

Abstract
We tested the combination of a broadly neutralizing HIV antibody with the latency reversal agent vorinostat (VOR). Eight participants received 2 month-long cycles of VRC07-523LS with VOR. Low-level viremia, resting CD4+ T-cell–associated HIV RNA (rca-RNA) was measured, and intact proviral DNA assay (IPDA) and quantitative viral outgrowth assay (QVOA) were performed at baseline and posttreatment. In 3 participants, IPDA and QVOA declines were accompanied by significant declines of rca-RNA. However, no IPDA or QVOA declines clearly exceeded assay variance or natural decay. Increased resistance to VRC07-523LS was not observed. This combination therapy did not reduce viremia or the HIV reservoir.
Clinical Trials Registration. NCT03803605.
Keywords: HIV latency, broadly neutralizing antibody, latency reversal
Histone deacetylases (HDAC) contribute to human immunodeficiency virus (HIV) persistence [1], and HDAC inhibitors can reverse HIV latency in vivo at the level of cell-associated HIV RNA expression [2–4]. Vorinostat (VOR) induces HIV expression sufficiently to allow viral clearance in vitro [5], and repeatedly engages host biomarkers with latency reversal in vivo [3, 6]. We measured the effect of administering a broadly neutralizing antibody with VOR to clear reactivated cells and decrease the frequency of latent infection.
A clonal relative of the human monoclonal antibody (mAb) VRC01, the sequence of VRC07-523LS is engineered to increase potency, breadth, and plasma half-life [7]. In prior studies a 40mg/kg infusion of VRC07-523LS maintained serum concentrations >10 µg/mL for over a month, a level inhibitory for a broad range of HIV species [7]. We assessed the safety of this combination, and its ability to reduce persistent HIV infection.
METHODS
This study (NCT03803605) was approved by the University of North Carolina Biomedical Institutional Review Board and registered at Clinicaltrials.gov. Informed consent was obtained from all participants and lymphocytes were obtained by leukapheresis. Resting CD4+ (rCD4+) T cells were isolated, and replication-competent virus measured by quantitative viral outgrowth assay (QVOA) as described [2]. Intact proviral DNA assay (IPDA) was performed with minor modifications [8].
Changes induced in rCD4+ T-cell–associated RNA (rca-RNA) in vivo in response to VOR and VRC07-523LS were measured [2] with minor modifications. Total RNA was isolated from 36 replicates of 1 million resting cells using the MagJet RNA Kit (Thermo Fisher Scientific). For HIV-1 gag RNA quantification, triplicate polymerase chain reaction (PCR) amplifications were performed using TaqMan Fast Virus 1-Step Master Mix, and published HIV-1 gag primers and probe [9]. For TBP RNA quantification, duplicate PCR amplifications were performed [10]. A standard curve was generated for each reaction using gag and TBP gBlocks (IDT). Low-level viremia was measured from ultracentrifuged plasma as described [2]. HIV gag RNA copy numbers were assessed using a quantitative PCR (qPCR) assay [11].
Latent reservoir viruses (LRVs) were isolated from QVOA supernatants. Supernatants from multiple wells with virus were pooled, generating a swarm representative of the latent reservoir. Primary human CD4+ T cells were isolated from cryopreserved peripheral blood mononuclear cells (Miltenyi Biotech) and infected as described [12], and 1.5 × 106 cells were infected using 0.25–1mL virus supernatant by spinoculation (1125g) for 2 hours at 20°C. After spinoculation, 2mL of R20 supplemented with interleukin 2 (IL-2) was added to each infection and left for 72 hours. Cells were plated in a 12-well plate at the concentration of 0.5 × 106 cells/mL in R20 plus 30 U/mL of IL-2.
For binding assays, 2 × 105 cells/well were incubated with 10 μg/mL mAb for 2 hours at 37°C followed by surface staining with anti-CD4-PerCP-Cy5.5 for 20 minutes at room temperature. Cells were resuspended in 100 μL/well Cytofix/Cytoperm, incubated for 20 minutes at 4°C, followed by staining with anti-p24 antibody and secondary fluorescein isothiocyanate (FITC)-conjugated antibody (goat anti-human IgG(H+L)-FITC, KPL) for 25 minutes at 4°C. Cells were washed and resuspended in 125 μL phosphate-buffered saline-1% paraformaldehyde. Samples were acquired within 24 hours using a BD Fortessa cytometer.
Infected cell elimination assays were performed as described [12] with minor modifications. Target/effector cell suspension was plated in V-bottom 96-well plates and cocultured with VRC07-523LS starting at 100 µg/mL with 6 subsequent dilutions at 1:5. Specific killing was determined by reduction in percent of p24-positive cells in the presence of mAbs after considering nonspecific killing, and calculated as:
The antibody mix (A32, 7B2, 2G12, and CH44 HIV Env Abs) was used as a positive control [12]. Antibody-dependent cellular cytotoxicity (ADCC) end point concentration (EC), defined as the lowest mAb concentration capable of mediating ADCC in our in vitro assay, was calculated by interpolation of the mAb concentration that intersected the cutoff of 15% specific killing. Statistical significance was calculated using Wilcoxon signed rank test.
Illumina MiSeq library preparation was performed as described [13]. Viral RNA was extracted from LRVs pools, cDNA synthesized using the env V3/V5 Primer ID primer (HXB2 positions 6976 to 7650). Sequences were sequentially processed using Illumina bcl2fastq and template consensus sequence pipeline software version 2.3.2 (https://primer-id.org/tcs) to correct sequencing errors and avoid resampling. Paired ends were joined using a consensus end joining script and phylogenetic trees constructed using Muscle version 3.8.31 (http://www.drive5.com/muscle) and rooted to HXB2.
RESULTS
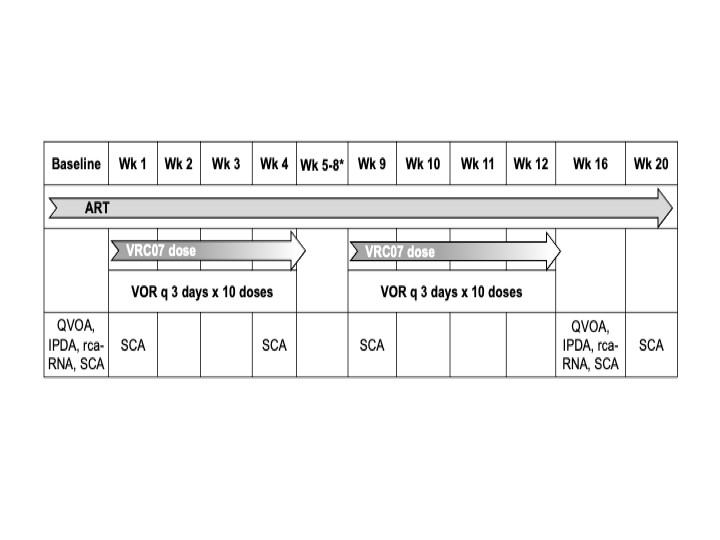
Fifteen antiretroviral therapy (ART)-treated, aviremic people with HIV enrolled. Entry criterion included baseline frequency of rCD4+ T-cell infection (RCI) >0.30 per million cells to allow a measurement of significant decline, given QVOA assay characteristics [14]. Seven of 15 participants did not receive study treatment as baseline RCI was <0.30 per million cells. Therefore, 6 men and 2 women, suppressed (<50 copies/mL) for at least 2.8 years (Supplementary Table 1), received VOR and VRC07-523LS (Supplementary Figure 1). In 5 of 8 participants with sufficient cells available, ex vivo exposure [2] to VOR resulted in a significant increase in rca-RNA in rCD4+ T cells (Supplementary Table 2). The day after VRC07-523LS 40mg/kg infusion, participants initiated VOR, 400mg orally every 72 hours for 30 days. ART was maintained. After a 4 to 8-week pause, participants received a second cycle of VRC07-523LS and VOR. The second cycle for 1 participant (V7-008) started 5 months after the first due to the coronavirus disease 2019 (COVID-19) pandemic.
VRC07-523LS and VOR were well tolerated. Grade 1 adverse events possibly associated with VOR (nausea, diarrhea, pruritis in 1 participant each) or VRC07-523LS (diaphoresis, arthralgia in 1 each, fatigue in 2) all resolved without treatment. Five participants had transient grade 3 hypertension at the time of leukapheresis, resolving without intervention.
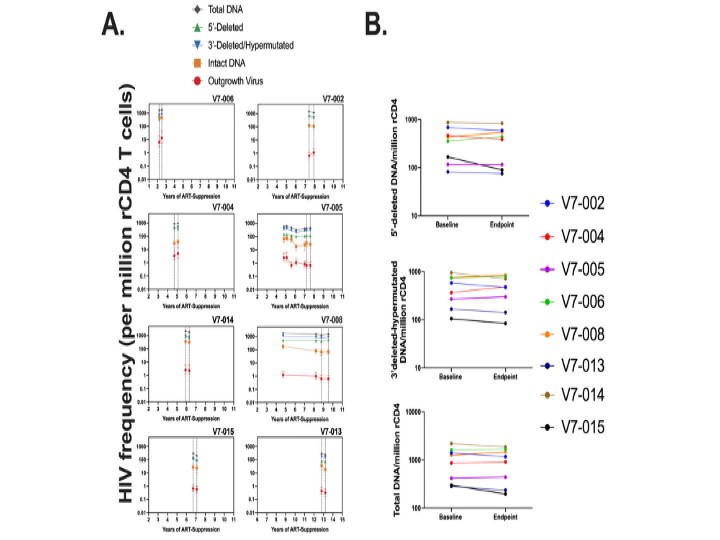
HIV gag rca-HIV RNA was measured at baseline and the end of study. As a biomarker of VOR effect [6], we validated serial host gene induction (Supplementary Figure 2). We observed quantitative increases in rca-RNA after the second cycle in 3 participants (V7-005, -006, and -008), consistent with serial VOR induction of rca-RNA expression. Four participants (V7-002, -013, -014, -015) showed a significant decrease in HIV gag rca-HIV RNA following the second cycle (Figure 1A).
Figure 1.

Impact of VOR and VRC07-523LS on HIV reservoir measurements. A, Changes in resting CD4 cell-associated HIV-1 gag RNA from baseline to week 16 after 2 cycles of VOR/VRC07-523LS. Declines (red) and increases (blue) were significant P<.0001 by Wilcoxon 2-sample test. At each time point 24–36 replicates of 1 × 106 rCD4+ T cells were evaluated. Mean and SD are shown. B, Linear scale frequency of replication-competent HIV in rCD4 T cells measured by quantitative viral outgrowth assay (red) and intact HIV DNA by IPDA (orange) before and after the study intervention. Dotted lines highlight the study period. Errors bars for QVOA represent the 95% CIs from the maximum likelihood method. Errors bars for IPDA measurements represent 95% CIs for the proviral frequency estimates using the total error from the QuantaSoft analysis software. C and D, Log scale changes in frequency of (C) replication-competent HIV by QVOA and (D) intact HIV DNA by IPDA. Abbreviations: CI, confidence interval; HIV, human immunodeficiency virus; IPDA, intact proviral DNA assay; QVOA, quantitative viral outgrowth assay; rCD4 T cell, resting CD4 T cell; VOR, vorinostat.
We also measured frequency of RCI by QVOA, and frequency of intact proviral genomes in rCD4+ T cells by IPDA (Figure 1B–1D). As the QVOA assay is robust and reproducible, declines of more than 50% on serial measures are infrequently seen and would be suggestive evidence for reservoir depletion [14]. While there was a numerical decline in the point estimate of IUPM in 3 participants (V7-013, -014, -015) who displayed a significant decrease in HIV gag rca-HIV RNA, no QVOA reductions exceeded the 50% threshold.
These samples were assayed in parallel by IPDA. We observed similar numerical reductions in intact proviruses (Figure 1B and 1D and Supplementary Figure 3) in 3 participants (V7-013, -014, -015) with parallel declines in rca-RNA and QVOA. IPDA reductions were also seen in 3 other participants (V7-002, -005, -006). Only 1 participant had a 50% decline of IPDA (intact HIV DNA per million rCD4+ T cells median −11, mean −20, range +9 to −78; percent decline in intact HIV DNA per million rCD4+ T cells median −14%, mean −11%, range +31% to −49%). No significant change was seen in total HIV DNA or nonintact proviral frequencies by IPDA (Supplementary Figure 3).
Low level viremia (LLV) has been linked to viral sequences recovered from the rCD4+ T cell reservoir. We measured LLV by SCA twice at baseline, twice during study interventions, and twice posttreatment (Supplementary Figure 1). We predicted that LLV could be dampened by activity of a broadly neutralizing antibody on cells producing HIV. Exposure to VRC07-523LS [7] 4 weeks after dosing appeared adequate (Supplementary Table 3). LLV was barely detectable in 7 of 8 participants, with no reduction in LLV during or after VRC07-523LS administration (Supplementary Table 4).
Insufficient RNA was available for sequence analysis in 7 participants with profound viral suppression and 1 participant (V7-006) with persistent LLV (25 to 36 copies/mL [c/mL] at 5 time points, median=32 c/mL). MiSeq deep sequencing with Primer ID [13] sequenced 2 regions of env associated with resistance to VRC01 (loop D and V5 [15]). As sequencing was performed on LRVs generated by pooling supernatants from QVOA wells containing multiple LRVs, we could not accurately estimate the frequency of LRV variants. However, sequences were genetically well mixed (Supplementary Figure 4), suggesting little change in diversity between the 2 time points. One potential exception was a viral lineage with an observed frequency of 18% at baseline that was absent at the end of study. The limited number of LRVs acquired prevented determination of the cause of the apparent loss of this lineage, due to inadequate sampling.
To confirm HIV Env epitopes expressed by reactivated latently infected cells could be recognized by VRC07-523LS, we performed an infected cells antibody binding assay [12] in 7 participants (V7-014 isolates were not assessable). VRC07-523LS recognized cells infected with LRVs obtained prior to infusions (baseline; Supplementary Figure 1) with the median frequency of 34.8% (range, 23%–50%) infected cells bound by VRC07-523LS at entry, and median 25.8% (range 11%–51.7%; Figure 2A) bound at the end of study. Median fluorescence intensity at baseline (514, range 300–1200) was unchanged at the end of study (476, range 148–1048; Figure 2B).
Figure 2.

VRC07-523LS binding to and elimination of LRV-infected cells. A, The frequency of infected cells with antibody bound to their surface (% p24+Ab+) and (B) MFI. CH65 antibody (Supplementary Reference 1) was used as negative control. C and D, End point concentrations (μg/mL), the concentration at which at least 15% target cell killing occurs. The data represent a single experiment with mAb starting with 100 µg/mL and subsequent 1:5 dilutions tested in duplicate. Statistical significance was calculated using Wilcoxon signed rank test. Horizontal lines indicate mean. Abbreviations: EC, end point concentration; LRV, latent reservoir virus; mAb, monoclonal antibody; MFI, mean fluorescence intensity; NS, not significant.
We assessed the sensitivity of persistent viruses to ADCC mediated by VRC07-523LS using an infected cell elimination assay. At the highest concentration of antibody tested (100 µg/mL), median LRV killing mediated by VRC07-523LS at baseline was 8% (range, 0%–28%) and median killing at the end of study was 16% (range, 0%–36%). The observed changes in ADCC susceptibility of LRVs from baseline to the end of study were not statistically significant (Figure 2C).
We also determined the lowest concentration of VRC07-523LS at which at least 15% specific ADCC was observed (EC). The median EC of VRC07-523 against baseline LRVs was 78 µg/mL, and median EC against end point LRVs was 17 µg/mL; 4 LRVs displayed increased susceptibility to VRC07-523LS at end point. No reductions in sensitivity to VRC07-523LS were seen (Figure 2D). Finally, standard neutralization assays (Supplementary Reference 2) showed no significant change in neutralization over time (Supplementary Table 5).
DISCUSSION
This study combined a latency reversing agent with a potent broadly neutralizing antibody. We hypothesized that VOR would stimulate expression of persistent proviral HIV from rCD4+ T cells, and VRC07-523LS might clear persistent infection while viral replication was inhibited by uninterrupted ART. We administered VOR every 72 hours, as this interval was shown to repeatedly induce rca-RNA expression without significant clinical toxicity or immune dysfunction [3].
VRC07-523LS infusions combined with VOR was safe and well tolerated, with only grade I treatment-related adverse events. Unfortunately, the combined treatment had no significant impact on the replication-competent reservoir as measured by QVOA. We observed a significant decline of rca-RNA in 4 of 8 participants, which could reflect depletion of cells producing full-length HIV RNA, a substantial proportion of which might not have been replication competent and therefore not result in a measurable decline in QVOA or IPDA. As the frequency of cells expressing HIV RNA is much larger than that of cells producing replication-competent HIV, a significant but modest decline of rca-RNA may not be measurable as a significant decline of RCI. Alternatively, as the rca-RNA assay has not been extensively characterized in longitudinal studies, this change could reflect slow natural decay of persistent infection [14]. Notably, all 4 participants with a significant decline of rca-RNA (V7-002, -013, -014, -015) also displayed a decline of IPDA, and 3 (V7-013, -014, -015) displayed a decline in QVOA.
We were surprised to see no effect of treatment on LLV in V7-006, with between 25 and 36 c/mL at all time points without evidence of viral resistance to VRC07-523LS. This raises the concern that, by itself, ADCC may be too inefficient to target and eliminate small populations of persistently infected cells, particularly with transient expression.
In summary, serial administration of VRC07-523LS and VOR was well tolerated but did not deplete markers of persistent infection. There are several limitations to this work, including the small sample size and lack of a control group. However, even if the modest trends towards latency reversal and clearance seen in rca-RNA, IPDA, and QVOA in 3 of 8 participants were validated in a larger, controlled study, we would still conclude that achieving an HIV cure requires more effective latency reversal coupled with efficacious immune interventions.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
Acknowledgments. We thank D. J. Hazuda and Merck Laboratories for the provision of vorinostat. We thank the Vaccine Production Program, Vaccine Immunology Program, Clinical Trials Program, and Office of Regulatory Science of the Vaccien Research Center and the Vaccine Clinical Materials Program/Leidos Biomedical Research for providing scientific support and in-vestigational Good Manufacturing Practice product (VRC07-523LS). We are grateful for the expert procedural care provided by Y. Park and the staff of the UNC Blood Bank, and for clinical support from the staff of the UNC Clinical and Translational Research Center. We thank R. Bosch of the Harvard Statistical Data Analysis Center for helpful discussions. Finally, we are grateful for the dedication of the participants who made this study possible.
Author contributions. C. L. G., M. T., S. D. F., S. B. J., J. D. K., G. F., J. J. E., N. M. A., and D. M. M. conceived and designed the studies. L. G., A. B. M., R. A. K., and J. R. M. provided VRC07-523LS and assays. M. T., S. B. J., K. S. J., S. D. F., S. L. M. R., M. C., M. J. M., B. A., J. L. K., M. C., R. J. G., M. F. M., J. D. K., J. J. E., and D. M. M. carried out the experiments and clinical study. K. S. J., C. L. G., S. D. F., N. M. A., and D. M. M. wrote the manuscript with input from all authors.
Disclaimer . The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of any trade names, commercial products, or organizations imply endorsement by the US government.
Financial support. This work was supported by National Institutes of Health (grant numbers U01 AI 117844 and UM1 AI 126619 to D. M. M., UL1TR002489 to the University of North Carolina (UNC) Translational and Clinical Sciences Institute (TRaCS) Institute, F30 AI 145588 to S. D. F. and AI50410 to the UNC Center for AIDS Research); and in part with federal funds from the National Cancer Institute, National Institutes of Health (contract numbers HSN261200800001E and 75N91019D00024).
Potential conflicts of interest. J. J. E. reports grants from Merck during the conduct of the study, personal fees from Merck, and personal fees from Gilead outside the submitted work. D. M. M. reports personal fees from Merck, and holds common stock in Gilead. C. L. G. reports research grants from Viiv and Gilead during the conduct of this study. All other authors report no potential conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Presented in part: 11th International AIDS Society Conference on HIV Science, Berlin, 18–21 July 2021.
References
- 1. Turner AW, Margolis DM.. Chromatin regulation and the histone code in HIV latency. Yale J Biol Med 2017; 90:229–43. [PMC free article] [PubMed] [Google Scholar]
- 2. Archin NM, Liberty AL, Kashuba AD, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012; 487:482–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Archin NM, Kirchherr JL, Sung JA, et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J Clin Invest 2017; 127:3126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elliott JH, Wightman F, Solomon A, et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog 2014; 10:e1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu G, Swanson M, Talla A, et al. HDAC inhibition induces HIV-1 protein and enables immune-based clearance following latency reversal. JCI Insight 2017; 2:e92901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maxwell JW, Falcinelli SD, Nefedov A, et al. Cellular gene modulation of HIV-infected CD4 T cells in response to serial treatment with the histone deacetylase inhibitor vorinostat. J Virol 2020; 94:e00351–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gaudinski MR, Houser KV, Doria-Rose NA, et al. ; VRC 605 Study Team. Safety and pharmacokinetics of broadly neutralising human monoclonal antibody VRC07-523LS in healthy adults: a phase 1 dose-escalation clinical trial. Lancet HIV 2019; 6:e667–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bruner KM, Wang Z, Simonetti FR, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019; 566:120–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ceriani C, Streeter GS, Lemu KJ, et al. Defining stable reference genes in HIV latency reversal experiments. J Virol 2021; 95:e02305-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Falcinelli SD, Shook-Sa BE, Dewey MG, et al. Impact of biological sex on immune activation and frequency of the latent HIV reservoir during suppressive antiretroviral therapy. J Infect Dis 2020; 222:1843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Somsouk M, Dunham RM, Cohen M, et al. The immunologic effects of mesalamine in treated HIV-infected individuals with incomplete CD4+ T cell recovery: a randomized crossover trial. PLoS One 2014; 9:e116306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tuyishime M, Garrido C, Jha S, et al. Improved killing of HIV-infected cells using three neutralizing and non-neutralizing antibodies. J Clin Invest 2020; 130:5157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou S, Jones C, Mieczkowski P, Swanstrom R.. Primer ID validates template sampling depth and greatly reduces the error rate of next-generation sequencing of HIV-1 genomic RNA populations. J Virol 2015; 89:8540–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crooks AM, Bateson R, Cope AB, et al. Precise quantitation of the latent HIV-1 reservoir: implications for eradication strategies. J Infect Dis 2015; 212:1361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lynch RM, Wong P, Tran L, et al. HIV-1 fitness cost associated with escape from the VRC01 class of CD4 binding site neutralizing antibodies. J Virol 2015; 89:4201–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.