

Abstract
A 4-year-old child was transferred to the paediatric intensive care unit with acute respiratory failure following 4 days of fever, nausea and vomiting. Chest X-ray on admission had an enlarged cardiac silhouette and transthoracic echo confirmed a large pericardial effusion. An emergent pericardiocentesis was performed at bedside which drained nearly 1000 mL of purulent fluid. Postdrainage course was complicated by acute systolic and diastolic heart failure, thrombocytopenia and acute renal failure. A chest CT and MRI were concerning for a diffuse mediastinal soft-tissue density, so the patient underwent interventional radiology-guided biopsy complicated by haemorrhage requiring mediastinal exploration and subtotal thymectomy. Histopathology revealed changes consistent with kaposiform lymphangiomatosis and MRI demonstrated involvement of the lumbar spine and right hip. Following a course of intravenous antibiotics, the patient was started on sirolimus and prednisolone and ultimately discharged home.
Keywords: pericardial disease, infectious diseases, paediatric intensive care
Background
Paediatric pericardial effusions are an uncommon occurrence that can be due to a variety of aetiologies. Most commonly in the USA, they are primarily due to malignancy (39%) or idiopathic (37%), while rheumatological (9%), renal (8%) or other diagnoses (5%) make up a minority of cases.1 2 Only rarely are they due to bacterial infections, although internationally infectious aetiologies are more common with one study from Northern India finding that tuberculosis, bacterial and viral causes were most common.3
Clinically, pericardial effusions present along a spectrum of illness with the most severe complication being cardiac tamponade, where diastolic filling is compromised by elevated pericardial pressures. Chronic effusions typically present with large volume effusions as the pericardium stretches to accommodate the volume until a critical point is reached, while acute effusions will rapidly progress to tamponade.1
With the advent of modern vaccinations and antibiotics, purulent pericardial effusions are extremely uncommon. Historically, Staphylococcus aureus, Haemophilus influenzae, Neisseria meningitidis and Streptococcus pneumoniae were the most common bacterial aetiologies. Currently, S. aureus and H. influenzae are the most common causes.4 5 Purulent pericardial effusions secondary to Group A Streptococcus (GAS) are extremely rare with less than 10 cases reported in the literature.6–11
While the overwhelming majority of GAS infections are mild, invasive GAS infections are a source of significant morbidity and mortality predominantly from sepsis, necrotising fasciitis and streptococcal toxic shock syndrome. These infections follow a bimodal distribution with children <2 and adults >65 making up a majority of cases, especially those patients with underlying medical conditions such as diabetes.12 The severity of infection is also influenced by the serotype of GAS involved, with various mutations contributing to the ability of the bacteria to evade the immune system and to invade deeper tissues.13–15
Case presentation
Our patient was a 4-year-old boy with no medical history who presented to an outside emergency department following 4 days of fever, fatigue and poor intake. His parents had initially managed this with acetaminophen at home but due to persistent fever and fatigue brought him to the emergency department.
In the emergency department, the patient was found to have respiratory distress and was placed on high-flow nasal cannula. Evaluation at that time was notable for a chest X-ray showing diffuse bilateral pulmonary infiltrates and a poorly defined cardiomediastinal silhouette. Initial labs were notable for a lactic acid 15 mmol/L, white cell count 5.6x109/L with a neutrophil predominance, platelets 28x109/L, and arterial blood gas pH 7.38, pCO2 31 mm Hg, pO2 65 mm Hg, HCO3 18 mmol/L. A rapid strep swab was positive, COVID-19 PCR was negative and blood cultures were drawn. He was given a normal saline bolus and treated with levalbuterol, ipratropium, ceftriaxone, dexamethasone and sodium bicarbonate. He was then transferred to our paediatric intensive care unit for evaluation.
The patient had significant dyspnoea and appeared fatigued. Initial vitals showed heart rate 146 beats/min, respiratory rate 31 breaths/min, oxygen saturation 99% on 2 L nasal cannula and blood pressure 99/67 mm Hg. Physical examination was notable for significant retractions, tachypnoea and diminished breath sounds in the left lower lobe of his lungs with coarse breath sounds throughout. His cardiac examination was remarkable for muffled heart sounds but with normal S1 and S2, no murmurs and no rubs. There was no pulsus paradoxus appreciated. His abdominal examination was unremarkable without hepatosplenomegaly. He was also noted to have diffuse petechiae on his trunk. Repeat chest X-ray showed a significantly enlarged cardiac silhouette and pulmonary infiltrates so a stat echocardiogram was performed which showed a large pericardial effusion with collapse of the right atrium and qualitatively normal function (figure 1, videos 1 and 2). Laboratory evaluation was notable for a white cell count of 4.3x109/L with 90.2% neutrophils, haemoglobin of 118 g/L and platelets 22x109/L, for which he was transfused a unit of platelets. A complete metabolic panel was notable for a creatinine of 0.88 mg/dL, Blood Urea Nitrogen of 45 mg/dL, glucose of 68 mg/dL, uric acid of 13.2 mg/dL, Lactate Dehydrogenasen of 724 unit/L. ECG showed sinus tachycardia with first-degree A-V block, low QRS voltages and non-specific ST-T wave abnormalities.
Figure 1.
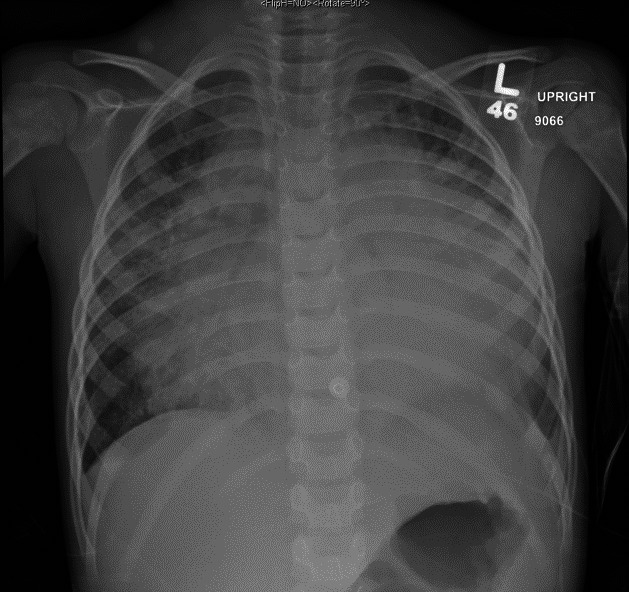
Chest radiography on presentation to our hospital with pronounced cardiomegaly and evidence prominent pulmonary vascularity, diffuse central airspace opacities and a small left pleural effusion.
Video 1.
Video 2.
The patient was intubated and underwent emergent pericardiocentesis with pig-tail drain placement which yielded 957 mL of cloudy, mixed bloody and purulent fluid. Pericardial cytology was reviewed and showed extensive Gram positive cocci in pairs and chains, and no blasts. The patient developed septic shock secondary to suspected disseminated bacterial infection requiring fluid resuscitation, stress dose hydrocortisone and epinephrine infusion.
His antibiotic regimen was broadened to include vancomycin and cefepime. A repeat echocardiogram showed improvement of the effusion but demonstrated moderately to severely depressed biventricular systolic dysfunction. Approximately 30 hours following pericardiocentesis, culture of the pericardial fluid grew GAS. An echocardiogram also showed a retroatrial mass prompting a CT scan of his chest which showed a heterogeneous attenuation of soft tissue density in the mediastinum. At the same time, a CT of his heart was performed which demonstrated normal structure with mild-moderate dilation of his right atrium and ventricle with bowing of the interatrial septum towards the left atrium (figure 2). The patient subsequently developed anuric renal failure requiring continuous renal replacement therapy. Over the course of several weeks his cardiac function and renal function improved and at the time of discharge his creatinine was normal at 0.32 mg/dL and his ejection fraction was 60%.
Figure 2.

Chest CT obtained after draining his pericardial effusion. (A) and (B) are obtained from a CT of the chest with contrast and show a relatively homogeneous soft tissue density within the mediastinum without discrete lymph nodes, rim-enhancing collection or enhancing mass. There are bilateral consolidative perihilar and central opacities.
The patient underwent an MRI of his chest when stable to further define the lesion and assess for resolution (figure 3). This showed a diffuse T2 hyperintensity of the entire mediastinum extending to the neck soft tissues superior with no enhancement or diffusion restriction. The visualised portion of the abdomen showed numerous T2 hyperintense foci in a miliary pattern through the entire splenic parenchyma.
Figure 3.

MRI obtained after chest CT showed a diffuse mediastinal mass. (A) Poorly defined T2 hyperintense mass in the anterior mid mediastinum. (B–D) are contrast-enhanced images, (B) arterial-phase contrast-enhanced imaging, (C) 5 min delayed vascular imaging, (D) interstitial-phase imaging. These demonstrate delayed-enhancement as is consistent with a diagnosis of kaposiform lymphangiomatosis.
The decision was then made to obtain an image-guided biopsy of his mediastinal mass. Following biopsy, he developed brisk bleeding and haemorrhagic shock. Cardiothoracic surgery emergently placed a chest tube that yielded 200 mL of bloody fluid. Emergent mediastinal exploration found a large haemothorax from an inflamed, edematous and fibrous thymus. There was also a fair amount of lymphatic drainage from the superior mediastinum. Histopathology of the mass showed numerous thin-walled vascular channels lined by endothelium positive for CD31 and D2-40. No significant cellular atypia was seen. On further examination, occasional cellular proliferation with focal spindle cell morphology, extravasation of erythrocytes and focal haemosiderin deposition were noted and the patient was diagnosed with kaposiform lymphangiomatosis (KLA) (figure 4).
Figure 4.

Pathology slides. (A) H&E stain at lower power (bar scale 500 um): the lesion is located in the thymic tissue and its vicinity and is represented by numerous thin-walled anastomosing vascular channels. (B) H&E stain at middle power view (bar scale 100 um): the lining cells are spindle without nuclear atypia. Right upper corner shows a Hassel Corpuscles, proving to be thymic tissue. (C) Immunohistochemical stain for CD31 (bar scale 100 um): CD31 antibody stained the spindle lining. (D) Immunohistochemical stain for D2-40 (bar scale 100 um): D2-40 antibody also stained the spindle lining, more specifically proving the lining spindle cells to be lymphatic endothelium.
During his stay, the patient developed lower extremity pain so a skeletal survey was performed. This identified sclerosis with metaphyseal changes most pronounced along the proximal right humerus and the bilateral distal femoral metaphyses, with a follow-up dedicated X-rays demonstrating patchy sclerosis of these bones. Follow-up MRI of these areas showed T2 enhancement with postcontrast enhancement of the metaphyses of the humerus, distal femoral metadiaphysis. In addition, a repeat MRI of his chest showed diffuse increased T2 signal in the L1 vertebral body with central focal enhancement. These findings were highly suggestive of KLA.
Investigations
His initial working diagnosis was group A Streptococcal pericarditis with sepsis. The most likely source of GAS infection was his history of pharyngitis. The large volume of the effusion suggested a chronic process with secondary infection. While it was clear his pericardial fluid was infected with GAS, there were several features of his illness that prompted further workup for the primary aetiology of his effusion. The large volume of his effusion and absence of complete haemodynamic collapse suggested a more chronic effusion. Given his thrombocytopenia, we considered a malignant cause, though cytological evaluation of his peripheral smear and the pericardial fluid showed no evidence of malignancy, which was further confirmed by negative flow cytometry. The rheumatology service was consulted and did not identify a rheumatological cause. Rheumatological workup revealed a normal C4 complement and low C3 complement of 51 mg/dL, his ANA, and anti-DNAse B were negative. Immunology was consulted for possible immunodeficiency predisposing him to an atypical infection. Immunological workup showed a poor response to pneumococcal vaccination. However, neutrophil oxidative burst testing, diphtheria, and tetanus antitoxoids and lymphocyte subset testing were all normal. These results were felt to be inconclusive and repeat testing was recommended following repeat pneumococcal vaccination.
A primary cardiac aetiology was also considered, especially with his cardiac dysfunction. A cardiomyopathy and myocarditis workup for common infectious, metabolic and autoimmune causes was negative, and his cardiac function improved with resolution of sepsis. Genetics was consulted for suspected dysmorphology and atypical illness. Arteriovenous panel testing for mutations of ACVL1, ENG, RASAI, SMAD4 was negative.
Over the course of evaluation, there was increasing evidence towards a lymphovascular anomaly. Lymphovascular anomalies may present as tumours such as kaposiform haemangioendothelioma or tufted angiomas; malformations, such as Gorham-Stout disease (GSD), generalised lymphatic anomaly (GLA), KLA; or channel anomalies.16 These all share overlapping features, which depend on the site of involvement, and definitive diagnosis may require tissue biopsy, as was the case in our patient. After our patient’s malformation was identified as KLA, it was thought to be the source of his pericardial effusion, as such effusions are commonly seen in patients with KLA. Per recent consensus guidelines on the management of complicated lymphatic anomalies, these diagnoses should be suspected when a lymphatic malformation is associated with lytic bone lesions, any non-malignant chylous effusion, or evidence of protein-losing enteropathy.17 The discussion below will cover the characteristics of our patient and examine them in relationship to other entities, with a focus on lymphatic malformations. Specific reviews are available for a broader discussion of vascular anomalies.16 18
Imaging
Recent consensus guidelines for management of complicated lymphatic anomalies recommend whole body MRI, skeletal survey and chest imaging including a chest radiography and ultrashort echo-time MRI or CT of the chest as the preferred imaging for suspected lymphatic anomalies.17
In our patient, both CT and MRI showed diffuse soft-tissue density throughout the mediastinum without discrete mass or lesion. This imaging finding is non-specific and can be associated with various lymphatic malformations including KLA, central conducting lymphatic anomaly (CCLA), and GLA. Moderate enhancement of the thickening on MRI/CT supports KLA over CCLA or GLA, especially when seen along the mediastinum and retroperitoneum.17 19 Towards the end of his hospitalisation, he had a repeat MRI of his chest which showed progressively enhancing T2 hyperintensity in the superior mediastinum extending to the bilateral supraclavicular and visualised cervical regions and encasing the mediastinal vasculature.
GSD, GLA, KLA can all present with osseous involvement. KLA and GLA will both have multiple lytic lesions sparing the cortex in non-contiguous sites, while GSD will have involvement of the cortex. On MRI, GSD, GLA and KLA can all demonstrate T2 enhancement and heterogeneous contrast enhancement, although the involvement of the cortex can help distinguish between GSD and GLA or KLA.20–22 Osseous lesions are a diagnostic criterion for GSD, while both GLA and KLA can present without osseous involvement.19–22
Haematologic abnormalities
Our patient initially presented with significant thrombocytopenia of 22x109/L. This responded appropriately to transfusion but he had recurrent thrombocytopenia that was thought to be due to disseminated intravascular coagulation (DIC) in the context of sepsis. His Prothrobin time was 1.57, activated Partial Thromboplastin Time 32.3 s, D-Dimer 19.16 µg/mL, fibrinogen 497 mg/dL. Patients with KLA often present with moderate thrombocytopenia with evidence of DIC even in the absence of acute illness.22–24 Thrombocytopenia is uncommon in GLA and GSA but kaposiform haemangioendothelioma can present with significant thrombocytopenia in the context of Kasabach-Merritt phenomenon (KMP), during which platelets and fibrinogen are consumed within the characteristic vascular lesions.25 26 The patient’s normal fibrinogen levels suggested against KMP being the source of his initial coagulopathy. He also had normalisation of his platelets and DIC labs prior to treatment of his KLA, which suggests that DIC was the predominant cause and not the KLA.
Pathology
While clinical examination, imaging features and haematological parameters can help distinguish between various lesions, tissue biopsy is often needed to make a definitive diagnosis. On gross pathology, lymphatic malformations are characterised by thin-walled vascular channels filled with pink proteinaceous material. On histopathology, round endothelial cells line the vessels and will stain with lymphatic markers PROX-1 and Podoplanin/D2‐40, as was seen in our patient. CD31 is a non-specific endothelial marker and can be positive in both lymphatic and vascular lesions. Both GLA and GSD have similar histological findings, including both osteoclasts and osteoblasts.21 KLA can be distinguished from these other lesions by the presence of spindled lymphatic endothelial cells. There also can be local haemorrhages, blood filled lymphatic channels, extravasation of erythrocytes and haemosiderin deposition.16 18 19 These findings can also be seen in kaposiform haemangioendothelioma, although this lesion tends to be isolated, rarely involves the thorax, and has cutaneous involvement. KLA is more diffuse, commonly involves the thorax and has less cutaneous involvement.25
Genetics
There is no syndromic association with KLA, although NRAS mutations have been implicated as discussed below. GLA is associated with a somatic mutation of the P1K3CA gene, which has also been implicated in a variety of overgrowth syndromes such as CLOVES (Congenital Lipomatous Overgrowth, Vascular malformation, Epidermal nevi, Scoliosis/skeletal/spinal anomalies)syndrome. A genetic cause of GSD has yet to be identified.18
Treatment
Our primary focus was relief of cardiac tamponade in order to restore adequate haemodynamics and supportive therapy for septic shock including antibiotics and renal replacement. After the identification of Streptococcus, antibiotic therapy was narrowed and clindamycin was added for toxic shock syndrome.
Once KLA was determined to be the most likely cause, the oncology service and the vascular anomalies team were consulted who recommended sirolimus and prednisolone. Given the severity of his initial presentation, treatment was deferred until he completed a 4-week course of antibiotics.
Outcome and follow-up
At his 6-month follow-up visit, the patient was doing well on sirolimus and had been weaned from prednisolone therapy. His lower extremity symptoms resolved and he had a normal activity level. Following administration of the 23-valent pneumococcal vaccine, repeat pneumococcal antigen testing showed appropriate humoral immune response. He continues to follow with orthopaedic surgery, the vascular anomalies team, pulmonology and cardiology. On subsequent follow-up visits with the haematology and vascular anomalies team and in discussion with his caretaker, the decision was made to stop sirolimus following a year of treatment with close monitoring thereafter.
Discussion
Our patient presented with many clinical features consistent with other documented cases of KLA. Patients often present in early childhood, with median ages at presentation 6.5 years (range, birth–44 years) and 4.4 years (range 1.1–9 years) per two case series of KLA.24 26 These series also cite a possible male predominance with four males and two females in a series of six patients and thirteen males and seven females of twenty patients. Patients often present with non-specific respiratory complaints, either acutely or with more chronic symptoms initially attributed to another cause. Haematological manifestations predominantly are thrombocytopenia and bleeding. Mediastinal soft tissue involvement is nearly universal frequently with associated effusions, pericardial effusions and/or pleural effusions at 70% and 85%, respectively. Subcutaneous masses occur in a minority of patients.24 26
The prognosis for patients with KLA is grim with mean 5-year survival reported at 51% and overall survival of 34%.24 The most commonly used treatment is sirolimus with or without steroids and vincristine, which is hypothesised to slow disease progression and improve symptoms in some patients, although many will continue to worsen and die.23 27 28 Several studies have identified activating NRAS mutations in KLA lesions and body fluids of these patients.29 30 The RAS/mitogen-activated protein kinase pathway signalling is essential for control of cell proliferation, differentiation and survival, with multiple abnormalities in this pathway associated with genetic syndromes, vascular and lymphatic malformations and malignancies.29–31 PI3K/mTOR signalling is activated by NRAS, which may help provide mechanistic support for this treatment in KLA. Other NRAS-mediated signalling pathways, such as RAF and ERK, will remain activated despite treatment with sirolimus which may help explain the variable response to this treatment. NRAS-specific therapeutics are being studied for treatment of melanoma and may eventually prove useful in the treatment of KLA.29 30 Bisphosphonates may also be used to stabilise osseous lesions and prevent further resorption of bone.18 22 23
Some cases of KLA and other lymphatic malformations may have discrete areas of obstruction that can be identified by Intranodal dynamic magnetic resonance lymphangiography or unipedal or bipedal lymphoscintigraphy using Tc 99 m sulphur colloid with and without single-photon emission CT. These obstructions may be relieved surgically via a lymphaticovenous anastomosis.17
Patient’s perspective.
As we grow up, our perception of a nightmare changes little by little. At 15, the worst thing a girl believes she could feel was the heartbreak as ‘the love of her life’ leaves her for a different girl. In that fleeting moment she truly believes her world is crumbling around her. At 18 its receiving the rejection letter from her dream college and then again at 21, the loss of their dream job. All of that, however, is manageable. We know long before all that comes to pass, that it’s survivable. Genuine pain isn’t a broken arm or some foolish break-up. Real pain lies in the silence as you stare out of a hospital window into the darkness; unable to sleep because every cell in your body, as a parent, is longing to fix what’s wrong with your child. Nothing life throws at you prepares you to stand in a room filled with machines. One to help his tiny heart beat or another to fill his lungs with air. The defeating feeling of millions of dollars of medical degrees and centuries of experience standing in front of me and no one knows what is wrong with my little boy. Nothing compares to that level of emptiness. Mommas are suppose to heal all the bumps and bruises and bellyaches a little boy gets, but this isn’t something that momma can fix. There is nothing I can do to make my little boy feel any better other than sit and hope I’ll hear my little boy’s voice again soon. How is a mother supposed to fill that void? Is it supposed to be anger? Sadness? Do I cry? Or do I hold strong because someone has to?
My son never hurt anyone. In fact, he did quite the opposite. All he wants is to smile and giggle and play. His day rose, was made, and set with love and compassion. Hard to believe that just days before this nightmare began he was running around the pumpkin patch to celebrate Halloween and flashing his goofy smile to every new friend he made as he ran and played.
I hide in the corner as I cry because if my baby boy hears me he’ll know just how scared I really am and he will definitely quickly follow. Anxiety attacks are starting to become a daily occurrence. Everyday I wake up sore and achy from the night before; Both emotionally and physically. In those morning moments when I again realize this isn’t a dream and everything around me is reality … I weep. Every morning it’s this outcry of sheer emptiness. I feel hollow. I don’t quite know the best way to describe how I’m feeling but it’s almost like a manic depression. One minute I’m fine, another I’m blistering angry at the laughter in the next room, and then another where I’m on the verge of breaking. This isn’t a fate I would wish on my worst enemy. Test after test and day after day with no definitive answers.
This is a MOTHER’S WORST NIGHTMARE!
To see her sweet little boy in front of her alive but not living … not really.
It’s now been 6 months since my son’s diagnosis and everyday has been an uphill battle. One half of his new version of life is relearning how to do basic tasks and skills, while the other is strapped into a carseat on the way to one of dozens of specialists. All of whom closely and attentively monitor his medications and well-being. We carry on with physical therapy and kindergarten, but the fear deep in my stomach remains. I constantly feel this dark cloud hover over me. I wait for the bottom to fall out with heartbreaking news that round two has begun and it’s time for us to fight again.
Learning points.
Large pericardial effusions may present with secondary infections with atypical organisms. Such presentations should prompt a workup to identify the underlying aetiology of the effusions.
Kaposiform lymphangiomatosis is aggressive and progressive lymphovascular malformation with a poor prognosis. It may present with a variety of symptoms including pericardial and pleural effusions.
Lymphatic malformations present with many overlapping characteristics and extensive evaluation including advanced imaging and biopsy may be required to make a definitive diagnosis.
The mainstay of treatment for kaposiform lymphangiomatosis (KLA) is sirolimus, with or without steroids or vincristine. Bisphosphonates can also be used to stabilise the osseous lesions.
Recent studies have identified NRAS mutations in multiple patients with KLA, this mutation may help explain the partial response to sirolimus in these patients, as the mTOR signalling pathway is mediated by NRAS but other signalling pathways are likely still activated.
Acknowledgments
The author are grateful to Dr Bihong Zhao for pathology slides and Dr A J Munoz for uploading visual data.
Footnotes
Correction notice: This article has been corrected since published online, the videos has been updated to remove the patient's details.
Contributors: TK prepared manuscript and collected all relevant data. GI was involved in clinical care, revision and submission of manuscript. Collaborating authors: VP and MDP helped with imaging studies.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Consent obtained from parent(s)/guardian(s)
References
- 1.Mou SS, McCrory MC. Inflammatory Heart Disease: Pericardial Effusion and Tamponade, Pericarditis, and Myocarditis. In: Critical heart disease in infants and children. 3rd ed. Philadelphia, PA: Elsevier, 2019: 351–64. https://www.clinicalkey.com/#!/content/book/3-s2.0-B9781455707607000280?scrollTo=%23hl0000404 [Google Scholar]
- 2.Kühn B, Peters J, Marx GR, et al. Etiology, management, and outcome of pediatric pericardial effusions. Pediatr Cardiol 2008;29:90–4. 10.1007/s00246-007-9014-1 [DOI] [PubMed] [Google Scholar]
- 3.Bagri NK, Yadav DK, Agarwal S, et al. Pericardial effusion in children: experience from tertiary care center in northern India. Indian Pediatr 2014;51:211–3. 10.1007/s13312-014-0378-z [DOI] [PubMed] [Google Scholar]
- 4.Feldman WE. Bacterial etiology and mortality of purulent pericarditis in pediatric patients. review of 162 cases. Am J Dis Child 1979;133:641–4. 10.1001/archpedi.1979.02130060081019 [DOI] [PubMed] [Google Scholar]
- 5.Flores-González JC, Rubio-Quiñones F, Hernández-González A, et al. Pneumonia and purulent pericarditis caused by Streptococcus pneumoniae: an uncommon association in the antibiotic era. Pediatr Emerg Care 2014;30:552–4. 10.1097/PEC.0000000000000186 [DOI] [PubMed] [Google Scholar]
- 6.Angoulvant F, Bellanger H, Magnier S, et al. Acute purulent pericarditis in childhood: don’t forget β-haemolytic group-A Streptococcus. Intensive Care Med 2011;37:1709–10. 10.1007/s00134-011-2259-4 [DOI] [PubMed] [Google Scholar]
- 7.Vigneswaran WT, Hardie R, Ferguson JC, et al. Cardiac tamponade due to Lancefield group A beta haemolytic streptococcal pericarditis. Thorax 1985;40:549–50. 10.1136/thx.40.7.549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Waili BR, Zacharias SK, Aslem E. Group A streptococcal pericarditis in a four-month-old infant: case report. Sultan Qaboos Univ Med J 2017;17:e241–3. 10.18295/squmj.2016.17.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pemira SM, Tolan RW. Invasive group A Streptococcus infection presenting as purulent pericarditis with multiple splenic abscesses: case report and literature review. Clin Pediatr 2012;51:436–41. 10.1177/0009922811430345 [DOI] [PubMed] [Google Scholar]
- 10.Bhaduri-McIntosh S, Prasad M, Moltedo J, et al. Purulent pericarditis caused by group A Streptococcus. Tex Heart Inst J 2006;33:519–22. [PMC free article] [PubMed] [Google Scholar]
- 11.Fry E, Urbanczyk J, Price J, et al. Streptococcus pyogenes Pericarditis with Resultant Pulmonary Trunk Compression Secondary to Mycotic Pseudoaneurysm. Case Rep Cardiol 2018;2018:e3514797:1–4 https://www.hindawi.com/journals/cric/2018/3514797/ 10.1155/2018/3514797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson GE, Pondo T, Toews K-A, et al. Epidemiology of invasive group A streptococcal infections in the United States, 2005-2012. Clin Infect Dis 2016;63:478–86. 10.1093/cid/ciw248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cole JN, Barnett TC, Nizet V, et al. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol 2011;9:724–36. 10.1038/nrmicro2648 [DOI] [PubMed] [Google Scholar]
- 14.Soderholm AT, Barnett TC, Sweet MJ, et al. Group A streptococcal pharyngitis: immune responses involved in bacterial clearance and gas-associated immunopathologies. J Leukoc Biol 2018;103:193–213. 10.1189/jlb.4MR0617-227RR [DOI] [PubMed] [Google Scholar]
- 15.Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 2000;13:470–511. 10.1128/CMR.13.3.470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams DM, Brandão LR, Peterman CM, et al. Vascular anomaly cases for the pediatric hematologist oncologists-An interdisciplinary review. Pediatr Blood Cancer 2018;65:e26716. 10.1002/pbc.26716 [DOI] [PubMed] [Google Scholar]
- 17.Iacobas I, Adams DM, Pimpalwar S, et al. Multidisciplinary guidelines for initial evaluation of complicated lymphatic anomalies—expert opinion consensus. Pediatr Blood Cancer 2020;67:e28036. 10.1002/pbc.28036 [DOI] [PubMed] [Google Scholar]
- 18.Adams DM, Ricci KW. Vascular anomalies. Hematol Oncol Clin North Am 2019;33:455–70. 10.1016/j.hoc.2019.01.011 [DOI] [PubMed] [Google Scholar]
- 19.Goyal P, Alomari AI, Kozakewich HP, et al. Imaging features of kaposiform lymphangiomatosis. Pediatr Radiol 2016;46:1282–90. 10.1007/s00247-016-3611-1 [DOI] [PubMed] [Google Scholar]
- 20.Kato H, Ozeki M, Fukao T, et al. Mr imaging findings of vertebral involvement in Gorham–Stout disease, generalized lymphatic anomaly, and kaposiform lymphangiomatosis. Jpn J Radiol 2017;35:606–12. 10.1007/s11604-017-0674-3 [DOI] [PubMed] [Google Scholar]
- 21.Lala S, Mulliken JB, Alomari AI, et al. Gorham-Stout disease and generalized lymphatic anomaly—clinical, radiologic, and histologic differentiation. Skeletal Radiol 2013;42:917–24. 10.1007/s00256-012-1565-4 [DOI] [PubMed] [Google Scholar]
- 22.Ozeki M, Fujino A, Matsuoka K, et al. Clinical features and prognosis of generalized lymphatic anomaly, Kaposiform lymphangiomatosis, and Gorham-Stout disease. Pediatr Blood Cancer 2016;63:832–8. 10.1002/pbc.25914 [DOI] [PubMed] [Google Scholar]
- 23.Crane J, Manfredo J, Boscolo E, et al. Kaposiform lymphangiomatosis treated with multimodal therapy improves coagulopathy and reduces blood angiopoietin‐2 levels. Pediatr Blood Cancer 2020;67:e28529. 10.1002/pbc.28529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Croteau SE, Kozakewich HPW, Perez-Atayde AR, et al. Kaposiform lymphangiomatosis: a distinct aggressive lymphatic anomaly. J Pediatr 2014;164:383–8. 10.1016/j.jpeds.2013.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji Y, Chen S, Peng S, et al. Kaposiform lymphangiomatosis and kaposiform hemangioendothelioma: similarities and differences. Orphanet J Rare Dis 2019;14:165. 10.1186/s13023-019-1147-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji Y, Chen S, Yang K, et al. Kaposiform hemangioendothelioma: current knowledge and future perspectives. Orphanet J Rare Dis. 2020;15. 10.1186/s13023-020-1320-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eng W, Al-Sayegh H, Ma C, et al. Kaposiform lymphangiomatosis: update on outcomes and use of sirolimus as a therapeutic intervention. Blood 2018;132:3734. 10.1182/blood-2018-99-110989 [DOI] [Google Scholar]
- 28.Adams DM, Trenor CC, Hammill AM, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics 2016;137:e20153257. 10.1542/peds.2015-3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barclay SF, Inman KW, Luks VL, et al. A somatic activating NRAS variant associated with kaposiform lymphangiomatosis. Genet Med 2019;21:1517–24. 10.1038/s41436-018-0390-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozeki M, Aoki Y, Nozawa A, et al. Detection of NRAS mutation in cell-free DNA biological fluids from patients with kaposiform lymphangiomatosis. Orphanet J Rare Dis 2019;14:215. 10.1186/s13023-019-1191-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aoki Y, Niihori T, Inoue S-ichi, et al. Recent advances in RASopathies. J Hum Genet 2016;61:33–9. 10.1038/jhg.2015.114 [DOI] [PubMed] [Google Scholar]