) based on their location at the start of their track, and classified as staying within their compartment or crossing into the adjacent compartment by comparing the entire track to tissue growth in the brightfield channel. The boundary between sub-epithelial and sub-mesothelial mesenchyme was between 2- and 3-cell bodies away from the mesothelium. Cells were classified as differentiating into airway smooth muscle based on their positions and their progressively elongating morphology.
Bulk RNA-seq analysis of Myocd-mutant lungs
Bulk RNA-seq data for 5 control and 5 mutant E13.5 lungs in which Myocd had been deleted from the mesenchyme were downloaded from GEO [accession number GSE143394 (Young et al., 2020)] and processed using the DESeq2 package (Love et al., 2014). Briefly, the DESeq2 package was used to import data, estimate size factors and dispersions, and then calculate differential expression of genes and significance using the Wald test. Log fold changes were shrunk according to the DESeq2 package guidelines. We then used volcano plots to visualize adjusted p-values and log2 fold changes for either genes that correlated positively (sets 1-4) or negatively (set 5) with DC1 in Figure 5D (Figure S8A) or smooth muscle cluster marker genes from Figures 1B and 1D (Figure S8C). Since most of the genes were not significantly differentially expressed, we also plotted adjusted p-values against the mean of regularized log transformed counts obtained using the DESeq2 package to confirm that these genes were detected at high enough levels in the bulk RNA-seq dataset and found that there was no bias of differential expression based on transcript abundance (Figures S8B and S8D).
Acknowledgments
We would like to acknowledge Dr. Wei Wang and the Genomics Core Facility of Princeton University. We would like to thank Dr. Wei Shi (Keck School of Medicine of USC) for generously providing us with the Tbx4-rtTA;tet-O-Cre mouse line and Jacques Drouin (Montreal Clinical Research Institute) for sharing the Pitx2 antibody. We would like to thank the Confocal Imaging Facility, a Nikon Center of Excellence, in the Department of Molecular Biology at Princeton University for instrument use and technical advice. We would also like to thank members of the Tissue Morphodynamics Group for helpful discussions and feedback on the article. This work was supported by the NIH (HD0990300 and HL120142) and an HHMI Faculty Scholars Award to C.M.N and a CIHR award (MOP 126115) to S.H. K.G. was supported in part by a postgraduate scholarship-doctoral (PGS-D) from the Natural Sciences and Engineering Research Council of Canada and the Dr. Margaret McWilliams Predoctoral Fellowship from the Canadian Federation of University Women. J.M.J. was supported in part by an NIH NRSA Fellowship (F30 HL139039).
Author contributions
K.G. and C.M.N. conceptualized the study, designed the experiments, interpreted the data, and wrote the article. K.G. performed the experiments and collected the data. J.M.J. carried out the motif discovery analysis. H.T., M.Z., and S.H. provided all reagents and technical assistance for obtaining VinTS results. All authors provided input on the final article.
Declaration of interests
The authors declare no competing interests.
Published: March 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103838.
Supplemental information
Data and code availability
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Al Alam D., Green M., Tabatabai Irani R., Parsa S., Danopoulos S., Sala F.G., Branch J., El Agha E., Tiozzo C., Voswinckel R., et al. Contrasting expression of canonical Wnt signaling reporters TOPGAL, BATGAL and Axin2(LacZ) during murine lung development and repair. PLoS One. 2011;6:e23139. doi: 10.1371/journal.pone.0023139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badri K.R., Zhou Y., Schuger L. Embryological origin of airway smooth muscle. Proc. Am. Thorac. Soc. 2008;5:4–10. doi: 10.1513/pats.200704-049VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriga E.H., Franze K., Charras G., Mayor R. Tissue stiffening coordinates morphogenesis by triggering collective cell migration in vivo. Nature. 2018;554:523–527. doi: 10.1038/nature25742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya D., Azambuja A.P., Simoes-Costa M. Metabolic reprogramming promotes neural crest migration via yap/tead signaling. Dev. Cell. 2020;53:199–211.e6. doi: 10.1016/j.devcel.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill E.W., Park J.S., Chung E., Chen F., Magella B., Potter S.S. Single cell dissection of early kidney development: multilineage priming. Development. 2014;141:3093–3101. doi: 10.1242/dev.110601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes A.N., Lefevre J.G., Wilson S., Hamilton N.A., Little M.H. Cap mesenchyme cell swarming during kidney development is influenced by attraction, repulsion, and adhesion to the ureteric tip. Dev. Biol. 2016;418:297–306. doi: 10.1016/j.ydbio.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Cotton J.L., Li Q., Ma L., Park J.S., Wang J., Ou J., Zhu L.J., Ip Y.T., Johnson R.L., Mao J. YAP/TAZ and hedgehog coordinate growth and patterning in gastrointestinal mesenchyme. Dev. Cell. 2017;43:35–47.e4. doi: 10.1016/j.devcel.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danopoulos S., Alonso I., Thornton M.E., Grubbs B.H., Bellusci S., Warburton D., Al Alam D. Human lung branching morphogenesis is orchestrated by the spatiotemporal distribution of ACTA2, SOX2, and SOX9. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018;314:L144–L149. doi: 10.1152/ajplung.00379.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Langhe S.P., Carraro G., Tefft D., Li C., Xu X., Chai Y., Minoo P., Hajihosseini M.K., Drouin J., Kaartinen V., Bellusci S. Formation and differentiation of multiple mesenchymal lineages during lung development is regulated by beta-catenin signaling. PLoS One. 2008;3:e1516. doi: 10.1371/journal.pone.0001516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Langhe S.P., Sala F.G., Del Moral P.M., Fairbanks T.J., Yamada K.M., Warburton D., Burns R.C., Bellusci S. Dickkopf-1 (DKK1) reveals that fibronectin is a major target of Wnt signaling in branching morphogenesis of the mouse embryonic lung. Dev. Biol. 2005;277:316–331. doi: 10.1016/j.ydbio.2004.09.023. [DOI] [PubMed] [Google Scholar]
- Eastman Q., Grosschedl R. Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr. Opin. Cell Biol. 1999;11:233–240. doi: 10.1016/s0955-0674(99)80031-3. [DOI] [PubMed] [Google Scholar]
- El Agha E., Kheirollahi V., Moiseenko A., Seeger W., Bellusci S. Ex vivo analysis of the contribution of FGF10(+) cells to airway smooth muscle cell formation during early lung development. Dev. Dyn. 2017;246:531–538. doi: 10.1002/dvdy.24504. [DOI] [PubMed] [Google Scholar]
- Freddie C.T., Ji Z., Marais A., Sharrocks A.D. Functional interactions between the Forkhead transcription factor FOXK1 and the MADS-box protein SRF. Nucleic Acids Res. 2007;35:5203–5212. doi: 10.1093/nar/gkm528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraddi R.R., Chung C.Y., Heinz R.E., Balcioglu O., Novotny M., Trejo C.L., Dravis C., Hagos B.M., Mehrabad E.M., Rodewald L.W., et al. Single-cell transcriptomes distinguish stem cell state changes and lineage specification programs in early mammary gland development. Cell Rep. 2018;24:1653–1666.e7. doi: 10.1016/j.celrep.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin K., Mao S., Guyomar T., Miller E., Radisky D.C., Kosmrlj A., Nelson C.M. Smooth muscle differentiation shapes domain branches during mouse lung development. Development. 2019;146:dev181172. doi: 10.1242/dev.181172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss A.M., Tian Y., Cheng L., Yang J., Zhou D., Cohen E.D., Morrisey E.E. Wnt2 signaling is necessary and sufficient to activate the airway smooth muscle program in the lung by regulating myocardin/Mrtf-B and Fgf10 expression. Dev. Biol. 2011;356:541–552. doi: 10.1016/j.ydbio.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grashoff C., Hoffman B.D., Brenner M.D., Zhou R., Parsons M., Yang M.T., Mclean M.A., Sligar S.G., Chen C.S., Ha T., Schwartz M.A. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263–266. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati G.S., Sikandar S.S., Wesche D.J., Manjunath A., Bharadwaj A., Berger M.J., Ilagan F., Kuo A.H., Hsieh R.W., Cai S., et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science. 2020;367:405–411. doi: 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaksma C.J., Schwartz R.J., Tomasek J.J. Myoepithelial cell contraction and milk ejection are impaired in mammary glands of mice lacking smooth muscle alpha-actin. Biol. Reprod. 2011;85:13–21. doi: 10.1095/biolreprod.110.090639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Buettner F., Theis F.J. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics. 2015;31:2989–2998. doi: 10.1093/bioinformatics/btv325. [DOI] [PubMed] [Google Scholar]
- Heer N.C., Martin A.C. Tension, contraction and tissue morphogenesis. Development. 2017;144:4249–4260. doi: 10.1242/dev.151282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huycke T.R., Miller B.M., Gill H.K., Nerurkar N.L., Sprinzak D., Mahadevan L., Tabin C.J. Genetic and mechanical regulation of intestinal smooth muscle development. Cell. 2019;179:90–105.e21. doi: 10.1016/j.cell.2019.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer A.M., Finley L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019;1:177–188. doi: 10.1038/s42255-019-0032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaslove J.M., Nelson C.M. Smooth muscle: a stiff sculptor of epithelial shapes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018;373:20170318. doi: 10.1098/rstb.2017.0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.Y., Pang M.F., Varner V.D., Kojima L., Miller E., Radisky D.C., Nelson C.M. Localized smooth muscle differentiation is essential for epithelial bifurcation during branching morphogenesis of the mammalian lung. Dev. Cell. 2015;34:719–726. doi: 10.1016/j.devcel.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N., Vu T.H. Parabronchial smooth muscle cells and alveolar myofibroblasts in lung development. Birth Defects Res. C Embryo Today. 2006;78:80–89. doi: 10.1002/bdrc.20062. [DOI] [PubMed] [Google Scholar]
- Kumar M.E., Bogard P.E., Espinoza F.H., Menke D.B., Kingsley D.M., Krasnow M.A. Mesenchymal cells. Defining a mesenchymal progenitor niche at single-cell resolution. Science. 2014;346:1258810. doi: 10.1126/science.1258810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.Y., Marston D.J., Walston T., Hardin J., Halberstadt A., Goldstein B. Wnt/Frizzled signaling controls C. elegans gastrulation by activating actomyosin contractility. Curr. Biol. 2006;16:1986–1997. doi: 10.1016/j.cub.2006.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie K.O., Mitchell J.J., Woodcock-Mitchell J.L., Low R.B. Alpha smooth muscle actin expression in developing and adult human lung. Differentiation. 1990;44:143–149. doi: 10.1111/j.1432-0436.1990.tb00547.x. [DOI] [PubMed] [Google Scholar]
- Li Q., Sun Y., Jarugumilli G.K., Liu S., Dang K., Cotton J.L., Xiol J., Chan P.Y., Deran M., Ma L., et al. Lats1/2 sustain intestinal stem cells and Wnt activation through TEAD-dependent and independent transcription. Cell Stem Cell. 2020;26:675–692.e8. doi: 10.1016/j.stem.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Rowan S.C., Liang J., Yao C., Huang G., Deng N., Xie T., Wu D., Wang Y., Burman A., et al. Categorization of lung mesenchymal cells in development and fibrosis. iScience. 2021;24:102551. doi: 10.1016/j.isci.2021.102551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke T.H., Rudat C., Wojahn I., Weiss A.C., Kleppa M.J., Kurz J., Farin H.F., Moon A., Christoffels V.M., Kispert A. Tbx2 and Tbx3 act downstream of Shh to maintain canonical Wnt signaling during branching morphogenesis of the murine lung. Dev. Cell. 2016;39:239–253. doi: 10.1016/j.devcel.2016.08.007. [DOI] [PubMed] [Google Scholar]
- Luo Y., El Agha E., Turcatel G., Chen H., Chiu J., Warburton D., Bellusci S., Qian B.P., Menke D.B., Shi W. Mesenchymal adenomatous polyposis coli plays critical and diverse roles in regulating lung development. BMC Biol. 2015;13:42. doi: 10.1186/s12915-015-0153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailleux A.A., Kelly R., Veltmaat J.M., De Langhe S.P., Zaffran S., Thiery J.P., Bellusci S. Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development. 2005;132:2157–2166. doi: 10.1242/dev.01795. [DOI] [PubMed] [Google Scholar]
- Menshykau D., Blanc P., Unal E., Sapin V., Iber D. An interplay of geometry and signaling enables robust lung branching morphogenesis. Development. 2014;141:4526–4536. doi: 10.1242/dev.116202. [DOI] [PubMed] [Google Scholar]
- Metzger R.J., Klein O.D., Martin G.R., Krasnow M.A. The branching programme of mouse lung development. Nature. 2008;453:745–750. doi: 10.1038/nature07005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger R.J., Krasnow M.A. Genetic control of branching morphogenesis. Science. 1999;284:1635–1639. doi: 10.1126/science.284.5420.1635. [DOI] [PubMed] [Google Scholar]
- Miller L.A., Wert S.E., Clark J.C., Xu Y., Perl A.K., Whitsett J.A. Role of Sonic hedgehog in patterning of tracheal-bronchial cartilage and the peripheral lung. Dev. Dyn. 2004;231:57–71. doi: 10.1002/dvdy.20105. [DOI] [PubMed] [Google Scholar]
- Moiseenko A., Kheirollahi V., Chao C.M., Ahmadvand N., Quantius J., Wilhelm J., Herold S., Ahlbrecht K., Morty R.E., Rizvanov A.A., et al. Origin and characterization of alpha smooth muscle actin-positive cells during murine lung development. Stem Cells. 2017;35:1566–1578. doi: 10.1002/stem.2615. [DOI] [PubMed] [Google Scholar]
- Neumann N.M., Perrone M.C., Veldhuis J.H., Huebner R.J., Zhan H., Devreotes P.N., Brodland G.W., Ewald A.J. Coordination of receptor tyrosine kinase signaling and interfacial tension dynamics drives radial intercalation and tube elongation. Dev. Cell. 2018;45:67–82 e6. doi: 10.1016/j.devcel.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen N.T.T., Contreras-Moreira B., Castro-Mondragon J.A., Santana-Garcia W., Ossio R., Robles-Espinoza C.D., Bahin M., Collombet S., Vincens P., Thieffry D., et al. RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res. 2018;46:W209–W214. doi: 10.1093/nar/gky317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt R., Gullotta F., Nusspaumer G., Unal E., Ivanek R., Zuniga A., Zeller R. Molecular signatures identify immature mesenchymal progenitors in early mouse limb buds that respond differentially to morphogen signaling. Development. 2019;146:dev173328. doi: 10.1242/dev.173328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman J., Schuyler W., Mcdonald J.A., Roser S. Heparin inhibits lung branching morphogenesis: potential role of smooth muscle cells in cleft formation. Am. J. Med. Sci. 1998;316:368–378. doi: 10.1097/00000441-199812000-00003. [DOI] [PubMed] [Google Scholar]
- Schildmeyer L.A., Braun R., Taffet G., Debiasi M., Burns A.E., Bradley A., Schwartz R.J. Impaired vascular contractility and blood pressure homeostasis in the smooth muscle alpha-actin null mouse. FASEB J. 2000;14:2213–2220. doi: 10.1096/fj.99-0927com. [DOI] [PubMed] [Google Scholar]
- Shyer A.E., Tallinen T., Nerurkar N.L., Wei Z., Gil E.S., Kaplan D.L., Tabin C.J., Mahadevan L. Villification: how the gut gets its villi. Science. 2013;342:212–218. doi: 10.1126/science.1238842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert H.J., Van der Flier L.G., Sato T., Van Es J.H., Van Den Born M., Kroon-Veenboer C., Barker N., Klein A.M., Van Rheenen J., Simons B.D., Clevers H. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Solon J., Levental I., Sengupta K., Georges P.C., Janmey P.A. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys. J. 2007;93:4453–4461. doi: 10.1529/biophysj.106.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J.D., Leek J.T., Bass A.J. 2019. Edge: Extraction of Differential Gene Expression. R package Version 2.16.0.https://github.com/StoreyLab/edge [Google Scholar]
- Sukonina V., Ma H., Zhang W., Bartesaghi S., Subhash S., Heglind M., Foyn H., Betz M.J., Nilsson D., Lidell M.E., et al. FOXK1 and FOXK2 regulate aerobic glycolysis. Nature. 2019;566:279–283. doi: 10.1038/s41586-019-0900-5. [DOI] [PubMed] [Google Scholar]
- Tao H., Zhu M., Lau K., Whitley O.K.W., Samani M., Xiao X., Chen X.X., Hahn N.A., Liu W., Valencia M., et al. Oscillatory cortical forces promote three dimensional cell intercalations that shape the murine mandibular arch. Nat. Commun. 2019;10:1703. doi: 10.1038/s41467-019-09540-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler S., Buttner M., Fischer D.S., Lange M., Bergen V., Lickert H., Theis F.J. Concepts and limitations for learning developmental trajectories from single cell genomics. Development. 2019;146:dev170506. doi: 10.1242/dev.170506. [DOI] [PubMed] [Google Scholar]
- Trowe M.O., Airik R., Weiss A.C., Farin H.F., Foik A.B., Bettenhausen E., Schuster-Gossler K., Taketo M.M., Kispert A. Canonical Wnt signaling regulates smooth muscle precursor development in the mouse ureter. Development. 2012;139:3099–3108. doi: 10.1242/dev.077388. [DOI] [PubMed] [Google Scholar]
- Ustiyan V., Bolte C., Zhang Y., Han L., Xu Y., Yutzey K.E., Zorn A.M., Kalin T.V., Shannon J.M., Kalinichenko V.V. FOXF1 transcription factor promotes lung morphogenesis by inducing cellular proliferation in fetal lung mesenchyme. Dev. Biol. 2018;443:50–63. doi: 10.1016/j.ydbio.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren S.C., Margineanu A., Alibhai D., Kelly D.J., Talbot C., Alexandrov Y., Munro I., Katan M., Dunsby C., French P.M. Rapid global fitting of large fluorescence lifetime imaging microscopy datasets. PLoS One. 2013;8:e70687. doi: 10.1371/journal.pone.0070687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A.C., Xu J., Yin Y., Smith C., Schmid G., Ornitz D.M. FGF9 and SHH signaling coordinate lung growth and development through regulation of distinct mesenchymal domains. Development. 2006;133:1507–1517. doi: 10.1242/dev.02313. [DOI] [PubMed] [Google Scholar]
- Yamada T., Suzuki E., Gejyo F., Ushiki T. Developmental changes in the structure of the rat fetal lung, with special reference to the airway smooth muscle and vasculature. Arch. Histol. Cytol. 2002;65:55–69. doi: 10.1679/aohc.65.55. [DOI] [PubMed] [Google Scholar]
- Yin Y., White A.C., Huh S.H., Hilton M.J., Kanazawa H., Long F., Ornitz D.M. An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev. Biol. 2008;319:426–436. doi: 10.1016/j.ydbio.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T., Kawai-Kowase K., Owens G.K. Forced expression of myocardin is not sufficient for induction of smooth muscle differentiation in multipotential embryonic cells. Arterioscler Thromb. Vasc. Biol. 2004;24:1596–1601. doi: 10.1161/01.ATV.0000137190.63214.c5. [DOI] [PubMed] [Google Scholar]
- Young R.E., Jones M.K., Hines E.A., Li R., Luo Y., Shi W., Verheyden J.M., Sun X. Smooth muscle differentiation is essential for airway size, tracheal cartilage segmentation, but dispensable for epithelial branching. Dev. Cell. 2020;53:73–85.e5. doi: 10.1016/j.devcel.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D., et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Menke D.B., Jiang M., Chen H., Warburton D., Turcatel G., Lu C.H., Xu W., Luo Y., Shi W. Spatial-temporal targeting of lung-specific mesenchyme by a Tbx4 enhancer. BMC Biol. 2013;11:111. doi: 10.1186/1741-7007-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M., Tao H., Samani M., Luo M., Wang X., Hopyan S., Sun Y. Spatial mapping of tissue properties in vivo reveals a 3D stiffness gradient in the mouse limb bud. Proc. Natl. Acad. Sci. U S A. 2020;117:4781–4791. doi: 10.1073/pnas.1912656117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.