

Abstract
Despite treatment with intensive chemotherapy, acute myeloid leukemia (AML) remains an aggressive malignancy with a dismal outcome in most patients. We found that AML cells exhibit an unusually rapid accumulation of the repressive histone mark H3K27me3 on nascent DNA. In cell lines, primary cells and xenograft mouse models, inhibition of the H3K27 histone methyltransferase EZH2 to de-condense the H3K27me3-marked chromatin of AML cells enhanced chromatin accessibility and chemotherapy-induced DNA damage, apoptosis, and leukemia suppression. These effects were further promoted when chromatin de-condensation of AML cells was induced upon S-phase entry after release from a transient G1 arrest mediated by CDK4/6 inhibition. In the p53-null KG-1 and THP-1 AML cell lines, EZH2 inhibitor and doxorubicin co-treatment induced transcriptional reprogramming that was, in part, dependent on de-repression of H3K27me3-marked gene promoters and led to increased expression of cell death-promoting and growth-inhibitory genes.
In conclusion, decondensing H3K27me3-marked chromatin by EZH2 inhibition represents a promising approach to improve the efficacy of DNA-damaging cytotoxic agents in AML patients. This strategy might allow for a lowering of chemotherapy doses with a consequent reduction of treatment-related side effects in elderly AML patients or those with significant comorbidities.
Keywords: Chromatin accessibility, EZH2, Epigenetics, Chemotherapy, Apoptosis
Introduction
AML arises from transformation of early hematopoietic progenitor cells (HPCs), leading to clonal expansion and differentiation arrest (1). AML is genetically heterogeneous (2–5), with distinct subclones coexisting in the same patient and creating obstacles for the development of effective molecularly-targeted therapies (6–8). Thus, the frontline therapy for most AML patients remains non-targeted intensive induction chemotherapy. This regimen is generally offered only to “fit” patients (young adult and/or individuals of high-performance status), and therefore effective therapies for elderly or unfit patients are needed.
Most conventional chemotherapeutic agents induce DNA damage and cell death by targeting tumor cells during DNA replication (9–11). However, chromatin structure influences the efficacy of DNA damaging agents. For example, compacted chromatin is more resistant to formation of double-strand breaks (DSBs) (12,13), and histone deacetylase (HDAC) inhibitors, which cause chromatin relaxation, increase sensitivity of cancer cells to ionizing radiation (14). Moreover, chemical profiling of DNA damage sites induced by cytotoxic drugs suggests that DNA damage induced by Topoisomerase II inhibitors Daunomycin and Etoposide is not random, but preferentially targets H3K36me3- and H3K9me1-marked transcriptionally active, de-condensed regions of the genome (15,16). By contrast, H3K27me3-marked, repressed genomic regions are relatively inaccessible to these cytotoxic agents (15,16). In euchromatin, H3K27me3-marked nucleosome arrays represent the most compact chromatin in the genome (17,18), suggesting that de-condensing its structure might enhance the efficiency of cytotoxic agents.
Using the chromatin assembly assay (CAA) to examine the loading of chromatin proteins on nascent DNA at single-cell resolution (19–22), we discovered that, upon induction of differentiation, embryonic stem cells (ESCs) and CD34+ HPCs exhibit a significant period when post-replicative chromatin is globally devoid of the repressive histone mark H3K27me3 (21,22). This transient phase of ‘open’ nascent chromatin allows the recruitment of lineage-determining transcription factors (TFs) onto nascent DNA to promote differentiation (21,22).
In the present study, we used the CAA to examine the recruitment of H3K27me3 EZH2 histone methyltransferase (HMT, a component of the Polycomb Repressive Complex 2) and the deposition of H3K27me3 to nascent DNA of CD34+ AML blasts. EZH2 and H3K27me3 were readily detected on nascent DNA of AML cell lines and blasts from newly diagnosed patients. These findings provided the rationale for combining EZH2 inhibitors and cytotoxic drugs with the goal of enhancing DNA damage and apoptosis of AML blasts. We show here that ‘opening’ the H3K27me3-marked chromatin of AML cells by EZH2 inhibition renders DNA more vulnerable to DNA damaging agents, leading to enhanced cell death ex vivo and leukemia suppression in mice. Such effects were further improved by enhancing the targeting of nascent chromatin through enrichment of S-phase AML cells. In the p53-null KG-1 and THP-1 cell lines, treatment with EZH2 inhibitor/Doxorubicin induced a transcriptional reprogramming that was dependent, in part, on de-repression of H3K27me3-marked promoters and led to increased expression of cell death and growth-inhibitory genes.
Materials and Methods
Cell lines, primary AML samples and cell cultures
The THP-1 line was purchased from ATCC. The KG-1 and KG-1a lines were provided by Dr. Kathrin M. Bernt (The Children’s Hospital of Philadelphia). Cells were cultured in Iscove’s Modified Dulbecco’s Medium (Gibco) supplemented with 10% heat-inactivated FBS (Biowest USA), 1% penicillin–streptomycin and 1% L-glutamine (Thermo Fisher Scientific) at 37°C, 5% CO2. Cell lines were tested for mycoplasma every 3 months as described (23). The lines carry a deletion or single nucleotide mutation in the p53 gene causing premature termination of p53 mRNA translation potentially generating truncated proteins (p53-176 and p53-226 in the THP-1 and KG-1 lines, respectively) (24). However, these cell lines can be considered p53-null since they do not express detectable levels of wild-type or truncated p53 proteins.
Primary AML samples were obtained after written informed consent in accordance with the Declaration of Helsinki and the Thomas Jefferson University IRB-approved protocol #17D.803 (PI-Palmisiano).
Molecular characteristics and clinical outcomes of the samples are described in Supplemental Table I. Primary cells were maintained in SFEM supplemented with StemSpan™ CC100 and recombinant thrombopoietin (TPO), all from Stem Cell Technologies.
Drugs
Doxorubicin, Ara-C, and Bleomycin were obtained from Cayman Chemical and resuspended in sterile water. Etoposide was purchased from Sigma-Aldrich and resuspended in DMSO.
For ex vivo studies, GSK126 and UNC1999 were purchased from Cayman Chemical; EPZ-6438 was purchased from Selleckchem and Palbociclib was obtained from LC Laboratories. They were resuspended in DMSO.
Immunofluorescence and Confocal Microscopy
1.5-2.0 x 104 cells were spun onto slides using a cytocentrifuge, fixed for 10 min in 3.7% paraformaldehyde at room temperature (RT) and washed with PBS. Cells were permeabilized with 0.25% Triton-X/PBS and blocked with the #WESTBL-RO solution (Roche) solution for one hour. Then, cells were incubated with primary antibodies (γ-H2AX, 1:200 Millipore #056361; RAD51, 1:100, Santa Cruz #8349) overnight at 4°C. Slides were washed with 1X PBS and incubated 1 hour at RT with secondary antibodies (Alexa Fluor 488 goat anti-mouse or Alexa Fluor 555 goat anti-rabbit (Thermo Fisher Scientific). Samples were mounted using DAPI Fluoromount-G (Southern Biotech; #0100-20). Image acquisition was performed using a confocal microscope Nikon C2 with lasers set on λ = 405-488-562 nm. Images were analyzed using Fiji ImageJ 2.0 (Cambridge Astronomical Survey Unit) and Adobe Photoshop 2020 (Adobe Systems) software.
Comet Assay
Neutral comet assays were performed on AML cell lines and primary samples as described (25). Images were captured on a Nikon Eclipse Ni (Nikon) with a 10X objective and analyzed using CometScore software. All samples were blinded and 80-120 cells per sample were scored.
Apoptosis
Cells were incubated at 100,000/ml with 1μl of the CellEvent Caspase 3/7 Green Detection Reagent (#C10427; Thermo Fisher) for 15 min at 37°C and analyzed by flow cytometry using a 488-nm excitation laser. Apoptosis was also assessed by Annexin V staining. For this assay, 100,000 cells were spun down and resuspended in 50 μL of Annexin V Binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4), containing 1 μl of Cy 5.5-Annexin V (#559935; BD Pharmigen) for 15 min at RT. Samples were analyzed by flow cytometry using a 640 nm laser.
Cell Cycle Analysis
For cell cycle analysis, cells were incubated for 5 min with sodium citrate 0.1%, Triton-X 0.1%, propidium iodide 50 μg/ml. DNA content was subsequently analyzed using a BD FACS Celesta flow cytometer using a 561 nm laser.
Chromatin Assembly Assay
CAA experiments were performed as described (21) with minor modifications. AML cells were pulse-labeled with 5 μM EdU for 15 min. Approximately 20,000 cells per each condition were harvested and cytospun (750 rpm for 5 min) onto polylysine-coated slides (Polyscience), fixed with 4% formaldehyde, permeabilized in 0.25% Triton-X, and subjected to Click-iT reaction (Invitrogen) to conjugate biotin to EdU. Cells were incubated with mouse antibody to biotin and rabbit antibodies to the protein of interest (H3K27me3: Millipore #07-449; EZH2: Cell Signaling Technology # 5246; H2AK119ub: Cell Signaling Technology #8240S; RING1b: Cell Signaling Technology #5694). Proximity ligation assay (PLA) reactions were performed according to the manufacturer (Olink), as described (21). Following PLA, EdU was detected by immunostaining with Alexa Fluor 488-conjugated anti-biotin mouse antibody.
RNA-sequencing
RNA-seq was performed on three independent biological replicates per condition. Samples from different conditions were processed together to avoid differences in batch preparation. For each sample, 2 x 106 cells were used to extract total RNA and generate RNA-seq libraries. KG-1 cells were plated at 1 x 106 cells/ml in SFEM supplemented with StemSpan™ CC100 (Stem Cell Technologies) and treated with Doxo (0.3 μM; 24 hours), GSK126 (5 μM; 12 hours pre-treatment plus 24 hours treatment), or the GSK126/Doxo combination (GSK126 5μM, 12 hours pre-treatment plus 24 hours treatment; Doxo (0.3 μM; 24 hours). Total RNA was isolated with the RNeasy Plus Mini Kit (#74134, Qiagen) and used to prepare libraries using NEBNext® Ultra™ II RNA Library Prep Kit for Illumina (New England Biolabs Inc., MA, USA), following the manufacturer’s protocol. Libraries were sequenced on a NextSeq 500 instrument using 75-bp single-end chemistry. Raw FASTQ sequencing reads were mapped against the reference human genome Ensembl Version GRCh38 utilizing additional information from the gene transfer format (.gtf) annotation from GENCODE version GRCH29 using RSEM. Total read counts and normalized Transcripts Per Million (TPM) were obtained using RSEM’s calculate-expression function. Data were deposited to SRA database (BioProjectID: PRJNA693321). Before determining differential expression, batch effects and sample heterogeneity were tested using iSeqQC (26). Differential gene expression was tested using the DESeq2 package in R/Bioconductor. Genes were considered differentially expressed (DE) if they had adjusted p ≤ 0.05 and absolute fold change ≥ 2. Plots were generated using R/Bioconductor. Canonical pathways modulated by drug treatments were identified by DAVID (27).
ChIP Sequencing and ChIP-seq analyses
Samples from different conditions were processed together to prevent batch effects. For each condition, 3 x 106 KG-1 cells were cross-linked with 1% formaldehyde for 5 min at RT, washed once in 1.25 M glycine/PBS and twice with 1X cold PBS. The pellet was resuspended in ChIP lysis buffer following a published protocol (28). Barcoded libraries were made with NEB ULTRA II DNA Library Prep Kit for Illumina, and sequenced on Illumina NextSeq 500, producing 75bp SE reads. Chip-seq analyses were performed as described in the Supplemental Methods. Data were deposited to SRA database (BioProjectID: PRJNA693).
Mice
For leukemogenesis assays, 2x106 THP-1 Luc cells or 1.5x106 cells from AML sample #040 were injected i.v. in 7- to 9-week-old NRG NOD-Rag1null IL2rgnull or NRG-SGM3 mice (The Jackson Laboratory).
GSK126 (Chemgood) was dissolved in 20% Captisol adjusted to pH 4.5 with 1 N acetic acid and administered i.p. every 24 hours for 7 days.
Mice were treated for five consecutive days with 5+3 chemotherapy consisting of: 3 days of i.v. co-injection of Ara-C (50 mg/kg; Sigma-Aldrich) and Doxorubicin (1.5 mg/kg; Sigma-Aldrich), followed by i.p. injection of Ara-C alone on days 4 and 5.
Palbociclib isethionate was purchased from LC Laboratories and mixed in the chow by Research Diets Inc at 800 mg/kg. This dosing was based on the average daily food intake of NRG mice to deliver 150 mg/kg/day of Palbociclib given at libitum for the duration of the experiment. Leukemia burden was monitored by imaging of mice injected with THP-1-Luciferase cells or by flow cytometry of peripheral blood human CD45+ cells in mice injected with primary AML cells. Animal experiments were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University under protocol number 00012.
Statistical Analyses
Data, expressed as mean ± S.D. of three experiments, were analyzed for statistical significance by unpaired, two-tailed Student’s t-test or One-way ANOVA. P<0.05 was considered statistically significant. Kaplan–Meier plots for mouse survival experiments were generated using the GraphPad Prism 8.0 software. Statistical significance of differences in survival was assessed by log-rank test.
ATAC-sequencing, Colony Formation Assays, Lentiviral Production and Cell Transduction, Protein Analysis, Quantitative PCR
Detailed protocols for these experimental procedures are described in Supplementary Materials and Methods and Supplemental Table II.
Results
The H3K27me3 repressive mark accumulates rapidly on nascent DNA of AML blasts.
We used our previously developed CAA (21) to examine the accumulation of H3K27me3 on nascent DNA of single AML blasts. In this assay, DNA is labeled in living cells in a pulse-chase manner with EdU which is then chemically conjugated with biotin. The proximity of a protein to nascent (EdU positive) DNA is then examined by the proximity ligation assay (PLA) using antibodies to biotin and to the protein of interest. We observed that H3K27me3 accumulates rapidly on nascent DNA of AML cell lines THP-1, KG-1 (Fig. 1A and B), KG-1a (Supplemental Fig. 1A) and of CD34+ blasts of several AML primary samples with recurrent molecular abnormalities (eg., FLT3-ITD/TKD, NPM1, DNMT3A, and NRAS mutations) (Fig. 1C–D and Supplemental Table I). These mutations have no direct effect on the expression/activity of H3K27 HMTs and demethylases. Instead, in CD34+ cells of AML sample #8, which carries a frameshift mutation of ASXL1 (G646fs*12) in ~40% of the cells causing decreased H3K27me3 levels (29), accumulation of H3K27me3 on nascent DNA was heterogeneous with several EdU-positive cells showing weak H3K27me3 loading (Fig. 1D).
Figure 1. Accumulation of H3K27me3 on nascent DNA of AML cells.
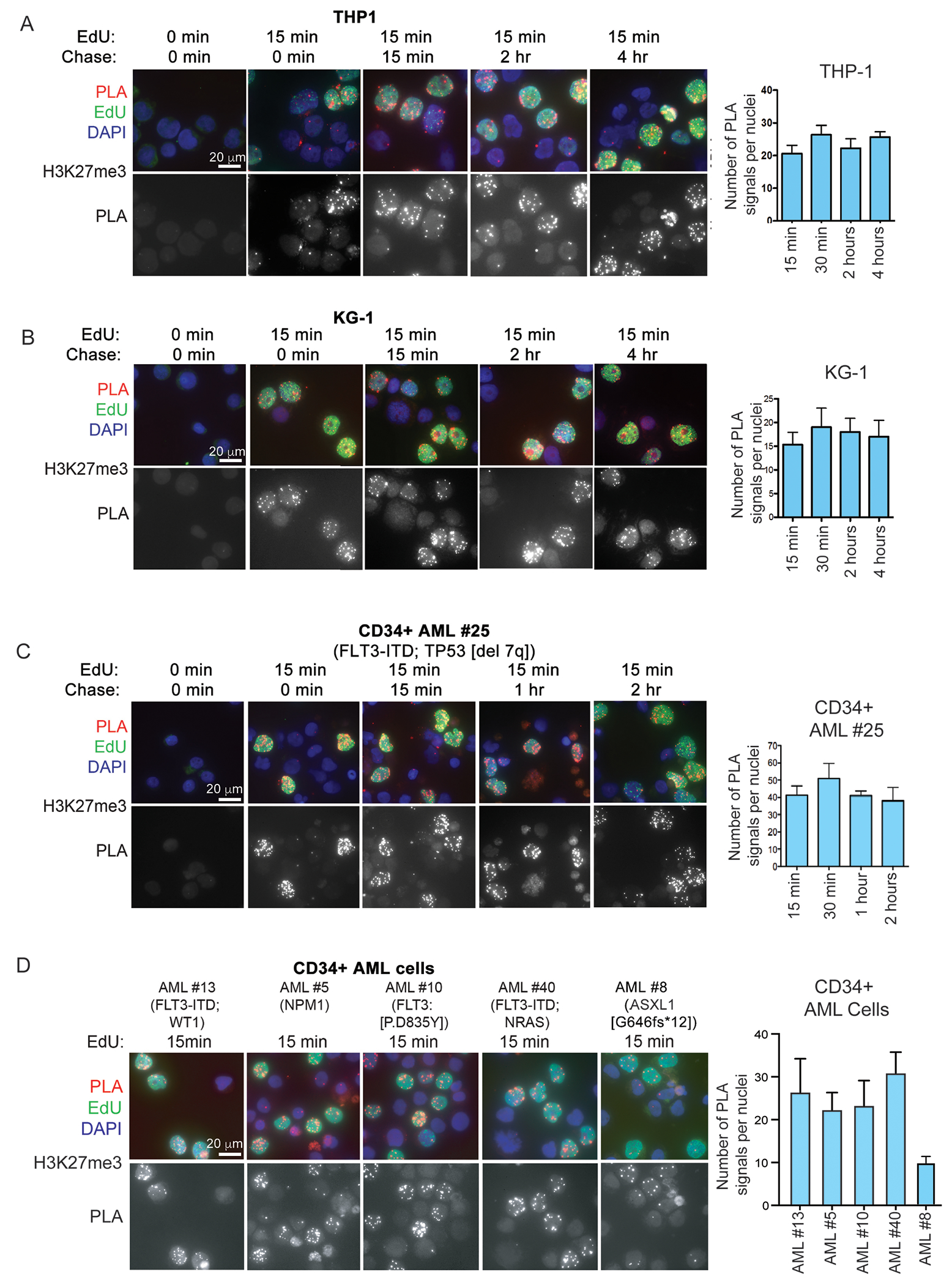
DNA of AML cell lines (A, B), and primary AML samples (C, D) was labeled with EdU for 15 min and chased for the indicated times (A, B, C) or processed without chase (D). After biotin conjugation, CAA was performed between nascent DNA (biotin) and H3K27me3. PLA (red), Edu (biotin; green), DAPI (blue). Lower panels show PLA signals only. Quantification of CAA experiments is shown to the right.
Since the H3K27me3 mark is associated with the most condensed nucleosome arrays in the genome (17,18, 30), the fast accumulation of H3K27me3 on nascent DNA of AML blasts indicates that these cells possess a “condensed” structure of nucleosomes at H3K27me3-marked genomic regions following DNA replication.
The level of H3K27me3 depends on the activity of EZH2 and, to a lesser extent, of EZH1 (31). We found that EZH2 is associated with nascent DNA of THP-1, KG-1, and KG-1a cell lines (Supplemental Fig. 2A) and of CD34+ blasts from several primary AML samples (Supplemental Fig. 2B), in line with high H3K27me3 levels on their nascent DNA.
Inhibition of EZH2 activity by GSK126 or EPZ-6438 treatment (32) of THP-1 or of primary cells of sample AML#40 markedly decreased H3K27me3 loading on nascent DNA (Supplemental Fig. 3A and B).
We assessed whether inhibiting EZH2 led to genome-wide changes in chromatin accessibility of THP-1 and KG-1 cells. Using the ATAC-seq assay, we identified an average of 11,823 and 16,198 new accessible sites in GSK126-treated THP-1 and KG-1 cells respectively, compared to the untreated counterparts (Supplemental Fig. 3C and D), consistent with enhanced chromatin accessibility. The vast majority of the newly accessible genomic sites correspond to regulatory regions including promoters and intronic/intergenic enhancers.
A component of the Polycomb Repressor Complex 1 (PRC1) the H2A119 ubiquitin ligase RING1B, and H2A119ub are also rapidly associated with EdU-labeled DNA in AML cell lines KG-1 and THP-1, and H2A119ub stably associates hereafter (Supplemental Fig. 4). These findings suggest that the compaction of H3K27me3-marked chromatin of AML cells is further ensured by rapidly establishing and maintaining a functional PRC1 on their nascent DNA.
De-condensing the H3K27m3-marked post-replicative chromatin of AML blasts enhances chemotherapy-induced DNA damage.
Having established that inhibiting EZH2 de-condenses ‘nascent’ H3K27me3-marked chromatin in AML blasts, we investigated whether this effect increases their sensitivity to DNA-damaging agents. DNA damage was examined by counting γ-H2AX foci (33) and DNA breaks using the comet assay (25). Compared to untreated cells, treatment of THP-1 cells with Doxorubicin (Doxo) caused a marked increase in the number of γ-H2AX foci and DNA breaks, and these numbers were further enhanced by a 16-hour pre-treatment with GSK126 to de-condense H3K27me3-marked chromatin followed by GSK126/Doxo co-treatment (Fig. 2A). Enhanced DNA damage was also observed in EZH2-silenced, Doxo-treated THP-1 cells compared to cells treated with Doxo only (Supplemental Fig. 5 A, B).
Figure 2. EZH2 inhibition enhances cytotoxic drug-induced DNA damage in Doxo-treated AML cells.
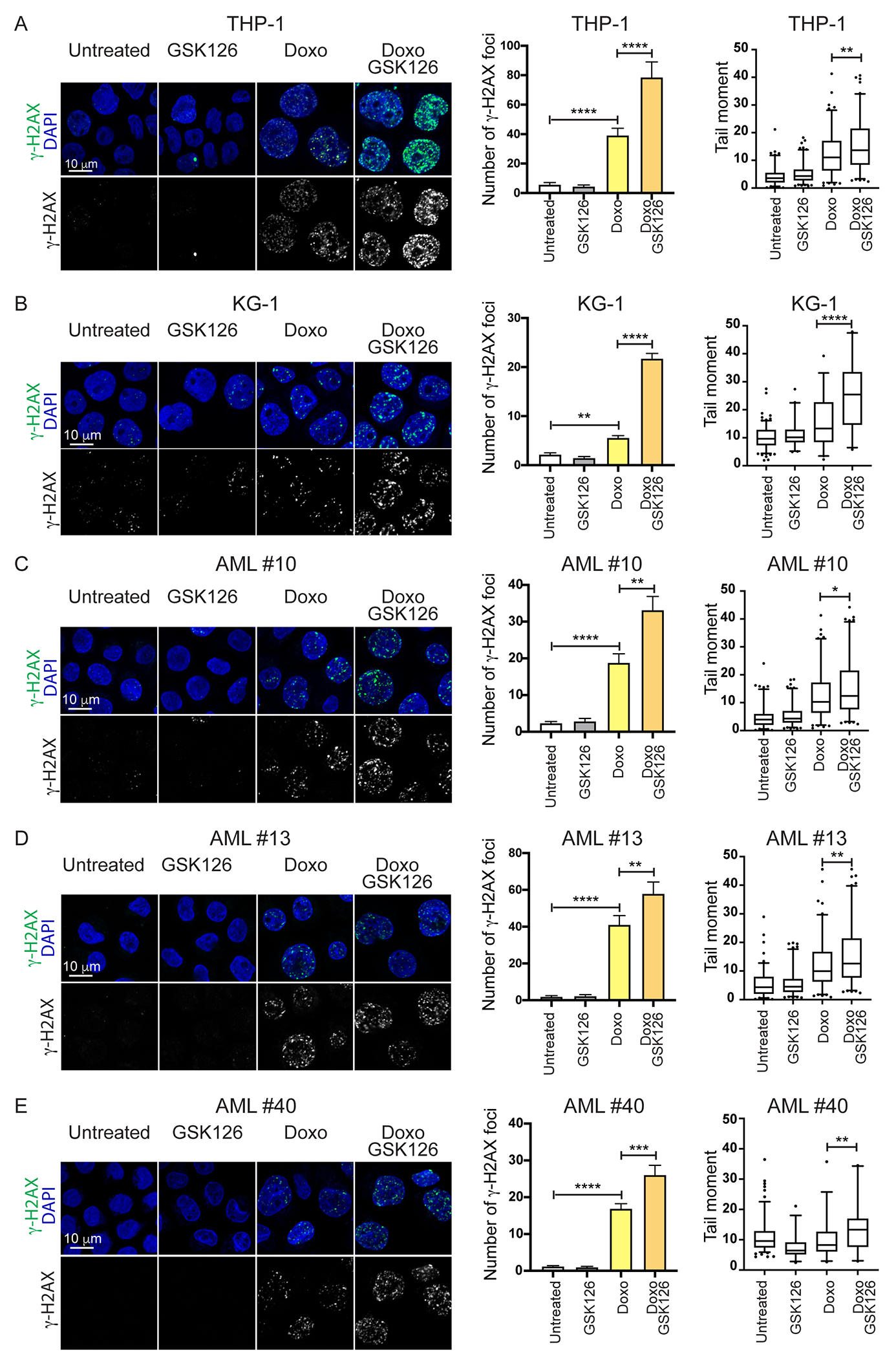
(A-E) Evaluation of DNA damage (immunofluorescence microscopy analysis of γ-H2AX foci and comet assay) in cytotoxic agent-treated AML cell lines THP-1 and KG-1 (A, B) or primary AML cells (samples #10, #13, and #40; C-E). Cells were treated with GSK126 (5 μM) alone, Doxo (0.1 μM) alone, or pre-treated with GSK126 and then treated with the GSK126/Doxo combination for 24 hr. Quantification of γ-H2AX foci plus SEM is shown in the middle. One-way Anova: *p<0.05, **p<0.01; ***p<0.001 ****p<0.0001. Comet assay in drug-treated AML cells is shown to the right; 80–120 cells were analyzed in each sample. One-way Anova; *p<0.05; **p<0.01; ***p<0.0001.
Higher levels of DNA damage were also detected in GSK126/Doxo-treated AML cell lines KG-1 (Fig. 2B) and KG-1a (Supplemental Fig. 1B), and in CD34+ cells of primary AML patient samples (Fig. 2C–E), as well as in THP-1 and primary AML samples co-treated with the dual EZH1/2 inhibitor UNC1999 (34) and Doxo (Supplemental Figure 6A–D).
Notably, GSK126 treatment did not increase Doxo-induced DNA damage in AML sample #8 (Supplemental Fig. 7). This sample exhibits low-levels of H3K27me3-marked nascent chromatin in many cells due to the ASXL1 mutation in 40% of the cells (Fig. 1D). AML sample #8 also has a loss-of-function mutation of the STAG2 gene in >90% of the cells (Supplemental Table I). AML cells with mutant STAG2 exhibit high sensitivity to DNA-damaging agents including doxorubicin (35). Thus, the combination of low-H3K27me3 levels associated with the ASXL1 mutation and the high sensitivity to DNA-damaging agents associated with STAG2 mutations might explain why EZH2 inhibition did not enhance further Doxo-induced DNA damage in AML sample #8. Significantly higher levels of DNA damage were also observed in THP-1 cells co-treated with GSK126 and Bleomycin (a drug that causes single-strand and double-strand breaks directly) or GSK126 and Etoposide (Supplemental Fig. 8)
The homologous recombination (HR) DNA repair pathway is frequently altered in AML cells (36). Thus, we asked whether the increase in DNA damage in GSK126/Doxo-treated AML cells was due to defective DNA damage repair. To this end, we analyzed the number of RAD51 foci (indicative of HR-DSB repair) and observed that they were not diminished by GSK126 treatment following Doxo-induced DNA damage in THP-1 cells (Supplemental Fig. 9A). Likewise, GSK126 treatment of THP-1 cells did not diminish the number of 53BP1 foci (indicative of NHEJ repair) compared to treatment with Doxo only (Supplemental Fig. 9B).
Together, these findings strongly suggest that, at least in THP-1 cells, defective DSB repair does not play an important role in the increase in Doxo-induced DNA damage.
De-condensing H3K27me3-marked chromatin enhances chemotherapy-induced apoptosis and inhibition of colony formation of AML blasts.
To determine whether the increased DNA damage in GSK126/Doxo-treated AML cells results in enhanced cell death, we assessed apoptosis by flow cytometry analysis of Caspase 3/7 activation and by Annexin V staining. Consistent with the increase in DNA damage, apoptosis was markedly enhanced in GSK126/Doxo-treated AML cell lines and CD34+ blasts from several AML patients, compared to treatment with Doxo only (Fig. 3A; Supplemental Fig. 10). Enhanced apoptosis was also observed in the THP-1 and KG-1 lines co-treated with the selective EZH2 inhibitor EPZ-6438 and Doxo and in primary AML samples co-treated with the dual EZH1/2 inhibitor UNC1999 and Doxo, compared to treatment with Doxo alone (Supplemental Figure 11).
Figure 3. EZH2 inhibition enhances apoptosis of cytotoxic agent-treated AML cells.
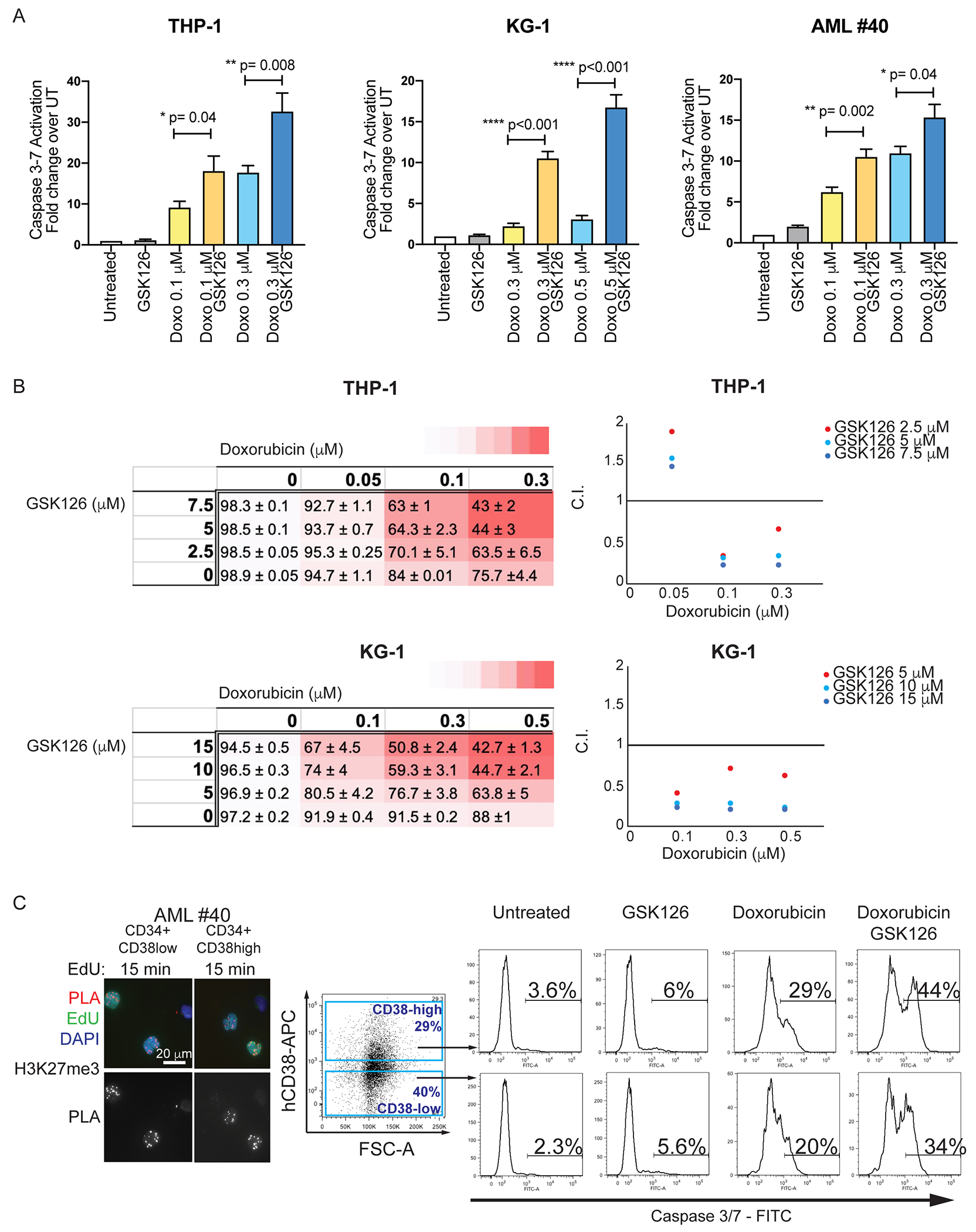
(A) THP-1, KG-1, and CD34+ AML primary cells (sample #40) were treated with Doxo alone (0.1 μM, 0.3 or 0.5 μM), or in combination with GSK126 (5 μM), for 48 hr. Apoptosis (Caspase 3/7 activity) was analyzed by flow cytometry. Graph bars represent fold change +/− SEM in the % of Caspase 3/7 activation over the untreated control. Unpaired t-test: p<0.05; **p<0.01; ***p<0.001; (B) Combination Indexes (C.I.) of GSK126/Doxorubicin-induced apoptosis in THP-1 and KG-1 cells. THP-1 or KG-1 cells were treated with three different concentrations of GSK126 and/or Doxorubicin. Left: Dose-response matrices showing % of viable cells (+/−SEM) after treatment with increasing doses of Doxorubicin, GSK126 and combinations of drug pair Doxo/GSK126. Colors in the dose-response matrices indicate different levels of responses (from white to red, decreasing viable cells) of THP-1 and KG-1 cells. Right: Viability data elaborated using CompuSyn software to calculate C.I. plotted on the Y-axis of a 2D graph. The X axes show increasing concentrations of Doxorubicin, while the different colors indicate increasing concentrations of GSK126 (see labels). (C.I.= 0.90-1.10 indicates additive effects; C.I.=0.1-0.9 indicates synergistic effects). (C) H3K27me3 accumulation on nascent DNA (left) and Caspase 3/7 activation in AML sample #40 CD34+CD38high or CD34+CD38low cells treated with GSK126 (5 μM), Doxorubicin (0.1 μM), or the two drugs in combination.
Apoptosis induced by the Doxo 0.1μM/GSK126 5μM co-treatment was similar to that induced by a 3-fold higher concentration of Doxo alone in THP-1 and AML #40 cells. In KG-1 cells, the Doxo 0.3μM/GSK126 10μM combination was markedly more effective than Doxo alone at 0.3 or 0.5 μM (Fig. 3A). Compared to single drug treatment, the GSK126/Doxo combination increased synergistically the number of apoptotic THP-1 and KG-1 cells at multiple doses as indicated by Chou-Talalay combination indices (Fig. 3B).
Blast cells of AML sample #40 are exclusively CD34+ and a significant portion of these cells consists of the more primitive CD34+CD38low subset, providing the opportunity to test whether this stem cell-enriched subset is also vulnerable to co-treatment with GSK126/Doxo. Both subsets exhibited a similar amount of H3K27me3-marked nascent chromatin and a comparable increase in apoptosis upon GSK126/Doxo co-treatment (Fig. 3C). Enhanced apoptosis susceptibility by GSK126/Doxo co-treatment was also observed in the CD34+CD38high and the CD34+CD38low subsets of two additional AML samples (Supplemental Fig. 12). Notice that AML sample #15 has a homozygous p53-null mutation in approximately 90% of the cells (Supplementary Table I). Enhanced apoptosis was also observed in EZH2-silenced, Doxo-treated THP-1 cells compared to cells treated with Doxo only (Supplemental Fig. 5C).
We also examined the effect of the GSK126/Doxo combination on colony formation of AML cells. Single treatments with Doxo or GSK126 caused moderate suppression of AML colonies; however, the GSK126/Doxo co-treatment almost completely eradicated clonogenic AML cells in the tested samples (Supplemental Fig. 13), suggesting that this cell subset may be particularly vulnerable to enhanced DNA damage promoted by chromatin de-condensation. Notably, the GSK126/Doxo combination was at least as effective as single Doxo treatments used at 2-3 fold higher concentrations.
De-condensing H3K27me3-marked chromatin of S-phase-enriched AML cells further increases Doxo-induced DNA damage and apoptosis.
Doxorubicin induces DNA damage during DNA replication by inhibiting DNA Topoisomerase II and by preventing re-ligation of DNA breaks (10). Consistent with this mode of action, inhibiting S-phase entry by treating THP-1 cells with the CDK4/6 inhibitor Palbociclib (Supplemental Fig. 14A) completely suppressed Doxo-induced DNA damage and apoptosis (Supplemental Fig. 14 B,C). Based on these findings, we hypothesized that our strategy of combining an EZH2 inhibitor with a S-phase-selective cytotoxic agent (Doxo) would further increase its efficiency when directed to replicating cells. Thus, THP-1 cells were first treated with Palbociclib to induce G1 arrest, and then allowed to re-enter the cell cycle after Palbociclib washout. This treatment significantly increased the number of S-phase cells (~54%; at 8-10 hours post-washout) compared to the unsynchronized counterpart (S-phase: ~24%) (Fig. 4A). Likewise, GSK126 treatment of S-phase-enriched THP-1 cells markedly increased their chromatin accessibility compared to GSK126-treated unsynchronized cells, as revealed by the detection of ~ 30,000 new ATAC-seq peaks in S-phase-enriched THP-1 cells (Fig. 4B). Consistent with these effects, GSK126/Doxo co-treatment of S phase-enriched THP-1 cells strongly increased the number of DNA damage foci and of apoptotic cells, compared to GSK126/Doxo-treated unsynchronized THP-1 cells (Fig. 4C).
Figure 4. GSK126 treatment of S phase-enriched AML cells further increases Doxo-induced DNA damage and apoptosis.
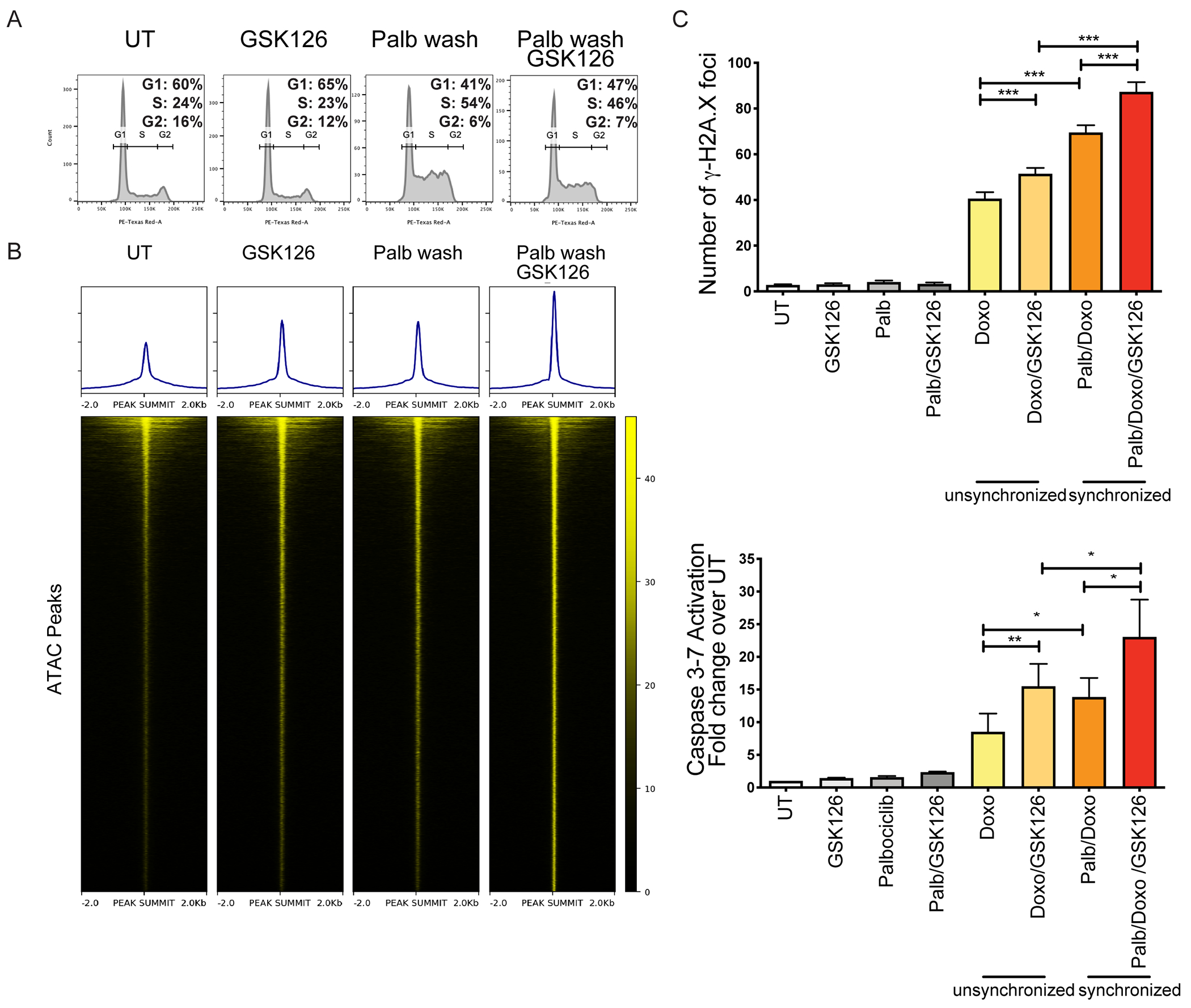
(A) DNA content analysis of THP-1 cells (untreated, treated with Palbociclib (24 h; 500 nM)/washed, and cultured for an additional 10 hours in medium without Palbociclib or without Palbociclib+GSK126. (B) Heatmaps of normalized chromatin accessibility reads from ATAC-seq experiments in untreated and GSK126-treated unsynchronized THP-1 cells and in cells released from Palbociclib-induced cell cycle arrest and left untreated or treated with GSK126; heatmaps are ordered by total ATAC signals and each row represents the same DNA segment in all conditions. (C) THP-1 cells were treated with Palbociclib (500 nM; 24 h) to induce G1 arrest. After washing, THP-1 cells re-entering S phase were treated with GSK126 (5 μM; 12 h), before adding Doxo (0.1 μM) or GSK126/Doxo. DNA damage and apoptosis were examined after 24 h and 48 h, respectively. Left, quantification of γ-H2AX foci +/− SEM. Anova ***p<0.001; Right, apoptosis (Caspase 3/7 activity) by flow cytometry. Mean +/− SEM. Anova, *p<0.05: **p<0.01.
Transcriptional activation of pro-apoptotic and growth-inhibitory genes in AML cells treated with the GSK126/Doxo combination
The increased DNA damage and apoptosis of GSK126/Doxo-treated AML cells is likely to correlate with changes in gene expression that may reflect the cumulative effects induced by Doxo and GSK126 treatment, a new transcription program modulated by the GSK126/Doxo combination, or both.
To investigate this, we performed RNA-seq analyses of untreated, Doxo- or GSK126- treated (24 hr) KG-1 cells, or of cells pre-treated with GSK126 (16 hr) and then co-treated with GSK126/Doxo for 24 hr. The p53-null KG-1 line was chosen because the GSK126/Doxo treatment was highly effective in inducing DNA damage and apoptosis (detected after 24 and 48-hour treatment, respectively), compared to single treatments (Fig. 2B and Fig. 3A). Of note, no apoptosis is detected in cells treated with GSK126/Doxo for 24 hr. Moreover, AML with p53 mutations is associated with chemoresistance and disease relapse (37), suggesting that findings in the p53-null KG-1 line may be translationally significant.
Compared to untreated cells, the GSK126/Doxo co-treatment was much more effective in modulating gene expression than treatment with Doxo or GSK126 alone (1,593 genes up-/down-regulated by GSK126/Doxo co-treatment versus 80 and 81 up-/down-regulated by GSK126 or Doxo treatments alone; FDR <5%) (Fig. 5A). Of the genes differentially expressed in GSK126/Doxo-treated vs. untreated cells, 1,508 were specific for the GSK126/Doxo co-treatment. Using DAVID (26) to perform functional analysis of these genes, cytokine-mediated signaling (p= 4.5 x 10−20), type I and IFN-γ response (p=7.1 x 10−16 and p=1.8 x 10−8, respectively) were the three top-ranked pathways. Fig. 5B shows the heat-map of selected genes included in these pathways that were up-regulated by the GSK126/Doxo co-treatment; qPCR analysis confirmed the selective GSK126/Doxo-induced upregulation of these genes, a subset of which is shown in Fig. 5C. These pathways include genes with the potential to induce apoptosis (e.g., STAT1, IRF-1, IRF-9, ISG-15, and ISG-20) (38–40). Several genes typically regulated by Doxo via a p53-dependent mechanism (eg., BBC3 (PUMA), CDKN1A (p21), TP53INP2, and TP53I11) were also markedly overexpressed in GSK126/Doxo-treated p53-null KG-1 cells (Fig. 5B and Fig. 5C). Increased expression of some of the genes activated by GSK126/Doxo co-treatment was also confirmed by western blot, concomitant with a marked increase in γ-H2AX levels (Fig. 5D).
Figure 5. Gene subset regulated by GSK126/Doxo treatment in the KG-1 cell line.
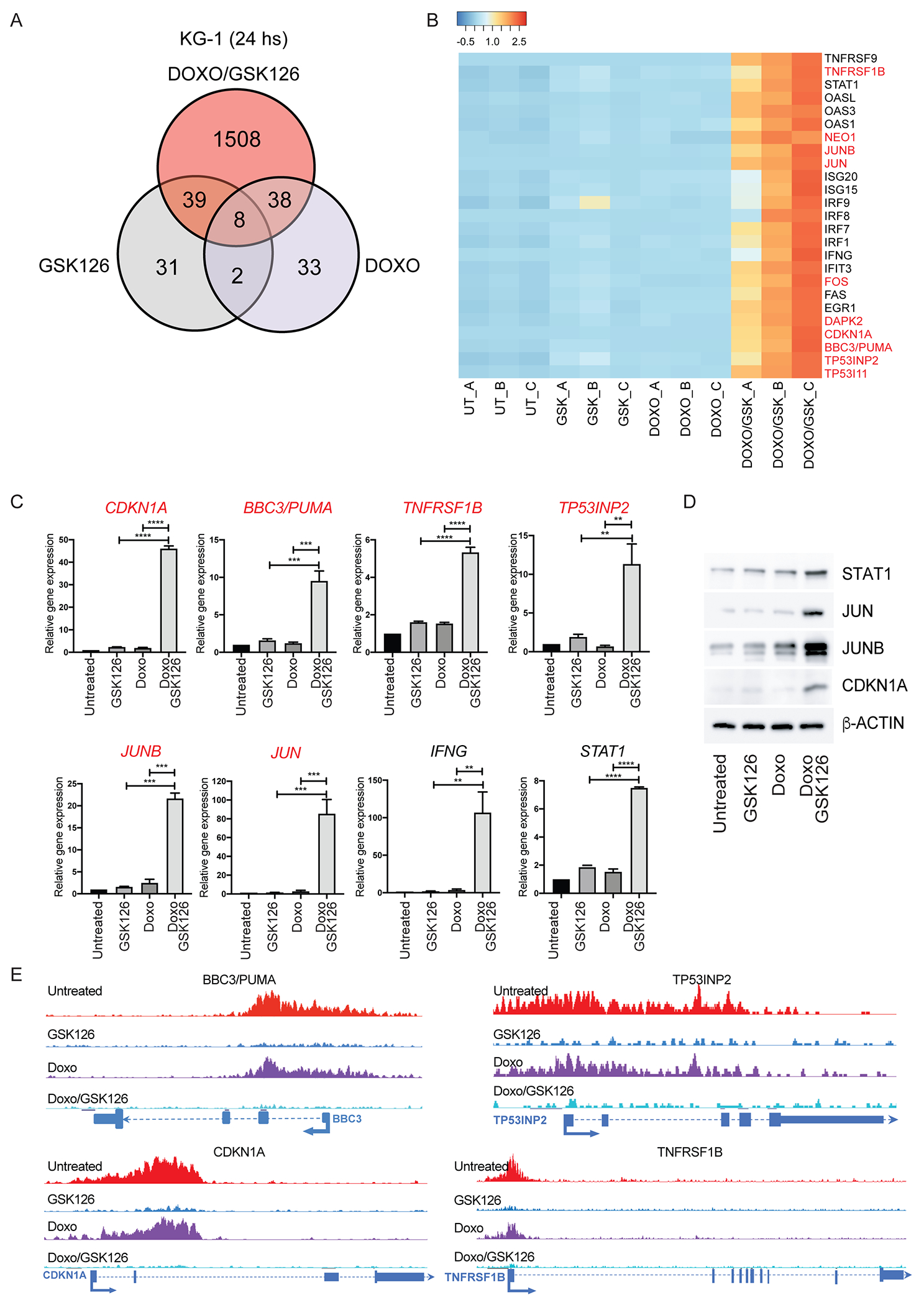
(A) Venn diagram of genes differentially expressed in GSK126/Doxo-treated vs. Doxo- or GSK126-treated KG-1 cells; (B) Heat-map of genes selectively regulated by GSK126/Doxo compared to untreated or single GSK126 or Doxo treatments; (C) qPCR analysis of selected genes differentially regulated by GSK126/Doxo treatment. Data represent mean + SD of three independent experiments. Statistical analysis: one way ANOVA with Bonferroni’s correction; **p<0.01, ***p<0.001, ****p<0.0001; (D) Western blot analysis of selected genes overexpressed in GSK126/Doxo-treated cells; (E) Examples of four genes from H3K27me3 ChIP-seq in KG-1 cells showing loss of H3K27me3 marking after GSK126 or GSK126/Doxo treatment. Genes labeled in red in the heat-map and in the q-PCR experiments (B, C, D) exhibit loss of H3K27me3 marking after GSK126 or GSK126/Doxo treatment.
The increased expression of selected genes induced by the GSK126/Doxo co-treatment may depend on de-methylation of H3K27me3-marked promoters, allowing chromatin “opening” and binding of TFs expressed/activated in GSK126/Doxo-treated AML cells.
Thus, we performed an anti-H3K27me3 ChIP-seq experiment in KG-1 cells under the same conditions used for the RNA-seq experiment. Approximately 27% of the genes exclusively up-regulated by the GSK126/Doxo treatment show strong H3K27me3 peaks at their promoters in untreated cells; this percentage is higher (35%), among genes with ≥ 4-fold increased expression. Both percentages are higher than expected by chance (Fisher’s Exact Test p < 2.2 x 10−16). Selected genes that lost the H3K27me3 mark in GSK126- and GSK126/Doxo-treated AML cells are labeled in red in Fig. 5B–C and shown in Fig. 5E.
The remaining genes lack the H3K27me3 mark in their promoters in untreated cells but are transcriptionally activated only after GSK126/Doxo treatment. This suggests that they became targets of TFs induced/activated in GSK126/Doxo-treated KG-1 cells.
Among the H3K27me3-marked genes activated by GSK126/Doxo treatment, CDKN1A is particularly interesting because its expression may be responsible for the slow cycling/late S-phase arrest of GSK126/Doxo-treated KG-1 cells, which contrasts with the rapid G2 arrest of KG-1 cells treated with Doxo only (Supplemental Fig. 15). The increase in the fraction of late S-phase cells induced by GSK126/Doxo treatment may enhance the cell population targeted by Doxo, possibly augmenting DNA damage (41).
Similar studies were also performed using the p53-null THP-1 cell line. In contrast to the KG-1 line in which changes in gene expression were mostly induced by the GSK126/Doxo co-treatment (Fig. 5A), the RNA-seq analysis of untreated or drug-treated (GSK126, Doxo, or both) THP-1 cells revealed that Doxo treatment alone modulated the expression of 765 genes compared to the 1217 genes regulated by the GSK126/Doxo co-treatment; of interest, EZH2 inhibition alone modulated the expression of 416 genes (Supplemental Fig. 16A). Of the genes differentially expressed in GSK126/Doxo-treated vs. untreated cells, 599 were specific for the GSK126/Doxo co-treatment (FDR <5%). However, some of the genes upregulated by Doxo alone exhibited increased expression upon GSK126/Doxo co-treatment of THP-1 cells. The heat-map of a subset of genes exhibiting selective or enhanced expression in GSK126/Doxo-treated cells is shown in Supplemental Fig. 16B. Several of these genes are involved in biological processes such as growth suppression/apoptosis (CDKN1A, DUSP4, GADD45A, INKA2, RASL10A, SMAD9, TNFRSF11A), and stress response (ATF3, CLU, ETV7, FOSB, FOXD3, SGK1) that may explain the increased apoptosis of GSK126/Doxo-treated THP-1 cells.
To assess whether changes in expression induced by the GSK126/Doxo co-treatment were associated with loss of H3K27me3 marking in the promoter of GSK126/Doxo-modulated genes, anti-H3K27me3 ChIP-seq experiments of THP-1 cells were performed under the same conditions used for the RNA-seq experiments. Approximately 32% of the genes exclusively up-regulated by the GSK126/Doxo treatment exhibit loss of the distinct H3K27me3 peaks at their promoters present in untreated cells; this percentage is similar to that (27%) observed in Doxo/GSK126-treated KG-1 cells. The H3K27me3 mark was also lost in the promoter of some genes that, compared to treatment with Doxo alone, exhibited a further increase in expression upon GSK126/Doxo co-treatment (eg., the ATF3 gene). Examples of genes that lost the H3K27me3 mark are labeled in Supplemental Fig. 16B (red) and shown in Supplemental Fig. 16C.
Together, these data strongly suggest that loss of H3K27me3 marking is involved in the transcriptional reprogramming induced by GSK126/Doxo co-treatment of AML cells.
Co-treatment with GSK126 enhances the effect of Doxo/Ara-C in suppressing AML burden in mice, especially after targeting nascent chromatin
Frontline therapy for de novo AML typically consists of a remission-inducing protocol that combines Ara-C (7 days) with Doxorubicin (3 days). We tested whether inhibiting EZH2 enhances the anti-leukemia effect of this therapy using preclinical models of human AML. To this end, NOD-Rag1null IL2rgnull (NRG) mice injected with Luciferase-expressing THP-1 cells were treated with vehicle (control), GSK126, Doxo/Ara-C (DA 5+3) (42), or DA + GSK126 (Fig. 6A). The therapeutic effect was assessed by monitoring leukemia burden by bioluminescence imaging of leukemic mice 1- and 2-weeks post-treatment and by determining their median survival. The DA/GSK126 combination was more effective than DA or GSK126 alone in suppressing leukemia burden (Fig. 6B) and in prolonging survival (median survival: 61 days vs. 55 and 54 days respectively; Fig. 6C).
Figure 6. Leukemia burden and survival of chemotherapy and/or GSK126-treated immunodeficient mice injected with AML cells.
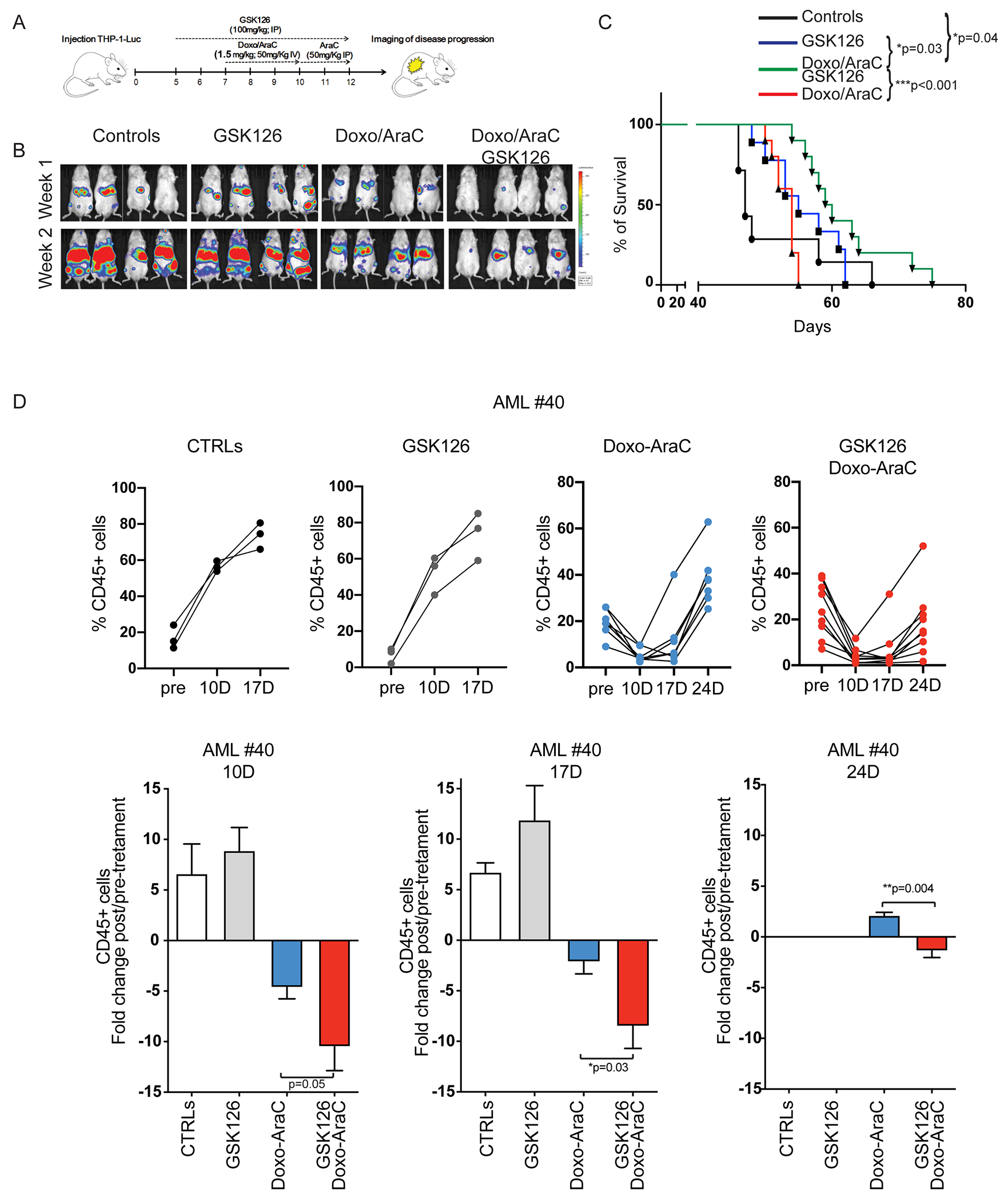
(A) Drug treatment and imaging protocol of NRG mice injected with THP-1 Luciferase+ cells (1 x106/mouse). 5 days post-injection, mice were left untreated (controls), treated with GSK126 alone for 7 days (GSK126 group), or pre-treated (2 days) with GSK126 and co-treated with GSK126 and chemotherapy for 5 additional days (Doxo/AraC/GSK126 group); the Doxo/AraC only group was treated for 5 days, starting 7 days post THP-1 cell injection. (B) Mouse serial bioluminescence images acquired 1 and 2 weeks after the end of treatment; drug treatments are indicated at top. (C) Kaplan-Meier survival curves for all four cohorts (control and three drug treatment regimens) of NRG mice injected with THP-1 cells. Long-rank test: *p<0.05, **p<0.01. (D) Leukemia burden of untreated and drug-treated NRG-SGM3 mice injected with primary AML cells. Mice were injected with AML sample #40 (1.5x 106 cells/mouse). When peripheral blood human CD45-positive cells were >10%, mice were divided in 4 groups and treated with vehicle, GSK126, Doxo/Ara-C, or the GSK126/Doxo/Ara-C combination; leukemia burden was assessed by measuring the percentage of peripheral blood leukemic cells by anti-human CD45-FITC flow cytometry at one-week interval starting 10 days after therapy cessation. Upper row, leukemia load shown as percentage of CD45+ cells in individual mice; lower row, fold changes of the percentage of leukemic cells in pre-treated vs treated mice in each cohort. Unpaired t-test *p<0.05, **p<0.01.
The effect of the chemotherapy/GSK126 combination was also assessed in NRG-SGM3 mice injected with AML sample #040. In this model, mice were treated with vehicle, GSK126 alone, Doxo/Ara-C alone, or the GSK126/Doxo/Ara-C combination when peripheral blood leukemic cells, detected by anti-CD45 flow cytometry, were ~10-35%. Doxo was given at a dose of 1.0 mg/Kg/3 days and Ara-C was given at 33.3 mg/Kg/5 days, which is one-third lower of that used in the THP-1 model. Leukemia burden was monitored by assessing the percentage of CD45+ leukemic cells three times at one week-interval, starting 10 days after termination of the therapy.
Vehicle- and GSK126-treated mice exhibited a similar increase in leukemia burden over the course of the experiment (Fig. 6D). As expected, treatment with chemotherapy alone suppressed AML growth. However, 24 days after therapy termination peripheral blood leukemic cells were more numerous than at the start of the treatment in 6/6 mice (Fig. 6D). By contrast, the combined GSK126/Ara-C/Doxo treatment was clearly more effective with a significant improvement over treatment with chemotherapy alone in suppressing AML growth at each time of analysis (Fig 6D, bottom). In particular, 24 days post-therapy termination only 1/8 mice had a leukemia burden (peripheral blood CD45+ cells) higher than at the start of the treatment (Fig. 6D). Notice that this AML sample is from a high-risk patient who had a rapidly fatal disease relapse after a short-lived complete remission (Supplemental Table I).
Potentiation of chemotherapy-induced DNA damage and apoptosis by de-condensing H3K27me3-marked chromatin was significantly enhanced in S-phase-enriched THP-1 cells (Fig. 4). We extended this strategy to our in vivo model of NRG mice injected with THP-1-Luciferase cells (Fig. 7A). We observed that, compared to the control group, leukemic mice temporarily given a Palbociclib-containing diet (72 hr), and analyzed 48 hours later, exhibited an increased number of bone marrow S-phase THP-1 cells (29.8% vs 13.7%, respectively; Fig. 7B), consistent with the effect seen in the ex vivo experiment (Fig. 4). De-condensing the H3K27me3-marked chromatin of S-phase enriched AML cells, followed by Doxo/Ara-C co-treatment was more effective than pre-treatment with Palbociclib followed by the Doxo/Ara-C combination in suppressing THP-1 leukemia burden (Fig. 7C) and in prolonging survival (median survival 81 days vs 75 days; Fig. 7D). Comparison of the Fig. 6 and Fig. 7 survival experiments strongly suggest that the GSK126/chemotherapy combination was more effective when given to mice pre-treated short-term with Palbociclib to increase the fraction of S-phase AML cells.
Figure 7. Leukemia burden and survival of NRG mice injected with THP-1-Luc cells, pre-treated with Palbociclib and then treated with chemotherapy and/or GSK126.
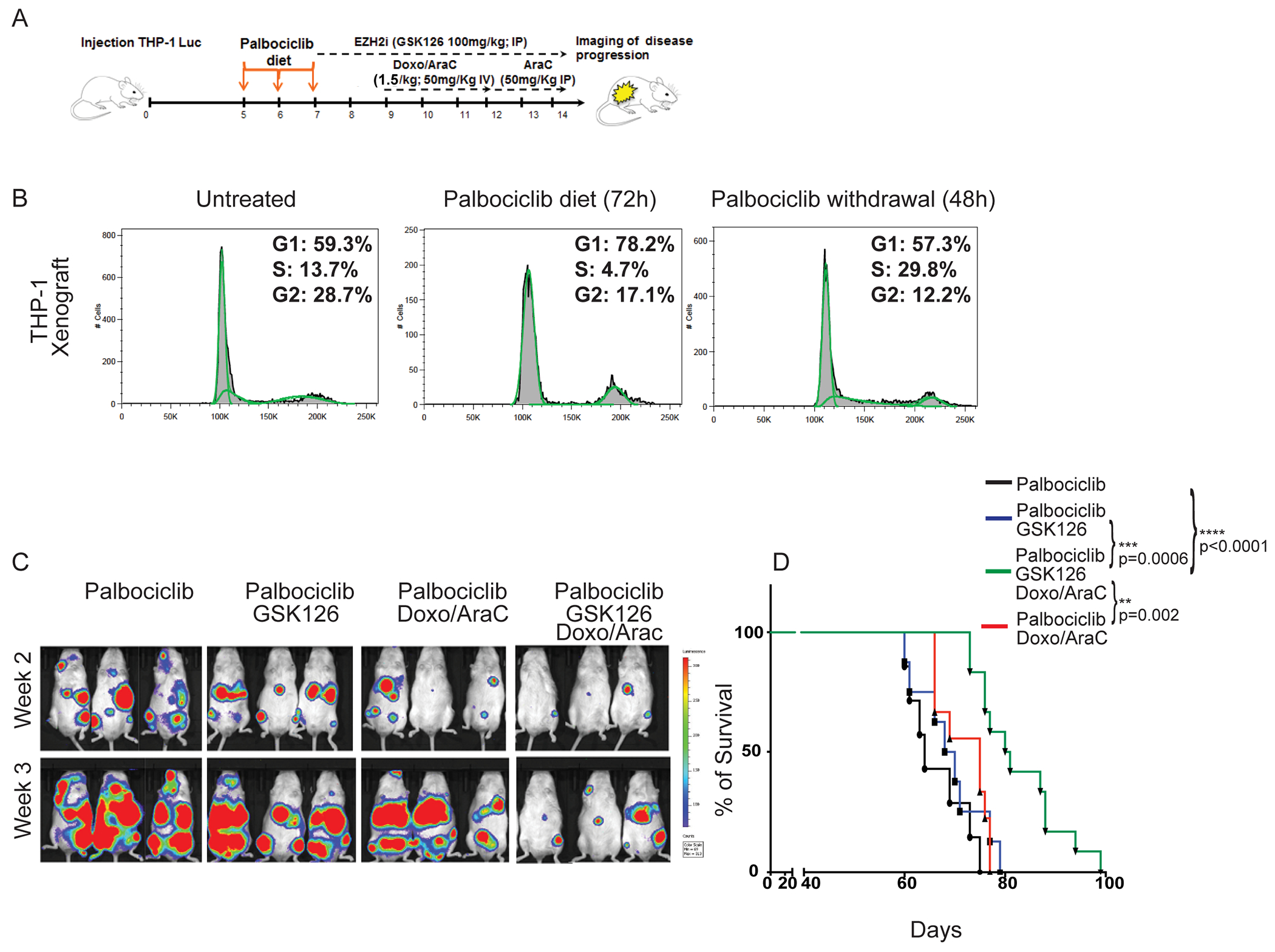
(A) NRG mice were injected with THP-1-Luciferase+ cells (1 x106/mouse). 5 days post-injection, mice were treated for 3 days with Palbociclib (150 mg/Kg in the diet). Mice were left untreated (Palb), treated with GSK126 alone for 7 days (Palb/GSK126 group), or pre-treated (2 days) with GSK126 and then co-treated with GSK126 and chemotherapy for 5 additional days (Palb/GSK126/Doxo/AraC group); the Palb/Doxo/AraC group was treated for 5 days, starting 10 days post-cell injection. (B) DNA content analysis of THP-1 cells purified from the bone marrow of NRG mice (untreated, fed with Palbociclib-supplemented chow for 72 hours, fed with Palbociclib-supplemented chow for 72 hours and then for 48 hours with Palbociclib-free chow) injected 4 weeks earlier with THP-1 cells (1 x 106 cells/mouse); (C) Bioluminescence images acquired 2 and 3 weeks after the end of treatment. (D) Kaplan-Meyer survival curves of mouse cohorts (control and drug-treated). Long-rank test: **p<0.01, ***p<0.001, ****p<0.0001
Discussion
We show here that blast cells from most AML patients exhibit rapid association of the H3K27me3 repressive mark with nascent DNA. This chromatin structure protects from cytotoxic agent-induced DNA damage, which typically occurs during DNA replication. In an effort to improve the current protocol of AML remission induction, we designed a novel therapy based on the sequential treatment with an EZH2 inhibitor to de-condense the H3K27me3-marked chromatin followed by chemotherapy. This approach significantly increased Doxorubicin-induced DNA damage and apoptosis ex vivo, and leukemia suppression in mice. Such effects were further improved by enhancing the targeting of chemotherapy to newly replicating de-condensed DNA after short-term CDK4/6 inhibition.
The structure of chromatin and chromatin-based epigenetic mechanisms are thought to play critical roles in cancer etiology/maintenance and thus are considered very attractive therapeutic targets (43–45). Significant effort has focused on developing inhibitors of histone-modifying enzymes, with the expectation that changing the structure of chromatin will affect overall transcription and thus will be beneficial in cancer treatment. While some success has been achieved, for example in the treatment of cutaneous T cell lymphoma (CTCL) with HDAC inhibitors (46), EZH2 inhibitors have shown limited clinical success (47). To some extent this is reflected in our findings that EZH2 inhibition by GSK126 treatment induced only a modest decrease in AML colony formation and a modest increase in the survival of mice injected with THP-1 cells.
Chromatin structure is also correlated to therapy response (48,49). In particular, increased chromatin accessibility is associated with response to HDAC inhibitors in CTCL (46), and appears to pre-determine glucocorticoid sensitivity in acute lymphoblastic leukemia (50). Moreover, high expression of PRC2 genes correlates with low chromatin accessibility and anthracycline resistance in breast cancer (49).
Chromatin structure is likely to influence the efficacy of anthracyclines, as these drugs induce DNA damage preferentially at de-condensed genomic regions (15,16), and promote histone eviction by incorporating more efficiently into open rather than compact chromatin (16).
However, this knowledge has not been exploited yet to design more effective therapies with currently used cytotoxic agents. Our approach is based on the novel concept of using EZH2 inhibitors in AML, and possibly in other malignancies, not as direct therapeutic agents but as modifiers of chromatin structure to enhance the effects of DNA-damaging agents.
We postulated that de-condensing the H3K27me3-marked chromatin of AML blasts by inhibiting EZH2 activity would render DNA more accessible to DNA damaging agents, causing enhanced cell death. We also hypothesized that enriching S-phase AML cells with open chromatin and targeting them with chemotherapy would further enhance DNA damage and cell death. Both hypotheses were validated by the ex vivo and in vivo studies reported here using AML cell lines and primary patients’ samples.
Our approach is applicable to most cases of AML, with the possible exception of those (approximately 10%) with gene mutations impairing the activity of the PRC2 complex (eg., loss-of-function mutations/deletions of EZH2, ASXL1, or BCOR). However, these mutations are often heterozygous and might be present in a minority of AML cells, raising the possibility that high levels of H3K27me3 may be also present in AML samples with these mutations.
EZH2 inhibition by GSK126 treatment alone induced a slight decrease in AML colony formation and a modest increase in the survival of mice injected with THP-1 AML cells. These findings are consistent with results of a study showing that EZH2 expression/activity is required for AML colony formation and disease maintenance in mice (51). However, AML suppression induced by EZH2 gene deletion was more effective than inhibition of EZH2 activity (51), suggesting the involvement of enzyme-independent mechanisms.
In our studies, the effect of EZH2 inhibition was initially designed as mostly auxiliary, since it promotes enhanced DNA damage and apoptosis by rendering the chromatin structure of AML cells more accessible to DNA damaging agents. However, chromatin de-condensation induced by EZH2 inhibition can also allow the re-activation of repressed genes if TFs expressed in GSK126/Doxo-treated AML cells bind to gene promoters not accessible in untreated cells due to H3K27me3 marking. In the KG-1 and THP-1 cell lines, this mechanism might be responsible for the increased expression of several cell death-promoting and growth-inhibitory genes. GSK126/Doxo treatment of AML cells also induced the expression of genes lacking the H3K27me3 mark in their promoter; we propose that in this case, H3K27me3 loss might have an indirect role, through re-activation and DNA binding of TFs which are repressed in untreated AML cells due to condensed chromatin at their promoters.
In summary, our approach of targeting cytotoxic agents to S-phase cells with de-condensed post-replicative chromatin has the potential to achieve a more effective killing of AML cells than presently possible due to the intrinsic ‘chemoresistance’ caused by their highly compacted chromatin structure. Our strategy could be especially beneficial in limiting chemotherapy side effects in elderly AML patients or those with significant comorbidities. For these patients, reduced doses of cytotoxic agents might achieve AML suppression comparable to that of standard chemotherapy, if combined with EZH2 inhibitors to enhance DNA accessibility of cytotoxic agents.
Most promisingly, our protocols for treating AML-bearing mice have the potential to be adopted in the clinic, since CDK4/6 and EZH2 inhibitors are currently approved and/or under evaluation for anti-cancer therapies.
Supplementary Material
Statement of Significance.
Pharmacological inhibition of EZH2 renders DNA of AML cells more accessible to cytotoxic agents, facilitating leukemia suppression with reduced doses of chemotherapy.
Acknowledgments
We thank the support of the Flow Cytometry Core Facility and Bioimaging Shared Resource of the Sidney Kimmel Cancer Center at Thomas Jefferson University, which are supported by the NCI Core grant (NCI 5 P30 CA-5603).
Financial Support:
This work was supported by NIH grants R01-HL127895 (B. Calabretta and A. Mazo), R01CA257251 (B. Calabretta), R01GM075141 (A. Mazo), R01CA226432 (C.M. Eischen) and the Herbert A. Rosenthal fund. DD was supported by Training grant T32CA236736.
Authors’ disclosures
Dr. Eischen discloses research funding from AbbVie unrelated to this manuscript. The other authors declare no potential conflicts of interest.
References
- 1.Thomas D, and Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017; 129:1577–1585, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The Cancer Genome Atlas Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dohner H, Weisdorf DJ, and Bloomfield C. Acute myeloid leukemia. N Engl J Med 2015; 373:1136–1152. [DOI] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Gerstung M, Bullinger L, Potter NE, Henser M, Thol F, Bolli N, Gundem G, Van Loo P, Martincorena I, Ganly P, Mudie L, McLaren S, O’Meara S, Reine K, Jones DR, Teague JW, Butler AP, Greaves MF, Ganser A, Dohner K, Schlenk RF, Dohner H, and Campbell PJ. Genome classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li S, Mason EC, and Melnick A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Current Opin Genet Dev 2016; 36:100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, Pignaux A, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015; 125:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirsch P, Zhang Y, Tang R, Joulin V, Boutroux H, Pronier E, et al. Genetic hierarchy and temporal variegation in the clonal history of acute myeloid leukaemia. Nat Commun 2016; 7:12475–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espinosa E, Zamora P, Feliue J, Gonzalez Boran M. Classification of anticancer drugs- a new system based on therapeutic targets. Cancer Treat Rev 2003; 29:515–523. [DOI] [PubMed] [Google Scholar]
- 10.Nitiss J DNA topoisomerase II and its growing repertoire of biological functions. Nature Rev Cancer 2009; 9:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung-Ong K, Glaever G, and Nislov C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol 2013; 20:648–659. [DOI] [PubMed] [Google Scholar]
- 12.Cann KL and Dellaire G. Heterochromatin and the DNA damage response: The need to relax. Biochem Cell Biol 2011; 89:45–60. [DOI] [PubMed] [Google Scholar]
- 13.Takata H, Hanafusa T, Mori T, Shimura M, Iida Y, Ishikawa K, et al. Chromatin compaction protects genomic DNA from radiation damage. PLoS One 2013; 8:e75622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karagiannis TC, Harikrishnan KN, and El-Osta A. Disparity of histone deacetylase inhibition on repair of radiation-induced DNA damage on euchromatin and constitutive heterochromatin compartments. Oncogene 2017; 26:3963–3971. [DOI] [PubMed] [Google Scholar]
- 15.Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun 2013; 4:1908–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pang B, de Jong J, Qiao X, Wessels LFA, and Neefjes J. Chemical profiling of the genome with anti-cancer drugs defines target specificities. Nature Chem Biol 2015; 11:472–481. [DOI] [PubMed] [Google Scholar]
- 17.Margueron R and Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature 2011; 469:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, et al. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 2012; 337:971–975. [DOI] [PubMed] [Google Scholar]
- 19.Petruk S, Sedkov Y, Johnston DM, Hodgson JW, Black KL, Kovermann SK, et al. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 2012; 150:922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petruk S, Black KL, Kovermann SK, Brock HW, Mazo A. Stepwise histone modifications are mediated by multiple enzymes that rapidly associate with nascent DNA during replication. Nat Commun 2013; 4:2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petruk S, Mariani SA, De Dominici M, Porazzi P, Minieri V, Cai J, et al. Structure of nascent chromatin is essential for hematopoietic lineage specification. Cell Rep 2017; 19:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petruk S, Cai J, Sussman R, Sun G, Kovermann SK, Mariani SA, et al. Delayed accumulation of H3K27me3 on nascent DNA is essential for recruitment of transcription factors at early stages of stem cell differentiation. Mol Cell 2017; 66:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Dominici M, Porazzi P, Soliera AR, Mariani SA, Addya S, Fortina P, et al. Targeting CDK6 and BCL2 Exploits the “MYB Addiction” of Ph+ Acute Lymphoblastic Leukemia. Cancer Res 2018; 78:1097–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugimoto K, Toyoshima H, Sakai R, Miyagawa K, Hugiwaka K, et al. Frequent mutations in the p53 gene in human myeloid leukemia cell lines. Blood 1992; 79: 2378–2383. [PubMed] [Google Scholar]
- 25.Collins AR. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol 2004; 26:249–261. [DOI] [PubMed] [Google Scholar]
- 26.Kumar G, Ertel A, Feldman G, Kupper J, and Fortina P. iSeqQC: a tool for expression-based quality control in RNA sequencing. BMC Bioinformatics 2020; 21,56. doi: 10.1186/s12859-020-3399-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang DW, Sherman BT, and Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 28.Barbieri E, Trizzino M, Welsh SA, Owens TA, Calabretta B, Carroll M, et al. Targeted Enhancer Activation by a Subunit of the Integrator Complex. Mol Cell 2018; 71:103–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inoue D, Kitaura J, Togami K, Nishimura K, Enomoto Y, Uchida T, et al. Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J Clin Invest 2013; 123:4627–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chittðck EC, Latwiel S, Miller TCR, and Muller CW. Molecular architecture of polycomb repressive complexes. Biochem Soc Trans 2017; 45:193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 2008; 32:503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012; 492(7427):108–112. [DOI] [PubMed] [Google Scholar]
- 33.Kuo LJ and Yang LX. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo 2008; 22:305–309. [PubMed] [Google Scholar]
- 34.Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 2015; 125:346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mondal G, Stevers M, Goode B, Ashworth A, and Solomon DA. A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nat Commun 2019; 10:1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esposito MT, and So CWE. DNA damage accumulation and repair defects in acute myeloid leukemia: implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 2014; 123:545–561. [DOI] [PubMed] [Google Scholar]
- 37.Barbosa K, Li S, Adams PD, and Deshpade AJ. The role of TP53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosomes Cancer 2019; 58:875–888. [DOI] [PubMed] [Google Scholar]
- 38.Kotredes KP and Gamero AM. Interferons as inducers of apoptosis in malignant cells. J Interferon Cytokine Research 2013; 33:162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao J, Senthil M, Ren B, Yan J, Xing Q, Yu J, et al. IRF-1 transcriptionally upregulates PUMA, which mediates the mitochondrial apoptotic pathway in IRF-1-induced apoptosis in cancer cells. Cell Death Differ 2010; 17:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomsen MK, Bakiri L, Hasenfuss SC, Hamacher R, Martinez L, and Wagner EF. JUNB/AP-1 controls IFN-γ during inflammatory liver disease. J Clin. Invest 2013;123:5258–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chao HX, Poovey CE, Privette AA, Grant GD, Chao HY, Cook JG, et al. Orchestration of DNA damage checkpoint dynamics across the human cell cycle. Cell Syst 2017; 5:445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wunderlich M, Mizukawa B, Chou F-S, Sexton C, Shrestha M, Sauntharajah Y, et al. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood 2013; 121:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Helin K, and Dhanak D. Chromatin proteins and modifications as drug targets. Nature 2013; 502:480–488. [DOI] [PubMed] [Google Scholar]
- 44.Feinberg AP, Koldobskiy MA, and Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 2016; 17:284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nichol JN, Dupéré-Richer D, Ezponda T, Licht JD, and Miller WH Jr. H3K27 Methylation: A Focal Point of Epigenetic Deregulation in Cancer. Adv Cancer Res 2016; 131:59–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qu K, Zaba LC, Satpathy AT, Giresi PG, Li R, Jin Y, et al. Chromatin Accessibility Landscape of Cutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors. Cancer Cell 2017; 32:27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eich M-L, Athar M, Ferguson III JE, and Varambally S. EZH2-targeted therapies in cancer: Hype or a reality. Cancer Res 2020; 80:5449–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. The chromatin accessibility landscape of primary human cancers. Science 2018; 362(6413). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seoane JA, Kirkland JG, Caswell-Jin JL, Crabtree GR, and Curtis C. Chromatin regulators mediate anthracycline sensitivity in breast cancer. Nat Medicine 2019; 25:1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jing D, Huang Y, Liu X, Sia KCS, Zhang JC, Tai X, et al. Lymphocyte-Specific Chromatin Accessibility Pre-determines Glucocorticoid Resistance in Acute Lymphoblastic Leukemia. Cancer Cell 2018; 34:906–921. [DOI] [PubMed] [Google Scholar]
- 51.Basheer F, Giotopoulos G, Meduri E, Yun H, Mazan M, Sasca D, et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J Exp Med 2019; 216:966–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.