

Abstract
Dominant dynamin 2 (DNM2) mutations are responsible for the autosomal dominant centronuclear myopathy (AD-CNM), a rare progressive neuromuscular disorder ranging from severe neonatal to mild adult forms. We previously demonstrated that mutant-specific RNA interference is an efficient therapeutic strategy to rescue the muscle phenotype at the onset of the symptoms in the AD-CNM knockin-Dnm2R465W/+ mouse model. Our objective was to evaluate the long-term benefit of the treatment along with the disease time course. We demonstrate here that the complete rescue of the muscle phenotype is maintained for at least 1 year after a single injection of adeno-associated virus expressing the mutant-specific short hairpin RNA (shRNA). This was achieved by a maintained reduction of the mutant Dnm2 transcript. Moreover, this long-term study uncovers a pathological accumulation of DNM2 protein occurring with age in the mouse model and prevented by the treatment. Conversely, a physiological DNM2 protein decrease with age was observed in muscles from wild-type mice. Therefore, this study highlights a new potential pathophysiological mechanism linked to mutant protein accumulation and underlines the importance of DNM2 protein expression level for proper muscle function. Overall, these results strengthen the allele-specific silencing approach as a robust, safe, and efficient therapy for AD-CNM.
Keywords: allele-specific silencing, gene therapy, dominant centronuclear myopathy, dynamin 2, RNA interference, in vivo long-term benefits
Graphical abstract
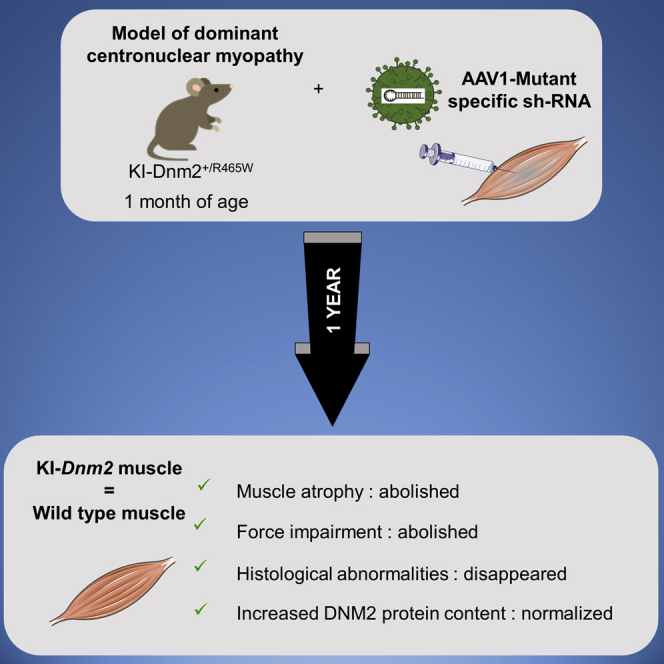
A single intramuscular injection of an adeno-associated virus expressing a mutant-specific shRNA induces a long-term protection of the muscle structure and function and prevents the DNM2 accumulation occurring with age in the knockin-Dnm2R465W/+ mouse model of autosomal dominant centronuclear myopathy.
Introduction
Autosomal dominant centronuclear myopathy (AD-CNM) (MIM: 160150) is a rare congenital myopathy characterized by progressive muscle weakness, mainly affecting limb and facial muscles. Age of onset and severity of symptoms are variable, ranging from severe-neonatal to mild-adult forms.1 Histological hallmarks in muscle biopsies consist of nuclear centralization in absence of regenerative process associated with atrophy and predominance of type 1 fibers and radial arrangement of sarcoplasmic strands radiating from the central nuclei.1 AD-CNM results from heterozygous mutations in the DNM2 gene, which encodes dynamin 2 (DNM2),2,3 and to date, 37 mutations (mainly missense) have been identified.2,4, 5, 6, 7, 8, 9, 10, 11 Dominant DNM2 mutations also cause rare cases of Charcot-Marie-Tooth peripheral neuropathy (CMT) (MIM: 606482)12 and hereditary spastic paraplegia.13 DNM2 belongs to the superfamily of large guanosine triphosphatases (GTPases) and is involved in endocytosis and intracellular vesicle trafficking through its role in deformation of biological membranes by oligomerization at the neck of membranes invagination, leading to the release of vesicles.14,15 Furthermore, several studies have highlighted the role of DNM2 as a regulator of actin and microtubule cytoskeletons.15,16
DNM2 mutations in AD-CNM are thought to lead to a gain of function and/or a dominant negative effect through the potential formation of abnormal stable DNM2 oligomers associated with mis-regulated GTPase activity.17,18 In addition, absence of haploinsufficiency in AD-CNM is supported by data from patients and absence of phenotype developed by heterozygous knockout mice expressing 50% Dnm2.19,20 Consequently, AD-CNM fulfills all the criteria for the development of allele-specific silencing therapy by RNA interference (RNAi) devoted to silence the mutated mRNA without affecting the normal transcript.21 Indeed, we have established the proof of concept of allele-specific RNA-silencing therapy in a knockin mouse model of DNM2-linked CNM (KI-Dnm2R465W/+) and patient-derived cells.22 The KI-Dnm2R465W/+ mouse model (thereafter called KI-Dnm2R465W) harbors the most frequent DNM2-CNM mutation (30% of AD-CNM patients) and progressively develops features of the human disease, including impairment of force generation, muscle atrophy, and morphological abnormalities of the muscle fibers.23 We showed that adeno-associated virus (AAV)-mediated allele-specific RNAi targeting the R465W mutant allele is able to completely prevent this muscle phenotype in young mice while partially restoring muscle function in older mice, probably due to a viral transduction defect.22 In the present study, we aim at establishing the long-term benefit of mutant-specific silencing after a single AAV-short hairpin RNA (shRNA) injection in young KI-Dnm2R465W mice and to improve the treatment efficacy in older mice.
Results
Therapeutic benefit on muscle phenotype is maintained long term
To assess the long-term maintenance of phenotype restoration, a single intramuscular injection of AAV1 expressing or not the R465W-mutant-specific shRNA (i.e., AAV1-sh9 and AAV1-control) was performed in tibialis anterior muscle (TA) of the KI-Dnm2R465W mice at 1 month of age (1011 viral genomes [vgs]/TA), and mice were sacrificed after 1 year. Heterozygous (HTZ) KI-Dnm2R465W mice received the therapeutic AAV1-sh9 in the right TA and AAV1-control in the contralateral muscle. The wild-type (WT) mice received the AAV1-control in the left muscle and PBS in the contralateral muscle.
An 18% reduction in muscle mass was observed in untreated HTZ muscle, while a single injection of AAV-sh9 restored muscle mass close to WT values (Figure 1A). Measurement of fiber diameter performed on transversal TA sections immunolabeled with Laminin antibody showed a reduced mean fiber size in HTZ muscle compared with WT, which was corrected in the treated muscle (Figure 1B). In addition, fiber size distribution in the treated muscle was similar to the WT with the exception of persistence of medium-sized fibers and a slightly lower number of large fibers (Figures 1C and S1). Muscle histopathology was studied on transversal TA sections stained with hematoxylin and eosin (H&E), succinate dehydrogenase (SDH), and reduced diphosphopyridine nucleotide diaphorase (DPNH) (Figures 1D and S2). The disease-specific histopathological abnormalities, i.e., central accumulation of oxidative cell compartments within fibers, were observed in 18% of fibers in HTZ control muscles and absent in AAV1-sh9-treated muscles (Figure 1E, shown by asterisks in Figure 1D). Moreover, global DPNH and SDH staining in entire muscle section showed an increased oxidative staining in the posterior region of the untreated HTZ muscles compared with WT, which was also abolished in AAV1-sh9-treated muscles (Figure S2). Absence of myofiber with central nuclei in H&E confirmed that the AAV-sh9 was not toxic and did not induce muscle degeneration (Figure 1D, lower panel). Transient activation of the ubiquitin-proteasome and autophagy pathways was previously demonstrated in the heterozygous KI model at 2 months of age.23 Therefore, we assessed the expression of Gabarapl1, p62, Atg 4, Murf1, and atrogin 1 in the TA muscle of the 13-month-old mice by qRT-PCR. With the exception of p62 expression, which slightly decreased in HTZ mice, we found similar expression of all genes in untreated WT and HTZ mice, whereas Murf1 and Gabarapl1 expression decreased in heterozygous treated mice (Figure S3). We have recently reported a reduction of the satellite cell content in muscle of KI-Dnm2R465W mice associated with a decreased expression of Pax7.24,25 Therefore, we analyzed Pax7 expression in TA muscles from WT, HTZ-control, and HTZ-sh9-treated mice by quantitative real-time PCR and immunostaining. We confirmed a decrease of ∼60% in Pax7 expression in HTZ-untreated muscle compared with the WT by qPCR and Pax7 expression was not improved in the AAV1-sh9-treated HTZ mice (Figures 1F and S4). Finally, force measurements showed that absolute and specific forces were significantly reduced in TA muscle from untreated HTZ mice (−30% and −20%, respectively) and were restored to normal values in treated HTZ mice (Figures 1G and 1H). Overall, these results showed a long-term benefit of allele-specific silencing therapy since muscle mass, morphology, and function were maintained 1 year after a single injection of the therapeutic AAV.
Figure 1.

Muscle phenotypic rescue persists at least 1 year long
(A) Muscle mass relative to body weight in milligrams and grams from WT and AAV-shRNA-injected mice (HTZ n = 23; WT n = 22). (B) Histogram of the mean Feret diameter (WT n = 6; HTZ n = 8). (C) Combined frequencies of fiber size in TAs (WT n = 6; HTZ n = 8 of each condition). (D) Histochemical staining of TA sections from WT and AAV-injected HTZ mice. HE, hematoxylin and eosin staining; SDH, succinate dehydrogenase staining. Asterisks indicate fibers with abnormal central accumulations. Scale bars represent 50 μm. (E) Quantification of histological abnormality. Scatterplot represents individual percentages of histological abnormality from heterozygous control or treated mice (HTZ-ctl n = 6; HTZ treated n = 4). (F) Relative Pax7 mRNA expression in WT, HTZ control, and HTZ muscles injected with AAV-sh9. WT was used as the normalizer sample (WT n = 11; HTZ n = 11). (G and H) Absolute maximal force (G; g, grams) and specific maximal force (H) developed by TAs (in G and H, HTZ n = 23 and WT n = 22). ns, not significant. In scatterplots (A, B, and E–H), bars represent mean ± SEM. Statistical analysis was performed using a two-tailed Mann-Whitney U test. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 compared with WT.
Viral genome persists in HTZ muscle after a 1-year treatment
We measured persistence of AAV in HTZ and WT muscles after 1 year through quantification of vgs per nanogram of genomic DNA. A similar amount of vgs was found in HTZ muscles injected with AAV1-sh9 and AAV1-control (960 vgs/ng; Figure 2A), also supporting absence of toxicity of the long-term expression of the sh9 RNA and a similar persistence of both viruses in HTZ muscles. In contrast, AAV1-control values were almost 10-fold higher in WT muscle (∼8,800 vgs/ng; Figure 2A). We looked at potential immune response induced in HTZ muscle by long-term expression of shRNA through the potential induction of interferon response by semi-quantitative RT-PCR. Similar expression of two interferon-induced genes (Oas1 and Stat1) was observed in HTZ-control, HTZ-sh9, and WT muscles (Figure 2B), ruling out abnormal immune response in HTZ muscles. This result, along with absence of fibrosis and signs of muscle necrosis regeneration in H&E-stained muscle sections (Figure 1D), confirmed safety of long-term expression of the therapeutic molecule. In addition, given that vg amount 1 month after AAV injection was comparable between 1-month-old WT and HTZ mice (Figure S5), our results also suggested a partial loss of vg in the HTZ muscle with time independent of muscle regeneration but without significant impact on the therapeutic benefit at 1 year.
Figure 2.

Viral amount and expression of interferon-induced genes in AAV-injected mice
(A) Quantification of the viral genomes (vgs) per nanogram of genomic DNA in mice injected with AAV-sh9 or control. WT muscles injected with PBS were included as negative control (HTZ n = 11; WT-control n = 6; WT-PBS n = 5). (B) Quantification of expression of the interferon-induced genes Oas1 and Stat1 mRNA relative to Gapdh mRNA by RT-PCR in muscle from 13-month-old mice treated at 1 month of age. Left panel: Oas1 expression is shown. Right panel: Stat1 expression is shown (HTZ-control n = 11; HTZ-control n = 11; WT-PBS n = 11). Scatterplot bars represent mean ± SEM. Statistical analysis was performed using a two-tailed Mann-Whitney U test. ∗p < 0.05.
Specific silencing of mutated allele is maintained over time
Expression of Dnm2 transcript was quantified by semi-quantitative RT-PCR normalized with ATPase transcript. A significant decrease in the Dnm2 mRNA content was observed in sh9-expressing HTZ muscle compared with untreated HTZ and WT muscles (−20%; Figure 3A) and confirmed by qPCR (Figure S6A). Allele-specific silencing was checked using Dnm2 RT-PCR and EcoNI enzymatic digestion to discriminate mutated (undigested) from WT allele (digested; Figure 3B). A significant decrease of mutant/WT ratio was observed in treated muscle (−30%) due to specific silencing of the mutant transcript and unchanged expression of the WT Dnm2 mRNA (Figure 3B). A second RT-PCR assay using primers designed for specific amplification of either WT or mutant alleles confirmed these results (Figure S6B). The DNM2 protein expression was then quantified using western blot and densitometric analysis. In untreated HTZ muscles, a 2-fold increase in DNM2 expression was observed compared with WT muscles, while DNM2 expression was similar in WT and HTZ-treated muscle (Figure 3C). Altogether, these data demonstrated that allele-specific silencing was maintained 1 year after a single AAV1-sh9 intramuscular injection. These results also highlighted an age-related accumulation of the DNM2 protein without increase in mRNA expression in muscle of HTZ mice at 1 year of age.
Figure 3.

Specific silencing on mutant allele and protein is maintained over time
(A) Semi-quantitative Dnm2 and Atpase RT-PCR products and quantification of Dnm2 expression normalized to Atpase (HTZ n ≥ 10; WT n = 12). (B) EcoNI digestion profile of Dnm2 PCR products and quantification of the mutant/WT, mutant/Atpase, and WT/Atpase ratios (HTZ and WT n = 11). (C) DNM2 western blot and quantification of signal by densitometry. GAPDH was used as loading control (HTZ and WT n = 8). In scatterplots (A–C), the bars are mean values and error bars indicate SEM. ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.1 two-tailed using a Mann-Whitney U test comparison. HTZ, heterozygous; Mut, mutant; WT, wild type.
Dynamin 2 protein is down-regulated with age in normal mouse muscle
The increased DNM2 protein expression in HTZ muscles compared with WT muscles of 13-month-old mice (Figure 3C) suggested a change of DNM2 expression with age. To test this hypothesis, we investigated the DNM2 expression in TA muscles from WT and HTZ KI-Dnm2R465W mice at 1 month, 3 months, and 6 months of age using western blot (Figure 4A). In WT muscle, the DNM2 content remained stable between 1 and 3 months and was then decreased by half at 6 months of age. In HTZ muscle, the DNM2 protein content was higher at 1 month of age compared with WT and returned to amount comparable with WT in 3-month-old mice, in accordance with previous observation at this age.26 Then, between 3 and 6 months of age, the DNM2 protein content remained stable in HTZ muscle, leading to a 2-fold-higher protein content compared with WT at 6 months of age. Together, these data support a physiological decrease in DNM2 protein content in WT muscle from 3 to 6 months of age, which was impaired in presence of the R465W DNM2 mutation.
Figure 4.

DNM2 protein expression in wild-type mouse muscle and non-muscle tissues with aging
(A) DNM2 western blot of total protein extracted from tibialis anterior at 1, 3, and 6 months of age and quantification of signal by densitometry. (B) GAPDH was used as loading control (n ≥ 3). Semi-quantitative Dnm2 and Atpase RT-PCR of mRNA extracted from tibialis anterior muscle at 9 months and quantification of Dnm2 expression normalized to Atpase are shown (n ≥ 3). (C) DNM2 and GAPDH western blot and total protein stain free extracted from WT brain, liver, quadriceps, gastrocnemius, and heart at 1 and 9 months of age. The histograms show quantification of DNM2 protein signal by densitometry using total protein loading (stain free) as loading control (WT-1M n = 4; WT-9M n ≥ 3). SF, protein stain free. In scatterplots, bars represent mean ± SEM. Statistical analysis was performed using a two-tailed Mann-Whitney U test. ∗p < 0.05.
We then investigated whether this physiological decrease of DNM2 occurred at transcriptional level by quantifying Dnm2 mRNA in TA in 1- and 9-month-old mice using semi-quantitative RT-PCR. As shown in Figure 4B, the level of Dnm2 mRNA was similar in these muscles, regardless of the age and genotype. Therefore, as for the accumulation of mutant protein observed in mutant muscle (Figure 3C), the physiological decrease of DNM2 protein content in WT muscle results from a post-translational event, which is altered in HTZ mice.
We then wondered whether the physiological decrease in DNM2 protein content with aging in WT mice was muscle specific. We investigated the DNM2 expression in several muscles (quadriceps, gastrocnemius, and heart) and non-muscle tissues (liver and brain) from WT mice at 1 and 9 months of age using western blot (Figure 4C). DNM2 protein content was quantified by densitometry and normalized with total protein detected by Stain-Free. In WT brain and liver, no significant decrease in DNM2 protein expression was highlighted with age (Figure 4C; p = 0.82 and 0.41, respectively). In muscle tissues, while the significance threshold is not reached (p = 0.057 for all of them), we observed a decrease in DNM2 protein amount between 1 and 9 months with a more important decrease in quadriceps and gastrocnemius muscles than in heart.
DNM2 protein content was also compared between WT and HTZ mice at the age of 9 months (Figure S7). No difference was found between the two genotypes in brain and heart, while an increase in protein content was observed in quadriceps and gastrocnemius but below the significant threshold (p = 0.057). Altogether, these results supported a skeletal-muscle-specific physiological decrease of DNM2 protein with age in WT mice that would be impaired in HTZ mice.
Dynamin 2 expression is also down-regulated with age in healthy human muscle
We then investigated the relevance of age-related variation in DNM2 protein in human. To this end, we assessed DNM2 expression in muscle biopsies from healthy subjects and CNM patients at various ages (ranging from 1 month to 60 years old) by western blot (Figure 5A). For analysis, we grouped the control samples as younger or older than 20 years old (n = 6 for both groups; Figure 5B). We observed that DNM2 expression was ∼30% lower in the older group compared with the younger (p < 0.05). Moreover, when plotting individual DNM2 expression level as a function of age, we observed a negative correlation (r = −0.7; p < 0.05) in controls samples. Considering CNM samples, Spearman correlation was non-significant (r = 0.70; p = 0.17), and the number of patients was not sufficient to constitute a group of age; however, at matched age, we observed a level of DNM2 that tends to be higher in CNM patients (Figures 5C and S8).
Figure 5.

DNM2 protein expression in healthy human muscle and CNM muscle
(A) Representative DNM2 western blot of total protein extracted from human muscle biopsies. GAPDH and total protein loading were used as loading control. (B) Histograms showing quantification of DNM2 protein signal by densitometry using total protein loading (stain free) as loading control (<20 years n = 6; >20 years n = 6). (C) Individual plot of densitometry of bands corresponding to DNM2 normalized to total protein stain free as a function of age. Control patients are represented by black plots (n = 12) and CNM patients by red plots (n = 5). Individual plot represents mean ± SEM of one to four technical duplicates. Statistical analysis was performed using Spearman correlation (∗p < 0.05).
High dose of therapeutic AAV does not improve the transduction defect occurring in old mice
We previously highlighted a viral transduction defect in 6-month-old mice compared with 1-month-old HTZ mice explaining the lower efficacy of the treatment in old mice.22 Therefore, we wanted to explore the possibility to improve treatment efficacy in old mice by using a 10-fold increase in therapeutic AAV1-sh9 compared with the dose used in our previous study (1011 vgs/TA). A single intramuscular injection was performed in TA of HTZ mice at 6 months of age (1012 vgs/TA). HTZ mice received an AAV1-sh9 in the right TA and PBS in the contralateral muscle. The WT mice received only PBS in both TAs since such amount of virus in a WT mouse muscle leads to muscle regeneration. Mice were sacrificed after 3 months to perform molecular analysis and force measurements. We did not show significant variation in the total Dnm2 transcript in treated mice and only a slight reduction of the mutant transcript (Figure 6B). As already reported with a lower dose,22 a significant improvement of contractile properties occurred, but muscle mass was unchanged (Figure 6A). We then evaluated the vg amount in HTZ muscles injected with high dose of virus (1012 vgs/TA) in comparison with HTZ muscle samples injected with low dose of virus (1011 vgs/TA). We found 382 vgs/ng of DNA in the latter muscles versus 1,055 vgs/ng in muscle injected with the 10-fold higher dose (Figure 6C). A virus loss subsequent to muscle regeneration due to potential toxicity was ruled out by absence of central nuclei observed in H&E staining (Figure 6D). Overall, despite improvement of muscle contractile properties, a 10-fold increase of therapeutic AAV1-sh9 was unable to bypass the transduction defect in 6-month-old KI-Dnm2 mice.
Figure 6.

High dose of therapeutic AAV-sh9 does not improve the benefit in old mice
(A) Muscle mass relative to body weight in milligrams and grams, absolute maximal force in grams, and specific maximal force (in grams/mg) developed by TAs from WT and AAV-injected HTZ mice (WT n = 8, HTZ-control n = 7, and HTZ-sh9 n = 4). (B) Quantification of Dnm2 expression normalized to Atpase from semi-quantitative Dnm2 and Atpase RT-PCR (left) and quantification of the mutant/WT ratio from EcoNI digestion profile of Dnm2 PCR (right; WT ≥ 4, HTZ-Ctl n = 4, and HTZ-sh9 = 4). (C) Quantification of the vgs per nanogram of DNA in HTZ 6-month-old mice injected with AAV1-sh9 at low (n = 4) or high dose (n = 4). (D) Representative H&E staining of TA sections from WT and AAV-injected HTZ mice. Scale bars represent 50 μm. In histogram and scatterplots (A–C), bars represent mean ± SEM. Statistical analysis was performed using Mann-Whitney U test. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Discussion
High efficacy and specificity of RNAi to silence morbid genes have been at the origin of numerous clinical trials. These two properties of RNAi have been also used to specifically target the mutated allele without affecting the normal allele in dominant inherited diseases, even when the two sequences only differed by 1 nt. Proof of concept of allele-specific RNAi (AS-RNAi) was achieved for almost 20 diseases in patient-derived cells and/or animal models, including the DNM2-linked dominant centronuclear myopathy.21,22 Here, we addressed a key point for preclinical development of AAV-mediated therapy for AD-CNM, which is the long-term maintenance of therapeutic benefit in vivo.
The long-term expression of transgene is one of the advantage of AAV delivery, as transgene expression has been detected up to 10 years after single injection in non-dividing tissues.27 However, a decrease of transgene expression starting from 7 months after AAV injection in mice has been also reported.28 To the best of our knowledge, only one study addressed the long-term maintenance of AAV-mediated AS-RNAi therapy in vivo, showing a decrease in treatment benefit with time in a mouse model of cardiomyopathy through an undetermined mechanism,29 highlighting a potential limitation for the preclinical development of this therapy. Here, we report a rescue of muscle mass, force, and histological phenotypes developed by the heterozygous KI-Dnm2R465W mice23 1 year upon a single AAV1-sh9 injection performed at the beginning of symptoms, demonstrating the long-term maintenance of the therapeutic benefit previously demonstrated after a 3-month treatment.22 Specific reduction of Dnm2 mutant is therefore able to revert the impairment of the muscle contractile properties and prevent the development of muscle atrophy and histological features. In addition, we confirmed in TA the decreased expression of the Pax7 satellite cells marker previously reported in gastrocnemius muscle,24 which was not restored after treatment. The capability of AAV vector to efficiently transduce satellite cells is controversial, with studies supporting absence of transduction30 or efficient transduction using serotypes 8 and 9 or even with serotype 1, despite a lower efficacy.31 Therefore, the absence of Pax7 rescue might result from limited transduction of satellite cells with AAV1 vector that may be improved in the future by other serotypes. Nevertheless, our results show that the correction of Pax7 expression is not a prerequisite to prevent fiber atrophy.
Our study identified a 2-fold increase in DNM2 protein content in heterozygous TA from 6-month-old mice not seen at mRNA level, suggesting that this post-transcriptional regulation results from mutant protein accumulation and absence of degradation. Of note, an increase in DNM2 protein without increase in mRNA expression has been also reported in X-linked centronuclear myopathy (XLCNM) disease.19 DNM2 protein accumulation was not observed at younger age in this KI-Dnm2R465W model (in neonates and 2-month-old mice),23,26 while intracellular accumulations have been already observed by immunofluorescence in isolated HTZ muscle fiber.23 However, in the recent model of severe CNM KI-Dnm2S619L/+, a 2-fold increase in DNM2 protein level was also demonstrated at 2 months of age.32 In accordance with these observations, transgenic zebrafish models expressing the R465W and S619L mutants showed a protein mislocalization and aggregation that correlate with the severity of the CNM mutations.33 Finally, the propensity of several DNM2 mutants to form abnormally stable oligomers compared with WT has been already reported in vitro.32 Autophagy and the ubiquitin-proteasome system are the two major intracellular quality control and recycling mechanisms that are responsible for degradation of misfolded or aggregated proteins.34 Interestingly, autophagy was shown to be directly impacted by Dnm2 mutation.35,36 Therefore, the DNM2 oligomers and polymers formed by the DNM2 R465W mutant could be initially efficiently handled by intracellular quality control systems that would become overloaded and/or mis-functional with time, leading to the increase in DNM2 protein level. However, we confirmed that the abnormal activation of the ubiquitin-proteasome and autophagy genes observed at 2 months of age in heterozygous mice that normalizes at 8 months23 remains normalized at 13 months. The decrease in Murf1 and Gabarapl1 expression observed in treated heterozygous mice could be due to a direct regulation of these genes by DNM2.
Altogether, the propensity to aggregate shared by several Dnm2 CNM mutation (and not seen in CMT-related mutation),33 the correlation between the degree of aggregation and the severity of the disease32 and this study, and our observation of increase with age in parallel of the disease progression argue in favor of their contribution to pathogenesis. Along the same lines, it is noteworthy that overexpression of WT DNM2 by itself could trigger muscle defects,37,38 underlying the importance of a tiny DNM2 level regulation for proper muscle development, maturation, and maintenance.
Our study also led to identification of a physiological decrease of DNM2 protein content in healthy muscle with age, and this age-related decrease seems to be muscle specific. Our results argue for the relevance of this DNM2 decrease with age in human muscle from healthy controls, while the level of DNM2 expression in CNM patients remains in the range of young controls, regardless of their age. In the literature, few studies have investigated the localization or the level of DNM2 in patients. Normal expression of dynamin 2 mRNA and protein has been reported in fibroblasts from CNM patients.39, 40, 41 In addition, aggregates have been reported in one late-onset patient with a normal protein amount,9 and recently, an abnormal western blot profile was reported for a severe young CNM patient.32 Altogether, these data may suggest that the level of DNM2 required for proper muscle function would vary according to muscle maturation stage, and this process would be impaired more or less early according to the DNM2 mutation, leading to unbalanced WT/mutant protein ratio with age and reinforcing under this postulate the asset of a mutant-specific approach.
AAVs transgene expression results from a complex multistep process, including AAV entry by endocytosis, microtubule-dependent endosomal trafficking, endosome escape, nuclear import, capsid uncoating allowing single-strand DNA release, and double-strand conversion.42 Given the known functions of DNM2, several steps of AAV processing could be additively affected by DNM2 dysfunction. Our study shows an AAV transduction defect in the KI-Dnm2R465W/+ model that is probably already present in young mice. Indeed, the amount of vg detected after 1 year in mice injected at 1 month of age is lower in HTZ than in WT mice, while this amount is comparable after 1 month. This argues for a higher rate of vg loss with time in HTZ muscles that would be independent of regeneration. However, the benefit on contractile properties despite undetectable silencing of the mutated allele in 6-month-old mice supports a low therapeutic threshold and is encouraging to persevere in the improvement of AAV transduction in our model. Notably, insulin treatment,43 removal of capsid phosphorylation sites,44 and stimulation of autophagy45 have been reported to improve transduction efficiency. Otherwise, other serotypes or non-viral delivery can be considered alone or in combination with AAV treatment. In the latter case, the non-viral delivery could transiently improve the muscle state in order to enhance the transduction capabilities at the time of AAV injection.
To date there is no curative treatment available for AD-CNM patients, despite a treatment with acetylcholinesterase inhibitor reported to improve muscle strength in few DNM2-CNM patients.46 We have previously succeeded to correct the DNM2 mRNA by trans-splicing but with a low efficiency.47 Non-allele-specific Dnm2 RNAi (i.e., targeting both alleles of Dnm2) mediated by antisense oligonucleotides was also reported to efficiently improve the progression of the disease in two AD-CNM mouse models (i.e., KI-Dnm2R465W/+ and KI-Dnm2S619L/+).26,32 Unfortunately, the long-term benefits have not been reported using this approach. The proof of concept of AS-RNAi therapy in vitro and in vivo22 and our current demonstration of long-term efficacy in an animal model enlarge the therapeutic possibilities for the disease. In addition to efficiency and specificity provided by RNAi, AS-RNAi introduces a third property of safety, since 50% expression is preserved from the healthy untargeted allele. This may be of particular importance in a context of physiological decrease with age of the DNM2 expression.
In conclusion, the demonstration that time does not impair the treatment benefit or the level of mutant silencing associated with the prevention of protein accumulation and absence of toxicity confirms allele-specific RNAi mediated by AAV delivery as a strategy of choice to treat AD-CNM due to dynamin 2 mutations.
Materials and methods
Data analysis and statistics
Graphics and statistical analyses were performed with GraphPad Prism software v.9. Values were expressed as means ± SEM. The number of samples (n), representing the number of independent biological replicates, was indicated in the figure legends. For molecular analysis, the experiments were repeated at least three times per biological replicate and averaged. We used nonparametric statistical tests to analyze our data since the normality could not be assumed or tested or the variances were not equal between groups. Statistical comparisons between two groups were performed using unpaired two-tailed Mann-Whitney U test as specified. Spearman correlation analysis was used to evaluate the strength of relationship between DNM2 protein content and age. Statistical tests applied are indicated in the figure legends. p < 0.05 was considered as statistically significant. In most of the figures, the gels are cropped for more conciseness, but samples presented were run on a same gel.
Total RNA extraction and cDNA analysis
Total RNAs were isolated from muscle by TRIZOL reagent (Life Technologies, France) following standard protocol after disruption using Fastprep Lysing Matrix D and Fastprep apparatus (MP Biomedical, France). Total RNA (1 μg) was submitted to reverse transcription using the Superscript III reverse transcriptase kit (Life Technologies) and oligo-dT primers. The cDNAs were amplified by PCR under the following conditions: 96°C for 5 min, cycles of 30 s at 96°C, 30 s at the appropriate temperature (58°C–62°C), 30 s at 72°C, and a final step of 7 min at 72°C. Semi-quantitative RT-PCR was used to determine the total Dnm2 expression; the appropriate number of PCR cycles has been selected in order to have the amplification in the exponential range (i.e., 23 cycles to amplify Gapdh and 28 cycles for Dnm2). Sequences of the primers were indicated previously.22 To quantify allele-specific silencing, an assay was developed using EcoNI restriction enzyme, allowing discrimination between WT and mutant cDNAs. Dnm2 PCR products (393 bp) containing the mutation were EcoNI digested and run on agarose gel. EcoNI digests only RT-PCR product amplified from the WT, leading to two fragments (247 and 146 bp), allowing discrimination between WT and mutated alleles. The densitometric ratio between the non-digested band (mutated) and the sum of the signal for the two digested bands (WT) is done and compared with untreated conditions where this ratio is set to 1 to estimate the efficiency of the small interfering RNA (siRNA) on the mutated allele. The digestion of murine Dnm2 amplicons was performed after 32 cycles at the end of the exponential phase of amplification. For this assay, half of the PCR products were digested using 2U of EcoNI (New England Biolabs, France) for 2 h at 37°C. Image acquisition of PCR products after agarose gel electrophoresis was performed using a Geni2 gel imaging system (Ozyme, France), and associated signal was quantified using ImageJ Software (NIH; http://rsbweb.nih.gov/ij).
Semi-quantitative RT-PCR was also used to determine the total expression of two interferon-induced genes (Oas1 and Stat1) with appropriate number of PCR cycles selected in order to have the amplification in the exponential range (i.e., 34 cycles to amplify OasI and 36 cycles to amplify Stat1). Sequences of the primers were indicated previously.22 Pax7, Gabarapl1, p62 Atg4, Murf1, and Atrogin 1 mRNA quantification was performed by real-time PCR using a Lightcycler 480 II (Roche, Switzerland) and 1× SYBR Green (Roche, Switzerland) reactions using 0.5 μM of forward and reverse primers and 5 μL of cDNA diluted at 1:10 in nuclease-free water. PCR cycles were a 5-min, 95°C pre-incubation step followed by 45 cycles with a 95°C denaturation for 10 s, 58°C or 60°C annealing for 15 s, and 72°C extension for 15 s. Relative expression was calculated as mean values of 2−ΔΔCT normalized using the Atpase housekeeping gene. Primers sequences are Pax7 Fw 5ʹ-GAG TTC GAT TAG CCG AGT GC-3ʹ, Rv 5ʹ-GTG TTT GGC TTT CTT CTC GC-3ʹ; Atpase Fw 5ʹ-CCCCCTGTTCAGGTCTACGG-3ʹ, Rv 5ʹ-GACGGTGCGCTTGATGTAGG-3ʹ; Atrogin1 Fw 5ʹ-GCCTTCAAAGGCCTCACG-3ʹ, Rv 5ʹ-CTGAGCACATGCAGGTCTGGG-3ʹ; Murf1 Fw 5ʹ-CGACCGAGTGCAGACGATCATCTC-3ʹ, Rv 5ʹ-GTGTCAAACTTCTGACTCAGC-3ʹ; Gabarap11 Fw 5ʹ-CATCGTGGAGAAGGCTCCTA-3ʹ, Rv 5ʹ-ATACAGCTGGCCCATGGTAG-3ʹ; Atg4 Fw 5ʹ-ACAGATGATCTTTGCCCAGG-3ʹ, Rv 5ʹ-TAGACTTGCCTTCGCCAACT-3ʹ; and P62 Fw 5ʹ-GCTCAGGAGGAGACGATGAC-3ʹ, Rv 5ʹ-AGGGGTCTAGAGAGCTTGGC-3ʹ.
Protein extraction and western blot
Frozen TAs and tissues were mechanically grinded in liquid nitrogen using a ceramic mortar and pestle precooled in dry ice. Powder was then weighed and homogenized in cold lysis buffer (200 μL per 30 mg of powder) containing 50 mM of Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, and NP40 1% supplemented with protease inhibitor cocktail 1% (Sigma-Aldrich, France). Samples were then incubated under agitation in a cold room for 4 h and sonicated on ice. After centrifugation (14,000 × g, 4°C, 15 min), protein concentration in the supernatant was determined with the BCA Protein Assay Kit (Thermo Scientific Pierce, France). Ten micrograms of proteins were mixed with loading buffer (50 mM Tris-HCl, SDS 2%, glycerol 10%, 2-mercaptoethanol 1%, and bromophenol blue) and denatured at 90°C for 5 min. For TA, protein extract samples were separated on SDS-PAGE 10% and transferred onto polyvinylidene fluoride (PVDF) membranes (0.45 μm pore size, Life Technologies) overnight at 100 mA at 4°C. The other protein extracts (i.e., mouse tissues and human muscles) were separated on 10% Mini-PROTEAN TGX Stain-Free (Bio-Rad), and the gel was transferred on nitrocellulose membrane using Transblot Turbo RTA transfer kit for 8 min at 2.5 A. Total protein loading was determined by stain-free staining. After transfer, membranes were blocked for 1 h at room temperature in PBS containing non-fat dry milk 5% and Tween 20 0.1% and then exposed to rabbit polyclonal anti-dynamin 2 antibody (ab3457, dilution 1:1,000 or ab65556 dilution 1:4,000, Abcam, UK) or rabbit polyclonal anti-GAPDH antibody (Sc-25778 dilution 1:2,000, Santa Cruz, France) in PBS-Tween-20 0.1%, milk 1% overnight at 4°C. Membranes were rinsed in PBS-Tween-20 0.1% and incubated 1 h at room temperature with horseradish-peroxidase-conjugated secondary antibody (anti-rabbit from Jackson ImmunoResearch, UK) in PBS-Tween-20 0.1%. Chemiluminescence was detected using ECL detection Kit (Merck-Millipore, Germany) in a Chemidoc Imaging Systems (Bio-Rad, France), and signal quantification was performed using ImageJ software.
AAV production
A cassette containing the shRNA under the control of H1 RNA polymerase III promoter has been inserted in a pSMD2 expression plasmid. The sh9RNA was generated from the Dnm2 19 nt siRNA targeting the mouse mutation (c.1393A>T, p.R465W) and harboring a mismatch with the WT sequence in position 9 of the sense strand. This siRNA has been previously selected for its efficacy and allele specificity.22 The shRNA maker function of the Serial Cloner software was used to generate shRNA sequences (available on request) for subsequent subcloning in the pH1-Super plasmid. Sense and antisense sh9-RNA (Eurogentec, Belgium) was annealed using annealing solution (pSilencer kit, Ambion) according to manufacturer's instructions and inserted in BglII-HindIII sites in pH1-Super plasmid. The cassette pH1-sh9RNA was then inserted in the pSMD2 between BamH1 and SalI sites. AAV vectors (serotype 1) were produced in HEK293 cells after transfection of the pSMD2-shRNA plasmid or empty pSMD2 plasmid, the pXX6 plasmid coding for viral helper genes required for AAV production, and the pRepCap plasmid (p0001) coding for AAV1 capsid, as described previously. Viral particles were purified on iodixanol gradients and concentrated on Amicon Ultra-15 100K columns (Merck-Millipore). The concentration of vgs (vgs/mL) was determined by quantitative real-time PCR on a LightCycler480 (Roche Diagnostics, France) by using TaqMan probe in the inverted terminal repeat (ITR) sequences. A control pSMD2 plasmid was 10-fold serially diluted (from 107 to 101 copies) and used as a control to establish the standard curve for absolute quantification. Sequences of primers and probes are available on request or indicated previously.22 AAV serotype 1 vectors containing shRNA specific of the mutant allele (AAV-sh9) and a control (AAV-control) were produced.
Mice and in vivo transduction
The dynamin 2 mutant mouse line was established on C57Bl/6 background at the Mouse Clinical Institute (MCI) (Illkirch, France; http://www-mci.u-strasbg.fr).23 All mice used in this study were housed on a 12-h light/dark cycle and received standard diet and water ad libitum in the animal facility of Sorbonne University (Paris, France). Male heterozygous KI-Dnm2R465W mice were injected at 1 and 6 months of age under isoflurane anesthesia. We chose male mice since the characterization of the model has been done on male; the WT littermates were used as controls. Two intramuscular injections of 30 μL within 24-h interval were performed using 29G needle in TAs. AAV-sh9 and AAV-control were injected at 1 month of age and at 6 months of age in order to achieve 1011 and 1012 vgs/muscle, respectively. Age-matched WT mice non-injected or injected with the AAV-control or PBS were used as control.
For 1-month-old mice injected for 1 year, 23 heterozygous mice were injected (i.e., 23 right TA with the AAV1-sh9 and 23 left TA with the AAV1-control) and 11 WT mice (i.e., 11 right TA with the AAV1-control and 11 left TA with PBS); all of them were analyzed for the muscle force. We decided to inject a high number of mice to prevent potential loss due to death of mice, considering the long period. Eleven heterozygous TAs of each condition and 14 WT TAs were then used for molecular biology, and nine heterozygous TA of each condition and eight WT were used for histology. For 6-month-old mice injected for 3 months with high dose of AAV1-sh9, five heterozygous mice were injected (i.e., four TAs with the AAV1-sh9 and six TAs with the PBS) and 11 WT mice (i.e., 11 TA with PBS); all of them were analyzed for the muscle force, molecular biology, and histology.
Muscle contractile properties
The isometric contractile properties of TAs were studied in situ on mice anesthetized with 60 mg/kg pentobarbital. The distal tendon of the TA was attached to a lever arm of a servomotor system (305B Dual-Mode Lever, Aurora Scientific). The sciatic nerve was stimulated by a bipolar silver electrode using a supramaximal (10 V) square wave pulse of 0.1-ms duration. Absolute maximal isometric tetanic force was measured during isometric contractions in response to electrical stimulation (frequency of 25–150 Hz; train of stimulation of 500 ms). All isometric contraction measurements were made at optimal muscle length. Force is expressed in grams (1 g = 9.8 mN). Mice were sacrificed by cervical dislocation, and TAs were weighted. Specific maximal force was calculated by dividing absolute force by muscle weight.
Histomorphological analyses
TAs were frozen in liquid-nitrogen-cooled isopentane. Transverse sections of TA (8 μm thick) were stained with H&E, SDH, and reduced DPNH by standard methods. Light microscopy was performed using an Axioscope Z1 Slide scanner (Zeiss, France) at 20× magnification. Exposure settings were identical between compared samples and viewed at room temperature. Histomorphological abnormalities were manually counted on SDH staining using cell counter plugin Fiji software;48 eight untreated and eight treated heterozygous TA sections from different animals were counted.
Immunocytochemistry
For immunocytochemistry, muscle cryosections (8 μm thick) were fixed in paraformaldehyde 4% (15 min at room temperature). After washing in PBS, cryosections were permeabilized in Triton X-100 0.5% in PBS for 10 min at room temperature and blocked in PBS-Triton X-100 0.1%, BSA 5%, and Donkey serum 5% for 30 min. Samples were incubated with rabbit anti-Laminin (Abcam ab11575) overnight at 4°C in PBS with Triton X-100 0.1% and BSA 1%. After PBS-Triton X-100 0.1% washes, samples were incubated with goat anti-rabbit Alexa Fluor 568 secondary antibody (Life Technologies, France) for 60 min at room temperature. For satellite cell staining, transverse 8-μm cross-sections were fixed with 4% paraformaldehyde, blocked in PBS containing 5% fetal calf serum (FCS) and 0.01% Triton X-100, and then incubated overnight with primary antibody against PAX7 (1:20; DSHB, USA) and labeled with Alexa Fluor 568 (1:300, Life Technologies, France). Slides were then stained with 4ʹ,6-diamidino-2-phenylindole (DAPI) for nuclei staining. The slides were mounted with Vectashield mounting medium (Vector Laboratories, UK). Fluorescent images acquisition was performed at 20× magnification on Axioscope Z1 Slide scanners (Zeiss, France) for Laminin and on a Nikon Ti2 microscope, driven by Metamorph Software (Roper), equipped with a motorized stage and a Yokogawa CSU-W1 spinning disk head coupled with a Prime 95 sCMOS camera (Photometrics) for PAX7. For quantification of fiber size, images of TA sections labeled with anti-Laminin antibody were used to automatically determine the Feret diameter of each fiber after manual segmentation using the Fiji software. The normal distribution of the values was plotted using Microsoft Excel software (eight heterozygous untreated, eight heterozygous treated, and six WT TA sections from different animals were analyzed). Satellite cells were counted using the "cell counter" plugin of ImageJ. TAs from each condition (WT [n = 6], untreated heterozygous [n = 5], and treated heterozygous [n = 5]) were imaged at 20× magnification, and at least five different fields were analyzed (corresponding to an average of 900 fibers).
Viral genome quantification
Viral infection efficacy was evaluated through quantification of vgs in injected muscles. Genomic DNA was extracted from mouse muscle sections using DNA purification kit (Promega, France) according to the manufacturer's protocol. Copy numbers of AAV genomes were quantified on 100 ng of genomic DNA by quantitative real-time PCR on a LightCycler480 (Roche Diagnostics, France) by using TaqMan probe. A linearized plasmid containing ITR sequences was used to establish the standard curve for absolute quantification. Sequences of primers also used for viral titration are available on request.
Human biopsies
Twelve control human muscle samples from patients ranging from ages 3 to 60 years old were obtained from the MyoBank-AFM bank of tissues for research, a partner in the EU network EuroBioBank, or from the Morphological Unit of the Institute of Myology (Paris, France) in accordance with European recommendations and French legislation. The five CNM-patient's biopsies were obtained from the Morphological unit and are ranging from age 1 month to 48 years and harbor different DNM2 missense mutations (i.e., the DNM2 E611K, R369W, A618T, R465W, and R522H).
Study approval
Animal studies conform to the French laws and regulations concerning the use of animals for research and were approved by an external ethical committee (APAFIS no. 21085-2019060719395546) delivered by the French Ministry of Higher Education and Scientific Research.
Acknowledgments
We thank the Histomics Platform from the Brain and Spinal Cord Institute (Paris, France) for light microscopy imaging, the MyoBank-AFM and the Histopathology Laboratory from the Institut de Myologie (Paris, France) for providing human muscle samples, and the Penn Vector Core, Gene Therapy Program (University of Pennsylvania, Philadelphia, USA) for providing pAAV1 plasmid (p0001). We would like to thank Catherine Coirault for fruitful discussions and comments. This work was supported by the Institut National de la Santé et de la Recherche Médicale (Inserm), Inserm Transfert (CoPoC grant), the Association Institut de Myologie (AIM), Sorbonne Université, and the Agence Nationale de la Recherche ANR Dynather (ANR-18-CE17-0006-02 to M. Bitoun).
Author contributions
D.T. and M. Bitoun conceived and designed the experiments. D.T., B.P., L.M., M.L., M. Beuvin, and M. Bitoun performed the experiments. L.J. and S.B.-Z. produced the AAV vector. M.T.B. and N.R. provided the human muscle samples and helpful discussions. D.T., L.M., and M. Bitoun analyzed the data. D.T. and M. Bitoun wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.02.009.
Contributor Information
Delphine Trochet, Email: d.trochet@institut-myologie.org.
Marc Bitoun, Email: m.bitoun@institut-myologie.org.
Supplemental information
References
- 1.Romero N.B. Centronuclear myopathies: a widening concept. Neuromuscul. Disord. 2010;20:223–228. doi: 10.1016/j.nmd.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Böhm J., Biancalana V., DeChene E.T., Bitoun M., Pierson C.R., Schaefer E., Karasoy H., Dempsey M.A., Klein F., Dondaine N., et al. Mutation spectrum in the large gtpase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum. Mutat. 2012;33:949–959. doi: 10.1002/humu.22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bitoun M., Maugenre S., Jeannet P.Y., Lacène E., Ferrer X., Laforêt P., Martin J.J., Laporte J., Lochmüller H., Beggs A.H., et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 2005;37:1207–1209. doi: 10.1038/ng1657. [DOI] [PubMed] [Google Scholar]
- 4.Biancalana V., Romero N.B., Thuestad I.J., Ignatius J., Kataja J., Gardberg M., Héron D., Malfatti E., Oldfors A., Laporte J. Some DNM2 mutations cause extremely severe congenital myopathy and phenocopy myotubular myopathy. Acta Neuropathol. Commun. 2018;6:93. doi: 10.1186/s40478-018-0593-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casar-Borota O., Jacobsson J., Libelius R., Oldfors C.H., Malfatti E., Romero N.B., Oldfors A. A novel dynamin-2 gene mutation associated with a late-onset centronuclear myopathy with necklace fibres. Neuromuscul. Disord. 2015;25:345–348. doi: 10.1016/j.nmd.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Catteruccia M., Fattori F., Codemo V., Ruggiero L., Maggi L., Tasca G., Fiorillo C., Pane M., Berardinelli A., Verardo M., et al. Centronuclear myopathy related to dynamin 2 mutations: clinical, morphological, muscle imaging and genetic features of an Italian cohort. Neuromuscul. Disord. 2013;23:229–238. doi: 10.1016/j.nmd.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen T., Pu C., Wang Q., Liu J., Mao Y., Shi Q. Clinical, pathological, and genetic features of dynamin-2-related centronuclear myopathy in China. Neurol. Sci. 2015;36:735–741. doi: 10.1007/s10072-014-2028-6. [DOI] [PubMed] [Google Scholar]
- 8.Fujise K., Okubo M., Abe T., Yamada H., Takei K., Nishino I., Takeda T., Noguchi S. Imaging-based evaluation of pathogenicity by novel DNM2 variants associated with centronuclear myopathy. Hum. Mutat. 2022;43:169–179. doi: 10.1002/humu.24307. [DOI] [PubMed] [Google Scholar]
- 9.Kierdaszuk B., Berdynski M., Karolczak J., Redowicz M.J., Zekanowski C., Kaminska A.M. A novel mutation in the DNM2 gene impairs dynamin 2 localization in skeletal muscle of a patient with late onset centronuclear myopathy. Neuromuscul. Disord. 2013;23:219–228. doi: 10.1016/j.nmd.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 10.Neto O.A., De Martins C.A., Carvalho M., Chadi G., Seitz K.W., Oliveira A.S.B., Conti Reed U., Laporte J., Zanoteli E. DNM2 mutations in a cohort of sporadic patients with centronuclear myopathy. Genet. Mol. Biol. 2015;38:147–151. doi: 10.1590/S1415-4757382220140238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reumers S.F.I., Erasmus C.E., Bouman K., Pennings M., Schouten M., Kusters B., Duijkers F.A.M., van der Kooi A., Jaeger B., Verschuuren-Bemelmans C.C., et al. Clinical, genetic, and histological features of centronuclear myopathy in The Netherlands. Clin. Genet. 2021;100:692–702. doi: 10.1111/cge.14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Züchner S., Noureddine M., Kennerson M., Verhoeven K., Claeys K., De Jonghe P., Merory J., Oliveira S.A., Speer M.C., Stenger J.E., et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat. Genet. 2005;37:289–294. doi: 10.1038/ng1514. [DOI] [PubMed] [Google Scholar]
- 13.Sambuughin N., Goldfarb L.G., Sivtseva T.M., Davydova T.K., Vladimirtsev V.A., Osakovskiy V.L., Danilova A.P., Nikitina R.S., Ylakhova A.N., Diachkovskaya M.P., et al. Adult-onset autosomal dominant spastic paraplegia linked to a GTPase-effector domain mutation of dynamin 2. BMC Neurol. 2015;15:223. doi: 10.1186/s12883-015-0481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonny B., Burd C., De Camilli P., Chen E., Daumke O., Faelber K., Ford M., Frolov V.A., Frost A., Hinshaw J.E., et al. Membrane fission by dynamin: what we know and what we need to know. EMBO J. 2016;35:2270–2284. doi: 10.15252/embj.201694613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferguson S.M., De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 2012;13:75–88. doi: 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durieux A.C., Prudhon B., Guicheney P., Bitoun M. Dynamin 2 and human diseases. J. Mol. Med. 2010;88:339–350. doi: 10.1007/s00109-009-0587-4. [DOI] [PubMed] [Google Scholar]
- 17.Kenniston J.A., Lemmon M.A. Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. 2010;29:3054–3067. doi: 10.1038/emboj.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L., Barylko B., Byers C., Ross J.A., Jameson D.M., Albanesi J.P. Dynamin 2 mutants linked to centronuclear myopathies form abnormally stable polymers. J. Biol. Chem. 2010;285:22753–22757. doi: 10.1074/jbc.C110.130013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cowling B.S., Chevremont T., Prokic I., Kretz C., Ferry A., Coirault C., Koutsopoulos O., Laugel V., Romero N.B., Laporte J. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J. Clin. Invest. 2014;124:1350–1363. doi: 10.1172/JCI71206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferguson S., Raimondi A., Paradise S., Shen H., Mesaki K., Ferguson A., Destaing O., Ko G., Takasaki J., Cremona O., et al. Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev. Cell. 2009;17:811–822. doi: 10.1016/j.devcel.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trochet D., Prudhon B., Vassilopoulos S., Bitoun M. Therapy for dominant inherited diseases by allele-specific RNA interference: successes and pitfalls. Curr. Gene Ther. 2015;15:503–510. doi: 10.2174/1566523215666150812115730. [DOI] [PubMed] [Google Scholar]
- 22.Trochet D., Prudhon B., Beuvin M., Peccate C., Lorain S., Julien L., Benkhelifa-Ziyyat S., Rabai A., Mamchaoui K., Ferry A., et al. Allele-specific silencing therapy for Dynamin 2-related dominant centronuclear myopathy. EMBO Mol. Med. 2018;10:239–253. doi: 10.15252/emmm.201707988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durieux A.C., Vignaud A., Prudhon B., Viou M.T., Beuvin M., Vassilopoulos S., Fraysse B., Ferry A., Lainé J., Romero N.B., et al. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet. 2010;19:4820–4836. doi: 10.1093/hmg/ddq413. [DOI] [PubMed] [Google Scholar]
- 24.Almeida C.F., Bitoun M., Vainzof M. Satellite cells deficiency and defective regeneration in dynamin 2-related centronuclear myopathy. FASEB J. 2021;35:1–14. doi: 10.1096/fj.202001313RRR. [DOI] [PubMed] [Google Scholar]
- 25.Fongy A., Falcone S., Lainé J., Prudhon B., Martins-Bach A., Bitoun M. Nuclear defects in skeletal muscle from a Dynamin 2-linked centronuclear myopathy mouse model. Sci. Rep. 2019;9:1580. doi: 10.1038/s41598-018-38184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buono S., Ross J.A., Tasfaout H., Levy Y., Kretz C., Tayefeh L., Matson J., Guo S., Kessler P., Monia B.P., et al. Reducing dynamin 2 (DNM2) rescues DNM2-related dominant centronuclear myopathy. Proc. Natl. Acad. Sci. U. S. A. 2018;115:11066–11071. doi: 10.1073/pnas.1808170115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchlis G., Podsakoff G.M., Radu A., Hawk S.M., Flake A.W., Mingozzi F., High K.A. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood. 2012;119:3038–3041. doi: 10.1182/blood-2011-09-382317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bish L.T., Morine K., Sleeper M.M., Sanmiguel J., Wu D., Gao G., Wilson J.M., Sweeney H.L. Adeno-associated virus (AAV) serotype 9 provides global cardiac gene transfer superior to AAV1, AAV6, AAV7, and AAV8 in the mouse and rat. Hum. Gene Ther. 2008;19:1359–1368. doi: 10.1089/hum.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang J., Wakimoto H., Seidman J.G., Seidman C.E. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342:111–114. doi: 10.1126/science.1236921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnett A.L., Konieczny P., Ramos J.N., Hall J., Odom G., Yablonka-Reuveni Z., Chamberlain J.R., Chamberlain J.S. Adeno-associated viral vectors do not efficiently target muscle satellite cells. Mol. Ther. Methods Clin. Dev. 2014;1:14038. doi: 10.1038/mtm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwon J.B., Ettyreddy A.R., Vankara A., Bohning J.D., Devlin G., Hauschka S.D., Asokan A., Gersbach C.A. In vivo gene editing of muscle stem cells with adeno-associated viral vectors in a mouse model of duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 2020;19:320–329. doi: 10.1016/j.omtm.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muñoz X.M., Kretz C., Silva-Rojas R., Ochala J., Menuet A., Romero N.B., Cowling B.S., Laporte J. Physiological impact and disease reversion for the severe form of centronuclear myopathy linked to dynamin. JCI Insight. 2020;5:e137899. doi: 10.1172/jci.insight.137899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao M., Smith L., Volpatti J., Fabian L., Dowling J.J. Insights into wild-type dynamin 2 and the consequences of DNM2 mutations from transgenic zebrafish. Hum. Mol. Genet. 2019;28:4186–4196. doi: 10.1093/hmg/ddz260. [DOI] [PubMed] [Google Scholar]
- 34.Kocaturk N.M., Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front. Cell Dev. Biol. 2018;6:128. doi: 10.3389/fcell.2018.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durieux A.C., Vassilopoulos S., Lainé J., Fraysse B., Briñas L., Prudhon B., Castells J., Freyssenet D., Bonne G., Guicheney P., et al. A centronuclear myopathy - dynamin 2 mutation impairs autophagy in mice. Traffic. 2012;13:869–879. doi: 10.1111/j.1600-0854.2012.01348.x. [DOI] [PubMed] [Google Scholar]
- 36.Puri C., Manni M.M., Vicinanza M., Hilcenko C., Zhu Y., Runwal G., Stamatakou E., Menzies F.M., Mamchaoui K., Bitoun M., et al. A DNM2 centronuclear myopathy mutation reveals a link between recycling endosome scission and autophagy. Dev. Cell. 2020;53:154–168.e6. doi: 10.1016/j.devcel.2020.03.018. [DOI] [PubMed] [Google Scholar]
- 37.Cowling B.S., Toussaint A., Amoasii L., Koebel P., Ferry A., Davignon L., Nishino I., Mandel J.L., Laporte J. Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am. J. Pathol. 2011;178:2224–2235. doi: 10.1016/j.ajpath.2011.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu N., Bezprozvannaya S., Shelton J.M., Frisard M.I., Hulver M.W., McMillan R.P., Wu Y., Voelker K.A., Grange R.W., Richardson J.A., et al. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J. Clin. Invest. 2011;121:3258–3268. doi: 10.1172/JCI46267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bitoun M., Durieux A.C., Prudhon B., Bevilacqua J.A., Herledan A., Sakanyan V., Urtizberea A., Cartier L., Romero N.B., Guicheney P. Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum. Mutat. 2009;30:1419–1427. doi: 10.1002/humu.21086. [DOI] [PubMed] [Google Scholar]
- 40.Echaniz-Laguna A., Nicot A.S., Carré S., Franques J., Tranchant C., Dondaine N., Biancalana V., Mandel J.L., Laporte J. Subtle central and peripheral nervous system abnormalities in a family with centronuclear myopathy and a novel dynamin 2 gene mutation. Neuromuscul. Disord. 2007;17:955–959. doi: 10.1016/j.nmd.2007.06.467. [DOI] [PubMed] [Google Scholar]
- 41.Koutsopoulos O.S., Kretz C., Weller C.M., Roux A., Mojzisova H., Böhm J., Koch C., Toussaint A., Heckel E., Stemkens D., et al. Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur. J. Hum. Genet. 2013;21:637–642. doi: 10.1038/ejhg.2012.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nonnenmacher M., Weber T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther. 2012;19:649–658. doi: 10.1038/gt.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carrig S., Bijjiga E., Wopat M.J., Martino A.T. Insulin therapy improves adeno-associated virus transduction of liver and skeletal muscle in mice and cultured cells. Hum. Gene. Ther. 2016;27:892–906. doi: 10.1089/hum.2016.073. [DOI] [PubMed] [Google Scholar]
- 44.Qiao C., Zhang W., Yuan Z., Shin J.-H., Li J., Jayandharan G.R., Zhong L., Srivastava A., Xiao X., Duan D. Adeno-associated virus serotype 6 capsid tyrosine-to-phenylalanine mutations improve gene transfer to skeletal muscle. Hum. Gene Ther. 2010;21:1343–1348. doi: 10.1089/hum.2010.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hösel M., Huber A., Bohlen S., Lucifora J., Ronzitti G., Puzzo F., Boisgerault F., Hacker U.T., Kwanten W.J., Klöting N., et al. Autophagy determines efficiency of liver-directed gene therapy with adeno-associated viral vectors. Hepatology. 2017;66:252–265. doi: 10.1002/hep.29176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gibbs E.M., Clarke N.F., Rose K., Oates E.C., Webster R., Feldman E.L., Dowling J.J. Neuromuscular junction abnormalities in DNM2-related centronuclear myopathy. J. Mol. Med. 2013;91:727–737. doi: 10.1007/s00109-013-0994-4. [DOI] [PubMed] [Google Scholar]
- 47.Trochet D., Prudhon B., Jollet A., Lorain S., Bitoun M. Reprogramming the dynamin 2 mRNA by spliceosome-mediated RNA trans-splicing. Mol. Ther. Nucleic Acids. 2016;5:e362. doi: 10.1038/mtna.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.