

SUMMARY
This study addresses the roles of nuclear receptor corepressor 2 (NCOR2) in prostate cancer (PC) progression in response to androgen deprivation therapy (ADT). Reduced NCOR2 expression significantly associates with shorter disease-free survival in patients with PC receiving adjuvant ADT. Utilizing the CWR22 xenograft model, we demonstrate that stably reduced NCOR2 expression accelerates disease recurrence following ADT, associates with gene expression patterns that include neuroendocrine features, and induces DNA hypermethylation. Stably reduced NCOR2 expression in isogenic LNCaP (androgen-sensitive) and LNCaP-C4–2 (androgen-independent) cells revealed that NCOR2 reduction phenocopies the impact of androgen treatment and induces global DNA hypermethylation patterns. NCOR2 genomic binding is greatest in LNCaP-C4–2 cells and most clearly associates with forkhead box (FOX) transcription factor FOXA1 binding. NCOR2 binding significantly associates with transcriptional regulation most when in active enhancer regions. These studies reveal robust roles for NCOR2 in regulating the PC transcriptome and epigenome and underscore recent mutational studies linking NCOR2 loss of function to PC disease progression.
In brief
Long et al. show that reduced levels of NCOR2 lead to accelerated prostate cancer recurrence during androgen withdrawal in a patient-derived xenograft model. NCOR2 reduction is characterized by incomplete response to androgen withdrawal, and recurrent tumors show increased neuroendocrine traits. These phenotypic changes are associated with hypermethylated enhancers.
Graphical abstract
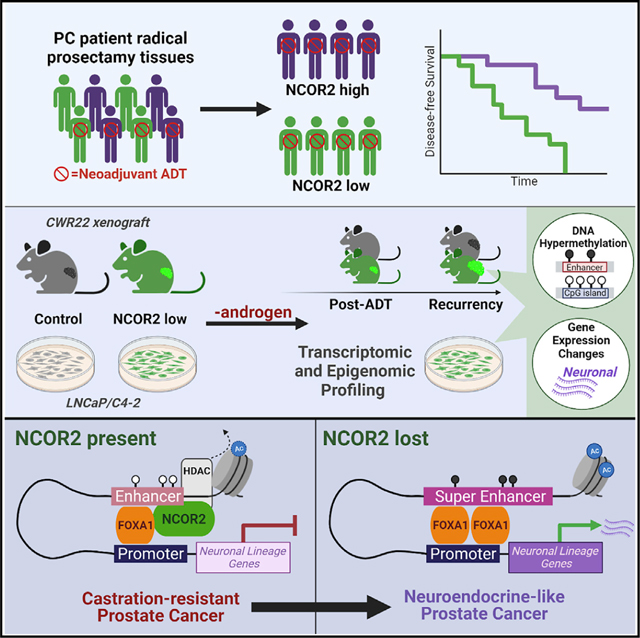
INTRODUCTION
Prostate cancer (PC) patients with advanced disease receive androgen deprivation therapy (ADT) but frequently experience treatment failure leading to ADT-resistant PC (ADT-RPC). Treatment failure arises because, in part, oncogenic events lead to corruption of androgen receptor (AR) signaling by several mechanisms, including structural variation and distorted interactions with coregulator proteins (Henzler et al., 2016, Chen et al., 2018, Viswanathan et al., 2018). These events combine with epigenetic switching, defined as alterations in histone modifications and/or DNA methylation that alter accessibility to transcription factors, whereby AR genomic binding is redirected (Roe et al., 2017, Pihlajamaa et al., 2014), including the recommissioning of embryonic enhancers (Pomerantz et al., 2020). As a result, AR signaling no longer regulates luminal differentiation, and alternative programs are enhanced, leading to novel lineages such as neuroendocrine prostate cancer (NEPC) (Beltran et al., 2019, Sheahan and Ellis, 2018, Ku et al., 2017). Understanding the mechanisms that change AR genomic interactions have the potential to be exploited to sustain and augment ADT.
Nuclear receptor corepressor 2/silencing mediator for retinoid and thyroid hormone receptors (NCOR2) has been identified as a frequently altered corepressor in PC and other cancers (Khanim et al., 2004, Girault et al., 2003). Notably, NCOR2 is among the top 5 mutated corepressors in the SU2C study of 444 men with advanced PC (Abida et al., 2019, Armenia et al., 2018). The current study addresses the role of NCOR2 in determining the effectiveness of ADT.
NCOR2 binds AR and other nuclear hormone receptors (NRs) (Hu and Lazar, 1999) and allosterically interacts with histone deacetylases (Li et al., 2000, Jin et al., 2018) to promote repressive histone marks such as histone H3 lysine 9 trimethylation (H3K9me3), which recruit the C–phosphate–G (CpG) methylation machinery (Hashimoto et al., 2010). Similarly, NCOR2 interacts with KAISO (Yoon et al., 2003) and with the long non-coding RNA (lncRNA) SHARP in both cases to trigger DNA methylation (McHugh et al., 2015). Therefore, in PC, we and others have reasoned that expression and mutation changes in NCOR2 disrupt its ability to regulate the epigenome and thus rewires AR-genomic interactions and impacts the duration and success of ADT (Khanim et al., 2004).
However, NCOR2 is not an obligate co-repressor. Murine transgenic approaches reveal histone deacetylase (HDAC)-independent roles for NCOR2 (Jepsen et al., 2000, Bhaskara et al., 2008, You et al., 2013). NCOR2 significantly accumulates at open chromatin and actively transcribed genomic regions (Long et al., 2015) and can actively enhance transcription by estrogen receptor-β (ERβ) (Peterson et al., 2007) and AR (Laschak et al., 2011). It also remains unclear how and where NCOR2 interacts with the genome, given the diversity of interacting transcription factors (Lee et al., 2000, Zhuang et al., 2018).
More broadly, it is challenging to define NCOR2-dependent cistrome-transcriptome relationships that are positive or negative given the non-linearity of gene-enhancer relationships (Roadmap Epigenomics Consortium et al., 2015, Jung et al., 2019), which are further shaped by topologically associated domains (TADs; reviewed in Campbell, 2019). Furthermore, it is unclear how NCOR2-triggered CpG methylation impacts transcription. Elevated DNA methylation at promoter regions of high CpG density results in transcriptional silencing (reviewed in Long et al., 2017) but in low-density CpG regions; for example, within enhancers, DNA methylation may selectively recruit transcription factor binding (Yin et al., 2017) and participate in allele-specific gene regulation (Song et al., 2019).
Thus, it is unknown how and where NCOR2 is recruited to the genome in PC progression, how this relates to DNA methylation, and how this positively or negatively impacts gene expression programs that are required for recurrence during ADT. We sought to define how NCOR2 expression impacts gene expression and determines androgen-dependent and androgen-independent transcriptional signaling in progression to ADT-RPC. The CWR22 xenograft model of PC progression was utilized to examine the role of NCOR2 in determining response to ADT in vivo and associations with molecular features of ADT-RPC in patients. Lastly, we utilized isogenic models of androgen-sensitive (LNCaP) and ADT-resistant (LNCaP-C4–2) PC cells to map the global regulatory functions of NCOR2 with regard to its cistrome, transcriptome, and DNA methylome.
RESULTS
Lower NCOR2 expression levels are associated with shorter disease-free survival
Genomic analyses in PC have identified frequent changes to NCOR2 (Robinson et al., 2015b), supporting a cancer-driver role for this co-repressor. We queried seven cohorts of PC clinical samples (Figure 1A) and found that NCOR2 expression and function is disrupted through a variety of mutations and alterations (Figure 1B). However, the functional significance of these clinically relevant NCOR2 alterations is unexplored. Although there are both copy number gains and amplifications, we have chosen to model loss of NCOR2 based on converging lines of evidence supporting an important role for loss of NCOR2 functionality. As shown in Figures 1B and 1C, negative disruptions of NCOR2 through shallow or deep deletions or mutations resulting in truncations or other predicted loss of function mutations are more common than copy number increases. Moreover, NCOR2 expression was examined in a 707-patient tissue microarray of radical prostatectomy (RP) samples by immunohistochemistry (Table S1; Figure 1D). Notably, a patient subset (n = 136) received adjuvant ADT before (n = 126) or following RP (n = 10). Univariate regression analyses identified significant associations of NCOR2 expression (H-score) in patients with race (decreased in African Americans), body mass index (BMI; decreased in overweight/obese), presurgical prostate-specific antigen (PSA; decreased with presurgical PSA > 4 ng/mL), and adjuvant ADT. Multivariate regression identified additional association with Gleason sum (decreased with Gleason sum 8+) (Table S2).
Figure 1. NCOR2 status in clinical samples.

(A) Percent of adenocarcinoma (Cancer Genome Atlas Research Network, 2015) (blue), metastatic (Abida et al., 2019, Robinson et al., 2015a, Armenia et al., 2018, Taylor et al., 2010) (red), and neuroendocrine (Beltran et al., 2016) (green) PC patient samples with NCOR2 genomic alterations.
(B) Proportion of NCOR2 mutation and alteration types from (A) (n = 2,291).
(C) Distribution of mutations along the human NCOR2 protein; RD, repression domain; SANT, SW13/ADA2/NCOR/TFIIB-like domain; DAD, deacetylase-activating domain; HID, histone-interacting domain; ID, protein interaction domain.
(D) Distribution of protein expression (H score) by IHC in the RPCCC PC TMA. The red line indicates median expression; NCOR2-low tumors, green; NCOR2-high tumors, purple. Representative tissue cores and H scores from six individual patients showing low and high nuclear staining of NCOR2 (below).
(E) BCR survival assessment of patients with high and low NCOR2 that did or did not receive adjuvant ADT with RP. Significance of Cox proportional hazards regression (log rank test); significant shifts in survival (HR, hazard ratio).
Univariate and multivariate Cox proportional hazards regression revealed relationships between clinical variables and time to biochemical recurrence (BCR) (Table S3). Gleason sum and pathologic stage were identified as significant indications of reduced BCR survival (Figure S1A). Patients receiving adjuvant ADT also had reduced survival but had a significantly skewed distribution of Gleason sum (Figure S1B), consistent with the fact that patients diagnosed with aggressive primary disease are more likely to be given adjuvant ADT (Siddiqui and Krauss, 2018).
Following normalization for either Gleason sum, pathologic stage, race, or BMI, NCOR2 levels (median cut-off) did not stratify survival of patients (Figures S1C and S1D). Strikingly, however, reduced NCOR2 significantly associated with worse BCR survival in patients receiving adjuvant ADT (Figure 1E). No such relationships were observed in patients who received surgery without ADT. These observations strongly support the concept that reduced NCOR2 dampens response to ADT in PC patients.
Reduced NCOR2 expression accelerates disease progression in vivo
We screened a panel of short hairpin RNAs (shRNAs) against NCOR2 and identified two that reduced NCOR2 mRNA and protein by a maximum of ~50% in the LNCaP and LNCaP-C4–2 cell lines (Figures S2A–S2D). Predictable growth responses toward R1881 and enzalutamide were found in both cell lines (Figure S2E), and dihydrotestosterone (DHT) exposure did not alter NCOR2 expression (Figure S2F).
We assessed the impact of reduced NCOR2 expression in the CWR22 xenograft model, which, like for the disease in men, requires androgens for primary growth and exhibits an initial strong response to ADT followed by recurrence over a 3- to 9-month window. A NCOR2-targeting shRNA (shNCOR2) or non-targeting shRNA control (shCTL) were introduced to digested CWR22 tissue via lentiviral infection under puromycin selection for 24 h before implantation. A total of 130 animals were inoculated with CWR22-shCTL or CWR22-shNCOR2 tissue (n = 65 per group), with 100 animals (n = 50 per group) designated for follow-up through to recurrence or end of study. Control and shNCOR2 CWR22 tumors were allowed to grow in the presence of androgens until they reached approximately 0.3 cm3 (two consecutive measurements of >0.3 cm3) before androgen withdrawal (Figures S3A and S3B). Tumors were collected at three time points: (1) once tumors had established, just before androgen withdrawal (PRE), (2) 7 days after androgen withdrawal (POST), and (3) recurrent tumors following regrowth to >150% of the tumor volume just before androgen withdrawal (RT).
Green fluorescent protein (GFP) detection of the shRNA constructs and reduced NCOR2 levels were confirmed at all stages of disease (Figures 2A and 2B; Figure S3C). Expression of androgen-regulated genes was acutely repressed by ADT but became reexpressed in recurrence concomitant with increased AR expression (Figure S3D) (Kim et al., 2002). Reduced NCOR2 expression did not impact the primary androgen-stimulated growth rate of tumors before ADT (Figure 2C). However, the initial ADT response, as measured by maximum tumor regression after androgen withdrawal, was significantly reduced in shNCOR2 tumors (Figure 2D). Thirty-six out of 46 (78%) control tumors achieved >40% reduction in tumor size post-AD (mean regression = 54%). However, only 27 out of 49 shNCOR2 tumors (55%) reached >40% regression (mean maximum regression = 45%) (chi-square = 0.027). Furthermore, while Ki-67 staining was starkly reduced 7 days after ADT in both control and shNCOR2 tumors, there was significantly more Ki-67 staining remaining in shNCOR2 tumors, indicative of a dampened ADT response (Figure 2E).
Figure 2. NCOR2 loss alters response to ADT in the CWR22 model of PC progression.

(A) Representative fluorescent imaging of xenograft tumors at point of recurrence >300 days post-ADT.
(B) Relative NCOR2 expression in select tumors pre-ADT (n = 5,5), post-ADT (n = 10,10), and in RT (n = 22,28); *p < 0.05 significant difference between respective distribution and that of shCTL pre-ADT tumors.
(C) Violin plots showing the distribution of tumor sizes at time of ADT (left) and time to reach ADT (right).
(D) Violin plots showing the distribution of maximum regression after androgen withdrawal for each tumor (left) and overall proportions of tumors that reached 40% regression (right). Data are summarized as boxplots representing lower, middle, and upper quartiles. Whiskers indicate maximum and minimum values, excluding outliers defined as 1.5× interquartile range (IQR) (B–D).
(E) Representative IHC (left) and quantification (right) of Ki-67 staining in select tumors pre-ADT (n = 2 each), post-ADT (n = 6 each), and in RT (n = 3 each); *p < 0.05. Data are reported as mean ± standard deviation (SD).
(F) Kaplan-Meier representation of the recurrence-free survival proportions in shCTL and shNCOR2 tumors post-ADT (n = 50 each).
Recurrence rates were determined in 50 xenografts each for shCTL and shNCOR2. Control tumor recurrence rates reflected previous findings (Su et al., 2013, Affronti et al., 2017, Seedhouse et al., 2016). However, as shown in Figure 2F, knockdown of NCOR2 significantly reduced the time to recurrence from a median of 232 days to 180 days (log rank = 0.021; hazard ratio = 1.74). Interestingly, significantly reduced NCOR2 expression was found in control recurrent tumors (RT) (Figure 2B), suggesting that reduced NCOR2 expression is a common event following ADT. Nevertheless, having reduced NCOR2 before ADT resulted in more rapid recurrence.
NCOR2 knockdown accelerates the molecular features of ADT resistance in vivo
Transcriptome and DNA methylome profiling was performed to assess the molecular impact of NCOR2 knockdown at various stages of disease progression (n = 5 each for shCTL and shNCOR2 at three stages of progression; PRE, POST, and RT). No NCOR2-associated expression changes were observed in tumors before ADT (Figure S3E). However, significant expression changes were observed in post-ADT (96 total differentially expressed genes [DEGs]; 50 upregulated and 46 downregulated) and RT (529 DEGs; 225 upregulated and 304 downregulated).
Gene set enrichment analysis (GSEA) confirmed stark suppression of proliferative pathways in POST relative to PRE tumors (Figure S3F), corroborating Ki-67 expression patterns (Figure 2E). Furthermore, when comparing recurrent tumors with or without knockdown of NCOR2, we found enrichment of gene sets related to interferon response and epithelial-mesenchymal transition (EMT) (Figure S4C). Assessment of gene sets previously used to identify ADT-resistant phenotypes (Bluemn et al., 2017) revealed a transient depletion of AR and fibroblast growth factor receptor (FGFR) signaling post-ADT that was reestablished in recurrence as expected (Figure 3A). These patterns were not affected by NCOR2 status. However, recurrent tumors with knockdown of NCOR2 (RT-shNCOR2) displayed increased enrichment for neuroendocrine signaling over shCTL counterparts (Figure 3A). This observation was typified by increased SYP, CHGB, and NTN1 and reduced AR mRNA expression as well as elevated SYP protein levels as assessed by immunohistochemistry (IHC) in RT-shNCOR2 tumors (Figures 3B and 3C).
Figure 3. NCOR2 loss accelerates the molecular features of ADT resistance during PC progression in vivo.

(A) Gene set variation analysis of established pathways associated with ADT-R in PC.
(B) Gene expression profiles for SYP, CHGB, NTN1, and AR in CWR22 groups.
(C) IHC showing representative examples of SYP expression in shCTL-RT and shNCOR2-RT samples (left); scale bars, 20 μm. Quantification of IHC in RT is shown on the right. Data are summarized as boxplots representing lower, middle, and upper quartiles. Whiskers indicate maximum and minimum values, excluding outliers defined as 1.5× IQR (A–C).
(D) Heatmap of scaled expression depicting the 41 most significant DEGs in NCOR2-low relative to NCOR2-high tumors within the SU2C cohort that were also identified in RT-shNCOR2 (left). Column annotations show grouped neuroendocrine scores (NE.quartiles) for each tumor. Kaplan-Meier curves of overall survival proportions between major tumor clusters are identified.
(E) Volcano plots depicting NCOR2-dependent DMPs (red = upregulated, blue = downregulated) determined at different stages of disease.
(F) Proportion of DMPs in each comparison; hypermethylated, red; hypomethylated, blue.
(G) Total number of NCOR2 and stage-specific DMPs in PRE-ADT and RT.
(H) Representative genomic view of the HOXA3 locus showing the average methylation levels within each CWR22 group; CpG island regions, purple; arrows, example DMPs; mean value, black dot; IQR, horizontal lines.
(I) Binning analysis depicting the median methylation calculated for genomic regions relative to TSS (top) and CpG island (bottom) loci. For TSS, each bin represents 100 bp, with the TSS centered at bin 50. For CpG islands, shore (orange) and shelf (blue) bins represent 200 bp, while islands (green) are variable depending on genomic length but centered on bin 30.
We investigated how NCOR2-dependent gene expression in CWR22 recurrence associated with outcome in patients receiving ADT in the SU2C cohort (Abida et al., 2019). Patient tumors (n = 270) were categorized by NCOR2-high or NCOR2-low expression (quartile) and DEGs identified for each subgroup with additional filtering (>1.5 Z scores, >20% of tumors) to identify genes most strongly associated with NCOR2 status. These patient-derived NCOR2-dependent genes were then overlapped with NCOR2-dependent RT-associated genes identified in CWR22, resulting in a 41-gene set that separated patients into two major tumor clusters that were significantly associated with neuroendocrine score (Abida et al., 2019) (upper versus lower quartile, chi-square = 0.014) (Figure 3D; Figure S4A). Furthermore, univariate Cox proportional hazards regression revealed tumor cluster membership significantly associated with risk of death following ADT (hazard ratio = 2.54; 95% confidence interval [95% CI] = 1.11 to 5.82; score (log rank) test = 0.021).
Given the links between NCOR2 and DNA methylation machinery, we examined changes in DNA methylation following NCOR2 knockdown and androgen withdrawal using the Infinium EPIC array platform. Differential methylation analyses (false discovery rate [FDR] < 0.05, 10% change in methylation) demonstrated that NCOR2 knockdown induced broad DNA hypermethylation in the PRE-ADT setting, which became less apparent 7 days POST-ADT but greatly enhanced after recurrence (Figure 3E). Importantly, recurrence itself, regardless of NCOR2 status, is a major driver of hypermethylation events with over 170,000 hypermethylated positions comparing shCTL-PRE with shCTL-RT (Figure 3F). However, NCOR2-associated differentially methylated positions (DMP) strongly overlapped with stage-specific DMPs (Figure 3G). We found that shNCOR2 tumors were significantly more hypermethylated than shCTL counterparts within each stage of disease (Figure S4B). Even before the stress of ADT, loss of NCOR2 leads to subtle increases in methylation at loci that gain high-level methylation following recurrence. For example, progressive hypermethylation was observed broadly at the HOXA3 locus (Figure 3H). Binning analyses revealed that progressive and NCOR2-dependent hypermethylation was enriched at regions distal to transcription start site (TSS) and CpG island loci (Figure 3I), and ChromHMM-defined chromatin states in LNCaP cells (Hamada et al., 2015, Ernst and Kellis, 2012, Valdés-Mora et al., 2017) indicated enrichment of DMPs at active and poised enhancer regions (Figure S4C). Gene-annotated differentially methylated region (DMR) analysis for NCOR2-dependent methylation in CWR22 recurrence revealed 32 genes that were also differentially expressed in the same comparison, including several associated with neuronal development (e.g., CHGA and NTN1).
Intriguingly, hypermethylation in ADT-RPC was observed in a pilot study of primary and unpaired ADT-RPC human tumor samples (local recurrences; Figure S5B). Genomic annotation identified ADT-RPC DMP enrichment at active and poised enhancer regions, reflecting the hypermethylation associated with CWR22 recurrence and with NCOR2 loss in vitro and in vivo (Figure S4D). This suggests that enhancer hypermethylation is a general phenomenon associated with ADT-RPC.
NCOR2 impacts DHT-dependent and DHT-independent transcriptomes
Given the relationships between reduced NCOR2 and ADT, we measured the impact of stable NCOR2 knockdown in the isogenic LNCaP and LNCaP-C4–2 (C4–2) cell lines on DHT-dependent (10 nM, 6 h) and DHT-independent transcriptomes via RNA sequencing (RNA-seq). Principal component analyses revealed that experimental conditions explained the majority of variation in expression (Figures S5A and S5B). DEGs (FDR < 0.05, fold change [FC] > 1.2) were determined as either DHT-dependent in CTL cells (Figure 4A, magenta), DHT-dependent in NCOR2-knockdown cells (green), or NCOR2-dependent in the absence of DHT (orange). There were more DHT-dependent DEGs in LNCaP cells (1,396 total; 917 upregulated and 479 downregulated) than in C4–2 cells (700 total; 423 upregulated and 277 downregulated) (Figures 4A and 4B). Conversely, the NCOR2-dependent transcriptome was strikingly larger in C4–2 cells (2,138 total; 1,331 upregulated and 807 downregulated) than in LNCaP cells (444 total; 252 upregulated and 192 downregulated).
Figure 4. Identifying NCOR2- and DHT-dependent gene expression patterns in LNCaP and C4–2 cells.

(A) Venn diagram depicting DEGs determined in each comparison; NCOR2 dependent (shNCOR2-EtOH/shCTL-EtOH), orange; DHT dependent in shCTL cells (shCTL+DHT/shCTL-EtOH), magenta; DHT dependent in shNCOR2 cells (shNCOR2+DHT/shNCOR2-EtOH), green.
(B) Heatmap of DHT- or NCOR2-dependent DEGs in LNCaP (left) and C4–2 (right).
(C) GSEA results of selected significantly (p adjusted ≤ 0.05) enriched gene sets related to androgen response, endocrine resistance, epigenetic regulation, neuronal differentiation, EMT, and cell cycle (RB/p53) programs; NS, non-significant.
(D) LISA analysis of DHT- and NCOR2-dependent DEGs. Each point represents a single ChIP-seq dataset queried from the CistromeDB. The top 20 most significant enrichments for both up- and downregulated genes are highlighted.
While loss of NCOR2 expression did not alter a majority of the DHT capacity (genes affected) or sensitivity (magnitude of change), there were, nevertheless, an additional 335 and 148 genes that gained DHT regulation, and, conversely, 465 and 194 genes lost DHT regulation. These findings indicate a selective skewing of transcriptional response to DHT stimulation following reduced NCOR2 levels.
To infer functional insight, GSEA was applied using a previously compiled ~4,000-gene set list from MSigDB and other prostate-specific gene sets (Table S4). Normalized enrichment scores of representative significantly enriched (p adjusted ≤ 0.05) gene sets from selected categories are shown in Figure 4C. Strong enrichment of androgen response pathways was induced by both DHT stimulation and NCOR2 knockdown, suggesting that reduced NCOR2 expression partially phenocopies DHT exposure. There was substantial commonality between NCOR2- and DHT-dependent pathways, including gene sets related to endocrine therapy resistance. However, a subset of functional gene sets showed distinct cell type enrichment. Gene sets related to epigenetic regulation were significantly enriched in LNCaP cells but not in C4–2 cells. Conversely, gene sets related to neuronal differentiation were positively enriched in C4–2 cells yet negatively enriched in LNCaP cells. Gene sets related to EMT and RB/p53 cell cycle programs displayed similar trends, further supporting AR-independent functions of NCOR2.
Transcriptional master regulator (MR) analysis of DEGs was applied using LISA (Qin et al., 2020) (Figure 4D). DHT-dependent DEGs enriched strongly for AR and the pioneer factor forkhead box (FOX) transcription factor FOXA1 in both cell lines. FOXA1 was also enriched in NCOR2-dependent transcriptomes as were a more diverse pool of regulators, including E2F and the methyl-binding factor MBD2. In C4–2 cells, several factors associated with neuroendocrine differentiation were identified, including FOXM1 and MYCN. These regulator enrichments were largely confirmed by MR analysis performed via iRegulon (Table S4).
These analyses strongly support the concept that reduced NCOR2 levels alter the DHT response by (1) skewing the capacity of the DHT-regulated transcriptome and by (2) mimicking a subset of the androgen transcriptional response in the absence of DHT stimulation. In C4–2 cells, the impact of reduced NCOR2 alone was significantly more extensive than in LNCaP cells and enriched for cell cycle and neuronal differentiation responses. These findings are consistent with increased neuroendocrine gene expression in CWR22 recurrent tumors that developed with knockdown of NCOR2 (Figures 3A and 3B).
Reducing NCOR2 expression induces hypermethylation at enhancer regions
We examined changes in DNA methylation following NCOR2 knockdown and DHT treatment in LNCaP and C4–2 cells. Differential methylation analyses (FDR < 0.05, 10% change in methylation; Figures S5C and S5D) revealed substantial hypermethylation following NCOR2 knockdown. NCOR2 knockdown induced DMPs in LNCaP cells (25,843 total DMPs; 98% hypermethylated) and C4–2 cells (184,663 total DMPs; 82% hypermethylated) (Figure 5A). Notably, C4–2 DMPs largely encompassed DMPs observed in LNCaP cells (50%) (Figure 5B). DHT treatment had no significant effect on DNA methylation in either cell line (Figure S5E).
Figure 5. NCOR2 loss results in a hypermethylation phenotype in LNCaP and C4–2 cells.

(A) Volcano plots representing DMPs following NCOR2 knockdown in LNCaP (left) and C4–2 (right) cells, hypomethylated, blue; hypermethylated, red.
(B) Venn diagram of DMPs identified in each cell.
(C) Binning analysis depicting the median methylation calculated for genomic regions relative to TSS (left) and CpG island (right) loci. For TSS, each bin represents 100 bp, with the TSS centered at bin 50. For CpG islands, shore (orange) and shelf (blue) bins represent 200 bp, while islands (green) are variable depending on genomic length but centered on bin 30.
(D) Relative proportions of all CpGs (top) or DMPs (middle, bottom) that annotate to ChromHMM regions.
(E) Peak centered densities of non-DMP CpG sites (black) or hypermethylated DMPs (red) centered at ChromHMM promoter or enhancer regions.
(F) Comparison of NCOR2 knockdown gene expression changes annotated with DMR promoters and/or DMR enhancers relative to genes not annotated. Distributions are compared by Kolmogorov-Smirnov (KS) test. Mean value (black dot) and IQR (horizontal lines) are shown.
(G) Representative genomic view of an NCOR2-dependent gene locus with annotated enhancer hypermethylation (GPR108). Tracks (from top), RefSeq gene (exons [red] and introns [black]); methylation detected in shCTL (black) or shNCOR2 C4–2 cells (green, blue); ChromHMM regions (color code is the same as shown in [D]); DMR regions (green); CpG island regions (purple).
Concordant with observations in the CWR22-shNCOR2 model, the NCOR2-dependent shifts in DMPs in both cell lines were most pronounced at sites distal to TSSs and CpG islands (Figure 5C). We exploited ChromHMM-defined chromatin states in LNCaP cells (Hamada et al., 2015, Ernst and Kellis, 2012, Valdé s-Mora et al., 2017) to reveal that hypermethylated DMPs (hyper-DMPs) were observed at a lower-than-expected rate at active promoter loci in both LNCaP and C4–2 cells and were conversely enriched in poised and active enhancer regions (21% background, 45% hyper-DMPs [LNCaP], 44% hyper-DMPs [C4–2]) (Figure 5D; Figure S6A). Peak centered densities of non-DMP CpG sites (black) or hypermethylated DMPs (red) centered at ChromHMM promoter or enhancer regions (Figure 5E) confirm the deenrichment of methylation changes at promoter regions but strong enrichment at enhancers.
More stringent definitions identified 929 and 7,845 DMRs (FDR < 0.05, Δ regional methylation > 5%) (Peters et al., 2015) in LNCaP and C4–2 cells, respectively, with >99% of these being hyper-DMRs (Figure S6B). A substantial proportion of DMRs mapped to at least one enhancer region (LNCaP = 37%; C4–2 = 29%), with fewer mapping to promoter regions (LNCaP = 30%; C4–2 = 21%). DMRs were also annotated to gene-proximal enhancers or promoters (enhancer region within±20 kb of a TSS; promoter region within ±2 kb of a TSS) (Figure S6C). In both cell lines, gene expression was significantly more affected by NCOR2 knockdown at genes with DMR-associated enhancers or promoters than genes without (Figure 5F). DMR enhancers tended to be distinct from promoter regions and exclusive of CpG islands, as shown at representative loci (Figure 3G; Figure S6D).
GSEA of NCOR2-dependent, DMR enhancer-associated genes did not enrich for androgen-regulated pathways (Figure S6E; Table S5) but rather for pathways associated with cancer and endocrine therapy resistance. DMR pathways unique to LNCaP cells were strongly enriched in interferon response, while those unique to C4–2 cells included neuronal and P53 pathways. Thus, both the DEGs and the DMRs that are dependent on NCOR2 knockdown in C4–2 cells affect pathways known to contribute to ADT resistance in men and are associated with increased neuroendocrine gene expression in CWR22 recurrent tumors that developed with knockdown of NCOR2 (Figures 3A and 3B).
The NCOR2 cistrome is regulated by DHT and associates with FOXA1
To gain further insight into why knockdown of NCOR2 led to more rapid recurrence during ADT and increased NEPC gene signature expression in recurrent tumors, we defined the NCOR2 cistromes in LNCaP and C4–2 cells in the presence or absence of DHT via NCOR2 chromatin immunoprecipitation with sequencing (ChIP-seq). The NCOR2 cistromes were largely distinct to each cell line and DHT stimulation (Figure 6A). In total, there were 1,258 and 1,908 robust NCOR2-binding peaks in untreated LNCaP and C4–2 cells, respectively. DHT exposure redistributed and increased the number of peaks to 1,473 and 2,080, respectively.
Figure 6. NCOR2 cistromes in LNCaP and C4–2 cells.

(A) Venn diagram of significant peaks identified in each condition.
(B) Relative genomic proportions of all ChromHMM regions (left) or NCOR2 cistromes that annotate to ChromHMM regions.
(C) Motif analysis of NCOR2 cistromes in LNCaP (left) and C4–2 (right) cells.
(D) Word cloud depicting the frequencies of factors observed in the top 200 most overlapping datasets from the complete CistromeDB with each NCOR2 cistrome. Factors with observed proportions near background (<1.2-fold enrichment) were removed.
(E) Heatmap of the mean normalized overlaps observed for each factor with each NCOR2 cistrome within the prostate subset of the CistromeDB (left). Peak centered densities of NCOR2 cistromes (LNCaP) against a publicly available LNCaP FOXA1 ChIP-seq dataset (Sunkel et al., 2016) (right). Mean normalized heatmap for histone mark datasets available within the prostate subset of the CistromeDB.
(F) Functional enrichment analysis of genes annotated to NCOR2 cistromes. Displayed are selected significantly enriched gene sets (q value < 0.05) related to androgen response, endocrine resistance, epigenetic regulation, neuronal differentiation, stem cell, EMT, and FOX transcription factor targets (FOX).
In LNCaP cells, almost half of peaks (45%) were within 3 kb of a TSS, which was modestly increased with DHT treatment (50%) (Figure 6B; Figures S7A and S7B). By contrast, the NCOR2 cistrome in C4–2 cells was more distal, with the highest proportions (~40%) of peaks falling between 10 and 100 kb of the closest TSS in basal and DHT-treated conditions. NCOR2 binding was enriched in enhancers and diminished in Polycomb-associated regions. NCOR2 binding was also enriched in promoter regions (Figure 6B). Motif analysis of NCOR2 peaks revealed significant enrichment for several NR response elements, including those of PPARs, RARs, THR, ERRα, GR, and PGR. Notably, NR motifs were more evident following DHT exposure, although some orphan NR (COUP-TFII, EAR2) enrichments were observed under basal conditions (Figure 6C; Figure S7C). Interestingly, in neither cell type nor under any condition were motifs for the AR half-site significantly enriched. NR motifs only represented a small proportion of significant enrichments, and, although the genomic sites of NCOR2 binding were largely unique across cell lines, the motif analyses revealed many similar enrichment patterns across conditions. FOX transcription factors (e.g., FOXA1, FOXO1) were significantly enriched in both LNCaP and C4–2 cells, regardless of DHT status, suggesting common interactions across cellular contexts. Similarly, ETS family members as well as CTCF and BORIS (CTCFL) were significantly enriched across conditions.
NCOR2 cistromes were queried against experimentally derived transcription factor-binding datasets included in the CistromeDB collection (>10,000 total ChIP-seq datasets across >1,100 factors) using GIGGLE (Layer et al., 2018) (Figures S7D and S7E; Table S6). Assessment of factors observed in the top 200 most enriched binding datasets supported co-accumulation of NCOR2 with factors consistent with motif analysis, for example, with strong enrichments for the AR-regulating pioneering factor FOXA1. Other commonly enriched factors included AR, MYC/MAX, E2F1, SUMO2, CREB1, and KMD5B. Filtering for datasets derived in prostate cell models similarly demonstrated significant NCOR2 binding overlap with FOXA1, CREB1, and SUMO2 as well as DAXX and ETV1 (Figure 6E). In a parallel analysis against histone modification profile datasets derived in prostate cells, NCOR2 displayed the most significant overlaps with H3K27ac and H3K4me2, consistent with a functional role for NCOR2 in regulating distal enhancer regions.
GSEA analyses of NCOR2 peak annotated genes reflected functions observed from analysis of transcriptome data, for instance, strong enrichment of androgen response pathways, particularly in LNCaP cells treated with DHT, that were either reduced or not significant in C4–2 cells (Figure 6F). HDAC activity and FOX function were commonly enriched among all conditions, including genes directly bound and regulated by FOXA2/FOXP3. Also, in line with observations from transcriptome and methylome functional pathway analyses, neuronal pathways were strongly enriched in C4–2 cells.
Integration of NCOR2-dependent transcriptomes, DNA methylomes, and cistromes
NCOR2-dependent omic integration was undertaken. In LNCaP cells, NCOR2-dependent gene expression changes significantly overlapped with expressed genes associated with NCOR2 binding and with DMRs at enhancers annotating to expressed genes (Figure 7A). In C4–2 cells, there were larger sets of overlaps between NCOR2-dependent DEGs and the NCOR2-bound genes and also enhancer methylated genes. Further, there was also significant overlap between the NCOR2-bound genes and genes associated with enhancer DMRs (Figure 7B).
Figure 7. Data integration identifies effects on SEs.

(A and B) Upset plot showing the intersection between DEGs upon NCOR2 knockdown, expressed genes associated with differentially methylated enhancer regions upon NCOR2 knockdown (DMR.enh), and expressed genes associated with NCOR2 binding found under basal (EtOH) conditions in both LNCaP (A) and C4–2 (B) cells. Significant overlaps are denoted by a red asterisk (*p < 0.05; **p < 0.01; ***p < 0.001).
(C) Overlaps of NCOR2 binding by ChIP-seq with identified SEs from LNCaP and C4–2 cells. Peak overlaps are shown for both basal (EtOH) and androgen-supplemented (DHT) conditions, and significance was determined by hypergeometric distribution.
(D) Functional enrichment analysis of genes annotated to NCOR2/SE shared regions from (C). The top 15 significant (p adjusted < 0.05) gene sets that related to the functional categories (cell differentiation, chromatin organization, proliferation, and metastasis) are displayed as counts within their respective functional category.
(E) Heatmap showing the expression levels of DEGs from CWR22 shNCOR2 recurrent tumors compared with control recurrent tumors (1,189) and the scaled expression (Z score) of those genes in mCRPC patient samples. The column annotations indicate pathology classification (Pathology), NEPC score, and AR signaling score (AR score). The row annotations indicate FC direction of CWR22 recurrent tumor DEGs (shNCOR2 compared with shCTL) and if that gene is regulated by an SE (identified in LNCaP and C4–2 cells).
Super enhancers (SEs) have gained recent attention in stem cell and cancer biology as reprogrammed enhancer “hubs” that drive expression of key genes implicated in lineage plasticity and therapeutic resistance (Adam et al., 2015, Ma et al., 2020, Whyte et al., 2013). Previous reports have shown that NCOR2 occupies lineage-specific SEs and serves as a key regulator of SE activation associated with cell reprogramming and differentiation (Zhuang et al., 2018, Siersbæk et al., 2017, Hnisz et al., 2013). Knockdown of NCOR2 in the CWR22 model led to accelerated recurrence with tumors exhibiting NEPC-like gene expression, suggesting that NCOR2 loss allows for greater plasticity. In the androgen-independent C4–2 cell line, NCOR2 loss associated with hypermethylation at enhancers, and the NCOR2 cistrome significantly overlapped with those enhancers that gain methylation. Thus, we asked if NCOR2’s role in regulation of SE-associated genes involved in lineage plasticity could explain these observations.
We identified SE regions in both LNCaP and C4–2 cells from previously published H3K27ac ChIP-seq datasets (Hnisz et al., 2013, He et al., 2021). Overall, we identified more SEs in C4–2 cells than in LNCaP cells. We overlapped these SEs with NCOR2-bound regions and found that there was significant overlap in all conditions (Figure 7C). To understand the biological relevance of these NCOR2-bound SEs, we queried these regions for known transcription factor-binding motifs (Figure S7F). We found significant enrichment for factors such as AR, FOXA1, and FOXA2 in LNCaP cells in both the DHT and basal (ethanol [EtOH]) conditions. However, in the androgen-independent C4–2 cells, there was enrichment for factors related to invasion and aggressive disease. The top enriched motifs in C4–2 (EtOH) cells associated with Wilms tumor 1 (WT1) binding, which has been shown to be upregulated in advanced PC and can promote hormone-independent tumor growth and metastasis (Devilard et al., 2006, Brett et al., 2013). We also observed enrichment for other factors shown to regulate SEs related to EMT and pluripotency (e.g., TGIF1) (Lee et al., 2015, Zhang et al., 2020) and androgen biosynthesis in castration-resistant PC (CRPC; e.g., NR5A2) (Zhou et al., 2021, Xiao et al., 2018).
We performed GSEA on genes annotated to NCOR2-bound SEs. From among the top 15 significantly enriched (p adjusted < 0.05) gene sets in each analysis, we defined six functional categories (neuronal differentiation, chromatin organization, hormone-dependent differentiation, invasion/metastasis, cell cycle/proliferation, and other cell differentiation) and displayed the number of significantly enriched genes sets that related to these functional categories (Figure 7D; Table S7). In LNCaP cells, there was enrichment for gene sets related to canonical NCOR2 function, such as chromatin organization. As expected, in the DHT-supplemented condition, there was enrichment for androgen response and hormone-dependent cell differentiation in both LNCaP and C4–2 cells. In both basal conditions, there was strong enrichment for genes involved in transforming growth factor-β (TGF-β) signaling and insulin-like growth factor (IGF)-related pathways that have been shown to promote tumor growth and metastasis. Unique to C4–2 basal conditions, we found enrichment for neuronal-related gene sets, which agrees with NCOR2’s role in the regulation of cellular differentiation of both neuronal and liver cells, and its downregulation can promote tumorigenesis in both contexts (Wu et al., 2019, Foley et al., 2011).
We next asked if the DEGs among CWR22 recurrent tumors with and without knockdown of NCOR2 (shNCOR2-RT versus shCTL-RT) could cluster metastatic CRPC (mCRPC) cases with high NEPC scores and low AR-signaling scores and how those DEGs associated with SEs found in LNCaP and C4–2 cells. We queried a dataset of 266 patients with mCRPC for which both RNA-seq and histological classification were available (Abida et al., 2019) and found that unsupervised clustering with the CWR22 DEGs segregate the cases into three major clusters (Figure 7E). Cluster 1 strongly enriched for cases with high NEPC scores, low AR scores, and small cell or adenocarcinoma with neuroendocrine features in comparison to clusters 2 and 3. Forty-six of the DEGs are annotated to SEs found in the LNCaP and/or C4–2 cell lines, and most were associated with genes that became upregulated following NCOR2 loss and recurrence.
DISCUSSION
Transcription factor co-regulators are drivers of progression in multiple hormone-responsive cancers (Girault et al., 2003, Khanim et al., 2004), including in the context of therapy resistance (Gong et al., 2018, Girault et al., 2003). Altered NCOR2 expression has consistently been reported in ADT-RPC (Abida et al., 2019, Robinson et al., 2015b). To date, ambiguity existed over whether gain or loss of NCOR2 functions were a PC driver (Girault et al., 2003, Khanim et al., 2004). The current study aimed to address this ambiguity with an integrative genomic approach exploiting in vitro, in vivo, and in silico resources covering the emergence of the ADT-RPC.
We exploited a patient TMA to establish that reduced NCOR2 was significantly associated with features of aggressive PC, such as higher PSA levels and shorter disease-free survival in patients who received adjuvant ADT. Furthermore, knockdown of NCOR2 in the CWR22 model of castration recurrence resulted in significantly more rapid recurrence with increased NEPC gene signatures after androgen withdrawal. While androgen withdrawal led to a dramatic decrease in proliferating cells in both control and NCOR2-knockdown xenografts 7 days after ADT, there were significantly more proliferating cells with knockdown of NCOR2 (Figure 2E). The combined observations of NCOR2 knockdown associating with decreased maximum regression and faster time to recurrence suggests that the reduction in NCOR2 makes some cells resistant to the effects of androgen depletion.
Supporting links between NCOR2 and androgen signaling, NCOR2 knockdown changed gene expression in a manner that significantly overlapped with DHT-responsive genes in both LNCaP and C4–2 cell lines. NCOR2-dependent gene expression patterns associated with both up- and downregulated genes, and although we cannot exclude indirect effects, this suggests that NCOR2 functions to modulate genes in both a positive and negative direction. NCOR2 cistrome analyses supported interactions with a diverse set of transcription factors and revealed evolution and expansion of binding choices in the C4–2 cells compared with LNCaP cells. The cistrome became more distal in C4–2 cells than in LNCaP cells, and binding regions were fairly evenly distributed between positive and negative relationships where NCOR2 appears to function either as a corepressor or a coactivator.
Surprisingly, reduced NCOR2 expression resulted in a profound increase in CpG methylation in both cell lines and in the CWR22 model. Changes in DNA methylation were enriched at enhancers and correlated with changes in gene expression both positively and negatively. This is compelling because NCOR2 has been well understood to act as a co-repressor of nuclear receptor transcriptional programs through its formation of large complexes that include HDACs, which, in turn, create a restrictive chromatin state ((Oberoi et al., 2011), (Chen and Evans, 1995), (Nagy et al., 1997), (Guenther et al., 2001), (Liao et al., 2003), (Yu et al., 2003)) or further recruitment of CpG methylation machinery (Cartron et al., 2013). Among the ChIP-seq enrichments associated with downregulated genes following NCOR2 knockdown were MBD2 in both LNCaP and C4–2 cells and MED1 in C4–2 cells (Figure 4D). Both MBD2 and MED1 are known to interact with DNA methylation at the level of reading and erasing, respectively, and suggest that NCOR2 may be part of a large complex that includes components of the DNA methylation regulatory system at enhancer regions. While the mechanism of how DNA methylation levels are affected in the context of reduced NCOR2 protein remains to be determined, our data suggest a complex relationship between CpG methylation and gene expression. The consequences of low-density CpG methylation at enhancer regions are not as well understood as high-density methylation of CpG islands at promoters. Enhancer methylation may contribute to stabilizing large, higher-order chromatin structures or altering transcription factor recruitment, both shown to play a role in lineage plasticity (Charlet et al., 2016, Song et al., 2019, Yin et al., 2017).
SEs play a key role in normal cellular differentiation, and recent evidence suggests that targeting SEs may be an alternative therapeutic strategy in a wide range of cancers that use lineage plasticity to escape therapeutic pressures (Ma et al., 2020, Bao et al., 2019). NCOR2 has been shown to be recruited to SEs by lineage-specific transcription factors along with HDACs to create an epigenetic barrier and repress transcription. Consequently, NCOR2 and other co-repressor complexes (Hnisz et al., 2013) may offer a safeguard to overactivation of SEs and aid in their complete shutdown during cell differentiation processes (Siersbæk et al., 2017, Zhuang et al., 2018). Because we see increased expression of neuroendocrine-related genes upon NCOR2 knockdown following androgen withdrawal in vivo and in vitro and a dampened response to ADT in patient samples and in our in vivo model, we rationalize that this is attributed to NCOR2’s role in regulation of SE-associated genes involved in lineage plasticity and therapy resistance.
We have shown that the NCOR2 cistrome in LNCaP and C4–2 cells enriches for active and poised enhancers and overlaps with many lineage-determining pioneer and transcription factors that have been associated with SEs involved in CRPC and NEPC, such as FOXA1 and MYC (Khan and Zhang, 2016, Lupien et al., 2008). Further, we demonstrated that within those NCOR2-bound regions, there was significant overlap with SEs. We found significant enrichment for binding motifs that associated with factors that regulate endocrine cell differentiation, such as AR and FOX family members (FOXA1/FOXA2) in LNCaP cells. Recently, FOXA1 has been implicated in the reprogramming of SEs that drive expression of neuronal-related genes that are required for NEPC progression (Baca et al., 2021). Interestingly, there was not enrichment for FOX family nor AR-binding motifs in presumably NCOR2-bound SEs identified in C4–2 cells. This suggests distinct SE landscapes between these two models. In C4–2 cells, there was enrichment for factors that have been shown to promote androgen-independent tumor growth and metastasis. This agrees with the phenotype of C4–2 cells as representing a more advanced and androgen-independent stage of PC progression than androgen-sensitive LNCaP cells. Further, when we queried gene sets related to NCOR2-SE annotated genes, we found strong enrichment for gene sets involved in neuronal differentiation, but that finding was unique to the C4–2 basal condition. This suggests that NCOR2 may co-occupy and regulate lineage-specific SEs in CaP that have the potential to drive androgen-independent proliferation and alternative lineages during therapeutic stress, such as ADT. Loss of NCOR2-mediated regulation at these SEs may increase the chances for stochastic or genetic-driven activation of transcriptional programs that drive survival and recurrence during androgen withdrawal. We speculate that without NCOR2’s role in regulating the precise recruitment of chromatin-modifying enzymes such as HDACs, these SEs are without a “shut-down” mechanism and can therefore become over-activated.
These studies indicate reduced NCOR2 function as a determinant of poor response to ADT and implicate that while NCOR2 indeed contributes to canonical androgen regulation and signaling through the AR, it additionally plays important roles in the epigenetic control of enhancer regions through a diverse set of factors, including FOX family members. Some NCOR2-bound enhancers may regulate lineage choice under the selective pressure of ADT, thereby contributing to more rapid recurrence. Interestingly, recurrent tumors in the CWR22 model from the shCTL group also exhibited reduced expression of NCOR2 following recurrence similar to the levels achieved experimentally in the shNCOR2 tumors, suggesting that selective pressure for loss of NCOR2 function may be a common characteristic for progression post-ADT. Indeed, neuroendocrine features were observed in RT-shCTL tumors and have been previously reported in wild-type CWR22 tumors, and increased molecular and morphological heterogeneity observed early in disease has a significant impact on progression (Huss et al., 2004, Seedhouse et al., 2016). Yet, the fact that RT-shNCOR2 exhibited increased neuroendocrine characteristics relative to shCTL counterparts suggests that releasing the epigenetic regulation of NCOR2 earlier in disease progression relative to the selective pressure of ADT is important in altering the potential for lineage plasticity ultimately required to develop recurrent tumors.
Limitations of the study
While this study utilized a large cohort (n = 50/group) to assess differences in rates of recurrence after androgen withdrawal in the CWR22 patient derived-xenograft, the study is limited by the use of a single xenograft model system. Therefore, it is unclear if these observations would be replicated in other prostate tumor models with different genetic backgrounds. In addition, the lack of a fully functioning immune system in the nu/nu hosts for the xenografts precludes determination of how NCOR2 loss might affect the tumor immune microenvironment. The hypermethylation of CpG dinucleotides observed in response to NCOR2 knockdown is limited to the ~850,000 loci covered by the Infinium MethylationEPIC BeadChip, which avoids genomic positions rich in repetitive elements. Therefore, we are unable to determine if the hypermethylation phenotype extends to repetitive elements.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dominic J Smiraglia, Roswell Park Comprehensive Cancer Center (dominic.smiraglia@roswellpark.org).
Materials availability
All cell lines and constructs generated in this study will be made available from the lead contact upon request.
Data and code availability
Publicly available PC cohort SNV, copy number, RNA-seq expression data, and clinical characteristics are available in cBioPortal and are detailed in the STAR KEY RESOURCES TABLE and cited in the main text. All raw and processed RNA-seq, ChIP-seq, and MethylationEPIC array data generated in this study have been deposited and made available as of the date of publication at NCBI-GEO under accession series GEO: GSE178820.
No unique code was generated in this study.
Any additional resources required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Anti-NCOR2, rabbit polyclonal | Abcam | Cat# ab24551, RRID:AB_2149134 |
| Anti-NCOR2, rabbit monoclonal | Cell Signaling | Cat# 62370, RRID:AB_2799628 |
| Anti-NCOR2, rabbit | Sigma-Aldrich | Cat# HPA001928, RRID:AB_1079454 |
| Normal Rabbit IgG antibody | Cell Signaling | Cat# 2729, RRID:AB_1031062 |
| Normal Rabbit IgG antibody | Santa Cruz | Cat# sc-2027, RRID:AB_737197 |
| Anti-AR, rabbit polyclonal | Millipore | Cat# 06-680 |
| Anti-TBP, rabbit polyclonal | Cell Signaling | Cat# 8515, RRID:AB_10949159 |
| Anti-GAPDH, rabbit monoclonal | Cell Signaling | Cat# 2118, RRID:AB_561053 |
| Anti-beta-actin, rabbit polyclonal | Abcam | Cat# ab8227, RRID:AB_2305186 |
| Rabbit anti-mouse immunoglobulins/HRP antibody | Agilent | Cat# P0161, RRID:AB_2687969 |
| Goat anti-rabbit immunoglobulins/HRP antibody | Agilent | Cat# P0448, RRID:AB_2617138 |
| Ki-67 antibody, rabbit polyclonal | Abcam | Cat# ab15580, RRID:AB_443209 |
| Synaptophysin antibody, rabbit monoclonal | Abcam | Cat# ab52636, RRID:AB_882786 |
|
| ||
| Biological samples | ||
|
| ||
| Prostate cancer patient-derived tissue microarrays | Roswell Park | Pathology Resource Network (PRN) and |
| and de-identified clinical information | Comprehensive Cancer Center | Data Bank Biorepository (DBBR) |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| TRIzol RNA Isolation Reagent | Thermo Fisher | Cat# 15596026 |
| AllPrep DNA/RNA/miRNA Universal Kit | QIAGEN | Cat# 80224 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| 5α-Dihydrotestosterone (DHT) solution | Millipore Sigma | Cat# D-073 |
| cOmplete, Mini Protease Inhibitor Cocktail | Millipore Sigma | Cat# 11836153001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| TruSeq Stranded Total RNA Library Prep Kit | Illumina | Cat# 20020596 |
| Infinium MethylationEPIC BeadChip Kit | Illumina | Cat# WG-317-1002 |
| ViaLightTM Plus Cell Proliferation and Cytotoxicity BioAssay Kit | Lonza | Cat# LT07-221 |
| TaqMan MicroRNA Reverse Transcription Kit | Applied Biosystems | Cat# 4366596 |
| Turbo DNA-Free kit | Invitrogen | Cat# AM2238 |
|
| ||
| Deposited data | ||
|
| ||
| Raw data | This study | GEO: GSE178820 series |
| Prostate Adenocarcinoma (TCGA, PanCancer Atlas) | TCGA | https://www.cbioportal.org/study/summary?id=prad_tcga_pan_can_atlas_2018 |
| Neuroendocrine Prostate Cancer (Multi-Institute, Nat Med 2016) | Beltran et al., 2016 | https://www.cbioportal.org/study/summary?id=nepc_wcm_2016 |
| The Metastatic Prostate Cancer Project | The Metastatic Prostate | https://www.cbioportal.org/study/summary?id=prad_mpcproject_2018 |
| (Provisional, November 2019) | Cancer Project | |
| Metastatic Prostate Cancer (SU2C/PCF Dream Team, Cell 2015) | (Robinson et al., 2015b) | https://www.cbioportal.org/study/summary?id=prad_su2c_2015 |
| Metastatic Prostate Adenocarcinoma (SU2C/PCF Dream Team, PNAS 2019) | Abida et al., 2019 | https://www.cbioportal.org/study/summary?id=prad_su2c_2019 |
| Prostate Adenocarcinoma (MSKCC, Cancer Cell 2010) | Tayior et al., 2010 | https://www.cbioportai.org/study/ summary?id=prad_mskcc |
| Prostate Adenocarcinoma (MSKCC/DFCI, Nature Genetics 2018) | Armenia et al., 2018 | https://www.cbioportai.org/study/ summary?id=prad_p1000 |
|
| ||
| Experimentai modeis: ceii lines | ||
|
| ||
| LNCaP-shControi | This study | N/A |
| C42-shControi | This study | N/A |
| LNCaP-shNCOR2-A | This study | N/A |
| LNCaP-shNCOR2-B | This study | N/A |
| C42-shNCOR2-A | This study | N/A |
| C42-shNCOR2-B | This study | N/A |
|
| ||
| Experimental modeis: organisms/strains | ||
|
| ||
| Hsd:Athymic Nude-Foxn1nu, male | Harlan/Envigo | 069 |
| CWR22 Prostate Cancer Xenograft Mouse (shControi, shNCOR2; V2LHS-251658) | This study | N/A |
|
| ||
| Oligonucieotides | ||
|
| ||
| NCOR2 (TaqMan Gene Expression Assay) | Thermo Fisher | Assay ID: Hs00196955_m1 |
| AR | Thermo Fisher | Assay ID: Hs00171172_m1 |
| GAPDH (TaqMan Gene Expression Assay) | Thermo Fisher | Assay ID:4326317E |
| DENND1B (forward: TTACACAATGAAAAGGGAGG) | IDT | N/A |
| TMPRSS2 (forward: ATGATGCTGCAGCCAGAACAG) | IDT | N/A |
| HERC3 (forward: AGGTTCTTGTAGGCTGTGGT) | IDT | N/A |
| CXCR7 (forward: GATGTGGGTTACAAAGCTG) | IDT | N/A |
| KLK3 (forward: ACTTCAGTGTGTGGACCTTGT) | IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| shNCOR2-A (Targeting) pGIPZ Lentiviral Construct | RPCCC Gene Modulation Resource | V2LHS-251658 |
| shNCOR2-B (Targeting) pGIPZ Lentiviral Construct | RPCCC Gene Modulation Resource | V2LHS-196739 |
| GIPZ Lentiviral shRNA Construct | Thermo Fisher | V2LHS |
|
| ||
| Software and aigorithms | ||
|
| ||
| Prism | GraphPad | Version 9.1.0 |
| R V3.6-4.0 | https://www.r-project.org/ | https://www.r-project.org/ |
| Salmon | Patro et al., 2017 | https://combine-lab.github.io/salmon/ |
| Bioconductor 3.2 | (Huber et al., 2015) | http://www.bioconductor.org |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| LISA | Qin et al., 2020 | http://lisa.cistrome.org/ |
| iRegulon | (Janky et al., 2014) | http://iregulon.aertslab.org/ |
| ChAMP | (Morris et al., 2014) | https://www.bioconductor.org/packages/release/bioc/html/ChAMP.html |
| DMRcate | Peters et al., 2015 | https://www.bioconductor.org/packages/release/bioc/html/DMRcate.html |
| Rsubread | (Liao et al., 2019) | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| csaw | Lun and Smyth, 2016 | https://bioconductor.org/packages/release/bioc/html/csaw.html |
| MotifDb | Shannon and Richards, 2021 | https://bioconductor.org/packages/release/bioc/html/MotifDb.html |
| HOMER v4.10 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| GIGGLE | Layer et al., 2018 | https://github.com/ryaniayer/giggie |
| GSEA v4.1.0 | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| MSigDB v7.4 | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/msigdb |
| ciusterProfiler | Wu et al., 2021 | https://bioconductor.org/packages/release/bloc/html/clusterProfiler.html |
| fgsea | Korotkevich et al., 2019 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Aperio Nuciear Aigorithm | Leica Biosystems | https://www.leicabiosystems.com/digital-pathology/analyze/ihc/aperio-nuclear-algorithm/ |
| ROSE: Rank Ordering of Super Enhancers | Whyte et al., 2013 | http://younglab.wi.mit.edu/super_enhancer_code.html |
|
| ||
| Other | ||
|
| ||
| GIBCO, RPMI 1640 Medium | Thermo Fisher | Cat# 11875093 |
| Magna ChIP Protein A+G Magnetic Beads | Sigma-Aidrich | Cat# 16-663 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and cell culture conditions
LNCaP cells were derived from a 50-year old male with PC who responded briefly to androgen deprivation therapy (Horoszewicz et al., 1980) and serve as a model for androgen sensitive PC. The C4–2 variant was derived in vivo from LNCaP using multiple rounds of selection in castrated mice and has metastatic potential, thus serving as an isogenic ADT resistant cell line model of aggressive PC (Wu et al., 1994). All cells were maintained at 37◦C and 5.0% CO2 using a cell culture incubator with UV contamination control. LNCaP and LNCaP-C4–2 cells were regularly maintained in RPMI 1640 Medium containing 10% FBS. All media was supplemented with 100 U/mL Penicillin-Streptomycin. Dihydrotestosterone (DHT, D-073–1ML, Sigma-Aldrich) was kept as 10mM EtOH stocks, and diluted to 1000x stocks prior to treatments. Prior to androgen treatment, cells were serum starved using charcoal stripped FBS (10%) for 72 hours. Cell lines were authenticated by STR profiling and confirmed mycoplasma free by RT-PCR in the RPCCC Genomics Shared Resource.
CWR22 model of PC progression
All animal experiments were carried out at the Department of Laboratory Animal Research at RPCCC in accordance with an Institutional Animal Care and Use Committee approved protocol. Male Athymic Nude BALB/c mice (Hsd:Athymic Nude-Foxn1nu) were purchased from Harlan/Envigo at approximately 2 months of age. Mice were allowed to reach approximately 3 months of age at which point they were surgically castrated and implanted with silastic tubing containing 12.5 mg of testosterone for sustained release 2 weeks prior to xenograft implantation. 1×106 CWR22 PC xenograft cells in a 1:1 mix of media to matrigel were injected subcutaneously on the right flank as previously described (Seedhouse et al., 2016). Total initial cohort size was 65 xenografts per group (shCTL, shNCOR2; V2LHS-251658), with 50 animals per group randomly designated for completion to recurrence. Tumor volumes were calculated from caliper measurements using the formula (length2 x width x 0.5234). Once tumors reached approximately 0.3 cm3 in size (two consecutive measurements > 0.3 cm3), androgen withdrawal was achieved by removal of the silastic tubing and tumor volumes were followed for a maximum of 336 days. Mice designated in recurrence group were sacrificed once tumors reached approximately 1.0 cm3, or if mice presented with ascites or were otherwise required by veterinary staff. At the time of sacrifice body and tumor weight were taken. Additionally, serum, tumor and in some cases liver, spleen and/or pancreas tissues with possible metastases were obtained and immediately flash frozen and stored at −80◦C. A tumor regression response to androgen withdrawal was defined as tumors that achieved a 40% loss in tumor size relative to size at withdrawal. A tumor was considered to be recurrent following androgen withdrawal once the primary subcutaneous tumor had reached a size that was 200% that of the original size of the tumor at withdrawal. Presence of shRNA targeting construct in recurrent tumors was verified by Sanger sequencing and bioluminescence imaging for select animals (n = 10 per group).
PC patient samples
The RPCCC prostate adenocarcinoma tissue microarray and associated de-identified clinical information was made available through the RPCCC Pathology Resource Network (PRN) and Data Bank and Bio-Repository (DBBR) core facilities. This collection includes tissue (3 distinct core samples from tumor and matching normal tissue) for 707 patients that underwent radical prostatectomy (RP) at RPCCC between 1993 and 2005. De-identified clinical annotations include patient characteristics (BMI, race, age, PSA), pathological information (Gleason sum, TNM), adjuvant therapy (ADT, radiation), and outcomes post-RP (biochemical recurrence, metastases, death) with maximum follow-up time of 18.6 years (mean = 8.8 years). Patients were considered to have received adjuvant ADT if given prior to surgery, or at any point post-surgery but before biochemical recurrence (BCR).
METHOD DETAILS
PC patient tissue microarray
Optimization and staining of NCOR2 (HPA001928, Sigma-Aldrich) in PC human tissue samples was performed by the RPCCC PRN. Quality assessment, nuclear identification and staining quantification (H-score) was performed using Aperio Nuclear v9 algorithm. Tissue cores were filtered for those with at least 20 detectable epithelial nuclei, and each individual core was pathologically examined to ensure tumor or normal involvement. Only patients with 2 or more cores that passed these criteria were retained for further analysis (564 patients available for NCOR2 analysis, and 463 for NCOR1). Univariate and multivariate linear regression was applied to examine relationships between extraneous clinical variables with NCORs protein expression. BCR survival separated on staining quantification (median cut-off) was assessed by Cox proportional hazards regression within clinical sub-groups to limit confounding variables, and statistical differences deemed by log-rank test.
Stable knockdown of NCOR2
Knockdown of NCOR2 in LNCaP and C4–2 cells was achieved by stable selection after transduction with lentiviral shRNA constructs targeting NCOR2. Two targeting constructs (V2LHS-251658 (shNCOR2-A), V2LHS-196739 (shNCOR2-B)) and one non-silencing control construct were selected from the V2LHS pGIPZ based lentiviral shRNA library (Thermo Fisher Scientific). Viral packaging and cellular infection (RPCCC shRNA Resource) yielded pGIPZ containing cells, which were maintained in media supplemented with puromycin (2μg/mL). For xenograft studies, NCOR2 targeting shRNA (V2LHS-251658) or non-targeting shRNA control were introduced to digested CWR22 tissue under puromycin selection for 24 hours prior to implantation.
RT-qPCR
Total RNA was isolated via TRIzol® reagent (Thermo Fisher Scientific) for candidate mRNA detection by use of the AllPrep DNA/RNA/miRNA Universal Kit (QIAGEN), following manufacturer’s protocols. Complementary DNA (cDNA) was prepared using iScriptTM cDNA Synthesis Kit (Bio-Rad), following manufacturer’s protocols. Relative gene expression was subsequently quantified via Applied Biosystems 7300 Real-Time PCR System (Applied Biosystems), for both TaqMan and SYBR Green (Thermo Fisher Scientific) applications. All targets were detected using either pre-designed TaqMan Gene Expression Assays (Thermo Fisher Scientific; AR, NCOR2, GAPDH, KRT8), pre-designed PrimeTime qPCR primers (IDT; KRT18, DENND1B, TMPRSS2, HERC3, CXCR7, FOS, KLK3) or custom designed qPCR primers (IDT) using a final primer concentration of 500nM. All primers for use with SYBR Green application were tested for specificity by melting curve analysis with subsequent product visualization on agarose gel. All RT-qPCR experiments were performed in biological triplicates, with at least technical duplicates. Fold changes were determined using the 2-ΔΔCt method as the difference between experimental group and respective control group. Significance of experimental comparisons was performed using Student’s t test.
Immunoblotting
Total cellular protein was isolated from exponentially growing cells for determination of target protein expression. Cells were harvested, then washed in ice cold PBS before lysing in ice cold RIPA buffer (50mM Tris-HCl pH 7.4, 150mM NaCl, 1% v/v Triton X-100, 1mM EDTA pH 8.0, 0.5% w/v sodium deoxychlorate, 0.1% w/v SDS) containing 1x cOmplete Mini Protease Inhibitor Tablets (Roche). Protein concentrations were quantified using the DC Protein Assay (Bio-Rad), following manufacturer’s protocols. Equal amounts of proteins (30–60μg) were separated via SDS polyacrylamide gel electrophoresis (SDS-PAGE) using precast 10% polyacrylamide gels (Mini-Protean TGX, Bio-Rad). Proteins were transferred onto polyvinylidene fluoride (PVDF) membrane (Roche) for 80V for 1.5 hours. Post transfer, membranes were blocked with 5% non-fat dry milk (NFDM) for 1 hour at room temperature. Blocked membranes were probed with primary antibody against NCOR2 (ab24551, Abcam; 62370, Cell Signaling), AR (PG-21, Millipore), IgG (2729S, Cell Signaling), TBP (8515S, Cell Signaling), GAPDH (2118, Cell Signaling), β—Actin (ab8227, Abcam), either overnight at 4◦C or for 3 hours at room temperature. Primary antibody was detected after probing for 1 hour with HRP-linked rabbit anti-mouse IgG (P0161, Dako) or goat anti-rabbit IgG (P0448, Dako) secondary antibody at room temperature using ECL Western Blotting substrate (Pierce). Signal and quantification was performed using the ChemiDoc XRS+ system (Bio-Rad).
Cell viability assay
Bioluminescent detection of cellular ATP as a measure of cell viability was undertaken using ViaLight® Plus Kit (Lonza Inc.) reagents. Cells were plated at optimal seeding density to ensure exponential growth (4×103 cells per well) in 96-well, white-walled plates. Wells were dosed with agents to a final volume of 100 μl. Dosing occurred at the beginning of the experiment, and cells were incubated for up to 120 hours. Luminescence was detected with Synergy™ 2 multi-mode microplate reader (BioTek® Instruments). Each experiment was performed in at least triplicate wells in triplicate experiments.
RNA-sequencing
RNA was extracted from LNCaP and C4–2 cells in the presence of DHT (10nM, 6hr) or EtOH. RNA was extracted at the indicated time points in the CWR22 xenograft model. To profile global transcriptional patterns, a minimum of biological triplicate samples (cell line studies; n = 3, xenograft studies; n = 5–6) per experimental condition were analyzed by RNA-seq. Approximately 30–50 mg of flash frozen tumor tissue was homogenized in 1 mL of TRIzol (for RNA) and 1 mL of Szak’s RIPA buffer containing 1x Protease Inhibitor (for Protein) using a Polytron PT 2100 tissue homogenizer. Approximately 10 ug of extracted RNA was then DNase treated using the TURBO DNA-free™ kit. RNA from tissue samples utilized for RNA-seq was isolated using the AllPrep DNA/RNA/miRNA Universal Kit (QIAGEN) as per manufacturer’s protocols.
Sequencing was performed at the RPCCC Genomics Shared Resource core facility, and sequencing libraries prepared with the TruSeq Stranded Total RNA kit (Illumina Inc), from 1ug total RNA. Quasi-alignment of raw sequence reads to the human transcriptome (hg19) and subsequent transcript abundance estimation was performed via Salmon (Patro et al., 2017). For CWR22 samples, alignments were first filtered to remove and reads aligning to mouse (GRCm38). Transcript abundance estimates were normalized and differentially expressed genes (DEGs) were identified using a standard DESeq2 (Love et al., 2014) pipeline. For cell line studies, final DEG determination was called after combining samples from multiple NCOR2 targeting shRNA into a single group. Transcriptional regulator analysis on DEGs was performed using LISA (Qin et al., 2020).
DNA methylation profiling
Cell lines were treated as for RNA-Seq. DNA was extracted from all samples using the AllPrep DNA/RNA/miRNA Universal Kit (QIAGEN), following manufacturer’s protocol. DNA methylation profiles were obtained using the Infinium MethylationEPIC BeadChip (EPIC array) platform (Pidsley et al., 2016), performed in the RPCCC Genomics Shared Resource. Data processing and quantification was accomplished using ChAMP (Morris et al., 2014). Briefly, detectible beta values for all probed CpG sites were initially compiled and filtered to remove those associated with multiple alignments and known SNPs, leaving reliable information for 791,398 CpG sites. To adjust for probe design bias (Infinium Type-I, Type-II), a beta-mixture quantile normalization method (BMIQ) was employed (Teschendorff et al., 2013). Additionally, cross-array batch effect was corrected using ComBat (Johnson et al., 2007). Differentially methylated Positions (DMPs) were determined using ChAMP and subsequently differentially methylated regions (DMR) identified using DMRcate (Peters et al., 2015).
Chromatin immunoprecipitation
ChIP was performed in LNCaP and C4–2 cells in the presence of DHT (10nM, 1hr) or EtOH in triplicate independent experiments. Briefly, approximately 20×106 cells were crosslinked with 1% formaldehyde solution, quenched with glycine (0.125 M) and harvested in cold PBS. Sonication of crosslinked chromatin was performed using a Bioruptor® UCD-200™ Sonicator (Diagenode) with optimized cycles for each cell type. Immunoprecipitation of sonicated material was performed with antibodies against NCOR2 (ab24551, Abcam) or IgG (sc-2027x, santa cruz) for 16 hours, and antibody/bead complexes isolated with Magna ChIPTM Protein A+G magnetic beads (Millipore). Complexes were washed, reverse crosslinked, and treated sequentially with RNase and proteinase K prior to DNA isolation. Sequencing (75bp single end, 49.1×106, 50.9×106 average reads/sample in LNCaP, C4–2 respectively) was performed at the RPCCC Genomics Shared Resource core facility. The NCOR2 cistrome was analyzed with Rsubread/csaw (Lun and Smyth, 2016), along with TF motif analyses (Shannon and Richards, 2021) (MotifDb). Peak density plots were performed using the annotatePeaks.pl tool available from the HOMER (Hypergeometric Optimization of Motif EnRichment) suite v4.10 (Heinz et al., 2010). In order to find potential transcription factor binding enrichment within NCOR2 cistromes, we utilized GIGGLE (Layer et al., 2018) to query the complete human transcription factor ChIP-seq dataset collection (10,361 and 10,031 datasets across 1,111 transcription factors and 75 histone marks, respectively) in Cistrome DB (Zheng et al., 2019). Prostate specific filtering limited analysis to 681 datasets across 74 TFs and 238 datasets across 19 HMs. For each query dataset, we determined the overlap of each NCOR2 cistrome. Putative co-enriched factors were identified by assessment of the number of time a given factor was observed in the top 200 most enriched datasets relative to the total number of datasets for that factor in the complete Cistrome DB (> 1.2 FC enrichment over background). For prostate specific analysis, overlaps across datasets were averaged for each factor.
Functional annotation of gene sets
Pathway enrichment analysis and gene set enrichment analysis (GSEA) were performed using gene sets from the Molecular signatures database (Subramanian et al., 2005) (MSigDB). Specifically, gene sets were compiled to assess enrichment of all BROAD Hallmark pathways, curated pathways (KEGG, BioCarta, Canonical, Reactome, Chemical/Genetic perturbations), and GO terms (Biological Processes). GSEA was implemented using the clusterProfiler (Wu et al., 2021) and fgsea (Korotkevich et al., 2019) packages in R. Master regulator analysis (MRA) was performed on select gene sets using iRegulon (Janky et al., 2014) implemented in Cytoscape.
Super-enhancer identification
Super-enhancers (SE) are distinguished from typical enhancers based on size, and the high density of activating histone modifications, as originally discovered by Whyte et al. (2013). Identified SE regions were obtained for LNCaP cells from Hnisz et al., 2013 and raw H3K27ac and IgG ChIP-seq data was obtained and used to call for SE’s in C4–2 cells from He et al., 2021 (GSE136130). In both LNCaP and C4–2, SE’s were defined based on the active enhancer mark H3K27ac ChIP-seq signals. SE’s were identified using ROSE (Rank Ordering of Super Enhancers), wherein enhancer-associated peaks are scored and ranked. A cluster of multiple enhancers that stretches a distance of 12.5kb were stitched together and scored based on H3K27ac peak signals. Scoring of super-enhancer peak signal intensity was calculated based on the total normalized reads minus the number of normalized reads from the IgG control. These defined regions are then sorted by ChIP signal score and normalized based on the amount of putative enhancer regions and the highest score. These enhancers are ranked based on enhancer enrichment. If the calculated score for a region has a slope greater than 1, it was deemed a super-enhancer.
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses were undertaken using the R platform for statistical computing (version 3.6.1 or later) and the indicated library packages implemented in Bioconductor, as detailed in the KEY RESOURCES TABLE. Details on statistical tests and parameters and significance cutoffs are listed in figure legends.
Supplementary Material
Highlights.
Reduced NCOR2 associates with faster biochemical recurrence in patients who received neoadjuvant ADT
NCOR2 knockdown in the CWR22 model accelerates recurrence during ADT
NCOR2 knockdown results in CpG hypermethylation enriched in predicted enhancers
The NCOR2 cistrome overlaps with super enhancers that may regulate lineage choice
ACKNOWLEDGMENTS
The following Shared Resources at Roswell Park Comprehensive Cancer Center are gratefully acknowledged: Genomics Shared Resource (sequencing), Gene Modulation Services (shRNA cell lines), Pathology Network Shared Resource (tissue microarray), Translational Imaging Shared Resource (in vivo imaging), and Small Molecule Screening Shared Resource (drug screen). At The James, Comprehensive Cancer Center The Proteomics Shared Resource, OSUCCC is gratefully acknowledged. Finally, the authors gratefully acknowledge thoughtful discussions on murine studies with Dr. Barbara A. Foster, Mr. Bryan Gillard, and Ms. Ellen Karasik.
M.J.C., D.J.S., and M.D.L. acknowledge support, in part, from the Prostate program of the Department of Defense Congressionally Directed Medical Research Programs (W81XWH-14-1-0608) and the National Cancer Institute (NCI) grant P30CA016056 involving the use of Roswell Park Comprehensive Cancer Center’s Shared Resources, and NCI Cancer Center Support Grant (P30CA016058) to the OSUCCC The James. M.J.C. and G.L. acknowledge support from the European Union-United States Atlantis Program (P116J090011). M.J.C. acknowledges support from the NCI grant awarded to the OSUCCC The James, CCSG, P30CA016058. M.D.L. acknowledges support from the National Institutes of Health (U24CA232979).
The graphical abstract was created with BioRender (https://BioRender.com).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110109.
REFERENCES
- Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen EM, et al. (2019). Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 116, 11428–11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS, Polak L, Kadaja M, Asare A, Zheng D, and Fuchs E. (2015). Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 521, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affronti HC, Long MD, Rosario SR, Gillard BM, Karasik E, Boerlin CS, Pellerite AJ, Foster BA, Attwood K, Pili R, et al. (2017). Dietary folate levels alter the kinetics and molecular mechanism of prostate cancer recurrence in the CWR22 model. Oncotarget 8, 103758–103774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, Coleman I, et al. ; PCF/SU2C International Prostate Cancer Dream Team (2018). The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 50, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca SC, Takeda DY, Seo JH, Hwang J, Ku SY, Arafeh R, Arnoff T, Agarwal S, Bell C, O’Connor E, et al. (2021). Reprogramming of the FOXA1 cistrome in treatment-emergent neuroendocrine prostate cancer. Nat. Commun. 12, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J, Li M, Liang S, Yang Y, Wu J, Zou Q, Fang S, Chen S, and Guo L. (2019). Integrated high-throughput analysis identifies super enhancers associated with chemoresistance in SCLC. BMC Med. Genomics 12, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, et al. (2016). Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Hruszkewycz A, Scher HI, Hildesheim J, Isaacs J, Yu EY, Kelly K, Lin D, Dicker A, Arnold J, et al. (2019). The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res. 25, 6916–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, and Hiebert SW (2008). Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 30, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, Bianchi-Frias D, Dumpit RF, Kaipainen A, Corella AN, et al. (2017). Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 32, 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett A, Pandey S, and Fraizer G. (2013). The Wilms’ tumor gene (WT1) regulates E-cadherin expression and migration of prostate cancer cells. Mol. Cancer 12, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ (2019). Tales from topographic oceans: topologically associated domains and cancer. Endocr. Relat. Cancer 26, R611–R626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2015). The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartron PF, Blanquart C, Hervouet E, Gregoire M, and Vallette FM (2013). HDAC1-mSin3a-NCOR1, Dnmt3b-HDAC1-Egr1 and Dnmt1-PCNA-UHRF1-G9a regulate the NY-ESO1 gene expression. Mol. Oncol. 7, 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JD, and Evans RM (1995). A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377, 454–457. [DOI] [PubMed] [Google Scholar]
- Chen Z, Wu D, Thomas-Ahner JM, Lu C, Zhao P, Zhang Q, Geraghty C, Yan PS, Hankey W, Sunkel B, et al. (2018). Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl. Acad. Sci. USA 115, 6810–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devilard E, Bladou F, Ramuz O, Karsenty G, Dales JP, Gravis G, Nguyen C, Bertucci F, Xerri L, and Birnbaum D. (2006). FGFR1 and WT1 are markers of human prostate cancer progression. BMC Cancer 6, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M. (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley NH, Bray I, Watters KM, Das S, Bryan K, Bernas T, Prehn JH, and Stallings RL (2011). MicroRNAs 10a and 10b are potent inducers of neuroblastoma cell differentiation through targeting of nuclear receptor corepressor 2. Cell Death Differ. 18, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault I, Lerebours F, Amarir S, Tozlu S, Tubiana-Hulin M, Lidereau R, and Bièche I. (2003). Expression analysis of estrogen receptor alpha coregulators in breast carcinoma: evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin. Cancer Res. 9, 1259–1266. [PubMed] [Google Scholar]
- Gong C, Man EPS, Tsoi H, Lee TKW, Lee P, Ma ST, Wong LS, Luk MY, Rakha EA, Green AR, et al. (2018). BQ323636.1, a Novel Splice Variant to NCOR2, as a Predictor for Tamoxifen-Resistant Breast Cancer. Clin. Cancer Res. 24, 3681–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Barak O, and Lazar MA (2001). The SMRT and N-CoR co-repressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol. 21, 6091–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada M, Ono Y, Fujimaki R, and Asai K. (2015). Learning chromatin states with factorized information criteria. Bioinformatics 31, 2426–2433. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Vertino PM, and Cheng X. (2010). Molecular coupling of DNA methylation and histone methylation. Epigenomics 2, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Wei T, Ye Z, Orme JJ, Lin D, Sheng H, Fazli L, Karnes RJ, Jimenez R, Wang L, et al. (2021). A noncanonical AR addiction drives enzalutamide resistance in prostate cancer. Nat. Commun. 12, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, Liu G, Coleman I, Lakely B, Li R, et al. (2016). Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun. 7, 13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horoszewicz JS, Leong SS, Chu TM, Wajsman ZL, Friedman M, Papsidero L, Kim U, Chai LS, Kakati S, Arya SK, and Sandberg AA (1980). The LNCaP cell line–a new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res. 37, 115–132. [PubMed] [Google Scholar]
- Hu X, and Lazar MA (1999). The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 402, 93–96. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss WJ, Gregory CW, and Smith GJ (2004). Neuroendocrine cell differentiation in the CWR22 human prostate cancer xenograft: association with tumor cell proliferation prior to recurrence. Prostate 60, 91–97. [DOI] [PubMed] [Google Scholar]
- Janky R, Verfaillie A, Imrichova H, Van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D, et al. (2014). iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 10, e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepsen K, Hermanson O, Onami TM, Gleiberman AS, Lunyak V, McEvilly RJ, Kurokawa R, Kumar V, Liu F, Seto E, et al. (2000). Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell 102, 753–763. [DOI] [PubMed] [Google Scholar]
- Jin X, Yan Y, Wang D, Ding D, Ma T, Ye Z, Jimenez R, Wang L, Wu H, and Huang H. (2018). DUB3 Promotes BET Inhibitor Resistance and Cancer Progression by Deubiquitinating BRD4. Mol. Cell 71, 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A. (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Jung I, Schmitt A, Diao Y, Lee AJ, Liu T, Yang D, Tan C, Eom J, Chan M, Chee S, et al. (2019). A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat. Genet. 51, 1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AZ, and Zhang X. (2016). dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 44, D164–D171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanim FL, Gommersall LM, Wood VH, Smith KL, Montalvo L, O’Neill LP, Xu Y, Peehl DM, Stewart PM, Turner BM, and Campbell MJ (2004). Altered SMRT levels disrupt vitamin D3 receptor signalling in prostate cancer cells. Oncogene 23, 6712–6725. [DOI] [PubMed] [Google Scholar]
- Kim D, Gregory CW, French FS, Smith GJ, and Mohler JL (2002). Androgen receptor expression and cellular proliferation during transition from androgen-dependent to recurrent growth after castration in the CWR22 prostate cancer xenograft. Am. J. Pathol. 160, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, and Sergushichev A. (2019). Fast gene set enrichment analysis. bioRxiv. http://biorxiv.org/content/early/2016/06/20/060012. [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbé DP, Gomez EC, Wang J, et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and anti-androgen resistance. Science 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laschak M, Bechtel M, Spindler KD, and Hessenauer A. (2011). Inability of NCoR/SMRT to repress androgen receptor transcriptional activity in prostate cancer cell lines. Int. J. Mol. Med. 28, 645–651. [DOI] [PubMed] [Google Scholar]
- Layer RM, Pedersen BS, DiSera T, Marth GT, Gertz J, and Quinlan AR (2018). GIGGLE: a search engine for large-scale integrated genome analysis. Nat. Methods 15, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Kim JH, Lee YC, Cheong J, and Lee JW (2000). Silencing mediator of retinoic acid and thyroid hormone receptors, as a novel transcriptional corepressor molecule of activating protein-1, nuclear factor-kappaB, and serum response factor. J. Biol. Chem. 275, 12470–12474. [DOI] [PubMed] [Google Scholar]
- Lee BK, Shen W, Lee J, Rhee C, Chung H, Kim KY, Park IH, and Kim J. (2015). Tgif1 Counterbalances the Activity of Core Pluripotency Factors in Mouse Embryonic Stem Cells. Cell Rep. 13, 52–60. [DOI] [PubMed] [Google Scholar]
- Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, and Wong J. (2000). Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 19, 4342–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MD, van den Berg PR, Russell JL, Singh PK, Battaglia S, and Campbell MJ (2015). Integrative genomic analysis in K562 chronic myelogenous leukemia cells reveals that proximal NCOR1 binding positively regulates genes that govern erythroid differentiation and Imatinib sensitivity. Nucleic Acids Res. 43, 7330–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G, Chen LY, Zhang A, Godavarthy A, Xia F, Ghosh JC, Li H, and Chen JD (2003). Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J. Biol. Chem. 278, 5052–5061. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W. (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MD, Smiraglia DJ, and Campbell MJ (2017). The Genomic Impact of DNA CpG Methylation on Gene Expression; Relationships in Prostate Cancer. Biomolecules 7, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, and Smyth GK (2016). csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 44, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, and Brown M. (2008). FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132, 958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Yang F, Mackintosh C, Jayani RS, Oh S, Jin C, Nair SJ, Merkurjev D, Ma W, Allen S, et al. (2020). Super-Enhancer Redistribution as a Mechanism of Broad Gene Dysregulation in Repeatedly Drug-Treated Cancer Cells. Cell Rep. 31, 107532. [DOI] [PubMed] [Google Scholar]
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, et al. (2015). The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, and Beck S. (2014). ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 30, 428–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, and Evans RM (1997). Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 89, 373–380. [DOI] [PubMed] [Google Scholar]
- Oberoi J, Fairall L, Watson PJ, Yang JC, Czimmerer Z, Kampmann T, Goult BT, Greenwood JA, Gooch JT, Kallenberger BC, et al. (2011). Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 18, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, Lord RV, Clark SJ, and Molloy PL (2015). De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 8, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TJ, Karmakar S, Pace MC, Gao T, and Smith CL (2007). The silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) corepressor is required for full estrogen receptor alpha transcriptional activity. Mol. Cell. Biol. 27, 5933–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, Van Djik S, Muhlhausler B, Stirzaker C, and Clark SJ (2016). Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlajamaa P, Sahu B, Lyly L, Aittomäki V, Hautaniemi S, and Jänne OA (2014). Tissue-specific pioneer factors associate with androgen receptor cistromes and transcription programs. EMBO J. 33, 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Qiu X, Zhu Y, Takeda DY, Pan W, Baca SC, Gusev A, Korthauer KD, Severson TM, Ha G, et al. (2020). Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 52, 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Fan J, Zheng R, Wan C, Mei S, Wu Q, Sun H, Brown M, Zhang J, Meyer CA, and Liu XS (2020). Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 21, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium; Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015a). Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 162, 454. [DOI] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015b). Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi K, et al. (2017). Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 170, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seedhouse SJ, Affronti HC, Karasik E, Gillard BM, Azabdaftari G, Smiraglia DJ, and Foster BA (2016). Metastatic phenotype in CWR22 prostate cancer xenograft following castration. Prostate 76, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, and Richards M. (2021). MotifDb: An Annotated Collection of Protein-DNA Binding Sequence Motifs (R package version 1.36.0). [Google Scholar]
- Sheahan AV, and Ellis L. (2018). Epigenetic reprogramming: A key mechanism driving therapeutic resistance. Urol. Oncol. 36, 375–379. [DOI] [PubMed] [Google Scholar]
- Siddiqui ZA, and Krauss DJ (2018). Adjuvant androgen deprivation therapy for prostate cancer treated with radiation therapy. Transl. Androl. Urol. 7, 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siersbæk R, Madsen JGS, Javierre BM, Nielsen R, Bagge EK, Cairns J, Wingett SW, Traynor S, Spivakov M, Fraser P, and Mandrup S. (2017). Dynamic Rewiring of Promoter-Anchored Chromatin Loops during Adipocyte Differentiation. Mol. Cell 66, 420–435.e5. [DOI] [PubMed] [Google Scholar]
- Song Y, van den Berg PR, Markoulaki S, Soldner F, Dall’Agnese A, Henninger JE, Drotar J, Rosenau N, Cohen MA, Young RA, et al. (2019). Dynamic Enhancer DNA Methylation as Basis for Transcriptional and Cellular Heterogeneity of ESCs. Mol. Cell 75, 905–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B, Gillard B, Gao L, Eng KH, and Gelman IH (2013). Src controls castration recurrence of CWR22 prostate cancer xenografts. Cancer Med. 2, 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkel B, Wu D, Chen Z, Wang CM, Liu X, Ye Z, Horning AM, Liu J, Mahalingam D, Lopez-Nicora H, et al. (2016). Integrative analysis identifies targetable CREB1/FoxA1 transcriptional co-regulation as a predictor of prostate cancer recurrence. Nucleic Acids Res. 44, 4105–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, and Beck S. (2013). A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdés-Mora F, Gould CM, Colino-Sanguino Y, Qu W, Song JZ, Taylor KM, Buske FA, Statham AL, Nair SS, Armstrong NJ, et al. (2017). Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat. Commun. 8, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, Haradhvala NJ, Freeman SS, Reed SC, Rhoades J, et al. ; PCF/SU2C International Prostate Cancer Dream Team (2018). Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 174, 433–447.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, and Chung LW (1994). Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int. J. Cancer 57, 406–412. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhou Y, Huan L, Xu L, Shen M, Huang S, and Liang L. (2019). LncRNA MIR22HG inhibits growth, migration and invasion through regulating the miR-10a-5p/NCOR2 axis in hepatocellular carcinoma cells. Cancer Sci. 110, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (N Y) 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Wang Y, Xu K, Hu H, Xu Z, Wu D, Wang Z, You W, Ng CF, Yu S, and Chan FL (2018). Nuclear Receptor LRH-1 Functions to Promote Castration-Resistant Growth of Prostate Cancer via Its Promotion of Intratumoral Androgen Biosynthesis. Cancer Res. 78, 2205–2218. [DOI] [PubMed] [Google Scholar]
- Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, et al. (2017). Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HG, Chan DW, Reynolds AB, Qin J, and Wong J. (2003). N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol. Cell 12, 723–734. [DOI] [PubMed] [Google Scholar]
- You SH, Lim HW, Sun Z, Broache M, Won KJ, and Lazar MA (2013). Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 20, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Li Y, Ishizuka T, Guenther MG, and Lazar MA (2003). A SANT motif in the SMRT corepressor interprets the histone code and promotes histone deacetylation. EMBO J. 22, 3403–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Song X, Zhang Z, Mao Q, Xia W, Xu L, Jiang F, and Dong G. (2020). Aberrant super-enhancer landscape reveals core transcriptional regulatory circuitry in lung adenocarcinoma. Oncogenesis 9, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, Chen CH, Brown M, Zhang X, Meyer CA, and Liu XS (2019). Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 47, D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wang Y, Wu D, Wang S, Chen Z, Xiang S, and Chan FL (2021). Orphan nuclear receptors as regulators of intratumoral androgen biosynthesis in castration-resistant prostate cancer. Oncogene 40, 2625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Q, Li W, Benda C, Huang Z, Ahmed T, Liu P, Guo X, Ibañez DP, Luo Z, Zhang M, et al. (2018). NCoR/SMRT co-repressors cooperate with c-MYC to create an epigenetic barrier to somatic cell reprogramming. Nat. Cell Biol. 20, 400–412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Publicly available PC cohort SNV, copy number, RNA-seq expression data, and clinical characteristics are available in cBioPortal and are detailed in the STAR KEY RESOURCES TABLE and cited in the main text. All raw and processed RNA-seq, ChIP-seq, and MethylationEPIC array data generated in this study have been deposited and made available as of the date of publication at NCBI-GEO under accession series GEO: GSE178820.
No unique code was generated in this study.
Any additional resources required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Anti-NCOR2, rabbit polyclonal | Abcam | Cat# ab24551, RRID:AB_2149134 |
| Anti-NCOR2, rabbit monoclonal | Cell Signaling | Cat# 62370, RRID:AB_2799628 |
| Anti-NCOR2, rabbit | Sigma-Aldrich | Cat# HPA001928, RRID:AB_1079454 |
| Normal Rabbit IgG antibody | Cell Signaling | Cat# 2729, RRID:AB_1031062 |
| Normal Rabbit IgG antibody | Santa Cruz | Cat# sc-2027, RRID:AB_737197 |
| Anti-AR, rabbit polyclonal | Millipore | Cat# 06-680 |
| Anti-TBP, rabbit polyclonal | Cell Signaling | Cat# 8515, RRID:AB_10949159 |
| Anti-GAPDH, rabbit monoclonal | Cell Signaling | Cat# 2118, RRID:AB_561053 |
| Anti-beta-actin, rabbit polyclonal | Abcam | Cat# ab8227, RRID:AB_2305186 |
| Rabbit anti-mouse immunoglobulins/HRP antibody | Agilent | Cat# P0161, RRID:AB_2687969 |
| Goat anti-rabbit immunoglobulins/HRP antibody | Agilent | Cat# P0448, RRID:AB_2617138 |
| Ki-67 antibody, rabbit polyclonal | Abcam | Cat# ab15580, RRID:AB_443209 |
| Synaptophysin antibody, rabbit monoclonal | Abcam | Cat# ab52636, RRID:AB_882786 |
|
| ||
| Biological samples | ||
|
| ||
| Prostate cancer patient-derived tissue microarrays | Roswell Park | Pathology Resource Network (PRN) and |
| and de-identified clinical information | Comprehensive Cancer Center | Data Bank Biorepository (DBBR) |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| TRIzol RNA Isolation Reagent | Thermo Fisher | Cat# 15596026 |
| AllPrep DNA/RNA/miRNA Universal Kit | QIAGEN | Cat# 80224 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| 5α-Dihydrotestosterone (DHT) solution | Millipore Sigma | Cat# D-073 |
| cOmplete, Mini Protease Inhibitor Cocktail | Millipore Sigma | Cat# 11836153001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| TruSeq Stranded Total RNA Library Prep Kit | Illumina | Cat# 20020596 |
| Infinium MethylationEPIC BeadChip Kit | Illumina | Cat# WG-317-1002 |
| ViaLightTM Plus Cell Proliferation and Cytotoxicity BioAssay Kit | Lonza | Cat# LT07-221 |
| TaqMan MicroRNA Reverse Transcription Kit | Applied Biosystems | Cat# 4366596 |
| Turbo DNA-Free kit | Invitrogen | Cat# AM2238 |
|
| ||
| Deposited data | ||
|
| ||
| Raw data | This study | GEO: GSE178820 series |
| Prostate Adenocarcinoma (TCGA, PanCancer Atlas) | TCGA | https://www.cbioportal.org/study/summary?id=prad_tcga_pan_can_atlas_2018 |
| Neuroendocrine Prostate Cancer (Multi-Institute, Nat Med 2016) | Beltran et al., 2016 | https://www.cbioportal.org/study/summary?id=nepc_wcm_2016 |
| The Metastatic Prostate Cancer Project | The Metastatic Prostate | https://www.cbioportal.org/study/summary?id=prad_mpcproject_2018 |
| (Provisional, November 2019) | Cancer Project | |
| Metastatic Prostate Cancer (SU2C/PCF Dream Team, Cell 2015) | (Robinson et al., 2015b) | https://www.cbioportal.org/study/summary?id=prad_su2c_2015 |
| Metastatic Prostate Adenocarcinoma (SU2C/PCF Dream Team, PNAS 2019) | Abida et al., 2019 | https://www.cbioportal.org/study/summary?id=prad_su2c_2019 |
| Prostate Adenocarcinoma (MSKCC, Cancer Cell 2010) | Tayior et al., 2010 | https://www.cbioportai.org/study/ summary?id=prad_mskcc |
| Prostate Adenocarcinoma (MSKCC/DFCI, Nature Genetics 2018) | Armenia et al., 2018 | https://www.cbioportai.org/study/ summary?id=prad_p1000 |
|
| ||
| Experimentai modeis: ceii lines | ||
|
| ||
| LNCaP-shControi | This study | N/A |
| C42-shControi | This study | N/A |
| LNCaP-shNCOR2-A | This study | N/A |
| LNCaP-shNCOR2-B | This study | N/A |
| C42-shNCOR2-A | This study | N/A |
| C42-shNCOR2-B | This study | N/A |
|
| ||
| Experimental modeis: organisms/strains | ||
|
| ||
| Hsd:Athymic Nude-Foxn1nu, male | Harlan/Envigo | 069 |
| CWR22 Prostate Cancer Xenograft Mouse (shControi, shNCOR2; V2LHS-251658) | This study | N/A |
|
| ||
| Oligonucieotides | ||
|
| ||
| NCOR2 (TaqMan Gene Expression Assay) | Thermo Fisher | Assay ID: Hs00196955_m1 |
| AR | Thermo Fisher | Assay ID: Hs00171172_m1 |
| GAPDH (TaqMan Gene Expression Assay) | Thermo Fisher | Assay ID:4326317E |
| DENND1B (forward: TTACACAATGAAAAGGGAGG) | IDT | N/A |
| TMPRSS2 (forward: ATGATGCTGCAGCCAGAACAG) | IDT | N/A |
| HERC3 (forward: AGGTTCTTGTAGGCTGTGGT) | IDT | N/A |
| CXCR7 (forward: GATGTGGGTTACAAAGCTG) | IDT | N/A |
| KLK3 (forward: ACTTCAGTGTGTGGACCTTGT) | IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| shNCOR2-A (Targeting) pGIPZ Lentiviral Construct | RPCCC Gene Modulation Resource | V2LHS-251658 |
| shNCOR2-B (Targeting) pGIPZ Lentiviral Construct | RPCCC Gene Modulation Resource | V2LHS-196739 |
| GIPZ Lentiviral shRNA Construct | Thermo Fisher | V2LHS |
|
| ||
| Software and aigorithms | ||
|
| ||
| Prism | GraphPad | Version 9.1.0 |
| R V3.6-4.0 | https://www.r-project.org/ | https://www.r-project.org/ |
| Salmon | Patro et al., 2017 | https://combine-lab.github.io/salmon/ |
| Bioconductor 3.2 | (Huber et al., 2015) | http://www.bioconductor.org |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| LISA | Qin et al., 2020 | http://lisa.cistrome.org/ |
| iRegulon | (Janky et al., 2014) | http://iregulon.aertslab.org/ |
| ChAMP | (Morris et al., 2014) | https://www.bioconductor.org/packages/release/bioc/html/ChAMP.html |
| DMRcate | Peters et al., 2015 | https://www.bioconductor.org/packages/release/bioc/html/DMRcate.html |
| Rsubread | (Liao et al., 2019) | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| csaw | Lun and Smyth, 2016 | https://bioconductor.org/packages/release/bioc/html/csaw.html |
| MotifDb | Shannon and Richards, 2021 | https://bioconductor.org/packages/release/bioc/html/MotifDb.html |
| HOMER v4.10 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| GIGGLE | Layer et al., 2018 | https://github.com/ryaniayer/giggie |
| GSEA v4.1.0 | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| MSigDB v7.4 | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/msigdb |
| ciusterProfiler | Wu et al., 2021 | https://bioconductor.org/packages/release/bloc/html/clusterProfiler.html |
| fgsea | Korotkevich et al., 2019 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Aperio Nuciear Aigorithm | Leica Biosystems | https://www.leicabiosystems.com/digital-pathology/analyze/ihc/aperio-nuclear-algorithm/ |
| ROSE: Rank Ordering of Super Enhancers | Whyte et al., 2013 | http://younglab.wi.mit.edu/super_enhancer_code.html |
|
| ||
| Other | ||
|
| ||
| GIBCO, RPMI 1640 Medium | Thermo Fisher | Cat# 11875093 |
| Magna ChIP Protein A+G Magnetic Beads | Sigma-Aidrich | Cat# 16-663 |