

Abstract
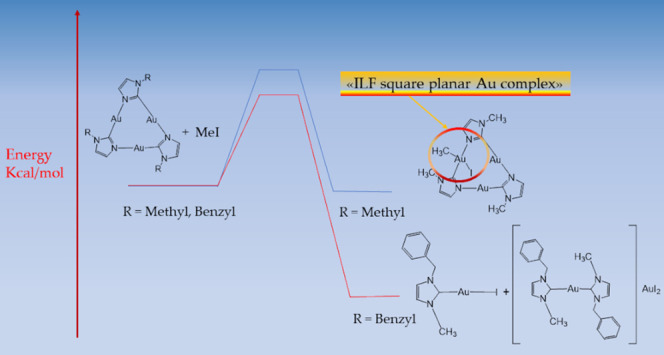
Coinage metal cyclic trinuclear compounds (CTCs) are an emerging class of metal coordination compounds that are valuable for many fine optoelectronic applications, even though the reactivity dependence by the different bridging ligands remains somewhat unclear. In this work, to furnish some hints to unravel the effect of substituents on the chemistry of Au(I) CTCs made of a specific class of bridging ligand, we have considered two imidazolate Au(I) CTCs and the effect of different substituents on the pyrrolic N atoms relative to classic metal oxidations with I2 or by probing electrophilic additions. Experimental suggestions depict a thin borderline between the addition of MeI to the N-methyl or N-benzyl imidazolyl CTCs, which afford the oxidized CTC in the former and the ring opening of the CTC and the formation of carbene species in the latter. Moreover, the reactions with iodine yield to the oxidation of the metal centers for the former and just of a metal center in the latter, even in molar excess of iodine. The analysis of the bond distances in the X-ray crystal structures of the oxidized highlights that Au(III)-C and Au(III)-N bonds are longer than observed for Au(I)–C and Au(I)–N bonds, as formally not expected for Au(III) centers. Computational studies converge on the attribution of these discrepancies to an additional case of inverted ligand field (ILF), which solves the question with a new interpretation of the Au(I)–ligand bonding in the oxidized CTCs, which furnishes a new interpretation of the Au(I)-ligand bonding in the oxidized CTCs, opening a discussion about addition/oxidation reactions. Finally, the theoretical studies outputs depict energy profiles that are compatible with the experimental results obtained in the reaction of the two CTCs toward the addition of I2, MeI, and HCl.
Short abstract
A revisitation of some classic oxidation reactions of gold centers in cyclic trinuclear compounds (CTCs) provides experimental results leading to the opportunity to delineate the effect of imidazole substituents in different outcomes from the reactions of CTCs with I2 or MeI. Moreover, with the match between experimental and theoretical results, a new interpretation of the oxidation states of tetracoordinate gold as cases of inverted ligand field (ILF) is discussed.
Introduction
Cyclic trinuclear complexes (CTCs) containing triangular d10 metal frame may be obtained with a series of angular ditopic anionic bridging ligands in combination with linearly coordinated M(I) cations from Group 11 elements; hence, ligands as pyrazolate (Pz), imidazolate (Im), 1,2,4-triazolate (Trz), pyridinate (Py), and carbeniate (Cb) have been employed to obtain this fascinating class of compounds.1,2 The attention toward these compounds was earlier focused on their elegant structures, which were later discovered to be associated with multipurpose and functional emissive properties.3,4 These observations were accompanied by the study of unique chemical properties due, for example, to π-acid/π-base features promoting them as potential materials for sensing, molecular recognition, in general, for optoelectronics application.4,5
Theoretical studies have shown that both the nature of the central metal and the ligand, as well as of the substituents on the ligands, modulates the acid–base properties of the CTCs, as well as their photophysical characteristics. For example, CTCs with carbeniates and imidazolates as ligands and Au(I) centers as metals feature superior π-basic properties.6 Therefore, gold(I) carbeniate or imidazolate CTCs, after their first preparation in the early 1970s7 and 1980s8 have been reconsidered for novel purposes as the preparation of stacked supramolecular compounds with electrophiles such as metal cations,9 π-acid organic molecules,10,11 or for the formulation of mixed metal Au/Ag or Au/Cu CTCs, whose peculiar emissive properties are strongly dependent on the heteronuclear M–M′ intertrimer interactions.12,13 In this latter work, the reaction between CTCs consisting of imidazole ligands with different alkyl or aryl substituents and Au(I) with a Cu(I) CTC, the [Cu-μ-N,N-3,5-(CF3)2-pyrazolate]3, allowed the formation of mixed-metal CTCs, consisting of Au2Cu or Cu2Au arrangements, depending on the stoichiometry of the reactions. These compounds are obtained with very high yields and they exhibit extraordinary stability in the solid state and strongly emission properties with quantum yields close to unity.13 By considering the acid/base scale build up according to theoretical calculations made by Tekarli,6 the imidazole-based Au(I) CTCs are π-basic while the Cu pyrazole based CTC is π-acid; therefore, the different outcomes of these reactions, yielding π–π adducts or mixed metal CTCs, highlighted that the mechanism is likely influenced by both the electronic and steric effects of the substituents on the imidazolate ligand.13 At a first glance, the steric effects may be considered as the key point but, to gain a deeper insight into the role of the substituents, additional studies are herein performed to highlight the reactivity of the gold centers belonging to the two different CTCs toward common oxidants. Therefore, the reactions of the [μ-C,N-1-methylimidazolate-Au]3, CTCMe, or [μ-C,N-1-benzylimidazolate-Au]3, CTCBz, with methyl iodide (MeI) and iodine (I2) were led. The reactions with halogens or MeI are a well-known method to analyze the tendency of metal centers to be oxidized. Iodine was already used as an oxidant for carbeniate and imidazolate Au(I) CTCs, where a stepwise iodine addition at the three Au(I) centers was observed for the former CTC14,15 but only the addition to one Au(I) center was reached for the benzylimidazolate system.16 On the other hand, the addition of MeI has been never attempted for this class of compounds. Iodine and MeI are different oxidants: Iodine, contrary to the other halogens, does not oxidize directly AuI to AuI3 (redox potential +1.41 V) but it requires a linear LAuI precursor (where L is a phosphane or a carbene);17,18 on the other hand, MeI is reported to activate the oxidation of Au(I) to Au(II) species in dinuclear compounds.19,20 In this work, to grasp the effect of N1-imidazole substituents on the reactivity of Au(I) CTCs, the addition of two classic oxidants as MeI or I2 to the [μ-C,N-1-methylimidazolate-Au]3 (labeled as CTCMe) and to [μ-C,N-1-benzylimidazolate-Au]3 (labeled as CTCBz) compounds were performed (see Scheme 1). The experimental results were compared to the ones reported in the literature and with the data obtained by computational methods.
Scheme 1. Au(I) CTCs with the Labeling of the Imidazole Atoms Quoted in the Manuscript.

The combined experimental/computational analysis revealed the different reactivity of this class of Au(I) CTC with the alternative substrates. In particular, the work not only highlights the non-innocent behavior of the imidazolate ligand but also reverts the electronic description of the formally depicted “Au(III)” square planar complexes by introducing the inverted ligand field (ILF) concept. In this view, a detailed analysis of the electronic structure revealed that the metal maintains the original d10 along the overall reaction pathways.
Results and Discussion
Au(I) cyclotrimers are a class of cyclic gold compounds where a triangular array of Au ions are linked by C,N or N,N bridging ligands. Following the reactions of different cyclotrimers, CTCs, with iodine and MeI are presented and discussed.
The Reactions of CTCMe and CTCBz with Iodine
The reaction of CTCMe with an oxidizing agent as iodine was performed with a molar excess of iodine in CH2Cl2, according to the method already used for CTCBz.16 The evaporation of the reaction mixture yielded a tarry solid, soluble in hot tetrahydrofuran (THF); upon cooling the THF solution, orange-red needles and a few red platelet crystals were obtained, but only the latter were suitable for X-ray crystal diffraction. The elemental analysis of the crystalline solid roughly corresponds to the fully oxidized CTCMeI6, but the separation of the two types of crystals allowed the characterization of both components. The most abundant crystals are the needles and, according to the elemental analysis, were characterized as the fully oxidized CTCMeI6, compound 1, while the platelets match with the partially oxidized CTCMeI4 • THF, where only two Au centers are bound to I atoms (compound 2). The MIR and FIR spectra of compounds 1 and 2 exhibit mild differences in intensity and energies (see Figures S1 and S2 in the Supporting Information), while the characterizations of compounds 1 and 2 in solution were complicated by their low solubility in organic solvents. Compound 1 upon dissolution affords to compound 2 in a large extent (see Figures S3 and S4 in the Supporting Information). This latter exhibits a 1H NMR spectrum with six doublets for the imidazole ligands (ranging between 7.66 and 7.07 ppm) and three signals for the N-methyl groups (3.84, 3.69, and 3.67 ppm). In the 13C NMR spectrum, only one signal was observed for the C2 of the imidazoles at 166.09 ppm, likely for the low intensity of the signals (see Figures S5 and S6 in the Supporting Information); interestingly, these 13C signals differ by <1 ppm from that of the C2 of the starting CTC, found at 167 ppm (see Figure S7 in the Supporting Information). The single-crystal X-ray diffraction (XRD) of the crystal platelets revealed a structure made of the CTCMe unit with two Au centers oxidized by the iodine and a molecule of THF.15 On the other hand, in the case of the reaction of CTCBz with iodine, a very soluble product was obtained in good yield, where only one Au center was bound with the I atoms.16
Computational Studies on the Reaction of CTCMe and CTCBz with Iodine
Although the addition of iodine is a well-known reaction with CTCs, a detailed analysis of the mechanism, an overall energetic/electronic rationalization of the full or partial oxidation of the metal centers, has never been provided. To fill this gap, theoretical calculations defined the mechanism involved in the addition of halogens to these systems and provided some useful hints on the evolution of the electronic structure during the reactivity.
Halogen Molecule Activation
The halogen molecule activation by Au(I) complexes have been already addressed in the literature and, in principle, the process is generally accompanied by the formation of a square planar complex with the coordination number around the metal augmented by two units for the formation of two new metal–I linkages.14−18,21 This, in principle, allows the oxidation of the metal center; thus, the gold achieves a formal oxidation state of +III, two units more than the starting +I, with the two halogen atoms being reduced to halides. To better understand the reactivity of the starting CTCs with diiodine, two molecules of the substrate have been taken into account given the potential involvement of triiodide anions.22 Thus, the first step of the computational analysis was the in silico isolation of an initial adduct between the trinuclear species, namely, CTCMe or CTCBz with the alternative methyl and benzyl substituents at the imidazole rings, and two I2 molecules. In both cases, the formation of the two initial adducts CTCMe*2I2 and CTCBz*2I2, shown in Figures 1a and 1b, is exergonic by −11.8 and −15.1 kcal mol–1, respectively.
Figure 1.

Optimized structure of the adducts: (a) CTCMe*2I2and (b) CTCBz*2I2. The phenyl rings in panel (b) are hidden for the sake of clarity.
Both structures display an already formed Au–I linkage since the original I1–I2 bonding elongates by ca. 0.3 Å, compared to the free I2 molecule. The I2–I3–I4 moiety achieves a linear arrangement, with an angle of 175°. The removal of the triiodide anion leaves a cationic intermediate with a gold center in a T-shape coordination. The formation of the cationic species CTCMeI+ and CTCBzI+, shown in Figure 2, has been estimated to be endergonic by +14.5 and +11.6 kcal mol–1 for the species with methyl and benzyl substituents, respectively. The computational analysis highlights a stepwise addition of I atoms to the Au center(s) with the formation of tricoordinated intermediates.
Figure 2.

Optimized structure of compound: (a) CTCMeI+ and (b) CTCBzI+. The phenyl rings in panel (b) are hidden for the sake of clarity.
The following coordination of the triiodide anion to the Au1 center, in trans position to I1, provides the square planar complex featuring two Au–I linkages, compound CTCMeI2 and CTCBzI2, with a free energy gain of −24.1 and −21.8 kcal mol–1, respectively. The overall calculated free energy associated to the reactivity between the isolated starting CTC and the first I2 molecule, up to the CTCI2, featuring a square planar complex, has been estimated to be −21.4 and −25.4 kcal mol–1 for the CTCMeI2 and CTCBzI2, respectively. The calculated structural CTCBzI2 data is well fitted with the experimental crystal structure reported in the literature.16
Further reactivity of the compounds CTCI2 with I2 molecules evolves similarly, although the cationic intermediates along the pathway feature bridging I between two adjacent Au centers with a slight positive NBO charge of +0.18. Once again, the freed triiodide can perform the attack to a metal center, providing the system with four iodide ligands, two for each metal center. A reasonable explanation of the different reactivity between methyl and benzyl substituents is related to a somewhat hindered attack of the triiodide moiety to the gold in the system with benzyl, while no steric constraints are encountered in the methyl case. The overall process of the activation of three I2 molecules with the consequent achievement of three square planar gold centers for the CTCMeI6, also labeled as compound 1 (overall free-energy gain of −51.2 kcal mol–1) is reported in detail in the SI (section 4.1, as well as summarized in Scheme S1 in the Supporting Information).
As aforementioned, traditionally, the activation of a halogen molecule by a transition-metal complex has been accepted to be accompanied by an increasing oxidation state by two units, thus, precedent publications assigned a formal +III oxidation state to the Au center in compounds related to the square planar CTCI2, shown in Figure 3a.
Figure 3.

Optimized structure of compound: (a) CTCBzI2 and (b) the lowest unoccupied molecular orbital (LUMO) of CTCI2 compounds.
According to the classic ligand field theory (LFT) description,23,24 the bonding in a square planar complex is assured by four electronic donations from populated combinations of the ligands into suitable empty metal orbitals. The classical LFT description is based on the assumption that the ligand-centered combinations lie lower in energy than those of the metal. Thus, for energy reasons, in a square planar d8 complex, the dx2–y2 metal orbital should be empty and the lowest unoccupied molecular orbital (LUMO) (antibonding feature), or one of the closer in energy molecular orbitals (MOs) of the system, should be an antibonding σ combination between the empty metal dx2–y2 and ligand’s orbitals with a stronger contribution from the metal. The corresponding bonding combination is filled at very low energy and mainly centered on the ligands. Some years ago, computational/experimental analysis of the electronic structure of a square planar complex of Cu“(III)”, [Cu(CF3)4]−,25,26 highlighted a reverse situation being one combination of the ligands empty and at higher in energy than the d-orbital; this suggests that the dx2–y2 metal orbital is populated and the metal electronic configuration could be better described as d10 more than a d8. The idea of Inverted Ligand Field (ILF) purposes an alternative bonding pattern in a square planar complex with three ligands to metal donations and the fourth interaction interpreted as a metal-to-ligands σ–donation. The presence of an empty ligand-centered combination influences the overall electronic structure with the LUMO now mainly centered on the ligands rather than on the metal. This description perfectly agrees with the bonding pattern in CTCI2 without any substantial variation due to the nature of the substituent at the imidazole rings. In this perspective, the LUMO, shown in Figure 3b, has a small contribution from the metal (28.5%) and 71.5% from the ligands, and 53.5% of this latter component comes from the iodide moieties. Thus, a formal +1 oxidation state could be reasonably assigned to the Au1 center, as well as in the Au2 and Au3 and the ILF description may be applied to all the products at any degree of iodinization, as well as to all tricoordinated intermediates encountered along the reaction pathway.
The ILF occurrence is also confirmed by the detailed NBO population analysis on compound CTCI2, revealing no significant difference in d-population of Au1, compared to the Au2 or Au3 center, with 9.42 d electrons associated with Au1, only 0.18 e– less than those at Au2 or Au3, once again confirming the ILF occurrence.
Another confirmation of the ILF could be found through structural comparison between the experimental/optimized structures of the starting compound and the product after the substrate activation. In this regard, the X-ray structures did not point out a shortening of the metal–ligand distances, as expected by an oxidative process. The occurrence of the ILF might be imputed to the specific reaction with I2 molecules given the very close electronegativity of the gold and iodine (2.54 vs. 2.66). In this regard, a computational analysis on the simple square planar [AuCl4]− anion revealed a similar behavior with the LUMO being an antibonding orbital mainly localized on the four Cl atoms (71%) and only 29% from the Au atoms. A reverse situation occurs for the σ-bonding counterpart (HOMO–15) well stabilized in energy and with a strong contribution (ca. 68%) from the metal. This allows concluding that the ILF is ubiquitous in the chemistry of square planar gold complexes also when associated with more electronegative elements, such as chlorine.
The Reactions of CTCMe and CTCBz with MeI
Afterward, the reactions of CTCMe and CTCBz with MeI were performed under mild experimental conditions, using MeI as the solvent and as the reactant. Methyl iodide readily reacts with several types of Au(I) compounds17−22 and recently it has been proven that MeI can oxidize gold metal to form the [CH3AuI] derivative.27 In practice, an excess of MeI was added to the solid Au(I) cyclotrimers upon magnetic stirring at room temperature under an inert atmosphere. After the dissolution of the CTCMe or CTCBz in MeI, the reaction mixtures were monitored by spectroscopies during the time. In the case of CTCMe, the starting cyclotrimer was largely persistent in the reaction mixture, even after 1 day of stirring; in fact, by monitoring the reaction mixture between CTCMe and MeI by 1H NMR spectroscopy, recording the appearance of the signal at 1.78 ppm attributed to a methyl bound to the Au(I),19 compound 3 was present at 5%–8%, with respect to the starting CTCMe after 3–5 h of mixing. However, the isolation of compound 3 as yellow crystals was obtained with a yield of 36% after 1 week at 5 °C by adding hexane to the reaction mixture. The crystals of compound 3 are not emissive upon irradiation at 366 nm, they exhibit rather good bench stability, which becomes low once in CDCl3 solution, with consequent return of the starting cyclotrimer and MeI in large extent (see Figure S8 in the Supporting Information). The 1H NMR and 13C NMR spectra of the mixture generated upon dissolution of compound 3 in CDCl3 are reported in Figures S8 and S9 in the Supporting Information. However, in the 13C NMR spectrum, three peaks were found in the region 160–170 ppm: the most intense due to CTCMe (168 ppm) and two small signals at 165 and 167 ppm, which might be tentatively assigned to compound 3 (Figure S9). The solid-state characterization by elemental analysis and IR spectra (see Figure S10 in the Supporting Information) support the evidence that compound 3 is made of the CTCMe and MeI, but the real nature of compound 3 was confirmed only after the results of the XRD analysis on the single crystals (see below).
The reactivity between CTCMe with different substrates is depicted in Scheme 2.
Scheme 2. Schematic View of the Reactions of CTCMe with (a) an Excess of Iodine, (b) an Excess of MeI, and the Corresponding Products.

In the case of the reaction of CTCBz and MeI, the monitoring of the reaction revealed the ready appearance of two new compounds, in addition to the starting cyclotrimer, one presumably being ionic. The crystallization of the reaction mixture of MeI and CTCBz provided a microcrystalline solid, intensively glowing in the yellow upon irradiation at 366 nm, consisting predominantly of two phases. The 1H NMR spectrum shows two sets of signals in a ratio that are dependent on the solvent used for the NMR (see Figure S11 in the Supporting Information). In contrast to what was observed for compound 3, the microcrystalline solid was not completely soluble in CDCl3. The 1H NMR spectrum recorded in DMSO-d6 exhibits only two signals for the N–CH3, at 3.78 and 3.81 ppm in a 2:1 integral ratio. The striking feature useful for the attribution of these peaks was rendered by the 13C NMR spectrum (see Figure S12 in the Supporting Information); in fact, it displays two sets of signals in a 2:1 ratio at 181 and 184 ppm, attributable to the C atoms in position 2 of the imidazole in a typical range of chemical shifts for carbene species.28,29 The elemental analysis of the microcrystalline solid was interpreted for a mixture of a monocarbene-gold(I) compound (4) and a bis-carbene-gold(I) compound (5) in a 1:1 ratio (see Scheme 3, path a). The FIR spectrum, recorded in the microcrystalline solid, displays an intense band at 199.95 cm–1, which was attributed to the antisymmetric vibrational mode (ν3) of the AuI2– anion in the microcrystalline solid, confirming the presence of this counterion (see Figure S13 in the Supporting Information).30
Scheme 3. Schematic View of the Reactions of CTCBz with (a) 3 mol of Methyl Iodide, R = Me and X = I (Compound 4), R = Me and X = AuI2 (Compound 5) and (b) 3 mol of Iodine.

Data taken from ref (16).
The formation of carbene species from the reaction of CTCBz with acyl or alkyl halides16,31 and with HCl is already known. Regarding the reaction with HCl, it was reported the formation of the cationic bis(l-benzylimidazolyl-2-yl)gold(I) chloride, by the treatment of the likely 1-benzyl-imidazolate-2yl-gold-triphenylphosphane derivative with an aqueous solution of HCl.31 Also, the straight reaction of CTCMe or CTCBz with an aqueous solution of HCl results in the formation of carbene species (section 1.1 in the Supporting Information).
In the attempt to understand the different reactivity attained with the two starting cyclotrimers, the 13C NMR data were considered. Table 1 reports the 13C NMR chemical shifts for the C2 of the compounds 1–5, compared with the respective starting cyclotrimers and carbene species (see section 1.1 in the Supporting Information). Compound 6, [(ImMe-2yl)2Au]Cl (where ImMe is the 1-methyl-2yl-imidazole), displays a 13C NMR signal for the C2 atom at 167 ppm in CDCl3, which is in the range reported for dialkyl-NHC mono- or bis-carbene gold(I) derivatives,28 and for the C2 of the corresponding CTCMe (167 ppm), while the C2 of the free 1-methyl imidazole falls at 137 ppm in CDCl3; the C2 of compound [(ImBz-2yl)2Au]Cl falls at 180 ppm in DMSO-d6,8 which is in the same range of those reported for compounds 4 and 5 and at rather higher frequencies, if compared to that of the starting CTCBz (167 ppm, while the free 1-benzyl imidazole falls at 137 ppm in CDCl3). Finally, although shifts of 30 ppm are observed for the 13C NMR signals of the C2 atoms upon cyclization and formation of the corresponding CTCs, only the C2 of the 1-benzylimidazole carbene species is additionally shifted to higher frequencies (180 ppm) after the formation of the carbene compound (see Table 1). Generally, the 13C NMR signal for the C2 atom in NHC-carbene compounds moves toward high frequencies to an extent that is dependent on the Lewis acidity of the metal center and on the ability of the positively polarized C2 to withdraw π-electron density from the imidazole ring.32 Hence, by considering the herein compounds the 13C chemical shift in compound 7, [(ImBz-2yl)2Au]Cl (where ImBz = 1-benzyl-2yl-imidazole), indicates that the C2 is slightly richer in electron density than those of compounds 4 and 5, classified as carbenes.8 Moreover, the electron density of C2 in compound 6 seems to be comparable to that of the corresponding C2 in CTCMe; in fact, the 13C signals fall at 167 ppm for both compounds: the carbene 6 and the CTCMe. From these data, the 1-methylimidazole seems to supply more electron density on the C2 and, thus, on the gold center, conversely, the 1-benzyl-imidazole provides larger stability to the corresponding carbene gold(I) complexes, likely as an effect of the larger aromatic delocalization, despite a lower electron density on the metal center.
Table 1. 13C NMR Chemical Shifts for the C2-Imidazole for Compounds 1–7 and Relative Reference Compoundsa.
| compound | 13C chemical shift (ppm, DMSO-d6) |
|---|---|
| ImMe | 137b |
| ImBz | 137b |
| CTCMe | 167,b 168c |
| CTCBz | 167b |
| compound 1, CTCMeI6 | 166 |
| compound 2, CTCMeI4 | 166 |
| compound 3, CTCMe-MeI | 165, 167c |
| compound 4, ImBz-2yl-AuI | 184 |
| compound 5, [(ImBz-2yl)2Au]AuI2 | 181 |
| compound 6, [(ImMe-2yl)2Au]Cl | 167d |
| compound 7, [(ImBz-2yl)2Au]Cl | 180b |
Legend of compounds: ImMe = 1 methyl-imidazole; ImBz = 1-benzyl-imidazole; CTCMe = [μ–Au-C2,N3-1-methyl-imidazolate]3; and CTCBz = [μ–Au-C2,N3-1-benzyl-imidazolate]3.
Data taken from ref (8).
Recorded in CDCl3 solution,
See electronic Supporting Information.
Moreover, Table 1 highlights only one signal for the C2 for the imidazoles in 2, even though in compound 2, two different chemical environments are expected: one for the carbon attached to the square planar Au, N–AuI2-C, and another for the linear N–Au–C; actually, this evidence might be explained right as a consequence of the ILF, as the electronic populations in both Au centers are similar, as discussed above.
Computational Studies on the Reaction of Methyl Iodide with CTCMe and CTCBz
The reactivity between the initial CTC and CH3I has highlighted the great influence of the substituents at the imidazole ring, since, with the CTCBz, the main products are the monocarbene and bis-carbene species. Otherwise, the reaction with CTCMe partially evolves to compound 3 featuring a Au center in a square planar arrangement bonded to the methyl and the iodide. The dissolution of the crystals of 3, with the reformation of the starting CTCMe, underlines the instability of the square planar system in solution. In view to provide a reasonable overview of the electronic factors ruling the reactivity with the methyl iodide, a computational analysis has been conducted, similarly to the precedent I2 case.
Methyl Iodide Activation
All the computational efforts to activate the CH3I molecule by the Au center, maintaining the cyclic structure of the CTC, failed, since all the relaxed scans, obtained via stepwise shortening of the Au–C or Au–I distances, provided no reasonable results or too high an energy barrier being higher than 40 kcal mol–1. Alternatively, the computational analysis revealed the non-innocent behavior of the imidazole in the activation of the methyl iodide, because of the presence near to the frontier of populated delocalized π-system of the imidazole able to interact with the incoming methyl iodide for the potential formation of a new N–C linkage. This is confirmed by the detection of a transition state, 8BzTS, shown in Figure 4a, featuring the initial formation of the N3–C bonding and the cleavage of the H3C···I one, being as large as 2.90 Å, 0.7 Å longer than in the pristine free substrate. This allows also the initial cleavage of the Au1–N bonding with a ca. 0.2 Å elongation. The associated free-energy barrier for the achievement of 8BzTS is +21.2 kcal mol–1, featuring a planarization of the transferred methyl group (I–C–H angle being 95.7°), in some way, the aromatic electrophilic substitution. The elongation of the Au–N linkage by ca. 0.2 Å associates with a redistribution of the electronic density and the formation of a carbene ligand bonding the Au2 center.
Figure 4.

Optimized structures of (a) transition state 8BzTS and (b) intermediate 8Bz.
After the transition state 8BzTS, compound 8Bz is obtained, featuring the new N–C bond and the N–Au1 one with the unsaturation at the Au1 immediately compensated by the iodide coordination. The overall free-energy gain, associated with the formation of 8Bz from 8BzTS, has been estimated to be as large as −44.5 kcal mol–1, double than the energy barrier required for the achievement 8BzTS. Once a side of the cycle is broken, the system may evolve toward the formation of three monocarbene Au(I) compounds 4, by stepwise cleaving the still-present Au–N linkages. In particular, the interaction between 8Bz with a second CH3I molecule evolves through the formation of a transition state, namely, 9BzTS (see Figure 5a) with a free-energy barrier of +23.7 kcal mol–1 after that the formation of a carbene/iodide linear gold(I) complex 4 occurs together with a dinuclear species 9Bz, shown in Figure 5b with a free-energy gain of −31.9 kcal mol–1.
Figure 5.

Optimized structures of (a) transition state 9BzTS and (b) dinuclear intermediate 9Bz.
The dinuclear unit 9Bz reacts with a third methyl iodide molecule and, after bypassing a free-energy barrier of 23.2 kcal mol–1 associated with the transition state 4TS, two new units of 4 are exergonically formed (−33.1 kcal mol–1). An overall free energy of −41.4 kcal mol–1 has been estimated for the formation of three isolated molecules of 4. The overall reaction pathway for the reaction between CTCBz and CH3I up to three molecules of 4 is summarized in Figure 6a.
Figure 6.

Energy diagrams showing the energy profiles for the proposed mechanisms for the electrophilic addition of MeI additions to (a) CTCBz and (b) CTCMe. The red pathway in panel (b) has been dismissed, given the too-high energy barrier.
A reasonable explanation for the experimental obtainment of a neutral compound 4 and the ionic pair compound [Au(1-benzyl-3-methyl-2-imidazolyl-2yl)2]AuI2, namely 5, is also provided. In this regard, similarly to the recent work of Gust, Podewitz et al.,33 a scrambling ligand mechanism may occur involving the interaction between two molecules of 4. In particular, compound 5 is originated with a total free energy gain of −13.8 kcal mol–1, after bypassing a free-energy barrier of 20.6 kcal mol–1. The pairing of two linear complexes of 4 through weak aurophilic interactions has been estimated to be exergonic by −12.5 kcal mol–1 with a calculated Au···Au distance of 3.70 Å.
Concerning the reaction between the CTCMe and CH3I, the experiments have been revealed the formation of a very small amount of compound 3 featuring square planar coordination around one Au center due to the formation of two new bonds with methyl and an iodide moiety. The computational analysis of the reactivity between CTCMe and CH3I highlights a striking difference with the benzyl analogue CTCBz, given the too-high free-energy barrier for the achievement of the transition state, 10MeTS (32 kcal mol–1), red pathway in Figure 6b. Other reaction processes have been also explored by maintaining the cyclic planar structure of the CTC, but all pathways have been discarded given the high energy barriers. In particular, a relaxed scan for the extraction of the methyl group directly by the involved metal center through an SN2-type reaction has highlighted an energy barrier of >30 kcal mol–1, consistent with some already reported pathways requiring some drastic conditions, such as high temperature.34 The only remaining reasonable process should involve the cleavage or at least the weakening of the Au–N linkage, followed by the interaction between the metal center and the incoming methyl iodide through the iodine. Also, this process is not straightforward being the formation of compound 11Me, shown in Figure S14 in the Supporting Information, endergonic by 26.3 kcal mol–1 compared to the isolated CTCMe and CH3I. The high free-energy cost is due to the cleavage of the Au–N bonding only slightly compensated by the interaction between the gold and iodine. Possibly, intermediate 11Me may evolve to the final compound 3 after the achievement of the T-shape compound 12Me, shown in Figure S15 in the Supporting Information, after the splitting of the C–I bonding. Compound 12Me is formed with a free-energy gain of −16.4 kcal mol–1 and then the system evolves toward the final compound 3 featuring square planar coordination around one Au center. The last process must involve an isomerization from the cis configuration to the trans one in 3. The overall reaction from CTCMe and the CH3I up to compound 3 has been estimated to be endergonic by +0.4 kcal mol–1 and the presence of high barriers along the pathway is consistent with the very low yield of the reaction of ca. 8% after 5 h in solution up to a maximum of 36% after a week as a solid.
Similar to the case of CTCI2, also in compound 3, the bonding pattern could be reasonably explained in the light of ILF theory. The LUMO, shown in Figure 7, is mainly localized on the ligands rather than on the metal (Au contribution is 34% vs. 66% from the ligands). Thus, also, in this case, it is reasonable to assign a d10 configuration to the metal rather than the expected d8.
Figure 7.

Plot of the LUMO orbital of compound 3.
As already described above, compound 3 displays experimental features needing an interpretation such as the dissociation of crystals of 3 to the MeI and the parent CTC in CHCl3 solution. The computational analysis revealed that the dissociation of the crystal to reform CTC could not be explained with a mechanism involving the ring opening. In this regard, ad-hoc relaxed scans, performed via stepwise weakening of the Au–N bonding, revealed too-high energy barriers (>25 kcal mol–1). Otherwise, the iodide ligand seems to be more easily displaced (energy cost of ca. 15 kcal mol–1), and, once freed, may move toward the methyl ligand for a nucleophilic attack, restoring the methyl iodide. Such a process is confirmed by a slight positive charge on the methyl ligand, with the calculated NBO charge being +0.17.35
X-ray Crystal Structure Description of Compounds 2 and 3
The ORTEP style view of compounds 2 and 3 are reported in Figures 8 and 9, respectively, together with their atomic labeling schemes. The most significant bond distances and angles are reported in the caption of the figures, while the crystal data are reported in Table 2.
Figure 8.

ORTEP style view of compound 2. Ellipsoids are drawn at their 50% probability level. Hydrogen atoms and THF solvent molecule are omitted for the sake of clarity. Selected bond distances: C1–N1, 1.338(5) Å; C1–Au2, 2.006(3) Å; C2–N3, 1.350(4) Å; C2–Au3, 1.989(3) Å; C3–N5, 1.332(4) Å; C3–Au1, 2.005(3) Å; Au1–N1, 2.070(3) Å; Au1–I2, 2.6121(3) Å; Au1–I1, 2.6244(3) Å; Au2–N3, 2.051(3) Å; Au2–I3, 2.6119(3) Å; Au2–I4, 2.6235(3) Å; Au3–N5, 2.055(3) Å. Selected bond angles: N1–C1–Au2, 124.4(2)°; N3–C2–Au3, 123.1(2)°; N5–C3–Au1, 120.9(2)°; C3–Au1–N1, 172.12(13)°; I2–Au1–I1, 165.056(10)°; C1–Au2–N3, 174.39(13)°; I3–Au2–I4, 170.404(10)°; C2–Au3–N5, 174.87(12)°; C1–N1–Au1, 122.6(2)°; C2–N3–Au2, 120.0(2)°; C3–N5–Au3, 124.2(2)°.
Figure 9.
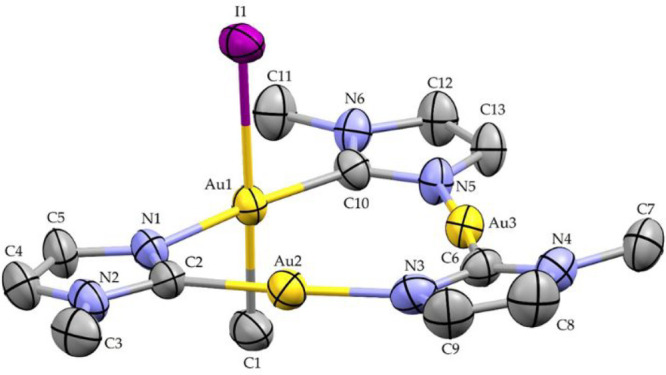
ORTEP style view of compound 3. Ellipsoids are drawn at their 50% probability level. Hydrogen atoms are omitted for the sake of clarity. Selected bond distances: C1–Au1, 2.151(8) Å; C2–N1, 1.349(10) Å; C2–Au2, 1.977(8) Å; C6–N3, 1.352(10) Å; C6–Au3, 1.984(7) Å; C10–N5, 1.340(9) Å; C10–Au1, 1.994(6) Å; Au1–N1, 2.046(6) Å; Au1–I1, 2.6957(6) Å; Au2–N3, 2.034(7) Å; Au3–N5, 2.036(6) Å. Selected bond angles (deg): C10–Au1–N1, 175.3(3)°; C1–Au1–I1, 175.7(3)°; C2–Au2–N3, 175.3(3)°; C6–Au3–N5, 174.7(3)°; C2–N1–Au1, 120.0(5)°; C6–N3–Au2, 122.2(5)°; C10–N5–Au3, 121.1(5)°; N1–C2–Au2, 123.6(5)°; N3–C6–Au3, 122.7(5)°; and N5–C10–Au1, 123.2(5)°.
Table 2. Crystal Data for Compounds 2 and 3.
| compound 2 · THF | compound 3 | |
|---|---|---|
| empirical formula | C12H15Au3I4N6·C4H8O | C13H18Au3IN6 |
| formula weight | 1413.90 | 976.14 |
| temperature | 200 K | 250 K |
| wavelength | 0.71073 Å | 0.71073 Å |
| crystal system | monoclinic | monoclinic |
| space group | P21/n | P21/c |
| Hall group | –P2yn | –P2ybc |
| unit-cell dimensions | a = 12.3839(4) Å | a = 13.7696(9) Å |
| b = 14.7473(4) Å | b = 9.6146(6) Å | |
| c = 15.7657(5) Å | c = 14.6662(10) Å | |
| β = 107.066(1)° | β = 95.941(2)° | |
| volume | 2752.49(15) Å3 | 1931.2(2) Å3 |
| Z | 4 | 4 |
| Density (calculated) | 3.412 g/cm3 | 3.357 g/cm3 |
| absorption coefficient, μ | 20.457 mm–1 | 24.339 mm–1 |
| F(000), F(000′) | 2472.0, 2443.69 | 1712.0, 1689.36 |
| Tmin, Tmax | 0.4586, 0.7460 | 0.546, 0.746 |
| Theta(max), data completeness | 30.087, 0.998 | 29.257, 0.999 |
| R (reflections) | 0.0184 (7785) | 0.0314(4666) |
| wR2 (reflections) | 0.0434 (8079) | 0.0877(5250) |
| S | 1.008 | 1.006 |
| Npar | 289 | 208 |
The molecular structure of compound 2 displays a skeletal core analogous to that of compound 3. Compound 2 consists of a trinuclear gold cyclic molecule, in which three imidazolate moieties bridge three metal centers through C and N atoms, forming an almost planar nine-membered ring. In compound 2, the nine-membered ring is deviated from planarity, especially atoms C2 and N3, which are at a distance of 0.262(2) and 0.187(2) Å, respectively, from the plane defined by the three gold atoms Au1, Au2, and Au3. Similar to compound 3, the imidazole rings also are sloped, with respect to the gold plane, with dihedral angles ranging from 7° to 15°. The Au(III) atoms are in a distorted square planar coordination environment. The I–Au–I bond angles significantly deviate from linearity with values of 165.06(1)° and 170.40(1)°. The Au···Au separations are 3.470(1), 3.532(1), and 3.594(1) Å, with the longer distance belonging to the oxidized Au(III) atoms and no aurophilic interactions, are observed. Noteworthy, regardless of what was observed for analogues, the crystal packing of compound 2 exhibits rather short intermolecular I···I interactions at 3.593 and 3.887 Å, resulting in the pairing of two by two molecules to form an extended chain of I···I–Au–I···I–Au–I···I· interactions with Au–I distances of 2.624 and 2.612 Å for the two Au–I bonds (see Figure S17 in the Supporting Information). This evidence may explain the lower solubility recorded for compound 2, with respect to the similar compounds reported in Table 3 and prepared by Balch.
Table 3. Selected Au–N and Au–C Bond Distances for Pristine CTCs Obtained with Different Ligandsa and Au–N and Au–C Bond Distances of the Relative Products Obtained by the Reaction of the CTCs with Iodine.
| CTCBzI2b | CTCcarbI2c | CTCcarbI4c | CTCcarbI6c | CTCMeI4 | |
|---|---|---|---|---|---|
| Au(III)–N | 1.913 (Au1–I) | 2.052 (Au1–I) | 2.072 (Au1–I) | 2.082 (Au1–I) | 2.051 (Au1–I) |
| Au(I)–N | 2.013 (Au2) | 2.072 (Au2) | 2.095 (Au2–I) | 2.086 (Au2–I) | 2.070 (Au2–I) |
| Au(I)–N | 2.063 (Au3) | 2.052 (Au3) | 2.012 (Au3) | 2.074 (Au3–I) | 2.054 (Au3) |
| Au(III)–C | 1.964 (Au1–I) | 2.032 (Au1–I) | 2.022 (Au1–I) | 2.032 (Au1–I) | 2.006 (Au1–I) |
| Au(I)–C | 2.024 (Au2) | 2.022 (Au2) | 2.012 (Au2–I) | 2.000 (Au2–I) | 2.004 (Au2–I) |
| Au(I)–C | 2.005 (Au3) | 1.972 (Au3) | 2.065 (Au3) | 2.065 (Au3–I) | 1.989 (Au3) |
| CTCBzd | CTCcarb | CTCMe | |||
| Au(I)–N | 2.042 | 2.032 | nde | ||
| 2.051 | |||||
| 2.036 | |||||
| Au(I)–C | 1.978 | 2.000 | nde | ||
| 1.998 | |||||
| 2.003 |
The core of the molecule in compound 3 resembles that of compounds already described.16,36,37 The two Au(I) atoms, namely Au2 and Au3, are at a distance of 3.507(2) Å, which is longer than those observed in the parent 9-membered ring reported in the literature [3.349 Å mean value found in the CCDC 2020 database]. Au2 and Au3 both show a nearly linear coordination environment, with C–Au–N bond angles of 175.3(3)° and 174.7(3)°, respectively, while the Au1 atom, displaying the bonding with the methyl and the iodine ligands, features square planar coordination. The Au–I and Au–C1 bond distances are 2.6957(6) and 2.151(8) Å, respectively. The Au–I bond distance is slightly longer than those observed in the derivatives obtained by the addition of iodine to CTCs.16,2 In compound 3, the intramolecular Au···Au separations are 3.471(1), 3.489(2), and 3.507 Å, respectively, with the longer distance belonging to the centers not involved in the bonding with the methyl and the iodine, the Au2···Au3, indicating a scalene triangular metal frame and no aurophilic interactions. The maximum deviation from the mean plane of the coordination around Au1 is observed for C1 atom [0.089(1) Å]. The 9-membered ring is almost planar. The dihedral angle between this mean plane and the coordination plane around Au1 is 85.36(1)°. The three imidazolate rings are slightly sloped, with respect to the mean plane of the 9-membered ring. The widest dihedral angle is observed for the imidazolate unit bridging the two Au(I) atoms: the dihedral angle between the C6,N3,C9,C8,N4 unit and the mean plane of the 9-membered ring is 7.04(1)°. Remarkably, the crystal packing exhibits neither aurophilic nor halogen interactions and it is built up through normal van der Waals interactions (see Figure S18 in the Supporting Information). The Au···Au separation between adjacent molecules is a minimum of 4.980(1) Å.
Notably, in compound 3, the Au1–C10 and Au1–N1 bond distances (1.994(6) Å and 2.046(6) Å, respectively) are slightly longer than those observed for the other two metal centers not connected in the Me–Au-I unit: the Au2–C2 and Au2–N3 bond distances are 1.977(8) and 2.034(7) Å, respectively, while the Au3–C6 and Au3–N5 distances are 1.984(7) Å and 2.036(6)Å, respectively. This trend of bond distances is also adopted in the products obtained by the stepwise oxidation by iodine of the N-methyl-C-methoxy-carbeniate gold(I) CTC,14 and it was found also in the case of the product obtained from the reaction of iodine with the 1-benzylimidazolate gold(I) CTC.16 Some representative Au–C and Au–N bond lengths of these compounds fully or partially oxidized with iodine CTCs are shown in Table 3. For compound 3, the crystal data of the starting cyclotrimer are not available, but by analyzing the bond lengths reported in Table 3, it indicates that the addition of iodine to the Au centers does not afford a shortening of the Au–C and Au–N bond distances, since it would be expected as a consequence of the oxidation of Au centers from a formal +1 oxidation state to a +3 oxidation state and the relative size contraction.
Conclusions
The reactivity of two gold CTCs, consisting of Au(I) centers and imidazole as bridging ligands having methyl or benzyl substituents at the N1 of the imidazole, has been considered toward probing reactants such as iodine, methyl iodide, and hydrochloric acid. Despite the close similarity of these starting CTCs systems, different experimental outcomes have been obtained. The results can be summarized as (i) the breakage of the cycles and the formation of monoligated or bis-ligated NHC Au(I) structures and (ii) the obtaining of CTC units with square planar tetra-coordinated Au centers. Remarkably, relative to the reaction of CTCs with MeI, different outcomes were attained: although in the case of the methyl-imidazole CTC, the square planar Au center slowly forms, for the benzyl imidazole CTC, the mono- and bis-NHC-gold(I) carbene structures are the only compounds formed and isolated. Following the same experimental conditions, only the presence of the two different substituents differentiates the outcomes. Theoretical studies provided essential help to unravel the enigma fixing the substituents at the N1 of the imidazole, as the activator or less of the other nitrogen, the N3, affording to two alternative mechanisms of addition: by the metal activation or by the π-aromatic imidazole interaction.
Although fully worth it, these are not the only remarkable results obtained from this work. Beyond the comparison of the 13C NMR imidazole C2 signal chemical shifts, whose interpretation is not straightforward, the analysis of the experimental Au–N and Au–C bond distances in the crystal structure of compounds 2 and 3 highlights very slight bond length contractions for the square planar Au centers, if compared to those of the bicoordinated Au centers. Computational calculations on these products attained electronic populations that are closer to d10 than to d8 configurations, opening a controversial issue on the oxidation of the Au centers.
An appropriate description of the bonding in this family of compounds could be obtained by evoking the ILF theory.25 This model reverts the theoretical optics and inverts the starting energy of the ligands concerning the central metal since the four ligands act as a 6e– donor rather than 8e– to the gold, as occurs in compound 2 or 3. In this regard, the fourth metal–ligand interaction could be better depicted as σ donation from the metal to the ligands. Thus, the metal constantly maintains unchanged its starting oxidation state of +1 never attains the classically expected Au(III). Even though the oxidation number is a formal assumption, in the case of CTCs, this new interpretation revises the reactivity toward oxidants of the linear gold(I) complexes in the perspective of the ILF theory.
Experimental Section
Materials
High-performance liquid chromatography (HPLC)-grade solvents, imidazoles, iodine (99%), and MeI (99%) were purchased from vendors. The reactions were led upon a flow of nitrogen and using dried solvents. The cyclotrimers were obtained by adding solid Ph3PAuCl to a −40 °C THF solution of the corresponding lithium imidazolate salt, followed by stirring, washing with water, with hexane, and crystallizing the raw product from CH2Cl2/hexane, following the procedure reported by Bonati et al.8
Crystals of HAuCl4·nH2O were obtained by storing, at 4 °C, a highly concentrated watery solution obtained by dissolving a chip of gold foil in aqua regia. The Ph3PAuCl was recovered as a microcrystalline solid from a suspension obtained by adding a double amount of PPh3 to a solution of HAuCl4 in ethanol.
Characterization
Elemental analyses (C, H, N, S) were performed in-house with a Fisons Instruments Model 1108 CHNS-O elemental analyzer. Melting points were obtained using a Model SMP3 Stuart Scientific instrument. IR spectra were recorded from 4000 cm–1 to 600 cm–1 with a PerkinElmer SPECTRUM ONE System FT-IR instrument. The following IR annotations were used: br = broad, m = medium, s = strong, sh = shoulder, vs = very strong, w = weak and vw = very weak. 1H and 13C NMR spectra were recorded on an Oxford-400 Varian spectrometer (400.4 MHz for 1H and 100 MHz for 13C). Chemical shifts, in ppm, for 1H and 13C NMR spectra are relative to internal Me4Si. The following NMR annotations were used: br = broad, d = doublet, dd = double doublet, t = triplet, m = multiplet, s = singlet. Electrospray mass spectra (ESI-MS) were obtained in positive- or negative-ion mode on a Series 1100 MSD detector HP spectrometer, using an acetonitrile or methanol mobile phase. The compounds were added to reagent-grade acetonitrile to give solutions of an approximate concentration of 0.1 mM. These solutions were injected (1 μL) into the spectrometer via an HPLC HP 1090 Series II system that was fitted with an autosampler. The pump delivered the solutions to the mass spectrometer source at a flow rate of 300 μL min–1, and nitrogen was employed both as a drying gas and a nebulizing gas. Capillary voltages were typically 4000 and 3500 V for the positive- and negative-ion mode, respectively. Confirmation of all major species in this ESI-MS study was aided by a comparison of the observed and predicted isotope distribution patterns, the latter of which was calculated using the IsoPro 3.0 computer program.
X-ray Structural Determination
The crystallographic data for compounds 2 and 3 were obtained by mounting a single crystal on glass fiber and transferring it to an APEX II Bruker CCD diffractometer. The APEX 3 program package38 was used to obtain the unit-cell geometrical parameters and for the data collection (30 s per frame scan time for a sphere of diffraction data). The raw frame data were processed using SAINT38 and SADABS39 to obtain the data file of the reflections. The structure was solved using SHELXT39 (intrinsic phasing method in the APEX 3 program). The refinement of the structures (based on F2 by full-matrix least-squares techniques) was performed using the SHELXTL-2014/7 program40 in the WinGX suite v.20142020.1.41 The H atoms were introduced in the refinement in defined geometry and refined “riding” on the corresponding carbon atoms. Crystallographic data were deposited with the Cambridge Crystallographic Data Centre as supplementary publication (CCDC reference code 2093399 for compound 2 and 2093397 for compound 3). Copies of the data can be obtained free of charge on application to the CCDC, 12 Union Road, Cambridge CB2 1EZ, U.K. (fax, (+44) 1223 336033; e-mail, deposit@ccdc.cam.ac.uk).
Computational Details
All the compounds were optimized at the DFT-B97D42 level of theory within the Gaussian16 package.43 All of the calculations were based on the CPCM model44 for the dichloromethane or iodomethane as the solvent, depending on the experimental conditions. The Triple Zeta basis set TZVP45 was used for all the atomic species, except for the Au and I atoms, for which the Stuttgart/Dresden (SDD) pseudo-potential46 was employed. All the optimized structures were validated as minima and/or transition states by computed vibrational frequencies. The contribution of each center to the molecular orbitals was estimated by using the AOMIX package.47 Cartesian coordinates, as well as the energetic features of all of the optimized structures, are reported in section 4 in the Supporting Information.
Reaction of [μ–Au-C2,N3-1-methylimidazolate]3 with Solid Iodine. Preparation of Compound 1 and 2
The [Au(μ-C2,N3-1-methyl-imidazolate)]3 (30 mg; 0.036 mmol) was dissolved in 2 mL of dry CH2Cl2, under nitrogen atmosphere, and solid iodine (45 mg; 0.178 mmol) was added under magnetic stirring at room temperature. The initial colorless solution turned to brown within 10 min. The solution was evaporated to dryness, the tarry solid was washed with hexane (6 × 2 mL) and dissolved in hot THF (10 mL) to obtain an orange solution. Upon slow cooling of the THF solution at room temperature, orange needle-shaped crystals were fastly formed while, from the mother liquor, needles mixed with some platelets were obtained; both types of crystals are sparingly soluble in organic solvents.
Characterization Needles, Compound 1·THF. Yield 38%
1H NMR (δ, room temperature, DMSO-d6): 7.74 (d, 3JH–H = 1.5 Hz, compound 1), 7.69 (m), 7.66 (d, 3JH–H = 1.5 Hz, compound 2), 7.62 (d, 3JH–H = 1.5 Hz, compound 2), 7.55 (d, 3JH–H = 1.5 Hz, compound 2), 7.40 (d,3JH–H = 1.5 Hz, compound 1), 7.38 (d, 3JH–H = 1.5 Hz, compound 2), 7.35 (d, 3JH–H = 1.5 Hz, compound 2), 7.07 (d, 3JH–H = 1.5 Hz, compound 2), 3.84 (s, compound 2), 3.81 (s, compound 1), 3.79 (s, compound 2), 3.71 (s), 3.67 (s, compound 2), 3.61 (m, 2H, THF), 1.77 (m, 2H, THF).
13C NMR (δ, room temperature, DMSO-d6): 166.1 (C2), 131.38, 130.88, 127.75, 125.91, 125.22, 122.95, 37.20, 36.78, 36.59.
MIR (cm–1): 3147 (w), 3126 (m), 2968 (m), 2936 (m), 2853 (m), 1554 (m), 1540 (m), 1461 (m), 1455 (m), 1436 (m, sh), 1409 (m), 1393 (m, sh), 1386 (m), 1361 (m), 1348 (m, sh), 1336 (w), 1323 (m), 1282 (m), 1159 (s), 1133 (m), 1084 (m), 1061 (m), 1028 (w), 965 (m), 903 (m), 861 (w), 825 (w), 725 (s).
FIR (cm–1): 692 (m), 667 (s), 648 (m, sh), 635 (m), 615 (m), 449 (s), 431 (m), 418 (w), 377 (w), 351 (w, sh), 338 (m), 311 (m), 301 (w, sh), 278 (m), 263 (w), 249 (m), 216 (w), 203 (m), 192 (s), 182 (m, sh), 174 (m), 164 (m), 154 (m), 149 (m), 140 (m), 133 (m), 116 (m).
Elemental analysis for C12H15Au3I6N6 + THF calcd %: C 11.52, H 1.39, N 5.04. Found %: C 12.00, H 1.32, N 5.24.
Characterization Platelets (Compound 2·THF). Yield 24%
1H NMR (δ, room temperature, DMSO-d6): 7.66 (d, 3JH–H = 1.5 Hz, 2H), 7.62 (d, 3JH–H = 1.5 Hz, 2H), 7.55 (d, 3JH–H = 1.5 Hz, 2H), 7.38 (d, 3JH–H = 1.5 Hz, 2H), 7.35 (d, 3JH–H = 1.5 Hz, 2H), 7.07 (d, 3JH–H = 1.5 Hz, 2H), 3.84 (s, 3H), 3.79 (s, 3H), 3.67 (s, 3H), 3.61 (m, 2H, THF), 1.77 (m, 2H, THF).
13C NMR (δ, room temperature, DMSO-d6): 166.09 (C2), 131.39, 130.90, 127.76, 125.92, 125.23, 122.96, 67.50 (THF), 37.20, 36.79, 36.60, 25.60 (THF).
MIR (cm–1): 3153 (w), 3125 (m), 3112 (m), 3073 (w), 3064 (w), 2969 (m), 2939 (m), 2924 (m), 2862 (m), 1586 (w), 1562 (w), 1554 (w), 1541 (m), 1456 (s), 1445 (m), 1403(m), 1382 (m), 1371 (m), 1356 (w), 1337 (w), 1316 (m), 1302 (w), 1280 (m), 1157 (s), 1134 (m, sh), 1081 (m), 1061 (m), 1025 (w), 961 (m), 920 (m), 897 (m), 844 (w), 743 (m, sh), 732 (s), 718 (m, sh).
FIR (cm–1): 692 (s), 686 (s), 670 (s), 658 (m, sh), 645 (m, sh), 618 (m), 609 (m), 446 (s), 428 (m, sh), 418 (w), 397 (w), 337 (m), 307 (m), 278 (m), 253 (m), 249 (m), 244 (m), 227 (w), 201 (s), 192 (s), 182 (m, sh), 174 (m), 164 (m), 154 (m), 149 (m), 140 (m), 133 (m), 116 (m).
Elemental analysis for C12H15Au3I4N6 + THF calcd %: C 13.59, H 1.64, N 5.94. Found %: C 13.05, H 1.32, N 5.54.
Reaction of [μ–Au-C2,N3-1-methylimidazolate]3 with an Excess of Methyl Iodide. Preparation of Compound 3
The [Au(μ-C2,N3-1-methyl-imidazolate)]3 (30 mg; 0.036 mmol) was dissolved in 2 mL of CH3I (excess), under a nitrogen atmosphere, and the solution was stirred at room temperature for 6 h in the darkness. After 3 h of stirring by monitoring the reaction by 1H NMR, the signal of compound 3 is 8% of those of the starting CTC. The pale yellow solution was layered with hexane and, upon storing at 5 °C for 1 week, yellow crystals were obtained. Yield = 36%.
MIR (cm–1): 3165 (w), 3141 (w), 3118 (w), 3007 (w), 2977 (w), 2933 (w), 2904 (w), 1657 (w), 1645 (w), 1628 (w), 1556 (w), 1536 (w), 1455 (m, sh), 1444 (s), 1400 (m), 1380 (m), 1317 (w), 1302 (w), 1282 (m), 1196 (m); 1154 (s), 1131 (m, sh), 1077 (m), 1024 (m), 968 (w), 828 (w), 816 (w), 733 (m, sh), 721 (s).
FIR (cm–1): 696 (s), 676 (s), 618 (w), 537 (w), 520 (w), 446 (s), 339 (m), 300 (m), 280 (m), 274 (m), 251 (m), 245 (m), 236 (m), 229 (m, sh), 218 (w), 205 (w), 199 (w), 192 (w), 177 (w), 152 (s), 141 (m, sh), 135 (m), 123 (m), 117 (w), 109 (m).
Elemental analysis for C13H18Au3IN6 calcd %: C 16.00, H 1.86, N 8.61. Found: C 16.51, H 1.79, N 8.51.
Reaction of [Au-μ-C2,N3-1-benzylimidazolate]3 with an Excess of Methyl Iodide. Preparation of Compounds 4 and 5
The [Au(μ-C2,N3-1-benzyl-imidazolate)]3 (30 mg; 0.028 mmol) was dissolved in 2 mL of CH3I (excess), under a nitrogen atmosphere, and the solution was stirred at room temperature for 6 h in the darkness. The pale yellow solution was concentrated to 1 mL and layered with hexane. A microcrystalline solid was obtained after 12 h at 5 °C and the microscope inspection revealed the presence of at least two different types of crystals. The solid was strongly emissive in the yellow range upon 366 nm irradiation.
1H NMR (δ, room temperature, DMSO): 7.63 (d, 3JH–H = 2 Hz, compound 4), 7.56 (d, 3JH–H = 2 Hz, compound 5), 7.53 (d, 3JH–H = 2 Hz, compound 4), 7.48 (d, 3JH–H = 2 Hz, 2H, compound 5), 7.39 (s, broad, 2H, compound 5), 7.38 (s, broad, 4H compound 5), 7.36–7.28 (m, benzyl groups, compound 4 and compound 5), 5.38 (s, 2H, compound 4), 5.35 (s, 4H, compound 5), 3.81 (s, N–CH3, compound 3), 3.78 (s, NCH3, compound 5).
13C NMR (δ, room temperature, acetone-d6): 184.4 (s, C2Im compound 4), 181.0 (s, C2Im, compound 5), 136.9 (s, Cypso, compound 4) 136.6 (s, Cypso, compound 5), 129 (s, compound 4), 128.8 (s, compound 4), 128.2, 127.9, 127.6, 123.5, 122.8, 122.2, 121, 54.1 (CH2–Bz, compound 4), 53.9 (2CH2–Bz, compound 5), 37.5 (NCH3, compound 4), 37.1 (2NCH3, compound 5).
MIR (cm–1): 3145 (w), 3114 (w), 3087 (w), 3069 (w), 3053 (w), 3028 (w), 3004 (w), 2954 (w), 2938 (w), 2886 (w), 2851 (w), 1687 (w), 1586 (w), 1557 (m), 1497 (m), 1468 (m), 1452 (m), 1404 (m), 1367 (m), 1330 (m), 1304 (m), 1216 (m), 1199 (m), 1155 (w), 1119 (m), 1078 (m), 1035 (w, sh), 1029 (m), 953 (w), 917 (w), 834 (m), 778 (m), 742 (s), 728 (s).
FIR (cm–1): 698 (s), 682 (w), 670 (w), 613 (m), 585 (m), 472 (m), 457 (m), 329 (w), 318 (w), 303 (w), 287 (w. sh), 278 (m), 259 (m), 250 (m, sh), 224 (w), 218 (w), 208 (m, sh), 201 (s), 185 (w), 174 (w), 168 (w), 160 (w), 151 (s) 131 (m), 124 (m), 113 (w).
ESI (−) (CH3OH, m/z, relative intensity): 450 (35) [AuI2]−. ESI (+) (CH3OH, m/z, relative intensity): 541 (100) [bis(1-benzyl-3-methyl-2yl-imidazolyl)-Au]+.
Elemental analysis for C30H36Au3I3N6 (compound 4 + compound 5 in 1:1 molar ratio) calcd %: C 26.63, H 2.44, N5.65. Found: C 26.82, H 2.50, N 5.51.
Acknowledgments
University of Camerino FAR Ateneo is acknowledged for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c03492.
Supplementary figures, Cartesian coordinates, and energy features of optimized structures (PDF)
Accession Codes
CCDC 2093397 and 2093399 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Galassi R.; Rawashdeh-Omary M. A.; Dias H. V. R.; Omary M. A. Homoleptic Cyclic Trinuclear d10 Complexes: From Self-Association via Metallophilic and Excimeric Bonding to the Breakage Thereof via Oxidative Addition, Dative Bonding, Quadrupolar, and Heterometal Bonding Interactions. Comments Inorg. Chem. 2019, 39, 287–348. 10.1080/02603594.2019.1666371. [DOI] [Google Scholar]
- Vickery J. C.; Olmstead M. M.; Fung E. Y.; Balch A. L. Solvent-Stimulated Luminescence from the Supramolecular Aggregation of a Trinuclear Gold(I) Complex That Displays Extensive Intermolecular Au ··· Au Interactions. Angew. Chem., Int. Ed. Engl. 1997, 36 (11), 1179–1181. 10.1002/anie.199711791. [DOI] [Google Scholar]
- Lu Z.; Yang Y. J.; Ni W. X.; Li M.; Zhao Y.; Huang Y. L.; Luo D.; Wang X.; Omary M. A.; Li D. Aggregation-Induced Phosphorescence Sensitization in Two Heptanuclear and Decanuclear Gold-Silver Sandwich Clusters. Chem. Sci. 2021, 12 (2), 702–708. 10.1039/D0SC05095D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Lu Z.; Wu K.; Ning G. H.; Li D. Coinage-Metal-Based Cyclic Trinuclear Complexes with Metal-Metal Interactions: Theories to Experiments and Structures to Functions. Chem. Rev. 2020, 120, 9675–9742. 10.1021/acs.chemrev.0c00011. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Yang H.; Xie M.; Li D. The π-Acidity/Basicity of Cyclic Trinuclear Units (CTUs): From a Theoretical Perspective to Potential Applications. Chem. Commun. 2019, 55 (50), 7134–7146. 10.1039/C9CC02969A. [DOI] [PubMed] [Google Scholar]
- Tekarli S. M.; Cundari T. R.; Omary M. A. Rational Design of Macrometallocyclic Trinuclear Complexes with Superior π-Acidity and π-Basicity. J. Am. Chem. Soc. 2008, 130 (5), 1669–1675. 10.1021/ja076527u. [DOI] [PubMed] [Google Scholar]
- Tiripicchio A.; Camellini M. T.; Minghetti G. The Crystal Structure of Tris-μ-[(Ethoxy)(N-p-Tolylimino)Methyl-N, C]Trigold(I), [(EtO)(MeC6H4N)CAu]3. J. Organomet. Chem. 1979, 171 (3), 399–406. 10.1016/S0022-328X(00)82664-9. [DOI] [Google Scholar]
- Bonati F.; Burini A.; Pietroni B. R.; Bovio B. Reactions of C-Imidazolyllithium Derivatives with Group Ib Compounds: Tris[μ-(1-Alkylimidazolato-N3,C2)]Tri-Gold(I) and -Silver(I). Crystal Structure of Bis(1-Benzylimidazolin-2-Yliden)Gold(I) Chloride. J. Organomet. Chem. 1989, 375 (1), 147–160. 10.1016/0022-328X(89)85094-6. [DOI] [Google Scholar]
- Burini A.; Fackler J. P.; Galassi R.; Grant T. A.; Omary M. A.; Rawashdeh-Omary M. A.; Pietroni B. R.; Staples R. J. Supramolecular Chain Assemblies Formed by Interaction of a π Molecular Acid Complex of Mercury with π-Base Trinuclear Gold Complexes. J. Am. Chem. Soc. 2000, 122 (45), 11264–11265. 10.1021/ja0024690. [DOI] [Google Scholar]
- Ghimire M. M.; Simon O. C.; Harris L. M.; Appiah A.; Mitch R. M.; Nesterov V. N.; Macchioni A.; Zuccaccia C.; Rabaâ H.; Galassi R.; Omary M. A. Binary Donor-Acceptor Adducts of Tetrathiafulvalene Donors with Cyclic Trimetallic Monovalent Coinage Metal Acceptors. Inorg. Chem. 2019, 58 (22), 15303–15319. 10.1021/acs.inorgchem.9b02294. [DOI] [PubMed] [Google Scholar]
- Rawashdeh-Omary M. A.; Omary M. A.; Fackler J.; Galassi R.; Pietroni B. R.; Burini A. Chemistry and Optoelectronic Properties of Stacked Supramolecular Entities of Trinuclear Gold(I) Complexes Sandwiching Small Organic Acids. J. Am. Chem. Soc. 2001, 123, 9689–9691. 10.1021/ja016279g. [DOI] [PubMed] [Google Scholar]
- Mohamed A. A.; Galassi R.; Papa F.; Burini A.; Fackler J. P. Gold(I) and Silver(I) Mixed-Metal Trinuclear Complexes: Dimeric Products from the Reaction of Gold(I) Carbeniates or Benzylimidazolates with Silver(I) 3,5-Diphenylpyrazolate. Inorg. Chem. 2006, 45 (19), 7770–7776. 10.1021/ic060792j. [DOI] [PubMed] [Google Scholar]
- Galassi R.; Ghimire M. M.; Otten B. M.; Ricci S.; McDougald R. N.; Almotawa R. M.; Alhmoud D.; Ivy J. F.; Rawashdeh A.-M. M.; Nesterov V. N.; Reinheimer E. W.; Daniels L. M.; Burini A.; Omary M. A. Cupriphication of Gold to Sensitize D10-D10 Metal-Metal Bonds and near-Unity Phosphorescence Quantum Yields. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (26), 201700890. 10.1073/pnas.1700890114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch A. L.; Doonan D. J. Mixed Valence Gold Chemistry: Stepwise Oxidation of a Cyclic Trigold(I) Complex. J. Organomet. Chem. 1977, 131 (1), 137–146. 10.1016/S0022-328X(00)91366-4. [DOI] [Google Scholar]
- Vickery J. C.; Balch A. L. X-Ray Crystallographic Studies of the Products of Oxidative Additions of Iodine to Cyclic Trinuclear Gold(I) Complexes: Directional Effects for Au–I···I–Au Interactions. Inorg. Chem. 1997, 36 (26), 5978–5983. 10.1021/ic970612t. [DOI] [PubMed] [Google Scholar]
- Bovio B.; Calogero S.; Wagner F. E.; Burini A.; Pietroni B. R. A 197Au Mössbauer Study of Reaction Products of Trimeric 1-Benzyl-2-Gold(I)-Imidazole Leading to AuI Carbene or AuI Imidazoline Complexes and Trinuclear AuIII Imidazolyl Derivatives. X-ray Crystal Structure of [{(μ-1-Benzylimidazolato-N3,C2)Au}3I2]. J. Organomet. Chem. 1994, 470 (1–2), 275–283. 10.1016/0022-328X(94)80179-7. [DOI] [Google Scholar]
- Scott V. J.; Labinger J. A.; Bercaw J. E. Mechanism of Reductive Elimination of Methyl Iodide from a Novel Gold(III)-Monomethyl Complex. Organometallics 2010, 29 (18), 4090–4096. 10.1021/om1006566. [DOI] [Google Scholar]
- Schneider D.; Schier A.; Schmidbaur H. Governing the Oxidative Addition of Iodine to Gold(I) Complexes by Ligand Tuning. Dalton Trans. 2004, 13, 1995–2005. 10.1039/b403005b. [DOI] [PubMed] [Google Scholar]
- Abdou H. E.; Mohamed A. A.; Fackler J. P. Oxidative Addition of Methyl Iodide to Dinuclear Gold(I) Amidinate Complex: Schmidbaur’s Breakthrough Reaction Revisited with Amidinates. Z. Naturforsch.–Sect. B: J. Chem. Sci. 2004, 59 (11–12), 1480–1482. 10.1515/znb-2004-11-1217. [DOI] [Google Scholar]
- Schmidbaur H.; Franke R. Organogold Chemistry. XVII. Synthesis and Reactions of the Gold(I)-Dimethylphosphonium-Bis-Methylid Dimer. Inorg. Chim. Acta 1975, 13 (C), 85–89. 10.1016/S0020-1693(00)90181-6. [DOI] [Google Scholar]
- Yang G.; Raptis R. G. Oxidation of Gold(I) Pyrazolates by Aqua Regia. X-Ray Crystal Structures of the First Examples of Trinuclear AuIII3 and AuIAuIII2 Pyrazolato Complexes. J. Chem. Soc., Dalton Trans. 2002, 268 (21), 3936–3938. 10.1039/b207600d. [DOI] [Google Scholar]
- Mealli C.; Ienco A.; Peruzzini M.; Manca G. The Atomic Level Mechanism of White Phosphorous Demolition by Di-Iodine. Dalton Trans. 2018, 47 (2), 394–408. 10.1039/C7DT04034B. [DOI] [PubMed] [Google Scholar]
- Schläfer H.; Figgis B. N.. Introduction to Ligand Fields. Ber. Bunsen. Phys. Chem. 1966, 70 ( (8), ), 932–933 10.1002/bbpc.19660700841. [DOI] [Google Scholar]
- Alvarez S. Orbital Interactions in Chemistry. 2nd Edition. By Thomas A. Albright, Jeremy K. Burdett and Myung Hwan Whangbo. Angew. Chem., Int. Ed. 2014, 53 (18), 4520–4521. 10.1002/anie.201311146. [DOI] [Google Scholar]
- Hoffmann R.; Alvarez S.; Mealli C.; Falceto A.; Cahill T. J.; Zeng T.; Manca G. From Widely Accepted Concepts in Coordination Chemistry to Inverted Ligand Fields. Chem. Rev. 2016, 116 (14), 8173–8192. 10.1021/acs.chemrev.6b00251. [DOI] [PubMed] [Google Scholar]
- Dimucci I. M.; Lukens J. T.; Chatterjee S.; Carsch K. M.; Titus C. J.; Lee S. J.; Nordlund D.; Betley T. A.; MacMillan S. N.; Lancaster K. M. The Myth of D8 Copper(III). J. Am. Chem. Soc. 2019, 141 (46), 18508–18520. 10.1021/jacs.9b09016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu S.; Wu X.; Chen M.; Zhou M.; Tsukuda T. Photoassisted Homocoupling of Methyl Iodide Mediated by Atomic Gold in Low-Temperature Neon Matrix. J. Phys. Chem. A 2017, 121 (44), 8408–8413. 10.1021/acs.jpca.7b08863. [DOI] [PubMed] [Google Scholar]
- de Frémont P.; Marion N.; Nolan S. P. Cationic NHC-Gold(I) Complexes: Synthesis, Isolation, and Catalytic Activity. J. Organomet. Chem. 2009, 694 (4), 551–560. 10.1016/j.jorganchem.2008.10.047. [DOI] [Google Scholar]
- Marchione D.; Izquierdo M. A.; Bistoni G.; Havenith R. W. A.; Macchioni A.; Zuccaccia D.; Tarantelli F.; Belpassi L. 13C NMR Spectroscopy of N-Heterocyclic Carbenes Can Selectively Probe σ Donation in Gold(I) Complexes. Chem. - Eur. J. 2017, 23 (11), 2722–2728. 10.1002/chem.201605502. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Litvinchuk A. P.; Lee H. G.; Guloy A. M. Crystal Structure and Vibrational Spectra of a New Viologen Gold(I) Iodide. Inorg. Chem. 1998, 37 (19), 4752–4753. 10.1021/ic980141q. [DOI] [PubMed] [Google Scholar]
- Bovio B.; Burini A.; Pietroni B. R. Reactions of Trimeric 1-Benzyl-2-Gold(I)Imidazole Leading to AuI Carbene Complexes. Crystal Structure of [1-Benzyl-3-Benzoyl-Imidazolin-2-Yliden]Chlorogold(I). J. Organomet. Chem. 1993, 452 (1–2), 287–291. 10.1016/0022-328X(93)83204-9. [DOI] [Google Scholar]
- Herrmann W. A.; Runte O.; Artus G. Synthesis and Structure of an Ionic Beryllium-”carbene” Complex. J. Organomet. Chem. 1995, 501 (1–2), C1–C4. 10.1016/0022-328X(95)05615-V. [DOI] [Google Scholar]
- Goetzfried S. K.; Gallati C. M.; Cziferszky M.; Talmazan R. A.; Wurst K.; Liedl K. R.; Podewitz M.; Gust R. N-Heterocyclic Carbene Gold(I) Complexes: Mechanism of the Ligand Scrambling Reaction and Their Oxidation to Gold(III) in Aqueous Solutions. Inorg. Chem. 2020, 59 (20), 15312–15323. 10.1021/acs.inorgchem.0c02298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. T.; van Rensburg J. M. J.; Axelsson M.; Ahlquist M. S. G.; Wendt O. F. Reactivity of NHC Au(i)–C σ-Bonds with Electrophiles. An Investigation of Their Possible Involvement in Catalytic C–C Bond Formation. Chem. Sci. 2011, 2 (12), 2373–2377. 10.1039/c1sc00428j. [DOI] [Google Scholar]
- Weinhold F.; Carpenter J. E. The Natural Bond Orbital Lewis Structure Concept for Molecules, Radicals, and Radical Ions. Struct. Small Mol. Ions 1988, 227–236. 10.1007/978-1-4684-7424-4_24. [DOI] [Google Scholar]
- Elbjeirami O.; Rashdan M. D.; Nesterov V.; Rawashdeh-Omary M. A. Structure and Luminescence Properties of a Well-Known Macrometallocyclic Trinuclear Au(i) Complex and Its Adduct with a Perfluorinated Fluorophore Showing Cooperative Anisotropic Supramolecular Interactions. Dalton Trans. 2010, 39 (40), 9465–9468. 10.1039/c0dt00736f. [DOI] [PubMed] [Google Scholar]
- White-Morris R. L.; Olmstead M. M.; Attar S.; Balch A. L. Intermolecular Interactions in Polymorphs of Trinuclear Gold(I) Complexes: Insight into the Solvoluminescence of AuI3(MeN = COMe)3. Inorg. Chem. 2005, 44 (14), 5021–5029. 10.1021/ic050381n. [DOI] [PubMed] [Google Scholar]
- APEX3 Software; Bruker: Madison, WI; available via the Internet at: https://www.bruker.com/en/products-and-solutions/diffractometers-and-scattering-systems/single-crystal-x-ray-diffractometers/sc-xrd-software/apex.html (accessed June 28, 2021).
- Krause L.; Herbst-Irmer R.; Sheldrick G. M.; Stalke D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48 (1), 3–10. 10.1107/S1600576714022985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71 (1), 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia L. J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45 (4), 849–854. 10.1107/S0021889812029111. [DOI] [Google Scholar]
- Grimme S. Semiempirical Hybrid Density Functional with Perturbative Second-Order Correlation. J. Chem. Phys. 2006, 124 (3), 034108. 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Gaussian 16, R. C. 0. (No Title). In The Structure of Small Molecules and Ions. Springer: New York, 2016, 10.1007/978-1-4684-7424-4. [DOI] [Google Scholar]
- Barone V.; Cossi M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102 (11), 1995–2001. 10.1021/jp9716997. [DOI] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100 (8), 5829. 10.1063/1.467146. [DOI] [Google Scholar]
- Dolg M.; Stoll H.; Preuss H.; Pitzer R. M. Relativistic and Correlation Effects for Element 105 (Hahnium, Ha). A Comparative Study of M and MO (M = Nb, Ta, Ha) Using Energy-Adjusted Ab Initio Pseudopotentials. J. Phys. Chem. 1993, 97 (22), 5852–5859. 10.1021/j100124a012. [DOI] [Google Scholar]
- Gorelsky S. I.; Lever A. B. P. Electronic Structure and Spectra of Ruthenium Diimine Complexes by Density Functional Theory and INDO/S. Comparison of the Two Methods. J. Organomet. Chem. 2001, 635 (1–2), 187–196. 10.1016/S0022-328X(01)01079-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.