

Abstract

The modification of metal nanoparticles (NPs) by incorporating additional metals is a key technique for developing novel catalysts. However, the effects of incorporating nonmetals into metal NPs have not been widely explored, particularly in the field of organic synthesis. In this study, we demonstrate that phosphorus (P)-alloying significantly increases the activity of precious metal NPs for the deoxygenation of sulfoxides into sulfides. In particular, ruthenium phosphide NPs exhibit an excellent catalytic activity and high durability against sulfur-poisoning, outperforming conventional catalysts. Various sulfoxides, including drug intermediates, were deoxygenated to sulfides with excellent yields. Detailed investigations into the structure–activity relationship revealed that P-alloying plays a dual role: it establishes a ligand effect on the electron transfer from Ru to P, facilitating the production of active hydrogen species, and has an ensemble effect on the formation of the Ru–P bond, preventing strong coordination with sulfide products. These effects combine to increase the catalytic performance of ruthenium phosphide NPs. These results demonstrate that P-alloying is an efficient method to improve the metal NP catalysis for diverse organic synthesis.
Keywords: alloys, deoxygenation, heterogeneous catalysis, phosphide, sulfoxide
Introduction
Incorporating additional metals is one of the key nanotechnologies used to impart unprecedented or significantly improved catalytic properties to single-metal nanoparticles (NPs). The resulting metal–metal nanoalloy catalysts have been widely studied and have delivered great benefits in diverse fields, including automobile exhaust cleaning, energy conversion, and industrially important reactions like petroleum reforming and fine chemical synthesis.1−4 Compared to metal–metal nanoalloys, the catalytic effect of incorporating nonmetals into metal NPs has not been widely explored. Among metal–nonmetal alloy catalysts, metal phosphide NPs have attracted increased attention as hydrotreating catalysts and electrocatalysts.5−8 However, despite their unique properties the use of metal phosphide catalysts in organic syntheses has rarely been investigated.9−13 Our group recently reported the unique catalytic properties exhibited by nonprecious metal phosphide (Ni2P and Co2P) NPs in selective liquid-phase molecular transformations, e.g., the transformation of biomass-derived molecules;14−18 the hydrogenation of nitriles, carbonyls, nitroarenes, and sulfoxides;19−22 the reductive amination of carbonyls;23 and the alkylation of oxindoles.24 These studies revealed the potential of nonprecious metal phosphide NPs to serve as a novel class of highly efficient heterogeneous catalysts for versatile liquid-phase reactions, outperforming conventional single nonprecious metal NPs. These results motivated us to study the catalytic properties of precious metal phosphides for organic synthesis. Owing to their rarity, studies on improving precious metal NP catalysts by alloying them with phosphorus are of great interest.25−29
The deoxygenation of sulfoxides to sulfides is one of the important reactions in organic chemistry.30,31 Recently, sulfoxides have served as effective directing groups in catalytic C–H functionalizations.32−34 Following functionalization, sulfoxide groups can be removed through deoxygenation, followed by desulfurization.35,36 Deoxygenation of sulfoxides also plays a crucial role in the pharmaceutical chemistry.37 For example, some sulfide-containing bioactive molecules, such as sulindac sulfide (anti-inflammatory drug) and ufiprazole (antiulcer drug), are synthesized by the deoxygenation of sulfoxide analogues.38,39 Molecular hydrogen (H2) is the most efficient reducing agent for deoxygenating sulfoxides to sulfides because water is formed as the sole byproduct. Although some precious metal NPs promote the deoxygenation of sulfoxides under mild conditions (e.g., atmospheric H2 pressure), catalysis using precious metal phosphides has not been reported to date.40−43 In this study, we investigated the effects of phosphorus (P)-alloying on NPs made of Pt, Pd, Rh, and Ru and found that the alloying significantly improved the catalytic activity of precious metal NPs for the deoxygenation of sulfoxides using H2. In particular, P-alloying increased the activity of Ru NPs by ten times; thus, they outperformed previously reported catalysts. Spectroscopic analyses, control experiments, kinetic studies, and density functional theory (DFT) calculations revealed that P-alloying causes two important effects. One is electron transfer from Ru to P, which facilitates the heterolytic dissociation of H2 to produce active hydrogen species for the deoxygenation of sulfoxides (ligand effect). The other is the modulation of the surface structure of Ru NPs by formation of the Ru–P bond, which suppresses the strong coordination with sulfide products (ensemble effect). This dual role of P-alloying significantly enhanced the catalytic activity and durability of Ru NPs (Figure 1).
Figure 1.
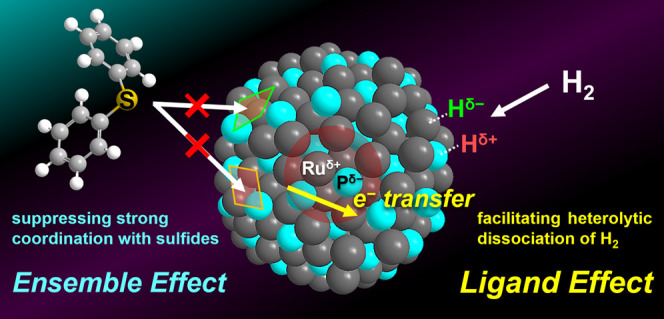
Effect of P-alloying on the catalytic properties of Ru NPs.
Results and Discussion
Catalyst Characterization
The SiO2-supported precious metal phosphide NPs (M–P/SiO2; M = Ru, Rh, Pd, or Pt) were synthesized by high-temperature pyrolysis using inorganic phosphorus reagents (see catalyst preparation in the Supporting Information).44 Initially, the metal precursors (RuCl3, RhCl3, Pd(NH3)4Cl2, or H2PtCl6) and ammonium hypophosphite (NH4H2PO2) were impregnated into SiO2. The as-obtained solid was heated at 823 K under flowing H2 gas, producing the SiO2-supported precious metal phosphide NPs (M–P/SiO2; M = Ru, Rh, Pd, or Pt). As references, unmodified SiO2-supported precious metal NPs (M/SiO2; M = Ru, Rh, Pd, or Pt) were prepared using a method identical to the aforementioned one except for the absence of NH4H2PO2.
The X-ray diffraction (XRD) patterns of M/SiO2 and M–P/SiO2 are shown in Figure 2a–d. After the phosphorus treatment, the XRD patterns of M/SiO2 corresponding to the metal nanoparticles changed to those of the metal phosphides. The peaks of Ru–P/SiO2, Rh–P/SiO2, Pd–P/SiO2, and Pt–P/SiO2 were attributed to Ru2P, Rh2P, Pd5P2, and PtP2, respectively.45−48 Representative images of M–P/SiO2 obtained by transmission electron microscopy (TEM) are depicted in Figure 2e–h. Ru–P/SiO2, Rh–P/SiO2, Pd–P/SiO2, and Pt–P/SiO2 have spherical NPs with mean diameters of 3.0, 2.9, 6.7, and 2.6 nm, respectively (Figure 2i–l). The distributions of metals, phosphorus, and silicon in M–P/SiO2 were determined by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) coupled with energy-dispersive X-ray spectroscopy (EDX) for elemental mapping (Figure S1). Elemental mapping revealed that the precious metals formed NPs, and the phosphorus atoms were homogeneously distributed within each NP. The NPs were also highly dispersed on the SiO2 support. TEM images of M/SiO2 confirmed the formation of spherical metal NPs of Ru, Rh, Pd, and Pt, with mean diameters of 3.7, 3.2, 9.3, and 3.0 nm, respectively (Figure S2).
Figure 2.

XRD patterns of (a) Ru–P/SiO2 and Ru/SiO2, (b) Rh–P/SiO2 and Rh/SiO2, (c) Pd–P/SiO2 and Pd/SiO2, and (d) Pt–P/SiO2 and Pt/SiO2. TEM images and size-distribution histograms of (e and i) Ru–P/SiO2, (f and j) Rh–P/SiO2, (g and k) Pd–P/SiO2, and (h and l) Pt–P/SiO2.
Evaluation of Catalytic Activity
The catalytic activities of M–P/SiO2 and M/SiO2 were assessed using the deoxygenation of diphenyl sulfoxide (1a) to diphenyl sulfide (2a) in H2 gas at atmospheric pressure and 373 K for 30 min (Figure 3). Interestingly, each of the metal phosphide NPs tested provided a higher yield of 2a than that obtained using the original metal NPs. Particularly, P-alloying of Ru NPs drastically enhanced their catalytic activity, resulting in a 77% yield of 2a that was approximately 10 times higher than the yield obtained using original Ru/SiO2 (8% yield) and the highest yield obtained using the M–P/SiO2 catalysts (the yields were less than 10% using Rh–P/SiO2, Pd–P/SiO2, and Pt–P/SiO2). The effect of the P/Ru molar ratio used in the preparation of Ru–P/SiO2 on the catalytic activity was also investigated (Scheme S1). The curve showing the dependence of the yield on the phosphorus amount had a volcano shape, with a maximum value at P/Ru = 0.86. To clarify the effect of P-alloying on NPs of metals other than Ru, the deoxygenation of 1a was performed by increasing the metal loadings and prolonging the reaction time from 30 min to 1 h (inset in Figure 3). Under these conditions, M–P/SiO2 NPs exhibited a catalytic activity 7–30 times higher than to their corresponding M/SiO2 counterparts. These results clearly demonstrate that P-alloying enhances the catalytic activity of metal NPs for the deoxygenation of sulfoxides.
Figure 3.

Catalytic performances of various SiO2-supported metal phosphide NPs for the deoxygenation of 1a compared to those of the unmodified single-metal NPs. Reaction conditions are as follows: catalyst (metal: 0.5 mol%), 1a (2.5 mmol), n-dodecane (5 mL), 30 min; for the inset, catalyst (metal: 2.5 mol%), 1a (0.5 mmol), n-dodecane (3 mL), 1 h. Yields were determined by gas chromatography–mass spectrometry (GC–MS) using an internal standard technique.
The scope of sulfoxides was investigated in the Ru–P/SiO2-catalyzed deoxygenation using H2 at atmospheric pressure (Scheme 1). Aryl, benzyl, and alkyl sulfoxides were efficiently converted to the corresponding sulfides with excellent yields (Scheme 12a–2f and 2n–2p). Ru–P/SiO2 also promoted the chemoselective deoxygenation of various functionalized sulfoxides. Aryl (2a–2c, 2e–2m, and 2s), benzyl (2c and 2d), carbonyl (2g and 2h), ether (2i), halogen (2j–2m), thioether (2q), amino acid (2r), and thiophosphate (2s) moieties were tolerated under the aforementioned reaction conditions, and high yields of their corresponding sulfides were obtained. Furthermore, this catalytic system could be applied for the preparation of some existing drug molecules; for example, structurally complex sulfoxides, sulindac (1t), omeprazole (1u), and oxfendazole (1v) were efficiently deoxygenated to produce the corresponding sulfide-containing bioactive molecules, namely sulindac sulfide (anti-inflammatory drug, 2t),37 ufiprazole (antiulcer drug, 2u),49 and fenbendazole (anthelmintic, 2v),50 in excellent isolated yields, demonstrating high utility of Ru–P/SiO2 for fine chemical synthesis. Ru–P/SiO2 was easily recovered by centrifugation after the reaction was complete and could be reused to afford 2a in a quantitative yield (Scheme 1). Ru–P/SiO2 was also applied to a gram-scale reaction, where 4.04 g of 1a was converted to 2a with a >99% yield (Scheme 2a). Based on the total number of Ru atoms in Ru–P/SiO2, the turnover number (TON) was 12 500 (32 000 based on the number of surface Ru atoms). Moreover, Ru–P/SiO2 operated well under 1 bar of H2 (Scheme 2b), and the TON reached 8900 (22 800 based on the number of surface Ru atoms). These TON values are greater than those of homogeneous and heterogeneous catalysts reported to date (Table S1 and Scheme S2).
Scheme 1. Deoxygenation of Various Sulfoxides Catalyzed by Ru–P/SiO2.

Reaction conditions are as follows: Ru–P/SiO2 (2.5 mol%), substrate (0.5 mmol), n-dodecane (3 mL), 373 K. Yields were determined by GC–MS using an internal standard technique.
Ru–P/SiO2 (0.5 mol%), substrate (2.5 mmol), n-dodecane (5 mL).
Second reuse.
Isolated yield.
H2O (3 mL).
Ru–P/SiO2 (10 mol%), 1s (0.125 mmol), THF (3 mL), 333 K.
THF (3 mL), 333 K.
Ru–P/SiO2 (10 mol%), 1u (0.125 mmol), anisole (5 mL), 353 K.
Ru–P/SiO2 (10 mol%), anisole (3 mL), 333 K.
Scheme 2. Gram-Scale Deoxygenation of 1a using Ru–P/SiO2.

Reaction conditions are as follows: (a) Ru–P/SiO2 (8 mmol%), 1a (4.04 g, 20 mmol); (b) Ru–P/SiO2 (10 mmol%), 1a (3.24 g, 16 mmol). The value in parentheses is the isolated yield.
To confirm that the sulfoxide deoxygenation is a heterogeneous catalytic system, Ru–P/SiO2 was removed by filtration at a ∼40% yield of 2a. Further treatment of the resulting filtrate under similar reaction conditions did not yield any product (Scheme S3). Additionally, inductively coupled plasma–atomic emission spectroscopy (ICP–AES, detection limit: 0.3 ppm) confirmed the absence of Ru species in the filtrate. These results demonstrated that the catalysis was not derived from leaching metal species. The TEM image and size-distribution histogram of the used Ru–P/SiO2 showed no significant aggregation or growth of ruthenium phosphide NPs after the reaction (Figure S3). These results are consistent with the durability of Ru–P/SiO2 in the gram-scale reactions and reuse experiments.
Evaluation of Durability
It is well-known that the sulfur atom strongly coordinates with the active sites of metals, significantly decreasing their catalytic performance.51,52 However, Ru–P/SiO2 exhibited remarkable durability against poisoning by sulfide products. To further assess the durability of Ru–P/SiO2, the influence of product poisoning on the catalytic activity of Ru–P/SiO2 was evaluated using Ru/SiO2 as a reference catalyst. Figure 4 depicts the time courses of the deoxygenation of 1a in the presence and absence of phenyl-p-tolyl sulfide using Ru–P/SiO2 and Ru/SiO2. Ru–P/SiO2 completed the deoxygenation of 1a in 1 h (red spheres). Even in the presence of 200 equiv of phenyl-p-tolyl sulfide per Ru, Ru–P/SiO2 quantitatively promoted the deoxygenation of 1a for 6 h, although the reaction rate decreased (red crosses). This catalytic behavior was in sharp contrast to that of Ru/SiO2. The initial reaction rate with Ru/SiO2 (33.5 mol molRu–1 h–1) was lower than that with Ru–P/SiO2 (218 mol molRu–1 h–1). Thereafter, the reaction rate decreased significantly at ∼30% conversion of 1a, and the reaction finally stopped at 45% conversion of 1a (blue spheres). Furthermore, the deoxygenation did not proceed at all in the presence of phenyl-p-tolyl sulfide (blue crosses), indicating strong poisoning of Ru/SiO2 by the sulfide product. These results clearly demonstrate that P-alloying significantly enhances the activity and durability of the Ru NPs for the deoxygenation of sulfoxides.
Figure 4.

Time courses of the deoxygenation of 1a using Ru–P/SiO2 and Ru/SiO2 in the presence or absence of phenyl-p-tolyl sulfide. Reaction conditions are as follows: catalyst (Ru: 0.5 mol%), 1a (2.5 mmol), phenyl-p-tolyl sulfide (2.5 mmol), n-dodecane (5 mL), H2 (1 bar), 373 K.
Ligand Effect by P-Alloying
The temperature dependences of the deoxygenation of 1a using Ru–P/SiO2 and Ru/SiO2 were further studied in the temperature range 333–363 K (Figure 5a). The initial reaction rates were determined at low conversions (<20%). The initial reaction rates increased with as the reaction temperature increased, and the Arrhenius plots for Ru–P/SiO2 and Ru/SiO2 showed good linearities. The apparent activation energy (Ea) calculated for Ru–P/SiO2 was 13.5 kJ mol–1, which was lower than that for Ru/SiO2 (21.1 kJ mol–1). The dependences of the initial reaction rates on the partial pressure of H2 and the concentration of 1a were also investigated. The initial reaction rates were dependent on the H2 partial pressure and independent of the concentration of 1a (Figure 5b and c). Furthermore, the H2/D2 kinetic isotope effects (KIEs) in the deoxygenation of 1a were observed using Ru–P/SiO2 (kH/kD = 1.6) and Ru/SiO2 (kH/kD = 1.7) (Figure 5d and e). These results suggest that the rate-determining step includes the reaction with a hydrogen species. To compare the H2 activation ability of Ru–P/SiO2 and Ru/SiO2, the H2–D2 exchange reaction was carried out (Figures 5f and S4). When Ru–P/SiO2 was used, the H2–D2 exchange reaction rate was 197 mol molRu–1 h–1, which was 1.8 times faster than that obtained when Ru/SiO2 (109 mol molRu–1 h–1) was used, indicating that the H2 activation ability of Ru–P/SiO2 is higher than that of Ru/SiO2. However, this difference in the H2–D2 exchange reaction rate cannot fully explain the activity enhancement of Ru NPs by P-alloying for sulfoxide deoxygenation because the difference in the initial reaction rate between Ru–P/SiO2 and Ru/SiO2 in sulfoxide deoxygenation (218 vs 33.5 mol molRu–1 h–1) is much larger than that in the H2–D2 exchange reaction rate. Based on the kinetic studies, we concluded that the rate-determining step in the deoxygenation of sulfoxides is the reaction process between the sulfoxide and the hydrogen species adsorbed on Ru NPs, which is enhanced by phosphorus-alloying. The results of the detailed kinetic analysis based on the Langmuir–Hinshelwood model agreed with the experimental data, which supports our conclusion well (Figure 5g and h. For details, see kinetic study in the Supporting Information).
Figure 5.

(a) Arrhenius plots of the deoxygenation of 1a using Ru–P/SiO2 (red spheres) and Ru/SiO2 (blue spheres). Reaction conditions are as follows: 1a (2.5 mmol), Ru catalyst (0.5 mol%), toluene (5 mL), H2 (1 bar), 333–363 K. Reaction times are as follows: 3–5 min (Ru–P/SiO2) or 15 min (Ru/SiO2). Dependence of the initial reaction rate on (b) the partial pressure of H2 and (c) the concentration of 1a. Reaction conditions are as follows: 1a (1.25–5.0 mmol), Ru–P/SiO2 (0.1–1.25 mol%), toluene (5 mL), H2 (0.5–3.0 bar), 373 K, 30 min. KIEs of the deoxygenation of 1a using (d) Ru–P/SiO2 and (e) Ru/SiO2. Reaction conditions are as follows: 1a (2.5 mmol), toluene (5 mL), 373 K. (f) H2–D2 exchange reaction using Ru–P/SiO2 and Ru/SiO2. Langmuir–Hinshelwood plots of (g) 1/√r0 vs 1/√[H2] and (h) √[Sulfoxide]/r0 vs [Sulfoxide]. Spheres (●) show the experimental data. These plots fit well with the linear transforms of the kinetic equation based on the Langmuir–Hinshelwood model.
To gain insight into the structure–activity relationships of Ru–P/SiO2, we performed X-ray absorption fine structure (XAFS) analysis. The Ru K-edge X-ray absorption near-edge structure (XANES) spectrum of Ru–P/SiO2 is depicted in Figure 6a, with Ru powder, RuO2, bulk-Ru2P, and Ru/SiO2 included for reference. The absorption edge energy of Ru–P/SiO2 (red line) is close to those of Ru powder (purple line) and Ru/SiO2 (blue line), suggesting that nearly zero-valent Ru species exist in Ru–P/SiO2.53Figure 6b shows the Fourier-transforms of the extended XAFS (FTs-EXAFS) spectra of Ru–P/SiO2, with Ru powder, RuO2, bulk-Ru2P, and Ru/SiO2 included for reference. The spectra of Ru–P/SiO2 and bulk-Ru2P exhibited two principal peaks at 1.8 and 2.6 Å, which were assigned to the Ru–P and Ru–Ru bonds, respectively.53,54 To further investigate the local structure of the Ru species in Ru–P/SiO2, curve-fitting was performed on the EXAFS spectra of Ru–P/SiO2 and bulk-Ru2P. The curve-fitted data are depicted in Figure S5, and the results are summarized in Table 1. The coordination number of the Ru–P shell in Ru–P/SiO2 (2.8) is nearly equal to that in bulk-Ru2P (2.6). In contrast, the coordination number of the Ru–Ru shell in Ru–P/SiO2 (2.8) is much smaller than that in bulk-Ru2P (4.9), indicating that Ru–P/SiO2 has coordinatively unsaturated Ru sites that may act as catalytic active sites for the deoxygenation of sulfoxides. We also characterized the used Ru–P/SiO2 after the deoxygenation of 1a using XAFS analysis. The Ru K-edge XANES spectrum and the result of the curve-fitting analysis of the EXAFS of the used Ru–P/SiO2 are similar to those of the fresh Ru–P/SiO2 (Figures S5 and S6 and Table S2), indicating that the electronic state and the local structure of Ru–P/SiO2 do not significantly change after the reaction.
Figure 6.

(a) Ru K-edge XANES spectrum of Ru–P/SiO2, with Ru powder, RuO2, bulk-Ru2P, and Ru/SiO2 included for reference. (b) Fourier-transforms (FTs) of the k3-weighted EXAFS spectra of Ru–P/SiO2, Ru powder, RuO2, bulk-Ru2P, and Ru/SiO2.
Table 1. Results of Curve-Fitting of Ru K-Edge EXAFS for Ru–P/SiO2, Bulk-Ru2P, and Ru/SiO2.
| sample | shell | CNa | r (Å)b | D.W.c | R factor (%) |
|---|---|---|---|---|---|
| Ru–P/SiO2 | Ru–P | 2.8 ± 0.6 | 2.30 ± 0.010 | 0.006 ± 0.0015 | 8.6 |
| Ru–Ru | 2.8 ± 0.9 | 2.80 ± 0.009 | 0.008 ± 0.0018 | ||
| bulk-Ru2P | Ru–P | 2.6 ± 0.4 | 2.32 ± 0.007 | 0.008 ± 0.0014 | 3.1 |
| Ru–Ru | 4.9 ± 0.5 | 2.80 ± 0.004 | 0.007 ± 0.0005 | ||
| Ru/SiO2 | Ru–Ru | 9.3 ± 0.5 | 2.67 ± 0.002 | 0.005 ± 0.0003 | 4.5 |
Coordination number.
Bond distance.
Debye–Waller factor.
Next, we analyzed Ru–P/SiO2 using X-ray photoelectron spectroscopy (XPS) to determine the electronic states of Ru and P. Figure 7a shows the Ru 3d XPS spectra of Ru–P/SiO2 and Ru/SiO2. The Ru 3d5/2 and 3d3/2 peaks of Ru–P/SiO2 were observed at 279.6 and 283.7 eV, respectively. These binding energies were similar to those of the metallic Ru species in Ru/SiO2.55 This result is consistent with that of the XANES analysis. The P 2p XPS spectrum of Ru–P/SiO2 showed two peaks (Figure 7b). The peak that appeared at 129.3 eV was close to that of elemental phosphorus (129.9 eV) and was assigned to the negatively charged phosphorus species (Pδ−).45 The other peak that appeared around 134.7 eV was ascribed to phosphate species (PO43–).56 We further investigated the electronic state of the surface Ru species by Fourier-transform infrared (FT-IR) spectroscopy. When CO was adsorbed onto the surface of Ru/SiO2, an absorption band was observed at 2007 cm–1, which was attributed to linearly adsorbed CO on the metallic Ru species (Figure 7c).57,58 In contrast, an absorption band corresponding to the linearly adsorbed CO on the surface of Ru–P/SiO2 appeared at 2060 cm–1 (Figure 7c). This blue-shift indicates the formation of positively charged Ru species (Ruδ+).29 XPS and FT-IR results disclose that the P-alloying of Ru NPs induces electron transfer from Ru to P (ligand effect).45,59 The polarized Ru–P bond dissociates H2 in a heterolytic manner to produce Hδ+ and Hδ−,11,12 which are active hydrogen species for the deoxygenation of sulfoxide.60,61 Therefore, the higher catalytic activity of Ru–P/SiO2 than that of Ru/SiO2, as shown in Figures 4 and 5a, can be ascribed to the ligand effect of phosphorus, which facilitates the heterolytic dissociation of molecular hydrogen.
Figure 7.
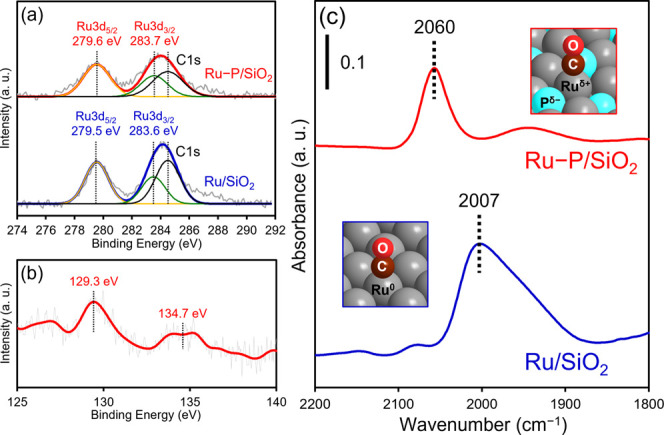
(a) Ru 3d XPS spectra of Ru–P/SiO2 and Ru/SiO2. The C 1s signal at 284.5 eV was derived from the adhesive carbon tape. (b) P 2p XPS spectrum of Ru–P/SiO2. (c) FT-IR spectra of CO adsorbed on the surface of Ru–P/SiO2 and Ru/SiO2.
Ensemble Effect by P-Alloying
We performed DFT calculations on the adsorption states of dimethyl sulfide on the ruthenium phosphide nanoalloy to investigate the origin of the high durability of Ru–P/SiO2. Based on the XRD patterns of Ru–P/SiO2 and Ru/SiO2 (Figure 2a), the crystal structures of orthorhombic Ru2P and hexagonal Ru were adopted, with the (210) and (0001) planes, respectively, being used for the adsorption of a sulfide molecule (Figures S7 and S8 and Table S3). The adsorption energies, ΔE = E(surface–sulfide) – E(surface) – E(sulfide), and the optimized adsorption structures on Ru2P(210) and Ru(0001) are summarized in Figure 8a–d. The dimethyl sulfide molecule was stably adsorbed at the Ru–Ru bridge site on both the Ru2P and Ru surfaces. Three types of bridge sites are present on the Ru2P surface: the b1 site is located between two Ru3 hollow sites (Figure 8a), the b2 site is located between a Ru3 hollow site and a Ru2P hollow site (Figure 8b), and the b3 site is located between two Ru2P hollow sites (Figure 8c). The adsorption energy (ΔE) of the b1 site, −51.6 kcal mol–1, was comparable to that of the bridge site of the unmodified Ru (Figure 8d; −50.3 kcal mol–1). In comparison, the adsorptions at the b2 and b3 sites, with ΔE = −37.1 and −31.8 kcal mol–1, respectively, were less stable. On-top-like adsorptions with average ΔE values of −44 and −41 kcal mol–1 were also observed for the Ru and Ru2P surfaces, respectively, as illustrated in the red frames in Figures S9 and S10. On the other hand, the hollow sites of the Ru and Ru2P surfaces and the Ru–P bridge site of Ru2P were less stable than the Ru–Ru bridge and on-top sites; the sulfide molecules moved to the Ru–Ru bridge or on-top sites during the geometry optimization calculations. These results indicated that strong adsorption of sulfide occurs preferentially at the b1 site of Ru2P, which is comparable to adsorption on the bridge site of the unmodified Ru. However, the b1 site accounts for only 12.5% of the total Ru–Ru bridge sites in Ru2P, as illustrated in Figures 8e and S11. Therefore, P-alloying significantly decreases the number of strong adsorption sites for sulfide and reduces the probability of strong adsorption of sulfide on the Ru surface, thus enhancing its durability against sulfur poisoning (ensemble effect). In summary, these results demonstrate that the high activity and durability of Ru–P/SiO2 are ascribed to the ligand and ensemble effects, respectively, caused by P-alloying.
Figure 8.

Optimized structures of dimethyl sulfide adsorbed onto Ru2P(210) and Ru(0001) surfaces: (a) b1, (b) b2, and (c) b3 bridge sites of Ru2P(210). (d) Bridge site of Ru(0001). Gray, light blue, yellow, brown, and light pink balls indicate Ru, P, S, C, and H, respectively. (e) Surface structures of Ru2P(210) and (inset) Ru(0001). Strong adsorption sites are marked in red.
Conclusion
In this paper, we report the effect of P-alloying on the catalytic properties and performance of precious metal NPs in the deoxygenation of sulfoxides to sulfides using H2. P-Alloying drastically improved the catalytic performance of Ru NPs in the deoxygenation of sulfoxides. The as-prepared ruthenium phosphide nanoalloy (Ru–P/SiO2) exhibited a high activity and selectivity for various sulfoxides, including sulfoxide-containing drug intermediates, to produce the corresponding sulfides in excellent yields when H2 was used as the reducing agent at ambient pressure. Furthermore, Ru–P/SiO2 was applicable to gram-scale reactions to achieve a TON of 12 500, which is the highest value reported to date. Ru–P/SiO2 was reusable without any significant loss in activity. Control experiments, kinetic studies, spectroscopic analyses, and DFT calculations revealed that the high activity and durability imparted by P-alloying are due to the ligand effect on the electron transfer from Ru to P, which facilitates the heterolytic dissociation of H2, and the ensemble effect on the formation of the Ru–P bond which prevents strong coordination with the sulfide products. This dual role of P-alloying greatly improved the catalytic performance of single ruthenium nanoparticle catalysts in the selective deoxygenation of sulfoxides. The results of this study establish P-alloying as a powerful method for the development of highly active and durable heterogeneous catalysts for a variety of organic syntheses.
Acknowledgments
We thank Ryo Ota of Hokkaido University for STEM analysis and Dr. Toshiaki Ina (SPring-8) for the XAFS measurements (2020A1487 and 2020A1640). A part of experimental analysis was supported by the “Nanotechnology Platform” Program at Hokkaido University (A-21-HK-0051) and the Nanotechnology Open Facilities in Osaka University (A-20-OS-0025) of the MEXT. The DFT calculations were performed on the Numerical Materials Simulator at NIMS.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00461.
General experimental details, catalyst preparation, typical reaction procedure, recycling experiments, gram-scale reaction, effect of molar ratio of phosphorus to ruthenium, comparison of activity of Ru–P/SiO2 with previously reported catalysts, hot filtration experiment, characterization, kinetic study, DFT calculation details, product identification, and 1H and 13C NMR spectra of products (PDF)
Author Contributions
T. Mitsudome conceived and directed the project. T. Mitsudome and H.I. cowrote the manuscript with input from all the authors. H.I. prepared the catalyst and conducted catalytic activity tests. A.N. performed the DFT calculations. S. Yamazoe performed the XAFS analysis. J.Y. performed the TEM measurements and analysis. S. Yamaguchi, K.N., and T. Mizugaki planned the experiments and analyzed the results. All authors commented critically on the manuscript and approved the final manuscript.
This work was supported by JSPS KAKENHI Grants 18H01790, 20H02523, 20H05879, and 21K04776; MEXT KAKENHI Grants 20H05878 and 20H05883 (hyper-ordered structures science); and JST PRESTO Grants JPMJPR21Q9 and JPMJPR20T4. This study was partially supported by JST-CREST Grant JPMJCR21L5 and the Cooperative Research Program of the Institute for Catalysis, Hokkaido University (21A1005).
The authors declare no competing financial interest.
Supplementary Material
References
- Zhou M.; Li C.; Fang J. Noble-Metal Based Random Alloy and Intermetallic Nanocrystals: Syntheses and Applications. Chem. Rev. 2021, 121, 736–795. 10.1021/acs.chemrev.0c00436. [DOI] [PubMed] [Google Scholar]
- Sankar M.; Dimitratos N.; Miedziak P. J.; Wells P. P.; Kiely C. J.; Hutchings G. J. Designing Bimetallic Catalysts for a Green and Sustainable Future. Chem. Soc. Rev. 2012, 41, 8099–8139. 10.1039/c2cs35296f. [DOI] [PubMed] [Google Scholar]
- Bing Y.; Liu H.; Zhang L.; Ghosh D.; Zhang J. Nanostructured Pt-Alloy Electrocatalysts for PEM Fuel Cell Oxygen Reduction Reaction. Chem. Soc. Rev. 2010, 39, 2184–2202. 10.1039/b912552c. [DOI] [PubMed] [Google Scholar]
- Wang D.; Li Y. Bimetallic Nanocrystals: Liquid-Phase Synthesis and Catalytic Applications. Adv. Mater. 2011, 23, 1044–1060. 10.1002/adma.201003695. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Zhang B. Recent Advances in Transition Metal Phosphide Nanomaterials: Synthesis and Applications in Hydrogen Evolution Reaction. Chem. Soc. Rev. 2016, 45, 1529–1541. 10.1039/C5CS00434A. [DOI] [PubMed] [Google Scholar]
- Xiao P.; Chen W.; Wang X. A Review of Phosphide-Based Materials for Electrocatalytic Hydrogen Evolution. Adv. Energy Mater. 2015, 5, 1500985. 10.1002/aenm.201500985. [DOI] [Google Scholar]
- Pei Y.; Cheng Y.; Chen J.; Smith W.; Dong P.; Ajayan P. M.; Ye M.; Shen J. Recent Developments of Transition Metal Phosphides as Catalysts in the Energy Conversion Field. J. Mater. Chem. A 2018, 6, 23220–23243. 10.1039/C8TA09454C. [DOI] [Google Scholar]
- Oyama S. T. Novel Catalysts for Advanced Hydroprocessing: Transition Metal Phosphides. J. Catal. 2003, 216, 343–352. 10.1016/S0021-9517(02)00069-6. [DOI] [Google Scholar]
- Carenco S.; Leyva-Pérez A.; Concepción P.; Boissière C.; Mézailles N.; Sanchez C.; Corma A. Nickel Phosphide Nanocatalysts for the Chemoselective Hydrogenation of Alkynes. Nano Today 2012, 7, 21–28. 10.1016/j.nantod.2011.12.003. [DOI] [Google Scholar]
- Chen Y.; Li C.; Zhou J.; Zhang S.; Rao D.; He S.; Wei M.; Evans D. G.; Duan X. Metal Phosphides Derived from Hydrotalcite Precursors toward the Selective Hydrogenation of Phenylacetylene. ACS Catal. 2015, 5, 5756–5765. 10.1021/acscatal.5b01429. [DOI] [Google Scholar]
- Gao R.; Pan L.; Wang H.; Zhang X.; Wang L.; Zou J.-J. Ultradispersed Nickel Phosphide on Phosphorus-Doped Carbon with Tailored d-Band Center for Efficient and Chemoselective Hydrogenation of Nitroarenes. ACS Catal. 2018, 8, 8420–8429. 10.1021/acscatal.8b02091. [DOI] [Google Scholar]
- Gao R.; Pan L.; Wang H.; Yao Y.; Zhang X.; Wang L.; Zou J.-J. Breaking Trade-Off between Selectivity and Activity of Nickel-Based Hydrogenation Catalysts by Tuning Both Steric Effect and d-Band Center. Adv. Sci. 2019, 6, 1900054. 10.1002/advs.201900054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Peng L.; Oveisi E.; Bulut S.; Sun D. T.; Asgari M.; Trukhina O.; Queen W. L. MOF-Derived Cobalt Phosphide/Carbon Nanocubes for Selective Hydrogenation of Nitroarenes to Anilines. Chem. - Eur. J. 2018, 24, 4234–4238. 10.1002/chem.201705400. [DOI] [PubMed] [Google Scholar]
- Fujita S.; Nakajima K.; Yamasaki J.; Mizugaki T.; Jitsukawa K.; Mitsudome T. Unique Catalysis of Nickel Phosphide Nanoparticles to Promote the Selective Transformation of Biofuranic Aldehydes into Diketones in Water. ACS Catal. 2020, 10, 4261–4267. 10.1021/acscatal.9b05120. [DOI] [Google Scholar]
- Ishikawa H.; Sheng M.; Nakata A.; Nakajima K.; Yamazoe S.; Yamasaki J.; Yamaguchi S.; Mizugaki T.; Mitsudome T. Air-Stable and Reusable Cobalt Phosphide Nanoalloy Catalyst for Selective Hydrogenation of Furfural Derivatives. ACS Catal. 2021, 11, 750–757. 10.1021/acscatal.0c03300. [DOI] [Google Scholar]
- Yamaguchi S.; Fujita S.; Nakajima K.; Yamazoe S.; Yamasaki J.; Mizugaki T.; Mitsudome T. Air-stable and Reusable Nickel Phosphide Nanoparticle Catalyst for the Highly Selective Hydrogenation of D-Glucose to D-Sorbitol. Green Chem. 2021, 23, 2010–2016. 10.1039/D0GC03301D. [DOI] [Google Scholar]
- Yamaguchi S.; Fujita S.; Nakajima K.; Yamazoe S.; Yamasaki J.; Mizugaki T.; Mitsudome T. Support-Boosted Nickel Phosphide Nanoalloy Catalysis in the Selective Hydrogenation of Maltose to Maltitol. ACS Sustainable Chem. Eng. 2021, 9, 6347–6354. 10.1021/acssuschemeng.1c00447. [DOI] [Google Scholar]
- Yamaguchi S.; Mizugaki T.; Mitsudome T. Efficient D-Xylose Hydrogenation to D-Xylitol over a Hydrotalcite-Supported Nickel Phosphide Nanoparticle Catalyst. Eur. J. Inorg. Chem. 2021, 2021, 3327–3331. 10.1002/ejic.202100432. [DOI] [Google Scholar]
- Mitsudome T.; Sheng M.; Nakata A.; Yamasaki J.; Mizugaki T.; Jitsukawa K. A Cobalt Phosphide Catalyst for the Hydrogenation of Nitriles. Chem. Sci. 2020, 11, 6682–6689. 10.1039/D0SC00247J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S.; Yamaguchi S.; Yamasaki J.; Nakajima K.; Yamazoe S.; Mizugaki T.; Mitsudome T. Ni2P Nanoalloy as an Air-Stable and Versatile Hydrogenation Catalyst in Water: P-Alloying Strategy for Designing Smart Catalysts. Chem. Eur. J. 2021, 27, 4439–4446. 10.1002/chem.202005037. [DOI] [PubMed] [Google Scholar]
- Fujita S.; Yamaguchi S.; Yamazoe S.; Yamasaki J.; Mizugaki T.; Mitsudome T. Nickel Phosphide Nanoalloy Catalyst for the Selective Deoxygenation of Sulfoxides to Sulfides under Ambient H2 Pressure. Org. Biomol. Chem. 2020, 18, 8827–8833. 10.1039/D0OB01603A. [DOI] [PubMed] [Google Scholar]
- Sheng M.; Yamaguchi S.; Nakata A.; Yamazoe S.; Nakajima K.; Yamasaki J.; Mizugaki T.; Mitsudome T. Hydrotalcite-Supported Cobalt Phosphide Nanorods as a Highly Active and Reusable Heterogeneous Catalyst for Ammonia-Free Selective Hydrogenation of Nitriles to Primary Amines. ACS Sustainable Chem. Eng. 2021, 9, 11238–11246. 10.1021/acssuschemeng.1c03667. [DOI] [Google Scholar]
- Sheng M.; Fujita S.; Yamaguchi S.; Yamasaki J.; Nakajima K.; Yamazoe S.; Mizugaki T.; Mitsudome T. Single-Crystal Cobalt Phosphide Nanorods as a High-Performance Catalyst for Reductive Amination of Carbonyl Compounds. JACS Au 2021, 1, 501–507. 10.1021/jacsau.1c00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S.; Imagawa K.; Yamaguchi S.; Yamasaki J.; Yamazoe S.; Mizugaki T.; Mitsudome T. A Nickel Phosphide Nanoalloy Catalyst for the C-3 Alkylation of Oxindoles with Alcohols. Sci. Rep. 2021, 11, 10673. 10.1038/s41598-021-89561-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupflin L. A.; Mormul J.; Lejkowski M.; Titlbach S.; Papp R.; Gläser R.; Dimitrakopoulou M.; Huang X.; Trunschke A.; Willinger M. G.; Schlögl R.; Rosowski F.; Schunk S. A. Platinum Group Metal Phosphides as Heterogeneous Catalysts for the Gas-Phase Hydroformylation of Small Olefins. ACS Catal. 2017, 7, 3584–3590. 10.1021/acscatal.7b00499. [DOI] [Google Scholar]
- Zhao M.; Ji Y.; Wang M.; Zhong N.; Kang Z.; Asao N.; Jiang W. J.; Chen Q. Composition-Dependent Morphology of Bi- and Trimetallic Phosphides: Construction of Amorphous Pd–Cu–Ni–P Nanoparticles as a Selective and Versatile Catalyst. ACS Appl. Mater. Interfaces 2017, 9, 34804–34811. 10.1021/acsami.7b08082. [DOI] [PubMed] [Google Scholar]
- Sharma A. K.; Joshi H.; Bhaskar R.; Singh A. K. Solvent-Tailored Pd3P0.95 Nano Catalyst for Amide–Nitrile Inter-Conversion, the Hydration of Nitriles and Transfer Hydrogenation of the C=O Bond. Dalton Trans. 2019, 48, 10962–10970. 10.1039/C8DT04667K. [DOI] [PubMed] [Google Scholar]
- Liu B.; Huang N.; Wang Y.; Lan X.; Wang T. Promotion of Inorganic Phosphorus on Rh Catalysts in Styrene Hydroformylation: Geometric and Electronic Effects. ACS Catal. 2021, 11, 1787–1796. 10.1021/acscatal.0c04684. [DOI] [Google Scholar]
- Furukawa S.; Matsunami Y.; Hamada I.; Hashimoto Y.; Sato Y.; Komatsu T. Remarkable Enhancement in Hydrogenation Ability by Phosphidation of Ruthenium: Specific Surface Structure Having Unique Ru Ensembles. ACS Catal. 2018, 8, 8177–8181. 10.1021/acscatal.8b02582. [DOI] [Google Scholar]
- Carreno M. C. Applications of Sulfoxides to Asymmetric Synthesis of Biologically Active Compounds. Chem. Rev. 1995, 95, 1717–1760. 10.1021/cr00038a002. [DOI] [Google Scholar]
- Madesclaire M. Reduction of Sulfoxides to Thioethers. Tetrahedron 1988, 44, 6537–6580. 10.1016/S0040-4020(01)90096-1. [DOI] [Google Scholar]
- Pulis A. P.; Procter D. J. C–H Coupling Reactions Directed by Sulfoxides: Teaching an Old Functional Group New Tricks. Angew. Chem., Int. Ed. 2016, 55, 9842–9860. 10.1002/anie.201601540. [DOI] [PubMed] [Google Scholar]
- Tang K.-X.; Wang C.-M.; Gao T.-H.; Chen L.; Fan L.; Sun L.-P. Transition Metal-Catalyzed C–H Bond Functionalizations by Use of Sulfur-Containing Directing Groups. Adv. Synth. Catal. 2019, 361, 26–38. 10.1002/adsc.201800484. [DOI] [Google Scholar]
- Zhu Y.-C.; Li Y.; Zhang B.-C.; Zhang F.-X.; Yang Y.-N.; Wang X.-S. Palladium-Catalyzed Enantioselective C–H Olefination of Diaryl Sulfoxides through Parallel Kinetic Resolution and Desymmetrization. Angew. Chem., Int. Ed. 2018, 57, 5129–5133. 10.1002/anie.201801146. [DOI] [PubMed] [Google Scholar]
- Nobushige K.; Hirano K.; Satoh T.; Miura M. Rhodium (III)-Catalyzed Ortho-Alkenylation through C–H Bond Cleavage Directed by Sulfoxide Groups. Org. Lett. 2014, 16, 1188–1191. 10.1021/ol5000605. [DOI] [PubMed] [Google Scholar]
- Takahashi F.; Nogi K.; Yorimitsu H. B2cat2-Mediated Reduction of Sulfoxides to Sulfides. Eur. J. Org. Chem. 2020, 2020, 3009–8833. 10.1002/ejoc.202000222. [DOI] [Google Scholar]
- Surur A. S.; Schulig L.; Link A. Interconnection of Sulfides and Sulfoxides in Medicinal Chemistry. Arch. Pharm. 2019, 352, 1800248. 10.1002/ardp.201800248. [DOI] [PubMed] [Google Scholar]
- Brunell D.; Sagher D.; Kesaraju S.; Brot N.; Weissbach H. Studies on the Metabolism and Biological Activity of the Epimers of Sulindac. Drug Metab. Dispos. 2011, 39, 1014–1021. 10.1124/dmd.110.037663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Guzmán P.; Mahecha-Mahecha C.; Gamba-Sánchez D. Electrophilic Chlorine from Chlorosulfonium Salts: A Highly Chemoselective Reduction of Sulfoxides. Chem. - Eur. J. 2020, 26, 10348–10354. 10.1002/chem.202001815. [DOI] [PubMed] [Google Scholar]
- Mitsudome T.; Takahashi Y.; Mizugaki T.; Jitsukawa K.; Kaneda K. Hydrogenation of Sulfoxides to Sulfides under Mild Conditions Using Ruthenium Nanoparticle Catalysts. Angew. Chem., Int. Ed. 2014, 53, 8348–8351. 10.1002/anie.201403425. [DOI] [PubMed] [Google Scholar]
- Touchy A. S.; Hakim Siddiki S. M. A.; Onodera W.; Kon K.; Shimizu K. Hydrodeoxygenation of Sulfoxides to Sulfides by a Pt and MoOx Co-Loaded TiO2 Catalyst. Green Chem. 2016, 18, 2554–2560. 10.1039/C5GC02806J. [DOI] [Google Scholar]
- Uematsu T.; Ogasawara Y.; Suzuki K.; Yamaguchi K.; Mizuno N. Platinum-Supporting Hollandite-Type Vanadium–Chromium Mixed Oxides as Efficient Heterogeneous Catalysts for Deoxygenation of Sulfoxides under Atmospheric H2 Pressure. Catal. Sci. Technol. 2017, 7, 1912–1920. 10.1039/C7CY00547D. [DOI] [Google Scholar]
- Kuwahara Y.; Yoshimura Y.; Haematsu K.; Yamashita H. Mild Deoxygenation of Sulfoxides over Plasmonic Molybdenum Oxide Hybrid with Dramatic Activity Enhancement under Visible Light. J. Am. Chem. Soc. 2018, 140, 9203–9210. 10.1021/jacs.8b04711. [DOI] [PubMed] [Google Scholar]
- Bowker R. H.; Smith M. C.; Pease M. L.; Slenkamp K. M.; Kovarik L.; Bussell M. E. Synthesis and Hydrodeoxygenation Properties of Ruthenium Phosphide Catalysts. ACS Catal. 2011, 1, 917–922. 10.1021/cs200238v. [DOI] [Google Scholar]
- Liu T.; Wang S.; Zhang Q.; Chen L.; Hu W.; Li C. M. Ultrasmall Ru2P Nanoparticles on Graphene: A Highly Efficient Hydrogen Evolution Reaction Electrocatalyst in Both Acidic and Alkaline Media. Chem. Commun. 2018, 54, 3343–3346. 10.1039/C8CC01166D. [DOI] [PubMed] [Google Scholar]
- Pu Z.; Amiinu I. S.; He D.; Wang M.; Li G.; Mu S. Activating Rhodium Phosphide-Based Catalysts for the pH-Universal Hydrogen Evolution Reaction. Nanoscale 2018, 10, 12407–12412. 10.1039/C8NR02854K. [DOI] [PubMed] [Google Scholar]
- Savithra G. H. L.; Bowker R. H.; Carrillo B. A.; Bussell M. E.; Brock S. L. Mesoporous Matrix Encapsulation for the Synthesis of Monodisperse Pd5P2 Nanoparticle Hydrodesulfurization Catalysts. ACS Appl. Mater. Interfaces 2013, 5, 5403–5407. 10.1021/am402003g. [DOI] [PubMed] [Google Scholar]
- Kang Q.; Li M.; Shi J.; Lu Q.; Gao F. A Universal Strategy for Carbon-Supported Transition Metal Phosphides as High-Performance Bifunctional Electrocatalysts towards Efficient Overall Water Splitting. ACS Appl. Mater. Interfaces 2020, 12, 19447–19456. 10.1021/acsami.0c00795. [DOI] [PubMed] [Google Scholar]
- Fan Z.; Shi J.; Bao X. Synthesis and Antimicrobial Evaluation of Novel 1,2,4-Triazole Thioether Derivatives Bearing a Quinazoline Moiety. Mol. Divers. 2018, 22, 657–667. 10.1007/s11030-018-9821-8. [DOI] [PubMed] [Google Scholar]
- Short C. R.; Flory W.; Hsieh L. C.; Barker S. A. The Oxidative Metabolism of Fenbendazole: a Comparative Study. J. Vet. Pharmacol. Ther. 1988, 11, 50–55. 10.1111/j.1365-2885.1988.tb00120.x. [DOI] [PubMed] [Google Scholar]
- Pinna F.; Menegazzo F.; Signoretto M.; Canton P.; Fagherazzi G.; Pernicone N. Consecutive Hydrogenation of Benzaldehyde over Pd Catalysts: Influence of Supports and Sulfur Poisoning. Appl. Catal., A 2001, 219, 195–200. 10.1016/S0926-860X(01)00685-8. [DOI] [Google Scholar]
- Badano J. M.; Quiroga M.; Betti C.; Vera C.; Canavese S.; Coloma-Pascual F. Resistance to Sulfur and Oxygenated Compounds of Supported Pd, Pt, Rh, Ru Catalysts. Catal. Lett. 2010, 137, 35–44. 10.1007/s10562-010-0336-x. [DOI] [Google Scholar]
- Ma R.; Yang T.; Gao J.; Kou J.; Chen J. Z.; He Y.; Miller J. T.; Li D. Composition Tuning of Ru-Based Phosphide for Enhanced Propane Selective Dehydrogenation. ACS Catal. 2020, 10, 10243–10252. 10.1021/acscatal.0c01667. [DOI] [Google Scholar]
- Cormier Z. R.; Andreas H. A.; Zhang P. Temperature-Dependent Structure and Electrochemical Behavior of RuO2/Carbon Nanocomposites. J. Phys. Chem. C 2011, 115, 19117–19128. 10.1021/jp206932w. [DOI] [Google Scholar]
- Aika K.; Ohya A.; Ozaki A.; Inoue Y.; Yasumori I. Support and Promoter Effect of Ruthenium Catalyst: II. Ruthenium/Alkaline Earth Catalyst for Activation of Dinitrogen. J. Catal. 1985, 92, 305–311. 10.1016/0021-9517(85)90265-9. [DOI] [Google Scholar]
- Yun G.; Guan Q.; Li W. The Synthesis and Mechanistic Studies of a Highly Active Nickel Phosphide Catalyst for Naphthalene Hydrodearomatization. RSC Adv. 2017, 7, 8677–8687. 10.1039/C7RA00250E. [DOI] [Google Scholar]
- Chin S. Y.; Williams C. T.; Amiridis M. D. FTIR Studies of CO Adsorption on Al2O3- and SiO2-Supported Ru Catalysts. J. Phys. Chem. B 2006, 110, 871–882. 10.1021/jp053908q. [DOI] [PubMed] [Google Scholar]
- Ikemiya N.; Senna T.; Ito M. Adlayer Structures of CO Adsorbed on Ru(0001) Electrode Studied by In-situ STM Combined with IRAS. Surf. Sci. 2000, 464, L681–L685. 10.1016/S0039-6028(00)00686-5. [DOI] [Google Scholar]
- Zhao R.; Liu C.; Zhang X.; Zhu X.; Wei P.; Ji L.; Guo Y.; Gao S.; Luo Y.; Wang Z.; Sun X. An Ultrasmall Ru2P Nanoparticles-Reduced Graphene Oxide Hybrid: An Efficient Electrocatalyst for NH3 Synthesis under Ambient Conditions. J. Mater. Chem. A 2020, 8, 77–81. 10.1039/C9TA10346E. [DOI] [Google Scholar]
- Yao K.; Yuan Z.; Jin S.; Chi Q.; Liu B.; Huang R.; Zhang Z. Efficient Hydrodeoxygenation of Sulfoxides into Sulfides under Mild Conditions Using Heterogeneous Cobalt–Molybdenum Catalysts. Green Chem. 2020, 22, 39–43. 10.1039/C9GC02465D. [DOI] [Google Scholar]
- Enthaler S.; Krackl S.; Irran E.; Inoue S. Deoxygenation of Sulfoxides to Sulfides in the Presence of Zinc Catalysts and Boranes as Reducing Reagents. Catal. Lett. 2012, 142, 1003–1010. 10.1007/s10562-012-0862-9. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.