

Abstract
The development of polymers that can replace engineered viral vectors in clinical gene therapy has proven elusive despite the vast portfolios of multifunctional polymers generated by advances in polymer synthesis. Functional delivery of payloads such as plasmids (pDNA) and ribonucleoproteins (RNP) to various cellular populations and tissue types requires design precision. Herein, we systematically screen a combinatorially designed library of 43 well-defined polymers, ultimately identifying a lead polycationic vehicle (P38) for efficient pDNA delivery. Further, we demonstrate the versatility of P38 in codelivering spCas9 RNP and pDNA payloads to mediate homology-directed repair as well as in facilitating efficient pDNA delivery in ARPE-19 cells. P38 achieves nuclear import of pDNA and eludes lysosomal processing far more effectively than a structural analogue that does not deliver pDNA as efficiently. To reveal the physicochemical drivers of P38’s gene delivery performance, SHapley Additive exPlanations (SHAP) are computed for nine polyplex features, and a causal model is applied to evaluate the average treatment effect of the most important features selected by SHAP. Our machine learning interpretability and causal inference approach derives structure–function relationships underlying delivery efficiency, polyplex uptake, and cellular viability and probes the overlap in polymer design criteria between RNP and pDNA payloads. Together, combinatorial polymer synthesis, parallelized biological screening, and machine learning establish that pDNA delivery demands careful tuning of polycation protonation equilibria while RNP payloads are delivered most efficaciously by polymers that deprotonate cooperatively via hydrophobic interactions. These payload-specific design guidelines will inform further design of bespoke polymers for specific therapeutic contexts.
Keywords: nonviral gene therapy, polymeric vehicles, CRISPR, pDNA delivery, ribonucleoprotein delivery, combinatorial design, machine learning, structure−activity relationships
Introduction
Nucleic acid therapeutics have transformed the treatment landscape for hereditary diseases such as sickle cell anemia,1 spinal muscular atrophy,2,3 Duchenne’s muscular dystrophy,4 and more broadly for acquired diseases with dysregulated gene expression patterns, such as cancer and diabetes.5,6 Clinicians currently rely almost exclusively on engineered viral vectors to navigate extracellular barriers such as payload protection from nuclease degradation, immune evasion, and targeting specific organs,7,8 and to overcome intracellular barriers such as cellular uptake, endosomal escape, payload unpackaging, and nuclear trafficking.9 Viral delivery is confronted with logistical, technological, and commercial obstacles in the form of limited cargo capacity,10 high manufacturing costs,11 significant regulatory burdens,12 and severe immune responses.13−15 To circumvent these challenges, biomaterials researchers have designed chemically defined synthetic delivery platforms such as polymers16 and lipids17 whose performance meets or exceeds benchmarks set by clinically deployed viral vectors.18,19
Exogenous nucleic acids can be delivered in the form of mRNA (mRNA), short interfering RNA (siRNA), plasmids (pDNA), antisense oligonucleotides (ASO), ribonucleoproteins (RNP), self-amplifying RNA or replicon RNA (saRNA or repRNA), and microRNA. Further, chemical modifications to ASO and siRNA payloads, such as the incorporation of 2′-fluoro, 2′-O-methyl, 2′-O-methoxyethyl, constrained ethyl, locked nucleic acid, and phosphorodiamidate functionalities can significantly alter hydrophobicity, serum stability, and immunostimulatory profiles.20 Existing biomaterial design frameworks seldom consider the stark biophysical contrasts between these varied nucleic acid modalities.21−25 Recognizing the limitations of a “one-size-fits-all” approach, various polymer design heuristics have been proposed to account for variations in the surface charge distribution, molecular size, morphology, flexibility, and hydrophobicity of nucleic acid payloads. In particular, polymer hydrophobicity,26−28 molecular architecture,29 and polymer length30,31 have been identified as the most pertinent design parameters in designing universally effective polymeric gene delivery vehicles.
Several studies have challenged the overarching assumption that the design requirements for polymeric vehicles are identical across different nucleic acid payloads. Blakney and co-workers reported that polymers optimized for siRNA and mRNA delivery could not be repurposed for saRNA payloads because of innate structural differences between these RNA modalities.32 The same group had earlier adopted a statistical design of experiments approach to identify the optimal polymer design space for pDNA, mRNA, and saRNA and concluded that saRNA delivery imposed the most exacting design requirements.33 Kaczmarek et al.(34) showed that polymers optimized for mRNA delivery could not be repurposed for pDNA delivery without making modular changes in monomer chemistry. Explorations of structure–function relationships for polymeric carriers are therefore indispensable to customize carrier properties for diverse therapeutic payloads, particularly for applications that involve codelivery of payloads with differing polymer design constraints. To date, the question of whether the design criteria for polymeric carriers of pDNA and RNP payloads overlap has neither been studied nor elucidated. Through combinatorial reversible addition–fragmentation transfer (RAFT) polymerization, high-throughput experimentation, and machine learning, we identify key differences in the physicochemical drivers of delivery performance, toxicity, and cellular uptake for pDNA and RNP payloads.
Recently, our group reported a chemically diverse library of well-defined statistical copolymers, accessing a broad range of physicochemical properties and intermolecular interactions with RNPs.35 In the present work (Figure 1), we study this multifactorial polymer library with the following objectives: (1) screen for polymers that facilitate efficient intracellular pDNA delivery, (2) understand whether the design constraints imposed by RNP payloads are applicable to pDNA payloads, (3) codeliver ribonucleoproteins and pDNA donors to facilitate homology-directed repair (HDR), and (4) translate these results to other targets such as mediating transgene expression in a challenging retinal transfection target cell type (ARPE-19) using the lead polymer P38 (p(DIPAEMA52-st-HEMA50)). P38 achieves higher nuclear import and is less likely to be entrapped within lysosomal compartments when compared to structural analogues that do not culminate in functional pDNA delivery. Having identified P38 as the lead structure for both RNP as well as pDNA delivery, we initially expected that the polymer design criteria for successful cellular delivery might be identical for both payloads. However, machine learning approaches such as SHapley Additive exPlanations (SHAP36) and causal inference reveal that structure–function relationships governing polymer-mediated intracellular delivery are payload-specific. While the degree of cooperativity during polymer deprotonation (parametrized by the Hill coefficient nHill) and the surface charge exert the greatest influence over RNP delivery, pDNA delivery efficiency is insensitive to the Hill coefficient and is instead controlled by polycation protonation equilibria (pKa). Our lead structure P38 conforms to two disparate sets of payload-dependent design specifications, establishing its utility and multifunctionality as a nonviral delivery platform that can be optimized toward clinical applications that demand functional delivery of multimodal cargoes.
Figure 1.

Polymers from a combinatorially designed library are assembled with pDNA payloads and polyplexes characterized thoroughly. Polyplex internalization, pDNA delivery efficiency, and toxicity are evaluated rapidly. Finally, interpretable machine learning approaches are applied to derive structure–function relationships.
Results and Discussion
Parallelized Screening Rapidly Identifies Lead pDNA Delivery Vehicle
RAFT is a highly versatile synthetic tool that realizes diverse polymer architectures, accommodates a variety of functional monomers, and obtains polymeric vehicles with tightly controlled molecular weight distributions and exquisitely tailored properties. We believe RAFT is particularly relevant to our work because it permits systematic investigation of polymer design attributes and identification of promising functionalities that can subsequently be deployed in other material platforms (such as poly(β-amino esters) and lipid nanoparticles). Our multiparametric copolymer library (Figure 2) incorporates cationic monomers of varying basicity wherein primary amines as well as tertiary amines with alkyl substituents of varying steric bulk and lipophilicity are represented. We targeted cationic incorporation levels of 100, 75, 50, and 25% while copolymerizing cationic monomers with neutral monomers of varying hydrophilicity. We posit that this combinatorial approach enables systematic variation of polymer pKa and hydrophobic–hydrophilic phase balance (Table 1). Through combinatorial polymerization and rapid screening, our previous work identified a polymeric carrier with outstanding RNP delivery characteristics.35 In the present work, we revisit this polymer library with the objective of identifying polymeric vehicles that realize efficient intracellular pDNA delivery. A total of 129 formulations, arising from the complexation of GFP-encoding pDNA with 43 polymers at three N/P ratios (the molar ratio of protonatable amines within the polymer to phosphate groups in the nucleic acid backbone) are characterized in detail via gel electrophoresis and DLS to determine pDNA binding affinity and polyplex size, respectively (Table 1).
Figure 2.

Polymer library synthesized via combinatorial RAFT polymerization. (A) Four cationic monomers of varying pKa values: 2-(diethylamino)ethyl methacrylate (DEAEMA), 2-aminoethylmethacrylamide hydrochloride (AEMA), 2-(diisopropylamino)ethyl methacrylate (DIPAEMA), and 2-(dimethylamino)ethyl methacrylate (DMAEMA) were studied. Three neutral monomers of varying hydrophilicities were used as comonomers: 2-methacryloyloxyethyl phosphorylcholine (MPC), poly(ethylene glycol) methyl ether methacrylate (PEGMEMA), and 2-hydroxyethyl methacrylate (HEMA). (B) For each pair of cationic and neutral monomers, we targeted cationic monomer incorporation levels from 0% to 100% in 25% increments, generating 43 polymers. The cationic incorporation was determined by 1H NMR and was used to calculate m and n values.
Table 1. Overview of Polymer and Polyplex Characterization and Machine Learning Model Descriptorsa.
|
Rh(nm) at N/P |
mobility
at N/P |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Mn (kDa) | % cat. | clogP | pKa | nHill | ζ (mv) | 5 | 10 | 20 | 5 | 10 | 20 |
| P1 | 18.4 | 100 | 9.2 | 6.7 | 4.2 | 12.4 | 873 | 638 | 1538 | F | F | F |
| P2 | 19.6 | 53 | 3.0 | - | - | –14.1 | 60 | 74 | 78 | N | N | N |
| P3 | 25.4 | 42 | –3.6 | - | - | 0.1 | 42 | 51 | 41 | N | N | N |
| P4 | 32.1 | 35 | –9.6 | - | - | –5.2 | 31 | 95 | 132 | N | N | N |
| P5 | 11.3 | 100 | –0.8 | 8.1 | 2.4 | 15.2 | 398 | 2238 | 373 | F | F | F |
| P6 | 12.9 | 65 | –10.8 | 8.0 | 1.6 | 12.8 | 338 | 124 | 106 | F | F | F |
| P7 | 22.3 | 46 | –14.4 | 7.9 | 1.6 | 2.9 | 721 | 95 | 111 | F | F | F |
| P8 | 21.6 | 24 | –23.3 | 6.8 | 3.0 | 6.9 | 172 | 248 | 458 | F | F | F |
| P9 | 17.4 | 100 | 11.7 | 6.0 | 9.5 | 20.8 | 310 | 223 | 1249 | F | F | F |
| P10 | 27.2 | 68 | 4.9 | 6.4 | 14.5 | 5.6 | 162 | 198 | 234 | N | P | F |
| P11 | 32.9 | 56 | –2.3 | 6.9 | 13.4 | 2.2 | 78 | 79 | 93 | N | N | P |
| P12 | 31.9 | 29 | –9.0 | - | - | –13.8 | 50 | 45 | 46 | N | N | N |
| P13 | 17.7 | 100 | 5.0 | 6.9 | 3.2 | 21.7 | 39 | 34 | 34 | F | F | F |
| P14 | 8.6 | 72 | –0.2 | 6.9 | 3.2 | 5.6 | 2510 | 3856 | - | P | F | F |
| P15 | 17.3 | 59 | –11.5 | 7.5 | 1.5 | 3.8 | 110 | 134 | 116 | P | P | P |
| P16 | 16.9 | 26 | –16.8 | 7.8 | 1.6 | 0.0 | 30 | 49 | 42 | P | P | P |
| P17 | 10.1 | 0 | –40.3 | - | - | –10.9 | 48 | 57 | 44 | N | N | N |
| P18 | 23.6 | 77 | 6.8 | 6.9 | 2.7 | 4.2 | 122 | 108 | 444 | N | N | F |
| P19 | 30.8 | 52 | 4.3 | 6.6 | 3.5 | –7.1 | 60 | 59 | 56 | N | N | N |
| P20 | 39.4 | 33 | 1.8 | 6.5 | 3.2 | –11.7 | 48 | 35 | 25 | N | N | N |
| P21 | 31.7 | 71 | –0.9 | 7.8 | 1.8 | 16.2 | 47 | 95 | 56 | F | P | F |
| P22 | 74.9 | 43 | –0.8 | 8.1 | 1.4 | 8.6 | 59 | 58 | 77 | F | F | F |
| P23 | 21.6 | 36 | –0.6 | 7.8 | 1.6 | 7.0 | 54 | 74 | 53 | F | F | F |
| P24 | 18.2 | 64 | 8.6 | 6.6 | 11.3 | 6.3 | 626 | 1013 | 512 | N | N | N |
| P25 | 22.9 | 47 | 5.5 | 6.8 | 5.1 | 2.3 | 72 | 669 | 170 | N | N | N |
| P26 | 43.7 | 25 | 2.4 | 6.9 | 2.3 | –9.4 | 35 | 180 | 13 | N | N | N |
| P27 | 8.7 | 75 | 3.6 | 7.0 | 1.8 | 8.3 | 26 | 30 | 36 | P | P | P |
| P28 | 8.6 | 50 | 2.1 | 7.0 | 1.9 | 18.5 | 58 | 86 | - | P | P | N |
| P29 | 35.3 | 25 | 0.7 | 6.8 | 2.0 | –0.1 | 18 | 14 | 5 | P | P | P |
| P30 | 3.6 | 0 | –0.7 | - | - | 6.8 | 79 | 123 | 54 | N | N | N |
| P31 | 14.1 | 74 | 7.2 | 7.5 | 4.6 | 15.2 | 822 | 673 | 850 | F | F | F |
| P32 | 15 | 50 | 5.2 | 7.6 | 3.8 | 9.4 | 1645 | 1541 | 1053 | F | F | F |
| P33 | 10.9 | 30 | 3.2 | 7.8 | 3.1 | 5.6 | 382 | 838 | 917 | F | F | F |
| P34 | 21.3 | 61 | –0.4 | 8.2 | 3.5 | 22.7 | 61 | 54 | 53 | F | F | F |
| P35 | 24.2 | 44 | 0.2 | 8.2 | 2.2 | 21.0 | 45 | 37 | 172 | F | F | F |
| P36 | 24 | 23 | 0.8 | 6.9 | 2.1 | 18.4 | 81 | 49 | 45 | F | F | F |
| P37 | 17.7 | 65 | 9.1 | 6.5 | 11.5 | 16.2 | 778 | 3633 | 572 | F | F | F |
| P38 | 17.9 | 51 | 6.4 | 7.3 | 16.1 | 12.8 | 708 | 968 | 820 | F | F | F |
| P39 | 16.3 | 25 | 3.8 | 6.4 | 8.5 | –0.7 | 250 | 616 | 768 | F | F | F |
| P40 | 8.5 | 60 | 4.0 | 7.2 | 3.0 | 4.8 | 2655 | 2429 | 2483 | F | F | F |
| P41 | 11.2 | 42 | 3.1 | 7.2 | 3.2 | 7.9 | 2034 | 859 | - | F | F | F |
| P42 | 29.3 | 31 | 2.1 | 7.3 | 3.1 | 2.5 | 601 | 346 | 925 | F | F | F |
| P43 | 3.6 | 0 | –0.7 | - | - | 6.8 | 50 | 56 | 56 | N | N | N |
The molecular weight (Mn) is determined via SEC-MALS and the cationic incorporation by 1H NMR. We also report clogP (calculated), pKa (titration), nHill (titration), and ζ-potential (capillary electrophoresis). The polyplex radius Rh (intensity-weighted Rhvia dynamic light scattering) and mobility of pDNA during gel electrophoresis are represented at N/P ratios of 5, 10, and 20. F indicates tight binding while N signifies migration comparable with free pDNA. Intermediate binding is denoted by P.
As shown in Table 1, pDNA binding affinity, as determined by gel electrophoretic mobility, is highly sensitive to the choice of neutral comonomer and the polymer pKa. For HEMA-based copolymers (P31 to P42), we observe strong binding between polymers and pDNA irrespective of polycation basicity. However, pDNA binding is considerably weaker for MPC-based copolymers (P1 to P16) and PEG-based copolymers (P18 to P29). It appears that the incorporation of highly hydrophilic PEG and MPC monomers hinders the formation of polyplexes by offering hydration repulsion, which is consistent with earlier reports.37−40 Interestingly, AEMA copolymers (P5 to P8, P21 to P23), which exhibit higher pKa values, are an exception to this trend and exhibit strong binding even when copolymerized with hydrophilic monomers. Unimodal populations with hydrodynamic radii (Rh) approaching 1 μm were formed when HEMA was used as the comonomer. In contrast, highly hydrophilic comonomers such as PEG and MPC which inhibit polymer–pDNA binding promote the formation of smaller polyplexes (<100 nm in Rh). Delivery efficiency screens with HEK293T cells reveal the proportion of GFP-positive cells within the transfected population (Figure 3) using flow cytometry. Interestingly, the hit polymer from our RNP delivery screening study,35 P38 displays the highest proportion of GFP-expressing cells and emerges as the lead candidate. Overall, by combining polyplex characterization data and the pDNA delivery screening data, we are able to unearth mechanistic insights and pDNA-specific structure–function relationships (vide infra).
Figure 3.

(A) Polyplexes are formulated at N/P ratios of 5, 10, and 20, and the proportion of cells expressing green fluorescent protein (GFP) evaluated via flow cytometry to identify top polymers. (B) Only N/P 20 formulations of top performers are denoted by white stars although GFP expression is substantial even at lower N/P ratios. Polyplexes formed from p(DIPAEMA52-st-HEMA50) or P38 effect the highest GFP expression.
P38 Polyplexes Evade Lysosomes and Import pDNA into Nuclei
The contrasts in the pDNA delivery performance between P38 and the rest of the library are probed through a library-wide evaluation of cellular internalization, followed by quantitative confocal microscopy. Cy5-labeled pDNA is complexed with each polymer at three N/P ratios (Figure 4A), and the Cy5 fluorescence intensity is measured via flow cytometry after 24 hours (Figure 4B). Unlike the universally high levels of cellular internalization of pDNA recorded across the polymer library, only three polymers (the hit polymer P38, P34, and P35) mediate substantial RNP internalization,35 indicating that cellular uptake constitutes a far greater challenge for the polymeric delivery of RNP payloads compared to pDNA payloads. P38 is not unique in facilitating highly efficient cellular internalization of pDNA polyplexes; for several other polymers, we observe Cy5 intensities significantly higher than P38 although their pDNA delivery performance does not approach P38. For example, the median Cy5 intensity of P41 polyplexes is 50% higher than that of P38 polyplexes. However, P41, a structural analogue of P38, is ineffectual for pDNA delivery (Figure 3). We hypothesize that polymers such as P41, which do not mediate functional pDNA delivery despite highly efficient cellular internalization, may adopt intracellular itineraries that do not culminate in nuclear import.
Figure 4.

(A) Polyplexes are formulated with Cy5-labeled pDNA and cellular internalization in HEK293T cells evaluated. (B) The geometric mean Cy5 intensity for each formulation is normalized to the highest value in the library. Unlike with RNP delivery, pDNA delivery is not inhibited by uptake.
Confocal imaging maps the intracellular distribution of Cy5-labeled polyplexes, providing estimates of the proportion of pDNA partitioned between the cytoplasmic and nuclear regions. The lead polymer P38, along with P41, a variant of P38 that produces near-zero levels of GFP expression despite exhibiting the highest levels of pDNA uptake, are both formulated with Cy5-labeled pDNA at an N/P ratio of 5. Twenty-four hours after transfection, cells are fixed, permeabilized, and stained with an AlexaFluor 546 conjugated antibody (to identify lysosome-associated membrane protein 2) and Hoechst 3342 (Figure 5). GFP expression is quite low in cells treated with P41 polyplexes whereas with P38 polyplexes, a much larger proportion of cells express GFP. Strikingly, we do not observe any differences in Cy5 intensity between P38 and P41, indicating comparable cellular uptake of Cy5-labeled polyplexes. This marked contrast in GFP expression, despite comparable levels of cellular uptake, strongly suggests that P38 and P41 polyplexes experience different retention times within lysosomal compartments. Colocalization analysis quantifies the Pearson’s correlation coefficient (PCC) between Cy5 signals from polyplexes and AlexaFluor 546 signals from lysosomes to estimate the likelihood of lysosomal entrapment. The mean PCC is slightly higher (0.20 ± 0.05) in P41 than the hit polymer P38 (0.12 ± 0.06), confirming that P41 is far more likely to be retained within lysosomal compartments than P38 (Figure S19).
Figure 5.

HEK293T cells transfected with P38 (hit polymer) and P41 (poor transfection despite high pDNA internalization) at an N/P ratio of 5. Various cellular compartments and intracellular polyplex distribution are visualized as follows: nuclei are stained with DAPI (blue), intracellular GFP expression (green), AlexaFluor 546 stained lysosomal compartments (magenta), Cy5-labeled pDNA payloads (orange), allowing quantification of colocalization. We observe poor transfection efficiencies in the P41 treatment group despite high levels of pDNA internalization. Colocalization analysis yields Pearson’s correlation coefficients (PCC), which reveal the higher propensity of P41 polyplexes to be entrapped within lysosomes compared to P38 polyplexes. Scale bar is 10 μm.
The spatial distribution of cytoplasmic pDNA (cyan) and nuclear pDNA (white) (Figure 6A,B) is studied to compare the nuclear accumulation of P38 and P41 polyplexes. Nuclei–pDNA distances for P38 and P41 polyplexes are plotted, assigning negative values to intranuclear pDNA, zero to pDNA at the periphery between the cytoplasmic and nuclear regions, and positive values to cytoplasmic polyplexes and extracellular polyplexes. Among GFP+ cells, pDNA from P38 formulations localize within closer proximity of nuclei as opposed to P41 formulations (Figure 6C). Quantile–quantile (Q–Q) plots compare the distributions of nuclei–pDNA distances for both P38 and P41, and the dissimilarity between P38 and P41 histograms is evident (Figure 6C). From the Q–Q plots, we see that the P41 distribution is skewed toward greater separation from nuclear peripheries, compared to the P38 distribution. The Kolmogorov–Smirnov test verifies this visual observation, establishing that the P38 and P41 distance histograms are not drawn from the same underlying distribution (p-value of 4.7 × 10–10). The propensity of pDNA, particularly to localize within close proximity of nuclei varies significantly between P38 and P41, with P41 polyplexes localizing further away from nuclear peripheries than P38. Finally, we observe a much higher proportion of nuclear polyplexes in the P38 treatment group (Figure 6D) than in P41. We conclude that the choice of polymeric vehicle dictates whether pDNA accumulates within cytoplasmic or nuclear regions. P38 polyplexes are also less likely to be colocalized within lysosomal compartments than P41, thereby protecting their payloads from lysosomal activity and steering pDNA into perinuclear regions.
Figure 6.

Three-dimensional reconstructions of GFP+ cells from (A) P38 and (B) P41 treatment groups. Cy5-labeled pDNA payloads were classified as cytoplasmic (cyan) or nuclear (gray). Scale bar is 5 μm. (C) From quantile–quantile (Q–Q) plots, we see that P41 nuclei–pDNA distances are shifted further to the right, indicating higher nuclear separation than P38. The Kolmagrov–Smirnov test (p-values shown inset) further confirms that the histograms are unlikely to be drawn from the same distribution. (D) Distribution of pDNA between nuclear and cytoplasmic regions for GFP+ cells. P38 polyplexes display higher nuclear accumulation than P41.
Machine Learning Identifies Differences in Design Criteria between RNP and pDNA Payloads
In this work, we apply machine learning (ML) to attribute predictive and causal importance to nine physicochemical variables that determine nucleic acid payload delivery. In contrast to predictive ML, we are motivated by interpreting and explaining the dependencies of biological outcomes on polyplex attributes. We are interested in identifying the dominance of polyplex attributes according to the nature of the cargo (pDNA vs RNP). For this purpose, we focus on building a comprehensive data set for pDNA and RNP delivery within our combinatorial library of 43 polymers, and we interpret the data set using machine learning methodologies. First, we use an ML interpretability method to unveil the predictive power of polyplex attributes on the delivery figures of merit (transfection efficiency, cellular uptake, and cellular toxicity). ML interpretability methods estimate the predictive importance of variables in nonlinear models, which are often appropriate for physical phenomena. Although the information we extract from this approach is useful, we also are interested in controlling for possible confounding between polyplex attributes (for instance polyplex size Rh is correlated with polyplex composition). For this purpose, we employ a causal inference approach.41 Causal inference aims to determine causal relationships from data, controlling for spurious correlations in data. We use these methods to decouple effects of known features on our data and determine which of the main predictive features have stand-alone causal effects all by themselves.
In contrast to our earlier study,35 where functional RNP delivery is observed mainly with P38 but to a lesser extent with P34 and P35, we identify additional polymers (P5, P21, P23, P34, P35, P36, and P37) where substantial levels of transgene expression are detected (Figure 3). This led us to hypothesize that structure–function relationships for RNP and pDNA payloads do not overlap. To delineate the physicochemical basis for RNP delivery performance, we had earlier applied random forest classifiers, an ensemble-based ML technique.35 However, the use of feature importance estimates from random forest classifiers to deduce structure–function trends has limitations. For instance, features that are highly correlated to truly influential features may be overselected, making it difficult to assess the true contribution of any given feature.42 Consequently, we might overestimate the importance of a given feature on model output or wrongly attribute effects to a noncausal feature that may be correlated with several causal features or confounder variables. To overcome the limitations of our earlier statistical modeling approach, we propose a combination of machine learning interpretability and causality modeling techniques. First, we apply SHapley Additive exPlanations (SHAP), a machine learning interpretability method that fairly attributes contributions from multiple features to the model output.36 This game-theoretic approach develops robust interpretations from predictive models trained on the data sets from the RNP and pDNA screening studies.43 For each of the three biological outputs (toxicity, efficiency, and uptake), we train a random forest model to binarily classify responses above or below the 90th percentile of the output variable and compute the relative importance of our nine polyplex descriptors (Table 1) via SHAP. As seen in Figure 7, pDNA delivery efficiency is primarily predicted by polycation protonation (pKa) while RNP delivery is correlated with attributes associated with hydrophobic interactions (nHill parametrizes hydrophobically driven cooperative deprotonation) along with electrostatic interactions (ζ-potential, pKa, % cationic incorporation). Our work is the first to employ statistical modeling to demonstrate that careful tuning of electrostatic interactions between pDNA and polymers by modulating the polymer pKa will enhance pDNA delivery efficiency.
Figure 7.
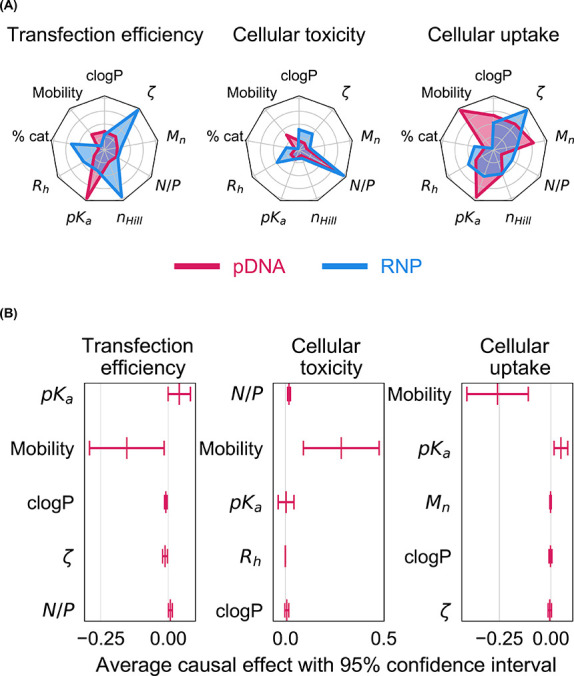
(A) The hydrophobicity (clogP), surface charge (ζ), length (Mn), composition (% cat.), and pKa of polymers were measured while polyplex formulations were described by their size (Rh) and the distance migrated by pDNA during gel electrophoresis (mobility). The contributions of these nine features to delivery efficiency, cellular toxicity, and uptake were computed for pDNA and RNP payloads using SHapley Additive exPlanations (SHAP). SHAP compares structure–function trends across RNP (blue) and pDNA (red) payloads. (B) Direct causal effects (in the form of average treatment effects) of the top five features from SHAP analysis were computed along with 95% confidence intervals. Positive and negative effects indicate protagonistic and antagonistic relationships, respectively.
Cellular toxicity and cellular internalization data sets from pDNA and RNP studies present contrasting trends. Other than N/P ratio, the polymer hydrophobicity (clogP) and polyplex size (Rh) are most predictive of toxicity among RNP polyplexes. For pDNA polyplexes, cellular toxicity is higher among polymers that inhibit pDNA migration during gel electrophoresis. Interestingly, the qualitative strength of polymer–pDNA binding (parametrized by pDNA mobility during gel migration assays) is the most impactful feature for both toxicity and delivery efficiency among pDNA polyplexes. Because polymer–pDNA binding is predictive of both toxicity and delivery, high delivery efficiencies will always be accompanied by low viability during pDNA delivery. In contrast, the structural basis for cytotoxicity and editing efficiency do not overlap for RNP polyplexes, suggesting that the trade-off between cytotoxicity and delivery performance is payload-dependent. Divergent trends are also observed in polyplex uptake between pDNA and RNP data sets; while only three polymers (P34, P35, and P38) promote substantial RNP internalization, the majority of the polymer library is able to shuttle pDNA payloads past cell membranes. Earlier, we observed that even among polyplexes where RNP payloads were not tightly bound to polymers, cellular internalization proceeded efficiently,35 establishing that RNP–polymer binding is not predictive of polyplex uptake. In contrast, pDNA polyplex uptake is primarily determined by whether polymers inhibit the migration of pDNA payloads during gel electrophoresis (Figure S4).
Subsequent to SHAP analysis, we quantified the causal effects (average treatment effect or ATE) of the top five SHAP-identified features.44 Although SHAP identifies features that are highly correlated to the model outputs (delivery efficiency, toxicity, and uptake), the actual causal effect of each polyplex feature might be masked by confounding effects. For instance, a dominant polyplex descriptor may control one or more nondominant descriptors causing us to misattribute their respective contributions. To correct for observed confounding effects caused by dominant polyplex descriptors, we estimate a linear conditional ATE model for each of the five top SHAP features controlling for all the other features. This model estimates a more realistic causal response for each polyplex feature than pure explainability models like SHAP. The ATEs for pDNA (Figure 7B) and RNP payloads (Figure S20) are plotted along with their 95% confidence intervals. For delivery efficiency, we found that pKa and pDNA migration, the top two features identified via SHAP have nonzero causal effects, albeit with large uncertainties. Similarly, pDNA mobility, the top SHAP contributor, also has a large causal effect on cellular uptake. Surprisingly, for cellular toxicity, the causal effect of the top SHAP feature (N/P) is far smaller than that of the second-ranked feature, DNA mobility. In contrast to SHAP, causal analysis reveals that polymer–pDNA binding is a dominant feature across all three biological responses (efficiency, toxicity, and uptake). Causal estimates are accompanied by confidence intervals, which illuminate uncertainties in our analysis and inform the design of polymer libraries that can minimize this uncertainty. For instance, given the large uncertainties associated with the causal effect of pDNA mobility, it would be more interesting to focus future synthetic efforts on polymers than span a broader range of pDNA binding affinities (at consistent and varied pKa values) to further understand the relationship between polymer–pDNA binding affinity and pDNA delivery performance. Because polymer–pDNA binding affinity has emerged as a critical design attribute, it may be necessary to substitute gel electrophoresis with alternative approaches (isothermal titration calorimetry or dye exclusion assays) in future studies to facilitate careful quantitative comparison of polymer−pDNA binding.
Although our screening results suggest overlapping design rules for pDNA and RNP delivery, data mining tools disprove this conjecture and establish that the physicochemical determinants of polymer-mediated pDNA delivery diverge from those of RNP delivery. Polymers that deprotonate cooperatively are more likely to succeed at intracellular RNP delivery while pDNA payloads require polymers with optimized polycation protonation equilibria and binding affinity. Despite RNP and pDNA imposing divergent constraints, it is fortuitous that P38 satisfies both sets of design criteria. This unique system proves to be a potent vector with the potential to codeliver RNP and pDNA payloads for homology-directed repair (detailed below).
P38 Mediates Homology-Directed Repair by Codelivering RNP and pDNA Payloads
Because P38 effectively delivers RNP and pDNA payloads, we evaluate the feasibility of codelivering RNPs with pDNA donors to achieve precise gene knock-in via HDR editing. Rational design of polyplexes for HDR editing requires optimization of the total nucleic acid dose,45,46 the proportion of sgRNA relative to the pDNA donor,4,47 and the polymer loading or the N/P ratio. We simultaneously examine the effects of (1) the total nucleic acid dose (1.5 and 2 μg/well); (2) payload composition, i.e., the weight ratio of sgRNA to pDNA (w/w ratios of 2:1, 1:1, 1:2, 1:3, 1:4, and 1:5 are evaluated); and (3) N/P ratio (1, 1.25, 1.5, 2). It is important to note that the payload composition is varied while keeping the total nucleic acid dose fixed at 1.5 or 2 μg per well for a 24-well plate. Taken together, 48 conditions are evaluated in this experimental matrix to identify the optimal conditions for HDR editing. We quantify the relative frequencies of NHEJ and HDR by measuring mCherry and GFP expression, respectively (Figure 8A). From these optimization efforts (Figure 8B), we conclude that both the rate of donor integration (quantified via GFP readouts) as well as the frequency of random indels (measured via mCherry expression) are highest when the maximum nucleic acid loading is selected (2 μg/well for a 24-well plate). Additionally, we note a nonmonotonic relationship between HDR frequency and the payload composition, wherein sgRNA-dominant payloads (2:1) and pDNA-dominant payloads (1:5) conditions both result in low HDR frequencies (<0.1%) while intermediate payload compositions (1:2 and 1:3 w/w) display the highest GFP expression (0.7%). Across all HDR payload compositions, P38 is able to encapsulate both RNP and donor pDNA completely (at an N/P ratio of 2). Gel electrophoresis studies of polyplex formulations of P38 and various molar ratios RNP and pDNA are furnished in the Supporting Information (Figure S8).
Figure 8.

(A) Schematic of NHEJ and HDR editing pathways. In cells engineered with the traffic light reporter system, the delivery of RNP alone results in imprecise gene editing via the NHEJ pathway (measured via mCherry expression), while codelivery of pDNA donor and RNP leads to gene knock-in via HDR (measured via GFP). (B) Optimization of formulation conditions for codelivering RNP and pDNA donor payloads. The total amount of nucleic acid is kept constant at either 1.5 or 2 μg per well while the weight ratio of single guide RNA (sgRNA) and pDNA donor is varied from 2:1 to 1:5. A formulation of 2 μg nucleic acid loading using a 1:2 w/w mixture of sgRNA and DNA maximizes HDR editing (quantified via GFP expression). (C) Fluorescent micrographs of HDR-edited cells treated with Lipofectamine 2000 or P38. Unpackaged payloads serve as negative controls. Scale bar is 100 μm. (D) Flow cytometry traces highlighting mCherry positive cell populations and GFP positive cells for the optimized formulation.
Subsequent to payload optimization, we benchmark the HDR performance of the hit polymer to commercial reagents at the optimized polyplex formulation conditions (2 μg total nucleic acid dose per well and a 1:2 w/w ratio of sgRNA and pDNA donor). The expression of mCherry and GFP, indicative of NHEJ and HDR editing, respectively, is measured in cells treated with P38 polyplexes at N/P ratios of 1.25, 1.5, 1.75, and 2. We also include Lipofectamine 2000 and JetPEI as positive controls (Figure 8C). While JetPEI results in almost no HDR-edited cells, Lipofectamine 2000 is the only reagent where more than 2% of the cell population is GFP-positive (Figure 8D). GFP expression does not exceed 0.7% when P38 is used to deliver HDR constructs, consistent with the results observed during payload optimization. We speculate that the causes underlying low HDR frequencies originate in cellular processes rather than polymeric design. For instance, we do not synchronize transfection with cell cycle,48,49 nor do we employ HDR-promoting drugs to bias editing pathways in favor of gene insertion.50 Even without the assistance of pharmacological additives, we obtain a substantial pool of HDR-edited cells, a population that can subsequently be sorted and expanded to meet therapeutic demands. Herein, we demonstrate the viability of P38 for HDR applications in this proof-of-concept study. Future research will focus on packaging covalently tethered RNP-donor payloads with P38 vehicles to boost HDR frequencies.51−53
P38 Mediates Functional Delivery pDNA to HEK293T and ARPE-19
Following screening studies in HEK293T (cells commonly used in vector and recombinant protein production), we perform additional experiments in this cell line to compare P38 with commercial pDNA transfection reagents. Further, we study differences in the pDNA delivery functionality of P38 (Figure 9A) between HEK293T and retinal pigment epithelia or ARPE-19 (a model for retinal gene delivery). Among HEK293T cells, both JetPEI and LPF 2000 achieve efficient pDNA delivery and promote GFP expression in 70–80% of the cell population. With P38 at an N/P ratio of 1, no GFP is detected, but GFP expression improves steadily at higher N/P ratios, climbing to 15% at an N/P ratio of 2.5 and about 60–80% at N/P ratios of 5 and 10. The GFP expression of P38 polyplexes at an N/P ratio of 10 is comparable to both JetPEI and Lipofectamine 2000, confirming that P38 is a highly effective pDNA delivery platform.
Figure 9.

(A) Summary of transfection and internalization efficiencies in HEK293T (black) and ARPE-19 (gray) cells. In HEK293T, P38 exhibits both high delivery efficiencies (measured by GFP expression) as well as high cellular uptake (measured by Cy5 intensity). In ARPE-19, we observe that delivery performance of P38 is inhibited by low levels of uptake, particularly at an N/P ratio of 10. (B) DLS and turbidity measurements reveal N/P-dependent trends in polyplex aggregation upon the addition of DMEM, with the N/P 10 formulation experiencing severe colloidal instability. We performed turbidimetric titrations in both D-PBS and in DMEM to understand the causes of N/P-dependent polyplex aggregation. Unlike in PBS, where polyplexes recover colloidal stability upon the addition of excess polymer and overcharging, aggregation is irreversible in DMEM because of the poor solubility of P38 in the media. DLS and turbidity measurements indicate that only lower N/P ratios permit colloidally stable polyplexes.
ARPE-19 is an important in vitro model for retinal delivery and a challenging transfection target because of its lower mitotic rates compared to HEK293T. Further, significant compositional differences exist between the cell membranes of HEK293T and ARPE-19; the retinal pigment epithelium’s role in the blood-retinal barrier endows ARPE-19 cells with several transporter proteins and efflux channels that may be absent in HEK293T cells.54 ARPE-19 resists transfection even when Lipofectamine 2000 and JetPEI are employed, both of which are only half as effective in ARPE-19 compared to HEK293T. This decrease in transfection performance when going from HEK293T to ARPE-19 cells is also observed in P38, where GFP expression is detected in 17.6%, 17.4%, and 21.3% of cells at N/P ratios of 2.5, 5, and 10, respectively (Figure 9A). Importantly, at an N/P ratio of 2.5, we observe slightly lower levels of cellular toxicity among P38-treated cells than with JetPEI, with a small loss of pDNA delivery efficacy (Figure S18). The improved cellular viability of P38 over JetPEI assumes relevance when repeated subretinal administration is necessary.
Seeking to unravel the reasons for the lower efficiency of P38 in ARPE-19, we compare the cellular uptake observed under these transfection conditions using the Cy5-labeled pDNA system previously described in Figure 4. Among HEK293T cells, cellular uptake increases gradually with increasing N/P ratios for P38, with the highest internalization efficiencies displayed by the N/P 10 formulation (Figure 9A). Unexpectedly, among ARPE-19 cells, cellular uptake peaks at an N/P ratio of 2.5 (75%) before declining rapidly to 60% and 20% for N/P ratios of 5 and 10, respectively. For the N/P 2.5 formulation, we observe nearly identical levels of cellular uptake for both cell types. At higher N/P ratios, however, the gap between HEK293T and ARPE-19 cellular internalization widens considerably. Only a third of the cells that internalize N/P 2.5 polyplexes express GFP in ARPE-19 cells, indicating that transfection is inhibited by endosomal release. Whereas among N/P 10 polyplexes, nearly all internalization events culminate in GFP expression in ARPE-19 cells, suggesting that excess polymer contributes to endosomal destabilization. At N/P ratios as high as 10, transgene expression in ARPE-19 cells is impeded by lowered cellular uptake, while at lower N/P ratios (5 and below), free P38 polymers that initiate endosomal leakage are scarcer, leading to inefficient intracellular delivery.
While HEK293T cells take up polyplexes promiscuously, resulting in high cellular uptake across the entire library (Figure 3), ARPE-19 cells internalize nanoparticles in a size-selective manner, with cellular uptake decreasing with increasing polyplex sizes.55,56 Previous reports suggest that ARPE-19 cells traffic larger lipoplexes via clathrin-mediated pathways, leading to longer entrapment within lysosomal compartments.57 Even in vivo, smaller polyplexes adopt trans-retinal pathways and undergo rapid internalization into retinal epithelia.58 We hypothesize that polyplex size differences might explain the trend of lower uptake with increasing N/P ratios among ARPE-19 cells. Through DLS measurements, we observed narrow size distributions ranging from 40–60 nm for all conditions (JetPEI, P38 N/P of 1, 2.5, 5, 10) when we formulate in water. Consistent with transfection protocols for ARPE-19, we formed polyplexes in water and then resuspended polyplexes in two volumes of serum-free DMEM and monitored aggregation over time (Figure 9B). While the hydrodynamic radii of polyplexes formed at N/P ratios of 2.5 and 5 plateau around 150–200 nm at the end of 40 min, the hydrodynamic radii of JetPEI and N/P 1 formulations approach 250–300 nm. However, severe aggregation and radii exceeding 1 μm are found in the highest N/P ratio studied (N/P of 10), indicating that excess polymer contributes to colloidal instability. We posit that the unexpectedly low uptake of P38 N/P 10 by ARPE-19 cells is attributable to the formation of micrometer-scale aggregates in cell culture media.
To identify the causes for severe aggregation in the N/P 10 formulation, we probe the phase behavior of P38 polyplexes across a dynamic range of N/P ratios using turbidimetric titrations. Titrations are performed in both PBS and in a 2:1 DMEM–water mixture (mimicking media composition during transfection) to monitor polyplex formation and stability as a function of N/P ratios and solvent environment. The pDNA (or polymer) solution is gradually titrated into the polymer (or pDNA) solution, while continuously recording changes in transmittance. Below a transmittance of 0.9, we observe the formation of white precipitates (shaded area in Figure 9B). In PBS, while adding polymer to DNA we observe a sharp decrease in solution transmittance as the N/P ratio approaches 1, indicating the loss of colloidal stability at charge neutrality. However, transmittance levels return to values close to 1 upon adding more polymer to induce overcharging and Coulombic repulsion of polyplexes. We observe similar behavior with the reverse sequence of addition (pDNA to polymer in PBS) although the zone of instability spans a much broader range of N/P ratios. Unlike P38 (random coil in solution), pDNA is semiflexible with a larger persistence length and therefore is not as effective in overcharging the polyplexes and restabilizing them.
Compared to D-PBS (pH 7), the DMEM–water mixture is much more alkaline (pH 8.4), leading to the deprotonation and phase separation of P38. Consequently, above N/P ratios of 0.3, we notice sharp decreases in transmittance, reflecting the onset of polyplex aggregation with increasing N/P ratio. Unlike in PBS, we do not recover colloidal stability via overcharging upon the addition of excess polymer; instead, we observe further decrease in transmittance with increasing N/P ratio, indicating that high N/P ratio polyplexes suffer an irreversible loss of colloidal stability when introduced to DMEM. Further, this inhomogeneous region spans a much larger N/P range in DMEM–water than in PBS. The size of polyplexes, their aggregation propensity in DMEM, and their N/P-dependent phase behavior all contribute to lowered cellular uptake and ultimately hinder P38-mediated intracellular pDNA delivery in ARPE-19 cells. We anticipate that orthogonal tuning of polyplex composition and size (enhancing colloidal stability) will improve cellular uptake, thereby promoting more efficient intracellular pDNA release in challenging cellular targets of transfection.
Conclusions
In this work, a lead structure (P38) that delivers pDNA efficiently and mediates high transgene expression emerged from the screening of a multiparametric polymer library. Because P38 was identified as a potent vector for RNP delivery in our previous screening campaign,35 we initially expected the polymer design criteria for pDNA and RNP payloads to be identical. To probe this conjecture, we applied SHapley Additive exPlanations (SHAP) to unravel the relationship between polymer attributes, payload type, and key biological outcomes. SHAP analysis established that the structural determinants of cellular uptake, toxicity, and delivery efficiency are payload-dependent, with RNP and pDNA payloads diverging in their design requirements. Unlike RNP delivery, which relies on both electrostatic and hydrophobic interactions to facilitate cytosolic RNP release, hydrophobic considerations are negligible for pDNA delivery. Our work is the first to apply machine learning to establish that pDNA delivery demands polymers with optimized polycation protonation equilibria and pDNA binding affinity. Through quantitative confocal microscopy, we analyzed the intracellular trajectories of polyplexes and observed lower lysosomal colocalization and higher nuclear import among polyplexes formed from the hit polymer P38, compared to a structural analogue of P38 that did not mediate pDNA delivery (P41). In our previous study, P38 outperformed four state-of-the-art commercial controls to deliver RNP payloads and mediate highly efficient genome editing.35 In this work, we find that P38 mediates functional delivery of pDNA payloads to HEK293T and ARPE-19 cells. Co-delivery of pDNA and RNP payloads by P38 results in significantly higher rates of homology-directed repair than JetPEI. Overall, our work establishes the utility and multifunctionality of P38, especially in applications that demand the codelivery of multiple payloads. Fundamental characterization of solution physics reveals that particle size and colloidal stabilization are important for improving cellular uptake in cell types reliant on caveolar endocytosis (requiring polyplex diameters within 60 nm). Overall, we demonstrate that exploration of chemically diverse polymer libraries uncovers novel polymeric vectors for multimodal delivery applications and creates a robust framework for the elucidation of payload-specific structure–function relationships.
Experimental Section
Experimental procedures for polymer synthesis and characterization (1H NMR, molecular weight determination, pKa and nHill estimation, and ζ-potential measurements) can be found in our earlier work.35 Experimental procedures for RNP polyplex formulation, RNP polyplex size distribution, surface charge, gel electrophoretic mobilities, and RNP delivery studies (toxicity, cellular uptake, and editing efficiency) can be found in our earlier work.35 The hit polymer P38 was resynthesized in two additional runs, and we obtained comparable molecular weight distribution and chemical composition, which bodes well for the reproducibility of RAFT.
Polyplex Characterization
The pDNA payload, pZsgreen (4708 bp), was purchased from Aldevron (Fargo, ND) and diluted in water to the desired concentration. Polymers were dissolved in ultrapure water to obtain a charge ratio of 15.15 nmol of ionizable amines per μL, and sterile-filtered. Polymer stock solutions were further diluted to the desired N/P ratio (5,10, and 20) prior to polyplex formation. Polyplexes were formed using an electronic multichannel pipet by controlled addition of polymer solution to an equal volume pDNA solution (0.02 μg/μL) in sterile water. The mixture was then incubated for 45 min at 23 °C. Polyplexes formulated at this concentration were used for DLS measurements, electrokinetic characterization, and transient transfection experiments.
Gel casting was done using a 0.6% agarose solution formed in TAE buffer. Ethidium bromide was used at a concentration of 0.017% v/v to visualize pDNA migration toward the positive electrode. Gel electrophoresis was performed at 80 V over 60 min and imaged using a transilluminator (Fotodyne, IL) under UV light. Polyplexes formulated for gel electrophoresis assays employed a higher concentration (0.05 μg/μL) of pDNA than what was used for biological studies (0.02 μg/μL) in order to facilitate clear visualization of the pDNA bands.
The Malvern Zetasizer (Malvern Instruments, MA) was used to evaluate the ζ-potential of P38 polyplexes at N/P ratios of 1, 2.5, 5, and 10. Measurements were performed under monomodal settings using the folded capillary measurement cell. A pDNA concentration of 0.02 μg/μL was employed. To characterize the surface potential of P38 in its unbound state, a concentration of 1 mg/mL was employed. Three to five measurements were acquired per treatment condition.
All DLS measurements in Figures 8 and S1–S3 were performed using the DynaPro plate reader III (Wyatt Instruments, CA). For DLS measurements in Figure 8, P38 polyplexes (N/P of 1, 2.5, 5, and 10) and JetPEI (N/P of 5) were formed in water, and 20 measurements were collected prior to the addition of Fluorbrite DMEM. To 100 μL of each polyplex, we added 200 μL of serum-free Fluorbrite DMEM (prefiltered to remove dust) and acquired DLS measurements at the rate of 7–8 acquisitions per minute to capture aggregation kinetics. Turbidimetric titrations were carried out in either D-PBS or Fluorbite DMEM–water mixtures (2:1 v/v) using procedures previously described by Jiang et al.(59)
For DLS measurements in Figures S1–S3, polyplexes were prepared at N/P ratios of 5, 10, and 20 in 10 mM PBS buffer using multichannel electronic pipettes. Polyplexes were incubated at 23 °C for 45 min prior to acquisition of measurements. Five acquisitions were collected per polyplex formulation (with an acquisition time of five seconds each), and the hydrodynamic radius was calculated as an average across five technical replicates. Noisy autocorrelation functions were filtered out using an automated baseline-filtering process, and the polyplex size distributions were computed using regularization fits. Intensity-weighted average hydrodynamic radii (Rh) were reported for all DLS data.
Cellular Assays
The HEK293T cell line engineered with a traffic light reporter system60 was used to assess both RNP and pDNA delivery by our polymer library. Cells were donated by the Osborne lab at the University of Minnesota, and subcloning was performed at the Genome Engineering Shared Resource at the University of Minnesota. Cells were seeded at 50 000 cells/mL in DMEM supplemented with 10% FBS in 48-well plates (Corning, MA). Cells were cultured for 24 h at 37 °C and 5% CO2 to allow the cells to adhere to the plate before performing pDNA transfection or gene editing via HDR payloads. Polyplexes were formed by adding 42.5 μL of polymer solution in water to an equal volume of pDNA solution in water and incubating for 45 min. At the time of transfection, cell culture media was aspirated and replaced with polyplexes suspended in two volumes of OptiMEM (170 μL). For transient transfection, the total volume of the polyplex solution added to each well was 150 μL (50 μL of polyplex solution and 100 μL of Opti-MEM). Manufacturer’s protocols were implemented for JetPEI (N/P of 5) and Lipofectamine 2000. After 4 h, wells were supplemented with a further 0.5 mL of FBS-supplemented DMEM. Twenty-four hours after transfection, the media was aspirated and replaced with fresh DMEM. Forty-eight hours after transfection, the cells were analyzed using flow cytometry. Although only one biological replicate was used in the ML analysis, a second biological replicate was performed to act as an independent control for efficiency, toxicity, and uptake across the library. We observed similar trends and results in the independent control. Screening studies for hit identification and toxicity measurements were performed on September 14, 2020 and October 11, 2020 as independent runs. Both biological replicates are furnished in the Supporting Information (section 9).
ARPE-19 cells were cultured in DMEM-F12 media supplemented with 10% FBS in a humidified incubator maintained at 37 °C and 5% CO2. The procedures for transfection, measurement of cellular uptake, and cytotoxicity were identical to the ones adopted for HEK293T cells with two deviations: (1) ARPE-19 cells were washed with D-PBS prior to the addition of polyplexes. (2) Polyplexes were resuspended in serum-free DMEM-F12 instead of in OptiMEM. For flow cytometry, cells were trypsinized and centrifuged at 1010g and 4 °C for 10 min. The supernatant was aspirated, and the cell pellet was resuspended in a 200 μL solution of PBS + 2% FBS + 400 nM Calcein violet AM (Thermo Fisher, Waltham, MA). Cells were incubated in ice for 20–30 min and vortexed prior to flow cytometry. For evaluating cell viability and GFP expression in transfected cells, the 405 and 488 nm laser lines (Biorad Inc., CA) were used. Single live cells were used for analysis, and gating schemes are furnished in the Supporting Information (Figures S11–S15). At least 10 000 events were collected per sample.
For homology-directed repair, HEK293T cells were cotransfected with a mixture of RNP and donor pDNA payloads. The total mass of nucleic acids, comprising sgRNA and the pDNA donor, was fixed at either 1.5 μg per well or 2 μg for a 24-well plate. However, their weight ratio was varied systematically from 2:1 to 1:5 in order to identify formulation conditions that would maximize the frequency of HDR events. We identified 2 μg of total nucleic acid loading per well and 1:2 w/w ratio of sgRNA: pDNA as the optimal condition. For N/P calculations, the phosphate groups in both the pDNA and sgRNA were considered. In a typical HDR experiment, RNP complexes were annealed by adding sgRNA solution to spCas9 solution in equal volumes. To assemble RNPs, spCas9 (Aldevron, ND) and sgRNA (Synthego, CA) solutions were prepared in PBS at concentrations of 0.019 and 0.39 mg/mL respectively, and ribonucleoproteins assembled through slow addition of sgRNA to spCas9 and annealing for 15 min. Within 15 min of RNP formation, an equal volume of the pDNA donor solution (at 0.04 mg/mL) was added and allowed to equilibrate for 5 min. The polymer solution (diluted to the desired N/P ratio in D-PBS) was slowly introduced into an equal volume of the payload mixture and incubated for 45 min at ambient temperature. Finally, this mixture was diluted in twice the volume of OptiMEM and added slowly to cells. Cells were plated 24 h prior to transfection at a density of 50 000 cells/mL. DMEM supplemented with 10% FBS was added 4 h after transfection, and cell culture was replaced 24 h after transfection. Cells were regularly passaged while approaching 80% confluency (roughly every 2 days) before being analyzed using flow cytometry on the seventh day after transfection. Cells were harvested for flow cytometry using procedures similar to the ones described above. The 405, 488, and 560 nm laser liens were used to detect Calcein Violet, GFP, and mCherry, respectively. At least 40 000 events were collected per sample.
For toxicity studies (Figures S16–S18), transfection was performed in 48-well plates according to procedures described previously. Two days after transfection, cell culture media was replaced with a 2% solution of CCK-8 (Dojindo) in Fluorbrite-DMEM. Thereafter, cells were incubated for 4 h at 37 °C and 5% CO2 and the absorbance of media measured at 480 nm at a gain of 90 using the Synergy H1 plate reader (Biotek, CA). Measurements of the CCK-8 solution without cells were collected, and this blank reading was subtracted from all data points. Absorbance values were normalized to untreated cells. Three to six wells were employed per condition.
To label pDNA payloads with Cy5, we followed the manufacturer’s protocols (Label IT Nucleic Acid Labeling Kit Cy5, Mirus, Madison, WI) and purified the labeled product through ethanol precipitation. The concentration of the final product was quantified via UV–vis spectrophotometry (Nanodrop, Thermo Fisher, Waltham, MA). HEK293T cells or ARPE-19 were transfected with polyplexes formulated with Cy5-labeled pDNA as described in earlier paragraphs. Twenty-four hours after transfection, cell culture media and polyplexes were removed and cells were trypsinized. Cell suspensions were transferred to V-bottomed 96 well plates. Samples were centrifuged at 1010g for 10 min at 4 °C. Cell pellets were washed with PBS and then resuspended in 200 μL/well Cell Scrub (Genlantis, San Diego, CA) for 10 min to remove uninternalized membrane-bound polyplexes. Cells were washed again with 300 μL/well PBS, centrifuged, and resuspended in a 100 μL solution of PBS + 2% FBS + 400 nM Calcein violet AM (Thermo Fisher, Waltham, MA). Samples were then analyzed on the ZE5 flow cytometer (Biorad Inc., CA) using the 633 nm laser line to detect Cy5, in addition to the 405 and 488 nm that detected Calcein violet and GFP, respectively. Five thousand events were collected per treatment condition. The geometric mean of Cy5 fluorescence intensities were computed and used for subsequent statistical analyses. Two biological replicates were performed (September 30, 2020 and November 29, 2020), and the first one was used for modeling. Both replicates are furnished in section 9 of the Supporting Information.
For confocal imaging, cells were seeded on sterilized gelatin-coated glass coverslips in 24-well plates at a concentration of 50 000 cells/mL a day before transfection. Twenty-four hours later, cells were fixed, and lysosomes labeled with the anti-LAMP2 primary antibody (Abcam catalog# ab25631, Cambridge, MA) and a secondary antibody (Invitrogen catalog# A11003) diluted to 1:200 and 1:1000, respectively. Antibodies were diluted in a solution of PBS containing 5% bovine serum albumin, 0.2% gelatin, and 0.1% Triton-X. Cells were counterstained with Hoechst 3342. After each antibody incubation step, cells were washed thrice with PBS/0.1% Triton-X for five minutes each. Coverslips were mounted on Prolong Glass (Thermo Fisher, Waltham, MA) and cured at room temperature in the dark for 2 days. Samples were imaged under an Olympus BX2 laser-scanning confocal microscope system equipped with an automated upright BX61 microscope base and PRIOR ProScanII motorized stage.
Imaris software (version 9.7.2, Bitplane) was utilized for all image processing and quantification. First, the background was automatically calculated and subtracted from all channels. The Imaris colocalization module was used to calculate Pearson’s correlation coefficients, and surface renderings of voxels containing both AlexaFluor568 and Cy5 signals were generated. Colocalization calculations for AlexaFluor568- and Cy5-containing voxels were performed inside the cytoplasmic compartments of GFP+ cells as well as in GFP– cells. Thresholds for both AlexaFluor568 and Cy5 were calculated in Imaris using the method described by Costes et al.,61 wherein correlation coefficients are calculated for all voxels containing both AlexaFluor568 and Cy5 signals. The threshold is reached when the correlation coefficient reaches zero.
Identification of Structure–Function Relationships
The structure–function relationships were estimated by a machine learning approach. We started by defining and measuring the nine polyplex descriptors. For both RNP and pDNA, we defined binary labels for each biological output of interest—efficiency, cellular toxicity, and uptake—using a 90th percentile variable-specific threshold. We found this threshold to be reasonable for our delivery goals and consistent with our previous study on RNPs. Next, we trained and evaluated various models (gradient boosting decision trees, logistic regression, random forest, balanced gradient boosting decision trees, and balanced random forest) using 5-fold cross validation and the scikit-learn and imblearn packages.62,63 Each fold was stratified to preserve the original class ratio in the data set. The best-performing model was a balanced random forest with 100 estimators. Figure S21 presents the final mean AUC across the 5 folds for each cargo. After the best-performing model was chosen, we retrained the model for each biological output using a 0.9–0.1 random train-test split. We use this trained model for interpretability via SHapley Additive exPlanations (SHAP).36 SHAP provides explanations for each feature by learning a local linear model with game-theoretic constraints. In particular, we apply the TreeSHAP algorithm and take the mean absolute SHAP value across all data points as our feature importance metric.43 This metric measures the power of each polyplex feature to predict whether a given data point will be above or below the 90th percentile of each biological output and cargo. Although SHAP is very useful to explain the predictive power of each feature, these explanations do not have causal interpretations because of observed and unobserved confounding effects. Thus, we cannot unambiguously determine the causal impact of a certain polyplex feature on a biological output. For this purpose, we train a linear causality model for each polyplex feature controlling for the observed confounding of all other features using the EconML package44 and approximate the conditional causal treatment effect of each feature over each biological output. The polyplex features can be ranked by average treatment effect (ATE) with a confidence interval computed for each feature. This causal ranking determines which polyplex features have a direct causal effect on the biological output, and which have effects due to potential confounding.
Acknowledgments
We acknowledge Nanite, Inc. and the Defense Advanced Research Projects Agency (DARPA) N660011824041 for funding. We acknowledge Craig Van Bruggen and Zhe Tan, Ph.D. for technical advice. We acknowledge the University Imaging Centers at the University of Minnesota. We gratefully acknowledge Dr. Guillermo Marques from the University Imaging Centers for sharing his expertise. We acknowledge Biorender.com.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00467.
Machine learning procedures and supplemental analyses; supplemental data from flow cytometry, DLS, gel migration, confocal imaging, and colocalization analyses (PDF)
3D rendering of HEK293T cells treated with P38 N/P = 5 pDNA polyplexes (MP4)
3D rendering of HEK293T cells treated with P41 N/P = 5 pDNA polyplexes [For both videos, lysosomes (orange), colocalized voxels of lysosome and pDNA in GFP-positive cells (magenta), and colocalized voxels of lysosome and pDNA in EGFP-negative cells (yellow) have been highlighted.] (MP4)
Author Present Address
# Chemical & Biological Engineering, Colorado School of Mines, 1613 Illinois Street, Golden, CO 80401, Unites States
Author Present Address
§ Bayramoglu Law, 1540 W. Warm Springs Road, Henderson, NV 89014, United States.
The authors declare the following competing financial interest(s): Theresa M. Reineke is one of the founders of Nanite, Inc. and has an equity interest. Nanite, Inc. is one of the sponsors of this research. This interest has been reviewed and managed by the University of Minnesota in accordance with its conflict-of-interest policy. Felipe Oviedo consults for Nanite, Inc. and has an equity interest.
Supplementary Material
References
- Newby G. A.; et al. Base Editing of Haematopoietic Stem Cells Rescues Sickle Cell Disease in Mice. Nature 2021, 595, 295–302. 10.1038/s41586-021-03609-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust K. D.; Wang X.; McGovern V. L.; Braun L.; Bevan A. K.; Haidet A. M.; Le T. T.; Morales P. R.; Rich M. M.; Burghes A. H. M.; Kaspar B. K. Rescue of the Spinal Muscular Atrophy Phenotype in a Mouse Model by Early Postnatal Delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mendell J. R.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- Lee K.; et al. Nanoparticle Delivery of Cas9 Ribonucleoprotein and Donor DNA in Vivo Induces Homology-Directed DNA Repair. Nat. Biomed. Eng. 2017, 1, 889–901. 10.1038/s41551-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anguela X. M.; High K. A. Entering the Modern Era of Gene Therapy. Annu. Rev. Med. 2019, 70, 273–288. 10.1146/annurev-med-012017-043332. [DOI] [PubMed] [Google Scholar]
- Mahajan A.; et al. Fine-Mapping Type 2 Diabetes Loci to Single-Variant Resolution Using High-Density Imputation and Islet-Specific Epigenome Maps. Nat. Genet. 2018, 50, 1505–1513. 10.1038/s41588-018-0241-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillmore J. D.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- Musunuru K.; et al. Vivo CRISPR Base Editing of PCSK9 Durably Lowers Cholesterol in Primates. Nature 2021, 593, 429–434. 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- Li C.; Samulski R. J. Engineering Adeno-Associated Virus Vectors for Gene Therapy. Nat. Rev. Genet. 2020, 21, 255–272. 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- Trapani I.; Tornabene P.; Auricchio A. Large Gene Delivery to the Retina with AAV Vectors: Are We There Yet?. Gene Ther. 2021, 28, 220–222. 10.1038/s41434-020-0174-4. [DOI] [PubMed] [Google Scholar]
- May M. Viral Vector Industry Boosts Product Quality and Quantity. Genet. Eng. Biotechnol. News 2020, 40, 42–44. 10.1089/gen.40.03.11. [DOI] [Google Scholar]
- Lapteva L.; Purohit-Sheth T.; Serabian M.; Puri R. K. Clinical Development of Gene Therapies: The First Three Decades and Counting. Mol. Ther. Methods Clin. Dev. 2020, 19, 387–397. 10.1016/j.omtm.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauletbekov D. L.; Pfromm J. K.; Fritz A. K.; Fischer M. D. In Retinal Degenerative Diseases; Bowes Rickman C., Grimm C., Anderson R. E., Ash J. D., LaVail M. M., Hollyfield J. G., Eds.; Springer International Publishing: Cham, 2019; pp 165–168. [Google Scholar]
- Nayak S.; Herzog R. W. Progress and Prospects: Immune Responses to Viral Vectors. Gene Ther. 2010, 17, 295–304. 10.1038/gt.2009.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Zhang L.; Liu H.; Cheng K. Delivery Strategies of the CRISPR-Cas9 Gene-Editing System for Therapeutic Applications. J. Controlled Release 2017, 266, 17–26. 10.1016/j.jconrel.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R.; Santa Chalarca C. F.; Bockman M. R.; Bruggen C. V.; Grimme C. J.; Dalal R. J.; Hanson M. G.; Hexum J. K.; Reineke T. M. Polymeric Delivery of Therapeutic Nucleic Acids. Chem. Rev. 2021, 121, 11527–11652. 10.1021/acs.chemrev.0c00997. [DOI] [PubMed] [Google Scholar]
- Kulkarni J. A.; Witzigmann D.; Thomson S. B.; Chen S.; Leavitt B. R.; Cullis P. R.; van der Meel R. The Current Landscape of Nucleic Acid Therapeutics. Nat. Nanotechnol 2021, 16, 630–643. 10.1038/s41565-021-00898-0. [DOI] [PubMed] [Google Scholar]
- Yin H.; et al. Structure-Guided Chemical Modification of Guide RNA Enables Potent Non-Viral in Vivo Genome Editing. Nat. Biotechnol. 2017, 35, 1179–1187. 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakney A. K.; McKay P. F.; Hu K.; Samnuan K.; Jain N.; Brown A.; Thomas A.; Rogers P.; Polra K.; Sallah H.; Yeow J.; Zhu Y.; Stevens M. M.; Geall A.; Shattock R. J. Polymeric and Lipid Nanoparticles for Delivery of Self-Amplifying RNA Vaccines. J. Controlled Release 2021, 338, 201–210. 10.1016/j.jconrel.2021.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts T. C.; Langer R.; Wood M. J. A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discovery 2020, 19, 673–694. 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K. Delivery of Magic Bullets: On the Still Rocky Road to Gene Therapy. Br. J. Pharmacol. 2009, 157, 151–152. 10.1111/j.1476-5381.2009.00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durymanov M.; Reineke J. Non-Viral Delivery of Nucleic Acids: Insight Into Mechanisms of Overcoming Intracellular Barriers. Front. Pharmacol. 2018, 9, 971. 10.3389/fphar.2018.00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead K. A.; Langer R.; Anderson D. G. Knocking Down Barriers: Advances in siRNA Delivery. Nat. Rev. Drug Discovery 2009, 8, 129–138. 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski P. S.; Rudra A.; Miao L.; Anderson D. G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. 10.1016/j.ymthe.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M.; Miller A. D.. Non-viral Gene Ther. Gene Des. Deliv.; Springer-Verlag Tokyo, 2005; pp 441–455. [Google Scholar]
- Liu Z.; Zhang Z.; Zhou C.; Jiao Y. Hydrophobic Modifications of Cationic Polymers for Gene Delivery. Prog. Polym. Sci. 2010, 35, 1144–1162. 10.1016/j.progpolymsci.2010.04.007. [DOI] [Google Scholar]
- Sunshine J. C.; Akanda M. I.; Li D.; Kozielski K. L.; Green J. J. Effects of Base Polymer Hydrophobicity and End-Group Modification on Polymeric Gene Delivery. Biomacromolecules 2011, 12, 3592–3600. 10.1021/bm200807s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurnani P.; Blakney A. K.; Terracciano R.; Petch J. E.; Blok A. J.; Bouton C. R.; McKay P. F.; Shattock R. J.; Alexander C. The In Vitro, Ex Vivo, and In Vivo Effect of Polymer Hydrophobicity on Charge-Reversible Vectors for Self-Amplifying RNA. Biomacromolecules 2020, 21, 3242–3253. 10.1021/acs.biomac.0c00698. [DOI] [PubMed] [Google Scholar]
- Bishop C. J.; Kozielski K. L.; Green J. J. Exploring the Role of Polymer Structure on Intracellular Nucleic Acid Delivery via Polymeric Nanoparticles. J. Controlled Release 2015, 219, 488–499. 10.1016/j.jconrel.2015.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbey W. T.; Wu K. K.; Mikos A. G. Size Matters: Molecular Weight Affects the Efficiency of Poly(ethylenimine) as a Gene Delivery Vehicle. J. Biomed. Mater. Res. 1999, 45, 268–275. . [DOI] [PubMed] [Google Scholar]
- Kim H. J.; Kim A.; Miyata K. Synthetic Molecule Libraries for Nucleic Acid Delivery: Design Parameters in Cationic/Ionizable Lipids and Polymers. Drug Metab. Pharmacokinet. 2022, 42, 100428. 10.1016/j.dmpk.2021.100428. [DOI] [PubMed] [Google Scholar]
- Blakney A. K.; Zhu Y.; McKay P. F.; Bouton C. R.; Yeow J.; Tang J.; Hu K.; Samnuan K.; Grigsby C. L.; Shattock R. J.; Stevens M. M. Big Is Beautiful: Enhanced saRNA Delivery and Immunogenicity by a Higher Molecular Weight, Bioreducible, Cationic Polymer. ACS Nano 2020, 14, 5711–5727. 10.1021/acsnano.0c00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakney A. K.; Yilmaz G.; McKay P. F.; Becer C. R.; Shattock R. J. One Size Does Not Fit All: The Effect of Chain Length and Charge Density of Poly(ethylene Imine) Based Copolymers on Delivery of pDNA, mRNA, and RepRNA Polyplexes. Biomacromolecules 2018, 19, 2870–2879. 10.1021/acs.biomac.8b00429. [DOI] [PubMed] [Google Scholar]
- Kaczmarek J. C.; Patel A. K.; Rhym L. H.; Palmiero U. C.; Bhat B.; Heartlein M. W.; DeRosa F.; Anderson D. G. Systemic Delivery of mRNA and DNA to the Lung Using Polymer-Lipid Nanoparticles. Biomaterials 2021, 275, 120966. 10.1016/j.biomaterials.2021.120966. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Le N.; Tan Z.; Brown M. E.; Jiang S.; Reineke T. M. Efficient Polymer-Mediated Delivery of Gene-Editing Ribonucleoprotein Payloads Through Combinatorial Design, Parallelized Experimentation, and Machine Learning. ACS Nano 2020, 14, 17626–17639. 10.1021/acsnano.0c08549. [DOI] [PubMed] [Google Scholar]
- Lundberg S. M.; Lee S.-I.. A Unified Approach to Interpreting Model Predictions. Proceedings of the 31st International Conference on Neural Information Processing Systems; 2017; pp 4768–4777.
- Dave K. M.; Han L.; Jackson M. A.; Kadlecik L.; Duvall C. L.; S Manickam D. DNA Polyplexes of a Phosphorylcholine-Based Zwitterionic Polymer for Gene Delivery. Pharm. Res. 2020, 37, 176. 10.1007/s11095-020-02899-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Lu X.; Jia F.; Tan X.; Sun X.; Cao X.; Wai F.; Zhang C.; Zhang K. Precision Tuning of DNA- and Poly(ethylene glycol)-Based Nanoparticles via Coassembly for Effective Antisense Gene Regulation. Chem. Mater. 2017, 29, 9882–9886. 10.1021/acs.chemmater.7b03520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Zeng F.; Jian H.; Wu S. Grafting Zwitterionic Polymer Chains onto PEI as a Convenient Strategy to Enhance Gene Delivery Performance. Polym. Chem. 2013, 4, 5810–5818. 10.1039/c3py00752a. [DOI] [Google Scholar]
- Sung S.-J.; Min S. H.; Cho K. Y.; Lee S.; Min Y.-J.; Yeom Y. I.; Park J.-K. Effect of Polyethylene Glycol on Gene Delivery of Polyethylenimine. Biol. Pharm. Bull. 2003, 26, 492–500. 10.1248/bpb.26.492. [DOI] [PubMed] [Google Scholar]
- Pearl J.; Glymour M.; Jewell N. P.. Causal Inference in Statistics: A Primer; John Wiley & Sons, 2016. [Google Scholar]
- Yu F.; Wei C.; Deng P.; Peng T.; Hu X. Deep Exploration of Random Forest Model Boosts the Interpretability of Machine Learning Studies of Complicated Immune Responses and Lung Burden of Nanoparticles. Sci. Adv. 2021, 7, eabf4130 10.1126/sciadv.abf4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg S. M.; Erion G. G.; Lee S.-I.. Consistent Individualized Feature Attribution for Tree Ensembles. arXiv 2019, 1802.03888 [cs, stat]. [Google Scholar]
- Chernozhukov V.; Chetverikov D.; Demirer M.; Duflo E.; Hansen C.; Newey W.; Robins J. Double/Debiased Machine Learning for Treatment and Structural Parameters. Econom. J. 2018, 21, C1–C68. 10.1111/ectj.12097. [DOI] [Google Scholar]
- Rozov S. M.; Permyakova N. V.; Deineko E. V. The Problem of the Low Rates of CRISPR/Cas9-mediated Knock-Ins in Plants: Approaches and Solutions. Int. J. Mol. Sci. 2019, 20, 3371. 10.3390/ijms20133371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. N.; Roth T. L.; Li P. J.; Chen P. A.; Apathy R.; Mamedov M. R.; Vo L. T.; Tobin V. R.; Goodman D.; Shifrut E.; Bluestone J. A.; Puck J. M.; Szoka F. C.; Marson A. Polymer-Stabilized Cas9 Nanoparticles and Modified Repair Templates Increase Genome Editing Efficiency. Nat. Biotechnol. 2020, 38, 44–49. 10.1038/s41587-019-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth T. L.; et al. Reprogramming Human T Cell Function and Specificity With Non-Viral Genome Targeting. Nature 2018, 559, 405–409. 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen J. M.; Chen X.; Liu J.; Gonçalves M. A. F. V. The Chromatin Structure of CRISPR-Cas9 Target DNA Controls the Balance Between Mutagenic and Homology-Directed Gene-Editing Events. Mol. Ther. Nucleic Acid 2019, 16, 141–154. 10.1016/j.omtn.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Staahl B. T.; Alla R. K.; Doudna J. A. Enhanced Homology-Directed Human Genome Engineering by Controlled Timing of CRISPR/Cas9 Delivery. Elife 2014, 3, e04766 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazi R.; Sghia-Hughes G.; Reid J. L.; Kubek S.; Haworth K. G.; Humbert O.; Kiem H.-P.; Adair J. E. Targeted Homology-Directed Repair in Blood Stem and Progenitor Cells With CRISPR Nanoformulations. Nat. Mater. 2019, 18, 1124–1132. 10.1038/s41563-019-0385-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson-Stevermer J.; Abdeen A. A.; Kohlenberg L.; Goedland M.; Molugu K.; Lou M.; Saha K. Assembly of CRISPR Ribonucleoproteins With Biotinylated Oligonucleotides via an RNA Aptamer for Precise Gene Editing. Nat. Commun. 2017, 8, 1711. 10.1038/s41467-017-01875-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird E. J.; Lovendahl K. N.; St. Martin A.; Harris R. S.; Gordon W. R. Increasing Cas9-Mediated Homology-Directed Repair Efficiency Through Covalent Tethering of DNA Repair Template. Commun. Biol. 2018, 1, 54. 10.1038/s42003-018-0054-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.; Mackley V. A.; Rao A.; Chong A. T.; Dewitt M. A.; Corn J. E.; Murthy N. Synthetically Modified Guide RNA and Donor DNA Are a Versatile Platform for CRISPR-Cas9 Engineering. eLife 2017, 6, e25312 10.7554/eLife.25312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkonen L.; Sato K.; Reinisalo M.; Kidron H.; Tachikawa M.; Watanabe M.; Uchida Y.; Urtti A.; Terasaki T. LC–MS/MS Based Quantitation of ABC and SLC Transporter Proteins in Plasma Membranes of Cultured Primary Human Retinal Pigment Epithelium Cells and Immortalized ARPE19 Cell Line. Mol. Pharmaceutics 2017, 14, 605–613. 10.1021/acs.molpharmaceut.6b00782. [DOI] [PubMed] [Google Scholar]
- Aukunuru J. V.; Ayalasomayajula S. P.; Kompella U. B. Nanoparticle Formulation Enhances the Delivery and Activity of a Vascular Endothelial Growth Factor Antisense Oligonucleotide in Human Retinal Pigment Epithelial Cells. J. Pharm. Pharmacol. 2010, 55, 1199–1206. 10.1211/0022357021701. [DOI] [PubMed] [Google Scholar]
- Kompella U. B.; Amrite A. C.; Pacha Ravi R.; Durazo S. A. Nanomedicines for Back of the Eye Drug Delivery, Gene Delivery, and Imaging. Prog. Retin. Eye Res. 2013, 36, 172–198. 10.1016/j.preteyeres.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo-Rodríguez A.; Delgado D.; Solinís M. A.; Gascón A. R.; Pedraz J. L. Solid Lipid Nanoparticles for Retinal Gene Therapy: Transfection and Intracellular Trafficking in RPE Cells. Int. J. Pharm. 2008, 360, 177–183. 10.1016/j.ijpharm.2008.04.023. [DOI] [PubMed] [Google Scholar]
- Bejjani R. A.; BenEzra D.; Cohen H.; Rieger J.; Andrieu C.; Jeanny J.-C.; Golomb G.; Behar-Cohen F. F. Nanoparticles for Gene Delivery to Retinal Pigment Epithelial Cells. Mol. Vis. 2005, 11, 124–132. [PubMed] [Google Scholar]
- Jiang Y.; Lodge T. P.; Reineke T. M. Packaging pDNA by Polymeric ABC Micelles Simultaneously Achieves Colloidal Stability and Structural Control. J. Am. Chem. Soc. 2018, 140, 11101–11111. 10.1021/jacs.8b06309. [DOI] [PubMed] [Google Scholar]
- Certo M. T.; Ryu B. Y.; Annis J. E.; Garibov M.; Jarjour J.; Rawlings D. J.; Scharenberg A. M. Tracking Genome Engineering Outcome at Individual DNA Breakpoints. Nat. Methods 2011, 8, 671–676. 10.1038/nmeth.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes S. V.; Daelemans D.; Cho E. H.; Dobbin Z.; Pavlakis G.; Lockett S. Automatic and Quantitative Measurement of Protein-Protein Colocalization in Live Cells. Biophys. J. 2004, 86, 3993–4003. 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaître G.; Nogueira F.; Aridas C. K. Imbalanced-learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning. J. Mach. Learn. Res. 2017, 18, 1–5. [Google Scholar]
- Pedregosa F.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.