


Keywords: synaptotagmin, somatodendritic, dopamine, exocytosis, neurotransmission
Abstract
Dopamine (DA) neurons can release DA not just from axon terminals, but also from their somatodendritic (STD) compartment through a mechanism that is still incompletely understood. Using voltammetry in mouse mesencephalic brain slices, we find that STD DA release has low capacity and shows a calcium sensitivity that is comparable to that of axonal release. We find that the molecular mechanism of STD DA release differs from axonal release with regard to the implication of synaptotagmin (Syt) calcium sensors. While individual constitutive knockout of Syt4 or Syt7 is not sufficient to reduce STD DA release, the removal of both isoforms reduces this release by approximately 50%, leaving axonal release unimpaired. Our work unveils clear differences in the mechanisms of STD and axonal DA release.
1. Introduction
Dopamine (DA) neurons of the mesencephalon play a key role in motor control, motivated behaviours and cognition [1,2]. DA neurons can release DA not only from axon terminals by a classical exocytotic mechanism [3,4], but also through their somatodendritic (STD) compartment, as demonstrated by multiple approaches including in vivo microdialysis, fast scan cyclic voltammetry (FSCV) and patch-clamp recordings of D2 receptor mediated currents in the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc) [5–9]. Although there is limited direct evidence, STD DA release is believed to be implicated in regulating the excitability of DA neurons through activation of STD D2 autoreceptors [5]. It has also been suggested to regulate motor behaviours [10,11], mainly by local activation of D1 receptors.
The molecular mechanism of STD DA release is still unclear. Reversal of the DA transporter (DAT) has been proposed [12], but other studies support the involvement of a vesicular exocytotic-like mechanism, in agreement with the fact that STD DA release is activity-dependent [5,13,14], reserpine-sensitive [5,15,16], calcium-dependent [5,13,15,17–19], dependent on active zone proteins [20,21], quantal in nature [22,23] and blocked by botulinum neurotoxins, which disrupt SNARE proteins [13,24,25]. Although large pools of DA-containing small clear synaptic vesicles are not found in the dendrites of DA neurons, this cellular compartment contains pleiomorphic vesicles that bear the vesicular monoamine transporter (VMAT2), suggesting that they could be sites of DA storage in dendrites [26]. Together, these findings suggest that, although there may be some fundamental differences between the mechanisms of terminal and STD DA release, both implicate a form of exocytosis.
Although STD DA release is calcium-dependent, conflicting results exist regarding the calcium sensitivity of STD DA release in comparison with axonal release. Previous studies performed in guinea pig reported that STD DA release persists at extracellular calcium concentrations as low as 0.5 mM, a concentration at which axonal release is typically abrogated [13,17]. By contrast, previous work performed with mouse tissue and indirectly detecting STD DA release using the patch-clamp technique and STD D2 receptor activation, reported that axonal and STD DA release display a similar calcium dependency [5,19,23,27–29]. Here, we reexamined this question in mouse brain slices after optimizing direct detection of STD DA using FSCV.
Finally, an important outstanding question is the identification of the molecular mechanism of STD DA release. Building on previous in vitro work suggesting possible roles of synaptotagmin (Syt) 4 (Syt4) and Syt7 [3], in the present study we tested the hypothesis that Syt4 and Syt7 play a key role in STD DA release in the intact brain by quantifying STD DA release in Syt4, Syt7 and Syt4/7 double constitutive knockout (KO) mice.
2. Material and methods
2.1. Animals
Male and female mice of 11–12 weeks were used in this study. For optogenetic experiments, B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J (Ai32, The Jackson Laboratory, stock 012569, USA) homozygote mice expressing a floxed H134R variant of the light-activated channelrhodopsin-2 were bred with homozygote B6.SJL-Slc6a3tm1.1(cre)Bkmn/J (DATIREScre, The Jackson Laboratory, stock 006660, USA) expressing the cre recombinase under control of the DAT promoter, allowing channelrhodospin-2 to be expressed selectively in DA neurons. Heterozygote DATIREScre mice were also used for experiments in which ChR2 was virally expressed. Constitutive KO mice for Syt4 (129S6.129X1(B6)-Syt4tm1Hahe/J, The Jackson Laboratory, stock #012400, USA) [30], Syt7 [31] and WT littermates were bred from heterozygous crosses or crossed with each other to obtain double KO mice. Genotyping for Syt4 KO mice was determined using specific primers to target the wild-type Syt4 sequence (primers Syt4WT-fwd and Syt4WT-rev) and the neomycin cassette within the mutated allele (primers neo-fwd and Syt4WT-rev)—Syt4WT-fwd: CACTTCCCTCACGTCAGAGGAG; Syt4WT-rev: GCAAGGAGAGCTCTTGGATGTG; neo-fwd: AACCACACTGCTCGACATTGGG. Genotyping for Syt7 KO mice was performed using specific primers to target the wild-type Syt7 sequence (Syt7WT-fwd: CATCCTCCACTGGCCATGAATG; Syt7WT-rev: GCTTCACCTTGGTCTCCAG) and the neomycin cassette within the mutated allele (neo-fwd: CTTGGGTGGAGAGGCTATTC; neo-rev: AGGTGAGATGACAGGAGATC), as provided by Jackson. Genotyping for Syt7 mutation in combined Syt4/7 KO mice was determined using another set of specific primers due to overlapping sequences within the neomycin cassette used in both the Syt4 and Syt7 mouse lines: neo-fwd: CTTGGGTGGAGAGGCTATTC and Syt7WTexon4: AGTGTCCAGGCTCCC. Experiments were performed blind with regard to animal genotype, with the exception of Syt4 KO mice, because these KO mice could be easily identified due to a neurodevelopmental alteration of the anterior commissure and corpus callosum (electronic supplementary material, figure S1C). Housing was at a constant temperature (21°C) and humidity (60%), under a fixed 12 h light/dark cycle, with food and water available ad libitum.
2.2. Stereotaxic injections
Six- to seven-week-old DATIREScre mice were anaesthetized with isoflurane (Aerrane; Baxter, Deerfield, IL, USA) and fixed on a stereotaxic frame (Stoelting,Wood Dale, IL, USA). A small hole was drilled in the exposed skull and a Hamilton syringe was used for the injections. For optogenetic experiments, an adeno-associated virus (AAV5-EF1a-DIO-hChR2(H134R)-EYFP, 4,2 × 1012 vg ml−1, UNC GTC Vector Core, USA) was injected bilaterally at the following injection coordinates [AP (anterior–posterior; ML (medial–lateral); DV (dorsal-ventral), from bregma], to infect neurons in the entire ventral mesencephalon: AP −3.0 mm; ML ± 1.0 mm; DV −4.5 mm. Animals recovered in their home cage and were closely monitored for 3 days. The animals were used one month after injection, allowing maximal expression of ChR2 in DA neurons. Success of the injection was visually validated each time during the slicing of the brains by visualizing the presence of the eYFP reporter.
2.3. Brain slice preparation and solutions
Acute brain slices from 11- to 12-week-old male or female mice were used for the FSCV recordings. When possible, matched pairs of WT and KO mice were used on each experimental day. The animals were anaesthetized with halothane, quickly decapitated and the brain harvested. Next, the brain was submersed in ice-cold oxygenated artificial cerebrospinal fluid (aCSF) containing (in mM): NaCl (125), KCl (2.5), KH2PO4 (0.3), NaHCO3 (26), glucose (10), CaCl2 (2.4), MgSO4 (1.3) and coronal VTA and/or striatal brain slices of 300 µm thickness were prepared with a VT1000S vibrating blade microtome. Once sliced, the tissue was transferred to oxygenated aCSF at room temperature and allowed to recover for at least 1 h. For recordings, slices were placed in a custom-made recording chamber superfused with aCSF at 1 ml min−1 and maintained at 32°C with a TC-324B single-channel heater controller (Warner Instruments, USA). All solutions were adjusted at pH 7.35–7.4, 300 mOsm kg−1 and saturated with 95% O2-5% CO2 at least 30 min prior to each experiment.
2.4. Fast scan cyclic voltammetry recordings
Optically or electrically evoked DA release was measured by FSCV using a 7 µm diameter carbon-fibre electrode placed into the tissue approximately 100 µm below the surface. A bipolar electrode (Plastics One, Roanoke, VA, USA) or an optical fibre connected to a 470 nm wavelength LED was placed approximately 200 µm away. Carbon-fibre electrodes were fabricated as previously described [32]. Briefly, carbon fibres (Goodfellow Cambridge Limited, UK) of 7 µm in diameter were aspirated into ethanol-cleaned glass capillaries (1.2 mm O.D., 0.68 mm I.D., 4 inches long; World Precision Instruments, FL, USA). The glass capillaries were then pulled using a P-2000 micropipette puller (Sutter Instruments, Novato, USA), dipped into 90°C epoxy for 30 s (Epo-Tek 301, Epoxy Technology, MASS, USA) and cleaned in hot acetone for 3 s. The electrodes were heated at 100°C for 12 h and 150°C for 5 days. Electrodes were polished and filed with potassium acetate at 4 M and potassium chloride at 150 mM. The protruding carbon fibres were cut using a scalpel blade under direct visualization to a length allowing to obtain maximal basal currents of 100 to 180 nA.
The electrodes were calibrated with 1 µM DA in aCSF before and after each recorded slice and the mean of the current values obtained were used to determine the amount of released DA. After use, electrodes were cleaned with isopropyl alcohol (Bioshop, Canada). The potential of the carbon-fibre electrode was scanned at a rate of 300 V s−1 according to a 10 ms triangular voltage wave (−400 to 1000 mV versus Ag/AgCl) with a 100 ms sampling interval, using a CV 203BU headstage preamplifier (Molecular Devices) and a Axopatch 200B amplifier (Molecular Devices, USA). Data were acquired using a Digidata 1440a analogue to digital converter board (Molecular Devices, USA) connected to a computer using Clampex (Molecular Devices, USA). Slices were left to stabilize for 20 min before any electrochemical recordings. After positioning of the bipolar stimulation electrode or the optical probe and carbon-fibre electrodes in the tissue, single pulses (400 µA or 30 mW, 1 ms) or pulse trains (30 pulses at 10 Hz) were applied to the tissue to trigger DA release. For evaluating the calcium dependency of axonal and STD release, variations of calcium concentrations in the aCSF (0, 0.5 and 2.4 mM) were compensated by changing the concentration of MgSO4 to keep divalent cation levels equivalent.
2.5. Immunohistochemistry
For Syt immunolabelling experiments, 40 µm brain slices from animals perfused with 4% paraformaldehyde (in PBS, pH-7.4) were cut with a cryostat (Leica CM 1800; Leica Canada) and used for immunohistochemistry (IHC). Because selective and specific Syt4, Syt7 and VMAT2 antibodies were all from the same host species (rabbit), a double labelling protocol adapted from Jackson ImmunoReseach (https://www.jacksonimmuno.com/technical/products/protocols/double-labeling-same-species-primary) with monovalent Fab fragments was used. After a PBS wash, the tissue was permeabilized, non-specific binding sites blocked (goat serum 5%) and incubated overnight at room temperature with the first primary antibody, rabbit anti-Syt1 (1/1000; #105002, Synaptic Systems, Germany), rabbit anti-Syt7 (1/1000; #105173, Synaptic Systems, Germany) or anti-Syt4 (1/1000; kind gift for Dr Mitsunori Fukuda, Japan), followed by 2 h with a first secondary antibody (rabbit Alexa Fluor-488–conjugated, 1 : 500, Invitrogen, Canada). A blocking step of antigenic sites from the first primary and secondary antibody combination was performed thereafter by a 3 h incubation with normal serum from the same species as the primary antibody, followed by a blocking solution (goat block: PBS, Triton X100 0.3%, bovine serum albumin 5%) with 50 µg ml−1 of unconjugated monovalent Fab fragments against the host of the primary antibody, overnight, at room temperature and under agitation. Slices were then washed, and a second labelling was performed with a second primary antibody (rabbit anti-VMAT2, 1 : 1000, gift of Dr Gary Miller, Columbia University), and a second secondary antibody (rabbit Alexa Fluor-546–conjugated, 1 : 500, Invitrogen). This VMAT2 antibody has been well-validated previously [33]. For each IHC staining, a control group was included, with the full protocol except for omission of the second primary antibody. A classical immunostaining protocol was used for the KO validation of Syt4 and Syt7 antibodies (electronic supplementary material, figure S1), using mouse anti-tyrosine hydroxylase (Millipore Sigma; 1 : 1000) and rabbit anti-Syt4 or anti-Syt7 primary antibodies (Synaptic Systems; 1 : 1000) subsequently detected using Alexa Fluor-488-conjugated and Alexa Fluor-546-conjugated secondary antibodies (Invitrogen; 1 : 500).
2.6. Confocal imaging
Images were acquired using an Olympus Fluoview FV1000 point-scanning confocal microscope (Olympus, Canada) with a 60 x oil-immersion objective (NA 1.35). Images acquired using 488 nm and 546 nm laser excitation were scanned sequentially to prevent non-specific bleed-through signal. All image analysis was performed using ImageJ (National Institutes of Health) software.
2.7. Reverse transcriptase-quantitative PCR
We used RT-qPCR to quantify the amount of mRNA encoding Syt1, 4, 5, 7 and 11 in brain tissue from P70 Syt4+/+ and Syt4−/− mice and P70 Syt7+/+ and Syt7−/− mice. Adult whole brains were harvested and homogenized in Trizol solution, then RNA extraction was performed using RNAeasy Mini Kit (Quiagen, Canada) according to the manufacturer's instructions. Total purified RNA (40 ng) was reverse-transcribed in a total of 20 µl including 1 µl of dNTP, 1 µl of random hexamer, 4 µl of 5 X buffer 5 X, 2 µl of dithiothreitol, 1 µl of RNAse-Out and 1 µl of the Moloney Murine Leukemia Virus reverse transcriptase enzyme (MML-V, Invitrogen). Quantitative PCR was carried out in a total of 15 µl consisting of 3 µl cDNA, 7.5 µl SYBER green PCR master mix (Quanta Biosciences, USA), 10 µM of each primer and completed up to 15 µl with RNA-free water. qPCR was performed on a Light Cycler 96 machine (Roche, Canada) using the following procedure: 10 min at 95°C; 40 cycles of 30 s at 95°C, 40 s at 57°C and 40 s at 72°C; 1 cycle of 15 s at 95°C, 15 s at 55°C and 15 s at 95°C. Results were analysed with Light Cycler 96 software and Excel. Primers used for qPCR were as follows: Syt1: 5′ GTGGCAAGACACTGGTGAT 3′ and 5′ CTCAGGACTCTGGAGATCG 3′; Syt4: 5′ CACTTCCCTCACGTCAGAGGAG 3′ and 5′ GCAAGGAGAGCTCTTGGATGTG 3′; Syt5: 5′ GTCCCATACGTGCAACTAGG 3′ and 5′ AACGGAGAGAGAAGCAGATG 3′; Syt7: 5′ CCAGACGCCACACGA 3′ and 5′ CCTTCCAGAAGGTCT 3′; Syt11: 5′ CTTGTATGGCGGGGTCTTGT 3′ and 5′ ATACGCCCCAGCTTTGATGA 3′ and GAPDH: 5′ GGAGGAAACCTGCCAAGTATGA 3' and 5' TGAAGTCGCAGGAGACAACC 3'.
2.8. Statistics
Data are presented as mean ± s.e.m. The level of statistical significance was established at p < 0.05 in one-way ANOVAs with appropriate post hoc tests and two-tailed t-tests, performed with Prism 8 software (GraphPad, ***p < 0.001, **p < 0.01, *p < 0.05, #p < 0.0001).
3. Results
3.1. D2 autoreceptors and the dopamine transporter limit the extent of somatodendritic dopamine overflow in mouse ventral tegmental area slices
Although detectable in other rodent models such as the guinea pig [6,7,9,28], STD DA release in mouse mesencephalic slices has previously been found to be challenging to reliably detect [19,28,29]. As mouse models are convenient for genetic manipulations, the difficulty to use this model has greatly slowed progress in better understanding the mechanisms and roles of this form of DA release. We therefore first aimed to optimize detection of STD DA release in mouse VTA slices by comparing different modes of stimulation and physiological parameters that may limit its extent.
We first compared optogenetic and electrical stimulation of DA neurons and examined whether blocking DA D2 autoreceptors and the DAT might increase extracellular DA levels and make STD DA easier to detect (figure 1). Previous studies performed in brain slices or in vivo typically triggered STD DA release using extracellular electrical stimulation [34–36]. In recent years, optical stimulation using channelrhodopsin-2 (ChR2) or other opsin variants has increasingly been used to obtain more selective activation of DA neuron axons [37]. However, to this date, this approach has not been used to selectively trigger STD DA release in FSCV experiments.
Figure 1.

Optogenetic and electrical stimulation trigger comparable levels of STD DA release in mouse VTA slices. (a) Animal models used for optogenetic experiments. We either used a mouse line expressing a floxed version of light-activated channelrhodopsin (ChR2) crossed with a DA-specific Cre driver line (DATIREScre) or performed stereotaxic injections of AAV5-EF1a-DIO-hChR2(H134R)-eYFP virus in the VTA of DATIREScre mice to selectively express ChR2 in DA neurons. (b) FSCV was used to monitor DA levels. A voltage ramp of −400 to 1000 mV versus Ag/AgCl at 300 V/s was used, with a 100 ms sampling interval. Recordings were made in coronal slices containing the VTA and DA release was triggered by either optical stimulation with a 470 nm blue light LED or with a bipolar stimulating electrode. (c) Representative traces (top) and voltammograms (bottom) of responses obtained in the VTA with 1 pulse (1 ms) of blue light (single pulse), in the presence of normal ACSF or a pulse train of stimulation (30 pulses of 1 ms at 10 Hz), in the presence of normal ACSF. (d) The same as (c), in the presence of ACSF + a DAT blocker (nomifensine, 5 µM) and an antagonist of D2 autoreceptors (sulpiride, 5 µM). (e) Representative traces (top) and voltammograms (bottom) of responses obtained in the VTA with 1 electrical pulse (1 ms, 400 µA) or a pulse train (30 electrical pulses of 1 ms at 10 Hz, 400 µA), in the presence of aCSF. (f) The same as (e), but in presence of aCSF + 5 µM nomifensine/sulpiride. (g–i) Effect of nomifensine/sulpiride on STD DA release measured by pulse-train electrical stimulation.
We compared single-pulse optical (1 ms, 470 nm) and electrical (1 ms, 400 µA) stimulation. Recordings were performed in the VTA of DATIREScre/Ai32 mice, in which ChR2 is conditionally expressed in all DA neurons, and in DATIREScre heterozygote mice injected in the VTA with a floxed hChR2-EYFP AAV construct. Neither stimulation conditions, either in normal aCSF (figure 1c,e) or in the presence of DAT (nomifensine, 5 µM) and D2 receptor blockade (sulpiride, 5 µM) (figure 1d,f) yielded detectable evoked elevations of extracellular DA (figure 1c,d). Previous works in guinea pig demonstrated that STD DA release is frequency dependent up to 10 Hz [9]. The use of pulse trains (30 pulses at 10 Hz) in the presence of nomifensine and sulpiride allowed reliable detection of STD DA release in VTA slices, both for electrical stimulation (average peak DA levels of 340 nM ± 28 nM in DATIREScre/Ai32 mice [n = 15] and 333 nM ± 32 nM in DATIREScre mice infected with ChR2 AAV [n = 10]) and for optical stimulation (average peak DA levels at 187 nM ± 17 nM in DATIREScre/Ai32 mice [n = 15] and 220 nM ± 37 nM in DATIREScre mice infected with ChR2 AAV [n = 10]) (figure 1d,f). Although peak levels of activity-dependent STD DA release in the two strains of mice tended to be higher with electrical compared to optical stimulation (compare bar graphs in figure 1d,f), the difference between the two modes of stimulation was significant only in the Ai32 mouse strain.
Reuptake through the DAT and the D2 autoreceptor are two well-known regulators of extracellular DA levels and DA release [38–40]. We next examined the effect of DAT and D2 receptor blockade individually to determine whether a combined block of reuptake and autoreceptor function was required to reliably detect STD DA release in response to electrical train stimulation (figure 1h,i). Each recording was performed after 15 min of nomifensine or sulpiride, or a combination of the two. Baseline levels of evoked DA in the absence of antagonist were small, but still reliably detected (49 nM ± 6 nM). Blockade of DAT or D2 receptors individually, caused a significant increase in the maximal amplitude of evoked STD DA release in the VTA (+296% ± 42% for nomifensine alone, n = 6; +210% ± 28% for sulpiride alone, n = 6), while a combination of the two drugs caused a cumulative increase of 500% ± 51% (n = 12), thus demonstrating that the two manipulations were mostly additive and that a combined blockade of both membrane proteins allowed to maximally increase the detected signal.
Because previous work evaluating STD DA release using FSCV in guinea pig brain slices reported the existence of notable stimulation-dependent attenuation [9], we also examined the stability of STD DA release in response to a series of seven consecutive stimuli with an interstimulus interval of 5 min. For these experiments, optical or electrical train stimulations were used in the presence of nomifensine and sulpiride. We found that while STD DA overflow evoked by optical stimulation showed a robust and progressive decrease in peak amplitude in response to repeated stimuli (electronic supplementary material, figure S2A), electrical stimulation failed to cause a similarly extensive rundown of STD DA, with only a modest, non-significant decrease of less than 20% detected by the last of seven stimuli (electronic supplementary material, figure S2B). Additional experiments would be required to identify the causes of this differential stimulation-dependent attenuation for the two modes of stimulation. All further experiments were performed with electrical stimulation.
3.2. Dopamine release in the ventral tegmental area and striatum exhibit similar calcium dependency
As we aimed to examine the role of Syt calcium sensors in STD DA release and in the face of conflicting previous results regarding the extent to which STD DA release depends upon extracellular calcium levels in comparison with axonal release [19,41], we next evaluated the release of DA in the VTA at 0, 0.5 and 2.4 mM of extracellular calcium (figure 2a). In comparison, we also examined axonal DA release in the dorsal striatum, a region that has been most closely examined in this respect. As expected, based on previous results [17,19], no release was detected in the striatum at 0 mM and 0.5 mM calcium, neither in response to single pulses or to trains (n = 13 slices/7 mice) (figure 2b).
Figure 2.

STD and axonal DA release exhibit a similar calcium dependency. (a) Protocol used for FSCV recordings. (b) Schematic representation of a striatal slice and average of [DA]o peaks obtained with single or pulse-train stimulations at 0, 0.5 and 2.4 mM of extracellular calcium in the aCSF, with or without the addition of 5 µM of nomifensine/sulpiride. (c) Schematic representation of a VTA slice and average [DA]o peaks obtained with pulse-train stimulations at 0, 0.5 and 2.4 mM of extracellular calcium in the aCSF containing 5 µM of nomifensine/sulpiride. (d) Average [DA]o peaks normalized to 2.4 mM of calcium obtained in the VTA and dorsal striatum (dStr) with pulse-train stimulation and aCSF containing nomifensine/sulpiride. Representative traces and voltammograms are shown on the right. Error bars represent ± s.e.m. The statistical analysis was carried out by a one-way ANOVA followed by a Dunnett test (ns, non-significant; ***p < 0.001; #p < 0.0001).
In the VTA, STD DA release, here again triggered in the presence of nomifensine/sulpiride (5 µM), was also undetectable at 0 mM extracellular calcium (n = 13 slices/7 animals), but readily detectable at 0.5 mM (n = 16 slices/10 mice) (figure 2c), as previously described in the guinea pig [19,41]. Evoked STD DA release at this concentration of calcium was, however, only 19% of the signal detected at 2.4 mM calcium (76 nM ± 10 nM, compared to 388 nM ± 39 nM) (figure 2c,d), which is compatible with previous results [19]. Recordings performed in striatal slices in the presence of nomifensine and sulpiride similarly revealed detectable DA release at 0.5 mM calcium (figure 2b) (1.1 µM ± 0.17 µM, n = 13 slices/7 mice). This represents 16% of the DA signal detected at 2.4 mM calcium (7 µM ± 0.87 µM) (figure 2b,d). Therefore, under the same experimental conditions, with no influence of DA uptake and D2 autoreceptor activation, evoked STD and axonal DA release show a similar calcium dependency (t-test, p = 0.2744). All further experiments were performed at 2.4 mM calcium.
3.3. Dopamine neurons in vivo express the calcium-sensors synaptotagmin 1, 4 and 7
Because Syt1 is the main calcium sensor of axonal DA release [3,42,43], and Syt4 and Syt7 were previously suggested to be critical for STD DA release based on in vitro experiments [3], we next evaluated the presence and subcellular localization of these Syt isoforms in DA neurons in vivo in the mouse brain.
IHC was used to test the hypothesis that Syt4 and Syt7 are present within the cell body and dendrites of DA neurons in close association with compartments containing VMAT2. Due to the impossibility to obtain suitable VMAT2 and Syt antibodies produced in different species, we took advantage of a double labelling protocol allowing the use of two primary antibodies from the same species (rabbit) (figure 3a). The approach was validated by the observation that in control experiments in which the second primary antibody was omitted, no signal was detected for the second antigen, demonstrating that the second secondary antibody was unable to bind to the first primary antibody after the blocking step. Immunoreactivity for Syt4 showed a clear somatic localization in DA neurons, with a notable overlap with VMAT2 (figure 3b), with little if any signal in terminals in the striatum. Confirming the specificity of the antibody, signal was absent from Syt4 KO DA neurons (electronic supplementary material, figure S1A). Syt7 immunoreactivity was found in both the STD region of DA neurons as well as in their terminal region in the striatum (figure 3c). Syt7 immunoreactivity was strongly reduced in Syt7 KO tissue, although some background signal was still detectable (electronic supplementary material, figure S1B), suggesting sub-optimal specificity. Finally, Syt1 was not readily detectable in the soma and dendrites of DA neurons (although present in local VMAT2-negative axonal-like varicosities), but highly expressed in the terminals in the striatum, as expected (figure 3d).
Figure 3.

DA neurons express the calcium-sensors Syt1, Syt4 and Syt7. (a) Protocol used for double immunostaining for two primary antibodies from the same host (adapted from a Jackson ImmunoResearch protocol). Use of normal rabbit serum and unconjugated Fab fragments for blocking after the first secondary. (b–d) IHC of midbrain and striatal slices of adult DATIREScre heterozygote mice showing colocalization of VMAT2 and either Syt1, Syt4 or Syt7 in DAergic neurons. Scale bar = 20 μm. (e) Control images were obtained using the full protocol without the use of the second primary antibody (in the midbrain).
3.4. Double knockout of Syt4 and Syt7 strongly reduces somatodendritic dopamine release
Considering the expression of Syt4 and Syt7 in DA neurons and their apparent localization in the STD compartment of these neurons, we hypothesized that evoked STD DA release should be reduced in constitutive Syt4 or Syt7 KO mice. We took advantage of existing mouse models in which the Syt4 and Syt7 genes were interrupted by the insertion of a neomycin cassette in the coding sequences of the calcium-binding domains [30,31]. These experiments were performed using electrical train stimulation, in the presence of nomifensine and sulpiride. To obtain a thorough understanding of the individual roles of Syt4 and Syt7, wild-type, heterozygous and KO littermates were compared, and recordings were performed for each mouse in the dorsal striatum, the ventral striatum (nucleus accumbens core and shell) and the VTA. These experiments revealed that axonal DA release in the striatum and STD DA release in the VTA were not significantly reduced in Syt4 or Syt7 KO mice (figure 4a,b).
Figure 4.
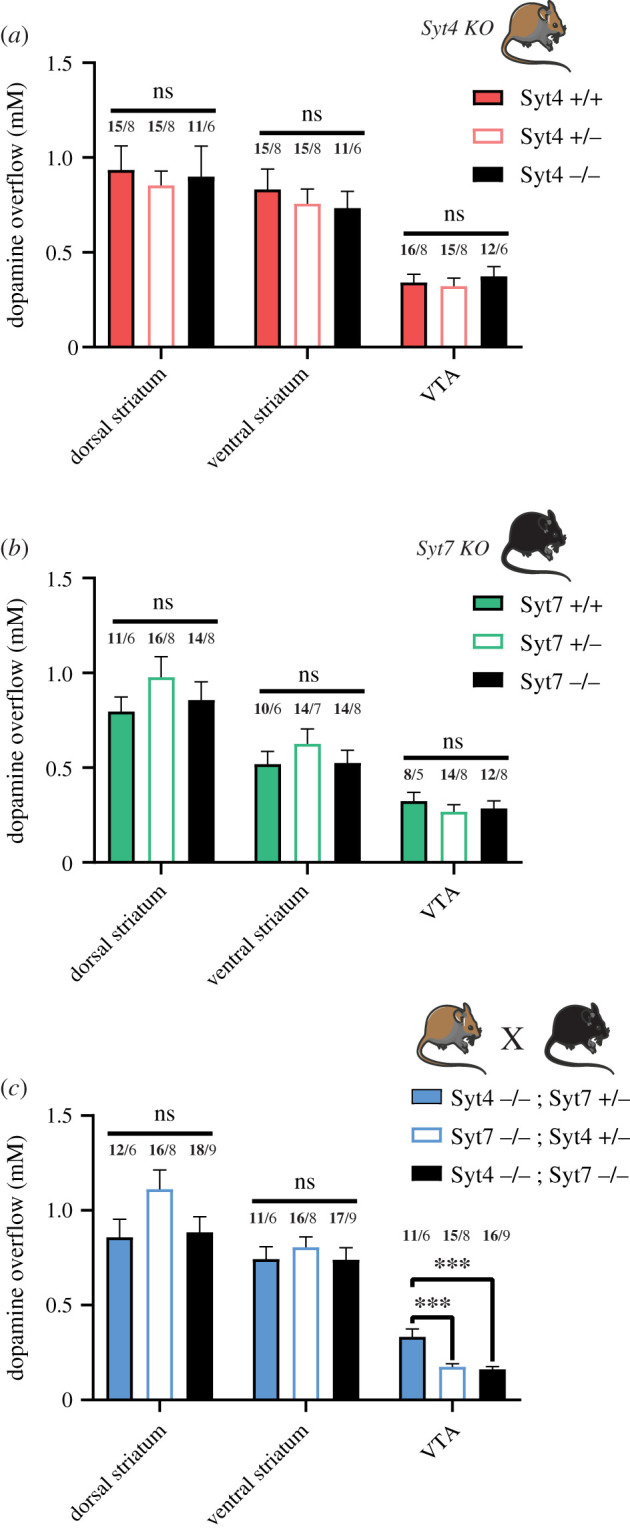
Double KO of Syt4 and Syt7 strongly reduces STD DA release. (a) Average [DA]o peaks (µM) obtained in the dorsal striatum, ventral striatum and VTA of Syt4 constitutive KO mice bred from heterozygous crosses. (b) Same for the constitutive Syt7 KO mice. (c) Same for double Syt4/Syt7 KO mice (Syt4−/−; Syt7−/−) and heterozygotes control animals (Syt4−/−; Syt7+/− and Syt7−/−; Syt4+/−). Each recording from the VTA was obtained in aCSF+5 μM nomifensine/sulpiride with two recording sites per slice, two slices per animal and pulse-train stimulation (30 pulses, 10 Hz, 400 µA). Each recording from the striatum was obtained in normal aCSF from six recording sites per slice, two slices per animal, for the dorsal striatum and four for the ventral striatum (nucleus accumbens core and shell), using single-pulse stimulation (1 ms, 400 µA). Error bars represent ± s.e.m. The statistical analysis was carried out by one-way ANOVA followed by a Tukey test (ns, non-significant; ***p < 0.001; #p < 0.0001). Numbers above bars represent the number of slices recorded (in bold) followed by number of animals used.
Because both isoforms are expressed in the STD domain of DA neurons, we aimed to determine if compensatory upregulation of the Syt isoforms occurred in individual KO mouse lines during development. Using qPCR and mRNA obtained from whole brain, we found that total levels of Syt4 mRNA were unchanged in Syt7 KO mice (electronic supplementary material, figure S1D). Similarly, the levels of Syt7 mRNA were unchanged in Syt4 KO mice (electronic supplementary material, figure S1E). To examine if functional compensation can explain the lack of change in STD DA release in the single KO mice, we next crossed these two mouse lines to generate a double Syt4 and Syt7 KO. As controls, Syt4−/−; Syt7+/− and Syt7−/−; Syt4+/− animals were also used for the FSCV recordings. Once again, no significant differences were found in the dorsal and ventral striatum (figure 4c). The evoked increase in extracellular DA concentration in the VTA of Syt4−/−; Syt7+/− (0.334 µM ± 0.04 µM; n = 6 mice) was similar to controls from the Syt4 and Syt7 individual KO mouse lines (respectively 0.341 µM ± 0.044 µM; n = 8 mice and 0.32 µM ± 0.046 µM; n = 5 mice). However, in Syt4−/−; Syt7−/− animals, we found a robust and significant approximately equal to 50% decrease of STD DA release (0.162 µM ± 0.014 µM; n = 9 mice). Interestingly, the Syt7−/−; Syt4+/− animals also showed a systematic decrease of STD DA release of about approximately 50% (0.175 µM ± 0.016 µM; n = 8 mice). Together these results argue that both Syt4 and Syt7 isoforms contribute to STD DA release, with functional compensation of one isoform by the other. These data also suggest a more critical role of Syt7 compared to Syt4 because the presence of only one Syt7 allele is sufficient to support STD DA release in the absence of Syt4.
4. Discussion
4.1. Characteristics of somatodendritic dopamine release
In the present study, we performed the first characterization of optically evoked STD DA release in the mouse mesencephalon using a combination of optogenetics and FSCV and compared its characteristics to release evoked by electrical stimulation. As previously reported by others, we found that the absolute levels of evoked DA overflow detected in this region were low compared to levels detected in the terminal region in the striatum. Furthermore, we found that a robust STD DA release signal could only be detected using pulse-train stimulation (figure 1). Blocking DA reuptake and D2 autoreceptor function using nomifensine and sulpiride caused a fivefold increase in peak signal amplitude, thus making detection of this signal straightforward and reproducible.
Using a repeated stimulation protocol, we found that repeated optical stimulation with a 5 min interval produces a strong rundown of STD DA release, whereas no such attenuation was seen with electrical stimulation (electronic supplementary material, figure S2). This finding is compatible with previous results reporting a similar rundown of optically evoked axonal DA release in the striatum [37]. Although a partial desensitization of ChR2 upon repeated stimulations could occur [44,45], a more likely explanation of the different rate of decrement obtained with optical and electrical stimulation is that in response to electrical stimulation, a decrement is not observed because this form of stimulation recruits neuromodulatory mechanisms that result from activation of afferent terminals releasing 5-HT, NE, acetylcholine, glutamate, GABA or neuropeptides onto DA neurons [37,46]. Further experiments will be required to test this hypothesis.
Our experiments comparing the impact of changes in extracellular calcium levels of STD and axonal DA release argue for the existence of a similar calcium dependency for both forms of release (figure 2). These findings are compatible with previous results obtained in mice, which were performed using patch-clamp recordings and the measurement of STD D2 receptor-mediated membrane currents [19]. It is possible that a different conclusion was reached in guinea pig brain slices because some aspect of the STD DA release mechanism is different in that species [17], as previously suggested [28]. Another possibility is that a component of axonal DA release is also included in the signal detected in the VTA. This possibility has been raised previously [41], but the available anatomical data actually suggests that DA-containing axonal varicosities are scarce in the VTA [47,48], except in the context of compensatory axonal sprouting associated with partial lesions [49]. It would nonetheless be useful to revisit this question in the future with additional anatomical and physiological work to provide more quantitative data.
4.2. Contribution of Syt4 and Syt7 to somatodendritic dopamine release
Finally, we examined the contribution of the Syt isoforms Syt4 and Syt7 to STD DA release. Both Syt4 and Syt7 have been linked to vesicular fusion. A role for Syt7 in asynchronous release was previously demonstrated [50]. This isoform has also been shown to synergize with Syt1 [51]. The function of Syt4 is more controversial because it has been proposed to act either as a negative regulator of vesicular exocytosis [52–54] or to promote some forms of neurotransmitter release [3,55,56], depending on the system investigated. In posterior pituitary nerve terminals, such positive and negative roles were reported to depend on calcium concentrations, suggesting that the function of Syt4 can be modulated according to physiological conditions [57].
In the dopaminergic system, acute downregulation of either isoform has previously been shown in vitro to severely reduce STD DA release, with no similar effect of Syt1 downregulation [3]. Although our present immunostaining results provide further support for the presence of these proteins in the STD compartment of DA neurons (figure 3), we failed to detect any significant decrease in evoked STD DA release in VTA slices prepared from individual constitutive Syt4 or Syt7 KO mice (figure 4). It is possible that, contrary to acute downregulation with siRNAs, constitutive gene deletion may lead to homeostatic compensation leading to elevated levels of Syt4 in Syt7 KO mice and vice versa. It is also possible that in vitro models lack homeostatic compensatory mechanisms that are recruited in vivo. Another possibility is that Syt4 and Syt7 play similar roles in supporting STD DA release and that one can compensate for the absence of the other in the context of constitutive gene deletion. The robust decrease in activity-dependent STD DA release in Syt4/Syt7 double KO mice supports this interpretation. In Syt4−/−; Syt7−/− mice or Syt7−/−; Syt4+/− mice, we observed a twofold decrease of STD DA release. This decrease was surprisingly not found in Syt4−/−; Syt7+/− mice, strongly suggesting that Syt7 plays a particularly important role and that a single allele of Syt7 is sufficient to sustain STD DA release in the absence of Syt4. This observation of a particularly important role of Syt7 in STD DA release could be linked to differential age-dependent expression of Syt7 and Syt4 in DA neurons. Indeed, the level of expression of Syt7 was previously demonstrated to increase as a function of postnatal age in mice [3]. Conversely, the expression of Syt4 in the brain has been shown to be maximal during the first postnatal week and to slowly decline thereafter [58,59]. Here, we conducted all experiments in adult animals, at a developmental stage where Syt7 may play a more important role in regulating STD DA release compared to Syt4. Further experiments comparing STD DA release in Syt4 and Syt7 double KO mice at different ages would thus be of interest in the future.
Considering that in the absence of both Syt4 and Syt7, approximately half of total evoked STD DA release remain, we also hypothesize that other calcium sensors or other forms of release are also involved. The main Syt isoform Syt1 might be of interest, as it was recently demonstrated as the main calcium sensor for fast striatal DA release in vivo [42], validating previous work showing an important role of Syt1 in axonal DA release in vitro [3]. Interestingly, evoked STD DA release measured by detecting D2-IPSCs was recently reported to be abolished in mice with conditional deletion of the active zone protein RIM, while spontaneous release remained intact [21]. However, the subcellular localization of RIM in the STD compartment of DA neurons is currently undetermined. Here, we have not found strong evidence for localization of Syt1 in the STD domain of DA neurons, but further examination of this possibility with higher resolution techniques would be relevant.
Our results bring to light an important role of Syt7 in both the dendritic and axonal compartment of DA neurons. Together with our observation that the two forms of DA secretion exhibit the same calcium dependency, our findings raise the interest of further evaluating a possible contribution of an axonal compartment to DA release within the mesencephalon, in line with previous speculations in the guinea pig model [60]. Further studies of STD and axonal DA release in mouse models defective for combinations of Syt1, Syt4 and Syt7 would seem warranted. Together our work provides a new perspective on the mechanism of STD DA release and raises new opportunities to disentangle the respective roles of axonal and STD DA release in DA-dependent physiological mechanisms and behaviours.
Acknowledgements
We thank Dr Wade G Regehr (Harvard Medical School) for providing the Syt7 constitutive KO mice, Dr Gary Miller (Columbia University) for providing the VMAT2 antibody and Marie-Josée Bourque for managing the mouse colonies and performing mouse genotyping.
Ethics
All procedures involving animals and their care were conducted in accordance with the Guide to care and use of Experimental Animals of the Canadian Council on Animal Care. The experimental protocols were approved by the animal ethics committees of the Université de Montréal.
Data accessibility
Primary data analysis files from this manuscript are available from the following doi:10.6084/m9.figshare.19083575. Data supporting this article have also been uploaded as part of the electronic supplementary material.
Authors' contributions
B.D.-L.: formal analysis, investigation, methodology, writing—original draft and writing—review and editing; C.D.: formal analysis, investigation and writing—review and editing; W.K.: formal analysis, investigation, methodology and writing—review and editing; N.F.: formal analysis and methodology; L.-E.T.: conceptualization, data curation, funding acquisition, project administration, resources, supervision, validation, writing—original draft and writing—review and editing.
All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Competing interests
We declare we have no competing interests.
Funding
This work was funded by the National Sciences and Engineering Research Council of Canada (NSERC, grant RGPIN-2020-05279) to L.-E.T. L.-E.T. also received support from the Krembil Foundation, the Brain Canada Foundation and the Henry and Berenice Kaufmann Foundation. B.D.-L. received a graduate student award from Parkinson Canada. C.D. received a studentship from the Fonds de la Recherche du Québec en Santé (FRQS).
References
- 1.Schultz W. 2007. Multiple dopamine functions at different time courses. Annu. Rev. Neurosci. 30, 259-288. ( 10.1146/annurev.neuro.28.061604.135722) [DOI] [PubMed] [Google Scholar]
- 2.Surmeier DJ, Graves SM, Shen W. 2014. Dopaminergic modulation of striatal networks in health and Parkinson's disease. Curr. Opin. Neurobiol. 29, 109-117. ( 10.1016/j.conb.2014.07.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendez JA, Bourque MJ, Fasano C, Kortleven C, Trudeau LE. 2011. Somatodendritic dopamine release requires synaptotagmin 4 and 7 and the participation of voltage-gated calcium channels. J. Biol. Chem. 286, 23 928-23 937. ( 10.1074/jbc.M111.218032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu C, Kaeser PS. 2019. Mechanisms and regulation of dopamine release. Curr. Opin. Neurobiol. 57, 46-53. ( 10.1016/j.conb.2019.01.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beckstead MJ, Grandy DK, Wickman K, Williams JT. 2004. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 42, 939-946. ( 10.1016/j.neuron.2004.05.019) [DOI] [PubMed] [Google Scholar]
- 6.Cragg S, Rice ME, Greenfield SA. 1997. Heterogeneity of electrically evoked dopamine release and reuptake in substantia nigra, ventral tegmental area, and striatum. J. Neurophysiol. 77, 863-873. ( 10.1152/jn.1997.77.2.863) [DOI] [PubMed] [Google Scholar]
- 7.Cragg SJ, Hawkey CR, Greenfield SA. 1997. Comparison of serotonin and dopamine release in substantia nigra and ventral tegmental area: region and species differences. J. Neurochem. 69, 2378-2386. ( 10.1046/j.1471-4159.1997.69062378.x) [DOI] [PubMed] [Google Scholar]
- 8.Elverfors A, Jonason J, Jonason G, Nissbrandt H. 1997. Effects of drugs interfering with sodium channels and calcium channels on the release of endogenous dopamine from superfused substantia nigra slices. Synap. N. Y. N 26, 359-369. () [DOI] [PubMed] [Google Scholar]
- 9.Rice ME, Cragg SJ, Greenfield SA. 1997. Characteristics of electrically evoked somatodendritic dopamine release in substantia nigra and ventral tegmental area in vitro. J. Neurophysiol. 77, 853-862. ( 10.1152/jn.1997.77.2.853) [DOI] [PubMed] [Google Scholar]
- 10.Andersson DR, Nissbrandt H, Bergquist F. 2006. Partial depletion of dopamine in substantia nigra impairs motor performance without altering striatal dopamine neurotransmission. Eur. J. Neurosci. 24, 617-624. ( 10.1111/j.1460-9568.2006.04953.x) [DOI] [PubMed] [Google Scholar]
- 11.Bergquist F, Shahabi HN, Nissbrandt H. 2003. Somatodendritic dopamine release in rat substantia nigra influences motor performance on the accelerating rod. Brain Res. 973, 81-91. ( 10.1016/S0006-8993(03)02555-1) [DOI] [PubMed] [Google Scholar]
- 12.Falkenburger BH, Barstow KL, Mintz IM. 2001. Dendrodendritic inhibition through reversal of dopamine transport. Science 293, 2465-2470. ( 10.1126/science.1060645) [DOI] [PubMed] [Google Scholar]
- 13.Fortin GD, Desrosiers CC, Yamaguchi N, Trudeau LE. 2006. Basal somatodendritic dopamine release requires snare proteins. J. Neurochem. 96, 1740-1749. ( 10.1111/j.1471-4159.2006.03699.x) [DOI] [PubMed] [Google Scholar]
- 14.Robertson GS, Damsma G, Fibiger HC. 1991. Characterization of dopamine release in the substantia nigra by in vivo microdialysis in freely moving rats. J. Neurosci. 11, 2209-2216. ( 10.1523/JNEUROSCI.11-07-02209.1991) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rice ME, Richards CD, Nedergaard S, Hounsgaard J, Nicholson C, Greenfield SA. 1994. Direct monitoring of dopamine and 5-HT release in substantia nigra and ventral tegmental area in vitro. Exp. Brain Res. 100, 395-406. ( 10.1007/bf02738400) [DOI] [PubMed] [Google Scholar]
- 16.Heeringa MJ, Abercrombie ED. 1995. Biochemistry of somatodendritic dopamine release in substantia nigra: an in vivo comparison with striatal dopamine release. J. Neurochem. 65, 192-200. ( 10.1046/j.1471-4159.1995.65010192.x) [DOI] [PubMed] [Google Scholar]
- 17.Chen BT, Rice ME. 2001. Novel Ca2+ dependence and time course of somatodendritic dopamine release: substantia nigra versus striatum. J. Neurosci. 21, 7841-7847. ( 10.1523/JNEUROSCI.21-19-07841.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yee AG, Forbes B, Cheung P-Y, Martini A, Burrell MH, Freestone PS, Lipski J. 2019. Action potential and calcium dependence of tonic somatodendritic dopamine release in the substantia nigra pars compacta. J. Neurochem. 148, 462-479. ( 10.1111/jnc.14587) [DOI] [PubMed] [Google Scholar]
- 19.Ford CP, Gantz SC, Phillips PEM, Williams JT. 2010. Control of extracellular dopamine at dendrite and axon terminals. J. Neurosci. 30, 6975-6983. ( 10.1523/JNEUROSCI.1020-10.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu C, Kershberg L, Wang J, Schneeberger S, Kaeser PS. 2018. Dopamine secretion is mediated by sparse active zone-like release sites. Cell 172, 706-718.e15. ( 10.1016/j.cell.2018.01.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson BG, Cai X, Wang J, Bunzow JR, Williams JT, Kaeser PS. 2019. RIM is essential for stimulated but not spontaneous somatodendritic dopamine release in the midbrain. eLife 8, e47972. ( 10.7554/eLife.47972) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaffe EH, Marty A, Schulte A, Chow RH. 1998. Extrasynaptic vesicular transmitter release from the somata of substantia nigra neurons in rat midbrain slices. J. Neurosci. 18, 3548-3553. ( 10.1523/JNEUROSCI.18-10-03548.1998) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gantz SC, Bunzow JR, Williams JT. 2013. Spontaneous inhibitory synaptic currents mediated by a G protein-coupled receptor. Neuron 78, 807-812. ( 10.1016/j.neuron.2013.04.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergquist F, Niazi HS, Nissbrandt H. 2002. Evidence for different exocytosis pathways in dendritic and terminal dopamine release in vivo. Brain Res. 950, 245-253. ( 10.1016/s0006-8993(02)03047-0) [DOI] [PubMed] [Google Scholar]
- 25.Ovsepian SV, Dolly JO. 2011. Dendritic SNAREs add a new twist to the old neuron theory. Proc. Natl Acad. Sci. USA 108, 19 113-19 120. ( 10.1073/pnas.1017235108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. 1996. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J. Neurosci. 16, 4135-4145. ( 10.1523/JNEUROSCI.16-13-04135.1996) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beckstead MJ, Ford CP, Phillips PEM, Williams JT. 2007. Presynaptic regulation of dendrodendritic dopamine transmission. Eur. J. Neurosci. 26, 1479-1488. ( 10.1111/j.1460-9568.2007.05775.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Courtney NA, Mamaligas AA, Ford CP. 2012. Species differences in somatodendritic dopamine transmission determine D2-autoreceptor-mediated inhibition of ventral tegmental area neuron firing. J. Neurosci. 32, 13 520-13 528. ( 10.1523/JNEUROSCI.2745-12.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ford CP, Phillips PEM, Williams JT. 2009. The time course of dopamine transmission in the ventral tegmental area. J. Neurosci. 29, 13 344-13 352. ( 10.1523/JNEUROSCI.3546-09.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferguson GD, Anagnostaras SG, Silva AJ, Herschman HR. 2000. Deficits in memory and motor performance in synaptotagmin IV mutant mice. Proc. Natl Acad. Sci. USA 97, 5598-5603. ( 10.1073/pnas.100104597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS, Andrews NW. 2003. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII–deficient mice. J. Cell Biol. 162, 543-549. ( 10.1083/jcb.200305131) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martel P, Leo D, Fulton S, Bérard M, Trudeau LE. 2011. Role of Kv1 potassium channels in regulating dopamine release and presynaptic D2 receptor function. PLoS ONE 6, e20402. ( 10.1371/journal.pone.0020402) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cliburn RA, et al. 2017. Immunochemical localization of vesicular monoamine transporter 2 (VMAT2) in mouse brain. J. Chem. Neuroanat. Monoamine Trans. Health Dis. 83–84, 82-90. ( 10.1016/j.jchemneu.2016.11.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson DL, Venton BJ, Heien MLAV, Wightman RM. 2003. Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clin. Chem. 49, 1763-1773. ( 10.1373/49.10.1763) [DOI] [PubMed] [Google Scholar]
- 35.Yang H, Michael AC. 2007. In vivo fast-scan cyclic voltammetry of dopamine near microdialysis probes. In Electrochemical methods for neuroscience, frontiers in neuroengineering (eds Michael AC, Borland LM). Boca Raton, FL: CRC Press/Taylor & Francis. [PubMed] [Google Scholar]
- 36.John CE, Jones SR. 2007. Fast scan cyclic voltammetry of dopamine and serotonin in mouse brain slices. In Electrochemical methods for neuroscience, frontiers in neuroengineering (eds Michael AC, Borland LM). Boca Raton, FL: CRC Press/Taylor & Francis. [PubMed] [Google Scholar]
- 37.O'Neill B, Patel JC, Rice ME. 2017. Characterization of optically and electrically evoked dopamine release in striatal slices from digenic knock-in mice with DAT-driven expression of Channelrhodopsin. ACS Chem. Neurosci. 8, 310-319. ( 10.1021/acschemneuro.6b00300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Congar P, Bergevin A, Trudeau LE. 2002. D2 receptors inhibit the secretory process downstream from calcium influx in dopaminergic neurons: implication of K+ channels. J. Neurophysiol. 87, 1046-1056. ( 10.1152/jn.00459.2001) [DOI] [PubMed] [Google Scholar]
- 39.Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, Strømgaard K, Gether U. 2011. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585-640. ( 10.1124/pr.108.000869) [DOI] [PubMed] [Google Scholar]
- 40.Lebowitz JJ, Khoshbouei H. 2020. Heterogeneity of dopamine release sites in health and degeneration. Neurobiol. Dis. 134, 104633. ( 10.1016/j.nbd.2019.104633) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen BT, Patel JC, Moran KA, Rice ME. 2011. Differential calcium dependence of axonal versus somatodendritic dopamine release, with characteristics of both in the ventral tegmental area. Front. Syst. Neurosci. 5, 39. ( 10.3389/fnsys.2011.00039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banerjee A, Lee J, Nemcova P, Liu C, Kaeser PS. 2020. Synaptotagmin-1 is the Ca2+ sensor for fast striatal dopamine release. eLife 9, e58359. ( 10.7554/eLife.58359) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delignat-Lavaud B, et al. 2021. The calcium sensor synaptotagmin-1 is critical for phasic axonal dopamine release in the striatum and mesencephalon, but is dispensable for basic motor behaviors in mice. bioRxiv. ( 10.1101/2021.09.15.460511) [DOI] [PMC free article] [PubMed]
- 44.Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. 2003. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl Acad. Sci. USA 100, 13 940-13 945. ( 10.1073/pnas.1936192100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Higgins J, Hermanns C, Malloy C, Cooper RL. 2017. Considerations in repetitive activation of light sensitive ion channels for long-term studies: channel rhodopsin in the Drosophila model. Neurosci. Res. 125, 1-10. ( 10.1016/j.neures.2017.07.001) [DOI] [PubMed] [Google Scholar]
- 46.Nair-Roberts RG, Chatelain-Badie SD, Benson E, White-Cooper H, Bolam JP, Ungless MA. 2008. Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience 152, 1024-1031. ( 10.1016/j.neuroscience.2008.01.046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bayer VE, Pickel VM. 1990. Ultrastructural localization of tyrosine hydroxylase in the rat ventral tegmental area: relationship between immunolabeling density and neuronal associations. J. Neurosci. 10, 2996-3013. ( 10.1523/JNEUROSCI.10-09-02996.1990) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deutch AY, Goldstein M, Baldino F, Roth RH. 1988. Telencephalic projections of the A8 dopamine cell group. Ann. N.Y. Acad. Sci. 537, 27-50. ( 10.1111/j.1749-6632.1988.tb42095.x) [DOI] [PubMed] [Google Scholar]
- 49.Fernandes Xavier FG, Doucet G, Geffard M, Descarries L. 1994. Dopamine neoinnervation in the substantia nigra and hyperinnervation in the interpeduncular nucleus of adult rat following neonatal cerebroventricular administration of 6-hydroxydopamine. Neuroscience 59, 77-87. ( 10.1016/0306-4522(94)90100-7) [DOI] [PubMed] [Google Scholar]
- 50.Bacaj T, Wu D, Burré J, Malenka RC, Liu X, Südhof TC. 2015. Synaptotagmin-1 and -7 are redundantly essential for maintaining the capacity of the readily-releasable pool of synaptic vesicles. PLoS Biol. 13, e1002267. ( 10.1371/journal.pbio.1002267) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tawfik B, Martins JS, Houy S, Imig C, Pinheiro PS, Wojcik SM, Brose N, Cooper BH, Sørensen JB. 2021. Synaptotagmin-7 places dense-core vesicles at the cell membrane to promote Munc13-2- and Ca2+-dependent priming. eLife 10, e64527. ( 10.7554/eLife.64527) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Littleton JT, Serano TL, Rubin GM, Ganetzky B, Chapman ER. 1999. Synaptic function modulated by changes in the ratio of synaptotagmin I and IV. Nature 400, 757-760. ( 10.1038/23462) [DOI] [PubMed] [Google Scholar]
- 53.Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER. 2009. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat. Neurosci. 12, 767-776. ( 10.1038/nn.2315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang G, Bai H, Zhang H, Dean C, Wu Q, Li J, Guariglia S, Meng Q, Cai D. 2011. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron 69, 523-535. ( 10.1016/j.neuron.2010.12.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson SL, et al. 2010. Synaptotagmin IV determines the linear Ca2+ dependence of vesicle fusion at auditory ribbon synapses. Nat. Neurosci. 13, 45-52. ( 10.1038/nn.2456) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Q, Fukuda M, Van Bockstaele E, Pascual O, Haydon PG. 2004. Synaptotagmin IV regulates glial glutamate release. Proc. Natl Acad. Sci. USA 101, 9441-9446. ( 10.1073/pnas.0401960101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Z, Bhalla A, Dean C, Chapman ER, Jackson MB. 2009. Synaptotagmin IV: a multifunctional regulator of peptidergic nerve terminals. Nat. Neurosci. 12, 163-171. ( 10.1038/nn.2252) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berton F, Iborra C, Boudier JA, Seagar MJ, Marquèze B. 1997. Developmental regulation of synaptotagmin I, II, III, and IV mRNAs in the Rat CNS. J. Neurosci. 17, 1206-1216. ( 10.1523/JNEUROSCI.17-04-01206.1997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ibata K, Fukuda M, Hamada T, Kabayama H, Mikoshiba K. 2000. Synaptotagmin IV is present at the Golgi and distal parts of neurites. J. Neurochem. 74, 518-526. ( 10.1046/j.1471-4159.2000.740518.x) [DOI] [PubMed] [Google Scholar]
- 60.Rice ME, Patel JC. 2015. Somatodendritic dopamine release: recent mechanistic insights. Phil. Trans. R. Soc. B 370, 20140185. ( 10.1098/rstb.2014.0185) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Primary data analysis files from this manuscript are available from the following doi:10.6084/m9.figshare.19083575. Data supporting this article have also been uploaded as part of the electronic supplementary material.