

Abstract
A DNA-based artificial metalloenzyme (ArM) consisting of a copper(II) complex of 4,4′-dimethyl-2,2′-bipyridine (dmbipy-Cu) bound to double-stranded DNA (dsDNA) as short as 8 base pairs with only 2 contiguous central pairs (G for guanine and C for cytosine) catalyzes the highly enantioselective Diels–Alder reaction, Michael addition, and Friedel–Crafts alkylation in water. Molecular simulations indicate that these minimal sequences provide a single site where dmbipy-Cu is groove-bound and able to function as an enantioselective catalyst. Enantioselective preference inverts when d-DNA is replaced with l-DNA. When the DNA is conjugated to a hydrophobic tail, the obtained ArMs exhibit enantioselective performance in a methanol–water mixture superior to that of non-amphiphilic dsDNA, and dsDNA-amphiphiles with more complex G•C-rich sequences.
Keywords: artificial metalloenzyme, DNA catalysis, asymmetric catalysis, catalysis in methanol−water, DNA-amphiphile
Artificial metalloenzymes (ArMs) can combine the chiral environment of biomolecular scaffolds with the catalytic properties of a transition metal.1−6 Since the introduction of DNA-based ArMs in 2005,7 enantioselective catalysis by metal ions or metal ion complexes assembled on a DNA scaffold has been demonstrated for Diels–Alder,7−13 Friedel–Crafts alkylation,14−18 Michael addition,19−22 and other reactions.23−30 Despite progress in correlating the catalytic performance of DNA-ArMs with their DNA sequence,8−12,15−18,21,24−28,30 pinpointing which of the many possible chiral microenvironments created by the interaction between the DNA and the bound metal, or metal complex, is responsible for enantioselective catalysis remains elusive. As a result, our ability to design catalysts based on minimal DNA sequences with localized catalytic sites is limited. Minimal DNA sequences may enable cost-effective implementation of DNA scaffolds as well as their facile incorporation in extended constructs, like DNA-amphiphiles, aiming at efficient catalysis in complex solvent environments. Here, it is demonstrated that DNA-ArMs consisting of a Cu(II) complex with 4,4′-dimethyl-2,2′-bipyridine (dmbipy-Cu) bound to minimal dsDNA sequences, containing only two contiguous G•C base pairs, can be as highly active and selective as ArMs based on more extended DNA sequences. Based on these findings and molecular simulations of dmbipy–Cu interactions with DNA, a single catalytic site is proposed. Moreover, to establish possible practical implications, it is shown that the minimal TTTGGTTT and AAACCAAA DNA sequences can be conjugated to hydrophobic tails to create DNA-amphiphile ArMs, which in methanol–water solvents outperform their corresponding DNA ArMs, as well as DNA-amphiphile ArMs based on the G-rich sequence HT21.
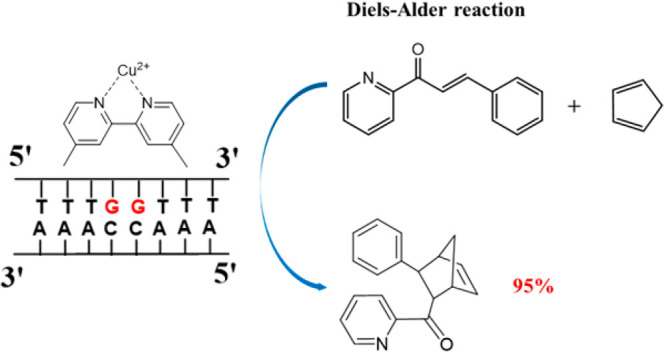
The Diels–Alder reaction between aza-chalcone (1a) and cyclopentadiene (2) was used to probe the catalytic performance of different dsDNA sequences (Figure 1 and Figure S1). There are four possible products for (3-phenylbicyclo[2.2.1]hept-5-en-2-yl)(pyridin-2-yl)methanone (3a): Re-endo, Si-endo, Re-exo, and Si-exo (Scheme S1) out of which the two endo enantiomers are dominant.31,32 The ability for enantioselectivity by the DNA-ArM catalyst is determined by the enantiomeric excess (ee) of the Si-endo versus Re-endo.6 Earlier reports show that both 4,4′-dimethyl-2,2′-bipyridine (dmbipy) and 2,2′-bipyridine (bipy) are groove binders to natural salmon testes DNA (st-DNA) with dmbipy exhibiting higher enantioselectivity and rate acceleration for the Diels–Alder reaction between (1a) and (2).33 Therefore, dmbipy was selected as the achiral ligand to bridge the Cu(II) catalytic center and the dsDNA scaffold.34 We include for comparison the performance of st-DNA and a G•C-rich dsDNA sequence composed of the 21 nucleotide G-rich human telomeric sequence HT21. The ArM catalysts based on st-DNA and HT21 exhibit high conversion and enantioselectivity with preference for the Si-endo (Figure 1) in agreement with expectations from a previous report.8
Figure 1.

Conversion and ee for Diels–Alder reaction catalyzed by different dsDNA sequences with dmbipy-Cu. ee% = (moles of Si-endo – moles of Re-endo)/(moles of Si-endo + moles of Re-endo) × 100%. All reactions were carried out in MOPS (20 mM, pH 6.5) at 4 °C for 3 h, st-DNA: 0.68 mg/mL (1.05 mM base pairs); synthetic dsDNA: 50 μM; [dmbipy-Cu]: 50 μM; aza chalcone (1a): 1 mM (i.e., 5% catalyst loading); cyclopentadiene: 5.6 μL (67 equiv). See experimental section (SI) for reaction procedure details. All data are averaged over three independent experiments. Parameters were determined for all displayed products by high-performance liquid chromatography (HPLC) analysis on a chiral stationary phase. The conversion and ee were not detectable in the absence of dsDNA under the conditions cited above (longer times were required to achieve detectable conversion).
dsDNA G•C-rich sequences have been verified as active and enantioselective catalysts for the Diels–Alder reaction of (1a) and (2).8 However, the presence of multiple secondary structures in samples of HT21 and its complementary sequence make efforts to identify catalytic sites a complicated task. Figure S2a shows that in the conditions of the reaction, multiple secondary structures, like antiparallel G-quadruplex, i-motif, and B-form,35 coexist. Moreover, variations among independently produced samples (i.e., prepared on different days using identical procedure) are evident. Our results are in agreement with reports from the literature where they showed that a completely hydrogen-bonded one-to-one complex from a double-stranded polyG•polyC sample had to be prepared enzymatically, as otherwise the sequences could also form self-complexes.36 Addition of 0.1 M NaCl, not present in the reaction condition, was required to stabilize the B-form (Figure S2b). Therefore, in order to narrow down the catalytically active chiral microenvironments in the DNA duplex and explain the origin of asymmetric catalysis achieved by the G•C-rich dsDNA-based ArMs, we tested a series of synthetic 21-base-pair dsDNA sequences including the “21” sequence, which consists of 21 base pairs of adenine (A) and thymine (T), the “21-6”, “21-6,7”, “21-6,7,8”, and “21-6,7,8,9” sequences, which contain one to four contiguous G•C pairs (see Table S1 and Scheme S2 for naming the DNA sequences used in this study). The CD results confirmed that all DNA sequences used in Figures 1 and S1 hybridized in the buffer used for catalysis and that the presence of dmbipy-Cu had no notable effect on the secondary structure of the dsDNA (Figures S4–S16). For comparison, the CD of st-DNA is shown in Figure S3. Sequence “21” showed four peaks between 245 and 285 nm (Figure S4), two maxima at 260 and 282 nm, and two minima at 248 and 267 nm, characteristic of the B′ heteronomous DNA-form adopted by poly(A)•poly(T) sequences.35,37 The presence of just one G has been shown to interrupt the A-track by decreasing the ellipticity at 260 nm, as shown in Figure S5.37,38
It has been stated earlier that three contiguous G are correlated with high ee for the Diels–Alder reaction of (1a) and (2).8 Indeed, while the “21” sequence (consisting only of A•T pairs) was almost inert toward this reaction, after introducing one G•C pair to the duplex, ee increased from 27% to 60%. When two contiguous G•C pairs are present in the “21” sequence, a notable increase in both conversion and enantioselectivity was observed for the “21-6,7” sequence (94% ee with preference for Si-endo at 80% aza-chalcone conversion). With the dsDNA sequences containing three or four G•C base pairs in a row, highly active hybrid catalysts were acquired, which provided the Si-endo isomer with excellent enantioselectivities (98% for both “21-6,7,8” and “21-6,7,8,9”). These results indicate that in order to obtain a highly enantioselective ArM catalyst for the Diels–Alder reaction of (1a) and (2), a minimum of two contiguous G•C pairs are at least required.
The position of the two G•C pairs is consequential. A comparison among 21-base-pair dsDNA sequences containing two G•C base pairs at different positions is shown in Figure S1. A “21-6,16” sequence with two separate G•C pairs provided only moderate enantioselectivity (68%) further emphasizing the importance for the G•C pairs to be contiguous. When the two contiguous G•C pairs were moved to the end of the sequence (“21-20,21”), the enantioselectivity dropped to 21%. However, after moving the two G•C pairs by two nucleotides inward (“21-18,19”), the ee was restored to 90%. By moving the two contiguous G•C pairs further toward the center zone, both conversion and enantioselectivity were increased again, with the best results given by the “21-10,11” sequence (84% conversion and 95% ee). The significant effect of the two G•C pairs’ position on the catalytic results indicates sensitivity of the enantioselectivity to the chiral microenvironment created by the binding position of the bipyridine complex to the dsDNA scaffold.
The simplicity of our system allowed us to propose the active site configuration by performing statistical mechanics-based modeling. Molecular dynamics (MD) simulations for the binding of dmbipy-Cu on two representative sequences: “21” and “21-10,11” in ionic aqueous solution were conducted (details in SI).39 The MD simulations show that the dmbipy-Cu complex (Figure S17) in both sequences binds to the minor groove of DNA without intercalating its double helix. As shown from the time evolution of the distance between the dmbipy-Cu and the DNA in Figure S18, the molecules remain bound along the entire equilibrated MD trajectory. Figure S19 presents the rise per base pair along the helix axis, namely, the distance between adjacent bases in the same DNA strand, computed over the entire MD trajectories. It is seen that, in both sequences, the average rise per base pair fluctuates around the crystallographic value denoting no increase, and thus no dmbipy-Cu intercalation. In Figure S20, the probability of finding dmbipy-Cu at various distances from the minor or major groove base atoms indicates that the dmbipy-Cu is a minor groove binder in both sequences. In particular, in “21”, the dmbipy-Cu complex resides in the minor groove covering five nucleotide pairs, from T8 to T12, while in “21-10,11”, it bounds in the vicinity of the G•C pairs covering a four-nucleotide sequence, from T7 to G10, as shown in Figure S21a depicting the minimum distance between the dmbipy-Cu and each of the 21 DNA nucleotide pairs. More specifically, the dmbipy-Cu forms close contacts with the electronegative minor groove atoms N2 and N3 of guanines and the N3 of adenine and O2 of thymine atoms residing in the proximity of the G•C pairs, as shown in Figure S21b (the notation of the base atoms follows the standard IUPAC-IUB numbering convention for the nucleic acid bases40). The computation of the electrostatic potential energy between the dmbipy-Cu and the DNA for both the “21” and the “21-10,11” systems (Figure S21c) shows that electrostatic interactions are more attractive in the “21-10,11”, indicating that electrostatics are a key parameter in the dmbipy-Cu binding of the DNA sequence containing the two contiguous G•C pairs.
The dmbipy-Cu to DNA binding pattern was further investigated by computing the probability of finding the copper atom or the pyridine methyl carbons at various distances from the minor groove base atoms, as depicted in Figure 2a and b, and Figure S22 (details in SI). In “21”, the position of the dmbipy-Cu was found to be highly localized, with both methyl carbons residing at short distances from the minor groove base atoms (cumulative probability p (0.3 ≤ s ≤ 0.5) approximately 0.93 and 0.80 for C15 and C9, respectively). Also, copper lies at large distances into the solvent (p (0.7 ≤ s ≤ 1.1) about 0.90). These findings indicate that the ligand orients both pyridine methyls into the groove as shown in the indicative configuration rendered from the MD trajectory (Figure 2c). This configuration is enabled by the quasi-planar structure of dmbipy-Cu which allows for the fit of the molecule in between the minor groove wall. By contrast, in “21-10,11”, the respective distributions are considerably wider denoting enhanced dmbipy-Cu mobility, in agreement with previous suggestions regarding dmbipy-Cu and G•C-rich st-DNA.33 We found that the pyridine methyls may be located in or out (p (0.5 ≤ s ≤ 1.2) approximately 0.70 and 0.80 for C15 and C9, respectively) of the minor groove. Also, the copper atom may reside in the groove or in the DNA hydration shell close to the edge of the groove (p (0.5 ≤ s ≤ 0.7) value about 0.55). The above findings show that the dmbipy-Cu may direct one of its pyridine rings in the groove, and the other in the solvent with the copper atom close to the minor groove edge (Figure 2d).
Figure 2.

Probability density functions of finding the copper atom (a) and pyridine methyl carbon C15 (b) of the dmbipy-Cu, at various distances from the minor groove base atoms in “21” (red) and “21-10,11” (blue) sequences, computed over the equilibrated MD trajectory (for a similar graph for pyridine methyl carbon C9, see Figure S22; the notation of carbon atoms is indicated in Figure S17). Indicative configurations rendered from the MD trajectory depicting the dmbipy-Cu binding to the DNA in “21” (c) and “21-10,11” (d). The reactant (1a), added by following the bidentate coordination pattern45 of the enone to the copper atom of the dmbipy-Cu, is shown in (e): the 5-coordinated triagonal bipyramidal complex has the N of (1a) and a N of dmbipy in the axial positions, while the three equatorial positions are occupied by the other N of pyridine, the O of (1a), and an O from water (not shown)). This configuration is depicted in (f) for “21” and (g,h) for “21-10,11”. (g, h) depict the same configuration in two rotated views for clarity. Water molecules in the rendered pictures are omitted for clarity; configurations including the water molecules are depicted in Figure S24. Color code for the dmbipy-Cu and reactant (1a): Cu (magenta), N (blue), C (cyan), H (white), O (red); color code for the DNA: guanine (green), cytosine (cyan), adenine and thymine (yellow).
The distinct configuration for dmbipy-Cu-dsDNA “21-10,11” is due to the presence of the G•C pairs, and the G•C-induced local widening of the minor groove, while on the contrary, the narrow minor groove of the pure A•T-sequence “21” does not enable such behavior (Figure S23). In other words, the groove size is sequence-dependent, and the widening is not induced by the dmbipy-Cu, as shown by calculations of the minor groove width for the two initial DNA structures (Figure S23). The above computations are in agreement with experiments showing that A•T-rich sequences exhibit narrower minor grooves than G•C-rich DNA.41,42
The dissimilar binding patterns of the dmbipy-Cu in “21” and “21-10,11” result in a different hydration level for copper, as revealed by the computed radial distribution functions shown in Figure S25a. In “21-10,11”, the coordination of water molecules around copper is partially restricted because of the “volume” occupied by the DNA, which reduces the accessible surface of the copper atom by the solvent; in “21-10,11” the Cu atom was found to coordinate with the oxygen atoms of the DNA phosphate groups with higher probability (cumulative probability p (s ≤ 0.3) approximately 0.7 and 0.4, respectively) than in the “21” sequence (see Figure S25b). Changes in the coordination of Cu in the presence of DNA have been suggested earlier.43
As shown in Figure 2e, the experimentally determined catalytic outcome requires a preferential acceleration of the cyclopentadiene attack through the Si-face of (1a) when attached to dmbipy-Cu bound to “21-10,11”, while no such acceleration is observed for “21”. We propose that this is accomplished by the distinct sequence-dependent binding patterns of the dmbipy-Cu in the sequences “21” and “21-10,11” as predicted by MD, which in turn can impose different orientations of the reactant (1a) with respect to DNA upon its bidentate coordination to the copper atom. Here, we do not undertake the task of simulating the coordination of (1a) with dmbipy-Cu in the presence of dsDNA in ionic aqueous solution, on the basis of density functional theory (DFT); such simulations are currently computationally extremely expensive because of the high number of atoms N involved; standard DFT algorithms scale with O(N3).44 Instead, we use the dmbipy-Cu and dsDNA configurations determined by MD and assume that the bidentate coordination of (1a) to dmbipy-Cu occurs with the two quasi-planar molecules orienting their planes to form a 5-coordinate trigonal bipyramidal complex, as predicted by previous DFT calculations in the absence of both the DNA and the solvent45 and shown in Figure 2e. We show schematically in Figure 2f and Figure 2g,h possible configurations of DNA-dmbipy-Cu-(1a) based on the above assumption (see also Figure S26 for alternative configurations). In the “21” sequence, both faces of the (1a) are away from the DNA double helix and in contact with the bulk solvent (Figure 2f), whereas in the sequence “21-10,11”, one face of the (1a) is close to the DNA double helix with the other face being in contact with the bulk solvent (Figure 2g,h). Consistent with the experimental findings, the “21” configuration cannot provide preference for Si-face vs Re-face attack of (2) to (1a). In the “21-10,11” configuration shown in Figure 2g and h, the Si-face is near the DNA. Therefore, in this configuration, the Si-face attack of (2) to (1a) will take place with (2) present in the pocket created between (1) and the DNA. In a recent study of the same reaction catalyzed by Cu2+-adenosine triphosphate, density functional theory gas-phase calculations suggested that the Si-face attack was favored due to hydrogen bonding of (2) with phosphate oxygen atoms leading to a reduced energy barrier for the cycloaddition.6 One possible explanation for our system is that DNA–(2) interactions in the pocket created between the Si-face of (1a) and dsDNA lead to a free energy transition state reduction causing the observed rate increase and high ee observed experimentally for “21-10,11”. An alternative explanation for the observed ee is that the relevant configuration for catalysis is with (1a) oriented as shown in Figure S26, i.e., with (1a) rotated by 180° compared to its configuration in Figure 2g and h. In this configuration, the Re-face of (1a) is near the DNA and is being shielded against Re-face attack. In this scenario, the rate acceleration observed experimentally can be attributed to an increase in local concentration as suggested in a previous study.43 At this point, we cannot provide definite support for one of the two aforementioned mechanisms, as they can both be consistent with the observed ee in favor of the Si-endo isomer.
The above experiments and simulations suggest that a catalytic site can be localized at two contiguous G•C pairs. Next, we investigated minimal sequences containing 12, 8, and 6 base pairs. As Figure 1 shows, the “12-6,7” and “8-4,5” sequences maintain high enantioselectivity. However, when the sequence was truncated to 6 base pairs (“6-3,4”), lower conversion (24%) and ee (78%) were observed. Thus, the catalytic results indicate that to maintain high enantioselectivity, the dsDNA sequence with two central contiguous G•C base pairs can be as short as 8 base pairs (“8-4,5”). We note that the highly enantioselective functionality of the “8-4,5” is obtained despite the increase in configurational disorder in 8 base pair sequences containing G•C pairs suggested by crystal structures and potential energy minimizations.46−48
The melting temperatures (Figure S27 and Table S6) of st-DNA, HT21, “8-4,5”, and “8” were measured at two concentrations (3 and 15 μM) which are lower than the 50 μM concentration used in the catalysis experiment. At 15 μM, all sequences exhibit melting temperatures that are higher than 4 °C. Results show that as the concentration increases the melting temperature increases in agreement with literature reports49 and the melting temperatures increase in the following order: “8” < “8-4,5” < HT21 < st-DNA.
Although the role of DNA sequence8,50,51 and length16 has been discussed before, to the best of our knowledge, the highly enantioselective performance of only two contiguous G•C base pairs in a sequence as short as eight nucleotides has not been demonstrated. Our short dsDNA sequences are comparable in size and performance with catalysts consisting of dmbipy-Cu and short (14-nucleotide) sequences that fold into hairpin structures containing a minimum of three G•C pairs.52
The binding constants of dmbipy-Cu based on DNA base pair concentrations are shown in Figure S28. The binding constant for st-DNA ((9.25 ± 0.75) × 103 M–1) is consistent with that reported by the Roelfes group (7.26 × 103 M–1).8 According to our results, the dmbipy-Cu can bind strongly to both “8” ((9.99 ± 0.79) × 103 M–1, corresponding to 78.3% of the dmbipy-Cu complex bound to “8”) and “8-4,5” ((10.34 ± 1.03) × 103 M–1, corresponding to 78.8% of the dmbipy-Cu complex bound to “8-4,5”) sequences, indicating that the enantioselective catalytic performance differences (i.e., rate acceleration in favor of the Si-endo isomer) are not due to differences in binding but differences in configuration. This is consistent with the MD simulations discussed above. We note that as stated in the caption of Figure 1, free dmbipy-Cu exhibits negligible activity under the experimental conditions we used.
The ability for inverting the enantioselectivity by switching from right-handed d-DNA to the left-handed l-DNA, introduced earlier,53,54 was confirmed for the “8-4,5” sequence (HPLC chromatograms are shown in Figure S29). The left-handed “8-4,5” sequence was confirmed by contrasting the CD spectra of l-DNA (Figure S30) with the CD spectra of the natural right-handed d-DNA (Figure S11). As shown in Table S7 (entry 1 and entry 2), by switching the duplex backbone of “8-4,5” from right-handed to left-handed and providing a microenvironment of opposite chirality, the enantioselectivity was reversed. When using d-DNA, cyclopentadiene addition from the Si-side is favored forming the Si-endo product, while for l-DNA, addition from the Re-side is favored to form the Re-endo product. By using equimolar amounts of l-DNA and d-DNA, a racemic endo product was obtained (Table S7, entry 3) as expected based on identical catalytic activities of the mirror images of the catalytic sites.
We also examined a series of 8-base-pair variations (Figure 3), which further confirmed the importance of two contiguous G•C pairs. Hybridization and the presence of the B-form conformation were verified for all of these dsDNA sequences by CD (Figures S11 and S31–S34). Activity and selectivity decreased by switching the G and C base groups of one of the two contiguous G•C pairs (group B, Figure 3). Further highlighting the importance of two contiguous G•C pairs, a sequence of 8 alternating G•C pairs (group C, Figure 3) also exhibited reduced activity and selectivity.
Figure 3.
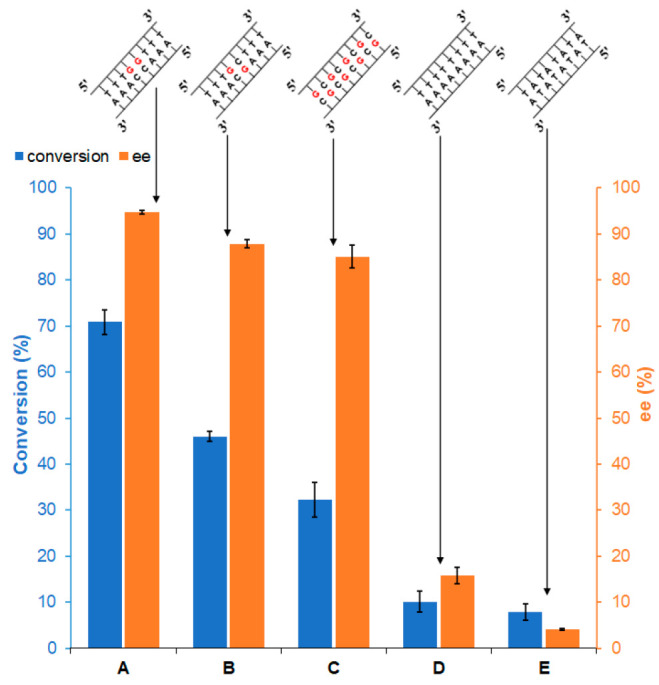
Conversion and ee for Diels–Alder reaction catalyzed by different 8-base pair dsDNA sequences with dmbipy-Cu. Reaction and conditions are the same as in Figure 1.
In addition to the Diels–Alder reaction, the “8-4,5” dsDNA sequence exhibits significantly enhanced enantioselectivity compared to the enantioselectivity achieved by the “8” dsDNA sequence for Friedel–Crafts alkylation (Figure 4a) and Michael addition (Figure 4b) in water. This finding further highlights the functionality of the short sequence with two contiguous G•C pairs.
Figure 4.
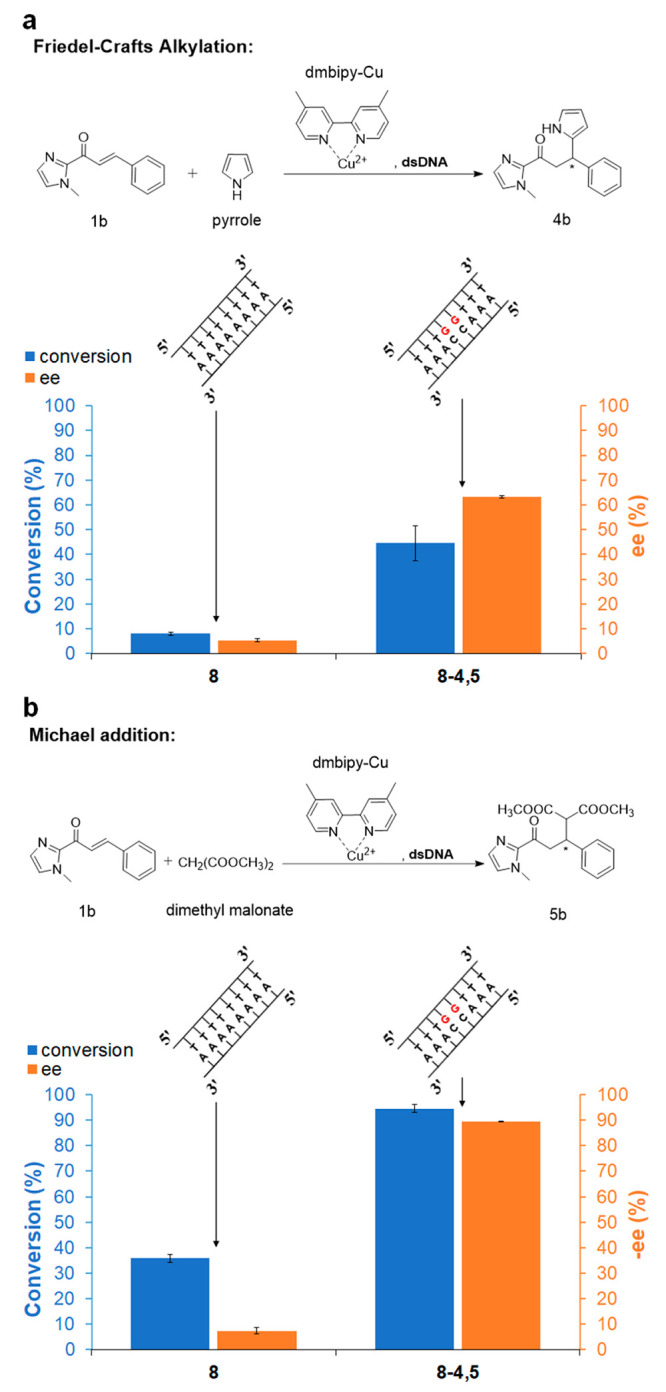
(a) Conversion and ee for Friedel–Crafts alkylation catalyzed by “8” or “8-4,5” with dmbipy-Cu. ee% = (moles of R enantiomer – moles of S enantiomer)/(moles of R enantiomer + moles of S enantiomer) × 100%.15 (b) Conversion and −ee for Michael addition reaction catalyzed by “8” or “8-4,5” with dmbipy-Cu. ee is negative for this reaction. −ee% = (moles of R enantiomer – moles of S enantiomer)/(moles of R enantiomer + moles of S enantiomer) × 100%.20 All reactions were carried out in MOPS (20 mM, pH 6.5) at 4 °C for 7 days; synthetic dsDNA: 50 μM; [dmbipy-Cu]: 50 μM; α,β-unsaturated 2-acyl imidazole (1b): 1 mM (i.e., 5% catalyst loading); pyrrole: 5 mM; dimethyl malonate: 11.4 μL (100 equiv). See Experimental Section (SI) for reaction procedure details. All data are averaged over three independent experiments. Parameters were determined for all displayed products by HPLC analysis on a chiral stationary phase.
The minimal sequences with a localized enantioselective site presented above are sufficient to match the performance of ArMs with more complicated sequences allowing for better assessment of the location of the catalytic site. Moreover, they may enable cost-effective utilization of DNA by complementing other approaches toward practical applications like the ones using bound DNA on solid matrices55,56 as well as exploring immobilization and imprinting approaches and hybrid catalysts.57−59 We demonstrate next a case where the performance of a minimal DNA sequence with 2 G•C pairs (“8-4,5” sequence) exceeds the performance of the HT21 sequence with 4 G•C triplets, highlighting the practical significance of the minimal sequences introduced herein. The “8-4,5” sequence and the HT21 sequence were used in a methanol–water mixture (v/v = 50/50) for the Diels–Alder reaction of (1a) and (2). As shown in Figure 5, both dsDNA sequences lost their enantioselective ability. This finding is in agreement with previous reports that the highly enantioselective DNA-based ArM has limited tolerance toward organic cosolvents.60−62
Figure 5.

(a) Chemical structures of “HT21”, “8-4,5”, “HT21-amph.”, and “8-4,5-amph.” (b) Conversion and ee of Diels–Alder reaction catalyzed by different dsDNA sequences with dmbipy-Cu in methanol–water mixture (v/v = 50/50). All reactions were carried out in MOPS (20 mM, pH 6.5) at 4 °C for 48 h, dsDNA: 15 μM; [dmbipy-Cu]: 15 μM; aza chalcone (1a): 1 mM (i.e., 1.5% catalyst loading); cyclopentadiene: 5.6 μL (67 equiv). See Experimental Section (SI) for reaction procedure details. All data are averaged over three independent experiments. Parameters were determined for all displayed products by HPLC analysis on a chiral stationary phase.
To recover the asymmetric catalytic performance of the synthetic dsDNA sequences in the methanol–water (v/v = 50/50) system, we introduce an additional driving force for assembly of the DNA strands. Earlier studies have demonstrated that conjugation of DNA to polyethylene glycol can improve its solubility, assembly, and catalytic function in organic solvents, albeit without showing enantioselectivity.63 A hydrophobic tail was synthesized64 and conjugated to both the 5′ end of the ssDNA sequence and the 3′ end of the complementary ssDNA sequence (Scheme S3).65 Successful conjugation was confirmed by matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (Table S3). CD spectra showed that tail conjugation did not have an effect on the secondary structure of the sequences in methanol–water (Figures S35–S36). “8-4,5” and its amphiphile showed B-form conformation. HT21 and its amphiphile in methanol–water had a strong positive peak at 291 nm and a negative peak around 260 nm, characteristic of antiparallel G-quadruplex and i-motif.35 However, the presence of the B form cannot be excluded for HT21, as the negative hump around 245 nm could result from cancellation of a negative peak from B-form and a positive peak from antiparallel G-quadruplex at this wavelength. The catalytic performance of the dsDNA amphiphiles in the methanol–water (v/v = 50/50) system is shown in Figure 5. A first observation is that the amphiphiles exhibit improved enantioselectivity compared to the free dsDNA sequences. This could be attributed to stabilization provided by the tails as supported by the increase in melting temperature for the amphiphiles. Figure S37 shows that the melting temperatures of “8-4,5” and “8-4,5-amph.” in methanol–water were 7.2 ± 0.6 °C and 35.0 ± 0.1 °C, respectively. In addition, we found that the enantioselectivity achieved by the “8-4,5-amph.” dsDNA amphiphiles (41%) was significantly higher than that given by the “HT21-amph.” (29%). Further work is required to decipher the structure of the amphiphiles, the distribution of solvents and reactants, and their effect on reaction rates and selectivities. However, this serves as a first example of an ArM based on a short dsDNA sequence with only two contiguous G•C pairs outperforming dsDNA with extra and more extended G-tracts in a catalytic system that benefits from contiguous G•C pairs. The findings reported here, and in earlier studies,52 justify further exploration of minimal sequences with localized catalytic sites incorporated in ArMs, from both the fundamental and practical standpoint.
Acknowledgments
This work was partially funded by the Whiting School of Engineering and the Bloomberg Distinguished Professorship program of the Johns Hopkins University. E.P. acknowledges a grant of Scientific Excellence by the Hellenic Foundation for Research and Innovation (Project: B.1003-ENGETACT). G.K.P. acknowledges support by the European Union through a fellowship under H2020-MSC-IF (Project: 796794-ENGEMED), and by the Greek Research and Technology Network for CPU-time provided in the National HPC facility “ARIS”. We would also like to thank the NMR core facility at the Department of Chemistry, the Center for Molecular Biophysics at Johns Hopkins University, and Dr. Katherine Tripp for technical assistance with CD spectra.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00513.
Experimental procedures, modeling details, additional catalytic results, CD, MS, NMR, HPLC details, including Schemes S1–S3, Tables S1–S7, and Figures S1–S37 (PDF)
Author Contributions
# J.G. and D.W. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Ringenberg M. R.; Ward T. R. Merging the best of two worlds: artificial metalloenzymes for enantioselective catalysis. Chem. Commun. 2011, 47 (30), 8470–8476. 10.1039/c1cc11592h. [DOI] [PubMed] [Google Scholar]
- Wang C.; Hao M.; Qi Q.; Dang J.; Dong X.; Lv S.; Xiong L.; Gao H.; Jia G.; Chen Y. Highly Efficient Cyclic Dinucleotide Based Artificial Metalloribozymes for Enantioselective Friedel-Crafts Reactions in Water. Angew. Chem., Int. Ed. 2020, 59 (9), 3444–3449. 10.1002/anie.201912962. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Roelfes G. Synergistic catalysis in an artificial enzyme by simultaneous action of two abiological catalytic sites. Nat. Catal. 2020, 3 (3), 289–294. 10.1038/s41929-019-0420-6. [DOI] [Google Scholar]
- Lu Y.; Yeung N.; Sieracki N.; Marshall N. M. Design of functional metalloproteins. Nature 2009, 460 (7257), 855. 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyster T. K.; Ward T. R. Genetic optimization of metalloenzymes: enhancing enzymes for non-natural reactions. Angew. Chem., Int. Ed. 2016, 55 (26), 7344–7357. 10.1002/anie.201508816. [DOI] [PubMed] [Google Scholar]
- Wang C.; Qi Q.; Li W.; Dang J.; Hao M.; Lv S.; Dong X.; Gu Y.; Wu P.; Zhang W.; Chen Y.; Hartig J. S. A Cu(II)-ATP complex efficiently catalyses enantioselective Diels-Alder reactions. Nat. Commun. 2020, 11 (1), 4792. 10.1038/s41467-020-18554-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfes G.; Feringa B. L. DNA-based asymmetric catalysis. Angew. Chem., Int. Ed. 2005, 44 (21), 3230–3232. 10.1002/anie.200500298. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Klijn J. E.; Feringa B. L.; Roelfes G. DNA-based asymmetric catalysis: sequence-dependent rate acceleration and enantioselectivity. J. Am. Chem. Soc. 2008, 130 (35), 11783–11790. 10.1021/ja803170m. [DOI] [PubMed] [Google Scholar]
- Rosati F.; Boersma A. J.; Klijn J. E.; Meetsma A.; Feringa B. L.; Roelfes G. A kinetic and structural investigation of DNA-based asymmetric catalysis using first-generation ligands. Chem. Eur. J. 2009, 15 (37), 9596–9605. 10.1002/chem.200900456. [DOI] [PubMed] [Google Scholar]
- Park S.; Okamura I.; Sakashita S.; Yum J. H.; Acharya C.; Gao L.; Sugiyama H. Development of DNA metalloenzymes using a rational design approach and application in the asymmetric Diels-Alder reaction. ACS Catal. 2015, 5 (8), 4708–4712. 10.1021/acscatal.5b01046. [DOI] [Google Scholar]
- Wang C.; Jia G.; Zhou J.; Li Y.; Liu Y.; Lu S.; Li C. Enantioselective Diels-Alder reactions with G-quadruplex DNA-based catalysts. Angew. Chem., Int. Ed. 2012, 51 (37), 9352–9355. 10.1002/anie.201204850. [DOI] [PubMed] [Google Scholar]
- Wilking M.; Hennecke U. The influence of G-quadruplex structure on DNA-based asymmetric catalysis using the G-quadruplex-bound cationic porphyrin TMPyP4· Cu. Org. Biomol. Chem. 2013, 11 (40), 6940–6945. 10.1039/c3ob41366g. [DOI] [PubMed] [Google Scholar]
- Roe S.; Ritson D. J.; Garner T.; Searle M.; Moses J. E. Tuneable DNA-based asymmetric catalysis using a G-quadruplex supramolecular assembly. Chem. Commun. 2010, 46 (24), 4309–4311. 10.1039/c0cc00194e. [DOI] [PubMed] [Google Scholar]
- García-Fernández A.; Megens R. P.; Villarino L.; Roelfes G. DNA-Accelerated Copper Catalysis of Friedel-Crafts Conjugate Addition/Enantioselective Protonation Reactions in Water. J. Am. Chem. Soc. 2016, 138 (50), 16308–16314. 10.1021/jacs.6b08295. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Feringa B. L.; Roelfes G. Enantioselective Friedel-Crafts reactions in water using a DNA-based catalyst. Angew. Chem., Int. Ed. 2009, 121 (18), 3396–3398. 10.1002/ange.200900371. [DOI] [PubMed] [Google Scholar]
- Park S.; Ikehata K.; Watabe R.; Hidaka Y.; Rajendran A.; Sugiyama H. Deciphering DNA-based asymmetric catalysis through intramolecular Friedel-Crafts alkylations. Chem. Commun. 2012, 48 (84), 10398–10400. 10.1039/c2cc35625b. [DOI] [PubMed] [Google Scholar]
- Park S.; Zheng L.; Kumakiri S.; Sakashita S.; Otomo H.; Ikehata K.; Sugiyama H. Development of DNA-based hybrid catalysts through direct ligand incorporation: Toward understanding of DNA-based asymmetric catalysis. ACS Catal. 2014, 4 (11), 4070–4073. 10.1021/cs501086f. [DOI] [Google Scholar]
- Wang C.; Li Y.; Jia G.; Liu Y.; Lu S.; Li C. Enantioselective Friedel-Crafts reactions in water catalyzed by a human telomeric G-quadruplex DNA metalloenzyme. Chem. Commun. 2012, 48 (50), 6232–6234. 10.1039/c2cc31320k. [DOI] [PubMed] [Google Scholar]
- Dey S.; Jäschke A. Tuning the stereoselectivity of a DNA-catalyzed Michael addition through covalent modification. Angew. Chem., Int. Ed. 2015, 54 (38), 11279–11282. 10.1002/anie.201503838. [DOI] [PubMed] [Google Scholar]
- Coquière D.; Feringa B. L.; Roelfes G. DNA-based catalytic enantioselective Michael reactions in water. Angew. Chem., Int. Ed. 2007, 46 (48), 9308–9311. 10.1002/anie.200703459. [DOI] [PubMed] [Google Scholar]
- Megens R. P.; Roelfes G. DNA-based catalytic enantioselective intermolecular oxa-Michael addition reactions. Chem. Commun. 2012, 48 (51), 6366–6368. 10.1039/c2cc31854g. [DOI] [PubMed] [Google Scholar]
- Punt P. M.; Langenberg M. D.; Altan O.; Clever G. H. Modular Design of G-Quadruplex MetalloDNAzymes for Catalytic C-C Bond Formations with Switchable Enantioselectivity. J. Am. Chem. Soc. 2021, 143 (9), 3555–3561. 10.1021/jacs.0c13251. [DOI] [PubMed] [Google Scholar]
- Fournier P.; Fiammengo R.; Jäschke A. Allylic amination by a DNA-diene-iridium (I) hybrid catalyst. Angew. Chem., Int. Ed. 2009, 48 (24), 4426–4429. 10.1002/anie.200900713. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Coquière D.; Geerdink D.; Rosati F.; Feringa B. L.; Roelfes G. Catalytic enantioselective syn hydration of enones in water using a DNA-based catalyst. Nat. Chem. 2010, 2 (11), 991–995. 10.1038/nchem.819. [DOI] [PubMed] [Google Scholar]
- Yum J. H.; Park S.; Hiraga R.; Okamura I.; Notsu S.; Sugiyama H. Modular DNA-based hybrid catalysts as a toolbox for enantioselective hydration of alpha, beta-unsaturated ketones. Org. Biomol. Chem. 2019, 17 (9), 2548–2553. 10.1039/C9OB00196D. [DOI] [PubMed] [Google Scholar]
- Hao J.; Miao W.; Cheng Y.; Lu S.-M.; Jia G.; Li C. Enantioselective Olefin Cyclopropanation with G-quadruplex DNA-based Biocatalysts. ACS Catal. 2020, 10, 6561. 10.1021/acscatal.0c01203. [DOI] [Google Scholar]
- Duchemin N.; Heath-Apostolopoulos I.; Smietana M.; Arseniyadis S. A decade of DNA-hybrid catalysis: from innovation to comprehension. Org. Biomol. Chem. 2017, 15 (34), 7072–7087. 10.1039/C7OB00176B. [DOI] [PubMed] [Google Scholar]
- Mansot J.; Vasseur J. J.; Arseniyadis S.; Smietana M. alpha, beta-Unsaturated 2-Acyl-Imidazoles in Asymmetric Biohybrid Catalysis. ChemCatChem 2019, 11, 5686. 10.1002/cctc.201900743. [DOI] [Google Scholar]
- Park S.; Sugiyama H. DNA as a Chiral Scaffold for Asymmetric Synthesis. Molecules 2012, 17 (11), 12792–12803. 10.3390/molecules171112792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Sugiyama H. DNA-based hybrid catalysts for asymmetric organic synthesis. Angew. Chem., Int. Ed. 2010, 49 (23), 3870–3878. 10.1002/anie.200905382. [DOI] [PubMed] [Google Scholar]
- Otto S.; Bertoncin F.; Engberts J. B. Lewis acid catalysis of a Diels- Alder reaction in water. J. Am. Chem. Soc. 1996, 118 (33), 7702–7707. 10.1021/ja960318k. [DOI] [Google Scholar]
- Otto S.; Engberts J. B. A systematic study of ligand effects on a Lewis-acid-catalyzed Diels- Alder reaction in water. Water-enhanced enantioselectivity. J. Am. Chem. Soc. 1999, 121 (29), 6798–6806. 10.1021/ja984273u. [DOI] [Google Scholar]
- Draksharapu A.; Boersma A. J.; Leising M.; Meetsma A.; Browne W. R.; Roelfes G. Binding of copper (II) polypyridyl complexes to DNA and consequences for DNA-based asymmetric catalysis. Dalton Trans. 2015, 44 (8), 3647–3655. 10.1039/C4DT02733G. [DOI] [PubMed] [Google Scholar]
- Roelfes G.; Boersma A. J.; Feringa B. L. Highly enantioselective DNA-based catalysis. Chem. Commun. 2006, (6), 635–637. 10.1039/b516552k. [DOI] [PubMed] [Google Scholar]
- Kypr J.; Kejnovska I.; Renciuk D.; Vorlickova M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009, 37 (6), 1713–1725. 10.1093/nar/gkp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray D. M.; Bollum F. A circular dichroism study of poly dG, poly dC, and poly dG: dC. Biopolymers: Original Research on Biomolecules 1974, 13 (10), 2087–2102. 10.1002/bip.1974.360131011. [DOI] [PubMed] [Google Scholar]
- Gudibande S. R.; Jayasena S. D.; Behe M. J. CD studies of double-stranded polydeoxynucleotides composed of repeating units of contiguous homopurine residues. Biopolymers 1988, 27 (12), 1905–1915. 10.1002/bip.360271205. [DOI] [PubMed] [Google Scholar]
- Brahms S.; Brahms J. G. DNA with adenine tracts contains poly(dA).poly(DT) conformational features in solution. Nucleic Acids Res. 1990, 18 (6), 1559–1564. 10.1093/nar/18.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantatosaki E.; Papadopoulos G. K. Binding Dynamics of siRNA with Selected Lipopeptides: A Computer-Aided Study of the Effect of Lipopeptides’ Functional Groups and Stereoisomerism. J. Chem. Theory Comput. 2020, 16 (6), 3842–3855. 10.1021/acs.jctc.9b01261. [DOI] [PubMed] [Google Scholar]
- Secrist J. A. III Nucleoside and Nucleotide Nomenclature. Curr. Protoc. Nucleic Acid Chem. 2000, 00 (1), A.1D.1–A.1D.3. 10.1002/0471142700.nca01ds00. [DOI] [PubMed] [Google Scholar]
- Rohs R.; West S. M.; Sosinsky A.; Liu P.; Mann R. S.; Honig B. The role of DNA shape in protein-DNA recognition. Nature 2009, 461 (7268), 1248–1253. 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon B. R.; Li Y.; Cote A.; Weirauch M. T.; Ding P.; Hughes T. R.; Navarre W. W.; Xia B.; Liu J. Structural basis for recognition of AT-rich DNA by unrelated xenogeneic silencing proteins. Proc. Natl. Acad. Sci. U.S.A. 2011, 108 (26), 10690–10695. 10.1073/pnas.1102544108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draksharapu A.; Boersma A. J.; Browne W. R.; Roelfes G. Characterisation of the interactions between substrate, copper (II) complex and DNA and their role in rate acceleration in DNA-based asymmetric catalysis. Dalton Trans. 2015, 44 (8), 3656–3663. 10.1039/C4DT02734E. [DOI] [PubMed] [Google Scholar]
- Bowler D. R.; Miyazaki T. O(N) methods in electronic structure calculations. Rep. Prog. Phys. 2012, 75 (3), 036503. 10.1088/0034-4885/75/3/036503. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; de Bruin B.; Feringa B. L.; Roelfes G. Ligand denticity controls enantiomeric preference in DNA-based asymmetric catalysis. Chem. Commun. 2012, 48 (18), 2394–2396. 10.1039/c2cc17350f. [DOI] [PubMed] [Google Scholar]
- Gardiner E. J.; Hunter C. A.; Packer M. J.; Palmer D. S.; Willett P. Sequence-dependent DNA structure: a database of octamer structural parameters. J. Mol. Biol. 2003, 332 (5), 1025–35. 10.1016/j.jmb.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Gardiner E. J.; Hunter C. A.; Lu X. J.; Willett P. A structural similarity analysis of double-helical DNA. J. Mol. Biol. 2004, 343 (4), 879–89. 10.1016/j.jmb.2004.08.092. [DOI] [PubMed] [Google Scholar]
- Il’icheva I. A.; Vlasov P. K.; Esipova N. G.; Tumanyan V. G. The Intramolecular Impact to the Sequence Specificity of c’A Transition: Low Energy Conformational Variations in AA/TT and GG/CC Steps. J. Biomol. Struct. Dyn. 2010, 27, 677–693. 10.1080/07391102.2010.10508581. [DOI] [PubMed] [Google Scholar]
- Mergny J. L.; Lacroix L. Analysis of thermal melting curves. Oligonucleotides 2003, 13 (6), 515–37. 10.1089/154545703322860825. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Megens R. P.; Feringa B. L.; Roelfes G. DNA-based asymmetric catalysis. Chem. Soc. Rev. 2010, 39 (6), 2083–2092. 10.1039/b811349c. [DOI] [PubMed] [Google Scholar]
- Amirbekyan K.; Duchemin N.; Benedetti E.; Joseph R.; Colon A.; Markarian S. A.; Bethge L.; Vonhoff S.; Klussmann S.; Cossy J.; Vasseur J.-J.; Arseniyadis S.; Smietana M. Design, Synthesis, and Binding Affinity Evaluation of Hoechst 33258 Derivatives for the Development of Sequence-Specific DNA-Based Asymmetric Catalysts. ACS Catal. 2016, 6 (5), 3096–3105. 10.1021/acscatal.6b00495. [DOI] [Google Scholar]
- Marek J. J.; Singh R. P.; Heuer A.; Hennecke U. Enantioselective Catalysis by Using Short, Structurally Defined DNA Hairpins as Scaffold for Hybrid Catalysts. Chem. Eur. J. 2017, 23 (25), 6004–6008. 10.1002/chem.201606002. [DOI] [PubMed] [Google Scholar]
- Wang J.; Benedetti E.; Bethge L.; Vonhoff S.; Klussmann S.; Vasseur J. J.; Cossy J.; Smietana M.; Arseniyadis S. DNA vs. mirror-image DNA: a universal approach to tune the absolute configuration in DNA-based asymmetric catalysis. Angew. Chem., Int. Ed. 2013, 125 (44), 11760–11763. 10.1002/ange.201306232. [DOI] [PubMed] [Google Scholar]
- Li Y.; Cheng M.; Hao J.; Wang C.; Jia G.; Li C. Terpyridine-Cu (II) targeting human telomeric DNA to produce highly stereospecific G-quadruplex DNA metalloenzyme. Chemical Science 2015, 6 (10), 5578–5585. 10.1039/C5SC01381J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti E.; Duchemin N.; Bethge L.; Vonhoff S.; Klussmann S.; Vasseur J.-J.; Cossy J.; Smietana M.; Arseniyadis S. DNA-cellulose: An economical, fully recyclable and highly effective chiral biomaterial for asymmetric catalysis. Chem. Commun. 2015, 51 (28), 6076–6079. 10.1039/C4CC10190A. [DOI] [PubMed] [Google Scholar]
- Park S.; Ikehata K.; Sugiyama H. Solid-supported DNA for asymmetric synthesis: a stepping-stone toward practical applications. Biomaterials Science 2013, 1 (10), 1034–1036. 10.1039/c3bm60134j. [DOI] [PubMed] [Google Scholar]
- Wight A.; Davis M. Design and preparation of organic- inorganic hybrid catalysts. Chem. Rev. 2002, 102 (10), 3589–3614. 10.1021/cr010334m. [DOI] [PubMed] [Google Scholar]
- Katz A.; Davis M. E. Molecular imprinting of bulk, microporous silica. Nature 2000, 403 (6767), 286–289. 10.1038/35002032. [DOI] [PubMed] [Google Scholar]
- Corma A.; Garcia H. Silica-bound homogenous catalysts as recoverable and reusable catalysts in organic synthesis. Adv. Synth. Catal. 2006, 348 (12–13), 1391–1412. 10.1002/adsc.200606192. [DOI] [Google Scholar]
- Megens R. P.; Roelfes G. Organic co-solvents in aqueous DNA-based asymmetric catalysis. Org. Biomol. Chem. 2010, 8 (6), 1387–1393. 10.1039/b921385f. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Shen K. G-quadruplex DNA-based asymmetric catalysis of michael addition: Effects of sonication, ligands, and co-solvents. Biotechnol. Prog. 2016, 32 (4), 891–898. 10.1002/btpr.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Shen K. DNA-based asymmetric catalysis: role of ionic solvents and glymes. RSC Adv. 2014, 4 (96), 54051–54059. 10.1039/C4RA10749G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H.; Abe N.; Shibata A.; Ito K.; Tanaka Y.; Ito M.; Saneyoshi H.; Shuto S.; Ito Y. Structure formation and catalytic activity of DNA dissolved in organic solvents. Angew. Chem. 2012, 124 (26), 6581–6585. 10.1002/ange.201201111. [DOI] [PubMed] [Google Scholar]
- Percec V.; Cho W.-D.; Ungar G.; Yeardley D. J. Synthesis and structural analysis of two constitutional isomeric libraries of AB2-based monodendrons and supramolecular dendrimers. J. Am. Chem. Soc. 2001, 123 (7), 1302–1315. 10.1021/ja0037771. [DOI] [Google Scholar]
- Kuang H.; Gartner T. E. III; Dorneles de Mello M.; Guo J.; Zuo X.; Tsapatsis M.; Jayaraman A.; Kokkoli E. ssDNA-amphiphile architecture used to control dimensions of DNA nanotubes. Nanoscale 2019, 11 (42), 19850–19861. 10.1039/C9NR03761F. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.