

Abstract
The different coordination behavior of the flexible yet sterically demanding, hemilabile P,N ligand bis(quinoline‐2‐ylmethyl)phenylphosphine (bqmpp) towards selected CuI, AgI and AuI species is described. The resulting X‐ray crystal structures reveal interesting coordination geometries. With [Cu(MeCN)4]BF4, compound 1 [Cu2(bqmpp)2](BF4)2 is obtained, wherein the copper(I) atoms display a distorted square planar and square pyramidal geometry. The steric demand and π‐stacking of the ligand allow for a short Cu⋅⋅⋅Cu distance (2.588(9) Å). CuI complex 2 [Cu4Cl3(bqmpp)2]BF4 contains a rarely observed Cu4Cl3 cluster, probably enabled by dichloromethane as the chloride source. In the cluster, even shorter Cu⋅⋅⋅Cu distances (2.447(1) Å) are present. The reaction of Ag[SbF6] with the ligand leads to a dinuclear compound (3) in solution as confirmed by 31P{1H} NMR spectroscopy. During crystallization, instead of the expected phosphine complex 3, a tris(quinoline‐2‐ylmethyl)bisphenyl‐phosphine (tqmbp) compound [Ag2(tqmbp)2](SbF6)2 4 is formed by elimination of quinaldine. The Au(I) compound [Au2(bqmpp)2]PF6 (5) is prepared as expected and shows a linear arrangement of two phosphine ligands around AuI.
Keywords: bisphosphine; cluster compounds; coinage metals; metal-metal interactions; N,P ligands
Flexible yet sterically demanding, hemilabile P,N ligands can enable versatile coordination geometries as shown through reactions of bis(quinoline‐2‐ylmethyl)phenylphosphine with selected CuI, AgI and AuI species. Among other interesting X‐ray crystal structures, a rarely observed Cu4Cl3 cluster with short Cu⋅⋅⋅Cu distances is presented.

1. Introduction
Multidentate P,N ligands stand out due to their hemilability, as they contain two electronically different coordination atoms (soft phosphorus and hard nitrogen). This versatile coordination behavior is very beneficial and needed in catalysis, for the preparation of MOFs or even luminescent materials.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 ] Hemilabile tertiary phosphines are preferably used due to their outstanding flexibility which can be modified by changing the substituents accordingly. The modification of the substituents can result in the adaptation of their steric and electronic properties, their bite angle and solubility, and even chiral phosphine ligands can be formed. These features can be used for the complexation of a wide range of metal ions; even the bridging of metal ions is possible with tertiary phosphines.[ 14 , 15 , 16 ] Although the bridging capabilities of phosphines are nowadays widely known in literature, they became popular only about 20 years ago.[ 17 , 18 , 19 ] In the meantime, further complexes have been synthesized, wherein the tertiary phosphine adopts a bridging position between two metal centers. In particular coinage metals ions in combination with P,N ligands are prone to the formation of polynuclear complexes which can even support metallophilic interactions. These short metal‐metal contacts can strongly influence the properties of the resulting complexes.[ 20 , 21 , 22 , 23 , 24 , 25 ] Nevertheless, conformation and composition of the complexes of coinage metals with P,N ligands are rather unpredictable due to the reactivity and diverse coordination capabilities of such ligands.[ 26 , 27 , 28 ] The newly synthesized CuI, AgI and AuI complexes of bis(quinolin‐2‐ylmethyl)phenylphosphine (bqmpp), presented in the following, complement the great variety of complexes of coinage metals and impressively demonstrate the unique coordination behavior of P,N ligands. The new complexes have been characterized by multinuclear (1H, 13C, 31P, 11B, 19F) 1D and 2D NMR spectroscopy as well as by single‐crystal X‐ray diffraction analysis, elemental analysis and mass spectrometry.
2. Results and Discussion
2.1. Synthesis and Crystal Structures
The synthesis of bis(quinolin‐2‐ylmethyl)phenylphosphine (bqmpp) has been previously reported by our group. [29] The crystal structure of bqmpp is now provided in the Supporting Information (Figure S1). For the synthesis of the copper(I) complex, bqmpp is dissolved in dry, degassed acetonitrile (MeCN) and the resulting mixture transferred to the equivalent amount of [Cu(MeCN)4]BF4 dissolved in MeCN with a cannula under inert gas. After stirring for 12 h at room temperature and following work‐up, the CuI compound 1 is obtained from the orange reaction solution (see experimental part) as an orange powder (Scheme 1). The compound is recrystallized by diffusion of diethyl ether into a solution of compound 1 in dichloromethane (DCM) at room temperature, yielding 74 % of orange crystals. Surprisingly, the so‐formed crystals of 1 are hardly soluble anymore in dichloromethane. The compound should be stored under inert gas in a sealed container to prevent any oxidation.
Scheme 1.

Synthesis of the copper(I) compounds 1 and 2 including the visualization of the binding mode in the crystal structures of the products.
The copper(I) complex 1 crystallizes in the monoclinic space group P21/c and a view of the molecular structure in the crystal is shown in Figure 1. Selected bond lengths and angles of compound 1 are listed in Table 1; crystal and structure refinement data can be found in the Supporting Information. In the cationic complex 1, two phosphine ligands bridge two copper(I) ions, similar to the silver(I) hexafluoroantimonate compound of the respective phosphine oxide previously reported by our group, where oxygen takes over the coordination task of phosphorus. [29] Similar to the previously reported phosphine oxide ligand, the structure of the ligand in 1 can be compared to a bird, where one ligand spreads both quinaldinyl substituents in an open‐wing arrangement, whereas the other ligand displays a closed‐wing arrangement with a N3⋅⋅⋅N4 distance of 3.082(5) Å. The Cu1 atom exhibits a distorted square pyramidal geometry with angles ranging from 83.3(1)–134.4(1)° and a τ 5 value of 0.17. [30] The second copper(I) atom is tetracoordinated by one nitrogen atom of one ligand and one nitrogen and phosphorus atom of the other ligand and adopts a distorted square planar geometry with angles ranging from 62.6(1)–154.1(1)° and a τ 4 value of 0.40. [31] Both copper atoms are very close to each other (2.588(9) Å); regardless, the shortest Cu⋅⋅⋅Cu distance in a phosphine complex was reported for a copper(I) hexafluorophosphate complex salt of bis(pyridine‐2‐ylmethyl)phenylphosphine with 2.488(12) Å. [32] The close proximity and the resulting π‐stacking interactions of the phenyl rings as well as the quinaldinyl rings of the closed‐wings ligand probably allow for such close copper contacts (see also Figure S2 of the Supporting Information). Furthermore, the Cu1−P1 (2.527(2) Å) and Cu2−P1 distances are not equidistant and differ by 0.281 Å. The Cu2−P2 distance of 3.333(1) Å is beyond a bonding distance. The Cu2−P1 distance of 2.246(1) Å is typical for this type of bond, and the same holds true for the Cu−N distances of 1.982(4)–2.133(4) Å.
Figure 1.

Molecular structure of the dinuclear complex cation of compound 1 in the crystal; view of the asymmetric unit. Thermal ellipsoids are drawn at 50 % probability level. Dichloromethane molecules and BF4 anions are omitted for clarity.
Table 1.
Selected bond lengths [Å] and angles [°] for compounds 1 and 2.
|
1 | |||
|
P1−Cu1 |
2.527(2) |
P2−C33 |
1.841(5) |
|
P1−Cu2 |
2.246(1) |
P2−C43 |
1.848(5) |
|
P2−Cu1 |
2.256(1) |
Cu1−N1 |
2.038(4) |
|
P1−C1 |
1.814(5) |
Cu1−N3 |
2.133(4) |
|
P1−C7 |
1.815(5) |
Cu2−N2 |
2.101(4) |
|
P1−C17 |
1.839(5) |
Cu2−N4 |
1.982(4) |
|
P2−C27 |
1.809(5) |
Cu1⋅⋅⋅Cu2 |
2.588(9) |
|
|
|
|
|
|
C1−P1−C7 |
107.0(2) |
P1−Cu2−N2 |
88.5(1) |
|
C1−P1−C17 |
102.8(2) |
P2−Cu1−N3 |
83.3(1) |
|
C7−P1−C17 |
98.0(2) |
P2−Cu1−N1 |
134.4(1) |
|
C27−P2−C33 |
105.7(2) |
Cu1−P1−Cu2 |
65.4(4) |
|
C27−P2−C43 |
103.7(2) |
N1−Cu1−N3 |
110.9(2) |
|
C33−P2−C43 |
101.8(2) |
N2−Cu2−N4 |
115.9(2) |
|
P1−Cu1−N1 |
83.6(1) |
|
|
|
2 | |||
|
P1−Cu1 |
2.377(2) |
Cu2−Cl1 |
2.294(2) |
|
P1−Cu2 |
2.393(2) |
Cu3−N3 |
2.020(5) |
|
P2−Cu3 |
2.314(2) |
Cu3−Cl2 |
2.382(2) |
|
P2−Cu4 |
2.452(2) |
Cu3−Cl3 |
2.380(2) |
|
P1−C1 |
1.815(8) |
Cu4−N4 |
2.019(5) |
|
P1−C7 |
1.828(6) |
Cu4−Cl1 |
2.279(2) |
|
P1−C17 |
1.843(6) |
Cu4−Cl2 |
2.415(2) |
|
Cu1−N1 |
2.005(5) |
Cl2⋅⋅⋅H15 |
2.849(1) |
|
Cu1−Cl2 |
2.435(2) |
Cl3⋅⋅⋅H25 |
2.850(2) |
|
Cu1−Cl3 |
2.383(2) |
Cl2⋅⋅⋅H51 |
2.801(2) |
|
Cu2−N2 |
2.021(5) |
Cu1⋅⋅⋅Cu2 |
2.451(1) |
|
Cu2−Cl3 |
2.424(2) |
Cu3⋅⋅⋅Cu4 |
2.447(1) |
|
|
|
|
|
|
C1−P1−C7 |
104.6(4) |
Cu1−P1−Cu2 |
61.9(5) |
|
C1−P1−C17 |
100.9(3) |
Cu3−P2−Cu4 |
61.7(5) |
|
C7−P1−C17 |
100.0(3) |
Cu1−Cl3−Cu2 |
61.3(5) |
|
C27−P2−C33 |
102.3(3) |
Cu3−Cl2−Cu4 |
61.3(5) |
|
C27−P2−C43 |
103.2(3) |
Cu2−Cl1−Cu4 |
93.7(6) |
|
C33−P2−C43 |
99.7(3) |
N1−Cu1−Cl2 |
114.1(2) |
|
P1−Cu1−N1 |
87.4(2) |
N2−Cu2−Cl1 |
117.5(2) |
|
P1−Cu2−N2 |
87.6(2) |
N3−Cu3−Cl2 |
128.5(2) |
|
P2−Cu3−N3 |
88.4(2) |
N4−Cu4−Cl1 |
117.9(4) |
|
P2−Cu4−N4 |
86.2(2) |
||
During the crystallization attempt of the latter copper(I) compound in DCM, another compound [Cu4Cl3(bqmpp)2]BF4 (2) with a different molecular structure formed in the crystallization apparatus. It crystallizes in the monoclinic space group P21/n. A view of the molecular structure of 2 in the crystal is shown in Figure 2. How the formation of 2 occurred is still unclear. A possible source for chloride atoms might come from the solvent dichloromethane. In fact, in the crystal structure of 2, additional electron density is observed, indicating the presence of disordered dichloromethane molecules. However, the disorder could not be resolved by an appropriate model (see Supporting Information). Selected bond lengths and angles of compound 2 are listed in Table 1, and crystal and structure refinement data can be found in the Supporting Information.
Figure 2.
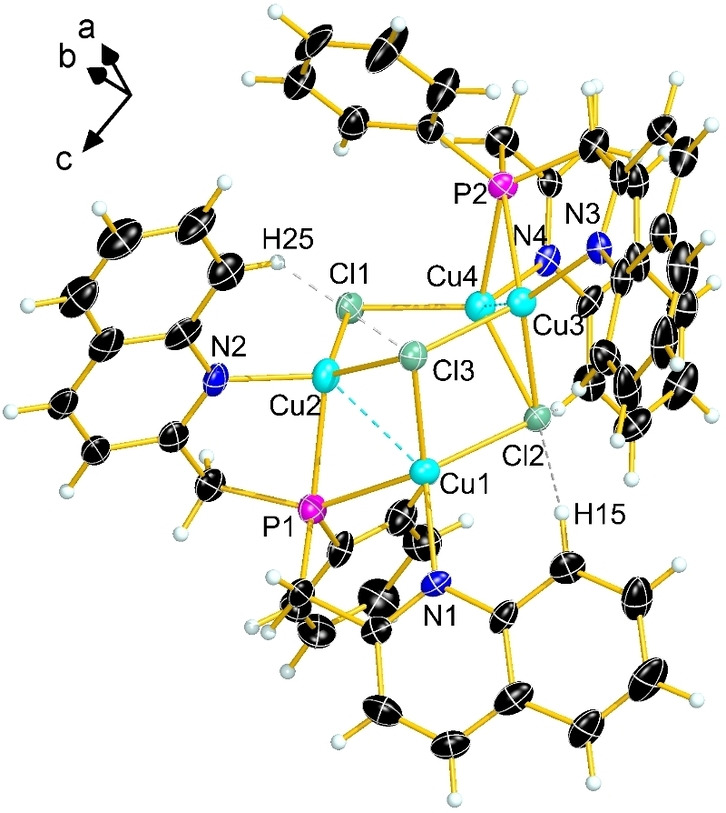
Molecular structure of cluster complex cation 2 in the crystal, view of the asymmetric unit. Thermal ellipsoids are drawn at 50 % probability level. Dichloromethane molecules and BF4 anions are omitted for clarity.
All copper(I) atoms in the crystal structure of 2 adopt a more or less distorted square pyramidal geometry with angles ranging from 86.2(2)–128.5(2)° and a τ 5 values between 0.04–0.20. [30] The metal‐metal distances (Cu1⋅⋅⋅Cu2 2.451(1) Å and Cu3⋅⋅⋅Cu4 2.447(1) Å) in 2 are very short, even shorter than in the previously mentioned copper(I) hexafluorophosphate complex salt of bis(pyridine‐2‐ylmethyl)phenylphosphine. [32] The coordination of both ligands in 2 occurs in an open‐wing manner. The Cu1−P1 and Cu2‐P1 distances are very similar and only differ by 0.016 Å, whereas the Cu3−P2 and Cu4−P2 distances differ by 0.138 Å. The Cu−N distances with an average value of 2.020 Å are very similar to each other, with the exception of Cu1−N1 (2.005 Å).
The highlight of this structure is the Cu4Cl3 distorted triangular prism with an additional vertex on one edge (Cl1 atom). Such Cu4Cl3 clusters are very rare, and a similar structure as observed in 2 has so far not been reported in the literature. [33] The distortion of the triangular prism results from the incorporation of chloride into the framework leading to diverse Cu−Cl and Cu⋅⋅⋅Cu distances. The Cu−Cl distances (2.279(2)–2.424(2) Å) are within the typical range for such distances. The different units of 2 are connected through attractive π‐interactions between the quinaldinyl rings of neighboring molecules in the crystal (see Figure S6 of the Supporting Information).[ 34 , 35 ]
An analogous crystallization attempt using dibromomethane in order to prepare the respective bromine analogue only yielded an orange, oily substance in the crystallization apparatus which was not further characterized.
For an attempted targeted synthesis of the complex unit in 2, phosphine bqmpp was dissolved in dry, degassed MeCN and the resulting mixture was transferred to two equivalents of CuCl dissolved in MeCN under inert gas using a cannula. After stirring for 12 h at room temperature, a yellow to orange powder was obtained. However, this resulting powder was found very poorly soluble and all recrystallization attempts failed. Additionally, powder diffraction data showed no agreement with simulated data of 2 (see Figure S3 of the Supporting Information).
The silver(I) compound 3 is synthesized by dissolving bqmpp in dry, degassed dichloromethane and transferring the resulting mixture to the equivalent amount of Ag[SbF6] dissolved in MeCN using a cannula. After stirring at room temperature for 12 h, the Ag(I) compound 3 is obtained as an orange oil after removal of all volatiles. Crystallization attempts to obtain a suitable single‐crystal of 3 for X‐ray diffraction have so far proven unsuccessful. However, during one of those attempts diffusing diethyl ether into a solution of compound 3 in MeCN, the crystal structure of the formed compound revealed a bisphosphine as the ligand (see Scheme 2 and Figure 3). Apparently, Ag[SbF6] can react with the phosphine in a side reaction whereby compound 4, containing silver(I) coordinating to tris(quinolin‐2‐ylmethyl)bisphenyl‐phosphine (tqmbp), is obtained. The formation of bisphosphines is very uncommon and a similar reaction behavior was not to be found in the literature. In this case the formation of the bisphosphine ligand involves the elimination of quinaldine, which is confirmed by NMR spectroscopy (see Supporting Information). Compound 4 is air‐sensitive and should be stored under inert gas.
Scheme 2.

Probable synthetic route for the formation of the silver(I) complexes 3 and 4. Visualization of the suspected binding of 3 (left) and the actual binding mode in the crystal structure of 4 (right).
Figure 3.

Molecular structure of the cationic complex of 4 in the crystal, view of the asymmetric unit. Thermal ellipsoids are drawn at 50 % probability level. Anions are omitted for clarity. Symmetry code: i=2=−x, 2=−y, 1=−z.
A targeted synthesis of tqmbp was attempted using different synthetical routes. Although NMR data indicates a successful synthesis of tqmbp in one case (δ=−14.1 ppm), so far none of the attempted strategies yielded a satisfactory result. A discussion on this topic can be found in the Supporting Information.
The silver(I) compound 4 crystallizes in the triclinic space group P=−1 and its molecular structure in the crystal is shown in Figure 3. Selected bond lengths and angles of compound 4 are listed in Table 2; crystal and structure refinement data can be found in the Supporting Information. The dinuclear cationic complex possesses an inversion center which lies in the middle of the Ag1−Ag1 i vector. The silver(I) atom Ag1 adopts a distorted square pyramidal geometry with angles ranging from 69.8(3)–152.4(2)° and a τ 5 value of 0.22. [30] The distance between both Ag atoms (3.221(2) Å) is shorter than the sum of van der Waals radii (3.44 Å), [36] but rather long compared to standard Ag−Ag distances, which are around 3.0 Å. In contrast, the Ag−N bonds are within the normal range (Ag1−N1 2.676(2) Å, Ag1−N2 2.635(1) Å). The Ag1−P1 and Ag1−P2 distances are very similar and only differ by 0.027 Å.
Table 2.
Selected bond lengths [Å] and angles [°] for compounds 4 and 5.
|
4 (symmetry code: i=2−x, 2−y, 1−z) | |||
|
P1−Ag1 |
2.410(4) |
P2−C27 |
1.818(1) |
|
P2−Ag1 |
2.437(3) |
P2−C33 |
1.835(1) |
|
P1−C1 |
1.845(1) |
Ag1−N1 |
2.676(2) |
|
P1−C7 |
1.821(1) |
Ag1−N2 |
2.635(1) |
|
P1−C17 |
1.865(1) |
Ag1⋅⋅⋅N3 |
3.871(8) |
|
P2−C17 |
1.861(1) |
Ag1⋅⋅⋅Ag1 i |
3.221(2) |
|
|
|
|
|
|
Ag1−P1−C1 |
114.7(4) |
P1−Ag1−N1 |
69.8(3) |
|
Ag1−P1−C7 |
106.3(4) |
P1−Ag1−N2 |
131.4(2) |
|
Ag1−P1−C17 |
123.5(4) |
N1−Ag1−N2 |
96.1(4) |
|
C1−P1−C7 |
100.5(6) |
P1−Ag1−Ag1 i |
79.9(9) |
|
C1−P1−C17 |
103.6(6) |
N1−Ag1−Ag1 i |
139.4(3) |
|
C7−P1−C17 |
105.7(5) |
P2−Ag1−Ag1 i |
91.8(9) |
|
P1−Ag1−P2 |
152.4(1) |
|
|
|
5 (symmetry code: i=−x, 1−y, −z) | |||
|
P1−C1 |
1.808(2) |
P1−Au1 |
2.306(5) |
|
P1−C7 |
1.830(2) |
Au1⋅⋅⋅N1 |
3.481(2) |
|
P1−C17 |
1.831(2) |
Au1⋅⋅⋅N2 |
3.610(2) |
|
|
|
|
|
|
C1−P1−C7 |
105.5(1) |
C7−P1−Au1 |
113.0(7) |
|
C1−P1−C17 |
106.1(1) |
C17−P1−Au1 |
114.8(8) |
|
C7−P1−C17 |
104.1(1) |
P1−Au1−P1 i |
180.0 |
|
C1−P1−Au1 |
112.6(7) |
||
The Ag1⋅⋅⋅N3 i distance of 3.871(8) Å and the Ag1⋅⋅⋅P1 i distance of 3.671(4) Å are beyond a bonding distance. The different units of 4 are connected through attractive π‐interactions between the quinaldinyl rings of neighboring molecules in the crystal with an averaged distance of 3.4 Å (Figure S4).[ 34 , 35 ]
For the synthesis of gold(I) complex 5, two equivalents of bqmpp were dissolved in dry, degassed dichloromethane and the resulting mixture was transferred to one equivalent of [AuCl(tht)] (tht=tetrahydrothiophene) under inert gas using a cannula. Solid potassium hexafluorophosphate was added to the mixture to remove chloride and to form the gold(I) compound 5 containing the non‐coordinating hexafluorophosphate anion (see Scheme 3 for details). After filtration, the remaining solvent was removed and gold(I) complex 5 was obtained as a yellow powder. Compound 5 can be recrystallized by diffusion of diethyl ether into a solution of compound 5 in dichloromethane yielding 80 % of crystalline 5. Compound 5 should be stored under inert gas and with exclusion of light.
Scheme 3.

Synthesis of the gold(I) compound 5 and visualization of the binding mode in the crystal structure of the product.
The gold(I) compound 5 crystallizes in the triclinic space group P=−1 with one formula unit in the unit cell (Figure 4). Selected bond lengths and angles of compound 5 are listed in Table 2; crystal and structure refinement data can be found in the Supporting Information. The cationic complex of 5 possesses an inversion center at Au1. The gold(I) atom is coordinated by the two phosphorus atoms in a linear arrangement. The Au1−P1 distance 2.306(5) Å falls within the normal range.
Figure 4.

Molecular structure of the complex cation of compound 5 in the crystal, view of the centrosymmetric molecule present in the unit cell. Thermal ellipsoids are drawn at 50 % probability level. The PF6 anion is omitted for clarity. Symmetry code: i==−x, 1=−y, =−z.
The Au1⋅⋅⋅N distances of 3.481(2) Å and 3.610(2) Å are beyond a bonding distance. The different units of 5 are connected through attractive π‐interactions between the quinaldinyl rings of neighboring molecules in the crystal with an averaged distance of 3.7 Å (Figure S5).[ 34 , 35 ]
2.2. Characterization of the Compounds by NMR Spectroscopy
Due to different solubilities, NMR spectra of the complexes were recorded in different solvents. The 31P{1H} NMR spectrum of 1 in CD3CN shows only one signal at −14.0 ppm. The coordination of the ligand seems not to affect δ31P of the latter (δ31P=−14.0 ppm) and only results in a strong broadening of the signal (Δν1/2=300 Hz). Coordination of the ligand to copper(I) becomes evident in the splitting pattern of the diastereotopic protons of the CH2 groups in the 1H NMR spectrum (Figure 5). The CH2 group in 1 shows larger 2 J PH couplings (6 and 8 Hz), which is in accordance with the increased coordination number of phosphorus, in comparison to the ligand, which shows a 2 J PH coupling of 1.4 Hz.
Figure 5.

Comparison of the signals for the CH2 group in the 1H NMR spectra of the free ligand (top) and 1 (bottom) in CD3CN.
The 31P{1H} NMR spectrum of 3 in CD3CN shows a phosphorus resonance at 12.2 ppm (Δν1/2=60 Hz) and is shifted to higher frequencies when compared to the free ligand (Δδ=22.8 ppm). However, the 31P{1H} NMR spectrum of 3 in CD2Cl2 shows a phosphorus resonance at 6.8 ppm (Δν1/2=400 Hz; shifted to higher frequencies when compared to the free ligand by Δδ=17.4 ppm). When lowering the temperature to −80 °C, the signal of the silver(I) complex in the 31P{1H} NMR in CD2Cl2 is split into a triplet (Figure 6, left). The splitting of the signal becomes already visible at RT (see Supporting Information) and is caused by the coupling of phosphorus to silver(I) with a coupling constant of 1 J PAg=250 Hz at −80 °C. The fine coupling to both NMR‐active nuclei of silver (107Ag and 109Ag) is not resolved; only the averaged signal is visible due to the dynamics of compound 3 in solution.
Figure 6.

31P NMR spectrum of 3 in CD2Cl2 at −80 °C visualizing the coupling of phosphorus to both silver(I) atoms (left) and a possible molecular structure of 3 in solution (right).
This silver‐phosphorus coupling constant indicates a tetracoordinated silver(I) cation in solution of which a possible structure is shown in Figure 6.[ 37 , 38 , 39 ]
The 31P{1H} NMR spectrum of 4 in CD3CN shows a phosphorus resonance at 11.4 ppm (Δν1/2=66 Hz; Δδ=22.8 ppm compared to the free ligand).
For the gold(I) compound 5 in CD2Cl2, the 31P{1H} NMR spectrum displays a phosphorus resonance at 36.4 ppm (Δν1/2=45 Hz). Compared to the free ligand, the signal is shifted to higher frequencies (Δδ=50.4 ppm). This change in chemical shift is common for gold(I) complexes of phosphines. [40]
All complexes show a broad phosphorus resonance in the 31P NMR spectrum at room temperature, indicating a dynamic behavior in solution similar to the complexes of the respective phosphine oxide published previously by our group. [29] The reason for the dynamics can be explained similarly and is most likely due to a rapid movement of the quinaldinyl rings in solution at room temperature. The corresponding variable temperature NMR spectra of compounds 1, 3 and 5 are shown in the Supporting Information.
A total dissociation of the metal complexes can be ruled out as no signal for the free ligand can be observed in the NMR spectra. However, the structure of the complexes in solution might not be the same as in the solid state. Interestingly, the line spacing of the isotopic patterns of 1 and 3 in the corresponding high‐resolution mass spectra suggests a mononuclear form for both compounds, respectively (Figures S4 and S5). In addition, the peaks for both complexes with one additional molecule of acetonitrile (for 1 [C26H21CuN2P]+: 496.10018 m/z; for 3 [C26H22 109AgN2P+MeCN]+: 540.07557 m/z) can be detected. This might indicate a mononuclear form for 1 and 3 in MeCN with solvent molecules occupying the free coordination sites. For the DCM solution of complex 3, however, a mononuclear form would imply a very unusual coordination geometry around the AgI core (CN of 2 or 3) with large repulsion between both quinaldinyl rings of the ligand. Therefore, the triplet in the 31P{1H} NMR spectrum of 3 in CD2Cl2 comes as no surprise.
3. Conclusion
The new compounds 1–5, that were formed utilizing CuI, AgI and AuI salts, demonstrate the coordination versatility of the bis(quinoline‐2‐ylmethyl)phenylphosphine (bqmpp) ligand towards coinage metals. With this ligand, very short Cu⋅⋅⋅Cu distances in the copper(I) complexes are supported. The steric demand and the possibility of π‐stacking of the quinaldinyl rings facilitate the existence of these short contacts. Compound 2 contains a rarely observed Cu4Cl3 cluster unit with a distorted trigonal prism, in which one edge is capped by a chlorido ligand. The formation of 2 is probably induced by the presence of DCM as the solvent. The unexpected formation of tris(quinoline‐2‐ylmethyl)bisphenyl‐phosphine (tqmbp) during the synthesis of the silver(I) compound 4 impressively shows the manifold reaction possibilities offered by the bqmpp ligand. A first step was made towards a targeted synthesis of the observed bisphosphine. In 4, a weak argentophilic interaction is observed between the two silver(I) ions. The Au(I) compound 5 completes the series of coinage group one complexes with bqmpp and shows the usually expected composition and molecular geometry of similar constituted gold(I) complexes.
Experimental Section
General Procedures: All compounds were handled under exclusion of moisture and oxygen in a protecting argon atmosphere using Schlenk techniques. Unless otherwise specified, all reagents and solvents were purchased from commercial sources and used as received. Complex [AuCl(tht)] [41] and ligand C26H21N2P [29] were prepared according to literature procedures. The solvents used were dried, freshly distilled and degassed prior to use. As drying agents SICAPENT® was used for acetonitrile and sodium for diethyl ether. DCM was distilled from CaH2 under a nitrogen atmosphere. The deuterated solvents CD2Cl2 and CD3CN were distilled and stored over molecular sieves under an argon atmosphere. NMR spectra were recorded with a Bruker Avance III spectrometer operating at 400.1 MHz (1H), 376.4 MHz (19F), 161.9 MHz (31P), 128.4 MHz (11B) and 100.6 MHz (13C). Chemical shifts are referenced to TMS (1H, 13C), 85 % H3PO4 (31P), CCl3F (19F) and BF3 in Et2O (11B) as external standards. All spectra were measured, if not mentioned otherwise, at 25 °C. The assignment of the signals is based on 2D (1H1H‐COSY, 1H13C‐HSQC and 1H13C‐HMBC) NMR experiments. Coupling constant (J) values are given in Hertz (Hz). The multiplicity of each resonance observed in the NMR spectra is reported as s=singlet; d=doublet; t=triplet; q=quartet; m=multiplet. ESI measurements were conducted using a Thermo Finnigan LTQ FT Ultra Fourier Transform Ion Cyclotron Resonance Mass Spectrometer.
Synthesis
[Cu2(bqmpp)2](BF4)2 (1): A solution of bis(quinoline‐2‐ylmethyl)phenyl‐phosphine (bqmpp) (0.286 g, 0.728 mmol) in dry acetonitrile (20 mL) was added to [Cu(MeCN)4]BF4 (0.229 g, 0.728 mmol) and stirred for 12 h. The solution turned orange. The solvent was removed and the remaining residue dissolved in dry, degassed dichloromethane. After removal of all volatiles, 1 was obtained as an orange powder. Crystallization from DCM yielded an orange microcrystalline solid. Crystalline yield: 0.291 g, 74 %.

1H NMR (CD3CN, 400 MHz): δ [ppm]=ABX spin system (A=B=H, X=P) 3.81 (dd, 2H, 2 J BX=5.8 Hz, 2 J AB=15.3 Hz, H11), 4.08 (dd, 2H, 2 J AX=8.0 Hz, 2 J AB=15.4 Hz, H11), 7.32 (d, 2H, 3 J HH=8.5 Hz, H9), 7.37 (t, 2H, 3 J HH=7.4 Hz, H2), 7.45 (m, 3H, H14, H2), 7.52 (t, 2H, 3 J HH=7.5 Hz, H1), 7.64 (t, 2H, 3 J HH=8.7 Hz, H13), 7.75 (dd, 2H, 3 J HH=8.1 Hz, 4 J HH=1.6 Hz, H3), 7.78 (d, 2H, 3 J HH=8.5 Hz, H6), 8.07 (d, 2H, 3 J HH=8.4 Hz, H10). 13C{1H} (CD3CN, 101 MHz) δ [ppm]=38.5 (d, 1 J CP=15.4 Hz, C11), 123.7 (d, 3 J CP=3.7 Hz, C9), 127.5 (s, C2), 127.9 (d, 2 J CP=1.3 Hz, C8), 128.9 (s, C3), 129.1 (s, C6), 129.9 (d, 3 J CP=9.1 Hz, C14), 131.1 (s, C1), 131.5 (d, 4 J CP=1.0 Hz, C15), 133.3 (d, 2 J CP=15.6 Hz, C13), 138.5 (s, C10), 147.5 (s, C5), 159.0 (s, C4). A signal for C12 could not be observed. 31P{1H} (CD3CN, 162 MHz) δ [ppm]= −14.0 (s). 11B{1H} (CD3CN, 128 MHz) δ [ppm]= −1.4 (s). +ESI HRMS (CH3CN/H2O) for [C26H21CuN2P]+: calcd. 455.0738 m/z; found 455.0733 m/z.
[Ag2(bqmpp)2](SbF6)2 (3): A solution of bqmpp (0.224 g, 0.570 mmol) in dry acetonitrile (15 mL) was added to solid AgSbF6 (0.196 g, 0.570 mmol) and the resulting mixture was stirred for 12 h. The solution turned orange and cloudy. After removal of all volatiles under vacuum, 3 was obtained as a light orange oil. Yield: 0.341 g, 84 %.

1H NMR (CD3CN, 400 MHz): δ [ppm]=ABX spin system (A=B=H, X=P) 4.17 (dd, 2H, 2 J BX=8.8 Hz, 2 J AB=14.4 Hz, H11), 4.26 (dd, 2H, 2 J AX=7.9 Hz, 2 J AB=14.4 Hz, H11), 7.21 (t, 2H, 3 J HH=7.4 Hz, H1), 7.35 (t, 2H, 3 J HH=7.2 Hz, H2), 7.48 (m, 3H, H14, H9), 7.55 (d, 3H, 3 J HH=7.8 Hz, H6, H15), 7.73 (d, 2H, 3 J HH=8.2 Hz, 4 J HH=1.1 Hz, H3), 7.82 (m, 2H, H13), 8.21 (d, 2H, 3 J HH=8.4 Hz, H10). 1H NMR (CD2Cl2, 400 MHz): δ [ppm]=ABX spin system (A=B=H, X=P) 4.33 (d, 2H, 2 J AB=13.8 Hz, H11), 4.49 (d, 2H, 2 J AB=13.0 Hz, H11), 7.11 (t, 2H, 3 J HH=7.3 Hz, H1), 7.32 (t, 2H, 3 J HH=7.7 Hz, H2), 7.49 (t, 2H, 3 J HH=6.6 Hz, H9), 7.65 (d, 2H, 3 J HH=8.2 Hz, H3), 7.70 (m, 2H, H6), 8.12 (d, 2H, 3 J HH=8.3 Hz, H10). 13C{1H} (CD3CN, 101 MHz) δ [ppm]=38.7 (d, 1 J CP=14.8 Hz, C11), 124.3 (d, 3 J CP=3.3 Hz, C9), 128.0 (d, 6 J CP=0.8 Hz, C2), 128.2 (d, 2 J CP=2.6 Hz, C8), 128.6 (d, 6 J CP=0.6 Hz, C6), 129.0 (d, 1 J CP=68.1 Hz, C12), 129.1 (d, 5 J CP=0.8 Hz, C3), 130.4 (d, 3 J CP=10.1 Hz, C14), 131.6 (s, C1), 133.0 (d, 4 J CP=1.9 Hz, C15), 134.1 (d, 2 J CP=16.3 Hz, C13), 140.0 (s, C10), 147.5 (s, C5), 158.1 (d, 4 J CP=1.2 Hz, C4). 13C{1H} (CD2Cl2, 101 MHz) δ [ppm]=39.6 (s, C11), 124.1 (s, C9), 127.6 (s, C2), 127.8 (s, C8), 127.9 (s, C6), 128.7 (s, C3), 128.9 (d, 1 J CP=17.4 Hz, C12), 130.2 (s, C14), 131.6 (s, C1), 133.1 (s, C15), 133.5 (s, C13), 140.4 (s, C10), 146.2 (s, C5), 157.1 (s, C4). 31P{1H} (CD3CN, 162 MHz) δ [ppm]=12.2 (s). 31P{1H} (CD2Cl2, 162 MHz) δ [ppm]=6.8 (t, 1 J PAg=218 Hz, 200 Hz). 19F{1H} (CD3CN, 376 MHz) δ [ppm]= −125.6 (m, 1 J FSb=1935 Hz). +ESI HRMS (CH3CN/H2O) for [C26H22 109AgN2P]+: calcd. 501.0490 m/z; found 501.0486 m/z. An EA measurement was not possible due to the presence of Sb.
[Ag2(tqmbp)2](SbF6)2 (4): 31P{1H} (CD3CN, 162 MHz) δ [ppm]=11.4 (s). +ESI HRMS (CH3CN/H2O) for [C42H33 107AgN3P2]+: calcd. 748.1201 m/z; found 748.1685 m/z. An EA measurement was not possible due to the presence of Sb.
[Au2(bqmpp)2]PF6 (5): [AuCl(tht)] was prepared according to literature procedures. [33] A solution of bqmpp (0.100 g, 0.250 mmol) in dry dichloromethane (2 mL) was added to [AuCl(tht)] (0.040 g, 0.125 mmol) dissolved in dichloromethane (1 mL). After stirring for 20 min, KPF6 (0.023 g, 0.125 mmol) was added to the solution to precipitate KCl. The reaction mixture was filtered and the solvent was removed in vacuo. A yellow solid was obtained. Crystallization from DCM yielded a yellow microcrystalline solid. Crystalline yield: 0.146 g, 80 %.

1H NMR (CD2Cl2, 400 MHz): δ [ppm]=ABX spin system (A=B=H, X=P) 3.97 (d, 2H, 2 J AB=15.1 Hz, H11), 4.26 (d, 2H, 2 J AB=15.1 Hz, H11), 7.09 (s, 2H, H9), 7.29 (t, 2H, 3 J HH=7.5 Hz, H2), 7.44 (m, 2H, H1), 7.49 (m, 2H, H13), 7.52 (s, 3H, H3, H15), 7.67 (m, 4H, H6, H14), 7.83 (d, 2H, 3 J HH=8.3 Hz, H10). 13C{1H} (CD2Cl2, 101 MHz) δ [ppm]=37.4 (s, C11), 122.4 (s, C9), 127.1 (d, 2 J CP=8.5 Hz, C8), 128.1 (s, C6), 128.6 (s, C15), 128.7 (s, C3), 129.5 (s, C2), 130.3 (s, C1), 132.6 (s, C13), 133.7 (s, C14), 137.2 (s, C10), 147.5 (s, C5), 154.7 (s, C4). A signal for C12 could not be observed. 31P{1H} (CD2Cl2, 162 MHz) δ [ppm]=36.4 (s), −143.8 (sept, 1 J PF=710 Hz, PF6 ‐). 19F{1H} (CD2Cl2, 376 MHz) δ [ppm]=−73.6 (d, 1 J PF=710 Hz). +ESI HRMS (CH3CN/H2O) for [C26H21AuN2P]+: calcd. 981.2550 m/z; found 981.2567 m/z.
X‐ray Crystallographic Studies: Detailed crystal and structure refinement data as well as additional figures are provided in the Supporting Information. Deposition Numbers href=https://www.ccdc.cam.ac.uk/services/structures?id=doi: 10.1002/open.202100224 2084633 (for bqmpp), 2072267 (for 1), 2072269 (for 2), 2072268 (for 3), 2072266 (for 5) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe href=http://www.ccdc.cam.ac.uk/structures Access Structures service.
Associated Content: Structure refinement data, NMR spectra of compounds 1, 3, 4 and 5.
Conflict of interest
The authors declare no conflict of interest.
4.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
Financial support by the German Federal Ministry for Economic Affairs and Energy on the basis of a decision by the German Bundestag (ZIM, Grant ZF4477702SL7) is gratefully acknowledged. Prof. Dr. T. M. Klapötke is thanked for his uninterrupted support over the years.
C. Kirst, J. Tietze, P. Mayer, H.-C. Böttcher, K. Karaghiosoff, ChemistryOpen 2022, 11, e202100224.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Fanfoni L., Meduri A., Zangrando E., Castillon S., Felluga F., Milani B., Molecules 2011, 16, 1804–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jaafar H., Li H., Misal Castro L. C., Zheng J., Roisnel T., Dorcet V., Sortais J.-B., Darcel C., Eur. J. Inorg. Chem. 2012, 2012, 3546–3550. [Google Scholar]
- 3. Munzeiwa W. A., Omondi B., Nyamori V. O., Beilstein J. Org. Chem. 2020, 16, 362–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wallesch M., Volz D., Zink D. M., Schepers U., Nieger M., Baumann T., Bräse S., Chem. A Eur. J. 2014, 20, 6578–6590. [DOI] [PubMed] [Google Scholar]
- 5. Artem'ev A. V., Davydova M. P., Berezin A. S., Ryzhikov M. R., Samsonenko D. G., Inorg. Chem. 2020, 59, 10699–10706. [DOI] [PubMed] [Google Scholar]
- 6. Rosario-Amorin D., Ouizem S., Dickie D. A., Wen Y., Paine R. T., Gao J., Grey J. K., De Bettencourt-Dias A., Hay B. P., Delmau L. H., Inorg. Chem. 2013, 52, 3063–3083. [DOI] [PubMed] [Google Scholar]
- 7. Chelucci G., Orrù G., Pinna G. A., Tetrahedron 2003, 59, 9471–9515. [Google Scholar]
- 8. Hung-Low F., Klausmeyer K. K., Polyhedron 2010, 29, 1676–1686. [Google Scholar]
- 9. Shankar B., Elumalai P., Shanmugam R., Singh V., Masram D. T., Sathiyendiran M., Inorg. Chem. 2013, 52, 10217–10219. [DOI] [PubMed] [Google Scholar]
- 10. Leca F., Lescop C., Rodriguez-Sanz E., Costuas K., Halet J. F., Réau R., Angew. Chem. Int. Ed. 2005, 44, 4362–4365; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 4436–4439. [Google Scholar]
- 11. Kirst C., Danaf N. A., Knechtel F., Arczynski T., Mayer P., Lamb D. C., Karaghiosoff K. L., J. Mater. Chem. C 2021, 9, 13366–13375. [Google Scholar]
- 12.E. I. Musina, A. S. Balueva, A. A. Karasik, in Organophosphorus Chem., Vol. 48 (Eds.: D. W Allen, D. Loakes, J. C. Tebby, 2019, pp. 1–63.
- 13. Musina E. I., Shamsieva A. V., Balueva A. S., Karasik A. A., Tertiary Phosphines: Preparation and Reactivity 2020. [Google Scholar]
- 14. Davydova M. P., Rakhmanova M. I., Bagryanskaya I. Y., Brylev K. A., Artem'ev A. V., J. Struct. Chem. 2020, 61, 894–898. [Google Scholar]
- 15. Davydova M. P., Berezin A. S., Samsonenko D. G., Artem'ev A. V., Inorg. Chim. Acta 2021, 521, 120347. [Google Scholar]
- 16. Hofbeck T., Niehaus T. A., Fleck M., Monkowius U., Yersin H., Molecules 2021, 26, 3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pechmann T., Brandt C. D., Werner H., Angew. Chem. Int. Ed. 2000, 39, 3909–3911; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 4069–4072. [Google Scholar]
- 18. Pechmann T., Brandt C. D., Röger C., Werner H., Angew. Chem. Int. Ed. 2002, 41, 2301–2303; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2398–2401. [Google Scholar]
- 19. Werner H., Angew. Chem. Int. Ed. 2004, 43, 938–954; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 956–972. [Google Scholar]
- 20. Schmidbaur H., Schier A., Angew. Chem. Int. Ed. 2015, 54, 746–784; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 756–797. [Google Scholar]
- 21. Scherbaum F., Grohmann A., Huber B., Krüger C., Schmidbaur H., Angew. Chem. Int. Ed. Engl. 1988, 27, 1544–1546; [Google Scholar]; Angew. Chem. 1988, 100, 1602–1604. [Google Scholar]
- 22. Dinda S., Samuelson A. G., Chem. A Eur. J. 2012, 18, 3032–3042. [DOI] [PubMed] [Google Scholar]
- 23. Ruan Z.-W., Zhang X., Pang A.-Y., Dai F.-R., Chen Z.-N., Inorg. Chem. Commun. 2020, 116, 107916. [Google Scholar]
- 24. Calhorda M. J., Ceamanos C., Crespo O., Gimeno M. C., Laguna A., Larraz C., Vaz P. D., Villacampa M. D., Inorg. Chem. 2010, 49, 8255–8269. [DOI] [PubMed] [Google Scholar]
- 25. Rong M. K., Holtrop F., Slootweg J. C., Lammertsma K., Coord. Chem. Rev. 2019, 382, 57–68. [Google Scholar]
- 26. Gibbons S. K., Hughes R. P., Glueck D. S., Royappa A. T., Rheingold A. L., Arthur R. B., Nicholas A. D., Patterson H. H., Inorg. Chem. 2017, 56, 12809–12820. [DOI] [PubMed] [Google Scholar]
- 27. Schäfer S., Gamer M. T., Lebedkin S., Weigend F., Kappes M. M., Roesky P. W., Chem. A Eur. J. 2017, 23, 12198–12209. [DOI] [PubMed] [Google Scholar]
- 28. Baranov A. Y., Pritchina E. A., Berezin A. S., Samsonenko D. G., Fedin V. P., Belogorlova N. A., Gritsan N. P., Artem'ev A. V., Angew. Chem. Int. Ed. 2021, 60, 12577–12584. [DOI] [PubMed] [Google Scholar]
- 29. Kirst C., Zoller F., Bräuniger T., Mayer P., Fattakhova-Rohlfing D., Karaghiosoff K., Inorg. Chem. 2021, 60, 2437–2445. [DOI] [PubMed] [Google Scholar]
- 30. Addison A. W., Rao T. N., Reedijk J., Van Rijn J., Verschoor G. C., Dalton Trans. 1984, 1349–1356. [Google Scholar]
- 31. Yang L., Powell D. R., Houser R. P., Dalton Trans. 2007, 955–964. [DOI] [PubMed] [Google Scholar]
- 32. Hung-Low F., Renz A., Klausmeyer K. K., Eur. J. Inorg. Chem. 2009, 2994–3002. [Google Scholar]
- 33. Peng R., Li M., Li D., Coord. Chem. Rev. 2010, 254, 1–18. [Google Scholar]
- 34. Janiak C., Dalton Trans. 2000, 3885–3896. [Google Scholar]
- 35. Jennings W. B., Farrell B. M., Malone J. F., Acc. Chem. Res. 2001, 34, 885–894. [DOI] [PubMed] [Google Scholar]
- 36. Batsanov S. S., Inorg. Mater. 2001, 37, 871–885. [Google Scholar]
- 37. Socol S. M., Verkade J. G., Inorg. Chem. 1984, 23, 3487–3493. [Google Scholar]
- 38. Attar S., Alcock N. W., Bowmaker G. A., Frye J. S., Bearden W. H., Nelson J. H., Inorg. Chem. 1991, 30, 4166–4176. [Google Scholar]
- 39. Bachman R. E., Andretta D. F., Inorg. Chem. 1998, 37, 5657–5663. [DOI] [PubMed] [Google Scholar]
- 40. Narayanaswamy R., Young M. A., Parkhurst E., Ouellette M., Kerr M. E., Ho D. M., Elder R. C., Bruce A. E., Bruce M. R. M., Inorg. Chem. 1993, 32, 2506–2517. [Google Scholar]
- 41. Uson R., Laguna A., Vicente J., J. Organomet. Chem. 1977, 131, 471–475. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.