

Abstract
Ageing, death, and potential immortality lie at the heart of biology, but two seemingly incompatible paradigms coexist in different research communities and have done since the nineteenth century. The universal senescence paradigm sees senescence as inevitable in all cells. Damage accumulates. The potential immortality paradigm sees some cells as potentially immortal, especially unicellular organisms, germ cells and cancerous cells. Recent research with animal cells, yeasts and bacteria show that damaged cell constituents do in fact build up, but can be diluted by growth and cell division, especially by asymmetric cell division. By contrast, mammalian embryonic stem cells and many cancerous and ‘immortalized’ cell lines divide symmetrically, and yet replicate indefinitely. How do they acquire their potential immortality? I suggest they are rejuvenated by excreting damaged cell constituents in extracellular vesicles. If so, our understanding of cellular senescence, rejuvenation and potential immortality could be brought together in a new synthesis, which I call the cellular rejuvenation hypothesis: damaged cell constituents build up in all cells, but cells can be rejuvenated either by growth and cell division or, in ‘immortal’ cell lines, by excreting damaged cell constituents. In electronic supplementary material, appendix, I outline nine ways in which this hypothesis could be tested.
Keywords: cellular senescence, cellular rejuvenation, damaged cell constituents, asymmetric cell division, cancer cells, stem cells
1. Introduction
Since the late nineteenth century, there have been two principal schools of thought about cellular ageing. One sees senescence as universal. Wear and tear are inevitable. The problem is to explain rejuvenation, without which all cells would die out. The other paradigm assumes that some cells are potentially immortal and immune to senescence, namely unicellular organisms, the germ cells of multicellular organisms and ‘immortal’ cell lines grown in laboratories. The problem is to explain senescence in the somatic cells of multicellular organisms. If senescence is optional, why did it evolve?
This debate was epitomized in the conflicting views of Émile Maupas (1842–1916) and August Weismann (1834–1914) and is still unresolved. Maupas, a French zoologist, argued that senescence is inherent in all cells, and that rejuvenation depends on sexual processes [1]. Weismann, a German professor of biology, proposed that unicellular organisms and germ cells are potentially immortal, but that somatic cells of multicellular organisms senesce. He assumed that animal germ cells inherited their potential immortality from unicellular organisms, which, in his own words, ‘carry the potentiality of unending life’. He continued, ‘The reproductive cells cannot lose that capacity for unlimited reproduction … But the somatic cells have lost this power… they became restricted to a fixed, though perhaps very large number of cell generations' [2].
These two approaches, which I call the ‘universal senescence paradigm’ (USP) and the ‘potential immortality paradigm’ (PIP), are still widely influential, but in different areas of biology. Within the last 15 years, the study of senescence and rejuvenation in bacteria and yeasts has led some researchers to argue that senescence through the accumulation of damage is inevitable in all forms of life, including unicellular organisms [3,4]. The origins of ageing and the origins of life itself may be intertwined [5].
By contrast, PIP is still taken for granted by many researchers on germ cells, embryonic stem cells (ESC) and cancer. This assumption is explicit in Kirkwood's [6] influential ‘disposable soma hypothesis', explicitly based on Weismann's ideas, which assumes that germ cells retain their youthfulness by virtue of energetically costly repair systems. Multicellular animals have evolved to allocate fewer resources to the repair and maintenance of somatic cells, which eventually die anyway. Error regulation is a ‘luxury’ that somatic cells cannot afford [7,8].
Both USP and PIP agree that damaged cell constituents (DCC) accumulate in somatic cells of multicellular animals. They disagree in that USP assumes that damage accumulates in all cells, whereas PIP assumes that some cells have almost infallible repair systems that are downregulated in normal somatic cells to save resources.
Here, I argue that recent advances in cell biology may enable this longstanding debate to be resolved through an expanded concept of rejuvenation.
I first look at cellular damage and repair systems at the molecular level, and then explore senescence, rejuvenation and potential immortality in all kingdoms of life, starting with multicellular animals, followed by plants, then bacteria and yeasts which, although they are unicellular organisms, are not immune to senescence, as Weismann assumed. I then discuss animal egg cells. Do they have near-perfect repair systems, as PIP supposes?
Finally, I consider the puzzle posed for USP by ‘immortal’ mammalian cell lines, including ESC and cancerous cells, such as the famous HeLa cells, initially isolated from the cervical cancer of an African-American woman, Henrietta Lacks, in 1951 [9] and still multiplying by symmetrical cell division in laboratories around the world. How have they acquired this immortality? Many cancers and ESC are now known to excrete large numbers of extracellular vesicles. I suggest that these cells rejuvenate themselves by excreting DCC in extracellular vesicles, an ability that most other animal cells lack. This hypothesis may provide a way of reconciling USP and PIP and point toward a unified understanding of senescence and rejuvenation applicable to all forms of life, which I call the cellular rejuvenation hypothesis. In electronic supplementary material, appendix, I outline nine ways in which this hypothess could be tested experimentally.
2. The repair of cellular damage
Some of the principal causes of molecular damage in cells are reactive oxygen species (ROS), highly reactive chemicals, such as peroxides, produced as a result of oxidative phosphorylation in mitochondria and other oxidative processes. ROS damage lipids by peroxidation, and proteins by carbonylation, introducing ketone or aldehyde groups into protein sidechains [10,11]. Lower levels of ROS generally lead to less accumulation of age-related damage [12]. This seems to be the main reason why caloric restriction in the diets of a wide variety of organisms, including yeasts, nematode worms, fruit flies, rodents and primates, prolongs lifespan and reduces the incidence of age-related diseases [13].
In addition to oxidative damage, various kinds of stress, including heat stress, lead to the unfolding of proteins, followed by misfolding and aggregation, for example in ‘stress granules’ made up of denatured ribonucleoproteins that accumulate when protein synthesis stalls [14].
Oxidative and stress damage are often reversible. Most carbonylated, misfolded and dysfunctional proteins are degraded by the ubiquitin–proteasome system, which is highly conserved in eukaryotic organisms. The first step is the tagging of misfolded or damaged proteins by covalent bonds to ubiquitin, a small protein found in almost all eukaryotic cells. These tags identify the damaged proteins as candidates for digestion by proteasomes, complex structures with tunnel-like internal cavities capable of degrading hundreds of different proteins [15].
Some aggregates of denatured proteins can be dissolved; their proteins are ubiquinated and degraded by proteasomes [16]. However, some resist disassembly and like other persistent aggregates can be dissolved only after being incorporated into autophagosomal vesicles [17], which have double membranes, and form around denatured protein complexes, dysfunctional mitochondria and other damaged structures, segregating the material from the rest of the cytoplasm. These vesicles fuse with lysosomes, whose hydrolytic enzymes digest most of the vesicular contents for recycling [18].
Damage to membranes can be overcome by several different repair systems [19]. Although protein, membrane and other repair systems [20] are highly effective, they are not infallible, at least in the somatic cells of animals. DCC accumulate as time goes on, sooner or later resulting in cellular senescence [21–23]. DCC include indigestible protein aggregates, defective mitochondria and lipofuscin granules, also known as ‘age pigment granules', which accumulate in lysosomes as ‘residual bodies’ [24,25]. These are all hallmarks of ageing, and increase with age in the cells of many mammalian tissues with harmful effects [26,27]. The accumulation of indigestible residues is exaggerated in pathological conditions like Alzheimer's disease, where denatured protein aggregates accumulate within neurons and plaques of denatured amyloid peptide build up between them [28].
Although some effects of ageing are reversible, the accumulation of DCC is irreversible. If cells stop growing and dividing, DCC continue to accumulate not only in mammalian cells, but in nematode worms, insects [29], plants [30], and in many other kinds of organisms.
In summary, not all cellular damage can be repaired. Cells can avoid senescence only if they get rid of DCC. How can they do so? I look first at the strategies employed by somatic cells in multicellular animals.
3. Cellular rejuvenation in multicellular animals
If somatic cells accumulate DCC as they grow older, how are some cells, especially stem cells, rejuvenated?
In an article in Nature entitled ‘The ageing, growth and death of cells' [31], I proposed the hypothesis that some cells escape senescence because they dilute DCC by growing and dividing. If they divide symmetrically, both daughters inherit similar amounts of DCC. If they divide asymmetrically and one daughter inherits most DCC, the other is rejuvenated, in the sense that it is freed from DCC (figure 1). Cell populations can grow quicker by symmetrical cell division, but DCC build up within them unless they keep on dividing fast enough. Mathematical models based on this hypothesis show a variety of outcomes depending on the rates of accumulation of DCC, the rates of cell division, the degree of asymmetrical partitioning of DCC, and the degree to which damage accumulation is autocatalytic. In symmetrically dividing cells, if DCC accumulate faster than cell division dilutes them, cell lines die out [32,33].
Figure 1.
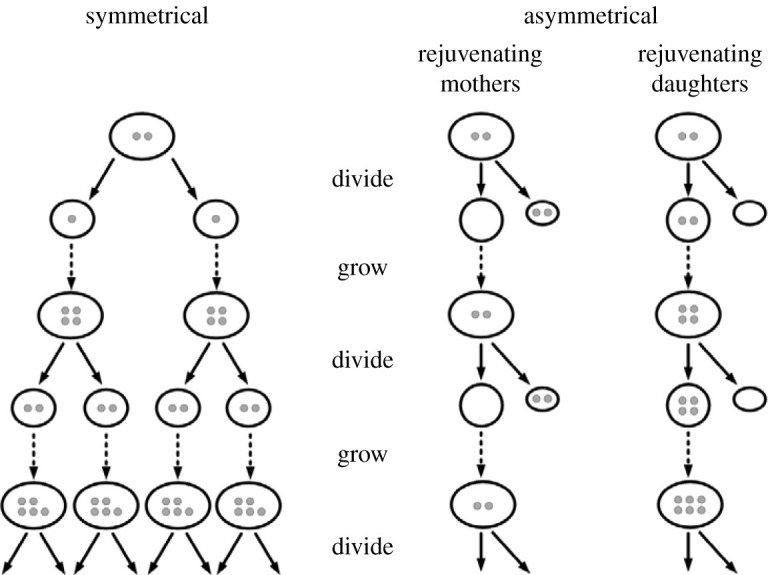
The distribution of damaged cell constituents (DCC) in symmetrical and asymmetrical cell division. DCC (small grey circles) build up in cells as time goes on and are inherited by the next generation through cell division. In symmetrical division they are distributed more or less equally between sisters. In asymmetrical division, the mother can be rejuvenated at the expense of the daughter, as in epithelial stem cells; or the daughter can be rejuvenated at the expense of the mother, as in budding yeasts.
Since around 2005 much evidence has built up for the asymmetrical partitioning of DCC in asymmetrical cell division (ACD) in animal stem cells [34], and also in bacteria and yeasts, as discussed below.
(a) . Stem cells
Stem cells usually divide asymmetrically and one of the ensuing cells is rejuvenated at the expense of the other. In adults, stem cells themselves are continually rejuvenated as they produce a succession of mortal daughters. In embryos, some stem cells work the other way round: their progeny are rejuvenated while the stem cells age, a pattern also seen in bacteria and yeasts, as discussed below (figure 1).
The stem cells in our own skin, bone marrow and intestinal crypts are continually rejuvenated, while their progeny differentiate and die [35,36]. In a study on people suffering from a degenerative disease in which mutant proteins accumulate in insoluble aggregates, the intestinal stem cells were free of these inclusions, even in elderly patients, while their short-lived progeny contained large amounts [37].
Stem cells are not entirely immune from ageing; some senesce as a result of genetic mutations, epigenetic changes and environmental damage [38]. When stem cells are killed by damage, nearby stem cells divide symmetrically to replace them [39], losing the rejuvenative advantage of ACD.
(b) . The partitioning of damaged cell constituents during asymmetric cell division
DCC aggregate in several ways. Denatured proteins accumulate in bodies near the nucleus, variously called aggresomes or JUxta-Nuclear Quality (JUNQ) control compartments. Meanwhile, Insoluble Protein Deposits (IPODs) [40] form in the peripheral cytoplasm.
One way in which DCC are differentially partitioned during ACD is through the linkage of aggresomes to one of the centrosomes of a dividing cell, resulting in their asymmetric distribution during cell division [41,42]. IPODs are partitioned asymmetrically by a different cytoskeleton-based system from aggresomes and JUNQs [34,43]. Damaged mitochondria are also partitioned asymmetrically into the daughter cells [44,45].
The result of these processes is that stem cells are rejuvenated and their daughters inherit ‘cargoes’ of DCC.
(c) . The 'Hayflick limit’ in symmetrically dividing cells
Weismann's idea about somatic cells having a limited capacity to divide was supported by the study of mammalian cells in tissue culture, such as fibroblasts, which divide symmetrically and die out after a limited number of divisions, sometimes called the ‘Hayflick limit’, which rarely exceeds 50–60 divisions [46,47]. At first sight, the senescence of these cell lines, which divide slower as they approach the limit of their lifespan, seems to support the idea of a build-up of DCC. So does the fact that oxidative stress accelerates their senescence [48]. However, DCC alone cannot explain the senescence of these lines, which also depends on the shortening of telomeres [49].
Telomeres are structures formed of repetitive DNA sequences at the ends of chromosomes, linked to a protein complex called shelterin [50]. In embryos, cells start with long telomeres which are usually shortened through successive divisions. Through a pre-programmed ‘count-down’ process, cell division stops when telomeres are too short. This system may have evolved in part to help prevent somatic cells proliferating cancerously [51]. Telomere-dependent ageing can be reversed if the telomerase enzyme system is activated and lengthens the telomeres, as occurs in ESC, some adult stem cells and in many cancer cells, enabling cell division to continue [52,53]. However, the effects of genetic manipulation may involve not only a lengthening of telomeres but also the activation of the c-Myc oncogene [54], which is a ‘master regulator’ of genetic changes characteristic of cancerous cells [55].
The reversibility of telomere-centred ageing seems to support PIP. So does another kind of epigenetic ageing, in which DNA and associated histone proteins are methylated progressively, making the cells less prone to divide as time goes on. The contrary process, demethylation, has a rejuvenating effect [56]. DNA and histones are demethylated in many cancers [53,57].
I return to a discussion of immortalized cell lines and cancer cells in §5.
4. How are cells rejuvenated in plants?
Whereas multicellular animals stop growing, senesce and die, some plants grow indefinitely, and their growing tips do not senesce. For example, trembling aspen trees propagate themselves vegetatively by root suckers forming clonal groves, some of which are more than 10 000 years old [58]. Some crops, like potatoes, are routinely propagated vegetatively.
All vascular plants, including ferns, conifers and flowering plants, contain meristems in their shoot and root tips, which are regions in which cells divide, and from which the stems, leaves, flowers, fruits and roots are derived. In the heart of these meristems are stem cells that divide by ACD. Their daughters undergo further divisions within young roots and shoots, then differentiate and sooner or later die [59].
ACD has been studied in the model plant Arabidopsis thaliana [60], but little is yet known about the partitioning of DCC. However, in the light of research on animal stem cells, bacteria and yeasts it seems likely that the stem cells are rejuvenated by ACD, while their mortal daughters inherit DCC. If so, plant stem cells would fit into a pattern of cellular senescence and rejuvenation found in other realms of life. This is a testable hypothesis, as discussed in electronic supplementary material, appendix.
(a) . Multiple fission
In many species of unicellular algae, including Chlorella, division takes place by multiple fission. A mother cell divides repeatedly to form 4, 8, 16 and up to 128 daughter cells [61,62] depending on the growth conditions [63], necessarily diluting DCC.
Many algae and plants, including mosses and ferns, reproduce vegetatively by spores, which are often produced in vast numbers by multiple fission. For example, in the sporangia on the fronds of the fern Marattia, each archesporial cell, itself produced by ACD, can give rise to 2656 spores [64]. Many fungi and protozoa also produce spores by multiple fission.
The patterns of cell division in plants are consistent with the idea that DCC build up in all cells over time, but some cells can be rejuvenated by ACD or by the dilution of DCC by multiple fission. Plants fit well within USP. Indeed, Weismann's PIP never applied to plants in the first place, because germ cells are not separated off from somatic cells at an early stage of embryology. Flowers develop from meristems in shoots.
5. Rejuvenation in bacteria and yeasts
Over the last 15 years there has been a widespread recognition that many bacteria senesce and undergo rejuvenation through growth and cell division [4,65]. Rejuvenation generally depends on ACD, even in cells that appear to divide symmetrically. The same is true of yeasts.
(a) . Bacteria
The aquatic bacterium Caulobacter crescentus has two kinds of cells: stalked cells rooted to a solid substratum through a tube-like stalk, and flagella-bearing swarmer cells (figure 2). Only stalked cells divide; at first, they give rise to swarmers in rapid succession, then produce them slower and slower, until they finally stop dividing and die. Through ACD, the stalked cells retain DCC and undergo a form of replicative ageing while the swarmer cells are rejuvenated [66].
Figure 2.
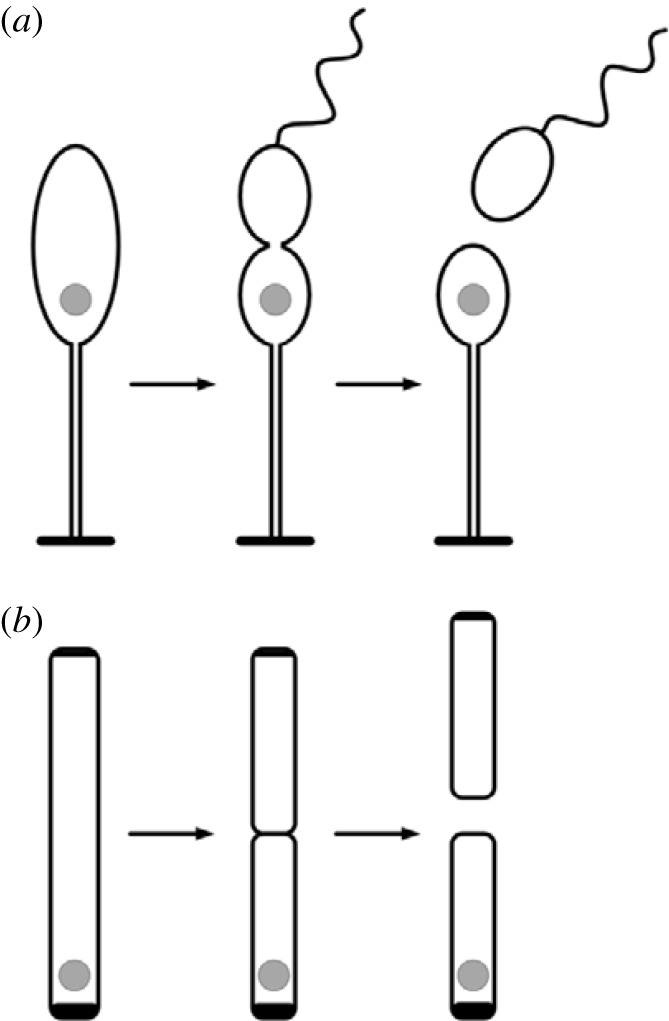
The partitioning of DCC in bacterial cell division. (a) A Caulobacter cell attached to a solid substratum by its stalk. When it divides DCC (grey circle) remain within the stalked cell, while its rejuvenated daughter, a swarmer cell, swims away, then settles down and becomes a stalked cell itself. Stalked cells divide repeatedly, accumulate DCC, and eventually die. (b) E. coli appears to divide symmetrically, but an aggregate of DCC located near the old pole of the cell is asymmetrically partitioned into the cell that inherits the old pole, while the other is rejuvenated.
Although most bacteria appear to divide symmetrically, this appearance is deceptive. The bacterium Escherischia coli divides into equal-sized daughters, but one inherits an old cell wall while the other receives a new one (figure 2). Daughters that inherit the old poles grow slower, produce fewer offspring and are more likely to die [67]. Aggregates of denatured proteins usually accumulate near the old pole. Sometimes new-pole cells inherit these inclusion bodies instead, and then they too divide slower [68]. In most cells that divide rapidly, there are no visible denatured protein aggregates, but cells with the old pole still seem to inherit DCC and divide slower than their sisters [69,70]. Thus in E. coli as in Caulobacter, one daughter cell is rejuvenated at the expense of the other.
(b) . Yeasts
Budding yeasts divide asymmetrically by budding off small daughter cells, which then grow and become mother cells themselves. In a classic study of the budding yeast Saccharomyces cerevisiae, used in brewing and baking for millennia, Mortimer & Johnston [71] found that mother cells gave rise to an average of 24 daughter cells; the rate of division slowed toward the last generation, and then the mother cells died.
Recent research on budding yeasts has revealed more details about the retention of denatured protein aggregates [72] and ageing mitochondria [73,74] by the mother cells while the daughters are freed from them. The cytoskeleton plays an essential role [75], as does the endoplasmic reticulum [76].
Unlike budding yeasts, fission yeasts, like Saccharomyce pombe, appear to divide symmetrically. However, as in E. coli, DCC in the form of protein aggregates are retained by one daughter cell while the other is freed from them and subsequently divides faster [33]. Under favourable growth conditions, in which cells grew and divided rapidly, few protein aggregates accumulated; many of the cells showed no signs of ageing, but a minority inherited denatured protein aggregates and died [77].
Yeasts are now among the most important model organisms for studies of senescence and rejuvenation at the cellular level [4,78]. Weismann's assumption that unicellular organisms are immune to senescence is not true, at least in the case of the most-studied yeasts and bacteria.
6. Are animal germ cells immune to senescence?
In PIP, there is no need to explain the rejuvenation of animal germ cells. They are immune to senescence ex hypothesi. By contrast, USP suggests that DCC are likely to accumulate in germ cells as they do in other cells. If so, how are germ cells rejuvenated?
Male gametes do not need to be rejuvenated. They are usually short-lived, and their cytoplasm plays little or no role in the new organism. In both animals and plants, they are formed by symmetrical cell divisions. In humans and other mammals, primary spermatocytes divide by meiosis to produce four viable sperm cells [79]. In flowers, the pollen mother cells divide by meiosis and give rise to four viable pollen grains [80].
By contrast, eggs are formed by ACD in both animals and plants [31]. They are surrounded by dying sisters. For example, in the fruit fly Drosophila, a cell called the oogonium goes through four cycles of cell division to produce 16 cells, of which only one, the oocyte, gives rise to the egg; the others, called nurse cells, nourish and sustain it and die [81]. The oocyte then undergoes meiosis, producing the egg and short-lived polar bodies [82].
In plants, the sisters of maturing egg cells die, just as they do in animals. For example, in the flowers of Arabidopsis, the megasporocyte, the cell from which the egg is ultimately derived, produces four cells by meiosis, three of which die [83].
To my knowledge, the partitioning of DCC in the production of egg cells in animals and plants has not yet been studied, nor has their partitioning between the oospheres and periplasm of fungi (see in electronic supplementary material, appendix).
7. How do ‘immortal’ cell lines and cancer cells avoid senescence?
PIP seems to be confirmed by ‘immortal’ mammalian and human cell lines that are not subject to the Hayflick limit, such as HeLa cells. Hundreds of immortal cell lines are commercially available and routinely used in research. Most show one or more of the hallmarks of cancer, including the activation of telomerase, enhanced energy production by glycolysis [84], and genetic changes, including increased mutation rates and chromosomal instability [85].
In cancers, not only do the cells escape from the controls that stop normal cells from dividing excessively, but they also need to avoid senescence, or else they would die out. One way in which they are protected from the build up of DCC is by a shift in energy production toward glycolysis, a feature of cancerous cells noted more than 80 years ago by the biochemist Otto Warburg and known as the Warburg effect [85]. Glycolysis produces fewer ROS than oxidative phosphorylation and hence less oxidative damage.
In some cancers, cells are rejuvenated by ACD in a similar way to normal stem cells; indeed many cancers arise from stem cells in the first place, and also contain stem cells that divide by ACD [86,87]. However, some cancerous cells grown in vitro divide symmetrically, like HeLa cells, without the kind of cryptic asymmetry that occurs in the divisions of E. coli and fission yeast cells ([88], their electronic supplementary material, figure S1).
This is where the paradigms come into head-on collision. For PIP, immortal cell lines pose no problem; they have near-perfect repair systems ex hypothesi. By contrast, these cell lines are problematic for USP. How can cancer cell lines that divide symmetrically and are not rejuvenated by ACD escape from senescence? The only possible answer seems to be that they have another way of getting rid of DCC, namely by excreting them in vesicles [31].
Many types of animal cell are now known to bud off membrane-bound vesicles into the extracellular space (figure 3). Some of these vesicles play an important role in intercellular communication, transferring DNA, RNA, proteins, lipids and virus particles from cell to cell [93]. Extracellular vesicles can be taken up by other cells either by fusing with their membranes, thus transferring their contents to the cytoplasm of the receptor cell, or by being engulfed by phagocytotic cells [94].
Figure 3.
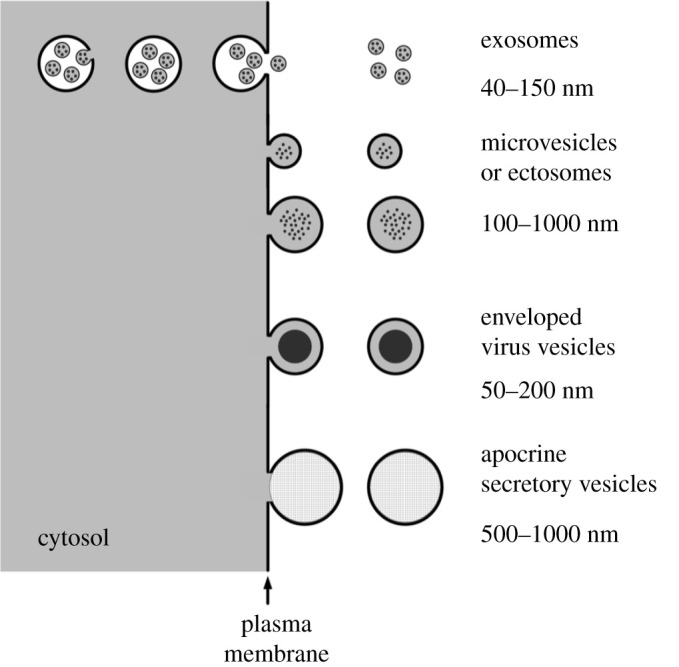
Some of the different kinds of membrane-bound extracellular vesicles secreted from mammalian cells. Exosomes are first budded off into intracellular vacuoles, called multivesicular bodies, before being released into the extracellular fluid when the multivesicular bodies fuse with the plasma membrane. Microvesicles of various types, also called ectosomes, are budded off the plasma membrane directly into the extracellular fluid [89,90]. Vesicles containing viruses, such as human immunodeficiency virus (HIV), are released within an envelope derived from the cell membrane [91]. Some glandular cells secrete vesicles, like the membrane-bound lipid globules produced by mammary glands as constituents of milk [92]. In addition, when dying cells break up they release membrane-bound ‘apoptotic bodies’ of varying sizes [90].
Some extracellular vesicles are known to contain DCC, including damaged DNA [95] and damaged proteins [89], including misfolded amyloid peptide aggregates produced in neurons affected by Alzheimer's disease [96,97]. Differentiating erythrocytes shed unwanted proteins in exosomes [98].
Many types of cancer cell release extracellular vesicles [93], and some amoeboid cancer cells shed unusually large ones, called oncosomes [99]. Extracellular vesicles carried in the blood are now used as diagnostic biomarkers for a wide range of cancers [100]. The infection of cells with cancer viruses changes the types of extracellular vesicles they produce [101], and the transformation from normal cells to immortalized cancer cells leads to a striking increase in the secretion of extracellular vesicles [102]. Vesicles from HeLa cells contain unfolded proteins, among other molecules [103].
A comparable process occurs in bacteria. Many species produce ‘minicells’ that lack chromosomes, cannot proliferate, and die. In E. coli, minicells are budded off from cell poles and contain aggregates of damaged proteins. Bacteria are rejuvenated by this ‘damage disposal mechanism’ and their daughters divide faster as a result [4,104]. This is a very underexplored area if research, and in electronic supplementary material, appendix, I suggest several new lines of enquiry.
8. How do embryonic stem cells escape senescence?
Mammalian ESC are capable of developing into any type of cell in the body. When cultured in vitro they divide symmetrically and are potentially immortal; not subject to the Hayflick limit [105]. They are the only normal cells that divide symmetrically and share potential immortality with immortalized and cancerous cell lines.
Under natural conditions, ESC are a short-lived phase at the beginning of embryology.
They are difficult to maintain in vitro under standard atmospheric conditions because they tend to differentiate spontaneously. They are best grown at oxygen levels of 2–5%, way below the usual 21% in the atmosphere [106,107]. These conditions resemble their hypoxic environment within pre-implantation embryos [108], which may protect them from the build-up of DCC by reducing damage from ROS. ESC also derive much of their energy from glycolysis, further reducing oxidative damage.
ESC prolifically produce extracellular vesicles, and vesicles derived from cultured ESC are now being tested for their therapeutic potential as a new version of stem cell therapy [109,110].
Four major marker genes are highly expressed in ESC, including c-Myc, each of which then activates gene complexes or modules. In many types of cancer, at least one of these ESC marker genes is activated, especially in the most aggressive tumours [111,112]. Although genes such as c-Myc are usually called oncogenes, as if they are specific to cancer, this is misleading. Cancer cells seem to reactivate key features of quintessentially youthful ESC. When cancer cells are grown under hypoxic conditions, like those under which ESC have evolved, further ESC genes are often activated and the cells become more prone to form aggressive tumours [113,114].
I suggest that one of the abilities that may be present in ESC and regained by cancerous cell lines may be the ability to excrete DCC in extracellular vesicles. In normal development, this ability is suppressed as cells differentiate. If somatic cells excreted DCC, intercellular spaces would become like open sewers, overwhelming the ability of phagocytes to clean up the debris. However, this excretory ability may be reactivated in immortalized cell lines and some cancers.
Thus many of the features of immortal cell lines and cancer cells may arise as a result of re-activating ESC gene modules. This similarity may include a rejuvenative system that enables the cells to get rid of DCC in vesicles.
9. Conclusion
Both USP and PIP are compatible with the dilution of DCC by growth and cell division, especially by ACD, in animals, yeasts and bacteria. Both paradigms predict similar processes in plants. Both paradigms are consistent with the dilution of DCC by multiple fission in plants, algae, fungi, protozoa and bacteria.
However, the paradigms conflict in relation to ESC and symmetrically dividing immortalized cell lines. USP assumes that DCC inevitably accumulates and that cells must somehow get rid of them. I propose that they do so by excreting DCC in extracellular vesicles.
PIP assumes near-perfect cellular repair systems that enable these cells to avoid the build-up of DCC. But can any repair system be perfect? If some DCC cannot be dissolved and recycled and hence accumulate, how could a repair system get rid of them? Almost the only possible answer is by expelling DCC from the cells by exocytosis.
Thus the two paradigms would converge if DCC are in fact expelled from ESC and cancerous cell lines in extracellular vesicles. For USP the expulsion of DCC would be seen as an excretory system, and for PIP a repair system. The effect would be the same.
After more than 130 years, it may soon be possible to resolve the debate between Maupas and Weismann. Maupas was right in thinking that senescence is inherent in all cells, and Weismann was right in thinking that some cell types in animals were special, able to avoid senescence in a way that is suppressed in most somatic cells. If this special repair system depends on the exocytosis of DCC, then we may soon arrive at a new synthesis that applies to all the kingdoms of life. I call this the cellular rejuvenation hypothesis: DCC build up in all cells as they age, but can be diluted either by growth and cell division, especially ACD and multiple fission or by the excretion of DCC in extracellular vesicles.
This cellular rejuvenation hypothesis is experimentally testable and in electronic supplementary material, appendix, I suggest nine new lines of empirical enquiry.
Acknowledgements
I thank Alex Gomez-Marin, Keith Roberts, Merlin Sheldrake and my anonymous referees for comments on drafts of this review, and Merlin Sheldrake for the diagrams.
Data accessibility
This article has no additional data.
Authors' contributions
A.R.S.: conceptualization, investigation, project administration, writing—original draft, writing—review and editing.
Competing interests
I declare I have no competing interests.
Funding
I am grateful to the Planet Heritage Foundation and the Watson Foundation for financial support.
References
- 1.Maupas A. 1888. Recherches expérimentales sur la multiplications des infusoires ciliés. Arch. Zool. Exp. Gén., 2 6, 165-177. [Google Scholar]
- 2.Weismann A. 1891. Essays upon heredity and kindred biological problems, Vol. I. Oxford, UK: Clarendon Press. [Google Scholar]
- 3.Gladyshev VN. 2013. The origin of aging: imperfectness-driven non-random damage defines the aging process and control of lifespan. Trends Genet. 29, 506-512. ( 10.1016/j.tig.2013.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moger-Reischer R, Lennon JT. 2019. Microbial ageing and longevity. Nat. Rev. Microbiol. 17, 679-690. ( 10.1038/s41579-019-0253-y) [DOI] [PubMed] [Google Scholar]
- 5.Currais A. 2016. The origin of life at the origin of ageing? Ageing Res. Rev. 35, 297-300. ( 10.1016/j.arr.2016.10.007) [DOI] [PubMed] [Google Scholar]
- 6.Kirkwood T. 1977. Evolution of aging. Nature 270, 301-304. ( 10.1038/270301a0) [DOI] [PubMed] [Google Scholar]
- 7.Kirkwood TBL, Cremer T. 1982. Cytogerontology since 1881: a reappraisal of August Weismann and a review of modern progress. Hum. Genet. 60, 101-121. ( 10.1007/BF00569695) [DOI] [PubMed] [Google Scholar]
- 8.Clegg RJ, Dyson RJ, Kreft JU. 2014. Repair rather than segregation of damage is the optimal unicellular aging strategy. BMC Biol. 12, 52. ( 10.1186/s12915-014-0052-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skloot R. 2019. The immortal life of Henrietta Lacks. London, UK: Picador. [Google Scholar]
- 10.Nystrom T. 2005. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 24, 1311-1317. ( 10.1038/sj.emboj.7600599) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iuchi K, Takai T, Hisatomi H. 2021. Cell death via lipid peroxidation and protein aggregation diseases. Biology 10, 399. ( 10.3390/biology10050399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finkel T, Holbrook NJ. 2000. Oxidants, oxidative stress and the biology of ageing. Nature 408, 239-246. ( 10.1038/35041687) [DOI] [PubMed] [Google Scholar]
- 13.Roth LW, Polotsky AJ. 2012. Can we live longer by eating less? A review of caloric restriction and longevity. Maturitas 71, 315-319. ( 10.1016/j.maturitas.2011.12.017) [DOI] [PubMed] [Google Scholar]
- 14.Protter DSW, Parker R. 2016. Principles and properties of stress granules. Trends Cell Biol. 26, 668-679. ( 10.1016/j.tcb.2016.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bard JAM, Ellen A, Goodall EA, Greene ER, Jonsson E, Dong KC, Martin A. 2016. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 87, 697-724. ( 10.1146/annurev-biochem-062917-011931) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maxwell BA, Gwon Y, Mishra A, Peng J, Nakamurake H, Zhang K, Kim HJ, Taylor JP. 2021. Ubiquitination is essential for recovery of cellular activities after heat shock. Science 372, eabc3593. ( 10.1126/science.abc3593) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gwon Y, Maxwell BA, Kolaitis RM, Zhang P, Kim HJ, Taylor JP. 2021. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science 372, eabf6548. ( 10.1126/science.abf6548) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaffagnini G, Martens S. 2016. Mechanisms of selective autophagy. J. Mol. Biol. 428, 1714-1724. ( 10.1016/j.jmb.2016.02.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jimenez AJ, Perez F. 2017. Plasma membrane repair: the adaptable cell life-insurance. Curr. Opin Cell Biol. 47, 99-107. ( 10.1016/j.ceb.2017.03.011) [DOI] [PubMed] [Google Scholar]
- 20.Igelmann S, et al. 2021. A hydride transfer complex reprograms NAD metabolism and bypasses senescence. Mol. Cell 81, 3848-3865. ( 10.1016/j.molcel.2021.08.028) [DOI] [PubMed] [Google Scholar]
- 21.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroeme G. 2013. The hallmarks of aging. Cell 153, 1194-1217. ( 10.1016/j.cell.2013.05.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santos AL, Sinha S, Lindner AB. 2018. The good, the bad, and the ugly of ROS: new insights on aging and aging-related diseases from eukaryotic and prokaryotic model organisms. Oxidative Med. Cell. Longev. 2018, 1941285. ( 10.1155/2018/1941285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogrodnik M, Salmonowicz H, Gladyshev VN. 2018. Integrating cellular senescence with the concept of damage accumulation in aging: relevance for clearance of senescent cells. Aging Cell 18, e12841. ( 10.1111/acel.12841) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunk UTV. 1989. On the origin of lipofuscin: the iron content of residual bodies, and the relation of these organelles to the lysosomal vacuome. A study on cultured human glial cells. Adv. Exp. Med. Biol. 265, 313-320. ( 10.1007/978-1-4899-5339-1_22) [DOI] [PubMed] [Google Scholar]
- 25.König J, Otta C, Hugoa M, Jung T, Bulteaub AL, Tilman Grunea T, Höhn A. 2017. Mitochondrial contribution to lipofuscin formation. Redox Biol. 11, 673-681. ( 10.1016/j.redox.2017.01.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunk UTV, Terman A. 2002. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 33, 611-619. ( 10.1016/S0891-5849(02)00959-0) [DOI] [PubMed] [Google Scholar]
- 27.Moreno-Garcia A, Kun A, Calero O, Medina M, Calero M. 2018. A overview of the role of lipofuscin in age-related neurodegeneration. Front. Neurosci. 12, 464. ( 10.3389/fnins.2018.00464) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giaccone G, Orsi L, Cupidi C, Tagliavini F. 2011. Lipofuscin hypothesis of Alzheimer's disease. Dement Geriatr. Cogn. Disord. 1, 292-296. ( 10.1159/000329544) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labuschagne CF, Brenkman AB. 2013. Current methods in quantifying ROS and oxidative damage in Caenorhabdytis elegans and other model organisms of aging. Ageing Res. Rev. 12, 918-930. ( 10.1016/j.arr.2013.09.003) [DOI] [PubMed] [Google Scholar]
- 30.Ciaka K, Tymiński M, Gniazdowska A, Krasuska U. 2020. Carbonylation of proteins—an element of plant ageing. Planta 252, 12. ( 10.1007/s00425-020-03414-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheldrake AR. 1974. The ageing, growth and death of cells. Nature 250, 381-385. ( 10.1038/250381a0) [DOI] [Google Scholar]
- 32.Hirsch HR. 1978. The waste-product theory of aging: waste dilution by cell division. Mech. Aging Dev. 8, 51-62. ( 10.1016/0047-6374(78)90006-4) [DOI] [PubMed] [Google Scholar]
- 33.Erjavec N, Cvijovic M, Klipp E, Nyström T. 2008. Selective benefits of damage partitioining in unicellular systems and its effect on aging. Proc. Natl Acad. Sci. USA 105, 18 764-18 769. ( 10.1073/pnas.0804550105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore DL, Jessberger S. 2017. Creating age asymmetry: consequences of inheriting damaged goods in mammalian cells. Trends Cell Biol. 27, 82-92. ( 10.1016/j.tcb.2016.09.007) [DOI] [PubMed] [Google Scholar]
- 35.Rando TA. 2006. Stem cells, ageing and the quest for immortality. Nature 441, 1080-1086. ( 10.1038/nature04958) [DOI] [PubMed] [Google Scholar]
- 36.Stern MM, Bickenbach JR. 2007. Epidermal stem cells are resistant to cellular aging. Aging Cell 6, 439-452. ( 10.1111/j.1474-9726.2007.00318.x) [DOI] [PubMed] [Google Scholar]
- 37.Rujano MA, et al. 2006. Polarized asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 4, e417. ( 10.1371/journal.pbio.0040417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodell MA, Rando TA. 2015. Stem cells and healthy aging. Science 350, 1199-1204. ( 10.1126/science.aab3388) [DOI] [PubMed] [Google Scholar]
- 39.Lopez-Garcia C, Klein AM, Simons BD, Winton DJ. 2010. Intestinal stem cell replacement follows a pattern of neutral drift. Science 330, 822-825. ( 10.1126/science.1196236) [DOI] [PubMed] [Google Scholar]
- 40.Johnston HE, Samant RS. 2021. Alternative systems for misfolding protein clearance: life beyond the proteasome. FEBS J. 288, 4464-4487. ( 10.1111/febs.15617) [DOI] [PubMed] [Google Scholar]
- 41.Fuentealba LC, Elvers E, Geissert G, Taelman V, De Robertis EM. 2008. Asymmetric mitosis: unequal segregation of proteins destined for degradation. Proc. Natl Acad. Sci. USA 105, 7732-7737. ( 10.1073/pnas.0803027105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamashita YM. 2009. The centrosome and asymmetric cell division. Prion 3, 84-88. ( 10.4161/pri.3.2.8821) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ogrodnik M, et al. 2014. Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin. Proc. Natl Acad. Sci. USA 111, 8049-8054. ( 10.1073/pnas.1324035111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katajisto P, et al. 2015. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348, 340-343. ( 10.1126/science.1260384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan Y, Finkel T. 2020. The mitochondrial regulation of stem cell aging. Mech. Aging Dev. 191, 111334. ( 10.1016/j.mad.2020.111334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayflick L. 1975. Cell biology of aging. Bioscience 25, 629-637. ( 10.2307/1297030) [DOI] [Google Scholar]
- 47.Shay JW, Wright WF. 2000. Hayflick, his limit and cellular ageing. Nat. Rev. Mol. Cell Biol. 1, 72-76. ( 10.1038/35036093) [DOI] [PubMed] [Google Scholar]
- 48.Balin AK, Fisher AJ, Anzelone M, Leong I, Allen RG. 2002. Effects of establishing cell cultures and cell culture conditions on the proliferative life span of human fibroblasts isolated from different tissues and donors of different ages. Exp. Cell Res. 274, 275-287. ( 10.1006/excr.2002.5485) [DOI] [PubMed] [Google Scholar]
- 49.Shay JW, Wright WF. 2019. Telomeres and telomerase: three decades of progress. Nat. Rev. Genet. 20, 299-309. ( 10.1038/s41576-019-0099-1) [DOI] [PubMed] [Google Scholar]
- 50.Schmutz I, de Lange T. 2016. Shelterin. Curr. Biol. 26, R397. ( 10.1016/j.cub.2016.01.056) [DOI] [PubMed] [Google Scholar]
- 51.Aubert G, Lansdorp PM. 2008. Telomeres and aging. Physiol. Rev. 88, 557-579. ( 10.1152/physrev.00026.2007) [DOI] [PubMed] [Google Scholar]
- 52.Donate LE, Blasco MA. 2011. Telomeres in cancer and ageing. Proc. R. Soc. B 366, 76-84. ( 10.1098/rstb.2010.0291) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu DH, et al. 2019. Lysine demethylase 2A promotes the progression of ovarian cancer by regulating the PI3 K pathway and reversing epithelial–mesenchymal transition. Oncol. Rep. 41, 917-927. ( 10.3892/or.2018.6888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Hannon GJ, Beach DH. 2000. Risky immortalization by telomerase. Nature 405, 755-756. ( 10.1038/35015674) [DOI] [PubMed] [Google Scholar]
- 55.Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. 2012. c-Myc and cancer metabolism. Clin. Cancer Res. 18, 5546-5553. ( 10.1158/1078-0432.CCR-12-0977) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huberman AD. 2020. Sight restored by turning back the epigenetic clock. Nature 588, 34-36. ( 10.1038/d41586-020-03119-1) [DOI] [PubMed] [Google Scholar]
- 57.Ehrlich M. 2009. DNA hypomethylation in cancer cells. Epigenomics 1, 239-259. ( 10.2217/epi.09.33) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding C, Schreiber SG, Roberts DR, Hamann A, Brouard JS. 2017. Post-glacial biogeography of trembling aspen inferred from habitat models and genetic variance in quantitative traits. Sci. Rep. 7, 4672. ( 10.1038/s41598-017-04871-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heidstra R, Sabatini S. 2014. Plant and animal stem cells: similar yet different. Nat. Rev. Mol. Cell Biol. 15, 301-312. ( 10.1038/nrm3790) [DOI] [PubMed] [Google Scholar]
- 60.Pillitteri LJ, Guo X, Dong J. 2016. Asymmetric cell division in plants: mechanisms of symmetry breaking and cell fate determination. Cell Mol. Life Sci. 73, 4213-4229. ( 10.1007/s00018-016-2290-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith GM. 1955a. Cryptogamic botany: algae and fungi, vol 1. New York, NY: McGraw-Hill. [Google Scholar]
- 62.Ivanov IN, Vítová M, Bišová K. 2019. Growth and the cell cycle in green algae dividing by multiple fission. Folia Microbiol. 64, 663-672. ( 10.1007/s12223-019-00741-z) [DOI] [PubMed] [Google Scholar]
- 63.Bišova K, Zachleder V. 2014. Cell-cycle regulation in green algae dividing by multiple fission. J. Exp. Bot. 65, 2585-2602. ( 10.1093/jxb/ert466) [DOI] [PubMed] [Google Scholar]
- 64.Smith GM. 1955b. Cryptogamic botany: bryophytes and pteridophytes, vol. 2. New York, NY: McGraw-Hill. [Google Scholar]
- 65.Steiner UK. 2021. Senescence in bacteria and its underlying mechanisms. Front. Cell Dev. Biol. 9, 688915. ( 10.3389/fcell.2021.668915) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ackermann M. 2008. Bacteria as a new model system for aging studies: investigations using light microscopy. Biotechniques 44, 564-567. ( 10.2144/000112829) [DOI] [PubMed] [Google Scholar]
- 67.Stewart EJ, Madden R, Paul G, Taddel F. 2005. Aging and death in an organism that reproduces by morphologically symmetric division. PLoS Biol. 3, e45. ( 10.1371/journal.pbio.0030045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lindner AB, Madden R, Demarez A, Stewart EJ, Tadei F. 2008. Assymmetric segregation of protein aggregates is associated with cellular aging and rejuvenation. Proc. Natl Acad. Sci. USA 105, 3076-3081. ( 10.1073/pnas.0708931105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lapinska U, Glover G, Capilla-Lasheras P, Young AJ, Pagliara S. 2019. Bacterial ageing in the absence of external stressors. Phil. Trans. R. Soc. B 374, 20180442. ( 10.1098/rstb.2018.0442) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Proenca AM, Rang CU, Qiu A, Shi C, Chao L. 2019. Cell aging preserves cellular immortality in the presence of lethal levels of damage. PLoS Biol. 17, e3000266. ( 10.1371/journal.pbio.3000266) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mortimer RK, Johnston JR. 1959. Life span of individual yeast cells. Nature 183, 1751-1752. ( 10.1038/1831751a0) [DOI] [PubMed] [Google Scholar]
- 72.Spokoini R, Moldavski O, Nahmias Y, England JL, Schuldiner M, Kaganovich D. 2015. Confinement of organelle-associated inclusion structures mediates asymmetric inheritance of aggregated protein in budding yeast. Cell Rep. 2, 738-747. ( 10.1016/j.celrep.2012.08.024) [DOI] [PubMed] [Google Scholar]
- 73.McFaline-Figueroa JR, Vevea J, Swayne TC, Zhou C, Liu C, Leung G, Boldogh IR, Pon LA. 2011. Mitochondrial quality control during inheritance is associated with lifespan and mother–daughter age asymmetry in budding yeast. Aging Cell 10, 885-895. ( 10.1111/j.1474-9726.2011.00731.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou C, et al. 2014. Organelle-based aggregation and retention of damaged proteins in asymmetrically dividing cells. Cell 159, 1-13. ( 10.1016/j.cell.2014.09.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Higuchi-Sanabria R, Pernice WMA, Vevea JD, Dana M, Wolken A, Boldogh IR, Pon LA. 2014. Role of asymmetric cell division in lifespan control in Saccharomyces cerevisiae. Yeast Res. 14, 1133-1145. ( 10.1111/1567-1364.12216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saarikangas J, Caudron F, Prasad R, Moreno DF, Bolognesi A, Aldea M, Barral Y. 2017. Compartmentalization of ER-bound chaperone confines protein deposit formation to the aging yeast cell. Curr. Biol. 27, 1-11. ( 10.1016/j.cub.2017.01.069) [DOI] [PubMed] [Google Scholar]
- 77.Coelho M, et al. 2013. Fission yeast does not age under favorable conditions, but does so after stress. Curr. Biol. 23, 1-9. ( 10.1016/j.cub.2013.07.084) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He C, Zhou C, Kennedy BK. 2018. The yeast replicative aging model. Biochem. Biophys. Acta 1864, 2690-2696. ( 10.1016/j.bbadis.2018.02.023) [DOI] [PubMed] [Google Scholar]
- 79.de Kretser DM, Loveland KL, Meinhardt A, Simorangkhir D, Wreford N. 1998. Spermatogenesis. Hum. Reprod. 13(Suppl. 1), 1-8. ( 10.1093/humrep/13.suppl_1.1) [DOI] [PubMed] [Google Scholar]
- 80.Wilson ZA, Zhang DB. 2009. From Arabidopsis to rice: pathways in pollen development. J. Exp. Bot. 60, 1479-1492. ( 10.1093/jxb/erp095) [DOI] [PubMed] [Google Scholar]
- 81.Bastock R, Johnston DS. 2008. Drosophila oogenesis. Curr. Biol. 18, R1082. ( 10.1016/j.cub.2008.09.011) [DOI] [PubMed] [Google Scholar]
- 82.Bufalino MR, DeVeale B, van der Kooy D. 2013. The asymmetric segregation of damaged proteins is stem cell-type dependent. J. Cell Biol. 201, 523-530. ( 10.1083/jcb.201207052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang WC, Shi DQ, Chen YH. 2010. Female gametophyte development in flowering plants. Annu. Rev. Plant Biol. 61, 89-108. ( 10.1146/annurev-arplant-042809-112203) [DOI] [PubMed] [Google Scholar]
- 84.Liberti MV, Locasale JW. 2016. The Warburg effect: how does it benefit cancer cell? Trends Biochem. Sci. 41, 211-218. ( 10.1016/j.tibs.2015.12.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144, 646-674. ( 10.1016/j.cell.2011.02.013) [DOI] [PubMed] [Google Scholar]
- 86.Reya T, Morrison SJ, Clarke MF, Weissman IL. 2001. Stem cells, cancer, and cancer stem cells. Nature 414, 105-111. ( 10.1038/35102167) [DOI] [PubMed] [Google Scholar]
- 87.Batile E, Clevers H. 2017. Cancer stem cells revisited. Nat. Med. 10, 1124-1134. ( 10.1038/nm.4409) [DOI] [PubMed] [Google Scholar]
- 88.Izumi H, Kaneko H. 2012. Evidence of asymmetric cell division and centrosome inheritance in human neuroblastoma cells. Proc. Natl Acad. Sci. USA 109, 18 048-18 053. ( 10.1073/pnas.1205525109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Desdin-Micó G, Mittelbrunn M. 2017. Role of exosomes in the protection of cellular homeostasis. Cell Adhes. Migr. 11, 127-134. ( 10.1080/19336918.2016.1251000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vagner T, Chin A, Mariscal J, Bannykh S, Engman DM, Di Vizio D. 2019. Protein composition reflects extracellular vesicle heterogeneity. Proteomics 19, 1800167. ( 10.1002/pmic.201800167) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Meckes DG, Raab-Traub N. 2011. Microvesicles and viral infection. J. Virol. 85, 12 844-12 854. ( 10.1128/JVI.05853-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wiking L, Stagsted J, Björk L, Nielsen JH. 2004. Milk fat globule size is affected by fat production in dairy cows. Int. Dairy J. 14, 909-913. ( 10.1016/j.idairyj.2004.03.005) [DOI] [Google Scholar]
- 93.Sheehan C, D'Souza-Schorey C. 2019. Tumor-derived extracellular vesicles: molecular parcels that enable regulation of the immune response in cancer. J. Cell Sci. 132, jcs235085. ( 10.1242/jcs.235085) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guenat D, Hermetet F, Prétet JL, Mougin C. 2017. Exosomes and other extracellular vesicles in HPV transmission and carcinogenesis. Viruses 9, v9080214. ( 10.3390/v9080211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takahashi A, et al. 2017. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 8, 15287. ( 10.1038/ncomms15287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. 2006. Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc. Natl Acad. Sci. USA 103, 11 172-11 177. ( 10.1073/pnas.0603838103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saman S, et al. 2012. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842-3849. ( 10.1074/jbc.M111.277061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Johnstone RM. 2005. Revisiting the road to the discovery of ectosomes. Blood Cells Mol. Dis. 14, 214-219. ( 10.1016/j.bcmd.2005.03.002) [DOI] [PubMed] [Google Scholar]
- 99.Minciacchia VR, Freeman MR, Di Vizio D. 2015. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 40, 41-51. ( 10.1016/j.semcdb.2015.02.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hoshino A, et al. 2020. Extracellular vesicle and particle biomarkers define multiple human cancers. Cell 182, 1044-1061. ( 10.1016/j.cell.2020.07.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kreger BT, Dougherty AL, Greene KS, Cerione RA, Antonyak MA. 2016. Microvesicle cargo and function changes upon induction of cellular transformation. J. Biol. Chem. 291, 19 774-19 785. ( 10.1074/jbc.M116.725705) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kanada M, Bachmann MH, Contag CH. 2016. Signaling by extracellular vesicles advances cancer hallmarks. Trends Cancer 2, 84-94. ( 10.1016/j.trecan.2015.12.005) [DOI] [PubMed] [Google Scholar]
- 103.Lin Y, Zhang C, Xiang P, Shen J, Sun W, Yu H. 2020. Exosomes derived from HeLa cells break down vascular integrity by triggering endoplasmic reticulum stress in endothelial cells. J. Extracell. Vesicles 9, 1722385. ( 10.1080/20013078.2020.1722385) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rang CU, Proenca A, Buetz C, Shi S. 2018. Minicells as a damage disposal mechanism in Escherischia coli. mSphere 3, e00428-18. ( 10.1128/mSphere.00428-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zeng X. 2007. Human embryonic stem cells: mechanisms to escape replicative senescence? Stem Cell Rev. 3, 270-279. ( 10.1007/s12015-007-9005-x) [DOI] [PubMed] [Google Scholar]
- 106.Prasad SM, et al. 2009. Continuous hypoxic culturing maintains activation of Notch and allows long-term propagation of human embryonic stem cells without spontaneous differentiation. Cell Prolif. 42, 63-74. ( 10.1111/j.1365-2184.2008.00571.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Keeley TP, Mann GE. 2019. Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiol. Rev. 161, 234. ( 10.1152/physrev.00041.2017) [DOI] [PubMed] [Google Scholar]
- 108.Kaneko KJ, DePamphilis ML. 2013. TEAD4 establishes the energy homeostasis essential for blastocoel formation. Development 140, 3680-3690. ( 10.1242/dev.093799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Han C, et al. 2016. Exosomes and their therapeutic potentials of stem cells. Stem Cells Int. 2016, 7653489. ( 10.1155/2016/7653489) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhu Q, et al. 2019. Embryonic stem cells-derived exosomes endowed with targeting properties as chemotherapeutics delivery vehicles for glioblastoma therapy. Adv. Sci. 6, 1801899. ( 10.1002/advs.201801899) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schoenhals M, Kassambara A, De Vos J, Hose D, Moreaux J, Klein B. 2009. Embryonic stem cell markers expression in cancers. Biochem. Biophys. Res. Commun. 383, 157-162. ( 10.1016/j.bbrc.2009.02.156) [DOI] [PubMed] [Google Scholar]
- 112.Kim J, Orkin SH. 2011. Embryonic stem cell-specific signatures in cancer: insights into genomic regulatory networks and implications for medicine. Genome Med. 3, 75. ( 10.1186/gm291) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mathieu J, et al. 2011. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 71, 4640-4652. ( 10.1158/0008-5472.CAN-10-3320) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pezzuto A, Carico E. 2018. Role of HIF-1 in cancer progression: novel insights. A review. Curr. Mol. Med. 6, 343-351. ( 10.2174/1566524018666181109121849) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.