

Abstract

Properly addressing the global issue of unsustainable plastic waste generation and accumulation will require a confluence of technological breakthroughs on various fronts. Mechanical recycling of plastic waste into polymer blends is one method expected to contribute to a solution. Due to phase separation of individual components, mechanical recycling of mixed polymer waste streams generally results in an unsuitable material with substantially reduced performance. However, when an appropriately designed compatibilizer is used, the recycled blend can have competitive properties to virgin materials. In its current state, polymer blend compatibilization is usually not cost-effective compared to traditional waste management, but further technical development and optimization will be essential for driving future cost competitiveness. Historically, effective compatibilizers have been diblock copolymers or in situ generated graft copolymers, but recent progress shows there is great potential for multiblock copolymer compatibilizers. In this perspective, we lay out recent advances in synthesis and understanding for two types of multiblock copolymers currently being developed as blend compatibilizers: linear and graft. Importantly, studies of appropriately designed copolymers have shown them to efficiently compatibilize model binary blends at concentrations as low as ∼0.2 wt %. These investigations pave the way for studies on more complex (ternary or higher) mixed waste streams that will require novel compatibilizer architectures. Given the progress outlined here, we believe that multiblock copolymers offer a practical and promising solution to help close the loop on plastic waste. While a complete discussion of the implementation of this technology would entail infrastructural, policy, and social developments, they are outside the scope of this perspective which instead focuses on material design considerations and the technical advancements of block copolymer compatibilizers.
Keywords: Recycling, Multiblock Copolymers, Compatibilization, Sustainability, Polymer Processing
1. Introduction
1.1. Motivation
In 1908, Jacques E. Brandenberger invented “cellophane” as the first clear polymer film and unknowingly launched what would soon become a multibillion-dollar industry.1,2 Today, single-use plastic packaging is ubiquitously found in everything from sterile medical supplies to individually wrapped fruits at the market. Thermoplastics—a class of polymers named for their ability to flow when sufficiently heated—have provided a low-cost solution to meet this growing demand for packaging. Unfortunately, less than 10% of all consumer generated plastic waste is recycled in the United States3 leading to an unsustainable accumulation of plastic waste in landfills and ecosystems.4 Not surprisingly, a wide variety of polymers comprise this plastic waste with the most common commercial polymers being highlighted in Figure 1.
Figure 1.

Structures, names, and recycling codes for common polymers discussed in this perspective.
Each of the polymers presented in Figure 1 differs in structure and/or chemical composition, resulting in a range of thermal and mechanical properties. Even polymers with the same repeat unit can have different properties depending on the molecular architecture, molar mass, and crystallinity, such as for high-density PE and low-density PE. While single polymers may be perfectly suited for a particular application and processing method, the performance demands of many products often require combinations. A prominant example is multicomponent products such as multilayer packaging where different polymers, and other materials like paper and foil, are combined to achieve the aggregate desired performance. For example, meeting food packaging requirements, including good containment, security, processability, durability, and low water/oxygen permeability, often necessitates between three and seven distinct layers.5 Films with nonpolymeric materials present an entirely separate set of challenges to recycling, but for simplicity the following discussions will only focus on purely polymeric multilayer packaging materials. Such packaging also typically introduces adhesives and other additives between and within different polymer layers.6 Whether combined before or after consumer use, the majority of thermoplastics end up as mixed plastic waste streams, and recovering valuable materials from this mixed waste stream has proven challenging.
Given that thermoplastics flow when heated, it would be convenient if mixed plastic waste streams could be simply mechanically recycled (i.e., melted and reprocessed) into a multicomponent plastic resin for use in other applications. However, due to the small entropy change upon mixing molecules of high molar mass, most polymers are thermodynamically immiscible.7 This is even true for some polymers with similar chemical structures; for example, PE and PP are both comprised entirely of hydrogen and aliphatic carbons, yet they are immiscible.8 For this reason, mechanical recycling typically does not result in a homogeneous material but instead produces large phase-separated domains of each component. The interfaces between these domains are typically weak due to sharp compositional gradients that preclude polymer chain entanglements and cocrystallization across the interface, compromising the mechanical performance. There is strong motivation to develop methods to “compatibilize” polymer blends to valorize the enormous quantities of otherwise unusable plastic waste. While “compatible” is sometimes used interchangeably with “miscible”, herein compatibilization refers specifically to a reduction of interfacial tension and domain size, and an increase in interfacial adhesion that leads to an improvement in polymer blend mechanical performance.9 Compatibilized blends, due to their distinct domains, retain thermal properties of each individual component, instead of amalgamation of thermal transitions often observed in miscible blends. Many research efforts have sought to address these issues by developing additives and processing approaches that not only strengthen interfaces by compatibilizing two or more disparate polymers, but also fit easily into existing infrastructure for mechanical recycling. In the following sections, we will briefly review the use of block copolymers (BCPs) for compatibilizing polymer blends before discussing recent investigations leveraging multiblock copolymers (MBCPs).
1.2. Conventional Approaches to Compatibilization
A conventional approach for compatibilizing a binary blend of polymers is to introduce an interfacially active polymer, termed a “compatibilizer”, that is often comprised of two polymer blocks, each miscible with one polymer blend component. The blocks can be chemically identical to or structurally similar to a blend component or can possess functionalities that enable chemical interactions with specific blend components, all promoting miscibility of one block into each polymer blend component.10,11 This can be realized via two distinct routes: (1) incorporating preformed BCP into the blend and (2) inducing reactions at polymer–polymer interfaces to form BCP in situ. Regardless of how they are generated, BCP compatibilizers are effective only by localizing at interfaces where they can interact with both homopolymer domains, thereby forming a “tether” across the interface.12 This idea is shown schematically in Figure 2. The mechanism of the BCP increasing interfacial adhesion between the homopolymers generally consists of entanglement formation or cocrystallization of the BCP with each homopolymer domain. The absence of these two mechanisms generally produces relatively weak interfacial adhesion where the interface fails by chain pullout.13
Figure 2.
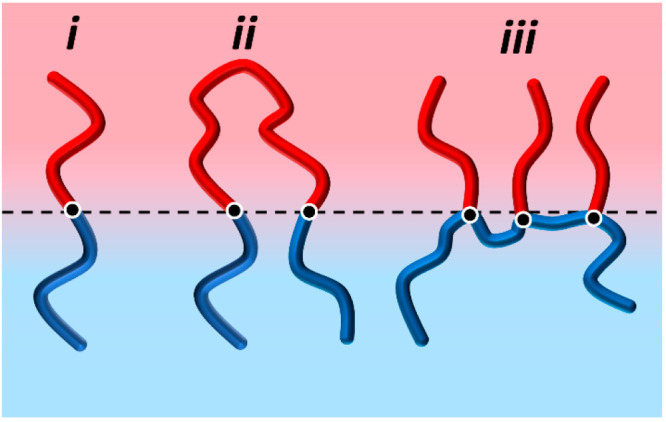
Illustration of a polymer–polymer interface being stabilized with a (i) diblock copolymer, (ii) triblock copolymer, and (iii) grafted copolymer.
The idea of using BCPs, both preformed and formed in situ, to compatibilize binary polymer blends has been actively studied since the 1970s.14−17 Compared to forming copolymers in situ, the use of premade diblock copolymers can afford more well-defined systems in terms of molecular structure and composition which allow for more methodical investigations of the structure–property relationships of BCP compatibilizers. For example, reactive formation of the PE-iPP diblock in situ is challenging because it requires installation of complementary functional groups on each block precursor; on the other hand, synthesizing a premade PE-iPP diblock can be readily achieved with good control of molecular architecture.18 While early reports suggested there might exist an optimal block length for compatibilization,19 later investigations revealed a more nuanced trade-off in molecular weight.15 Lower molecular weight copolymers show enhanced diffusivity and can more easily localize at the interface, but they do not provide the same stabilizing effect as higher molecular weight analogues.20 The reduced performance of lower molecular weight blocks has been attributed to being less capable of spanning the interface and less effective “anchoring” in the homopolymer domains by cocrystallization and/or entanglements. However, at higher BCP molecular weights, the critical micelle concentration (CMC) is substantially reduced for the BCP in each homopolymer and BCP micelles diffuse to the interface more slowly, all promoting a less effective compatibilizer.15,21−23 Therefore, while the concept of an “optimal” molecular weight is enticing, the reality is that compatibilizer design requires consideration of the specific material system. Beyond these material considerations, there is also the added complexity of differences in processing that can dictate system performance (e.g., stage mixing before compounding often improves localization to the interface).24,25
Similar questions have been posed regarding the necessity of a BCP architecture for stabilizing interfaces. Using a PS-PMMA system, Lee et al. demonstrated that, while random copolymers can localize to the blend interface, they do not achieve similar stabilizing effects supporting the notion that sufficiently large blocks are required in order to achieve compatibilization.26 It should be noted that many copolymers (including random copolymers) can reduce the dispersed particle size in polymer blends but do not necessarily stabilize the disperse phase from coalescing during subsequent processing.27 Efficient compatibilization is only realized when both domain size reduction and stabilization occur. Interestingly, Fayt et al. showed that a “tapered” transition between the blocks of a copolymer could further improve compatibilization relative to analogues with two distinct blocks.28 Other investigations in the area, both experimentally29 and computationally,30,31 have confirmed that interfacial activity and phase separation are strongly effected by subtle differences in block structure (e.g., degree of tapering). These encouraging results suggest that developing architectural complexity beyond a simple diblock could have significant impact on blend performance and is still an active area of investigation.32,33
The emphasis on diblock compatibilizers stems from both synthetic convenience and by analogy to traditional surfactants as well as early assertions that the diblock architecture outperformed other polymer architectures (i.e., graft copolymers and linear triblock copolymers).11,28,34 Early studies made claims that increasing the number of blocks had little, even detrimental, impact on interfacial adhesion of incompatible polymers; however, the polymers used in these studies were limited to triblocks.28 More recent studies investigating MBCP compatibilizers with four or more blocks—enabled by synthetic advancements—have revealed that MBCPs actually outperform traditional diblocks.18,35−37 These and other recent advances are discussed in depth in later sections of this perspective.
The other main strategy for introducing compatibilizers is to form them in situ via coupling reactions at the blend interface.38 This can be achieved in a variety of ways but typically includes a homopolymer with reactive handles which can couple with complementary functionalities of another homopolymer. All of the polymer architectures given in Figure 2 could be generated in situ if the polymer precursors are appropriately designed to participate in a coupling reaction. Fortuitously, compounding supplies the required heat and mixing to drive the chemical reaction, while mixing also disperses the phases during the mechanical recycling process, allowing for copolymer formation and blend compatibilization to occur in a single step. Therefore, unlike premade BCP compatibilizers which require additional synthesis and isolation prior to addition, reactive graft copolymers can often be implemented more readily. Intuitively, optimal copolymer formation can be achieved if the homopolymers being blended intrinsically contain complementary functional groups, but this approach must be implemented during the original production of the virgin materials and is therefore not generally applicable to the compatibilization of mixed waste streams.
Compatibilizers formed in situ have several other influences on the kinetics of compatibilization compared to premade compatibilizers. Since premade compatibilizers must localize to the blend interface—with no chemical reaction necessary for their formation—the primary influence on how fast the system can compatibilize is the diffusivity of the compatibilizer to the blend interface. For in situ formed compatibilizers, the transport of the reactive precursors and rate of formation at the interface both influence the rate at which the system can compatibilize. For example, if the reactive groups can form the compatibilizer significantly faster than the rate of droplet breakup during processing, then the result is less efficient dispersion of the minority phase.39 The predominant lesson being that the reaction rate must be balanced with the rate of growth of interfacial area (i.e., reduction in droplet size) during processing to optimize in situ blend compatibilization.
Leveraging functional groups inherent to the polymer (e.g., amine end-groups on nylons) to drive reactions with a complementary functionalized additive (e.g., maleic anhydride-grafted polymers) has been demonstrated to effectively compatibilize polymer blends.17,40 The most prevalent of these coupling chemistries is the amine-anhydride reaction due to its high reactivity and well-established synthetic methods for generating polymers with the required functionalities.38,41 Reactive compatibilization has been implemented industrially to compatibilize recycled mixed plastic at concentrations between 5 to 20 wt % of reactive resin.42−44 Extensive efforts have been made to quantify the reactivity of various other reactive handles by monitoring the coupling reactions as graft or block compatibilizers are formed,45 as well as quantifying the difference in reactivity of end-functionalized versus pendant-functionalized polymers.46 Because reactive compatibilization can result in various types of copolymers, developing a more rigorous understanding of the consequences and performance of copolymer architecture (i.e., graft vs linear) is necessary. Similarly, graft copolymers have structural parameters such as grafting density, graft distribution, and molecular weight of grafted chains that all influence their performance as compatibilizers, and more work is necessary to elucidate the underlying structure-performance relationships.46−48
2. Synthetic Methods
The discovery of living polymerizations by Szwarc et al. enabled access to well-defined block and graft copolymers that catalyzed decades of innovation.49 The further development and refinement of these methods led to various technological advances, including thermoplastic elastomers50 and, later on, BCP compatibilizers.6 In the time since, the combined contribution of many researchers have expanded the range of accessible polymer architectures enormously,51 but our understanding of the effect of architecture on compatibilizer performance has lagged behind. In Figure 3, a series of increasingly complex architectures that can be formed with only two distinct blocks is shown to illustrate the immense variety within this space. The discrepancy between our extensive synthetic capability and physical understanding poses an exciting opportunity to investigate and potentially implement new polymer architectures as novel compatibilizers.
Figure 3.

Diverse range of possible copolymer architectures using only two distinct blocks.
Several reviews exist that thoroughly discuss recent innovations in living polymerization chemistry,52−54 MBCP synthesis,55 and coupling reactions,56,57 so herein we will only highlight several key examples of how synthetic developments have enabled advancements in the field of blend compatibilization. Early studies largely focused on amorphous polymer compatibilizers (as those polymers could be directly attained from anionic polymerization), but the development of improved catalysts and chemistries have enabled the study of semicrystalline blends which are far more industrially relevant. For example, a synthetic strategy developed by Lee et al. to generate multiblock PLA-PB copolymers via a condensation polymerization of two hydroxy-terminated macromonomers with a diacyl chloride58 inspired a recent method by Nomura et al. that generated PET-PE MBCP compatibilizers.36 While PET-PE MBCPs have previously been generated in situ,59−61 the establishment of a robust synthetic method for their ex situ synthesis allows for a more methodical study of the structure–property relationship of these materials.36 Similarly, Wang et al. demonstrated the first synthesis of well-defined PE-g-aPP copolymers (with molar mass dispersities, Đ < 2).62 This advancement was made possible by the synthesis of a cyclooctene (COE)-terminated aPP macromonomer, which allowed for a grafting-through copolymerization with COE; a subsequent hydrogenation step transformed the PCOE-g-aPP into the desired PE-g-aPP.62 While similar syntheses have been demonstrated,63,64 the use of a COE-terminated macromonomer allowed for a graft-through approach that gave researchers unprecedented control over side chain length and grafting density in a well-defined PE-g-aPP copolymer.62 Beyond these academic developments, ongoing efforts at Dow have developed a method for generating olefinic BCPs using a known metallocene catalyst combined with an innovative “chain shuttling agent”65 to be used as compatibilizers66 and tie layers.67 From these examples, it is clear that synthetic innovations do not necessarily stem from a dramatic or singular discovery but instead usually arise from the refinement and creative application of previous syntheses.
Because of the incredible diversity in structure, number, and connectivity of blocks in BCPs, investigation in this field is guided primarily by (1) available synthetic routes for achieving the desired molecular architecture and (2) predictive theoretical models that enable rational molecular design.27 With the increased accessibility of polymer architectures, questions arise regarding how to make appropriate comparisons between them. For example, what would be the appropriate diblock copolymer comparison for a multiblock grafted copolymer? How can performance metrics between structurally dissimilar architectures be standardized to allow for relevant comparison? Seemingly small structural changes, like the addition of a block or a difference in block connectivity or sequence, can have repercussions on properties. While it has been shown that molecular weight dispersity can drive large changes in bulk self-assembly,68−70 only recently has polydispersity been investigated in the context of blend compatibilization.71 Similar investigations are now taking place around the use of MBCP compatibilizers and the results show great promise.18,35,36,58
3. Linear Multiblock Copolymer Compatibilizers
When considering systems beyond the conventional diblock, an immediately evident possibility is the MBCP architecture. The simplest MBCP architecture is an extension of the conventional diblock copolymer, where blocks are connected linearly to generate a linear multiblock copolymer (lMBCP). One can imagine that lMBCPs could similarly segregate to an interface to span and reinforce two homopolymer domains; rather than crossing the interface a single time as with a diblock copolymer, lMBCPs can cross the interface multiple times, effectively “stitching” the domains together. Assuming localization to the interface and appropriate segregation of the multiblock components to their respective homopolymer domains, the number of interface crossings scales with number of blocks (n) in the copolymer. This is shown schematically in Figure 4.
Figure 4.

Depiction of several MBCPs (n > 2) stabilizing a polymer–polymer interface, highlighting the increased number of interface crossings per molecule with an increased number of blocks.
Eagan and Xu et al. synthesized both tetrablock and hexablock PE-iPP copolymers directly from ethylene and propylene monomers using a hafnium-based living catalyst.18,35 To simulate recycling, HDPE/iPP blends were prepared by melt blending with lMBCP additives in a single microcompounding step. As shown in Figure 5, the blends achieved a strain-at-break of ∼500% with only 1 wt % of the lMBCP compatibilizer, a significant improvement from the ∼10% strain-at-break for neat blends prepared without compatibilizer.18 Also, the interfacial adhesion between HDPE and iPP films was significantly strengthened when a PE-iPP lMBCP was employed as a tie layer in multilayer laminates (peel strength > 6 vs < 0.5 N/mm for HDPE/iPP films). In the same work, researchers found that a traditional diblock copolymer with comparable block length performs worse as both a compatibilizer (Figure 5, strain at break 90% vs 500% for lMBCP) and a tie layer (peel strength ∼ 1 vs > 6 N/mm for lMBCP). For these diblock copolymers to show comparable performance they must be higher in molecular weight and concentration (ca. 5 wt %) than analogous MBCPs. Similarly, Nomura et al. synthesized PET-PE MBCPs for compatibilizing a PET/LLDPE system. They found that 2 wt % addition of lMBCP could convert brittle 80/20 PET/LLDPE blends (strain at break ∼10%) to ductile compatibilized blends (strain at break ∼400%). In contrast, analogous blends containing triblock copolymer with comparable block length exhibited strain at break of only ∼13%. In addition, as an interfacial tie layer, lMBCP increased the interfacial adhesion between PET and LLDPE over two orders of magnitude, while the triblock copolymer counterpart only increased adhesion by four times, compared to without a tie layer. These results also indicate that lMBCPs outperform triblock copolymers at comparable block length.36 It is clear from these findings that lMBCPs compatibilizers pose a competitive alternative to traditional diblock copolymers, but more work remains to understand the underlying mechanisms of their enhanced performance as well as the limits of the architectural advantage (e.g., the trade-off between anchoring ability and diffusivity when increasing block number and molecular weight).
Figure 5.

Uniaxial tensile elongation of PE/iPP materials and blends. Materials were melt-blended at 190 °C without BCPs (black) or with 1 wt % diblock (green), 1 wt % tetrablock (orange), or 5 wt % tetrablock copolymers (purple). These materials were then compression molded into tensile specimens at 180 °C and tensile tested at a rate of 100%/min (Adapted with permission from the work of Eagan et al.35 Copyright 2017 AAAS).
3.1. Mechanisms of Enhanced Compatibilizer Performance
In diblock copolymers, the performance is determined largely by two factors: areal density (which scales with localization at the interface) and molecular weight (which dictates the polymer–polymer interaction in the homopolymer domains).19,72 As shown in Figure 4, lMBCPs can cross the interface between two homopolymers multiple times. In a conventional diblock copolymer, a sufficiently high block length can result in entanglement of the blocks with the homopolymer domains, thereby strengthening the interface and reducing the possibility of failure by chain pullout. Based on theories concerning chain entanglements in homopolymers, a critical molecular weight (Mc), determined to be two to three times the molecular weight between entanglements (Me), is often required to observe the effects of entanglement.73 When considering entanglement across an interface, a shift in interfacial failure mechanism can be observed at four to five times the molecular weight of entanglement.72 In the case of lMBCP, entanglements can also be formed with midblocks which results in the possibility of “trapped entanglements”, instead of only end-blocks as for diblock copolymers, shown schematically in Figure 6. Eastwood et al. experimentally probed this question of midblock entanglement in lMBCP compatibilizers using diblocks and pentablocks and found that the failure mechanism changed at much lower molecular weights for midblocks than for chain ends (100 kg/mol for diblocks vs 30 kg/mol for pentablocks), suggesting that the conformational constraints imposed by molecular self-assembly characteristics at the interface improve midblock entanglement.73 This phenomenon has been predicted by Monte Carlo simulations of MBCP systems.74 While it is well understood that the chain ends of BCPs can fail by chain-pullout, the behavior of entangled midblocks (e.g., relaxation time and failure mechanism) and the molecular weight dependence of these phenomena remain opportunities for further investigation.73 Quantifying how entanglements scale with lMBCP structure will prove key to designing optimized MBCP to compatibilize complex polymer blends.
Figure 6.

Schematics of lMBCP forming trapped entanglements and cocrystallizing with homopolymers.
For semicrystalline polymers (e.g., PE, iPP, PET), another anchoring mechanism is available between the lMBCP and the homopolymers: cocrystallization (Figure 6). The block molecular weight necessary for cocrystallization to occur is typically higher than that for trapped entanglements because (1) polymer chains must fold into the crystal structure and (2) the BCP needs to be long enough to span the amorphous interfacial width to colocate with the crystalline homopolymer domains.18 When using a PE-iPP lMBCP with sufficiently large blocks as a tie layer between iPP and HDPE films, Xu et al. observed crystal formation near the interfaces between lMBCP and the homopolymers and proposed cocrystallization as contributing to the observed toughening.18 However, further experimentation is needed to definitively confirm that the lMBCP is incorporated into the crystalline domains of the homopolymers, possibly through polymer labeling and high spatial-resolution analytical techniques. With cocrystallization, it has been shown for polyethylene and its copolymers containing small amounts of acetate or ethyl branches that the temperature at which the crystallite melts can become intermediary to that of the two individual components.75 This effect, while small, in compatibilized blends where the vast majority of crystallites are usually composed of homopolymer, could provide an interesting property to study further in the presence of cocrystallizable MBCPs. Further, when crystallization occurs near an interface between two polymer domains with sufficient differences in crystallization rates, the volume contraction associated with crystallization can effectively pull material across the interface (a process known an “local crystallization”).76 This process can presumably occur concurrent to cocrystallization but has only been demonstrated at noncompatibilized polymer interfaces. Investigating this phenomenon with lMBCP compatibilized interfaces could give insight into toughening mechanisms.
MBCPs have achieved successful compatibilization at impressively low concentrations (ca. 0.5 wt % or lower)36 relative to the current industrially utilized reactive compatibilizers (5–20 wt %).42−44 This is a very encouraging indication that this technology could be realized industrially as it would reduce the amount of required additive and therefore lower the cost of implementation. A possible explanation for the increased efficiency of lMBCPs is that they can more easily localize to the interface (thereby outcompeting micellization). Micellization of diblock copolymers in homopolymers has been rigorously investigated. It has been observed that diblock copolymers readily form micelles in the major blend components—a feature that reduces compatibilization—and the tendency of micelle formation further increases with higher molecular weight. In contrast, there have been few studies on the micellization behavior of lMBCP in homopolymers. To help bridge this gap in our understanding, more systematic investigations on micellization as a function of MBCP physical parameters (e.g., block asymmetry, block molecular weight, number of blocks) are needed. A clear understanding of the design space of MBCPs will help understand the apparent high efficiency displayed by many MBCP compatibilizers and translate this technology to recycling solutions.
3.2. Future Work and Challenges
With the recently demonstrated successes of lMBCP as polymer blend compatibilizers, these materials are quickly gaining interest as a research area rife with opportunity. As discussed previously, trapped entanglements and cocrystallization of an lMBCP with a homopolymer can occur simultaneously to synergistically strengthen interfaces, but future experiments could work toward isolating either mechanism to better understand its contribution to overall performance. For example, polymer crystallinity is strongly process- and molecular architecture-dependent; therefore, with strategic material design and clever manipulation of processing conditions, the two mechanisms may be decoupled. This would enable a great opportunity for investigating their interplay in defining blend performance. Studies in this direction have revealed the limitations of current characterization techniques (e.g., peel tests)18 suggesting that development in both the materials of study and methods of study will be necessary to elucidate a clearer understanding of the system. Further, the use of amorphous model blends entirely eliminates the possibility of cocrystallization, thereby isolating the contribution of trapped entanglements. Strategic investigation of an amorphous system could yield many insights into the nature of trapped entanglements in MBCP-compatibilized blends.
Beyond these opportunities, many challenges still exist to further develop a theoretical framework for guiding material design of lMBCP compatibilizers. In blend compatibilization, vigorous melt mixing aids transport and migration of compatibilizer to the interface. Preliminary work seems to suggest that lMBCPs are more effective than diblock copolymers at localizing to the interface, but direct observation of the relative transport through homopolymers to interfaces has not yet been realized. Further, a complete physical understanding of factors driving the difference in localization is lacking yet would be incredibly beneficial for molecular (re)design of optimal compatibilizers. For example, factors like micelle formation and segregation strength affect the ability to localize to an interface but are strongly coupled and require careful study to deconvolute.77 Importantly, there is a wealth of theory and simulation studies aimed at understanding the behavior of diblock copolymers in homopolymers blends,32,74,78−81 but fewer have extended these investigations to lMBCPs.82−84 Some simulation work has been done on MBCPs in solution,85,86 highlighting some of their interesting assembly behavior, and applying similar efforts to MBCPs in homopolymer could be highly informative in the field for guiding experimental work.
Translating a mechanistic understanding into an efficient processing technique is also critical for further developing this technology. For example, if lMBCPs segregate efficiently to polymer–polymer interfaces, should they be incorporated directly into homopolymers as latent compatibilizers for ready to recycle opportunities after their initial use? Alternatively, given their competitive performance as adhesive tie layers, should lMBCPs be incorporated into products as tie layers in place of conventional adhesives such that they can readily compatibilize the resulting mixed polymer waste stream after use? Already there are innovative commercial products exploring these ideas with olefinic BCPs, but numerous opportunities for industrial and academic study still remain.
4. Grafted Multiblock Copolymer Compatibilizers
Another polymer architecture that has garnered interest for its potential use as a compatibilizer is the grafted multiblock copolymer (gMBCP). Recalling Figure 2iii, these molecules consist of a polymer backbone that is miscible with one phase of the blend and has grafted side chain polymers that are miscible with the other polymer in the blend. While these polymers can also be considered MBCPs owing to the multiple blocks that comprise them, they differ from lMBCP in their block interconnectivity. In contrast to lMBCP compatibilizers that cross back and forth across the interface, gMBCP compatibilizers have multiple grafted chains which can only cross the interface once, with the chain-ends of one block type located in one of the blend components. As a result of this architectural difference, only the backbone of gMBCP compatibilizers can form trapped entanglements, as opposed to both block types in lMBCPs. However, because gMBCPs have multiple grafted blocks, a single gMBCP may stitch the interface more than once.87 This effect seems to be supported by other studies that have shown increased compatibilization with increasing grafting density, paralleling the argument of increased blocks in lMBCPs.64 Another important consideration in the case of gMBCPs is the choice of which polymer should compose the backbone block versus grafted blocks because of the difference in their interactions with the homopolymer. The backbone of a gMBCP is constrained nearer to the interface and presents the opportunity for trapped entanglements, whereas the gMBCP grafted blocks consist of a single chain-end with more conformational freedom (shown in Figure 7). As a result of this asymmetry, differences arise when the backbones are included in the major phase versus the minor phase of a blend that should be considered when designing gMBCP. Though the aforementioned studies focused on preformed gMBCPs, these molecules are often generated in situ. While it is certainly more convenient for processing to form gMBCPs in situ, it also makes it more challenging to elucidate structure–performance relationships because the compatibilizers are not being isolated and rigorously characterized. For this reason, many investigations use polymer syntheses that allow for precise architectural control to help probe the variable space thereby informing future design of reactive systems.
Figure 7.

Schematic of reactively formed graft copolymers. Reactive groups must both be present at the interface to form the compatibilizer.
4.1. Premade Versus Reactive Graft Multiblock Copolymer Compatibilizers
Like linear copolymers, graft copolymer compatibilizers can either be premade prior to melt mixing or formed in situ through reactions between functional groups on each polymer (Figure 7). Premade graft copolymers can be synthesized through a variety of methods such as graft-to (i.e., reacting an end-functionalized chain with dispersed functionalities on a backbone)46 or by graft-from (i.e., using functional groups along the backbone to initiate the growth of the pendant chains).88 With recent advances in catalyst technology, it is now also possible to synthesize macromonomers containing the desired graft chain, and copolymerize these macromonomers with a graft-through approach.55 Premade graft copolymers and the controlled polymerization techniques used to make them provide the framework for systematic studies of the variables that affect compatibilization (e.g., backbone/side chain length and grafting density/spacing). Recent work in this field has shown that the addition of PE-g-iPP (gMBCP) to a 70/30 blend of HDPE/iPP increased strain at break from 18 to ∼900% and more than halved the average minority component droplet size.64 Moreover, it was shown that increasing the number and length of grafted chains—thereby increasing the number and strength of “anchors” afforded by entanglements and/or cocrystallization on each side of the interface—led to improved mechanical performance. Studies like this have started to explore the structural parameter space that can be accessed through new synthetic methods and are crucial for developing the necessary understanding to allow for rational design of compatibilizers in the future.
The insight gained from these investigations on premade gMBCPs can be applied to systems that reactively form gMBCPs. The precursors to these reactively formed copolymers are typically functionalized versions of an unreactive phase (often a polyolefin like PE) which has had functional groups incorporated into the backbone by either copolymerization or grafting techniques.6 One advantage of using these functionalized polymers to form graft copolymers in situ is that there is already a library of these functional polymers to choose from due to their use as tie layers in multilayer films.6 Moreover, we believe that forward-thinking design of multilayer systems to incorporate these functionalized polymers as tie layers that can function downstream as compatibilizers can open up new and exciting recycling pathways. This idea of integrating compatibilizers into tie layers is already being employed industrially to allow multilayer packaging films to enter recycling streams.67,89
Along with the design factors associated with premade graft copolymers, reactive systems bring an added layer of complexity in the form of reaction rates for generating in situ gMBCPs. In a 2001 study, Orr et al. characterized a wide array of functional pairs commonly used for in situ compatibilization.45 They found that an amine/anhydride had by far the fastest reaction rate, with the next fastest reaction measured (acid/epoxy) being almost two orders of magnitude slower. These results explain why anhydride functionalized polymers are utilized in most multilayer materials. However, we believe that by understanding the other underlying factors controlling reactivity, compatibilizers derived from slower coupling chemistries may become more viable. Functional groups with reduced reactivity tend to be more shelf-stable and less expensive, making them more attractive. The intrinsic reactivity of a functional group pair is thermodynamically driven, but the apparent reaction rate is largely governed by kinetic factors. Therefore, less reactive pairs can be accelerated by increasing the availability of functional groups at the interface. It has been found that droplet size distributions decrease when a reactive backbone is mixed directly with its complementary phase (i.e., mixing it in the phase with complementary reactive groups) as compared to premixing it with the phase it matches (i.e., putting it in the miscible phase and hoping it finds its way to the interface to react).43 Likewise, aggressive mixing can help mitigate slow intrinsic reaction rates, having been shown to increase effective reaction rates by almost two orders of magnitude compared with static annealing.46 These results suggest that reactive gMBCP performance is determined not just by the final polymer structure but by the processing conditions used to drive the copolymer formation. These preliminary findings indicate that gMBCPs could offer competitive performance to lMBCPs, but further investigations are necessary to understand and optimize the design of these compounds.
4.2. Future Work and Challenges
Despite their prevalence as compatibilizers and tie layers, a systematic understanding of the factors surrounding the compatibilization efficiency of gMBCPs has not yet been established. Current studies with tightly controlled graft architecture have had the copolymer backbone miscible with the majority component of the blend, but could similar improvements in performance be gained with the backbone in the minority phase and grafted side chains extending into the matrix? Similarly, how would performance be affected if both variants of the grafted copolymers (backbone in majority phase and backbone in minority phase) were concurrently introduced to a blend? It has been shown that, when the grafted side chains are miscible with the dispersed phase (and the backbone with the matrix), compatibilization decreases due to chain pullout of the grafts,90 so more deeply exploring the effects of this asymmetry is imperative for understanding gMBCP function. This asymmetry can be taken a step further by introducing grafted chains that are copolymers themselves. Leveraging insights gained from investigations of BCP compatibilizers, random copolymer grafted side chains could be introduced to increase the width of the interfacial region; tapered copolymer grafted side chains could also be studied for their previously demonstrated improvements to compatibilization.
More questions arise as to how transport of these graft polymers can be manipulated to maximize the amount of polymer that localizes to the interface. Specifically, it would be interesting to explore the effect of polymer miscibility (of both the backbone and the side chains) on localization behavior as well as differences in micellization behavior of gMBCPs formed in situ versus ex situ. In reactive systems requiring small molecule catalysts, it was seen that the most effective catalysts were those with solubility parameters in between the two polymers being compatibilized, causing them to localize at the interface (an analogous trend to what has been observed with diblock copolymers).91 Could strategies for tethering catalysts be used to improve reaction rates and circumvent solubility issues? Similarly, it has been observed that higher molecular weight polymer backbones can improve segregation to the interface and toughening of the interface (through previously described entanglement/cocrystallization mechanisms) but can hinder diffusion through the bulk. While it is known that higher molecular weight can promote micellization in diblock copolymers, it is not yet clear if the same is true for gMBCPs nor how limiting micellization of gMBCPs might be for compatibilization. Is there an optimal backbone length and/or grafting density to maximize localization to the interface? Or is it a nuanced relationship that must consider the identity of the backbone and the immiscible phase? Encouraged by preliminary studies, we believe the opportunities for gMBCP compatibilizers are plenty, but to achieve them we must first take a step back and closely consider the roles of the building blocks of the system.
5. Beyond Binary Blends
As the value of BCPs becomes more and more apparent for use as compatibilizers in mixed waste stream recycling, questions arise as to future directions and commercial implementation of these materials. To this point, the majority of studies have focused mostly on two phase systems (often some mixture of a polyolefin and either another polyolefin,18,35,92 a polyurethane,93−95 a polyester,36,96,97 or a polyamide24,98,99). However, a simple examination of the contents of any recycling bin reveals a wide array of materials that may at some point end up in a solid plastic waste stream together. Though current separation techniques (e.g., flotation, physical sorting, etc.) can enrich streams toward a single material, remaining impurities are highly variable in time and by source and difficult to predict. The net result of these difficulties is a highly inefficient process. This dilemma highlights a major need for advancing recycling technology. To date, significant advances have been made with model, virgin polymers, and their blends, but it is as of yet unclear how these will translate to real waste streams. The establishment of a series of models for more realistic plastic waste stream standards would allow researchers to study more relevant systems while enabling comparisons across laboratories. However, this is a formidable task as it is difficult to imagine what a prototypical realistic model waste stream might look like. Is it majority polyolefin? Are ternary blends or higher order mixtures most appropriate? These are complex questions from a materials standpoint that are necessarily intertwined with societal behaviors, industrial practices, and waste collection infrastructure which complicate an already convoluted problem.100
Importantly, strategies are emerging to address blends that are beyond binary. Recent work has demonstrated gMBCP compatibilizers can be effective for ternary PP/PA6/PS101 (as shown in Figure 8) and PP/PA6/SEBS102 blends. These blend compositions were specifically chosen to contain three mutually immiscible components because if any two components were miscible then it could be approximated as a binary blend and not a true ternary blend. Further, the blend choice encompasses the three major divisions of commercial consumer plastics: polyolefins, engineering plastics, and styrene-based polymers. This work represents an ambitious new direction in the field of using highly tailored MBCP compatibilizers to address more complex multicomponent blends that better simulate real-life waste streams. The compatibilizer precursor that is dispersed into the ternary blend is a polypropylene backbone grafted with side chains of maleic anhydride-styrene copolymers (PP-g-(MAH-co-PS)); the maleic anhydride can react in situ with amine groups on PA6 to generate the final gMBCP structure (PP-g-((MAH-g-PA6)-co-PS)). This synthetic approach of having reactive grafting groups within a grafted side chain shows great promise and could theoretically be extended to blends with even more than three components. As shown in Figure 8, when compatibilizing ternary systems there are many more interactions that must be taken into consideration. While this inherently complicates the design of compatibilizers, MBCPs offer an efficient materials platform for encoding various interactions and parameters into a single molecule.
Figure 8.

Interplay between phases and compatibilizer in preliminary ternary system studies (Adapted with permission from ref (101). Copyright 2011 Elsevier Ltd.).
Considering the aforementioned demonstrations of reactive gMBCP compatibilizers for ternary blends, the question naturally arises about whether a premade MBCP compatibilizer (linear or graft) with three or more distinct polymer chemistries could also be effective for multicomponent blends. As shown in Figure 3, there exists a wide range of potential connectivity for diblock copolymers, but for MBCPs the potential connectivity and order of the blocks expands exponentially.51 For example, moving from a diblock to a triblock expands the potential unique connectivities from one to three (i.e., while a linear diblock will always be an AB copolymer, a linear triblock can exist as ABC, ACB, and CAB copolymers). Moving from a linear polymer to more complex architectures (e.g., grafted copolymer and star copolymer) further confounds the possibilities. Given the enormity of possible polymer and architecture combinations, discovering effective compatibilizers will require strategic and targeted investigations. One particularly interesting architecture is the miktoarm star copolymer (where mikto means “mixed”). One can imagine a single star copolymer with a separate arm for each of several major recyclable polymer classes working as a “one-size-fits-all” compatibilizer. Similarly, a graft copolymer system with various arms of distinct polymer types (i.e., a “mikto”-grafted MBCP) could be developed to address multicomponent blends. And there is no reason to limit these systems to homopolymer side chains; a star or grafted MBCP could be composed of diblock, gradient, or even random copolymer side chains to improve interactions with specific interfaces. While it is unknown whether these molecular designs could outperform conventional compatibilizers, they illustrate the numerous opportunities for impactful investigation on polymer architecture in this field. With the many developments in polymer synthesis, newly accessible structures could bring us closer to a “universal compatibilizer”, but first, the basic principles surrounding the effectiveness of these new and exciting architectures must be understood.
6. Outlook
In recent years, lMBCP and gMBCP architectures have been used to compatibilize polymer blends and multilayer films. In the absence of compatibilizers, polymer blends have poor mechanical performance due to poor stress transfer across the interface. However, MBCP compatibilizers have been shown to effectively mitigate this issue by improving interfacial adhesion, at concentrations lower than those necessary for diblock copolymer compatibilizers. These results demonstrate the great potential of MBCPs for opening new pathways to recycle mixed plastic waste. Mechanical mixing has historically been viewed as “downcycling” because the resulting blends generally had inadequate properties due to degradation, additives, and molecular weight variability. With the development and optimization of modern compatibilizers, this old paradigm is quickly shifting. Properly compatibilized blends can have properties intermediate to their constituent parts and well within range of some commercial materials. Just like any other materials selection process in product design, we need to recognize that compatibilizers are not a one-size-fits all solution for all mixtures, rather successful outcomes require strategic implementation derived from leaning on knowledge of polymer chemistry, physics, and processing. Ultimately, the combination of compatibilization techniques and appropriate choice of application could valorize much of the solid plastic waste that is currently being lost to the environment, burned for energy, or deposited in landfills.
This is an area rich with many fundamental questions that need to be answered. For both lMBCPs and gMBCPs, we need to develop a better understanding of their diffusion, micellization, and localization behavior, both in homopolymer and in mixed blends, as well as the underlying mechanisms of their stabilization. Simultaneously, we must develop effective methods for incorporating compatibilizers into real life waste streams. In the same vein, extending fundamental research efforts from binary blends into the reality of multicomponent waste streams will be crucial to their successful implementation. Not only will these streams have multiple plastic components, but also various additives, such as pigments, dyes, stabilizers, nucleating agents, and particulates, that will complicate compatibilization. MBCPs offer a potentially exciting material solution to these issues, but our nascent understanding of their use as compatibilizers requires further development. With the multitudinous synthetic advancements of recent years, researchers are granted enormous freedom and flexibility in the design of these molecules which greatly enables new studies. Overall, progress in the field of BCP compatibilizers is guiding us toward a path for bringing plastic waste into a circular economy, but many exciting challenges lie ahead before we can close the loop.
Acknowledgments
We acknowledge our principal funding source, the National Science Foundation Center for Sustainable Polymers at the University of Minnesota, which is a National Science Foundation supported Center for Chemical Innovation (CHE-1901635).
Author Contributions
‡ J.L.S., A.J.Z., X.P., and W.R.L. made equal contributions. The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
References
- Wagner J. R. J.; Mark S. B.. Chapter 1–Introduction. In Multilayer Flexible Packaging; William Andrew, 2009; pp 3–11. [Google Scholar]
- Alexander H. Tullo. The Cost of Plastic Packaging. C&EN Glob. Enterp. 2016, 94 (41), 32–37. 10.1021/cen-09441-cover. [DOI] [Google Scholar]
- Environmental Protection Agency . Advancing Sustainable Materials Management: 2018 Fact Sheet; 2020. [Google Scholar]
- Geyer R.; Jambeck J. R.; Law K. L. Production, Use, and Fate of All Plastics Ever Made. Sci. Adv. 2017, 3 (7), e1700782 10.1126/sciadv.1700782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anukiruthika T.; Sethupathy P.; Wilson A.; Kashampur K.; Moses J. A.; Anandharamakrishnan C. Multilayer Packaging: Advances in Preparation Techniques and Emerging Food Applications. Compr. Rev. Food Sci. Food Saf. 2020, 19 (3), 1156–1186. 10.1111/1541-4337.12556. [DOI] [PubMed] [Google Scholar]
- Morris B. A.Adhesion. In The Science and Technology of Flexible Packaging; Elsevier, 2017; pp 351–400. 10.1016/B978-0-323-24273-8.00010-1. [DOI] [Google Scholar]
- Higgins J. S.; Lipson J. E. G.; White R. P. A Simple Approach to Polymer Mixture Miscibility. Philos. Trans. A. Math. Phys. Eng. Sci. 2010, 368 (1914), 1009. 10.1098/rsta.2009.0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh J. W.; Rudin A.; Keung J. C. A Review of Polyethylene–Polypropylene Blends and Their Compatibilization. Adv. Polym. Technol. 1994, 13 (1), 1–23. 10.1002/adv.1994.060130101. [DOI] [Google Scholar]
- Paul D. R. Interfacial Agents (“Compatibilizers”) for Polymer Blends. Polym. Blends 1978, 35–62. 10.1016/B978-0-12-546802-2.50008-7. [DOI] [Google Scholar]
- Traugott T. D.; Barlow J. W.; Paul D. R. Mechanical Compatibilization of High Density Polyethylene–Poly(Ethylene Terephthalate) Blends. J. Appl. Polym. Sci. 1983, 28 (9), 2947–2959. 10.1002/app.1983.070280922. [DOI] [Google Scholar]
- Fayt R.; Jérôme R.; Teyssié P. Molecular Design of Multicomponent Polymer Systems. I. Emulsifying Effect of Poly(Hydrogenated Butadiene-b-Styrene) Copolymers in LDPE/PS Blends. J. Polym. Sci. Polym. Lett. Ed. 1981, 19 (2), 79–84. 10.1002/pol.1981.130190207. [DOI] [Google Scholar]
- Creton C.; Kramer E. J.; Hui C. Y.; Brown H. R. Failure Mechanisms of Polymer Interfaces Reinforced with Block Copolymers. Macromolecules 1992, 25 (12), 3075–3088. 10.1021/ma00038a010. [DOI] [Google Scholar]
- Creton C.; Kramer E. J.; Brown H. R.; Hui C.-Y. Adhesion and Fracture of Interfaces Between Immiscible Polymers: From the Molecular to the Continuum Scale. Adv. Polym. Sci. 2001, 156, 53–136. 10.1007/3-540-45141-2_2. [DOI] [Google Scholar]
- Barentsen W. M.; Heikens D.; Piet P. Effect of Addition of Graft Copolymer on the Microstructure and Impact Strength of PS/LDPE Blends. Polymer 1974, 15 (2), 119–122. 10.1016/0032-3861(74)90012-3. [DOI] [Google Scholar]
- Riess G.; Jolivet Y. Rubber-Modified Polymers. Location of Block Copolymers in Two-Phase Materials. Advances in Chemistry 1975, 142, 243–256. 10.1021/ba-1975-0142.ch022. [DOI] [Google Scholar]
- Barentsen W. M.; Heikens D. Dynamic Mechanical Properties of Polystyrene/Low Density Polyethylene Blends. Polymer 1973, 14 (11), 579–583. 10.1016/0032-3861(73)90143-2. [DOI] [Google Scholar]
- Ide F.; Hasegawa A. Studies on Polymer Blend of Nylon 6 and Polypropylene or Nylon 6 and Polystyrene Using the Reaction of Polymer. J. Appl. Polym. Sci. 1974, 18 (4), 963–974. 10.1002/app.1974.070180402. [DOI] [Google Scholar]
- Xu J.; Eagan J. M.; Kim S.-S.; Pan S.; Lee B.; Klimovica K.; Jin K.; Lin T.-W.; Howard M. J.; Ellison C. J.; LaPointe A. M.; Coates G. W.; Bates F. S. Compatibilization of Isotactic Polypropylene (iPP) and High-Density Polyethylene (HDPE) with iPP–PE Multiblock Copolymers. Macromolecules 2018, 51 (21), 8585–8596. 10.1021/acs.macromol.8b01907. [DOI] [Google Scholar]
- Gaylord N. G. Compatibilization Concepts in Polymer Applications. Advances in Chemistry 1975, 142, 76–84. 10.1021/ba-1975-0142.ch007. [DOI] [Google Scholar]
- Macosko C. W.; Guégan P.; Khandpur A. K.; Nakayama A.; Marechal P.; Inoue T. Compatibilizers for Melt Blending: Premade Block Copolymers. Macromolecules 1996, 29 (17), 5590–5598. 10.1021/ma9602482. [DOI] [Google Scholar]
- Noolandi J.; Hong K. M. Interfacial Properties of Immiscible Homopolymer Blends in the Presence of Block Copolymers. Macromolecules 1982, 15 (2), 482–492. 10.1021/ma00230a054. [DOI] [Google Scholar]
- Galloway J. A.; Jeon H. K.; Bell J. R.; Macosko C. W. Block Copolymer Compatibilization of Cocontinuous Polymer Blends. Polymer 2005, 46 (1), 183–191. 10.1016/j.polymer.2004.10.061. [DOI] [Google Scholar]
- Chang K.; Macosko C. W.; Morse D. C. Interfacial Tension Measurement and Micellization in a Polymer Blend with Copolymer Surfactant: A False Critical Micelle Concentration. Macromolecules 2015, 48 (22), 8154–8168. 10.1021/acs.macromol.5b01268. [DOI] [Google Scholar]
- Willis J. M.; Favis B. D. Processing-morphology Relationships of Compatibilized Polyolefin/Polyamide Blends. Part I: The Effect of an Lonomer Compatibilizer on Blend Morphology. Polym. Eng. Sci. 1988, 28 (21), 1416–1426. 10.1002/pen.760282111. [DOI] [Google Scholar]
- Zhang C. L.; Feng L. F.; Gu X. P.; Hoppe S.; Hu G. H. Efficiency of Graft Copolymers as Compatibilizers for Immiscible Polymer Blends. Polymer 2007, 48 (20), 5940–5949. 10.1016/j.polymer.2007.07.042. [DOI] [Google Scholar]
- Lee M. S.; Lodge T. P.; Macosko C. W. Can Random Copolymers Serve as Effective Polymeric Compatibilizers?. J. Polym. Sci., Part B: Polym. Phys. 1997, 35 (17), 2835–2842. 10.1002/(SICI)1099-0488(199712)35:17<2835::AID-POLB8>3.0.CO;2-P. [DOI] [Google Scholar]
- Sundararaj U.; Macosko C. W. Drop Breakup and Coalescence in Polymer Blends: The Effects of Concentration and Compatibilization. Macromolecules 1995, 28 (8), 2647–2657. 10.1021/ma00112a009. [DOI] [Google Scholar]
- Fayt R.; Jerome R.; Teyssié P. Molecular Design of Multicomponent Polymer Systems, 13. Control of the Morphology of Polyethylene/Polystyrene Blends by Block Copolymers. Die Makromol. Chemie 1986, 187 (4), 837–852. 10.1002/macp.1986.021870414. [DOI] [Google Scholar]
- Lefebvre M. D.; Dettmer C. M.; McSwain R. L.; Xu C.; Davila J. R.; Composto R. J.; Nguyen S. T.; Shull K. R. Effect of Sequence Distribution on Copolymer Interfacial Activity. Macromolecules 2005, 38 (25), 10494–10502. 10.1021/ma0509762. [DOI] [Google Scholar]
- Lefebvre M. D.; Olvera de la Cruz M.; Shull K. R. Phase Segregation in Gradient Copolymer Melts. Macromolecules 2004, 37 (3), 1118–1123. 10.1021/ma035141a. [DOI] [Google Scholar]
- Shull K. R. Interfacial Activity of Gradient Copolymers. Macromolecules 2002, 35 (22), 8631–8639. 10.1021/ma020698w. [DOI] [Google Scholar]
- Levine W. G.; Seo Y.; Brown J. R.; Hall L. M. Effect of Sequence Dispersity on Morphology of Tapered Diblock Copolymers from Molecular Dynamics Simulations. J. Chem. Phys. 2016, 145 (23), 234907. 10.1063/1.4972141. [DOI] [PubMed] [Google Scholar]
- Von Tiedemann P.; Blankenburg J.; Maciol K.; Johann T.; Müller A. H. E.; Frey H. Copolymerization of Isoprene with P-Alkylstyrene Monomers: Disparate Reactivity Ratios and the Shape of the Gradient. Macromolecules 2019, 52 (3), 796–806. 10.1021/acs.macromol.8b02280. [DOI] [Google Scholar]
- Fayt R.; Jérôme R.; Teyssié P. Interface Modification in Polymer Blends. Multiphase Polymers: Blends and Ionomers 1989, 58, 38–66. 10.1021/bk-1989-0395.ch002. [DOI] [Google Scholar]
- Eagan J. M.; Xu J.; Di Girolamo R.; Thurber C. M.; Macosko C. W.; LaPointe A. M.; Bates F. S.; Coates G. W. Combining Polyethylene and Polypropylene: Enhanced Performance with PE/ i PP Multiblock Polymers. Science 2017, 355 (6327), 814–816. 10.1126/science.aah5744. [DOI] [PubMed] [Google Scholar]
- Nomura K.; Peng X.; Kim H.; Jin K.; Kim H. J.; Bratton A. F.; Bond C. R.; Broman A. E.; Miller K. M.; Ellison C. J. Multiblock Copolymers for Recycling Polyethylene-Poly(Ethylene Terephthalate) Mixed Waste. ACS Appl. Mater. Interfaces 2020, 12 (8), 9726–9735. 10.1021/acsami.9b20242. [DOI] [PubMed] [Google Scholar]
- Klimovica K.; Pan S.; Lin T. W.; Peng X.; Ellison C. J.; Lapointe A. M.; Bates F. S.; Coates G. W. Compatibilization of iPP/HDPE Blends with PE-g-iPP Graft Copolymers. ACS Macro Lett. 2020, 9 (8), 1161–1166. 10.1021/acsmacrolett.0c00339. [DOI] [PubMed] [Google Scholar]
- Macosko C. W.; Jeon H. K.; Hoye T. R. Reactions at Polymer-Polymer Interfaces for Blend Compatibilization. Prog. Polym. Sci. 2005, 30 (8–9), 939–947. 10.1016/j.progpolymsci.2005.06.003. [DOI] [Google Scholar]
- Majumdar B.; Keskkula H.; Paul D. R. Effect of the Nature of the Polyamide on the Properties and Morphology of Compatibilized Nylon/Acrylonitrile-Butadiene-Styrene Blends. Polymer 1994, 35 (25), 5468–5477. 10.1016/S0032-3861(05)80010-2. [DOI] [Google Scholar]
- Borggreve R. J. M.; Gaymans R. J. Impact Behaviour of Nylon-Rubber Blends: 4. Effect of the Coupling Agent, Maleic Anhydride. Polymer 1989, 30 (1), 63–70. 10.1016/0032-3861(89)90384-4. [DOI] [Google Scholar]
- Xanthos M.; Dagli S. S. Compatibilization of Polymer Blends by Reactive Processing. Polym. Eng. Sci. 1991, 31 (13), 929–935. 10.1002/pen.760311302. [DOI] [Google Scholar]
- DOW . Polymer Compatibilizer for Recycling. https://corporate.dow.com/en-us/news/press-releases/polymer-compatibilizer-for-recycling.html (accessed Jan 4, 2022).
- Utracki L. A.Reactive Compatibilization. In Comercial Polymer Blends; Chapman & Hall, 1998; pp 94–97. [Google Scholar]
- Quirk R. P.Compatibilization of Polymer Blends. US Patent 5264491, November 23, 1993.
- Orr C. A.; Cernohous J. J.; Guegan P.; Hirao A.; Jeon H. K.; Macosko C. W. Homogeneous Reactive Coupling of Terminally Functional Polymers. Polymer 2001, 42 (19), 8171–8178. 10.1016/S0032-3861(01)00329-9. [DOI] [Google Scholar]
- Jeon H. K.; Macosko C. W.; Moon B.; Hoye T. R.; Yin Z. Coupling Reactions of End- vs Mid-Functional Polymers. Macromolecules 2004, 37 (7), 2563–2571. 10.1021/ma030581n. [DOI] [Google Scholar]
- Kim S.; Kim J. K.; Park C. E. Effect of Molecular Architecture of in situ Reactive Compatibilizer on the Morphology and Interfacial Activity of an Immiscible. Polyolefin/Polystyrene Blend 1997, 38 (8), 1809–1815. 10.1016/S0032-3861(96)00714-8. [DOI] [Google Scholar]
- Yin Z.; Koulic C.; Pagnoulle C.; Jé Rô Me R. Reactive Blending of Functional PS and PMMA: Interfacial Behavior of in situ Formed Graft Copolymers. Macromolecules 2001, 34, 5132–5139. 10.1021/ma001798+. [DOI] [Google Scholar]
- Szwarc M. ‘Living’ Polymers. Nature 1956, 178 (4543), 1168–1169. 10.1038/1781168a0. [DOI] [Google Scholar]
- Whelan D.Thermoplastic Elastomers. In Brydson’s Plastics Materials; Elsevier, 2017; Vol. 26, pp 653–703. 10.1016/B978-0-323-35824-8.00024-4. [DOI] [Google Scholar]
- Bates F. S.; Hillmyer M. A.; Lodge T. P.; Bates C. M.; Delaney K. T.; Fredrickson G. H. Multiblock Polymers: Panacea or Pandora’s Box?. Science 2012, 336 (6080), 434–440. 10.1126/science.1215368. [DOI] [PubMed] [Google Scholar]
- Wang W.; Lu W.; Goodwin A.; Wang H.; Yin P.; Kang N.-G.; Hong K.; Mays J. W. Recent Advances in Thermoplastic Elastomers from Living Polymerizations: Macromolecular Architectures and Supramolecular Chemistry. Prog. Polym. Sci. 2019, 95, 1–31. 10.1016/j.progpolymsci.2019.04.002. [DOI] [Google Scholar]
- Chen Y.; Zhang L.; Jin Y.; Lin X.; Chen M. Recent Advances in Living Cationic Polymerization with Emerging Initiation/Controlling Systems. Macromol. Rapid Commun. 2021, 42 (13), 2100148. 10.1002/marc.202100148. [DOI] [PubMed] [Google Scholar]
- Grubbs R. B.; Grubbs R. H. 50th Anniversary Perspective: Living Polymerization - Emphasizing the Molecule in Macromolecules. Macromolecules 2017, 50 (18), 6979–6997. 10.1021/acs.macromol.7b01440. [DOI] [Google Scholar]
- Beyer V. P.; Kim J.; Remzi Becer C. Synthetic Approaches for Multiblock Copolymers. Polymer Chemistry 2020, 11, 1271–1291. 10.1039/C9PY01571J. [DOI] [Google Scholar]
- Hu L.; Vuillaume P. Y. Reactive Compatibilization of Polymer Blends by Coupling Agents and Interchange Catalysts. Compat. Polym. Blends Micro Nano Scale Phase Morphol. Interphase Charact. Prop. 2020, 205–248. 10.1016/B978-0-12-816006-0.00007-4. [DOI] [Google Scholar]
- Geng Z.; Shin J. J.; Xi Y.; Hawker C. J. Click Chemistry Strategies for the Accelerated Synthesis of Functional Macromolecules. J. Polym. Sci. 2021, 59, 963–1042. 10.1002/pol.20210126. [DOI] [Google Scholar]
- Lee I.; Panthani T. R.; Bates F. S. Sustainable Poly(Lactide- b -Butadiene) Multiblock Copolymers with Enhanced Mechanical Properties. Macromolecules 2013, 46 (18), 7387–7398. 10.1021/ma401508b. [DOI] [Google Scholar]
- Pawlak A.; Morawiec J.; Pazzagli F.; Pracella M.; Galeski A. Recycling of Postconsumer Poly(Ethylene Terephthalate) and High-Density Polyethylene by Compatibilized Blending. J. Appl. Polym. Sci. 2002, 86 (6), 1473–1485. 10.1002/app.11307. [DOI] [Google Scholar]
- Zhang Y.; Zhang H.; Guo W.; Wu C. Effects of Different Types of Polyethylene on the Morphology and Properties of Recycled Poly(Ethylene Terephthalate)/Polyethylene Compatibilized Blends. Polym. Adv. Technol. 2011, 22 (12), 1851–1858. 10.1002/pat.1683. [DOI] [Google Scholar]
- Todd A. D.; McEneany R. J.; Topolkaraev V. A.; Macosko C. W.; Hillmyer M. A. Reactive Compatibilization of Poly(Ethylene Terephthalate) and High-Density Polyethylene Using Amino-Telechelic Polyethylene. Macromolecules 2016, 49 (23), 8988–8994. 10.1021/acs.macromol.6b02080. [DOI] [Google Scholar]
- Wang H.; Onbulak S.; Weigand S.; Bates F. S.; Hillmyer M. A. Polyolefin Graft Copolymers through a Ring-Opening Metathesis Grafting through Approach. Polym. Chem. 2021, 12 (14), 2075–2083. 10.1039/D0PY01728K. [DOI] [Google Scholar]
- Rose J. M.; Mourey T. H.; Slater L. A.; Keresztes I.; Fetters L. J.; Coates G. W. Poly(Ethylene-Co-Propylene Macromonomer)s: Synthesis and Evidence for Starlike Conformations in Dilute Solution. Macromolecules 2008, 41 (3), 559–567. 10.1021/ma702190c. [DOI] [Google Scholar]
- Klimovica K.; Pan S.; Lin T. W.; Peng X.; Ellison C. J.; Lapointe A. M.; Bates F. S.; Coates G. W. Compatibilization of iPP/HDPE Blends with PE-g-iPP Graft Copolymers. ACS Macro Lett. 2020, 9 (8), 1161–1166. 10.1021/acsmacrolett.0c00339. [DOI] [PubMed] [Google Scholar]
- Arriola D. J.; Carnahan E. M.; Hustad P. D.; Kuhlman R. L.; Wenzel T. T. Catalytic Production of Olefin Block Copolymers via Chain Shuttling Polymerization. Science 2006, 312 (5774), 714–719. 10.1126/science.1125268. [DOI] [PubMed] [Google Scholar]
- Shan C. L. P.; Walton K. L.; Marchand G. R.; Carnahan E. M.; Karjala T.. Crystalline Block Composites as Compatibilizers. US Patent US8822599B2, 2014.
- Hu Y.; Conley B.; Walton K. L.; Shan C. L. P.; Marchand G. R.; Patel R. M.; Kupsch E.-M.; Walther B. W.. Multilayered Polyolefin-Based Films. US Patent US9511567B2, 2013.
- Listak J.; Jakubowski W.; Mueller L.; Plichta A.; Matyjaszewski K.; Bockstaller M. R. Effect of Symmetry of Molecular Weight Distribution in Block Copolymers on Formation of “Metastable” Morphologies. Macromolecules 2008, 41 (15), 5919–5927. 10.1021/ma800816j. [DOI] [Google Scholar]
- Lynd N. A.; Meuler A. J.; Hillmyer M. A. Polydispersity and Block Copolymer Self-Assembly. Prog. Polym. Sci. 2008, 33 (9), 875–893. 10.1016/j.progpolymsci.2008.07.003. [DOI] [Google Scholar]
- Hillmyer M. A. Polydisperse Block Copolymers: Don’t Throw Them Away. J. Polym. Sci., Part B: Polym. Phys. 2007, 45 (24), 3249–3251. 10.1002/polb.21323. [DOI] [Google Scholar]
- Chen H.; Ginzburg V. Reaction: Size Distribution in Olefin Block Copolymers: Not Bad at All. Chem. 2019, 5 (3), 491–492. 10.1016/j.chempr.2019.02.018. [DOI] [Google Scholar]
- Creton C.; Kramer E. J.; Brown H. R.; Hui C.-Y. Adhesion and Fracture of Interfaces Between Immiscible Polymers: From the Molecular to the Continuum Scal. Adv. Polym. Sci. 2001, 156, 53–136. 10.1007/3-540-45141-2_2. [DOI] [Google Scholar]
- Eastwood E. A.; Dadmun M. D. Multiblock Copolymers in the Compatibilization of Polystyrene and Poly(Methyl Methacrylate) Blends: Role of Polymer Architecture. Macromolecules 2002, 35 (13), 5069–5077. 10.1021/ma011701z. [DOI] [Google Scholar]
- Gersappe D.; Harm P. K.; Irvine D.; Balazs A. C. Contrasting the Compatibilizing Activity of Comb and Linear Copolymers. Macromolecules 1994, 27 (3), 720–724. 10.1021/ma00081a015. [DOI] [Google Scholar]
- Alamo R. G.; Glaser R. H.; Mandelkern L. The Cocrystallization of Polymers: Polyethylene and Its Copolymers. J. Polym. Sci., Part B: Polym. Phys. 1988, 26 (10), 2169–2195. 10.1002/polb.1988.090261011. [DOI] [Google Scholar]
- Yuan B.-L.; Wool R. P. Strength Development at Incompatible Semicrystalline Polymer Interfaces. Polym. Eng. Sci. 1990, 30 (22), 1454–1464. 10.1002/pen.760302206. [DOI] [Google Scholar]
- Schnell R.; Stamm M.; Rauch F. Segregation of Diblock Copolymers to the Interface between Weakly Incompatible Polymers. Macromol. Chem. Phys. 1999, 200 (7), 1806–1812. 10.1002/(SICI)1521-3935(19990701)200:7<1806::AID-MACP1806>3.0.CO;2-9. [DOI] [Google Scholar]
- Shull K. R.; Kramer E. J.; Hadziioannou G.; Tang W. Segregation of Block Copolymers to Interfaces between Immiscible Homopolymers. Macromolecules 1990, 23 (22), 4780–4787. 10.1021/ma00224a006. [DOI] [Google Scholar]
- Semenov A. N. Theory of Diblock-Copolymer Segregation to the Interface and Free Surface of a Homopolymer Layer. Macromolecules 1992, 25 (19), 4967–4977. 10.1021/ma00045a024. [DOI] [Google Scholar]
- Kim S. H.; Jo W. H. A Monte Carlo Simulation of Polymer/Polymer Interface in the Presence of Block Copolymer. I. Effects of the Chain Length of Block Copolymer and Interaction Energy. J. Chem. Phys. 1999, 110 (24), 12193–12201. 10.1063/1.479156. [DOI] [Google Scholar]
- Cho D.; Hu W.; Koberstein J. T.; Lingelser J. P.; Gallot Y. Segregation Dynamics of Block Copolymers to Immiscible Polymer Blend Interfaces. Macromolecules 2000, 33 (14), 5245–5251. 10.1021/ma981699k. [DOI] [Google Scholar]
- Balazs A. C.; Siemasko C. P.; Lantman C. W. Monte Carlo Simulations for the Behavior of Multiblock Copolymers at a Penetrable Interface. J. Chem. Phys. 1991, 94 (2), 1653–1663. 10.1063/1.460715. [DOI] [Google Scholar]
- Liu D.; Gong K.; Lin Y.; Bo H.; Liu T.; Duan X. Effects of Repulsion Parameter and Chain Length of Homopolymers on Interfacial Properties of An/Ax/2BxAx/2/Bm Blends: A DPD Simulation Study. Polymers (Basel) 2021, 13 (14), 2333. 10.3390/polym13142333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Gong K.; Lin Y.; Liu T.; Liu Y.; Duan X. Dissipative Particle Dynamics Study on Interfacial Properties of Symmetric Ternary Polymeric Blends. Polymers (Basel) 2021, 13 (9), 1516. 10.3390/polym13091516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindy M. E.; Prud’homme R. K.; Panagiotopoulos A. Z. Phase Behavior and Structure Formation in Linear Multiblock Copolymer Solutions by Monte Carlo Simulation. J. Chem. Phys. 2008, 128, 164906. 10.1063/1.2905231. [DOI] [PubMed] [Google Scholar]
- Halperin A. On the Collapse of Multiblock Copolymers. Macromolecules 1991, 24 (6), 1418–1419. 10.1021/ma00006a033. [DOI] [Google Scholar]
- López-Barrón C. R.; Tsou A. H. Strain Hardening of Polyethylene/Polypropylene Blends via Interfacial Reinforcement with Poly(Ethylene- Cb -Propylene) Comb Block Copolymers. Macromolecules 2017, 50 (7), 2986–2995. 10.1021/acs.macromol.7b00264. [DOI] [Google Scholar]
- Datta S.; Lohse D. J. Graft Copolymer Compatibilizers for Blends of Isotactic Polypropylene and Ethene-Propene Copolymers. 2. Functional Polymers Approach. Macromolecules 1993, 26 (8), 2064–2076. 10.1021/ma00060a040. [DOI] [Google Scholar]
- Embree K.DOW’s Breakthrough Barrier Film Technology Makes Stand-Up Pouches Recyclable. Plastics Today, June 21, 2016; https://www.plasticstoday.com/packaging/dows-breakthrough-barrier-film-technology-makes-stand-pouches-recyclable.
- Charoensirisomboon P.; Inoue T.; Weber M. Pull-out of Copolymer in situ-Formed during Reactive Blending: Effect of the Copolymer Architecture. Polymer 2000, 41 (18), 6907–6912. 10.1016/S0032-3861(00)00025-2. [DOI] [Google Scholar]
- Thurber C. M.; Xu Y.; Myers J. C.; Lodge T. P.; Macosko C. W. Accelerating Reactive Compatibilization of PE/PLA Blends by an Interfacially Localized Catalyst. ACS Macro Lett. 2015, 4 (1), 30–33. 10.1021/mz500770y. [DOI] [PubMed] [Google Scholar]
- Graziano A.; Jaffer S.; Sain M. Review on Modification Strategies of Polyethylene/Polypropylene Immiscible Thermoplastic Polymer Blends for Enhancing Their Mechanical Behavior. J. Elastomers Plast. 2019, 51 (4), 291–336. 10.1177/0095244318783806. [DOI] [Google Scholar]
- Song J.; Ewoldt R. H.; Hu W.; Craig Silvis H.; Macosko C. W. Flow Accelerates Adhesion between Functional Polyethylene and Polyurethane. AIChE J. 2011, 57 (12), 3496–3506. 10.1002/aic.12551. [DOI] [Google Scholar]
- Dogan S. K.; Reyes E. A.; Rastogi S.; Ozkoc G. Reactive Compatibilization of PLA/TPU Blends with a Diisocyanate. J. Appl. Polym. Sci. 2014, 131 (10), 40251. 10.1002/app.40251. [DOI] [Google Scholar]
- Lu Q.-W.; Hoye T. R.; Macosko C. W. Reactivity of Common Functional Groups with Urethanes: Models for Reactive Compatibilization of Thermoplastic Polyurethane Blends. J. Polym. Sci. Part A Polym. Chem. 2002, 40 (14), 2310–2328. 10.1002/pola.10310. [DOI] [Google Scholar]
- Abdul Razak N. C.; Inuwa I. M.; Hassan A.; Samsudin S. A. Effects of Compatibilizers on Mechanical Properties of PET/PP Blend. Compos. Interfaces 2013, 20 (7), 507–515. 10.1080/15685543.2013.811176. [DOI] [Google Scholar]
- Papadopoulou C. P.; Kalfoglou N. K. Comparison of Compatibilizer Effectiveness for PET/PP Blends: Their Mechanical, Thermal and Morphology Characterization. Polymer 2000, 41 (7), 2543–2555. 10.1016/S0032-3861(99)00442-5. [DOI] [Google Scholar]
- Holsti-Miettinen R.; Seppälä J.; Ikkala O. T. Effects of Compatibilizers on the Properties of Polyamide/Polypropylene Blends. Polym. Eng. Sci. 1992, 32 (13), 868–877. 10.1002/pen.760321306. [DOI] [Google Scholar]
- Shi D.; Ke Z.; Yang J.; Gao Y.; Wu J.; Yin J. Rheology and Morphology of Reactively Compatibilized PP/PA6 Blends. Macromolecules 2002, 35 (21), 8005–8012. 10.1021/ma020595d. [DOI] [Google Scholar]
- Eriksen M. K.; Pivnenko K.; Faraca G.; Boldrin A.; Astrup T. F. Dynamic Material Flow Analysis of PET, PE, and PP Flows in Europe: Evaluation of the Potential for Circular Economy. Environ. Sci. Technol. 2020, 54 (24), 16166–16175. 10.1021/acs.est.0c03435. [DOI] [PubMed] [Google Scholar]
- Wang D.; Li Y.; Xie X. M.; Guo B. H. Compatibilization and Morphology Development of Immiscible Ternary Polymer Blends. Polymer 2011, 52 (1), 191–200. 10.1016/j.polymer.2010.11.019. [DOI] [Google Scholar]
- Li H.; Xie X. M. Morphology Development and Superior Mechanical Properties of PP/PA6/SEBS Ternary Blends Compatibilized by Using a Highly Efficient Multi-Phase Compatibilizer. Polymer 2017, 108, 1–10. 10.1016/j.polymer.2016.11.044. [DOI] [Google Scholar]