

Abstract
We report the first enantioselective addition of pyrazoles to 1,3-dienes. Secondary and tertiary allylic pyrazoles can be generated with excellent regioselectivity. Mechanistic studies support a pathway distinct from previous hydroaminations: a Pd(0)-catalyzed ligand-to-ligand hydrogen transfer (LLHT). This hydroamination tolerates a range of functional groups and advances the hydrofunctionalization of dienes.
Keywords: pyrazole; 1,3-diene; hydroamination; Pd-catalysis; enantioselectivity
Graphical Abstract
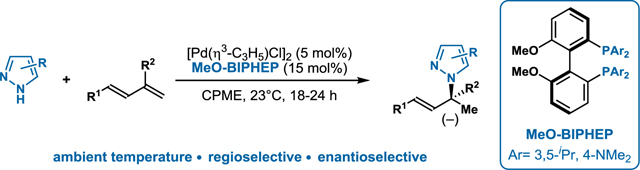
Nitrogen-containing heterocycles, such as pyrazoles, represent valuable scaffolds for drug discovery and thus remain an inspiration for synthetic methods (Figure 1A).1 The direct addition of a pyrazole to a double bond represents an attractive and atom-economical approach for forging C-N bonds. With regards to the coupling partner, conjugated dienes are ideal building blocks,2 with many being raw materials for various industrial applications, including polymerizations.3,4 Within the asymmetric hydroamination of dienes, there exists methods using anilines (Hartwig),5 secondary amines (our lab and Malcolmson),6,7 and primary amines (Mazet).8 In comparison to previously studied amines (with nucleophilicities N = 13–18 on Mayr scale9), pyrazoles present a challenge and opportunity because of their lower nucleophilicity (N = 9.6). Given the two reactive nitrogen atoms: a pyrrolic and a pyridinic nitrogen, the coupling of pyrazoles with unsymmetrical dienes can provide 32 isomers (Figure 1B). With both Rh and Pd-catalysts, Breit achieved enantioselective hydroamination of allenes using pyrazoles (Figure 1C).10 Concurrent with our studies, Chen and coworkers were independently pursuing the Pd-catalyzed hydroamination of isoprene. With indazoles and select pyrazoles, they were able to generate either achiral or racemic products.11 In this communication, we showcase the first asymmetric addition of pyrazoles to dienes. This mild hydroamination tolerates a variety of functional groups and occurs via a mechanism distinct to those previously proposed for diene hydroamination.
Figure 1.

Inspiration for asymmetric hydroamination of 1,3-dienes with pyrazoles.
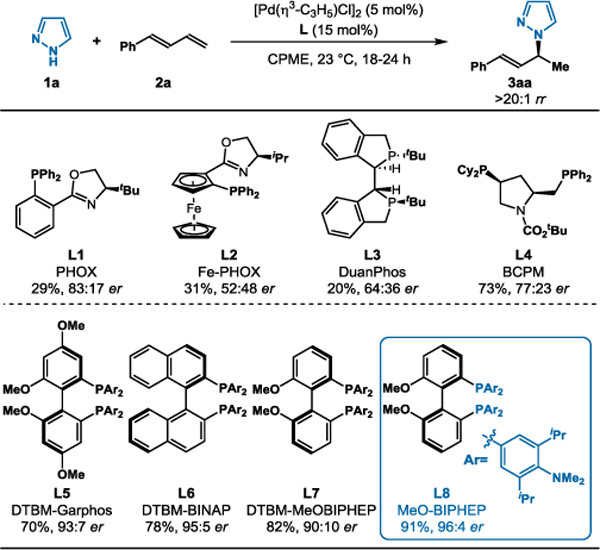
On the basis of our previous hydroaminations of dienes, we initiated investigations with Rh-catalysts.6,12 We chose pyrazole (1a) and 1-phenyl-butadiene (2a) as the model substrates and observed no desired reactivity (see SI). In contrast, under Pd-catalysis, the desired allylated pyrazole 3aa was obtained when using a range of achiral bisphosphine ligands (see SI). In search of an asymmetric variant (L1–L8), we found that atropoisomeric bisphosphine ligands gave the most promising results (Table 1). Thus, we focused on this ligand family to achieve enantioselective catalysis. The DTBM analogs L5–L7 afford desired pyrazole 3aa in 70–82% yield and good to excellent selectivity (>20:1 rr, 90:10–95:5 er). Substitution on the aryl groups most likely enhances reactivity by promoting ligand-substrate dispersion interactions in the transition state, a concept in accordance with literature observations by Buchwald and others.13 We observed similar ligand trends in Rh-catalyzed hydrofunctionalizations of alkynes.14 With further optimization15, we found commercially available MeO-BIPHEP ligand L8 afforded 3aa in 91% yield with >20:1 rr and 96:4 er.
Table 1.
Ligand Effects on Asymmetric Pyrazole Hydroamination
|
Reaction conditions: 1a (0.1 mmol), 2a (0.5 mmol), [Pd(η3-C3H5)Cl]2 (5 mol%), Ligand (15 mol%), CPME (0.4 mL), 23 °C, 18–24 h. Isolated yields. Regioselectivity determined by 1H NMR analysis of the unpurified reaction mixture. Enantioselectivity determined by chiral SFC. DTBM = 3,5-di-tert-butyl-4-methoxyphenyl.
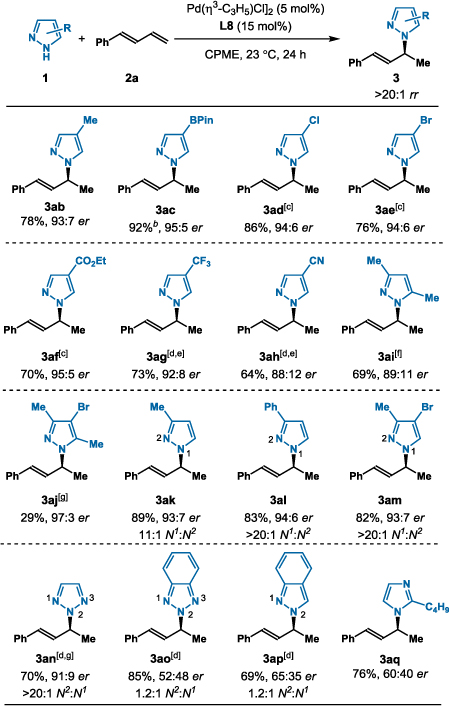
With L8 in hand, we examined the addition of various pyrazoles 1 to diene 2a (Table 2). Generally, allylated pyrazoles 3ab–3aj form with high enantioselectivity (88:12–97:3 er) and >20:1 rr in the coupling of diene 2a with pyrazoles 1b–1j. The electronic properties of the pyrazoles show negligible impact on enantioselectivity and regioselectivity. However, electron-withdrawing substituents show more sluggish reactivity and require extended reaction times (3ad–3af, 3aj) or higher temperature (3ag, 3ah) to obtain good yields (64–86%). Halogenated products (3ad, 3ae) are tolerated despite the potential for competing oxidative addition into the aryl halide bond; no side products from oxidative addition are observed. Electron-donating substituents allow for facile reactivity and shorter reaction times (3ai, 69%, 89:11 er). Other unsymmetrical pyrazoles provide moderate to excellent N1:N2 regioselectivity (3ak–3am, 11:1–20:1 rr). The N1:N2 regioselectivity favors allylation at the less sterically-congested nitrogen atom. Pyrazole tautomerization is known to occur in solution; for example, 5-Me pyrazole (1k) exists in a nearly 1:1 ratio of the 3-and 5-substituted pyrazoles on the basis of NMR spectroscopy.15 Despite the presence of tautomers, we observe high selectivity for formation of 3ak, which indicates tautomerization occurs faster than C–N bond formation. Together, these results represent the first enantioselective hydroamination of 1,3-dienes with azoles.16
Table 2.
Hydroamination of 1,3-Diene (2a) with Various Azoles[a]
|
Reaction conditions: 1 (0.2 mmol), 2a (1.0 mmol), [Pd(η3-C3H5)Cl]2 (5 mol%), MeO-BIPHEP L8 (15 mol%), CPME (0.8 mL), 23 °C, 24 h. Isolated yields. Regioselectivity determined by 1H NMR analysis of the unpurified reaction mixture. Enantioselectivity determined by chiral SFC.
NMR yield.
48 h
60 °C
12 h
8 h
36 h.
In addition to pyrazole substrates, several other azoles show promising reactivity under the standard conditions. The addition of 1H-1,2,3-triazole (3an) gives 70% yield and 91:9 er with high regioselectivity (>20:1 rr, >20:1 N2:N1).17 The coupling of 1H-benzotriazole, 1H-indazole, and 2-butyl-1H-imidazole with diene 2a provides the corresponding allylated azoles (3ao, 3ap, and 3aq) with promising reactivity and chemoselectivity, however, further optimization is needed.18 In stark contrast, pyrrole showed no reactivity, a result that supports our hypothesis on the mechanism (vida infra).
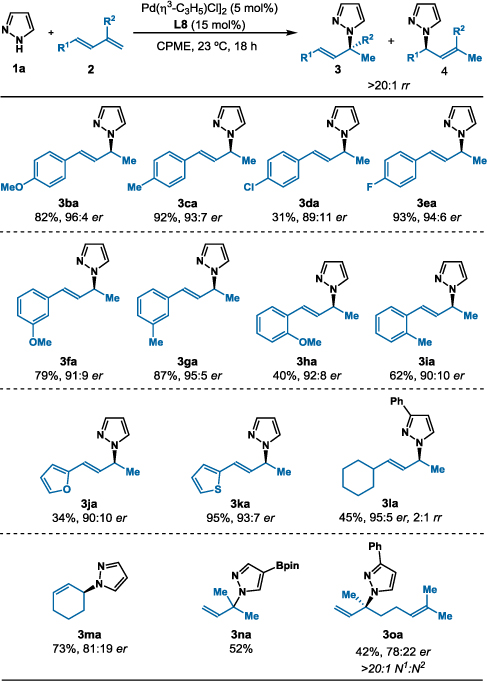
Next, we studied the hydroamination of fourteen different 1,3-dienes 2 with pyrazole 1a (Table 3). Varying substitution on the aryl dienes results in a range of chiral allylated pyrazoles 3ba–3ka (31–95% yield, >20:1 rr, 89:11–96:4 er). Both electron-rich methoxy-substituted (3ba, 3fa, and 3ha) and electron-poor fluoro-substituted (3ea) 1,3-dienes react well. In contrast, chloro-substituted phenyl diene 2d affords 3da in only 31% yield due to competing oxidative addition into the C–Cl bond. Sterically encumbered ortho-substituted dienes undergo addition to 3ha and 3ia in 40% and 62% yield, respectively. The protocol transforms heterocyclic substituted dienes 2j (R1 = 2-furyl) and 2k (R1 = 2-thiophene) into 3ja in 34% yield, 90:10 er, and >20:1 rr, and 3ka in 95% yield, 93:7 er, and >20:1 rr. Hydroamination of alkyl substituted 1,3-diene 2l yields the allylic pyrazole 3la in 45% yield, 95:5 er, and 2:1 rr. Cyclic dienes such as 1,3-cyclohexadiene (2m) couple with pyrazole (1a) to generate 3ma in 71% yield and >20:1 rr, albeit with a lower enantioselectivity of 81:19 er. Hydroamination of feedstocks, isoprene and myrcene, provide the tertiary allylic amines (3na, 3oa) as single structural isomers.
Table 3.
Hydroamination of Various 1,3-Dienes with Pyrazole (1a)
|
Reaction conditions: 1a (0.1 mmol), 2 (0.5 mmol), [Pd(η3-C3H5)Cl]2 (5 mol%), MeO-BIPHEP L8 (15 mol%), CPME (0.4 mL), 23 °C, 18 h. Isolated yields. Regioselectivity determined by 1H NMR analysis of the unpurified reaction mixture. Enantioselectivity determined by chiral SFC.
Electronic circular dichroism (ECD) is a powerful technique to determine absolute configuration.19 By using this method, we elucidated the absolute stereochemistry of the chiral allylated azoles. Comparing theoretical calculations and experimental data, a qualitative match (i.e., similar shapes) enabled assignment of the absolute configuration.19 TDDFT calculations produced the ECD spectra of (S)-3aa and (S)-3ab. Qualitative comparison to the experimental results suggests that the major enantiomer bears the (S)-configuration as drawn (see SI).
Previous reports on the hydrofunctionalization of dienes, including Chen’s hydroamination of isoprene, feature mechanisms that occur by Pd(II)–H catalysis.4,11 In these scenarios, alkene insertion into a Pd–H forges the new carbon-hydrogen bond and these transformations occur at elevated temperatures. On the basis of recent reports and our own observations, we propose that our ambient hydroamination occurs via the mechanism depicted in Figure 3. The palladium pre-catalyst interacts with a bisphosphine ligand to form active Pd(0) catalyst I. Both pyrazole 1 and 1,3-diene 2 bind to complex I to generate palladium intermediate II. Given Pd(II)’s preference to adopt a square planar geometry, we reason that diene coordinates to the Pd in an η2 fashion.11,20,21 From here, we imagine that the hydrogen atom is transferred directly from pyrazole 1 to 1,3-diene 2 through ligand-to-ligand hydrogen transfer (LLHT). Ionization of intermediate III followed by outer-sphere nucleophilic attack with pyrazole anion on the C3 carbon affords the desired allylic pyrazole 3 and regenerates Pd(0) complex I.
Figure 3.
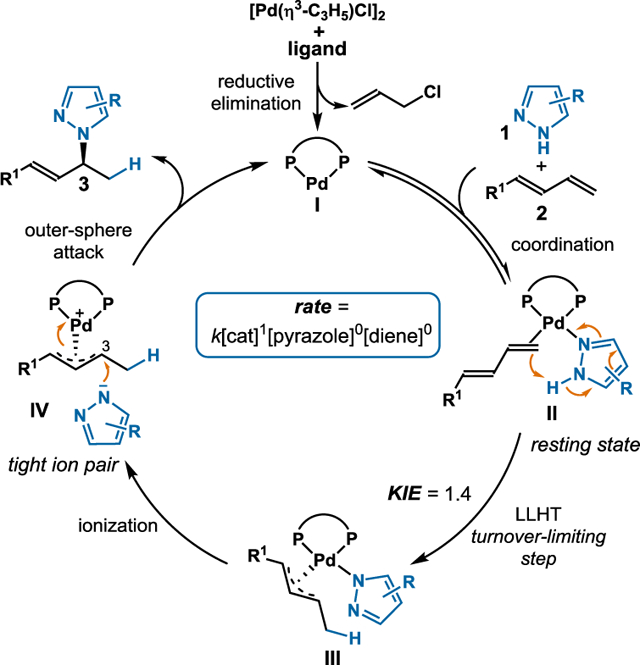
Proposed mechanism.
In support of a Pd(0) pathway, Huang22 and Rutjes’23 computations have shown that the formation of the Pd(II)–H complex from [Pd(η3-C 3H5)Cl]2 is kinetically infeasible at temperatures up to 80°C. In line with this, we do not observe Pd–H when studying a mixture of [Pd(η3-C3H5)Cl]2 with ligand in d8-toluene. Additionally, alternative Pd(0) precursors, including Pd(PPh3)4 and Pd(PtBu3)2, afford the allylic pyrazole albeit in lower yields and selectivity (see SI). In these cases, there is no acid additive, which makes Pd–H unlikely. By using Burés’ variable time normalization analysis (VTNA) method,24 we studied the kinetic profile and observed first order in catalyst and zero order in both the pyrazole (1) and diene (2). This rate law supports coordination of diene and pyrazole to Pd to generate intermediate II as the catalyst resting state.
In Zi’s study on hydrosulfonylation of dienes, theoretical calculations show that diene migratory insertion into Pd(II)–H is energetically unfavorable compared to LLHT.20 In analogy, we propose an LLHT that is the turnover-limiting step (Figure 3). Comparing the initial rates of deuterated pyrazole d-1a against 1H-pyrazole 1a in parallel, we observe a KIE of 1.4 (Figure 4A) which is similar with previous LLHT examples.25 When we subjected deuterium-labeled pyrazole d-1a to the standard conditions (Figure 4B), we see quantitative deuterium incorporation at the C4 position of d-3aa; the recovered diene shows no deuterium labelling. Together, these results suggest that hydrogen transfer is highly selective and irreversible. Of note, in Malcolmson’s hydroamination, the analogous experiment demonstrated deuterium scrambling.7 Additionally, hydroamination of diene 1a with pyrrole shows no reactivity. In comparison to pyrazole, the pyrrole lacks a second nitrogen atom. We reason the second nitrogen coordinates to Pd to provide the geometry needed for LLHT.
Figure 4.

Mechanistic studies.
Next, we performed a crossover study by adding pyrazole 1b to (S)-3aa in the presence of the Pd-catalyst (Figure 4C, entry 1). The crossover product (S)-3ab was generated where the major isomer possessed the same absolute configuration as the (S)-3aa starting material. A similar crossover experiment using a racemic mixture of 3aa was performed (Figure 4C, entry 2). After 18 h, the crossover product (S)-3ab (60:40 er) is afforded along with an enantioenriched mixture of (R)-3aa (34:66 er). Enantioenrichment of the R-enantiomer suggests that (S)-3aa reacts faster, transforming into (S)-3ab in these crossover experiments. These experiments suggest that Pd insertion into the C–N bond can be reversible, especially under conditions where pyrazole is present in large excess (see SI). We found that the use of excess diene affords better results and reason that this stoichiometry prevents reversible product formation. Based on these experiments, we favor a mechanism that involves outer sphere nucleophilic attack of pyrazole to complex III. In line with Tsuji–Trost transformations, the ionization of allylic pyrazole (S)-3 with Pd-catalyst would invert the configuration at the reactive center (Figure 4C). To afford the S-enantiomer of the crossover product, the nucleophile must attack through an SN2-like mechanism on the face of the olefin opposite to Pd. Our proposed mechanism fits with the convention of classifying nucleophilic attack on η3-Pd-π-allyl intermediates. Pyrazole, which is considered a “soft” nucleophile (pKa~19.8), would be expected to proceed through this outer-sphere pathway.26
In our and Malcolmson’s independently reported Pd-catalyzed diene hydrofunctionalizations, a competition experiment was performed using a mixture of E and Z dienes. In their studies, both (Z)- and (E)-1-phenylbutadiene (2a) converged to the same major enantiomer.7,27 Moreover, deuterium scrambling into the diene was observed. These results supported a Pd–H mechanism where hydropalladation is reversible. In this pyrazole study, however, we find when using a mixture of (Z)- and (E)-1-phenylbutadiene (2a) isomers, only the (E)-2a transforms to allylic pyrazole (3aa) (50% yield brsm), while the (Z)-2a is recovered (Figure 4D). These contrasting results point to a mechanism which differs from those previously invoked. Here, we reason that the (Z)-diene does not transform due to increased steric strain in the LLHT step.
Hydroamination represents an attractive way to transform dienes into nitrogen-containing building blocks. By using Pd-catalysis, we achieved the first enantioselective hydroamination of dienes with aromatic heterocycles. The allylation tolerates a broad range of substituted pyrazoles and dienes, and both secondary and tertiary allylated pyrazoles are obtained in good to excellent yields with high regio- and enantioselectivities. Insights from this study will guide the development of related couplings that feature heterocycles, which represent motifs pertinent in drug discovery.
Supplementary Material
Acknowledgements
Funding provided by UC Irvine, the National Institutes of Health (1R35GM127071) and the National Science Foundation (CHE-1465263). X.-H. Y. thanks the Beijing Institute of Technology Teli Young Fellow Project. We thank Solvias AG for donating commercial MeO-BIPHEP L8. We thank Lisa M. Nogle, David A. Smith, Adam Beard, Miroslawa Darlak, and Mark Pietrafitta for reversed phase purifications (Merck & Co., Inc.). We thank Donovon Adpressa for assistance with NMR structure elucidation (Merck & Co., Inc.). This work utilized the infrastructure for high-performance and high-throughput computing, research data storage and analysis, and scientific software tool integration built, operated, and updated by the Research Cyber infrastructure Center (RCIC) at the University of California, Irvine (UCI). The RCIC provides cluster-based systems, application software, and scalable storage to directly support the UCI research community. https://rcic.uci.edu.
References
- (1).Vitaku E, Smith DT, Njardson JT, J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (2).(a) For selected reviews on 1,3-dienes as building blocks, see: Reymond S, Cossy J, Chem. Rev. 2008, 108, 5359–5406; [DOI] [PubMed] [Google Scholar]; (b) Zhu Y, Cornwall RG, Du H, Zhao B, Shi Y, Acc. Chem. Res. 2014, 47, 3665–3678; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen J-R, Hu X-Q, Lu L-Q, Xiao W-J, Chem. Rev. 2015, 115, 5301–5365. [DOI] [PubMed] [Google Scholar]
- (3).(a) For selected reviews on polymerization of 1,3-dienes, see: Ricci G, Sommazzi A, Masi F, Ricci M, Boglia A, Leone G, Coord. Chem. Rev. 2010, 254, 661; [Google Scholar]; (b) Valente A, Mortreux A, Visseaux M, Zinck P, Chem. Rev. 2013, 113, 3836–3857; [DOI] [PubMed] [Google Scholar]; (c) Takeuchi D, Stereoselective Polymerization of Conjugated Dienes. in Encyclopedia of Polymer Science and Technology; Wiley: New York, 2013; pp 1–25. [Google Scholar]
- (4).For review on hydrofunctionalizations of 1,3-dienes, see: Adamson NJ, Malcolmson SJ, ACS Catal. 2020, 10, 1060–1076. [Google Scholar]
- (5).Löber O, Kawatsura M, Hartwig JF, J. Am. Chem. Soc. 2001, 123, 4366–4367. [DOI] [PubMed] [Google Scholar]
- (6).Yang X-H, Dong VM, J. Am. Chem. Soc. 2017, 139, 1774–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Adamson NJ, Hull E, Malcolmson SJ, J. Am. Chem. Soc. 2017, 139, 7180–7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tran G, Shao W, Mazet C, J. Am. Chem. Soc. 2019, 141, 14814–14822. [DOI] [PubMed] [Google Scholar]
- (9).For Mayr’s Database of Reactivity Parameters, see: https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/fe/.
- (10).Haydl AM, Xu K, Breit B, Angew. Chem. Int. Ed. 2015, 54, 7149–7153; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7255–7259; [Google Scholar]; (b) Hilpert LJ, Sieger SV, Haydl AM, Breit B, Angew. Chem. Int. Ed. 2019, 58, 3378–3381; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3416–3419. [Google Scholar]
- (11).Jiang W-S, Ji D-W, Zhang W-S, Zhang G, Min X-T, Hu Y-C, Jiang X-L, Chen Q-A, Angew. Chem. Int. Ed. 2021, 60, 8321–8328; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 8402–8409. [Google Scholar]
- (12).Yang X-H, Lu A, Dong VM, J. Am. Chem. Soc. 2017, 139, 14049–14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Lu G, Liu RY, Yang Y, Fang C, Lambrecht DS, Buchwald SL, Liu P, J. Am. Chem. Soc. 2017, 139, 16548–16555; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wagner JP, Schreiner PR, Angew. Chem. Int. Ed. 2015, 54, 12274–12296; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12446–12471; [Google Scholar]; (c) Ahlquist MSG, Norrby P-O, Angew. Chem. Int. Ed. 2011, 50, 11794–11797; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11998–12001; [Google Scholar]; (d) Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W, J. Am. Chem. Soc. 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Cruz FA, Dong VM, J. Am. Chem. Soc. 2017, 139, 1029–1032; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cruz FA, Zhu Y, Tercenio QD, Shen Z, Dong VM, J. Am. Chem. Soc. 2017, 139, 10641–10644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Aguilar-Parrilla F, Cativiela C, de Villegas MDD, Elguero J, Foces-Foces C, Laureiro JIG, Cano FH, Limbach H-H, Smith JAS, Toiron C, J. Chem. Soc. Perkin Trans. 2, 1992, 2, 1737–1742. [Google Scholar]
- (16).For Rhodium-catalyzed hydroarylation of 1,3-dienes with indoles for C-allylation, see: Marcum JS, Roberts CC, Manan RS, Cervarich TN, Meek SJ, J. Am. Chem. Soc. 2017, 139, 15580–15583. [DOI] [PubMed] [Google Scholar]
- (17).Nitrogen labeling of pyrazole products is based on the product, i.e., the major product is the N1 product. Triazole, indazole, and benzotriazole are labeled based on their naming conventions, i.e., the major products are the N2 products. [Google Scholar]
- (18).To buffer the conditions as demonstrated by Malcomson,7 we added base (Et3N) to the benzotriazole reaction, however, we saw no marked improvement, only decreased N2:N1 selectivity of 1:1. [Google Scholar]
- (19).(a) Furche F, Ahlrichs R, Wachsmann C, Weber E, Sobanski A, Vögtle F, Grimme S, J. Am. Chem. Soc. 2000, 122, 1717–1724; [Google Scholar]; (b) Berova N, Di Bari L, Pescitelli G, Chem. Soc. Rev, 2007, 36, 914–931. [DOI] [PubMed] [Google Scholar]
- (20).In the cited mechanism, dissociation of one of the phosphine arms occurs, which is reasonable to consider. For more information, see: Zhang Q, Dong D, Zi W, J. Am. Chem. Soc. 2020, 142, 15860–15869. [DOI] [PubMed] [Google Scholar]
- (21).Benn R, Jolly PW, Joswig T, Mynott R, Schick K-P, Naturforsch Z. 1986, 41b, 680–691. [Google Scholar]
- (22).Wu Z, Zhang M, Shi Y, Huang G, Org. Chem. Front. 2020, 7, 1502–1511. [Google Scholar]
- (23).Bernar I, Fiser B, Blanco-Ania D, Gómez-Bengoa E, Rutjes FPJT, Org. Lett. 2017, 19, 4211–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Burés J, Angew. Chem. Int. Ed. 2016, 55, 16084–16087; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16318–16321. [Google Scholar]
- (25).Zell D, Bursch M, Müller V, Grimme S, Ackermann L, Angew. Chem. Int. Ed. 2017, 56, 10378–10382; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10514–10518. [Google Scholar]
- (26).(a) Sha S-C, Zhang J, Carroll PJ, Walsh PJ, J. Am. Chem. Soc. 2013, 135, 17602–17609; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, van Vranken DL, Chem. Rev. 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]
- (27).Nie S-Z, Davison RT, Dong VM, J. Am. Chem. Soc. 2018, 140, 16450–16454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.