

Abstract
BINding ANAlyzer (BINANA) is an algorithm for identifying and characterizing receptor/ligand interactions and other factors that contribute to binding. We recently updated BINANA to make the algorithm more accessible to a broader audience. We have also ported the Python3 codebase to JavaScript, thus enabling BINANA analysis in the web browser. As proof of principle, we created a web-browser application so students and chemical-biology researchers can quickly visualize receptor/ligand complexes and their unique binding interactions.
Introduction
Many biochemical processes depend on the association of specific receptors (e.g., proteins) with their small-molecule ligands. The process by which a receptor recognizes its ligand (“molecular recognition”) is primarily determined by the noncovalent atomic interactions that form between the two, including hydrogen bonds, π–π stacking, cation−π interactions, electrostatic attraction and repulsion, and hydrophobics. These interactions contribute to the overall binding affinity of the receptor/ligand association, and their geometric configuration plays a role in determining specificity (i.e., the tendency to bind the receptor target but not other off-target receptors). Characterizing receptor/ligand interactions thus yields essential insights into the biological mechanisms underlying many processes (e.g., signaling, enzymatic catalysis, etc.). In the context of drug discovery, accurately characterizing receptor/ligand interactions allows medicinal chemists to assess whether a ligand merits further study and pharmaceutical development.
When assessing a single receptor/ligand complex, researchers often rely on manual inspection using visualization software such as VMD,1 PyMOL,2 or Chimera.3 But many use cases require the assessment of many—sometimes thousands—of predicted ligand poses. The BINding ANAlyzer (BINANA) algorithm (first released in 2011) addresses this challenge by automating ligand-pose analysis,4 enabling the characterization of far more receptor/ligand complexes than can be manually inspected. For example, McCarthy et al.5 used BINANA to identify novel inhibitors of KRAS, a GTPase protein activated via mutation in 15% of human cancers. After performing a high-throughput virtual screen to evaluate six million compounds for potential KRAS inhibition, BINANA was used to identify the top predicted ligands that formed reasonable interactions with the protein receptor. These efforts ultimately led to an experimentally validated KRAS inhibitor. In a second study, Poli et al. used BINANA to identify inhibitors of monoacylglycerol lipase (MAGL),6 a protein involved in the pathogenesis of neurodegenerative, cancer, inflammatory, and chronic-pain diseases. They docked approximately 14,000 molecules into the MAGL binding pocket and used BINANA to identify 17 compounds predicted to form critical interactions with the receptor. Subsequent experiments ultimately revealed three new compounds that inhibited MAGL activity; one even inhibited the proliferation of breast- and ovarian-cancer cell lines.
Several groups (including our own) have used BINANA to generate training data for machine-learning models (“scoring functions”) designed to identify ligands that merit more careful human scrutiny. Our NNScore2 algorithm7 leverages BINANA descriptors (among other metrics) to predict ligand binding strength. NNScore2 has been used to help identify novel inhibitors of haloalkane dehalogenase,8 VEGFR-2,9 and aromatase,10 among others. The DLSCORE scoring function11 similarly uses BINANA descriptors to predict binding. BINANA has also been incorporated into several other programs (e.g., HBonanza12 and POVME313), has inspired similar approaches,14,15 and has been included in the Open Drug Discovery Toolkit.16
These examples of broad adoption aside, the original BINANA implementation has some notable limitations. It runs only from the command line and provides no built-in visualization of the identified interactions, instead requiring separate visualization software. From the perspective of tool developers, BINANA 1.0 is also challenging because (1) its codebase organization does not allow for modular import into other Python scripts, (2) it is written in a now unmaintained programming language (Python2), and (3) its output is difficult to parse, complicating efforts to process BINANA analyses in other programs.
We developed BINANA 2 to address these challenges. The updated version can still run from the command line, but many users will benefit from our new web-browser implementation, which provides built-in molecular visualization that simplifies analysis. Tool-development researchers will benefit from updates to the Python codebase. We refactored the original implementation using a more modular programming approach that allows developers to integrate BINANA functions more easily into their projects (e.g., by importing individual modules as needed). We also added JSON- and CSV-formatted output for easy processing by other computational tools and rewrote the code to be compatible with Python3 and JavaScript transpilation. To further encourage broad adoption and integration, we release BINANA 2 under a more permissive license than previous versions (Apache License, Version 2.0). Users can download the source code free of charge from http://durrantlab.com/binana-download/ or access the browser app at http://durrantlab.com/binana/.
BINANA Python Codebase
Improving Modularity and Python3 Compatibility
We split the BINANA codebase into modules (separate files) to enable access as a Python library from other scripts. BINANA 1.0 was designed solely as a stand-alone application (i.e., its codebase was contained in a single file, and its functions were not organized into modules), but several other groups have nevertheless incorporated BINANA code into their software projects.11,13,16 To further enable such use, we refactored the BINANA codebase so other software can more easily import BINANA’s essential functions, including (1) loading PDBQT and PDB files containing receptor and bound-ligand structures, (2) analyzing those structures to identify specific receptor/ligand interactions, and (3) saving BINANA analyses in various formats.
In refactoring the BINANA code, we also updated the codebase to make it compatible with Python3. The original version of BINANA was written in the now discontinued Python2 language.
Documentation and Tutorials
We created a documentation website to further improve BINANA 2 usability: http://durrantlab.com/apps/binana/docs/. The website (1) describes how to use the stand-alone command-line BINANA program, (2) links to a video tutorial that shows how to use the BINANA browser app, (3) catalogs the extensive docstrings associated with each public function so tool developers can quickly learn how to access the application programming interface (API), and (4) provides a copy of a Jupyter notebook demonstrating how to use BINANA as a Python library. The BINANA download includes the same Python notebook in an “examples” directory, as well as a notebook and simple HTML file showing how to use the JavaScript version of the library.
JSON and CSV Output
The original version of BINANA saved binding-pose analyses to a PDB file or a VMD state file (for visualization using the popular program Visual Molecular Dynamics1). The new version of BINANA retains these features and further allows data export to the machine-readable JSON and CSV formats. Many researchers have used BINANA to automatically assess the binding poses of large compound sets (e.g., in the context of virtual screens5,6). To extract the data from these many analyses for subsequent processing, they have had to parse the BINANA-log text files directly. Now that BINANA outputs to JSON and CSV, this process will be much simplified. To further enable external analyses of BINANA-detected interactions, the JSON/CSV output files also include the bond distances and angles (where applicable) of each detected interaction.
Identifying Receptor/Ligand Interactions and Other Characterizations
To analyze a given receptor/ligand complex, the user provides BINANA with molecular models of the receptor and bound ligand in the PDBQT (recommended) or PDB format. BINANA then considers the positions and angles of various chemical groups to identify common interactions and otherwise characterize the complex.
BINANA 2 identifies the same receptor/ligand interactions and characterizations that previous versions identified. These include close (<4.0 Å by default) and closest (<2.5 Å) contacts as well as hydrophobic, salt-bridge, π–π, T-stacking, cation−π, and hydrogen-bond interactions. BINANA also tallies the number of times a ligand atom comes near the backbone or side chain of an α helix, beta sheet, or “other” secondary-structure amino acid. If the user provides models in the PDBQT format (which includes AutoDock atom types and Gasteiger partial charges17), BINANA also tallies the electrostatic energies between proximate receptor/ligand atoms, the ligand atom types, and the number of ligand rotatable bonds. Full details can be found in the original BINANA manuscript.4
The BINANA 2 interaction criteria are identical to the original version, with a few notable exceptions. For example, previously, the close and closest contacts were mutually exclusive (i.e., those receptor/ligand atom pairs that were close enough to be categorized as “closest” were not also considered to be “close”). In BINANA 2, all closest contacts are also close. We have also updated the hydrogen-bond definition so that sulfur atoms can serve as hydrogen-bond donors and acceptors.
BINANA can also now detect halogen bonds. The algorithm for detecting halogen bonds is the same as for hydrogen bonds, except an iodine, bromine, chlorine, or fluorine atom takes the places of the hydrogen atom. Additionally, a carbon atom can serve as a halogen-bond donor but not a hydrogen-bond donor (e.g., the iodine of an iodopyrimidinyl moiety can participate in a halogen bond18). Halogen bonds tend to be longer than hydrogen bonds, so BINANA accepts a separate user-defined halogen-bond cutoff length (via the halogen_bond_dist_cutoff parameter, which defaults to 5.5 Å). The donor-halogen-acceptor and donor-hydrogen-acceptor angle cutoffs are the same (specified via the renamed hydrogen_halogen_bond_angle_cutoff parameter, which defaults to 40°, the maximum allowed deviation from a straight line).
BINANA now also detects metal-coordination bonds whenever select metal cations come within a user-defined distance of nitrogen, oxygen, sulfur, iodine, bromine, chlorine, and fluorine atoms (specified via the metal_coordination_dist_cutoff parameter, which defaults to 3.5 Å).
Finally, we substantially improved how BINANA analyzes receptor/ligand complexes that do not include hydrogen atoms. In the case of protein receptors, BINANA 2 now identifies hydrogen bond donors/acceptors and charged groups based on the residue and atom names (assuming standardized PDB nomenclature). In the case of the ligand, it makes rudimentary assumptions about protonation based on the geometry of the heavy atoms. To avoid relying on these assumptions, we encourage users to add hydrogen atoms to their protein receptors using programs such as Open Babel19 and MolProbity20 and to their small-molecule ligands using Open Babel,19 Avogadro,21 and Gypsum-DL.22
BINANA JavaScript Codebase and Browser-App Implementation
Porting BINANA to JavaScript
To broaden the impact of our BINANA algorithm, we transpiled the Python code to JavaScript using a software tool called Transcrypt (transcrypt.org). Transpilation rewrites or “translates” computer code written in one language (e.g., Python) into another (e.g., JavaScript). The resulting JavaScript library, BINANA.js, has the same functionality as the Python version but can be easily accessed from web apps running in any modern web browser.
BINANA Browser App
To help noncomputationalists better engage with the library, we integrated BINANA.js into a user-friendly browser-based application that detects and visualizes receptor/ligand interactions.
Designing and Compiling the Browser-App User Interface
The BINANA browser app provides an interactive graphical user interface (GUI). We designed the BINANA GUI using the same approach described elsewhere.23,24 In brief, the GUI was written in the TypeScript programming language, which compiles to JavaScript. We used Vue.js, an open-source web application framework, to compose reusable GUI components (e.g., text fields, buttons, etc.), and the BootstrapVue library to style all GUI components consistently according to the Bootstrap4 framework. We also used a custom molecular-visualization Vue.js component that leverages the 3Dmol.js JavaScript library25 to display macromolecular and small-molecule structures in the browser, as required for visualizing BINANA-predicted receptor/ligand interactions.
To compile these components and the BINANA.js library itself into a single web app, we used Webpack, an open-source module bundler, to manage the organization and composition of our source libraries and files. Webpack copies required files, combines files where possible, removes unneeded code, and more. The build process also used Google’s Closure Compiler to optimize the file size and performance of the TypeScript-compiled JavaScript code.
Browser-App Usage
Advanced Parameters
The “Advanced Parameters” button appears at the top of the BINANA browser-app interface (Figure 1A). When clicked, a series of text fields appears that allows the user to modify the BINANA-library parameters. These fields initially contain the default values used by the BINANA command-line tool and Python library. We expect most users will wish to leave them unchanged, so they are hidden by default.
Figure 1.

Illustration of the BINANA browser-app interface. (A) The “Advanced Parameters” button allows users to specify custom BINANA parameters. (B) The “Input Files” panel allows users to load their receptor/ligand structures into the browser’s memory, either from their own computer or directly from the Protein Data Bank. (C) The “Molecular “Viewer” panel shows the detected interactions. (D) A legend below the viewer describes how the interactions are represented. (E) The “Save” button saves the results to the user’s local disk.
Input Files
The “Input Files” section allows users to load a ligand or receptor PDBQT or PDB file into their browser’s memory (Figure 1B). Users can specify a file on their local computer or a PDB ID to load a structure directly from the Protein Data Bank.26 The structures are never uploaded to any third-party server, helping to ensure data privacy. Users can also click the “Video Tutorial” button to learn more about basic use and the “Use Example Files” button to run BINANA with built-in example receptor and ligand files.
Molecular Viewer
The receptor/ligand complex appears in the “Molecular Viewer” section of the browser app (Figure 1C), which also presents three general categories of interactions: “Common,” “Contacts,” and “Aromatic.” Clicking on the corresponding button opens a drop-down menu so users can choose which specific interactions to visualize. Under “Common,” users can select “Hydrogen Bonds,” “Halogen Bonds,” “Hydrophobic,” “Salt Bridge,” and “Metal Coordination” interactions. Under “Contacts,” users can select “Close” or “Closest” interactions. Under “Aromatic,” users can select “π–π Stacking,” “T Shaped,” or “Cation−π” interactions.
Interaction Viewer
The identified receptor/ligand interactions appear in the molecular viewer (Figure 1C), and a legend below the viewer describes how each interaction is represented (Figure 1D). In brief, hydrogen and halogen bonds are solid black and green arrows, respectively, that point from the donor to the acceptor. Salt-bridge, metal-coordination, π–π stacking, T-stacking, and cation−π interactions are red, orange, blue, aqua, and navy dashed lines, respectively. Atoms that participate in hydrophobic, close-contact, and closest-contact interactions are marked with gray, purple, and fuchsia spheres, respectively.
The browser app also provides a “Save” button (Figure 1E) that allows users to save a zip file containing (1) a copy of the receptor/ligand structures, (2) thorough descriptions of all BINANA.js-detected interactions in the TXT, JSON, and CSV formats, (3) a VMD state file to enable more customized visualization using Visual molecular dynamics,1 and (4) a PNG image file of the molecular viewer for reference
Examples of Use
To test the web version of BINANA, we selected two receptor/ligand complexes and visualized them in the browser.
M2 Muscarinic Acetylcholine Receptor
Muscarinic acetylcholine receptors (mACHhRs) are G protein-coupled receptors (GPCRs) activated by acetylcholine.27 As of 2017, approximately 34% of FDA-approved drugs targeted GPCRs,28 so studying GPCR/ligand complexes is useful for structure-based drug discovery and design.28 Despite sharing between 64% and 82% sequence similarity, the five mACHhR subtypes differ in tissue distribution and GTP-binding protein partners.27 The M2 muscarinic receptor, for example, is expressed in peripheral tissues and regulates heart rate.27 Clinically approved small-molecule drugs that modulate M2 activity can effectively treat bradycardia (e.g., atropine29), urinary incontinence (e.g., tolterodine29), and more.
We used the BINANA web app to visualize a structure of M2 bound to LY2119620, a small-molecule, positive allosteric M2 modulator (Figure 2A). Allosteric muscarinic-receptor ligands are significant because they may enable improved receptor selectivity. All muscarinic receptors bind acetylcholine, so their orthosteric (primary) binding pockets are in many ways chemically similar. Identifying orthosteric ligands that bind to only one receptor subtype is thus challenging. In contrast, allosteric pockets may be more varied. Although LY2119620 also binds M4 muscarinic receptors and so is not strictly receptor specific,30 in principle, allostery enables the design of ligands with improved specificity. We downloaded a PDB file of the receptor/ligand complex (PDB 6OIK(27)), added hydrogen atoms to the structure and ligand using MolProbity,20,31,32 and loaded the resulting model into the BINANA web app.
Figure 2.
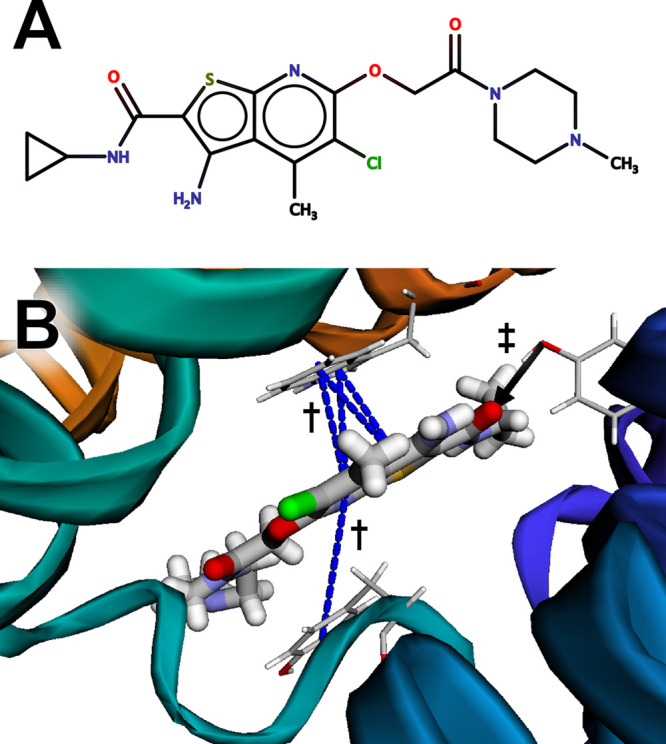
The muscarinic acetylcholine receptor M2 bound to LY2119620, an allosteric ligand. (A) A schematic of the ligand, created using MarvinSketch 18.24.0, ChemAxon (https://www.chemaxon.com). (B) The ligand is predicted to participate in π-stacking interactions with TRP422 and TYR177 (marked with daggers) and a hydrogen bond with TYR80 (marked with a double dagger).
BINANA visualization revealed that the aromatic LY2119620 bicyclic moiety forms π–π stacking interactions with TRP422 and TYR177 (Figure 2B, blue dashed lines, marked with daggers), and the LY2119620 carbonyl oxygen atom forms a hydrogen bond with the side-chain hydroxyl group of TYR80 (Figure 2B, black arrow, marked with a double dagger).
Pseudomonas aeruginosa Peptidyl-tRNA Hydrolase
Peptidyl tRNA hydrolase (Pth) is a potential drug target found in multiple species of bacteria, including Escherichia coli, Mycobacterium tuberculosis, Mycobacterium smegmatis, and Pseudomonas aeruginosa. The process of mRNA translation produces a peptidyl-tRNA intermediate, but ribosomes often release this intermediate when mRNA translation stalls. Pth separates peptidyl-tRNA into free tRNA and peptide by cleaving the ester bond between the C-terminus of the peptide and the 2′ or 3′ hydroxyl group at the 3′ end of the tRNA.33 This cleavage frees the tRNA and peptide for reuse. In the absence of Pth activity, peptidyl-tRNAs cannot be recycled, ultimately resulting in bacterial death.33 Small-molecule Pth inhibitors thus have potential as antibacterial therapeutics.
To demonstrate the BINANA browser app applied to docked (predicted) ligand poses, we performed a virtual screen targeting the Pth active site. In brief, we prepared a model of the Pth receptor from Pseudomonas aeruginosa based on the 4QBK crystal structure.34 We used PDB2PQR35−37 to add hydrogens atoms to the protein, OpenBabel19 to convert the PQR file to PDB, and MGLTools38 to convert the PDB file to PDBQT. Because Pth binds peptide-based compounds, we also prepared a virtual library of approximately 60,000 easy to synthesize dipeptides provided by the Distributed Drug Discovery (D3) program.22,39−42 We used Gypsum-DL to generate 3D models of these compounds and to enumerate alternate protonation, chiral, and tautomeric states.22 The dipeptide files were also converted to the PDBQT format using OpenBabel and MGLTools.
We performed an initial docking run using Webina,23 a browser-app version of the docking program AutoDock Vina.43 We used this initial run to determine appropriate coordinates and dimensions for the docking box and to confirm that our docking protocol could generally recapture the crystallographic pose of a known ligand (PDB 4QBK(34)). Having identified acceptable parameters, we docked all compounds (approximately 60,000) using command-line Vina running on resources provided by the University of Pittsburgh’s Center for Research Computing (default parameters).
The two best-scoring compounds both had Vina scores of −9.0 kcal/mol. We loaded one of these, (2S)-2-[(2R)-2-amino-3-(anthracen-9-yl)propanamido]-3-(quinolin-2-yl)propanoic acid, into the BINANA web app (Figure 3A). The visualization suggests that the ligand participates in π-stacking interactions with TYR68 (Figure 3C, blue dashed lines, marked with daggers) and hydrogen bonds with ASN116 (Figure 3B, black arrows, marked with double daggers).
Figure 3.
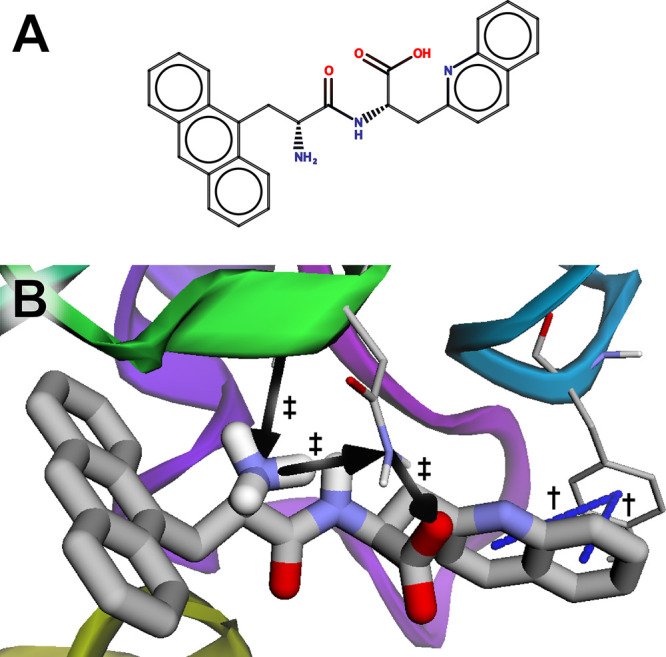
The peptidyl-tRNA hydrolase bound to a predicted ligand identified in a virtual screen. (A) A schematic of the ligand, created using MarvinSketch 18.24.0, ChemAxon (https://www.chemaxon.com). (B) The ligand is predicted to participate in π-stacking interactions with TYR68 (marked with daggers) and hydrogen bonds with ASN116 (marked with double daggers).
Related Programs
Several freely available programs can also detect some intramolecular interactions, including Arpeggio,44 PLIP,45 nAPOLI,46 UCSF Chimera,3 VMD,1 and Open-Source PyMOL.2 All these programs are open source except nAPOLI, which appears to be accessible only as a web server (Table 1). However, aside from BINANA, only Open-Source PyMOL is released under a permissive free software license. Arpeggio and PLIP are copyleft licensed, meaning that all derivative works must be similarly licensed (potentially limiting use in commercial settings), and UCSF Chimera and VMD are free only for noncommercial use. Because BINANA 2 is released under the terms of the permissive open-source Apache License, Version 2.0, others can incorporate BINANA code into their own programs without restrictions, even in commercial settings.
Table 1. Comparison of Freely Available Programs for Detecting Intramolecular Interactions. “PyMOL” refers to Open-Source PyMOL, not the commercial Incentive PyMOL. “Online” indicates whether the program can be accessed through a browser-based interface. “JavaScript” and “Python” indicate whether the program is available as an “importable” JavaScript or Python module, respectively. “Pure Python” indicates whether the program relies on third-party Python packages beyond those in the Python standard library. “Interactions” indicates whether the program can detect many interaction types or only the most frequent types.
| BINANA | Arpeggio | PLIP | nAPOLI | Chimera | VMD | PyMOL | |
|---|---|---|---|---|---|---|---|
| Open source | Yes | Yes | Yes | No | Yes | Yes | Yes |
| License | Apache 2 | GPLv3 | GPLv2 | – | Free for nonprofit | Free for nonprofit | BSD-like |
| Online | Yes | Yes | Yes | Yes | No | No | No |
| JavaScript | Yes | No | No | N/A | No | No | No |
| Python | Yes | Yes | Yes | N/A | Third party | Third party | Yes |
| Pure Python | Yes | No | No | N/A | No | No | No |
| Interactions | Many | Many | Many | Many | Few | Few | Few |
Users can access BINANA, Arpeggio, PLIP, and nAPOLI through convenient web-based interfaces (Table 1). But BINANA alone is available as a JavaScript library (BINANA.js), enabling easy integration into third-party browser apps. BINANA.js allows apps to detect intermolecular interactions in the browser itself, without requiring users to upload their (possibly proprietary) structures to a third-party system. Consequently, BINANA.js-powered browser apps do not require an extensive remote computing infrastructure where calculations take place “in the cloud.” Instead, a simple web server sends the BINANA.js library to users’ browsers to detect interactions locally on their own machines.
Except for nAPOLI, the programs listed in Table 1 can also be accessed as Python modules. BINANA, Arpeggio, and PLIP are written in Python, and others have made UCSF Chimera47 and VMD48 Python accessible. Although PyMOL is written predominantly in C and C++, it has extensive Python integration built in. Python is the most popular programming language for creating computational chemical-biology tools, and seamless Python integration makes it easy for researchers to incorporate external functionality into their own software. Of note, only BINANA is written in “pure Python,” meaning it does not rely on any third-party packages (e.g., NumPy) beyond those included in the Python standard library. This arguably makes BINANA-powered Python scripts somewhat easier to install and distribute. Programmers can simply copy the BINANA library into their projects, and it will work on any operating system that supports Python itself.
Not all the programs listed in Table 1 can detect the same broad range of intramolecular interactions. Like BINANA, the Arpeggio, PLIP, and nAPOLI programs can detect many interaction types, ranging from those that are frequent (e.g., hydrogen bonds) to those that are less frequent (e.g., T-shaped, π–π, and cation−π interactions). In contrast, although UCSF Chimera, VMD, and Open-Source PyMOL provide many broad and useful features, to the best of our knowledge, they detect only a few of the most frequent interaction types (e.g., hydrogen bonds).
Finally, we note that several commercial tools such as MOE (chemcomp.com), Discovery Studio (accelrys.com), SAMSON (samson-connect.net), Proasis4 (desertsci.com), and Small Molecule Drug Discovery Suite (schrodinger.com) include features for detecting intramolecular interactions. Although powerful, many of these commercial tools are expensive and otherwise have limitations on use.
Broad Compatibility
We have tested the BINANA Python and JavaScript libraries on the operating systems, Python versions, and browsers listed in Table 2. The software depends on no external Python or JavaScript libraries, so we do not anticipate compatibility issues on other untested setups.
Table 2. Operating System, Python, and Web-Browser Compatibility.
| Operating System | Python | Browser (JavaScript) |
|---|---|---|
| Ubuntu (Linux) 20.04.2 LTS | Python 3.9.1 | Chrome 95.0.4638.54 |
| Firefox 94.0 | ||
| macOS Mojave 10.14.6 | Python 3.6.7 | Chrome 97.0.4692.20 |
| Firefox 94.0.2 | ||
| Safari 14.1.2 | ||
| Microsoft Windows 11 Pro | Python 3.10.0 | Chrome 96.0.4664.45 |
| Firefox 94.0.2 | ||
| Edge 96.0.1054.34 | ||
| Android 11 | N/A | Chrome 96.0.4664.45 |
| Firefox 94.1.2 | ||
| iOS 15.1 | N/A | Safari 15.1 |
Conclusion
BINANA 2 retains the core functionality of the original version in that it can run as a stand-alone, command-line program. But it now also serves as a Python library that others can incorporate into their Python-based computational-biology tools. We also ported the BINANA library to JavaScript, enabling use in the web browser. To demonstrate how to incorporate BINANA.js into browser-based applications, we created the BINANA browser app. This app can be accessed online, enabling easy access and visualization without requiring command-line use.
Data Software and Availability
Users can download the BINANA 2 source code—including the Python3/JavaScript libraries and the web-app graphical user interface—free of charge from http://durrantlab.com/binana-download/. We release BINANA 2 under the terms of the open-source Apache License, Version 2.0. Users can also freely access the BINANA browser app at http://durrantlab.com/binana/, and the API documentation at http://durrantlab.com/apps/binana/docs/.
Acknowledgments
We thank the University of Pittsburgh’s Center for Research Computing for providing helpful computer resources. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health [R01GM132353 to J.D.D.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.1c01461.
SMILES.csv file: names and SMILES strings of ligands depicted in Figures 2 and 3 (TXT)
The authors declare no competing financial interest.
Supplementary Material
References
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graph 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.Pymol: An Open-Source Molecular Graphics Tool. In CCP4 Newsletter on Protein Crystallography; CCP4, 2002, Vol. 40, pp 82–92.
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. Ucsf Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. BINANA: A Novel Algorithm for Ligand-Binding Characterization. J. Mol. Graph Model 2011, 29, 888–893. 10.1016/j.jmgm.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy M. J.; Pagba C. V.; Prakash P.; Naji A. K.; van der Hoeven D.; Liang H.; Gupta A. K.; Zhou Y.; Cho K. J.; Hancock J. F.; Gorfe A. A. Discovery of High-Affinity Noncovalent Allosteric Kras Inhibitors That Disrupt Effector Binding. ACS Omega 2019, 4, 2921–2930. 10.1021/acsomega.8b03308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli G.; Lapillo M.; Jha V.; Mouawad N.; Caligiuri I.; Macchia M.; Minutolo F.; Rizzolio F.; Tuccinardi T.; Granchi C. Computationally Driven Discovery of Phenyl(Piperazin-1-Yl)Methanone Derivatives as Reversible Monoacylglycerol Lipase (Magl) Inhibitors. J. Enzyme Inhib Med. Chem. 2019, 34, 589–596. 10.1080/14756366.2019.1571271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. NNScore 2.0: A Neural-Network Receptor-Ligand Scoring Function. J. Chem. Inf Model 2011, 51, 2897–2903. 10.1021/ci2003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buryska T.; Daniel L.; Kunka A.; Brezovsky J.; Damborsky J.; Prokop Z. Discovery of Novel Haloalkane Dehalogenase Inhibitors. Appl. Environ. Microbiol. 2016, 82, 1958–1965. 10.1128/AEM.03916-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaballah M. Y.; Serya R. A. T.; Saad N.; Khojah S. M.; Ahmed M.; Barakat K.; Abouzid K. A. M. Towards Discovery of Novel Scaffold with Potent Antiangiogenic Activity; Design, Synthesis of Pyridazine Based Compounds, Impact of Hinge Interaction, and Accessibility of Their Bioactive Conformation on Vegfr-2 Activities. J. Enzyme Inhib Med. Chem. 2019, 34, 1573–1589. 10.1080/14756366.2019.1651723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianov A. M.; Nikolaev G. I.; Kornoushenko Y. V.; Usanov S. A. Click Chemistry in Silico, Docking, Quantum Chemical Calculations, and Molecular Dynamics Simulations to Identify Novel 1, 2, 4-Triazole-Based Compounds as Potential Aromatase Inhibitors. SN Applied Sciences 2019, 1, 1–16. 10.1007/s42452-019-1051-x. [DOI] [Google Scholar]
- Hassan M.; Mogollon D. C.; Fuentes O.. Dlscore: A Deep Learning Model for Predicting Protein-Ligand Binding Affinities. ChemRxiv Preprint, 2018.
- Durrant J. D.; McCammon J. A. HBonanza: A Computer Algorithm for Molecular-Dynamics-Trajectory Hydrogen-Bond Analysis. J. Mol. Graph Model 2011, 31, 5–9. 10.1016/j.jmgm.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. R.; Sorensen J.; Hensley N.; Wong C.; Zhu C.; Perison T.; Amaro R. E. POVME 3.0: Software for Mapping Binding Pocket Flexibility. J. Chem. Theory Comput 2017, 13, 4584–4592. 10.1021/acs.jctc.7b00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenegro Rabello M.; Rolim L. A.; Rolim Neto P. J.; Hernandes M. Z. Cyclomolder Software: Building Theoretical Cyclodextrin Derivatives Models and Evaluating Their Host: Guest Interactions. Journal of Inclusion Phenomena and Macrocyclic Chemistry 2019, 93, 301–308. 10.1007/s10847-019-00880-3. [DOI] [Google Scholar]
- Wu Z.; Ramsundar B.; Feinberg E. N.; Gomes J.; Geniesse C.; Pappu A. S.; Leswing K.; Pande V. Moleculenet: A Benchmark for Molecular Machine Learning. Chem. Sci. 2018, 9, 513–530. 10.1039/C7SC02664A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcikowski M.; Zielenkiewicz P.; Siedlecki P. Open Drug Discovery Toolkit (Oddt): A New Open-Source Player in the Drug Discovery Field. J. Cheminform 2015, 7, 26. 10.1186/s13321-015-0078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger J.; Marsili M. Iterative Partial Equalization of Orbital Electronegativity—a Rapid Access to Atomic Charges. Tetrahedron 1980, 36, 3219–3228. 10.1016/0040-4020(80)80168-2. [DOI] [Google Scholar]
- Lange A.; Gunther M.; Buttner F. M.; Zimmermann M. O.; Heidrich J.; Hennig S.; Zahn S.; Schall C.; Sievers-Engler A.; Ansideri F.; Koch P.; Laemmerhofer M.; Stehle T.; Laufer S. A.; Boeckler F. M. Targeting the Gatekeeper Met146 of C-Jun N-Terminal Kinase 3 Induces a Bivalent Halogen/Chalcogen Bond. J. Am. Chem. Soc. 2015, 137, 14640–14652. 10.1021/jacs.5b07090. [DOI] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An Open Chemical Toolbox. J. Cheminform 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. J.; Headd J. J.; Moriarty N. W.; Prisant M. G.; Videau L. L.; Deis L. N.; Verma V.; Keedy D. A.; Hintze B. J.; Chen V. B.; Jain S.; Lewis S. M.; Arendall W. B. 3rd; Snoeyink J.; Adams P. D.; Lovell S. C.; Richardson J. S.; Richardson D. C. Molprobity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform 2012, 4, 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropp P. J.; Spiegel J. O.; Walker J. L.; Green H.; Morales G. A.; Milliken K. A.; Ringe J. J.; Durrant J. D. Gypsum-DL: An Open-Source Program for Preparing Small-Molecule Libraries for Structure-Based Virtual Screening. J. Cheminform 2019, 11, 34. 10.1186/s13321-019-0358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochnev Y.; Hellemann E.; Cassidy K. C.; Durrant J. D. Webina: An Open-Source Library and Web App That Runs Autodock Vina Entirely in the Web Browser. Bioinformatics 2020, 36, 4513–4515. 10.1093/bioinformatics/btaa579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H.; Durrant J. D. Deepfrag: An Open-Source Browser App for Deep-Learning Lead Optimization. J. Chem. Inf Model 2021, 61, 2523–2529. 10.1021/acs.jcim.1c00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego N.; Koes D. 3dmol.Js: Molecular Visualization with Webgl. Bioinformatics 2015, 31, 1322–1324. 10.1093/bioinformatics/btu829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose P. W.; Prlic A.; Altunkaya A.; Bi C.; Bradley A. R.; Christie C. H.; Costanzo L. D.; Duarte J. M.; Dutta S.; Feng Z.; Green R. K.; Goodsell D. S.; Hudson B.; Kalro T.; Lowe R.; Peisach E.; Randle C.; Rose A. S.; Shao C.; Tao Y. P.; Valasatava Y.; Voigt M.; Westbrook J. D.; Woo J.; Yang H.; Young J. Y.; Zardecki C.; Berman H. M.; Burley S. K. The RCSB Protein Data Bank: Integrative View of Protein, Gene and 3D Structural Information. Nucleic Acids Res. 2017, 45, D271–D281. 10.1093/nar/gkw1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S.; Qu Q.; Robertson M. J.; Skiniotis G.; Kobilka B. K. Structures of the M1 and M2 Muscarinic Acetylcholine Receptor/G-protein Complexes. Science 2019, 364, 552–557. 10.1126/science.aaw5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schioth H. B.; Gloriam D. E. Trends in Gpcr Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov 2017, 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C. P.; Nahorski S. R.; Challiss R. A. Constitutive Activity and Inverse Agonism at the M2 Muscarinic Acetylcholine Receptor. J. Pharmacol Exp Ther 2006, 316, 279–288. 10.1124/jpet.105.094383. [DOI] [PubMed] [Google Scholar]
- Croy C. H.; Schober D. A.; Xiao H.; Quets A.; Christopoulos A.; Felder C. C. Characterization of the Novel Positive Allosteric Modulator, Ly2119620, at the Muscarinic M(2) and M(4) Receptors. Mol. Pharmacol. 2014, 86, 106–115. 10.1124/mol.114.091751. [DOI] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. 3rd; Snoeyink J.; Richardson J. S.; Richardson D. C. Molprobity: All-Atom Contacts and Structure Validation for Proteins and Nucleic Acids. Nucleic Acids Res. 2007, 35, W375–383. 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. Molprobity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das G.; Varshney U. Peptidyl-Trna Hydrolase and Its Critical Role in Protein Biosynthesis. Microbiology (Reading) 2006, 152, 2191–2195. 10.1099/mic.0.29024-0. [DOI] [PubMed] [Google Scholar]
- Singh A.; Kumar A.; Gautam L.; Sharma P.; Sinha M.; Bhushan A.; Kaur P.; Sharma S.; Arora A.; Singh T. P. Structural and Binding Studies of Peptidyl-Trna Hydrolase from Pseudomonas Aeruginosa Provide a Platform for the Structure-Based Inhibitor Design against Peptidyl-Trna Hydrolase. Biochem. J. 2014, 463, 329–337. 10.1042/BJ20140631. [DOI] [PubMed] [Google Scholar]
- Unni S.; Huang Y.; Hanson R. M.; Tobias M.; Krishnan S.; Li W. W.; Nielsen J. E.; Baker N. A. Web Servers and Services for Electrostatics Calculations with Apbs and PDB2PQR. J. Comput. Chem. 2011, 32, 1488–1491. 10.1002/jcc.21720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky T. J.; Czodrowski P.; Li H.; Nielsen J. E.; Jensen J. H.; Klebe G.; Baker N. A. PDB2PQR: Expanding and Upgrading Automated Preparation of Biomolecular Structures for Molecular Simulations. Nucleic Acids Res. 2007, 35, W522–525. 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky T. J.; Nielsen J. E.; McCammon J. A.; Baker N. A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–667. 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. Autodock4 and Autodocktools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott W. L.; Alsina J.; Audu C. O.; Babaev E.; Cook L.; Dage J. L.; Goodwin L. A.; Martynow J. G.; Matosiuk D.; Royo M.; Smith J. G.; Strong A. T.; Wickizer K.; Woerly E. M.; Zhou Z.; O’Donnell M. J. Distributed Drug Discovery, Part 2: Global Rehearsal of Alkylating Agents for the Synthesis of Resin-Bound Unnatural Amino Acids and Virtual D(3) Catalog Construction. J. Comb Chem. 2009, 11, 14–33. 10.1021/cc800184v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott W. L.; Audu C. O.; Dage J. L.; Goodwin L. A.; Martynow J. G.; Platt L. K.; Smith J. G.; Strong A. T.; Wickizer K.; Woerly E. M.; O’Donnell M. J. Distributed Drug Discovery, Part 3: Using D(3) Methodology to Synthesize Analogs of an Anti-Melanoma Compound. J. Comb Chem. 2009, 11, 34–43. 10.1021/cc800185z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott W. L.; O’Donnell M. J. Distributed Drug Discovery, Part 1: Linking Academia and Combinatorial Chemistry to Find Drug Leads for Developing World Diseases. J. Comb Chem. 2009, 11, 3–13. 10.1021/cc800183m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott W. L.; Denton R. E.; Marrs K. A.; Durrant J. D.; Samaritoni J. G.; Abraham M. M.; Brown S. P.; Carnahan J. M.; Fischer L. G.; Glos C. E.; Sempsrott P. J.; O’Donnell M. J. Distributed Drug Discovery: Advancing Chemical Education through Contextualized Combinatorial Solid-Phase Organic Laboratories. J. Chem. Educ. 2015, 92, 819–826. 10.1021/ed500135n. [DOI] [Google Scholar]
- Trott O.; Olson A. J. Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubb H. C.; Higueruelo A. P.; Ochoa-Montano B.; Pitt W. R.; Ascher D. B.; Blundell T. L. Arpeggio: A Web Server for Calculating and Visualising Interatomic Interactions in Protein Structures. J. Mol. Biol. 2017, 429, 365–371. 10.1016/j.jmb.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin S.; Schreiber S.; Haupt V. J.; Adasme M. F.; Schroeder M. Plip: Fully Automated Protein-Ligand Interaction Profiler. Nucleic Acids Res. 2015, 43, W443–447. 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassio A. V.; Santos L. H.; Silveira S. A.; Ferreira R. S.; de Melo-Minardi R. C. nAPOLI: A Graph-Based Strategy to Detect and Visualize Conserved Protein-Ligand Interactions in Large-Scale. IEEE/ACM Trans Comput. Biol. Bioinform 2019, 17, 1317–1328. 10.1109/TCBB.2019.2892099. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Guerra Pedregal J.; Marechal J. D. Pychimera: Use Ucsf Chimera Modules in Any Python 2.7 Project. Bioinformatics 2018, 34, 1784–1785. 10.1093/bioinformatics/bty021. [DOI] [PubMed] [Google Scholar]
- Betz R.Eigenstate/Vmd-Python: Installable VMD as a Python Module. GitHub. https://github.com/Eigenstate/vmd-python (accessed November 23, 2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.