

Abstract
Background
Posttraumatic stress disorder (PTSD) is a prevalent and disabling disorder. Evidence that PTSD is characterised by specific psychobiological dysfunctions has contributed to a growing interest in the use of medication in its treatment.
Objectives
To assess the effects of medication for reducing PTSD symptoms in adults with PTSD.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL; Issue 11, November 2020); MEDLINE (1946‐), Embase (1974‐), PsycINFO (1967‐) and PTSDPubs (all available years) either directly or via the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR). We also searched international trial registers. The date of the latest search was 13 November 2020.
Selection criteria
All randomised controlled trials (RCTs) of pharmacotherapy for adults with PTSD.
Data collection and analysis
Three review authors (TW, JI, and NP) independently assessed RCTs for inclusion in the review, collated trial data, and assessed trial quality. We contacted investigators to obtain missing data. We stratified summary statistics by medication class, and by medication agent for all medications. We calculated dichotomous and continuous measures using a random‐effects model, and assessed heterogeneity.
Main results
We include 66 RCTs in the review (range: 13 days to 28 weeks; 7442 participants; age range 18 to 85 years) and 54 in the meta‐analysis.
For the primary outcome of treatment response, we found evidence of beneficial effect for selective serotonin reuptake inhibitors (SSRIs) compared with placebo (risk ratio (RR) 0.66, 95% confidence interval (CI) 0.59 to 0.74; 8 studies, 1078 participants), which improved PTSD symptoms in 58% of SSRI participants compared with 35% of placebo participants, based on moderate‐certainty evidence.
For this outcome we also found evidence of beneficial effect for the noradrenergic and specific serotonergic antidepressant (NaSSA) mirtazapine: (RR 0.45, 95% CI 0.22 to 0.94; 1 study, 26 participants) in 65% of people on mirtazapine compared with 22% of placebo participants, and for the tricyclic antidepressant (TCA) amitriptyline (RR 0.60, 95% CI 0.38 to 0.96; 1 study, 40 participants) in 50% of amitriptyline participants compared with 17% of placebo participants, which improved PTSD symptoms. These outcomes are based on low‐certainty evidence. There was however no evidence of beneficial effect for the number of participants who improved with the antipsychotics (RR 0.51, 95% CI 0.16 to 1.67; 2 studies, 43 participants) compared to placebo, based on very low‐certainty evidence.
For the outcome of treatment withdrawal, we found evidence of a harm for the individual SSRI agents compared with placebo (RR 1.41, 95% CI 1.07 to 1.87; 14 studies, 2399 participants). Withdrawals were also higher for the separate SSRI paroxetine group compared to the placebo group (RR 1.55, 95% CI 1.05 to 2.29; 5 studies, 1101 participants). Nonetheless, the absolute proportion of individuals dropping out from treatment due to adverse events in the SSRI groups was low (9%), based on moderate‐certainty evidence. For the rest of the medications compared to placebo, we did not find evidence of harm for individuals dropping out from treatment due to adverse events.
Authors' conclusions
The findings of this review support the conclusion that SSRIs improve PTSD symptoms; they are first‐line agents for the pharmacotherapy of PTSD, based on moderate‐certainty evidence. The NaSSA mirtazapine and the TCA amitriptyline may also improve PTSD symptoms, but this is based on low‐certainty evidence. In addition, we found no evidence of benefit for the number of participants who improved following treatment with the antipsychotic group compared to placebo, based on very low‐certainty evidence. There remain important gaps in the evidence base, and a continued need for more effective agents in the management of PTSD.
Plain language summary
Medication for posttraumatic stress disorder
Why is this review important?
Posttraumatic stress disorder (PTSD) occurs after exposure to significant trauma and results in enormous personal and societal costs. Although it has traditionally been treated with psychotherapy, medication treatments have proven effective in PTSD treatment.
Who will be interested in this review?
‐ People with PTSD. ‐ Families and friends of people who suffer from PTSD. ‐ General practitioners, psychiatrists, psychologists, and pharmacists.
What question does this review aim to answer?
‐ Is pharmacotherapy effective for reducing PTSD symptoms in adults with PTSD?
Which studies were included in the review?
We included studies comparing medication with placebo or a control, or both, for the treatment of PTSD in adults. We included 66 trials in the review, with a total of 7442 participants.
What does the evidence from the review tell us?
There was evidence of a beneficial effect that selective serotonin reuptake inhibitors (SSRIs) improve PTSD symptoms compared to placebo, based on moderate‐certainty evidence. There was also evidence of a benefit for the noradrenergic and specific serotonergic antidepressant (NaSSA) mirtazapine and the tricyclic antidepressant (TCA) amitriptyline, in improving PTSD symptoms, based on low‐certainty evidence. We also found no evidence of benefit for the number of participants who improved following treatment with the antipsychotic group compared to placebo, based on very low‐certainty evidence. For the remaining medication classes, we did not observe evidence of a benefit for improving PTSD symptoms.
There was evidence of a harm that more people taking individual SSRI agents dropped out due to side effects than did those taking placebo, but absolute withdrawal rates were low for the SSRI groups.
What should happen next?
Most evidence for pharmacotherapy efficacy is related to SSRIs for acute treatment. There is an ongoing need to develop new pharmacotherapeutic treatments of PTSD.
Summary of findings
Summary of findings 1. Comparison 1: Alpha‐blockers versus placebo for posttraumatic stress disorder (PTSD).
| Comparison 1: Alpha‐blockers versus placebo for posttraumatic stress disorder (PTSD) | ||||||
|
Population: adults (aged 18‐85)
Settings: multi‐centre trials
Intervention: alpha‐blocker
Comparison: placebo Follow‐up: not specified | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With alpha‐blockers | |||||
| Treatment efficacy ‐ treatment response, as measured by the Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | N/A | N/A | N/A | N/A | N/A | We found no studies that looked at the number of participants who responded to prazosin compared to placebo. |
| Treatment tolerability, as measured by Dropouts due to adverse events (acute phase) | Study population | RR 0.99 (0.91 to 1.08) | 304 (1 study) | ⊕⊕⊕⊝ moderatea | No evidence of a difference in dropout rates were found in the alpha‐blocker group (13%) and placebo group (12%) | |
| 118 per 1000 | 117 per 1000 (108 to 128) | |||||
| Moderate | ||||||
| 118 per 1000 | 117 per 1000 (107 to 127) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval;RR: risk ratio. N/A: Not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded by one level due to serious risk of bias (concerns with randomisation procedures).
Summary of findings 2. Comparison 2: Antipsychotics versus placebo for posttraumatic stress disorder (PTSD).
| Comparison 2: Antipsychotics versus placebo for posttraumatic stress disorder (PTSD) | ||||||
|
Population: adults (aged 18‐85)
Settings: single and multi‐centre trials
Intervention: antipsychotics
Comparison: placebo Follow‐up: not specified | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With antipsychotics | |||||
| Treatment efficacy ‐ treatment response, as measured by the Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.51 (0.16 to 1.67) | 43 (2 studies) | ⊕⊝⊝⊝ very lowa,b,c | There was no evidence of a benefit of the number of participants in the antipsychotic groups (71%) compared to the placebo groups (37%) who responded and improved on the CGI‐I scale | |
| 368 per 1000 | 188 per 1000 (59 to 615) | |||||
| Moderate | ||||||
| 443 per 1000 | 226 per 1000 (71 to 740) | |||||
| Treatment tolerability, as measured by Dropouts due to adverse events (acute phase) | Study population | RR 0.98 (0.92 to 1.05) | 348 (5 studies) | ⊕⊕⊝⊝ lowa,b | Twice as many participants withdrew from the antipsychotic groups (16%) compared to the placebo groups (7%), but no important difference in dropout rates was found | |
| 71 per 1000 | 66 per 1000 (60 to 73) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded by one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded by one level due to serious imprecision (wide confidence intervals). cDowngraded by one level due to moderate heterogeneity (I2 of 50%).
Summary of findings 3. Comparison 3: MAOIs versus placebo for posttraumatic stress disorder (PTSD).
| Comparison 3: MAOIs versus placebo for posttraumatic stress disorder (PTSD) | ||||||
|
Population: adults (aged 18‐85)
Settings: multi‐centre trial
Intervention: MAOI
Comparison: placebo Follow‐up: not specified | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With MAOIs | |||||
| Treatment efficacy ‐ treatment response, as measured by the Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | N/A | N/A | N/A | N/A | N/A | We found no studies that looked at the number of participants who responded to phenelzine versus placebo. |
| Treatment tolerability, as measured by Dropouts due to adverse events (acute phase) | Study population | RR 1.14 (0.90 to 1.43) | 37 (1 study) | ⊕⊕⊝⊝ lowa,b | More participants dropped out from the placebo group (17%) compared to the MAOI group (5%), but we found no difference in dropout rates | |
| 167 per 1000 | 190 per 1000 (150 to 238) | |||||
| Moderate | ||||||
| 167 per 1000 | 190 per 1000 (150 to 239) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval;RR: risk ratio. N/A: Not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded by one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded by one level due to serious imprecision (wide confidence intervals).
Summary of findings 4. Comparison 4: NaSSAs versus placebo for posttraumatic stress disorder (PTSD).
| Comparison 4: NaSSAs versus placebo for posttraumatic stress disorder (PTSD) | ||||||
|
Population: adults (aged 18‐85)
Settings: single‐centre trial
Intervention: NaSSA
Comparison: placebo Follow‐up: not specified | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With NaSSAs | |||||
| Treatment efficacy ‐ Treatment response, as measured by the Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.45 (0.22 to 0.94) | 26 (1 study) | ⊕⊕⊝⊝ lowa,b | There was evidence of a benefit for the number of participants with PTSD who responded to treatment (65%) compared to placebo (22%). This is also indicated by the Risk Ratio of 0.45 which indicates that there is a statistically significantly greater number of people in the NaSSA group compared to the placebo group who improved on the CGI‐I scale | |
| 222 per 1000 | 100 per 1000 (49 to 209) | |||||
| Moderate | ||||||
| 222 per 1000 | 100 per 1000 (49 to 209) | |||||
| Treatment tolerability, as measured by Dropouts due to adverse events (acute phase) | Study population | RR 0.87 (0.68 to 1.11) | 36 (1 study) | ⊕⊕⊝⊝ lowa,b | The proportion of dropouts due to adverse events was high in participants receiving the NaSSA (18%) relative to placebo (5%), but there was no evidence of a harm between the numbers of participants that dropped out due to adverse events |
|
| 53 per 1000 | 176 per 1000 (20 to 1000) | |||||
| Moderate | ||||||
| 53 per 1000 | 178 per 1000 (20 to 1000) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; CAPS: Clinically Administered PTSD Scale;RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded by one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded by one level due to serious imprecision (wide confidence intervals).
Summary of findings 5. Comparison 5: SNRIs versus placebo for posttraumatic stress disorder (PTSD).
| Comparison 5: SNRIs versus placebo for posttraumatic stress disorder (PTSD) | ||||||
|
Population: adults (aged 18‐85)
Settings: multi‐centre trials
Intervention: SNRIs
Comparison: placebo Follow‐up: for one study at week 24 or at the time of discontinuation if before week 24; the remaining study did not specify follow‐up | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With SNRIs | |||||
| Treatment efficacy ‐ treatment response, as measured by the Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | N/A | N/A | N/A | N/A | N/A | We found no studies that looked at the number of participants who responded to venlafaxine versus placebo. |
| Treatment tolerability, as measured by Dropouts due to adverse events (acute phase) | Study population | RR 0.98 (0.88 to 1.10) | 687 (2 studies) | ⊕⊝⊝⊝ very lowa,b | Dropout rates due to adverse events were low in the SNRI (4%) and placebo groups (3%) | |
| 26 per 1000 | 25 per 1000 (23 to 29) | |||||
| Moderate | ||||||
| 27 per 1000 | 26 per 1000 (24 to 30) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval;RR: risk ratio. N/A: Not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded by one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded by two levels due to considerable heterogeneity (I2 of 92%).
Summary of findings 6. Comparison 6: SSRIs versus placebo for posttraumatic stress disorder (PTSD).
| Comparison 6: SSRIs versus placebo for posttraumatic stress disorder (PTSD) | ||||||
|
Population: adults (aged 18‐85)
Settings: single and multi‐centre trials
Intervention: SSRIs
Comparison: placebo Follow‐up: for 1 study at week 10 and for another study 14 days after the last dose of study drug; the remaining studies did not specify follow‐up | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With SSRIs | |||||
| Treatment efficacy ‐ treatment response, as measured by the Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.66 (0.59 to 0.74) | 1078 (8 studies) | ⊕⊕⊕⊝ moderatea | There was evidence of a benefit for the number of participants with PTSD who responded to treatment for the SSRI group (58%) compared to the placebo group (35%). This is also indicated by the Risk Ratio of 0.66 which indicates that there is a statistically significantly greater number of people in the SSRI group compared to the placebo group who improved on the CGI‐I scale | |
| 348 per 1000 | 229 per 1000 (205 to 257) | |||||
| Moderate | ||||||
| 328 per 1000 | 216 per 1000 (194 to 243) | |||||
| Treatment tolerability, as measured by Dropouts due to adverse events (acute phase) | Study population | RR 0.98 (0.96 to 1.00) | 2399 (14 studies) | ⊕⊕⊕⊝ moderatea | A similar proportion withdrew due to treatment adverse events (9% versus 7%) | |
| 66 per 1000 | 65 per 1000 (64 to 66) | |||||
| Moderate | ||||||
| 50 per 1000 | 49 per 1000 (48 to 50) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded by one level due to serious risk of bias (concerns with randomisation procedures).
Summary of findings 7. Comparison 7: TCAs versus placebo for posttraumatic stress disorder (PTSD).
| Comparison 7: TCAs versus placebo for posttraumatic stress disorder (PTSD) | ||||||
|
Population: adults (aged 18‐85)
Settings: single and multi‐centre trials
Intervention: TCAs
Comparison: placebo Follow‐up: not specified | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With TCAs | |||||
| Treatment efficacy ‐ treatment response, as measured by the Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.60 (0.38 to 0.96) | 40 (1 study) | ⊕⊕⊝⊝ lowa,b | There was evidence of a benefit for the number of participants with PTSD who responded to treatment in the TCA group (50%) compared to the placebo group (17%). This is also indicated by the Risk Ratio of 0.60, which indicates that there is a statistically significantly greater number of people in the TCA group compared to the placebo group who improved on the CGI‐I scale | |
| 167 per 1000 | 100 per 1000 (63 to 160) | |||||
| Moderate | ||||||
| 167 per 1000 | 100 per 1000 (63 to 160) | |||||
| Treatment tolerability, as measured by Dropouts due to adverse events (acute phase) | Study population | RR 0.92 (0.81 to 1.05) | 141 (3 studies) | ⊕⊕⊕⊝ moderatea | We found no evidence of a difference in dropout rates between the TCA groups (23%) and placebo groups (18%) | |
| 182 per 1000 | 167 per 1000 (147 to 191) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale;RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded by one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded by one level due to serious imprecision (wide confidence intervals).
Background
Description of the condition
Although the phenomenon of posttraumatic stress disorder (PTSD) has long been recognised (for example as "shell shock" or "combat neurosis"), this disorder was only officially recognised in the psychiatric nomenclature in 1980 (APA 1980). Diagnostic criteria for PTSD provided by the 3rd edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM‐III) encouraged research on the epidemiology, psychobiology, and treatment of PTSD. The latest version of the Diagnostic and Statistical Manual (i.e. DSM‐5) has made a number of notable revisions. First, PTSD is now classified in a new category, Trauma‐ and Stressor‐Related Disorders, in which the onset of every disorder has been preceded by exposure to a traumatic or otherwise adverse environmental event (Friedman 2016). Second, a fourth cluster of symptoms has been included (i.e. negative cognitions and mood) (APA 2013).
DSM‐5 criteria for PTSD are as follows. The "A" criterion states that a person must be exposed to a catastrophic event involving actual or threatened death or injury, or a threat to the physical integrity of him/herself or others (for example, sexual violence) for the event to be regarded as a trauma. The "B" criterion (i.e. intrusive recollection) includes symptoms that are distinctive and readily identifiable, like panic, terror, dread, grief, or despair. These symptoms manifest during the daytime as intrusive images, traumatic nightmares, and flashbacks. The "C" criterion (i.e. avoidance criterion) consists of behavioural strategies used by people with PTSD to reduce trauma‐related events. Symptoms included in the "D" criterion reflect negative cognitions and moods that have developed after exposure to the traumatic event (i.e. blame, anger, guilt, or shame). Symptoms included in the "E" criterion are from alterations in arousal or reactivity such as hypervigilance or paranoia. The "F" or duration criterion stipulates that symptoms must persist for at least one month before PTSD may be diagnosed. Within the "G" criterion (i.e. functional significance criterion) the survivor must experience significant social, occupational, or other distress as a result of these symptoms, and in the "H" criterion (or exclusion criterion), these symptoms cannot be due to medication use, substance use, or other illnesses (APA 2013).
Further, epidemiological research using DSM criteria for PTSD has determined that the disorder is highly prevalent in a wide range of settings, particularly in those people who have been exposed to significant traumas (Breslau 1991; Kessler 1995; Koenen 2017). Estimates from the National Comorbidity Survey Replication indicate lifetime PTSD prevalence rates of 3.6% and 9.7%, among men and women in the USA, respectively (Kessler 2005). In the World Mental Health Survey (WMHS), prevalence rates are high in low‐, middle‐, and high‐income countries, with differences in gender prevalence again seen (Koenen 2017). Higher rates of PTSD have been reported in post‐conflict settings (De Jong 2001).
There is growing evidence that PTSD results in enormous personal and societal costs; this is based on chronicity of symptoms, high comorbidity of psychiatric and medical disorders, marked functional impairment, and estimations of economic costs (Brunello 2001; Koenen 2017; Solomon 1997). Furthermore, PTSD may be characterised by specific psychobiological dysfunctions mediated by neurobiological mechanisms, which may in turn be targeted by specific preventive and therapeutic pharmacological interventions (Amos 2014; Bernardy 2017; Charney 1993). There is certainly growing evidence in PTSD for specific dysregulations of neurotransmitter systems (including the serotonin, noradrenaline, and dopamine systems) and neuroendocrine systems (including the hypothalamus‐pituitary‐adrenal axis), as well as for structural and functional neuroanatomical abnormalities (Bremner 2004; Canive 1997; Charney 1993; Connor 1998; Sherin 2011; Yehuda 1995).
Prior psychological trauma plays a causal role in PTSD, and psychotherapy has been widely deployed in its management. Although psychodynamic psychotherapy has long been the mainstay of treatment, there have been few controlled studies of this modality (Brom 1989; Gersons 2000; Gilboa‐Schechtman 2010). The value of so‐called psychological debriefing in the immediate aftermath of trauma remains to be proven (Rose 1998; Rose 2002). Indeed, there is evidence that acute post‐trauma debriefing may worsen PTSD symptoms, prompting guidelines to advise against its use (Gist 2015). Nevertheless, there is a growing body of evidence demonstrating that cognitive‐behavioural and similar psychotherapies are indeed effective in the treatment of PTSD (Bisson 2007; Bradley 2005; Harvey 2003; NICE 2018; Watkins 2018).
Whereas in older models medications might be valuable primarily as an adjunct to psychotherapy techniques in post‐traumatic reactions (Sargent 1940), contemporary psychobiological theory speculates that comorbid substance use in PTSD may represent an attempt at 'self‐medication' and that prescribed medication may be able to play a primary role in preventing or reversing the dysfunctions of PTSD (Charney 1993; Charney 2004; Ressler 2018). PTSD frequently includes comorbid disorders such as major affective disorders, dysthymia, alcohol or substance abuse disorders, anxiety disorders, or personality disorders (Friedman 2016). Certain of these comorbid conditions are known to respond to medication (Kessler 1995; Kessler 2005). Indeed, the position that medication treatment may be useful in PTSD seems to have gained gradually increasing acceptance (Asnis 2004; Baldwin 2014; Connor 1998; Cyr 2000; Davidson 2000; Foa 1999; Marshall 1996; Marshall 1998a; Marshall 2000; Ursano 2004; Shalev 1996).
Description of the intervention
Early reports of the pharmacotherapy of PTSD focused on the tricyclic antidepressants (TCAs) and the irreversible monoamine oxidase inhibitors (MAOIs) (Basoglu 1992; Bleich 1986; Burstein 1984; Chen 1991; Davidson 1987; Davidson 1989; Falcon 1985; Frank 1988; Hogben 1981; Irwin 1989; Kosten 1991; Lerer 1987; Olivera 1990; Milanes 1984; Reist 1989; Rubin 1993; Shestatzky 1988; White 1983). More recent work has focused on the selective serotonin reuptake inhibitors (SSRIs) (Brady 2000; Davidson 2001b; Hertzberg 2000; Marshall 1998b; Martenyi 2002a; Smajkic 2001; Tucker 2001; Tucker 2003; Zohar 2002), selective serotonin and norepinephrine reuptake inhibitors (SNRIs) (Davidson 2006a; Davidson 2006b) and the serotonin antagonists and reuptake inhibitors (SARIs) (Davis 2004; Hertzberg 1996; Hertzberg 1998; Hidalgo 1999; Liebowitz 1989).
Several other antidepressants have also been studied (Baker 1995; Canive 1998; Connor 1999b; Davidson 1998; Davidson 2003; Davis 2000; Davis 2008; Hamner 1998; Katz 1994; Neal 1997). In addition, benzodiazepines (Braun 1990; Dunner 1985; Lowenstein 1988), beta‐blockers (Famularo 1988; Kolb 1984), buspirone (Duffy 1992; Duffy 1994; LaPorta 1992; Fichtner 1994; Simpson 1991; Wells 1991), clonidine (Harmon 1996; Kolb 1984; Kinzie 1989), guanfacine (Davis 2008; Horrigan 1996), cyprohepadine (Brophy 1991; Gupta 1998), d‐cycloserine (Heresco‐Levy 2002), inositol (Kaplan 1996), mood‐stabilisers (Fesler 1991; Fichtner 1990; Ford 1996; Forster 1994; Hertzberg 1999; Keck 1992; Looff 1995; Szymanski 1991), typical (Bleich 1986; Dillard 1993) and atypical neuroleptics (Butterfield 2001; Burton 1999; Hamner 1996; Izrayelit 1998; Leyba 1998), opioids (Glover 1993), and the alpha‐blocker prazosin (Raskind 2018) have also received attention.
How the intervention might work
SSRIs and SNRIs are considered first‐line agents for the treatment of PTSD (Baldwin 2014; Bandelow 2008; Ipser 2012). These antidepressants increase serotonin output by blocking the serotonin transporter (SERT) and are effective in reducing symptoms of anxiety and fear (Stahl 2013). There are currently six SSRIs globally available for the treatment of PTSD symptoms: namely, sertraline, paroxetine, fluoxetine, fluvoxamine, citalopram, and escitalopram. Only two (sertraline and paroxetine) are FDA‐approved (Ravindran 2009). Similarly, the serotonin 1A (5HT1A) partial agonist, buspirone, has potential anxiolytic actions. These could theoretically be due to this agent's 5HT1A partial agonist actions at both presynaptic and postsynaptic 5HT1A receptors, with actions at both sites resulting in enhanced serotonergic activity in projections to the amygdala, prefrontal cortex, striatum, and thalamus. SSRIs and SNRIs theoretically operate using similar mechanisms. Since the onset of anxiolytic action for buspirone is delayed, this has led to the belief that 5HT1A agonists exert their therapeutic effects by adaptive neuronal events and receptor events, rather than simply by the acute occupancy of 5HT1A receptors. In this way, the presumed mechanism of action of 5HT1A partial agonists is analogous to the SSRIs, which also demonstrate delayed onset of action, and are also presumed to act by adaptations in neurotransmitter receptors. These delayed medication effects can be compared to the relatively rapid effect of the benzodiazepine anxiolytics, which act relatively acutely by occupying benzodiazepine receptors (Stahl 2013). The serotonin antagonist and reuptake inhibitor, nefazodone, is an older antidepressant that is thought to work through post‐synaptic 5‐HT2A receptor antagonism, inhibition of presynaptic serotonin and norepinephrine (NE) reuptake, and through blocking α1 receptors. Nefazodone is now rarely used, due to concerns of hepatotoxicity (Ravindran 2009). Liebowitz 1989 reviewed the clinical efficacy of trazodone (50 – 250 mg/day) in treating 22 patients with a dual diagnosis of substance abuse and anxiety symptoms. A substantial number of people suffered from symptoms of posttraumatic stress disorder (PTSD). Improvement was noted in all participants within the first month of treatment, and most reported symptomatic improvement after each dose of trazodone, resulting in an as‐needed pattern of usage.
Benzodiazepines, as a class, work on the central nervous system (CNS) through their effects on the GABAA receptors. Activation at the special benzodiazepine receptor site on the GABAA receptor promotes enhanced activity of the inhibitory neurotransmitter γ‐aminobutyric acid (GABA), thus resulting in various effects including: anxiolysis, sedation, muscle relaxation, cognitive effects, and anticonvulsant actions. These functions, and particularly the first two, would seem to have benefits for PTSD (Ravindran 2009).
Anti‐adrenergic agents or beta‐blockers or both target putative noradrenergic alterations seen in PTSD. Clonidine, for example, is commonly used as an antihypertensive agent, and is a centrally‐acting α2 adrenergic agonist that works to decrease sympathetic tone. As such, it was theorised to have potential effects on the hyperarousal symptoms seen in PTSD. Guanfacine, another α2 adrenergic agonist with a similar mechanism of action, has also been investigated, but no benefit has been reported (Davis 2008; Neylan 2006). The use of β‐adrenergic antagonists has also been investigated in PTSD, but primarily for a role in the secondary prevention of this disorder. Cahill 1994 demonstrated that a single dose of propranolol administered to healthy humans impaired subsequent recall of an emotionally‐arousing story but not for an emotionally‐neutral one, thus lending support for the theory that memory for emotional experiences involved the β‐ adrenergic system. Based on this, it was theorised that administration of a β‐adrenergic antagonist in the peritraumatic period might have a beneficial effect in blocking consolidation of the traumatic memory and thus prevent development of PTSD (Ravindran 2009).
Anticonvulsant medications, with their putative anti‐kindling effects, have been investigated for PTSD, although most of the evidence for use of these agents (e.g. carbamazepine and divalproex) is not well researched (Ravindran 2009; Ressler 2018).
TCAs are non‐specific in their actions on specific neurotransmitters, with their primary mechanism of action involving varying degrees of serotonin and NE reuptake inhibition. MAOIs, on the other hand, work by irreversibly inhibiting the enzyme monoamine oxidase, normally involved in metabolism of serotonin and NE. Both medication classes are generally considered second‐ or third‐line treatments due to their adverse event profile and need for dietary restriction (i.e. MAOIs) (Ravindran 2009).
Only a small number of controlled trials have investigated the adjunctive use of antipsychotic agents (which include olanzapine, risperidone, quetiapine, ziprasidone, and aripiprazole) in PTSD, and even fewer have explored their use as monotherapy (Ravindran 2009). Other agents like D‐cycloserine (DCS), a partial agonist at the NMDA receptor, have also been investigated as a potential treatment for PTSD. Arguing that the presence of flashbacks and intrusive memories in PTSD may be a function of extinction failure, and that learning and memory are both glutamate‐dependent processes, Heresco‐Levy 2002 and colleagues argued that enhancing glutamate transmission could facilitate the learning of new memories to replace the traumatic ones (as cited in Ravindran 2009). The addition of cyproheptadine, taken orally at night, has also been shown to control and decrease the intensity and frequency of nightmares (Brophy 1991; Gupta 1998). Similarly, 18 chronic posttraumatic stress disorder combat veterans who received the opioid nalmefene showed a favourable response, with a marked decrease of emotional numbing and other symptoms of PTSD, including startle response, nightmares, flashbacks, intrusive thoughts, rage and vulnerability as the dosage increased (Glover 1993). There was also evidence reported by studies assessing the mood‐stabilisers carbamazepine (Lipper 1986), valproate (Fesler 1991), lithium (Forster 1994), lamotrigine (Hertzberg 1999), and prazosin (Raskind 2018). There was, however, no significant difference found in the improvement score for inositol compared to placebo (Kaplan 1996).
Why it is important to do this review
A systematic review of studies of pharmacotherapy for PTSD is useful in tackling several questions for the field. First, is pharmacotherapy in fact an effective form of treatment in PTSD? Given the preponderance of psychological models and evidence for the efficacy of certain forms of psychotherapy in PTSD (Bisson 2013), the role of pharmacotherapy remains debatable for many. In a recently‐published guideline for the treatment of PTSD, the National Institute for Health and Care Excellence (NICE) recommends that preference be given to trauma‐focused psychological therapy over pharmacotherapy as a routine first‐line treatment for this disorder (NICE 2018).
Second, are certain medication classes more effective in the treatment of symptoms and/or more acceptable to the patient in terms of adverse events than others? The use of novel agents (such as prazosin) for PTSD in recent years raises the question of how these compare with older agents. Early recommendations, such as the expert consensus guideline series for the treatment of posttraumatic stress disorder (Foa 1999), suggested that the SSRIs, the serotonin modulator nefazodone, and the SNRI venlafaxine are first‐line medications for the treatment of PTSD, with benzodiazepines and mood‐stabilisers having a role in people with certain kinds of symptoms. More recent recommendations have highlighted paroxetine, mirtazapine, amitriptyline and phenelzine (NICE 2018). Support for such recommendations requires ongoing assessment of the literature on RCTs.
Third, can a systematic review of RCTs provide information about the most important factors affecting pharmacotherapy response? Clinical factors, such as the kind of pre‐existing trauma (e.g. combat‐related or not) and the presence of comorbid depression have all been suggested to play a role (Davidson 1993; Davidson 2000; Marshall 1998b; Van der Kolk 1994). It is possible that the database of RCTs in PTSD may include information about these variables.
Several reviews of the pharmacotherapy of PTSD have indeed been published in recent years (Albucher 2002; Asnis 2004; Hoskins 2015; Ipser 2012). These reviews have been useful in summarising the existing research, pointing to methodological flaws, and outlining areas for future research. Nevertheless, not all reviews have employed a systematic search strategy, and it has been suggested that even MEDLINE searches may miss over half of all RCTs in specialised health care journals (Hopewell 2002). Furthermore, not all studies have provided estimates of the effects of medication (Davidson 1997a; Penava 1996). Finally, not all reviews in this area have adhered to Cochrane Collaboration (Mulrow 1997) or similar (Moher 1999) guidelines for systematic identification of trials, investigation of sources of heterogeneity, measurement of methodological quality, and estimation of the effects of intervention.
The authors therefore updated a systematic review of RCTs of the pharmacotherapy of PTSD in adults, previously published in 2000 and 2006 (Stein 2000; Stein 2006), following Cochrane guidelines and software (Higgins 2011; RevMan 2020).
Objectives
To assess the effects of medication for reducing PTSD symptoms in adults with PTSD.
Methods
Criteria for considering studies for this review
Types of studies
We considered randomised controlled trials (placebo‐controlled and comparative trials) for inclusion. We also considered unpublished reports, abstracts, and brief and preliminary reports. We did not use differences between trials (for example, sample size, trial duration or language) to exclude studies. We also included cluster‐randomised controlled trials, cross‐over trials and multiple treatment trials in the analyses.
Types of participants
Participant characteristics
We included all studies of adult participants (aged 18 to 85 years) diagnosed with PTSD (as determined by the study author), irrespective of diagnostic criteria and measure, duration and severity of PTSD symptoms, and gender. These descriptors were, however, tabulated in order to address the question of their possible impact on the effects of medication.
We included adults on concomitant medications in the review, but we excluded adults on concomitant psychotherapy.
Comorbidities
We placed no restrictions on the presence of comorbid disorders secondary to PTSD.
Setting
We placed no restrictions by setting.
Subsets of participants
We excluded studies that reported a subset of participants that met the review inclusion criteria, to preserve randomisation.
Types of interventions
The review focuses only on medication treatments, in which the comparator was a placebo (active or non‐active) or other medication (i.e. control group). A parallel review of the psychotherapy of PTSD has been completed by a Cochrane team (Bisson 2007; Bisson 2013). More recently, a Cochrane review of RCTs of medication prophylaxis for PTSD has been published (Amos 2014).
This review classifies medications based on their putative mechanisms of action (taken from CCMD antidepressant classification map) (Davies 2015), and they do not necessarily map onto the drug classification schemes used in other reviews.
Experimental interventions
We grouped specific pharmacological interventions by medication class, listed below:
Alpha‐blockers (e.g. prazosin)
Anticonvulsants (e.g. tiagabine, divalproex, lamotrigine, and topiramate)
Antihistamines (e.g. hydroxyzine)
Antipsychotics (e.g. olanzapine, risperidone and quetiapine)
Benzodiazepines (e.g. alprazolam)
Dopamine beta‐hydroxylase inhibitors (e.g. nepicastat)
Hypnotics (e.g. eszopiclone)
Mono‐amine oxidase inhibitors (MAOIs, e.g. phenelzine)
NK‐1 receptor antagonists (e.g. orvepitant)
NMDA receptor antagonists (e.g. ketamine)
Norepinephrine and dopamine reuptake inhibitors (NDRIs, bupropion SR)
Noradrenaline reuptake inhibitors (NARs, e.g. reboxetine)
Noradrenergic and specific serotonergic antidepressants (NaSSAs, e.g. mirtazapine)
Other medications (e.g. ganaxolone, GR205171, GSK561679)
Reversible inhibitor of monoamine oxidase A (RIMA, e.g. brofaromine)
Second messenger system precursors (e.g. inositol)
Selective serotonin reuptake inhibitors (SSRIs, e.g. paroxetine, fluvoxamine, sertraline, fluoxetine, and citalopram)
Serotonin and norepinephrine reuptake inhibitor (SNRI, e.g. venlafaxine)
Serotonin antagonist and reuptake inhibitors (SARI, e.g. nefazodone)
Tricyclic antidepressants (TCAs, e.g. amitriptyline, desipramine and imipramine)
Comparator interventions
Placebo (active or non‐active)
Medication (control)
We placed no restrictions on timing, dosage, duration, or co‐interventions.
Types of outcome measures
Primary outcomes
-
Treatment efficacy
Treatment response (responders versus non‐responders) was determined from the Clinical Global Impressions Scale ‐ Improvement Item (CGI‐I). The CGI‐I ranges from 1 (normal, not at all ill) to 7 (among the most extremely ill patients). In this review, responders were defined on the CGI‐I as those with a score of 1 = "very much" or 2 = "much" improved (Guy 1976). Given that the CGI‐I is a widely‐used global outcome measure in RCTs of PTSD, this instrument served as a robust measure of the clinical value of treatment in PTSD (Davidson 1997b).
-
Treatment tolerability ‐ dropouts due to treatment‐emergent adverse events
The total proportion of participants who withdrew from the RCTs due to treatment‐emergent adverse events was included in the analysis as a surrogate measure of medication acceptability, in the absence of other more direct indicators of acceptability.
Secondary outcomes
-
Reduction of PTSD symptoms
Reduction in PTSD symptoms was determined from the total score on the Clinician Administered PTSD Scale (CAPS) (Blake 1990), a symptom‐severity measure that is increasingly used in RCTs of PTSD. The CAPS is designed to make a categorical PTSD diagnosis which corresponds to the DSM‐IV criteria. A "1, 2" rule is used to determine a diagnosis, whereby a frequency score of 1 (scale 0 = "none of the time" to 4 = "most or all of the time") and an intensity score of 2 (scale 0 = "none" to 4 = "extreme") is required in order to meet specific symptom criteria (Weathers 1999, as cited in the International Society for Traumatic Stress Studies).
PTSD symptom reduction was assessed for those trials which used other continuous measures of symptom severity besides the CAPS, as well as from summary statistics from self‐rated scales such as the Impact of Events Scale (IES) (Horowitz 1979), and the Davidson Trauma Scale (DTS) (Davidson 1997c). The IES evaluates the distress that is caused by traumatic events and is centred around two subscales (i.e. intrusion and avoidance). The IES is a 22‐item self‐report scale in which respondents choose options from 1 to 5. The DTS is a 17‐item self‐report measure that assesses the 17 DSM‐IV symptoms of PTSD. Items are rated on a 5‐point frequency (0 = "not at all" to 4 = "every day") and severity scale (0 = "not at all distressing" to 4 = "extremely distressing"). The scores range from 0 to 136.
Self‐rated scales were frequently the only outcome measures used in older trials and may continue to have a role in clinical practice. The efficacy of medication in alleviating symptoms within the three symptom clusters characteristic of PTSD (re‐experiencing/intrusion, avoidance/numbing, and hyperarousal) was determined using the CAPS‐B, CAPS‐C, and CAPS‐D subscales of the CAPS, as well as the relevant subscales of the self‐rated outcome measures.
-
Reduction in depressive symptoms
The reduction of comorbid symptoms was measured by depression scales, such as the Beck Depression Inventory (BDI) (Beck 1961), the Hamilton Depression scale (HAM‐D) (Hamilton 1960), and the Montgomery‐Asberg Depression Rating Scale (MADRS) (Montgomery 1979).
The Beck Depression Inventory (BDI) is a 21‐question multiple‐choice self‐report, one of the most widely used psychometric tests for measuring the severity of depression. A score of 0 to 9 indicates minimal depression, 10 to 18 mild depression, 19 to 29 moderate depression, and 30 to 63 severe depression. The Hamilton Depression scale (HAM‐D) is a multiple‐item questionnaire with 17 to 29 items (depending on the version). Patients are rated on a 3‐ or 5‐point scale. A score of 0 to 7 is normal and a score of 20 or higher moderate, severe, or very severe. The MADRS is a 10‐item diagnostic questionnaire which psychiatrists use to measure the severity of depressive episodes in patients with mood disorders. A higher MADRS score indicates more severe depression, and each item yields a score of 0 to 6. The overall scores range from 0 to 60. Usual cut‐off points are 0 to 6 ‐ normal/symptom absent; 7 to 19 ‐ mild depression; 20 to 34 ‐ moderate depression; and more than 34 ‐ severe depression.
-
Reduction in anxiety symptoms
The reduction of comorbid symptoms was measured by anxiety scales, such as the Covi Anxiety Scale (CAS) (Covi 1984) and the Hamilton Anxiety scale (HAM‐A) (Hamilton 1959).
The Covi Anxiety Scale is a three‐item scale along with three separate dimensions: verbal report, behaviour, and somatic symptoms of anxiety, and each dimension is rated on a 1‐to‐5 spectrum (1 = “not at all” to 5 = “very much”). The HAM‐A consists of 14 items and measures both psychic anxiety (mental agitation and psychological distress) and somatic anxiety (physical complaints related to anxiety). Each item is scored on a scale of 0 (not present) to 4 (severe), with a total score range of 0 to 56, where less than 17 indicates mild severity, 18 to 24 mild‐to‐moderate severity, and 25 to 30 moderate‐to‐severe.
-
Functional disability
Functional disability as measured by the Sheehan Disability Scale (SDS), which includes subscales to assess work, social and family‐related impairment (Sheehan 1996), was also included, when provided, to address the question of medication effectiveness. The Sheehan Disability Scale is a composite of three self‐rated items designed to measure the extent to which three major sectors in the patient’s life are impaired by panic, anxiety, phobic, or depressive symptoms. The patient rates the extent to which his or her 1) work, 2) social life or leisure activities, and 3) home life or family responsibilities are impaired by his or her symptoms on a 10‐point visual analogue scale. The numerical ratings of 0‐to‐10 can be translated into a percentage if desired. The three items may be summed into a single dimensional measure of global functional impairment that ranges from 0 (unimpaired) to 30 (highly impaired).
-
Treatment tolerability ‐ dropouts due to any cause
Dropout rates due to any cause were also compared in order to provide some indication of treatment effectiveness.
Main comparisons
We assessed the following outcomes and grouped them by specific pharmacological interventions according to medication class:
Comparison 1: Alpha‐blockers versus placebo.
Comparison 2: Anticonvulsants versus placebo.
Comparison 3: Antipsychotics versus placebo.
Comparison 4: Benzodiazepines versus placebo.
Comparison 5: Dopamine beta‐hydroxylase inhibitors versus placebo.
Comparison 6: Ganaxolone versus placebo.
Comparison 7: GR205171 versus placebo.
Comparison 8: GSK561679 versus placebo.
Comparison 9: Hypnotics versus placebo
Comparison 10: MAOIs versus placebo.
Comparison 11: NaSSAs versus placebo.
Comparison 12: NK‐1 receptor antagonists versus placebo.
Comparison 13: RIMAs versus placebo.
Comparison 14: SARIs versus placebo.
Comparison 15: SNRIs versus placebo.
Comparison 16: SSRIs versus placebo.
Comparison 17: TCAs versus placebo.
Comparison 18: Total effect of medication versus placebo.
Comparison 19: Head‐to‐head comparisons.
Comparison 20: Subgroup analyses ‐ Methodological criteria.
Comparison 21: Subgroup analyses ‐ Clinical criteria.
Comparison 22: Sensitivity analyses.
Comparison 23: Publication bias.
Search methods for identification of studies
Cochrane Specialised Register (CCMDCTR) The Cochrane Common Mental Disorders Group maintains a comprehensive, specialised register of randomised controlled trials, the CCMDCTR (to 13 November 2020). The register contains over 39,000 reference records (reports of RCTs) for anxiety disorders, depression, bipolar disorder, eating disorders, self‐harm and other mental disorders within the scope of this Group. The CCMDCTR is a partially studies‐based register with more than 50% of reference records tagged to about 12,500 individually PICO‐coded study records. Reports of trials for inclusion in the register were collated from (weekly) generic searches of MEDLINE (1950‐), Embase (1974‐) and PsycINFO (1967‐), quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. Reports of trials were also sourced from international trial registries, drug companies, the handsearching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses. Details of CCMD’s core search strategies (used to identify RCTs) can be found on the Group’s website with an example of the core MEDLINE search displayed in Appendix 1.
Electronic searches
An information specialist with the Cochrane Common Mental Disorders Group searched the following databases using keywords, subject headings and search syntax appropriate to each resource. The date of the latest, full search is 13 November 2020.
Cochrane Central Register of Controlled Trials (CENTRAL; 2020, issue 11) in the Cochrane Library (searched 13 November 2020);
Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR) (all years to June 2016);
Embase Ovid (1974 to 2020 Week 46);
MEDLINE ALL, Ovid (1946 to November 13, 2020);
PsycINFO Ovid (all years to November Week 46 2020);
PTSDPubs Proquest (previously PILOTS: Published International Literature On Traumatic Stress) (all years to 16 November 2020);
ClinicalTrials.gov (https://clinicaltrials.gov/);
WHO International Clinical Trials Registry Platform (ICTRP) (http://apps.who.int/trialsearch/).
The searches have been through a number of iterations over the years, initially concentrating on terms for population and intervention (+ RCT filter), across the main bibliographic databases either directly or via the Cochrane Common Mental Disorders Controlled Trials Register (Appendix 1).
As the number of different pharmacological interventions used to treat PTSD expanded beyond the traditional psychotropic drugs, an information specialist ran searches for population only, RCTs (CCMDCTR, all available years; key bibliographic databases, 2014 onwards) (Appendix 2). The search results were pre‐sifted (in duplicate), to weed out the non‐RCTs, prior to passing on the records on to the author team. The population only search, was designed to capture studies for a number of reviews within the CCMD group, for the treatment or prevention of PTSD.
We (the author team) undertook our own searches, initially using a broad strategy to find not only RCTs, but also open‐label trials, as well as journal and chapter reviews of the pharmacotherapy of PTSD in adults (Appendix 3).
Searching other resources
Reference Lists
We sought additional RCTs in reference lists of the retrieved articles.
Personal communication
We also obtained published and unpublished trials from key researchers, who were identified by the frequency with which they were cited in the bibliographies of RCTs and open‐label studies.
Data collection and analysis
We used Review Manager 5 (RevMan 5) to perform all analyses reported in this review (RevMan 2020).
Selection of studies
RCTs identified from the search were independently assessed for inclusion by three review authors (TW, JI and NP), based on information included in the abstract or the main body of the trial report, or both. We collated RCTs which we regarded as satisfying the inclusion criteria specified in the Data extraction and management and Criteria for considering studies for this review. Studies for which additional information was required in order to determine their suitability for inclusion in the review have been listed in the Characteristics of studies awaiting classification table, pending the availability of this information. We resolved any disagreements in assessment and collation by discussion with a fourth review author (DS).
Data extraction and management
We designed spreadsheet forms to record descriptive information, summary statistics of the outcome measures, the quality scale ratings, and associated commentary.
We collated the following information from each trial (additional information can be found in the Characteristics of included studies tables, but not all extracted data are reported in this review):
Description of the trials, including the primary researcher, the year of publication, and the source of funding.
Characteristics of the interventions, including the number of participants randomised to the treatment and control groups, the number of total dropouts per group as well as the number that dropped out due to adverse effects, the dose of medication and the period over which it was administered, and the name and class of the medication (e.g. SSRIs, TCAs, MAOIs and 'other medication).
Characteristics of trial methodology, including the diagnostic (e.g. DSM‐IV (APA 1994)) and exclusion criteria used, the screening instrument used (e.g. the Structured Clinical Interview for DSM‐IV (SCID) (Spitzer 1996)) for both the primary and comorbid diagnoses, the presence of comorbid major depressive disorders (MDDs), the use of a placebo run‐in or of a minimal severity criterion, the number of centres involved, and the trial's methodological quality (see below).
Characteristics of participants, including gender distribution and mean and range of ages, mean length of time with PTSD symptoms, whether they had been treated with the medication in the past (treatment naïvety), the number of participants in the sample with MDD, the number who experienced combat trauma, and the baseline severity of PTSD, as assessed by the trial's primary outcome measure or another commonly‐used scale.
Outcome measures employed (primary and secondary), and summary continuous (means and standard deviations (SD)) and dichotomous (number of responders) data. Additional information included whether data reflected the intention‐to‐treat (ITT) with last observation carried forward (LOCF) or mixed methods (MM) sample, or whether a completer/observed cases (OC) sample was reported.
Where information was missing, the review authors contacted investigators by email to obtain this information.
Assessment of risk of bias in included studies
We assessed the risks of bias of each included study using the Cochrane risk of bias tool (Higgins 2011), based on consideration of the following six domains:
Random sequence generation: did investigators use a random‐number table or a computerised random‐number generator?
Allocation concealment: was the medication sequentially numbered, sealed, and placed in opaque envelopes?
Blinding of participants, personnel, and outcome assessors for each main outcome or class of outcomes: was knowledge of the allocated treatment or assessment adequately blinded during the study?
Incomplete outcome data for each main outcome or class of outcomes: were missing or excluded outcome data adequately addressed?
Selective outcome reporting: were the reports of the study free of suggestion of selective outcome reporting? Such a judgement could only be made based on the availability of the protocol.
Other sources of bias: was the study apparently free of other problems that could put it at a high risk of bias?
Three independent review authors (TW, JI and NP) assessed and extracted the risks of bias for the included studies. We discussed any disagreements with a fourth review author (DS). Where necessary, we contacted the authors of the studies for further information. The review authors made a judgement on the risk of bias for each domain within and across studies, based on the following three categories: ’low’ risk of bias, ’unclear’ risk of bias, and ’high’ risk of bias. All risk of bias data are graphically presented and described in the text.
Measures of treatment effect
Categorical data
Relative risk (RR) of failure to respond to treatment was used as the summary statistic for the dichotomous outcome of interest (CGI‐I or related measure). We used the RR instead of the odds ratio, as odd ratios tend to underestimate the size of the treatment effect when the occurrence of the adverse outcome of interest is common (as was the case in this review, with an anticipated non‐response greater than 20%) (Deeks 2003; Deeks 2011), and because of the greater ease with which this statistic can be interpreted.
Continuous data
We calculated weighted mean differences (WMDs) for continuous summary data obtained from studies that used the CAPS. Alternatively, in cases in which a range of scales were used, such as in the assessment of symptom severity on the self‐rated IES and DTS scales, we determined the standardised mean difference (SMD). This method of analysis standardises the differences between the means of the treatment and control groups in terms of the variability observed in the trial.
In the case of data from trials using multiple fixed doses of medication, we avoided the bias introduced through comparing the summary statistics for multiple groups against the same placebo control by pooling the means and standard deviations across all the treatment arms as a function of the number of participants in each arm. In addition, when including summary statistics from the self‐rated scales, we preferred data from the DTS over the IES in trials which used both scales, given the inclusion in the former of a subscale assessing the hyperarousal symptom cluster (Weiss 1997), and concerns about the psychometric properties of the IES subscales (Creamer 2003).
Unit of analysis issues
Cluster‐randomised trials
In cluster‐randomised trials, groups of individuals, rather than individuals themselves, are randomised to different interventions. Analysing treatment response in cluster‐randomised trials without taking these groupings into account could be problematic, as participants within any one cluster often tend to respond in a similar manner, and thus analyses cannot assume that participants’ data are independent of the rest of the cluster. Cluster‐randomised trials also face additional risk‐of‐bias issues including recruitment bias, baseline imbalance, loss of clusters, and non‐comparability with trials randomising individuals (Higgins 2011). No cluster‐randomised trials were eligible for inclusion in this review.
Cross‐over trials
Cross‐over trials were only included in the calculation of summary statistics when it was (a) possible to extract medication and placebo/comparator data from the first treatment period alone, or (b) when the inclusion of data from both treatment periods was justified through a wash‐out period of sufficient duration as to minimise the risk of carry‐over effects (a minimum of two weeks or longer in the case of trials assessing the efficacy of agents with extended half‐lives, such as the SSRI, fluoxetine (Gury 1999)). In the latter case, data from both periods were only included when it was possible to determine the correlation between participants' responses to the interventions in the different phases (Elbourne 2002).
Multiple treatment groups
A few trials included in this review compared more than two intervention groups or multiple doses of the same medication against placebo. Including data from the same placebo group for these studies repeatedly in the same comparison would result in a unit‐of‐analysis error (Higgins 2011). To prevent these errors for trials comparing multiple dosages of the same agent to placebo, we averaged the mean and standard deviation of the outcome of interest across dosage groups. We included outcome data from multiple treatment arms in the same comparison if the agents tested were from different medication classes. We turned off the subtotals of the outcome if the placebo group appeared twice in the analysis to accommodate the second medication. In the case of trials testing multiple agents from the same classes, and in calculating the total effect across all medication classes, we restricted data from multi‐arm RCTs to the agent that was least represented in the database.
Dealing with missing data
All analyses of dichotomous data were intention‐to‐treat (ITT) and data from trials providing information on the original group size (prior to dropouts) were included in the analyses of treatment efficacy. We preferred within studies the inclusion of summary statistics for continuous outcome measures derived from mixed‐effects models, followed by last observation carried forward (LOCF) and observed cases (OC) summary statistics, in that order. This is in line with evidence that mixed‐effects methods are more robust to bias than LOCF analyses (Verbeke 2000).
Assessment of heterogeneity
We assessed heterogeneity of treatment response, i.e. whether the differences between the results of trials were greater than would be expected by chance alone, visually from the forest plot of the RR. It was also determined by means of the Chi2 test of heterogeneity, with a significance level of less than 0.10 interpreted as evidence of heterogeneity, given the low power of the Chi2 statistic when the number of trials is small (Deeks 2003).
In addition, we used the I2 heterogeneity statistic reported by RevMan to test the robustness of the Chi2 statistic to differences in the number of trials included in the groups being compared within each subgroup analysis (Higgins 2003). Differences in treatment response on the CGI‐I were determined by whether the confidence intervals for the effect sizes of the subgroups overlapped. We chose this method in preference to the stratified test, due to inaccuracies in the calculation in RevMan of the Chi2 statistic for dichotomous measures (Deeks 2003; Deeks 2011).
Thresholds for the interpretation of I2 can be misleading, since the importance of inconsistency depends on several factors. The review follows a rough guide for interpretation:
0% to 40%: might not be important.
30% to 60%: may represent moderate heterogeneity.
50% to 90%: may represent substantial heterogeneity.
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
Funnel plots provide a graphic illustration of the effect estimates of an intervention from individual studies against some measure of the precision of that estimate. We determined publication bias by visual inspection of a funnel plot of treatment response, with the consideration of confounding selection bias, poor methodological quality, true heterogeneity, artefact, and chance. Given that this calculation is dependent on having 10 trials per outcome, we could only calculate this for the SSRIs.
Data synthesis
We obtained summary statistics for categorical and continuous measures from a random‐effects model; this includes both within‐study sampling error and between‐studies variation in determining the precision of the confidence interval (CI) around the overall effect size, whereas the fixed‐effect model takes only within‐study variation into account. The summary statistics were expressed as an average effect size for each subgroup, as well as by means of 95% CIs.
Subgroup analysis and investigation of heterogeneity
We conducted subgroup analyses (Thomson 1994) in order to assess the degree to which methodological differences between trials might have systematically influenced differences observed in the primary treatment outcomes.
We grouped the trials according to the following methodological sources of heterogeneity:
The involvement of participants from a single centre or multiple centres. Single‐centre trials are more likely to be associated with lower sample size but with less variability in clinician ratings.
Whether or not trials were industry‐funded. In general, published trials which are sponsored by pharmaceutical companies appear more likely to report positive findings than trials which are not supported by for‐profit companies (Als‐Nielsen 2003; Baker 2003).
In addition, we used the following criteria to assess the extent of clinical sources of heterogeneity:
Whether or not the sample included combat veterans (this subgroup has been regarded as more resistant to treatment and is arguably more likely to have more chronic and severe symptoms, to have comorbid depression, and to be male). For the purposes of this review, those trials for which 10% or fewer of the sample consisted of war veterans were classified as non‐combat veteran RCTs.
Whether or not the sample included participants diagnosed with major depression. Such an analysis might assist in determining the extent to which the efficacy of a medication agent in treating PTSD is independent of its ability to reduce symptoms of depression, an important consideration given the classification of many of these medications as antidepressants.
Sensitivity analysis
We conducted sensitivity analyses, which determine the robustness of the review authors' conclusion and methodological assumptions made in conducting the meta‐analysis. Sensitivity analyses were conducted to determine whether treatment response on the CGI‐I differed as a result of:
Treatment response versus non‐response as the unit of comparison in determining medication efficacy. This comparison is regarded as necessary, given concerns that the former may result in less consistent summary statistics than the latter (Deeks 2002).
The exclusion of participants who were lost to follow‐up (LTF). This was determined through a 'worst case/best case' scenario (Deeks 2003). In the worst case, all the missing data for the treatment group were recorded as non‐responders, whereas in the best case, all missing data in the control group were treated as non‐responders (In the case of the one SSRI (Marshall 2007) and MAOI trial (Baker 1995) which only reported total LTF, the ratio of participants who dropped out in the medication and placebo groups was determined from the average ratio between these groups for those RCTs in the respective classes which did provide this information). Should the conclusions about treatment efficacy not differ between these two comparisons, we can assume that missing data in trial reports do not have a significant influence on outcome.
Summary of findings and assessment of the certainty of the evidence
The review authors compiled summary of findings tables to present the evidence for the primary outcomes of the review (i.e. number of responders and dropouts due to treatment‐emergent side effects). In addition, the following comparisons were prioritised and are presented in the summary of findings tables:
Comparison 1: Alpha‐blockers versus placebo.
Comparison 2: Antipsychotics versus placebo.
Comparison 3: MAOIs versus placebo.
Comparison 4: NaSSAs versus placebo.
Comparison 5: SNRIs versus placebo.
Comparison 6: SSRIs versus placebo.
Comparison 7: TCAs versus placebo.
We used the following six elements (Higgins 2011) to report this:
A list of all important outcomes, both desirable and undesirable;
A measure of the typical burden of these outcomes (e.g. illustrative risk, or illustrative mean, on control intervention);
Absolute and relative magnitude of effect (if both are appropriate);
Numbers of participants and studies addressing these outcomes
A grade of the overall quality of the body of evidence for each outcome
Space for comments.
The downgrading of the evidence rating for outcomes was based on five factors. Reasons for downgrading the evidence were classified as ’serious’ (downgrading the quality rating by one level) or ’very serious’ (downgrading the quality grade by two levels).
Limitations in the design and implementation of the trial.
Indirectness of evidence.
Unexplained heterogeneity or inconsistency of results.
Imprecision of results.
High probability of publication bias.
The review authors classified the quality of evidence for each outcome according to the following categories:
High quality: further research is very unlikely to change our confidence in the estimate of effect;
Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate;
Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate;
Very low quality: we are very uncertain about the estimate.
Results
Description of studies
Results of the search
We found a total of 23,309 studies through the search process (CCMDCTR 3660; MEDLINE 2489; PsycINFO 6222; ClinicalTrials.org 2371; Embase 1984; PILOTS 792; Proquest PTSDpubs 194; PubMed 5597). After the removal of 3052 duplicates, we scanned 20,257 titles and abstracts (if provided) for eligibility, and of these, we ruled out 20,126 studies. One hundred and thirty one studies initially seemed relevant, but after independent review of the full‐text studies, 65 failed to meet our inclusion criteria (see Characteristics of excluded studies), leaving 66 RCTs eligible for inclusion in the review (see Characteristics of included studies and Figure 1). Of the 66 trials, the review included 54 RCTs in the meta‐analysis. Eighteen studies are awaiting classification, and one study is ongoing (see Characteristics of studies awaiting classification and Characteristics of ongoing studies).
1.
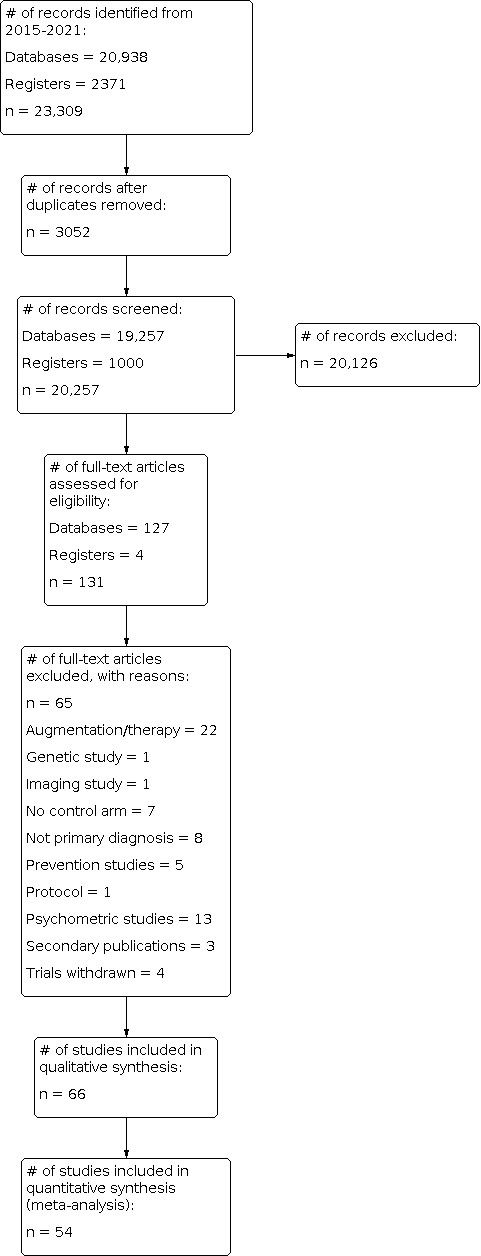
Study flow diagram.
Included studies
The review includes 66 RCTs of PTSD (7442 participants, age range: 18 to 82 years), four of which contained a maintenance component (Davidson 2001a, Marshall 2007, Martenyi 2002a, Van der Kolk 2007). Of the 66 trials, 58 were published, and all of these publications were in English. Pharmaceutical companies contributed funding for 35 of these trials. Twenty‐one studies were single‐centre trials, and 38 studies took place in multiple‐centres. There was insufficient information to determine the setting for the remaining seven trials. A placebo comparison group was used in all but six of the trials (McRae 2004 and Saygin 2002 compared nefazodone with the SSRI sertraline, while Smajkic 2001 compared the efficacy of the SSRIs sertraline, paroxetine and the SNRI venlafaxine). Chung 2004 assessed the efficacy and tolerability of mirtazapine against that of sertraline, Feder 2014 compared ketamine to midazolam (fixed 0.045 mg), and Spivak 2006 compared reboxetine (fixed 8 mg) to fluvoxamine (fixed 150 mg)). Of the remaining 60 RCTs, 26 of the trials included a SSRI treatment arm (one citalopram, dose range: 20 to 50 mg; eight fluoxetine, dose range: 10 to 80 mg; six paroxetine, dose range: 10 to 60 mg; 11 sertraline, dose range: 15 to 200 mg), two trials an alpha‐blocker intervention (prazosin, dose range: 1 to 15 mg; one of the trials included the antihistamine hydroxyzine, dose range: 10 to 100 mg), seven trials an anticonvulsant intervention (e.g. two tiagabine, dose range: 2 to 16 mg; two divalproex, dose range: 500 to 3000 mg; one lamotrigine, dose range: 25 to 500 mg; two topiramate, dose range: 25 to 400 mg), five trials an antipsychotic intervention (e.g. two olanzapine, dose range: 5 to 20 mg; two risperidone, dose range: 0.5 to 6 mg; one quetiapine, dose range: 25 to 800 mg), and three trials an experimental intervention (one ganaxolone, dose range: 200 to 600 mg; one GR205171, fixed 5 mg; one GSK561679, fixed 350 mg). The review also includes two trials with a MAOI intervention (phenelzine, dose range: 15 to 75 mg), one trial with a NaSSA intervention (mirtazapine, dose range: 15 to 50 mg); two trials with a RIMA intervention (brofaromine, dose range: 50 to 150 mg), one trial with a SNRI intervention (venlafaxine, dose range: 75 to 300 mg), and three trials with a TCA intervention (one amitriptyline, dose range: 50 to 200 mg; one desipramine, dose range: 50 to 200 mg; one imipramine, dose range: 50 to 300 mg). Single trials included a benzodiazepine (alprazolam, dose range: 1.5 to 6 mg) treatment arm, a dopamine beta‐hydroxylase inhibitor (nepicastat, dose range: 100 to 800 mg) treatment arm, a NK‐1 receptor antagonist (orvepitant, fixed 60 mg) treatment arm, a hypnotic (eszopiclone, dose 3 mg) treatment arm, and a second messenger system precursor (inositol, fixed 12 grams) treatment arm. The review also includes one trial investigating the norepinephrine and dopamine reuptake inhibitor bupropion SR (dose range: 100 to 300 mg).
None of the six RCTs which used a cross‐over design (Braun 1990; Feder 2014; Kaplan 1996; Rasmusson 2017; Reist 1989; Shestatzky 1988) provided enough information for inclusion of data in the meta‐analysis. These trials were all small and of poor quality, so their inclusion/exclusion is unlikely to have had a significant effect on the primary outcomes of the meta‐analysis. In brief, Braun 1990 compared an intervention of alprazolam with placebo using a sample of 16 outpatients over a period of 12 weeks. Feder 2014 compared ketamine and midazolam over 13 days amongst 41 participants. Kaplan 1996 investigated the efficacy of inositol for a sample of 13 participants from two different outpatient clinics. Rasmusson 2017 included 112 participants in an open‐label trial followed by a double‐blind trial investigating the experiential medication ganaxolone. Reist 1989 conducted a cross‐over trial of the TCA desipramine, in treating 27 combat veterans over a period of 10 weeks. Shestatzky 1988 assessed the efficacy of the MAOI phenelzine in a 12‐week trial of 13 PTSD outpatients.
In determining the long‐term effects of medication, Marshall 2007 conducted a 10‐week trial of paroxetine and found continued reduction in PTSD symptoms after an additional maintenance period of 12 weeks, while Van der Kolk 2007 assessed the efficacy of fluoxetine in 59 participants over five weeks followed by a six‐month maintenance phase. In a relapse‐prevention trial of sertraline, Davidson 2001b set out to determine whether 50 participants who were randomised to a placebo control for 28 weeks were more likely to experience clinical deterioration or relapse than the 46 randomised to a sertraline intervention for the same period. Participants in the relapse‐prevention phase of this trial had completed a 12‐week RCT of sertraline, and had also met responder criteria following the subsequent open‐label administration of this medication for a period of six months. In another relapse‐prevention trial, Martenyi 2002a re‐randomised 131 responders to a 12‐week short‐term RCT of fluoxetine to an additional 24 weeks of placebo or medication.
We noted a great deal of clinical heterogeneity across participants in the RCTs of this review. Of the 53 trials that provided information on the nature of the index trauma for PTSD, eight were composed exclusively of war veterans (Chung 2004; Davidson 1990; Hertzberg 2000; Kosten 1991; NCT00659230; Pfizer589; Raskind 2018; Reist 1989), 15 contained individuals exposed to 'civilian' traumas, such as car accidents, earthquakes, domestic violence and child molestation (Ahmadpanah 2014; Becker 2007; Brady 2000; Connor 1999a; Marshall 2007; McRae 2004; NCT01000493; Padala 2006; Pfizer588; Reich 2004; Saygin 2002; Smajkic 2001; Spivak 2006; Tucker 2007; Yeh 2011) and 30 contained people exposed to both combat‐related and civilian traumas (Baker 1995; Braun 1990; Butterfield 2001; Davidson 2001a; Davidson 2003; Davidson 2005; Davidson 2006a; Davis 2004; Davis 2008; Davis 2020; Dowd 2020; Dunlop 2017; Feder 2014; Hertzberg 1999; Kaplan 1996; Katz 1994; Li 2017; Marshall 2001; Martenyi 2002a; Martenyi 2007; Mathew 2011; Panahi 2011; Rasmusson 2017; Shestatzky 1988; Tucker 2001; Tucker 2003; Van der Kolk 1994; Van der Kolk 2007; Villarreal 2016; Zohar 2002). Of the 36 RCTs that provided information on comorbid psychopathology, 32 reported that the participants in the trials were diagnosed with other anxiety disorders besides PTSD, as classified according to DSM III, DSM III‐R, DSM‐IV DSM‐IV‐TR or DSM‐5 criteria (Ahmadpanah 2014; Becker 2007; Brady 2000; Braun 1990; Butterfield 2001; Carey 2012; Davidson 1990; Davidson 2001a; Davidson 2003; Davidson 2007; Davis 2004; Davis 2020; Dowd 2020; Dunlop 2017; Hamner 2008; Hertzberg 2000; Kaplan 1996; Li 2017; Marshall 2001; Marshall 2007; Mathew 2011; McCall 2018; Panahi 2011; Pfizer589; Raskind 2018; Rasmusson 2017; Reich 2004; Saygin 2002; Shestatzky 1988; Tucker 2001; Tucker 2003; Tucker 2007). The most commonly‐reported comorbid anxiety was panic disorder, with or without agoraphobia, which was reported for nine of the 13 studies that distinguished between individual anxiety disorder diagnostic categories (Braun 1990; Butterfield 2001; Davidson 1990; Davis 2004; Davis 2020; Marshall 2001; Marshall 2007; Saygin 2002; Tucker 2003). Twenty‐two studies included individuals with major depressive disorder (MDD), while MDD was an exclusion criterion for seven trials. We were unable to determine whether individuals with MDD could take part in the remaining trials, due to insufficient information.
Twenty trials assessed our primary efficacy outcome – the number of participants with PTSD who responded to treatment – using the CGI–I Scale (Brady 2000; Carey 2012; Chung 2004; Davidson 1990; Davidson 2001a; Davidson 2005; Davidson 2007; Friedman 2007; GSK 29060 627; Marshall 2001; Marshall 2007; Panahi 2011; Pfizer588; Pfizer589; Raskind 2018; Saygin 2002; Shestatzky 1988; SKB627; Tucker 2001; Zohar 2002), whilst 16 other trials assessed this outcome as a secondary measure using the same scale (Baker 1995; Butterfield 2001; Connor 2006; Davis 2004; Davis 2020; Feder 2014; Hamner 2008; Katz 1994; Martenyi 2002a; Martenyi 2002b; Mathew 2011; Raskind 2018; Rasmusson 2017; Tucker 2007; Villarreal 2016; Yeh 2011). Forty‐seven trials used the CAPS to assess the reduction in PTSD symptoms, while 23 used the CGI‐S, 25 used the DTS and 14 used the IES. The most common measures used to assess depression were the HAM‐D (n = 22) and the MADRS (n = 16). Eighteen trials assessed the reduction of anxiety symptoms with the HAM‐A (Braun 1990; Connor 2006; Davidson 1990; Davidson 2001a; Davis 2004; Davidson 1990; Friedman 2007; Kaplan 1996; Kosten 1991; Marshall 2007; Martenyi 2002a; Martenyi 2002b; McRae 2004; Padala 2006; Reist 1989; Spivak 2006; Tucker 2007; Villarreal 2016). Only 13 trials assessed functional disability using the SDS (Carey 2012; Connor 1999a; Davidson 2006a; Davidson 2006b; Davidson 2007; Davis 2020; GSK 29060 627; Marshall 2001; Mathew 2011; McRae 2004; SKB650; Tucker 2001; Tucker 2007). Post‐treatment follow‐up assessments ranged from two weeks in GSK 29060 627 to six months in Van der Kolk 2007. See Characteristics of included studies for more details.
Excluded studies
We excluded 65 studies from the review (see Characteristics of excluded studies). The most common reason for trials being considered ineligible was the augmentation of medication or psychotherapy within groups, or both (Aerni 2004; Back 2016; Bartzokis 2005; Batki 2014; Bountress 2018; Brunet 2018; Buhmann 2018; Feder 2021; Hamner 1997; Hamner 2003; Heresco‐Levy 2002; Monnelly 2003; Neylan 2006; NCT01325168; NCT01449955; NCT01477762; Otto 2003; Ramaswamy 2017; Raskind 2003; Raskind 2007; Stein 2002; Suris 2017). Psychometric studies were also excluded (Bremner 1997; Coupland 1997; Hamner 1999; Kanter 2001; Kellner 2000; Morgan 1995; Pitman 1990; Randall 1995; Reist 1995; Reist 2001; Southwick 1997; Vaiva 2003; Van der Kolk 1989). We excluded eight trials with a primary diagnosis other than PTSD (Angelidis 2019; Back 2018; Flanagan 2019; Jacobs‐Rebhun 2000; Naylor 2013; Petrakis 2006; Roache 2017; Verplaetse 2019), and seven trials with the absence of a control comparator (Eftekhari 2004; Frommberger 1998; Graham 2018; Inslicht 2018; Le 2018; Pollack 2011; Ramaswamy 2016). Boroughs 2018, Frank 1988 and Raskind 2013 were excluded because these trials were secondary analyses of previous studies, Mello 2009 because this study is a protocol, Flanagan 2018 because it is an imaging study, and Pape 2018 a genetic study. PTSD prevention studies were also excluded (Borrelli 2019, Frijling 2017; Gelpin 1996; Pitman 2002; Schelling 2004). Four trials have been withdrawn and are no longer in effect due to difficulty in achieving target enrolment numbers (NCT00557622; NCT00648375; NCT00706173; NCT01664260) and were therefore also excluded. See Characteristics of excluded studies for more details.
Studies awaiting classification
Eighteen trials are awaiting classification (Davis 2003; Fluoxetine, Davidson; Fluoxetine, Lilly; Nagy 1996; NCT00413296; NCT00672776; NCT01517711; NCT02637895; NCT02709018; NCT02933606; Nefazodone 1, BMS; Nefazodone 2, BMS; Olanzapine, Davidson; Paroxetine 2 ‐ SB; Paroxetine 3 ‐ SB; Sertraline 2, Pfizer; Sertraline 3, Pfizer; Sertraline 4, Pfizer). NCT00413296 conducted a study on the effectiveness of levetiracetam in 16 participants with PTSD. NCT00672776 assessed paroxetine compared to placebo in participants with PTSD. PET measurement of brain activation before and after paroxetine or placebo treatment also took place in NCT00672776. NCT01517711 assessed tramadol and placebo in 40 participants with PTSD, as measured by the CAPS, CGI‐S and various other secondary scales, and NCT02637895 investigated vortioxetine for posttraumatic stress disorder. Two trials NCT02709018 and NCT02933606 assessed losartan and BNC210 compared to placebo in posttraumatic stress disorder, respectively. There was insufficient information provided on the remaining studies awaiting classification. See Characteristics of studies awaiting classification for more details.
Ongoing studies
One study is ongoing (NCT01221792). NCT01221792 will compare carvedilol versus placebo using the DTS and the CAPS to determine levels of PTSD severity in people with PTSD. See Characteristics of ongoing studies.
Risk of bias in included studies
Risk of bias was assessed using the Cochrane risk of bias tool for allocation concealment, blinding, incomplete outcome data, selective reporting and other potential sources of bias. Sixteen of the included studies were rated as being at high risk for at least one type of bias (see Figure 2 and Figure 3).
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Randomisation
The randomisation procedure was described in 22 trials (Ahmadpanah 2014; Becker 2007; Carey 2012; Connor 1999a; Davidson 2001a; Davidson 2005; Davidson 2006a; Davis 2004; Davis 2008; Dunlop 2017; Friedman 2007; Kaplan 1996; Li 2017; Martenyi 2002a; Mathew 2011; Panahi 2011; Rasmusson 2017; Spivak 2006; Tucker 2007; Van der Kolk 2007; Villarreal 2016; Yeh 2011). Computer‐generated random codes were used in 10 of these trials (Connor 1999a; Davidson 2001a; Davis 2008; Friedman 2007; Li 2017; Martenyi 2002a; Spivak 2006; Tucker 2007; Villarreal 2016; Yeh 2011). Permuted blocks were generated by computer programmes for seven trials (Carey 2012; Davidson 2006a; Dunlop 2017; Mathew 2011; McCall 2018; Rasmusson 2017; Van der Kolk 2007) and prearranged random codes were used by Kaplan 1996, as well as a preassigned set of random numbers by Davidson 2005. The review classified all these studies as being at low risk for selection bias, while risk of bias was designated as unclear in the remaining studies.
Allocation concealment
Of the 66 RCTs, only 13 trials described adequate allocation concealment (Ahmadpanah 2014; Carey 2012; Connor 1999a; Davidson 2006a; Davis 2004; Davis 2008; Dowd 2020; Dunlop 2017; Feder 2014; Li 2017; Martenyi 2002a; McRae 2004; Rasmusson 2017) and were classified as low for risk of bias. A pharmacist or sponsor who prepared and supplied the study medication maintained allocation concealment in 11 trials (Ahmadpanah 2014; Carey 2012; Connor 1999a; Davidson 2006a; Davis 2004; Davis 2008; Dowd 2020; Dunlop 2017; Feder 2014; Li 2017; McRae 2004). One study maintained concealment with the use of sealed envelopes (Martenyi 2002a), and another with bottles with numbers and codes generated by a computer programme (Rasmusson 2017). Two trials were classified as being at high risk of bias because of no allocation concealment information (Chung 2004; Smajkic 2001). The remaining trials were classified as unclear because the trials did not provide enough information to determine the adequacy of the concealment used.
Blinding
Blinding of participants and personnel
Nine studies included in the review were classified as being at low risk for performance bias, as participants and personnel were explicitly described as blinded in the study report (Ahmadpanah 2014; Davis 2004; Davis 2020; Feder 2014; Kosten 1991; Li 2017; Mathew 2011; Raskind 2018; Yeh 2011). Two trials were classified as being at high risk of bias because neither trial blinded their participants and personnel (Chung 2004; Smajkic 2001). The review classified risk of bias for the remaining trials as unclear, despite investigators describing them in the study reports as double‐blinded, as they did not specify the actual parties blinded.
Blinding of outcome assessors
Although most of the trials were described as "double‐blind", only 13 of the trials explicitly described the assessment of outcome as blinded (Braun 1990; Connor 1999a; Davis 2008; Feder 2014; Li 2017; Marshall 2007; McCall 2018; Panahi 2011; Raskind 2018; Rasmusson 2017; Shestatzky 1988; Spivak 2006; Van der Kolk 2007). The extent to which blinding was preserved in those flexible‐dose trials which adjusted dosage based on tolerability is unclear. Indeed, the outcome was assessed independently of medication administration and side‐effect evaluation in only one RCT (Marshall 2007). Two comparative RCTs did not use any form of blinding (Chung 2004; Smajkic 2001), and it is not clear whether blinding was undertaken in another (Saygin 2002). The remaining studies were classified as being at unclear risk.
Incomplete outcome data
Thirty‐three studies failed to provide sufficient information to determine whether the medication and placebo groups were comparable for dropout proportions, or for the demographic and clinical characteristics of those who withdrew (Ahmadpanah 2014; Baker 1995; Braun 1990; Carey 2012; Chung 2004; Connor 1999a; Davidson 2001a; Davidson 2003; Davidson 2005; Davidson 2006b; Davis 2020; Dunlop 2017; Hamner 2008; Hertzberg 2000; Kaplan 1996; Marshall 2001; Marshall 2007; Martenyi 2002a; Mathew 2011; McCall 2018; McRae 2004; NCT01000493; Padala 2006; Pfizer588; Pfizer589; Raskind 2018; Rasmusson 2017; Reist 1989; Saygin 2002; Shestatzky 1988; SKB627; SKB650; Zohar 2002). The risk of attrition bias was rated as unclear for these trials. Substantial differences in the proportion of study dropouts between the medication and placebo groups for 11 trials were observed (Butterfield 2001; Connor 2006; Dowd 2020; Hertzberg 1999; Kosten 1991; Martenyi 2002b; NCT01681849; Reich 2004; Smajkic 2001; Spivak 2006; Van der Kolk 1994), justifying a rating of high risk. The review rated the remaining 22 studies as being at low risk for this domain, because of comparable dropout rates in each comparison group and no difference in the demographic characteristics. Intention‐to‐treat data for all outcomes were assessed in 33 studies, using LOCF (n = 33), completers (n = 19), observed cases (n = 2) and mixed‐effect models (n = 3) to account for missing data. Three studies did not specify how they addressed the issue of missing data.
Selective reporting
It was unclear whether selective reporting took place in 57 trials because the study protocols were not available for the study. Six studies were rated as being at low risk for selective reporting because authors reported on all outcomes as specified in their respective trial protocols (Davis 2020; Dunlop 2017; NCT00659230; NCT01000493; NCT01681849; Raskind 2018). In Davis 2008, Feder 2014, and Villarreal 2016, the prespecified secondary outcomes in the protocol did not appear in the study report, meriting a classification of high risk.
Other potential sources of bias
Twenty‐one studies were classified as being at low risk for other potential sources of bias (Ahmadpanah 2014; Baker 1995; Braun 1990; Connor 1999a; Davidson 1990; Davidson 2005; Davis 2020; Dowd 2020; Dunlop 2017; Feder 2014; Hertzberg 2000; Li 2017; McCall 2018; NCT00659230; NCT01681849; Panahi 2011; Raskind 2018; Rasmusson 2017; Saygin 2002; Van der Kolk 2007; Yeh 2011). The review rated the risk of other bias for the remaining studies as unclear due to industry involvement, whether through funding or provision of study medication.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7
See summary of findings tables for the main comparisons prioritised based on primary outcomes of medication classes compared to placebo for treating PTSD in adults: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7. Please refer to Types of outcome measures for a description of the scoring systems used in included studies.
Comparison 1: Alpha‐blockers versus placebo
Primary outcome measures
We found no studies that looked at the number of participants who responded to prazosin compared to placebo.
There was also no evidence of a harm for the number of participants who withdrew due to treatment‐emergent side effects (risk ratio (RR) 0.99, 95% confidence interval (CI) 0.91 to 1.08; 1 study, 304 participants; see Analysis 1.1; Raskind 2018), based on moderate‐certainty evidence.
1.1. Analysis.

Comparison 1: Alpha‐blockers versus placebo, Outcome 1: Dropout rate due to treatment‐emergent adverse effects
Secondary outcome measures
There was no evidence of a benefit of prazosin compared to placebo for the reduction of PTSD symptoms on the CAPS total (mean difference (MD) 0.10 points, 95% CI −5.13 to 5.33; 1 study, 271 participants; see Analysis 1.2; Raskind 2018). No evidence of a benefit for prazosin versus placebo was found for one trial that provided data for the reduction of PTSD symptoms on the self‐rated PTSD Checklist–Military Version (PCL‐M): (standardised mean difference (SMD) −0.18 points, 95% CI −0.41 to 0.06; 1 study, 271 participants; Analysis 1.3; Raskind 2018).
1.2. Analysis.

Comparison 1: Alpha‐blockers versus placebo, Outcome 2: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
1.3. Analysis.

Comparison 1: Alpha‐blockers versus placebo, Outcome 3: Self‐rated scales
There was no evidence of a benefit of prazosin in reducing depression symptoms on the Patient Health Questionnaire 9‐item depression (PHQ‐9) scale in participants with PTSD compared to placebo (MD −0.60 points, 95% CI −2.11 to 0.91; 1 study, 271 participants; see Analysis 1.4; Raskind 2018).
1.4. Analysis.

Comparison 1: Alpha‐blockers versus placebo, Outcome 4: Depression scale (typically Hamilton Depression)
A similar proportion of participants withdrew from the prazosin group (20%) compared to the placebo group (19%) (see Analysis 1.5; Raskind 2018).
1.5. Analysis.

Comparison 1: Alpha‐blockers versus placebo, Outcome 5: Dropout rate due to any cause
Comparison 2: Anticonvulsants versus placebo
Primary outcome measures
There was no evidence of a benefit that lamotrigine, tiagabine or topiramate improves treatment efficacy in participants with PTSD compared to placebo (RR 0.87, 95% CI 0.63 to 1.21; 4 trials, 315 participants (Davidson 2007; Hertzberg 1999; Tucker 2007; Yeh 2011).
Dropout rates due to adverse events were low in RCTs of divalproex, lamotrigine, tiagabine and topiramate, and were comparable to those observed in the placebo arms (11% versus 9%, respectively) (RR 0.98, 95% CI 0.92 to 1.05; 5 trials, 348 participants; see Analysis 2.2; Davidson 2007; Hamner 2008; Hertzberg 1999; Tucker 2007; Yeh 2011).
2.2. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 2: Dropout rate due to treatment emergent adverse effects
Secondary outcome measures
We assessed data on the reduction of PTSD symptoms, measured by the CAPS (total score), in five studies comparing divalproex, tiagabine and topiramate to placebo (MD −0.35 points, 95% CI −6.54 to −5.84; 5 trials, 411 participants; see Analysis 2.3; Davidson 2007; Davis 2008; Hamner 2008; Tucker 2007; Yeh 2011). We found no evidence of beneficial effect (P = 0.91). Divalproex and topiramate were not superior to placebo on either the re‐experiencing/intrusion subscale (MD 0.20 points, 95% CI −3.74 to 4.13; see Analysis 2.3), avoidance/numbing subscale (MD −0.37 points, 95% CI −8.65 to 7.91; see Analysis 2.3), or hyperarousal subscale (MD 0.21 points, 95% CI −2.57 to 2.99; see Analysis 2.3) of the CAPS in three trials with 141 participants (Davis 2008; Hamner 2008; Yeh 2011). We noted substantial and considerable heterogeneity for the re‐experiencing/intrusion and avoidance/numbing subscales, respectively (Chi2 = 4.19, df = 2 (P = 0.12); I2= 52%; Chi2= 9.87, df = 2 (P = 0.007); I2 = 80%).
2.3. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 3: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
Furthermore, we found no evidence of a benefit for divalproex, lamotrigine, tiagabine or topiramate versus placebo for the reduction of PTSD symptoms on the other symptom severity scales such as the CGI‐S and DGRP (SMD 0.15 points, 95% CI −0.31 to 0.60; 5 trials, 180 participants; see Analysis 2.4; Connor 2006; Davis 2008; Hamner 2008; Hertzberg 1999; Tucker 2007), or on the self‐rated DTS and IES (SMD −0.06 points, 95% CI −0.75 to 0.64; 3 trials, 84 participants; see Analysis 2.5; Connor 2006; Hamner 2008; Tucker 2007), with substantial heterogeneity found for the self‐rated measures (Chi2 = 4.86, df = 2 (P = 0.09); I2 = 59%).
2.4. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 4: Symptom severity: Other measures
2.5. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 5: Self‐rated scales
Similarly, there was no evidence of a benefit of divalproex, tiagabine or topiramate for the reduction in depression symptoms on the BDI, HAM‐D and MADRS for participants with PTSD compared to placebo (SMD −0.22 points, 95% CI −0.57 to 0.12; 3 trials, 131 participants; see Analysis 2.6; Connor 2006; Davis 2008; Yeh 2011). There was also no evidence of a benefit of divalproex and tiagabine compared to placebo on the reduction of anxiety symptoms on the HAM‐A (MD −0.66 points, 95% CI −3.66 to 2.35; 3 trials, 138 participants; see Analysis 2.7; Connor 2006; Davis 2008; Tucker 2007). We found no evidence of beneficial effect for the reduction of anxiety symptoms for ‘other’ measures of anxiety (SMD 0.55 points, 95% CI −0.21 to 1.31; 1 trial, 28 participants; see Analysis 2.8; Hamner 2008). In addition, there was no evidence of a benefit of tiagabine on the reduction of functional disability measured by the SDS in participants with PTSD compared to placebo (MD 0.41 points, 95% CI −1.48 to 2.30; 2 trials, 270 participants; see Analysis 2.9) (Davidson 2007; Tucker 2007).
2.6. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 6: Depression scale (typically Hamilton Depression)
2.7. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 7: Anxiety ‐ Hamilton Anxiety Scale
2.8. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 8: Anxiety ‐ Other scales
2.9. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 9: Sheehan Disability Scale
Similar dropout rates due to any cause were observed in the medication (28%) and placebo (32%) arms for the anticonvulsants (see Analysis 2.10; Connor 2006; Davidson 2007; Davis 2008; Hamner 2008; Hertzberg 1999; Tucker 2007; Yeh 2011).
2.10. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 10: Dropout rate due to any cause
Comparison 3: Antipsychotics versus placebo
Primary outcome measures
There was no evidence of a benefit of effect for olanzapine in participants with PTSD compared to placebo (RR 0.51, 95% CI 0.16 to 1.67; 2 trials, 43 participants; see Analysis 3.1; Butterfield 2001; Carey 2012), based on very low‐certainty evidence. We noted moderate heterogeneity across these trials (Chi2 = 1.99, df = 1 (P = 0.16); I2 = 50%).
3.1. Analysis.

Comparison 3: Antipsychotics versus placebo, Outcome 1: Clinical Global Impressions Scale Improvement Item (CGI‐I)
Twice as many participants withdrew due to treatment‐emergent side effects from the antipsychotic intervention arm (16%) compared to the placebo arm (7%), but we found no evidence of a harm between groups (RR 0.94, 95% CI 0.85 to 1.03; 5 trials, 174 participants; see Analysis 3.2; Butterfield 2001; Carey 2012; Padala 2006; Reich 2004; Villarreal 2016). The evidence available for this outcome was of low‐certainty.
3.2. Analysis.

Comparison 3: Antipsychotics versus placebo, Outcome 2: Drop‐out rate due to treatment emergent adverse effects
Secondary outcome measures
There was evidence of a benefit for the reduction PTSD symptoms following treatment with olanzapine, risperidone or quetiapine compared to placebo for the CAPS total (MD −14.10 points, 95% CI −22.57 to −5.62; 3 trials, 129 participants; see Analysis 3.3; Carey 2012; Reich 2004; Villarreal 2016). There was evidence of a benefit for the reduction PTSD symptoms for this outcome on the re‐experiencing/intrusion subscale (MD −4.73 points, 95% CI −7.55 to −1.90; see Analysis 3.3) and hyperarousal subscale (MD −5.14 points, 95% CI −8.16 to −2.11; see Analysis 3.3) of the CAPS (Carey 2012; Reich 2004; Villarreal 2016). There was, however, no evidence of a benefit for the reduction of PTSD symptoms on the avoidance/numbing subscale (MD −6.01 points, 95% CI −13.72 to 1.70; see Analysis 3.3) of the CAPS (Carey 2012; Reich 2004; Villarreal 2016), which is confirmed by the substantial heterogeneity found for this subscale (Chi2 = 5.73, df = 2 (P = 0.06); I2 = 65%). The substantial heterogeneity for this outcome is partially reflected in the unusually large and important medication effect observed in Carey 2012 (MD −15.10 points, 95% CI −24.47 to −5.73). Removing this trial resulted in a substantially reduced and statistically non‐significant treatment effect (MD −2.50 points, 95% CI −6.93 to 1.93), with no heterogeneity observed across the effects for the remaining trials (Chi2 = 0.05, df = 1 (P = 0.82); I2 = 0%).
3.3. Analysis.

Comparison 3: Antipsychotics versus placebo, Outcome 3: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
Furthermore, we found evidence of a beneficial effect of olanzapine and quetiapine compared to placebo in the reduction of PTSD symptoms on the symptom severity CGI‐S scale (SMD −0.55 points, 95% CI −0.94 to −0.17; 2 trials, 108 participants; see Analysis 3.4; Carey 2012; Villarreal 2016), with no evidence of a benefit on the self‐rated DTS (SMD −0.47 points, 95% CI −1.06 to 0.11; 3 trials, 119 participants; see Analysis 3.5; Butterfield 2001; Carey 2012; Villarreal 2016), including the re‐experiencing/intrusion subscale (MD 0.51 points, 95% CI −0.58 to 1.61; 1 trial, 15 participants; see Analysis 3.5), avoidance/numbing subscale (MD −0.30 points, 95% CI −1.38 to 0.78; 1 trial, 15 participants; see Analysis 3.5) and hyperarousal subscale (MD −0.06 points, 95% CI −1.13 to 1.01; 1 trial, 15 participants; see Analysis 3.5) of the DTS (Butterfield 2001). We also found moderate heterogeneity for the total score on the self‐rated measures (Chi2 = 3.75, df = 2 (P = 0.15); I2 = 47%).
3.4. Analysis.

Comparison 3: Antipsychotics versus placebo, Outcome 4: Symptom severity: Other measures
3.5. Analysis.

Comparison 3: Antipsychotics versus placebo, Outcome 5: Self‐rated scales
There was evidence of a benefit of olanzapine and quetiapine compared to placebo in the reduction of depression symptoms (SMD −0.62 points, 95% CI −1.01 to −0.23; 2 trials, 108 participants; see Analysis 3.6; Carey 2012; Villarreal 2016) on the HAM‐D and MADRS. There was no evidence of a beneficial effect of quetiapine on the reduction of anxiety symptoms in participants with PTSD compared to placebo (MD −3.03 points, 95% CI −6.21 to 0.15, 1 trial, 80 participants; see Analysis 3.7; Villarreal 2016), as measured by the HAM‐A. Similarly, we found no evidence of a beneficial effect of olanzapine compared to placebo for the reduction of functional disability on the Sheehan Disability Scale (SDS) (MD −6.57 points, 95% CI −14.74 to 1.61; 2 trials, 43 participants; see Analysis 3.8; Butterfield 2001; Carey 2012). Substantial heterogeneity was also observed for the two trials investigating functional disability measured by the SDS (Chi2 = 2.53, df = 1 (P = 0.11); I2 = 61%).
3.6. Analysis.

Comparison 3: Antipsychotics versus placebo, Outcome 6: Depression Scale (typically Hamilton Depression)
3.7. Analysis.

Comparison 3: Antipsychotics versus placebo, Outcome 7: Anxiety ‐ Hamilton Anxiety Scale
3.8. Analysis.

Comparison 3: Antipsychotics versus placebo, Outcome 8: Sheehan Disability Scale
Although differences in dropout rates due to any cause in the medication (38%) and placebo (51%) arms were evident, no evidence of a difference was found across the four trials with 133 participants (see Analysis 3.9; Butterfield 2001; Padala 2006; Reich 2004; Villarreal 2016).
3.9. Analysis.

Comparison 3: Antipsychotics versus placebo, Outcome 9: Drop‐out rate due to any cause
Comparison 4: Benzodiazepines versus placebo
Primary outcome measures
We found no studies that looked at the number of participants who responded to alprazolam versus placebo.
No participants dropped out due to treatment‐emergent side effects in the alprazolam and placebo groups (see Analysis 4.1; Braun 1990). There is therefore no evidence of a harm in dropouts due to adverse events.
4.1. Analysis.

Comparison 4: Benzodiazepines versus placebo, Outcome 1: Drop‐out rate due to treatment emergent adverse effects
Secondary outcomes measures
We found no information that assessed most of the additional outcomes; but the dropout rate was similar for the alprazolam (43%) and placebo groups (33%) (see Analysis 4.2; Braun 1990).
4.2. Analysis.

Comparison 4: Benzodiazepines versus placebo, Outcome 2: Drop‐out rate due to any cause
Comparison 5: Dopamine beta‐hydroxylase inhibitors versus placebo
Primary outcome measures
We found no studies that looked at the number of participants who responded to nepicastat versus placebo.
There was no evidence of a harm in the number of participants who withdrew due to treatment‐emergent side effects for the nepicastat (63%) and placebo groups (57%), even though the dropout rates were substantial in both groups (RR 1.10, 95% CI 0.77 to 1.58; 1 trial, 91 participants; see Analysis 5.1; NCT00659230).
5.1. Analysis.

Comparison 5: Dopamine beta‐hydroxylase inhibitors versus placebo, Outcome 1: Drop‐out rate due to treatment emergent adverse effects
Secondary outcome measures
We found no evidence of a benefit of nepicastat on CAPS total symptom severity (MD −5.27 points, 95% CI −16.72 to 6.18; see Analysis 5.2), or on the re‐experiencing/intrusion subscale (MD 2.63 points, 95% CI −1.67 to 6.93; see Analysis 5.2), avoidance/numbing subscale (MD 0.58 points, 95% CI −4.57 to 5.73; see Analysis 5.2), or hyperarousal subscale (MD 2.05 points, 95% CI −1.82 to 5.92; see Analysis 5.2) of the CAPS for reducing PTSD symptoms, in one trial with 86 participants (NCT00659230).
5.2. Analysis.

Comparison 5: Dopamine beta‐hydroxylase inhibitors versus placebo, Outcome 2: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
Overall there were 12 (24%) dropouts in the nepicastat group and nine (18%) dropouts in the placebo group (see Analysis 5.3; NCT00659230).
5.3. Analysis.

Comparison 5: Dopamine beta‐hydroxylase inhibitors versus placebo, Outcome 3: Drop‐out rate due to any cause
Comparison 6: Ganaxolone versus placebo
Primary outcome measures
We found no studies that looked at the number of participants who responded to ganaxolone versus placebo.
There was no evidence of a harm for the number of participants who withdrew due to treatment‐emergent side effects (RR 0.60, 95% CI 0.10 to 3.45; 1 trial, 112 participants; see Analysis 6.1; Rasmusson 2017).
6.1. Analysis.

Comparison 6: Ganaxolone versus placebo, Outcome 1: Drop‐out rate due to treatment emergent adverse effects
Secondary outcome measures
No evidence of a benefit was observed for ganaxolone medication on CAPS total symptom severity (MD −2.50 points, 95% CI −10.92 to 5.92; 1 trial, 99 participants; see Analysis 6.2; Rasmusson 2017), or on re‐experiencing/intrusion subscale (MD −0.80 points, 95% CI −4.09 to 2.49; see Analysis 6.2), avoidance/numbing subscale (MD −1.60 points, 95% CI −5.74 to 2.54; see Analysis 6.2), or hyperarousal subscale (MD −1.10 points, 95% CI −3.96 to 1.76; see Analysis 6.2) on the CAPS for reducing PTSD symptoms. Similarly, no evidence of a benefit of ganaxolone versus placebo was observed for the reduction in PTSD symptoms on the PCL scale (self‐rated measure) (SMD −0.17 points, 95% CI −0.59 to 0.25; 1 trial, 86 participants; see Analysis 6.3; Analysis 6.4; Rasmusson 2017).
6.2. Analysis.

Comparison 6: Ganaxolone versus placebo, Outcome 2: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
6.3. Analysis.

Comparison 6: Ganaxolone versus placebo, Outcome 3: Symptom severity: Other measures
6.4. Analysis.

Comparison 6: Ganaxolone versus placebo, Outcome 4: Self‐rated scales
In addition, we found no evidence of a difference in dropouts due to any cause for the ganaxolone intervention arm (33%) compared to the placebo arm (17%) (see Analysis 6.5; Rasmusson 2017), even though more dropouts were observed in the medication arm.
6.5. Analysis.

Comparison 6: Ganaxolone versus placebo, Outcome 5: Drop‐out rate due to any cause
Comparison 7: GR205171 versus placebo
Primary outcome measures
There was no evidence of a benefit of GR205171 compared to placebo for the number of participants with PTSD who responded to treatment (RR 0.78, 95% CI 0.42 to 1.44; 1 trial, 39 participants; see Analysis 7.1; Mathew 2011).
7.1. Analysis.

Comparison 7: GR205171 versus placebo, Outcome 1: Clinical Global Impressions Scale Improvement Item (CGI‐I)
No participants dropped out due to treatment‐emergent side effects in the GR205171 and placebo groups (see Analysis 7.2; Mathew 2011).
7.2. Analysis.

Comparison 7: GR205171 versus placebo, Outcome 2: Drop‐out rate due to treatment emergent adverse effects
Secondary outcome measures
There was no evidence of a benefit for GR205171 compared to placebo in the reduction of PTSD symptoms on the CAPS total (MD −7.60 points, 95% CI −22.59 to 7.39; 1 trial, 39 participants; see Analysis 7.3; Mathew 2011).
7.3. Analysis.

Comparison 7: GR205171 versus placebo, Outcome 3: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
Twice as many participants dropped out due to any cause from the placebo arm (48%) compared to the GR205171 intervention arm (18%) (see Analysis 7.4; Mathew 2011).
7.4. Analysis.

Comparison 7: GR205171 versus placebo, Outcome 4: Drop‐out rate due to any cause
Comparison 8: GSK561679 versus placebo
Primary outcome measures
There was no evidence of a benefit of GSK561679 on treatment efficacy in participants with PTSD compared to placebo (RR 1.08, 95% CI 0.88 to 1.31; 1 trial, 128 participants; see Analysis 8.1; Dunlop 2017).
8.1. Analysis.

Comparison 8: GSK561679 versus placebo, Outcome 1: Clinical Global Impressions Scale Improvement Item (CGI‐I)
We found no studies that looked at the number of participants who dropped out due to treatment‐emergent side effects.
Secondary outcome measures
We found no studies that looked at the secondary outcome measures of GSK561679 compared to placebo.
Comparison 9: Hypnotics versus placebo
Primary outcome measures
We found no studies that looked at the number of participants who responded to eszopiclone versus placebo.
We found no studies that looked at the number of participants who dropped out due to treatment‐emergent side effects.
Secondary outcome measures
There was no evidence of a benefit of eszopiclone in reducing PTSD symptoms on the CAPS total (MD −1.00 points, 95% CI −19.57 to 17.57; 1 trial, 16 participants; see Analysis 9.1; Dowd 2020) or on the CGI‐S (SMD −0.09 points, 95% CI −1.08 to 0.90; 1 trial, 16 participants; see Analysis 9.2; Dowd 2020).
9.1. Analysis.

Comparison 9: Hypnotics versus placebo, Outcome 1: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
9.2. Analysis.

Comparison 9: Hypnotics versus placebo, Outcome 2: Symptom severity: Other measures
There was also no evidence of a benefit of eszopiclone compared to placebo for the reduction in depression symptoms on the MADRS (SMD −2.67 points, 95% CI −12.68 to 7.34; 1 trial, 16 participants; see Analysis 9.3; Dowd 2020).
9.3. Analysis.

Comparison 9: Hypnotics versus placebo, Outcome 3: Depression Scale (typically Hamilton Depression)
We found no evidence of a difference for dropouts due to any cause for the eszopiclone (46%) and placebo (25%) group, even though dropouts were higher in the medication arm (see Analysis 9.4; Dowd 2020).
9.4. Analysis.

Comparison 9: Hypnotics versus placebo, Outcome 4: Drop‐out rate due to any cause
Comparison 10: MAOIs versus placebo
Primary outcome measures
We found no studies that looked at the number of participants who responded to phenelzine versus placebo.
More participants dropped out from the placebo group (17%) compared to the phenelzine group (5%) due to treatment‐emergent side effects, although we found no evidence of a harm in the dropout rates (see Analysis 10.1; Kosten 1991). This outcome was based was on low‐certainty evidence.
10.1. Analysis.

Comparison 10: MAOIs versus placebo, Outcome 1: Drop‐out rate due to treatment emergent adverse effects
Secondary outcome measures
There was evidence of a benefit of the MAOI phenelzine compared to placebo for reducing total PTSD symptoms on the IES (SMD −1.06 points, 95% CI −1.75 to −0.36; see Analysis 10.2) for 1 trial with 37 participants (Kosten 1991). This benefit was also apparent for the re‐experiencing/intrusion subscale (SMD −1.07 points, 95% CI −1.77 to −0.38; 1 trial, 37 participants; see Analysis 10.2; Kosten 1991) and the avoidance/numbing subscale (SMD −0.81 points, 95% CI −1.49 to −0.14; 1 trial, 37 participants; see Analysis 10.2; Kosten 1991) on the IES.
10.2. Analysis.

Comparison 10: MAOIs versus placebo, Outcome 2: Self‐rated scales
There was no evidence of a benefit of phenelzine compared to placebo on the reduction in depression symptoms on the HAM‐D (MD −2.80 points, 95% CI −7.15 to 1.55; 1 trial, 37 participants; see Analysis 10.3). There was evidence for the reduction in anxiety symptoms on the Covi Anxiety Scale (SMD −0.67 points, 95% CI −1.34 to −0.01; 1 trial, 37 participants; see Analysis 10.4; Kosten 1991), however, based on a marginally significant effect (P = 0.05).
10.3. Analysis.

Comparison 10: MAOIs versus placebo, Outcome 3: Depression Scale (typically Hamilton Depression)
10.4. Analysis.

Comparison 10: MAOIs versus placebo, Outcome 4: Anxiety ‐ Other scales
We found no evidence of a difference for dropouts due to any cause for the phenelzine (21%) and placebo (11%) groups (see Analysis 10.5; Kosten 1991).
10.5. Analysis.

Comparison 10: MAOIs versus placebo, Outcome 5: Drop‐out rate due to any cause
Comparison 11: NaSSAs versus placebo
Primary outcome measures
There was evidence of a benefit of mirtazapine in improving PTSD symptoms compared to placebo (RR 0.45, 95% CI 0.22 to 0.94; 1 trial, 26 participants (Davidson 2003); see Analysis 11.1). This was based on low‐certainty evidence.
11.1. Analysis.

Comparison 11: NaSSAs versus placebo, Outcome 1: Clinical Global Impressions Scale Improvement Item (CGI‐I)
There was no evidence of a harm in the number of participants who withdrew due to treatment‐emergent side effects for the mirtazapine (18%) and placebo groups (5%) (RR 0.87, 95% CI 0.68 to 1.11; 1 trial, 36 participants; see Analysis 11.2; Davidson 2003), based on moderate‐certainty evidence.
11.2. Analysis.

Comparison 11: NaSSAs versus placebo, Outcome 2: Drop‐out rate due to treatment emergent adverse effects
Secondary outcome measures
There was no evidence of beneficial effect of mirtazapine for reducing PTSD symptoms on the CAPS total (MD −3.20 points, 95% CI −14.74 to 8.34; 1 trial, 61 participants; see Analysis 11.3; Davis 2020), or on the re‐experiencing/intrusion subscale (MD −2.50 points, 95% CI −6.32 to 1.32; see Analysis 11.3), on avoidance/numbing subscale (MD −0.50 points, 95% CI −6.22 to 5.22; see Analysis 11.3), or hyperarousal subscale (MD −0.20 points, 95% CI −3.51 to 3.11; see Analysis 11.3) on the CAPS for reducing PTSD symptoms. Similarly, we found no evidence of a benefit of an effect for reducing PTSD symptoms on the self‐rated DTS (SMD −0.31 points, 95% CI −1.02 to 0.41; 2 trials, 87 participants; see Analysis 11.5; Davidson 2003; Davis 2020). We also detected moderate heterogeneity on the self‐rated DTS measures (Chi2 = 2.26, df = 1 (P = 0.13); I2 = 56%). There was, however, evidence of a benefit of mirtazapine for reducing PTSD symptoms on the SPRINT and CGI‐S (SMD −0.43 points, 95% CI −0.87 to −0.00; 2 trials, 87 participants; see Analysis 11.4; Davidson 2003; Davis 2020).
11.3. Analysis.

Comparison 11: NaSSAs versus placebo, Outcome 3: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
11.5. Analysis.

Comparison 11: NaSSAs versus placebo, Outcome 5: Self‐rated scales
11.4. Analysis.

Comparison 11: NaSSAs versus placebo, Outcome 4: Symptom severity: Other measures
There was no evidence of a benefit of mirtazapine compared to placebo for the reduction of depression symptoms on the HADS‐D and MADRS (SMD −0.26 points, 95% CI −1.39 to 0.88; 2 trials, 87 participants; see Analysis 11.6; Davidson 2003; Davis 2020). There was evidence of a benefit of mirtazapine for reducing anxiety symptoms on the HADS‐A scale (SMD −0.88 points, 95% CI −1.77 to −0.00; 1 trial, 25 participants; see Analysis 11.7; Davidson 2003), however, based on a marginally significant effect (P = 0.05).
11.6. Analysis.

Comparison 11: NaSSAs versus placebo, Outcome 6: Depression Scale (typically Hamilton Depression)
11.7. Analysis.

Comparison 11: NaSSAs versus placebo, Outcome 7: Anxiety ‐ Other scales
There was no evidence of a difference in the number of participants who withdrew due to any cause for the mirtazapine (17%) and placebo (19%) groups (see Analysis 11.8; Davidson 2003; Davis 2020; Chung 2004).
11.8. Analysis.

Comparison 11: NaSSAs versus placebo, Outcome 8: Drop‐out rate due to any cause
Comparison 12: NK‐1 receptor antagonists versus placebo
Primary outcome measures
We found no studies that looked at the number of participants who responded to orvepitant versus placebo.
We found no studies that looked at the number of participants who dropped out due to treatment‐emergent side effects.
Secondary outcome measures
We found no studies that looked at most secondary outcomes measures of orvepitant compared to placebo, but the proportion of dropouts was high in participants receiving orvepitant (78%) and placebo (65%), but no evidence of a difference was found (RR 0.58, 95% CI 0.32 to 1.04; 1 trial, 129 participants; see Analysis 12.1; NCT01000493).
12.1. Analysis.

Comparison 12: NK‐1 receptor antagonists versus placebo, Outcome 1: Drop‐out rate due to any cause
Comparison 13: RIMAs versus placebo
Primary outcome measures
There was no evidence of a benefit of brofaromine on treatment efficacy in participants with PTSD compared to placebo (RR 0.80, 95% CI 0.44 to 1.44; 2 trials, 178 participants; see Analysis 13.1; Baker 1995; Katz 1994). We noted moderate heterogeneity for this outcome (Chi2 = 2.33, df = 1 (P = 0.13); I2 = 57%).
13.1. Analysis.

Comparison 13: RIMAs versus placebo, Outcome 1: Clinical Global Impressions Scale Improvement Item (CGI‐I)
There was also no evidence of a harm in the number of participants who withdrew due to treatment‐emergent side effects for the brofaromine group (5%) compared to the placebo group (2%) (see Analysis 13.2; Baker 1995; Katz 1994).
13.2. Analysis.

Comparison 13: RIMAs versus placebo, Outcome 2: Drop‐out rate due to treatment emergent adverse effects
Secondary outcome measures
There was no evidence of a benefit of a reduction in PTSD symptoms following treatment with brofaromine compared to placebo for CAPS total (MD −5.06 points, 95% CI −15.93 to 5.81, 2 trials, 178 participants; see Analysis 13.3; Baker 1995; Katz 1994).
13.3. Analysis.

Comparison 13: RIMAs versus placebo, Outcome 3: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
There was also no evidence of a difference for the number of participants who withdrew due to any cause from the brofaromine (30%) and placebo groups (29%) (see Analysis 13.4; Katz 1994).
13.4. Analysis.

Comparison 13: RIMAs versus placebo, Outcome 4: Drop‐out rate due to any cause
Comparison 14: SARIs versus placebo
Primary outcome measures
We found no studies that looked at the number of participants who responded to nefazodone versus placebo.
More participants dropped out of the nefazodone group due to treatment‐emergent side effects (19%) compared to placebo group (7%), but no evidence of a harm in dropout rates was observed (Analysis 14.1; Davis 2004).
14.1. Analysis.

Comparison 14: SARIs versus placebo, Outcome 1: Drop‐out rate due to treatment emergent adverse effects
Secondary outcome measures
The review found no evidence of a benefit of nefazodone on total CAPS symptom severity (MD −5.60 points, 95% CI −21.26 to 10.06; 1 trial, 41 participants; see Analysis 14.2; Davis 2004), as well as for the re‐experiencing/intrusion subscale (MD −1.30 points, 95% CI −6.80 to 4.20; 1 trial, 41 participants; see Analysis 14.2; Davis 2004), avoidance/numbing subscale (MD −1.60 points, 95% CI −9.23 to 6.03; 1 trial, 41 participants; see Analysis 14.2; Davis 2004), or hyperarousal subscale (MD −2.90 points, 95% CI −8.40 to 2.60; 1 trial, 41 participants; see Analysis 14.2; Davis 2004) on the CAPS.
14.2. Analysis.

Comparison 14: SARIs versus placebo, Outcome 2: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
More participants dropped out of the nefazodone group due to any cause (32%) compared to the placebo group (13%) (see Analysis 14.3; Davis 2004; Saygin 2002).
14.3. Analysis.

Comparison 14: SARIs versus placebo, Outcome 3: Drop‐out rate due to any cause
Comparison 15: SNRIs versus placebo
Primary outcome measures
We found no studies that looked at the number of participants who responded to venlafaxine versus placebo.
There was no evidence of a harm for the number of participants who withdrew due to treatment‐emergent side effects for the venlafaxine (4%) versus placebo (3%) groups (see Analysis 15.1; Davidson 2006a; Davidson 2006b), based on very low‐certainty evidence. The smaller of the two studies contributed to this. We also noted considerable heterogeneity, however, for this outcome (Chi2 = 12.98, df = 1 (P < 0.001); I2 = 92%).
15.1. Analysis.

Comparison 15: SNRIs versus placebo, Outcome 1: Drop‐out rate due to treatment emergent adverse effects
Secondary outcome measures
There was evidence of a benefit of the SNRI venlafaxine compared to placebo for reducing total PTSD symptoms on the CAPS total (MD −8.11 points, 95% CI −12.29 to −3.92; 2 trials, 687 participants; see Analysis 15.2; Davidson 2006a; Davidson 2006b), as well as for the re‐experiencing/intrusion subscale (MD −1.95 points, 95% CI −3.36 to −0.54; 2 trials, 687 participants; see Analysis 15.2; Davidson 2006a; Davidson 2006b), avoidance/numbing subscale (MD −3.42 points, 95% CI −5.23 to −1.61; 2 trials, 687 participants; see Analysis 15.2; Davidson 2006a; Davidson 2006b), or hyperarousal subscale (MD −2.29 points, 95% CI −3.67 to −0.91; 2 trials, 687 participants; see Analysis 15.2; Davidson 2006a; Davidson 2006b) of the CAPS. We also found evidence of a benefit of venlafaxine compared to placebo for reducing PTSD symptoms on the CGI‐S (SMD −0.31 points, 95% CI −0.46 to −0.16; 2 trials, 687 participants; see Analysis 15.3; Davidson 2006a; Davidson 2006b). Moreover, we found evidence of a benefit of venlafaxine on total DTS symptom severity (SMD −0.26 points, 95% CI −0.47 to −0.05; 1 trial, 358 participants; see Analysis 15.4; Davidson 2006b), as well as for the re‐experiencing/intrusion subscale (MD −0.23 points, 95% CI −0.44 to −0.02; 1 trial, 358 participants; see Analysis 15.4; Davidson 2006b), avoidance/numbing subscale (MD −0.23 points, 95% CI −0.44 to −0.02; 1 trial, 358 participants; see Analysis 15.4; Davidson 2006b), or hyperarousal subscale (MD −0.24 points, 95% CI −0.45 to −0.03; 1 trial, 358 participants; see Analysis 15.4; Davidson 2006b) on the DTS.
15.2. Analysis.

Comparison 15: SNRIs versus placebo, Outcome 2: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
15.3. Analysis.

Comparison 15: SNRIs versus placebo, Outcome 3: Symptom severity: Other measures
15.4. Analysis.

Comparison 15: SNRIs versus placebo, Outcome 4: Self‐rated scales
Furthermore, there was no evidence of a benefit of venlafaxine compared to placebo on the reduction in depression symptoms on the HAM‐D (SMD −0.20 points, 95% CI −0.42 to 0.02; 1 trial, 329 participants; see Analysis 15.5; Davidson 2006a). There was, however, evidence of a benefit of venlafaxine on the reduction in functional disability measured with the SDS (MD −2.01 points, 95% CI −3.30 to −0.72; 2 trials, 687 participants; Analysis 15.6; Davidson 2006a; Davidson 2006b)) .
15.5. Analysis.

Comparison 15: SNRIs versus placebo, Outcome 5: Depression Scale (typically Hamilton Depression)
15.6. Analysis.

Comparison 15: SNRIs versus placebo, Outcome 6: Sheehan Disability Scale
We found no evidence of a difference in the number of participants who withdrew due to any cause (see Analysis 15.7; Davidson 2006a), which is confirmed by the similar dropout rates in both groups (30% versus 33%).
15.7. Analysis.

Comparison 15: SNRIs versus placebo, Outcome 7: Drop‐out rate due to any cause
Comparison 16: SSRIs versus placebo
Primary outcome measures
There was evidence of a benefit of fluoxetine, paroxetine and sertraline compared to placebo in participants with PTSD (RR 0.66, 95% CI 0.59 to 0.74; 8 trials, 1078 participants; see Analysis 16.1; Brady 2000; Connor 1999a; Hertzberg 2000; Marshall 2001; Marshall 2007; Panahi 2011; Tucker 2001; Zohar 2002), based on moderate‐certainty evidence. Fluoxetine, paroxetine and sertraline therefore improved PTSD symptoms. We also noted evidence of a benefit for the number of participants who responded to treatment for the individual SSRI paroxetine (RR 0.64, 95% CI 0.55 to 0.74; 3 trials. 728 participants; see Analysis 16.2; Marshall 2001; Marshall 2007; Tucker 2001), for sertraline (RR 0.68, 95% CI 0.56 to 0.81; 3 trials, 285 participants; see Analysis 16.2; Brady 2000; Panahi 2011; Zohar 2002), but not for fluoxetine (RR 0.73, 95% CI 0.19 to 2.82; 2 trials, 65 participants; see Analysis 16.2; Connor 1999a; Hertzberg 2000). The failure to detect a treatment effect for fluoxetine may reflect the small samples, and hence the low power of this comparison.
16.1. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 1: Clinical Global Impressions Scale Improvement Item (CGI‐I)
16.2. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 2: CGI: Individual SSRI agents
The proportion of dropouts due to adverse events was higher amongst participants receiving the SSRIs fluoxetine, paroxetine and sertraline (9%) compared to placebo (7%) (see Analysis 16.3; Brady 2000; Connor 1999a; Davidson 2001a; Davidson 2006b; GSK 29060 627; Hertzberg 2000; Li 2017; Marshall 2001; Marshall 2007; Martenyi 2002a; NCT01681849; Panahi 2011; Tucker 2001; Zohar 2002), based on moderate‐certainty evidence. In addition, a difference in treatment‐emergent side effects was evident for the separate SSRI paroxetine (RR 1.55, 95% CI 1.05 to 2.29; 5 trials, 1101 participants; see Analysis 16.4; GSK 29060 627; Marshall 2001; Marshall 2007; NCT01681849; Tucker 2001), but not for the SSRI fluoxetine (RR 0.86, 95% CI 0.25 to 2.96; 3 trials, 367 participants; see Analysis 16.4; Connor 1999a; Hertzberg 2000; Martenyi 2002a) or for the SSRI sertraline (RR 1.34; 95% CI 0.87 to 2.05; 6 trials, 931 participants; see Analysis 16.4; Brady 2000; Davidson 2001a; Davidson 2006b; Li 2017; Panahi 2011; Zohar 2002).
16.3. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 3: Drop‐out rate due to treatment emergent adverse effects
16.4. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 4: Drop‐out rate due to treatment emergent adverse effects: Individual SSRI agents
Secondary outcomes measures
There was evidence of a benefit of the SSRIs compared to placebo for reducing total PTSD symptoms on the CAPS (MD −4.91 points, 95% CI −7.48 to −2.34, 15 trials, 2615 participants; see Analysis 16.5; Brady 2000; Davidson 2001a; Davidson 2006b; GSK 29060 627; Marshall 2001; Marshall 2007; NCT01681849; Pfizer588; Pfizer589; SKB627; SKB650; Tucker 2001; Tucker 2003; Van der Kolk 2007; Zohar 2002). However, there was considerable heterogeneity in effect size estimates for this outcome (Chi2 = 29.51, df = 14 (P = 0.009); I2 = 53%). The SSRIs were also superior to placebo on both the re‐experiencing/intrusion subscale (MD −1.86 points, 95% CI −2.82 to −0.90; see Analysis 16.5), avoidance/numbing subscale (MD −3.68 points, 95% CI −4.82 to −2.54; see Analysis 16.5), and hyperarousal subscale (MD −2.45 points, 95% CI −3.30 to −1.59; see Analysis 16.5) on the CAPS in 8 trials with 1813 participants (Brady 2000; Davidson 2001a; Davidson 2006b; GSK 29060 627; Marshall 2001; Marshall 2007; Tucker 2001; Tucker 2003).
16.5. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 5: Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms
The pattern of symptom severity on the CAPS for the separate SSRI medications was similar to that observed for treatment efficacy, with both paroxetine MD −6.32 points, 95% CI −11.49 to −1.14; 6 trials, 1375 participants; see Analysis 16.6; GSK 29060 627; Marshall 2001; Marshall 2007; SKB627; SKB650; Tucker 2001) and sertraline (MD −4.45 points, 95% CI −7.17 to −1.73; 6 trials, 1133 participants; see Analysis 16.6; Brady 2000; Davidson 2001a; Davidson 2006b; Pfizer588; Pfizer589; Zohar 2002), demonstrating evidence of a benefit of a reduction in PTSD symptoms. We also noted considerable heterogeneity for the individual SSRI paroxetine (Chi2 = 21.87, df = 5 (P < 0.001); I2 = 77%). There was once again insufficient evidence to determine whether fluoxetine was effective in reducing total PTSD (MD −0.90 points, 95% CI −12.31 to 10.51; 1 trial, 59 participants; see Analysis 16.6; Van der Kolk 2007), as well as citalopram (MD 4.78 points, 95% CI −15.95 to 25.51; 1 trial, 35 participants; see Analysis 16.6; Tucker 2003), compared to placebo.
16.6. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 6: CAPS: Individual SSRI agents
There was evidence of a benefit of the SSRIs fluoxetine, paroxetine and sertraline compared to placebo for the reduction of PTSD symptoms on the CGI‐S (SMD −0.32 points, 95% CI −0.55 to −0.09; 6 trials, 862 participants; see Analysis 16.7; Connor 1999a; Davidson 2006b; GSK 29060 627; Hertzberg 2000; Li 2017; Panahi 2011). There was considerable heterogeneity in effect size estimates for this outcome (Chi2 = 10.24, df = 5 (P = 0.07); I2 = 51%). There was no evidence of a benefit of the SSRIs compared to placebo for a reduction in total PTSD symptoms on the DTS (SMD 0.11, 95% CI −0.50 to 0.72; 10 trials, 1698 participants; see Analysis 16.8; Brady 2000; Connor 1999a; Davidson 2005; Davidson 2006b; GSK 29060 627; Hertzberg 2000; Li 2017; Panahi 2011; SKB650; Tucker 2003), and this is confirmed by the substantial heterogeneity found between trials (Chi2 = 295.12, df = 9 (P < 0.001); I2 = 97%). We did however observe evidence of a benefit of four trials of sertraline for the re‐experiencing/intrusion subscale (MD −0.30 points, 95% CI −0.58 to −0.03; 5 trials, 986 participants; see Analysis 16.8; Brady 2000; Davidson 2006b; GSK 29060 627; Li 2017; Panahi 2011), and avoidance/numbing subscale (MD −0.31 points, 95% CI −0.58 to −0.04; 5 trials, 985 participants; see Analysis 16.8; Brady 2000; Davidson 2006b; GSK 29060 627; Li 2017; Panahi 2011), but not for the hyperarousal subscale (MD −0.48 points, 95% CI −0.98 to 0.01; 4 trials, 805 participants; see Analysis 16.8; Davidson 2006b; GSK 29060 627; Li 2017; Panahi 2011) on the DTS. Substantial heterogeneity was also found for each of the subscales (I2 = 75%, I2 = 74%, I2 = 90% respectively).
16.7. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 7: Symptom severity: Other measures
16.8. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 8: Self‐rated scales
There was no evidence of a benefit of citalopram, paroxetine or sertraline compared to placebo on the reduction in depression symptoms on the BDI‐R and HAM‐D (SMD −0.20 points, 95% CI −0.44 to 0.04; 3 trials, 270 participants; see Analysis 16.9; Brady 2000; Marshall 2007; Tucker 2003). Similarly, there was no evidence of a benefit of paroxetine or sertraline compared to placebo on the reduction in anxiety symptoms on the HAM‐A (MD −0.22 points, 95% CI −1.84 to 1.41; 3 trials, 606 participants; see Analysis 16.10; Davidson 2001a; Davidson 2006b; Marshall 2007). In contrast, we found evidence of a benefit of fluoxetine, paroxetine and sertraline compared to placebo on the reduction in functional disability measured by the SDS (MD −2.10 points, 95% CI −3.00 to −1.21; 6 trials, 1336 participants; see Analysis 16.11; Connor 1999a; Davidson 2006b; GSK 29060 627; Hertzberg 2000; Marshall 2001; Tucker 2001).
16.9. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 9: Depression Scale (typically Hamilton Depression)
16.10. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 10: Anxiety ‐ Hamilton Anxiety Scale
16.11. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 11: Sheehan Disability Scale
We observed similar dropout rates in the medication (33%) and placebo arms (28%); see Analysis 16.12. We also observed substantial heterogeneity in effect size estimates for this outcome (Chi2 = 74.38, df = 20 (P < 0.001); I2 = 73%).
16.12. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 12: Drop‐out rate due to any cause
Finally, there was no evidence of a benefit of the SSRI fluoxetine compared to placebo for a reduction in PTSD symptoms on the CAPS after long‐term treatment (MD 9.62 points; 95% CI −3.53 to 22.77; 1 trial, 16 participants; see Analysis 16.13; Marshall 2007).
16.13. Analysis.

Comparison 16: SSRIs versus placebo, Outcome 13: Continuation trials
Comparison 17: TCAs versus placebo
Primary outcome measures
There was evidence of a benefit of amitriptyline on treatment efficacy in participants with PTSD compared to placebo (RR 0.60, 95% CI 0.38 to 0.96; 1 trial, 40 participants; see Analysis 17.1; Davidson 1990) based on low‐certainty evidence.
17.1. Analysis.

Comparison 17: TCAs versus placebo, Outcome 1: Clinical Global Impressions Scale Improvement Item (CGI‐I)
There was no evidence of a harm for the number of participants who withdrew due to treatment‐emergent side effects in the amitriptyline, desipramine and imipramine groups (23%) compared to the placebo groups (18%) (see Analysis 17.2; Davidson 1990; Kosten 1991; Reist 1989), based on moderate‐certainty evidence.
17.2. Analysis.

Comparison 17: TCAs versus placebo, Outcome 2: Drop‐out rate due to treatment emergent adverse effects
Secondary outcomes measures
We found no evidence of a benefit for amitriptyline and imipramine compared to placebo for reducing total IES symptom severity (SMD −0.54 points, 95% CI −1.18 to 0.10; 2 trials, 74 participants; see Analysis 17.3; Davidson 1990; Kosten 1991), as well as for the re‐experiencing/intrusion subscale (SMD −0.44 points, 95% CI −0.99 to 0.11; 2 trials, 74 participants; see Analysis 17.3; Davidson 1990; Kosten 1991) and avoidance/numbing subscale (SMD −0.23 points, 95% CI −1.51 to 1.04; 2 trials, 74 participants; see Analysis 17.3; Davidson 1990; Kosten 1991) on the IES. We noted moderate heterogeneity for the total IES symptom severity outcome (Chi2 = 1.83, df = 1 (P = 0.18); I2 = 45%) and substantial heterogeneity for the avoidance/numbing subscale of the IES (Chi2 = 7.16, df = 1 (P = 0.007); I2 = 86%).
17.3. Analysis.

Comparison 17: TCAs versus placebo, Outcome 3: Self‐rated scales
There was no evidence of a benefit of amitriptyline or imipramine compared to placebo on the reduction in depression symptoms on the HAM‐D (SMD −0.67 points, 95% CI −1.58 to 0.25; 2 trials, 74 participants (Davidson 1990; Kosten 1991) see Analysis 17.4) . There was also substantial heterogeneity for this outcome (Chi2 = 3.60, df = 1 (P = 0.06); I2 = 72%). We did, however, observe evidence of a benefit of amitriptyline on a reduction of anxiety symptoms on the HAM‐A (MD −7.60 points, 95% CI −12.74 to −2.46; 1 trial, 33 participants; see Analysis 17.5; Davidson 1990). We found no evidence of a benefit for the TCA imipramine on a reduction in anxiety symptoms on the Covi Anxiety Scale (SMD −0.46 points, 95% CI −1.08 to 0.17; 1 trial, 41 participants; see Analysis 17.6; Kosten 1991).
17.4. Analysis.

Comparison 17: TCAs versus placebo, Outcome 4: Depression Scale (typically Hamilton Depression)
17.5. Analysis.

Comparison 17: TCAs versus placebo, Outcome 5: Anxiety ‐ Hamilton Anxiety Scale
17.6. Analysis.

Comparison 17: TCAs versus placebo, Outcome 6: Anxiety ‐ Other scales
In addition, more participants dropped out of the amitriptyline and imipramine groups (34%) due to any cause (see Analysis 17.7; Davidson 1990; Kosten 1991).
17.7. Analysis.

Comparison 17: TCAs versus placebo, Outcome 7: Drop‐out rate due to any cause
Comparison 18: Total effect of medication versus placebo
There was evidence of a benefit across all medication classes (RR 0.74, 95% CI 0.64 to 0.85; 19 trials, 1822 participants; see Analysis 18.1). There was also evidence that medication class explained a substantial proportion of variability in treatment efficacy when assessed across all trials providing data on this outcome (Chi2 = 19.59, df = 6 (P = 0.003); I2 = 69.4%; see Analysis 18.1).
18.1. Analysis.

Comparison 18: Total effect of medication versus placebo, Outcome 1: Clinical Global Impressions ‐ Improvement (CGI‐I) or similar) scale: no. of responders
We found no evidence of a harm for the proportion of dropouts due to adverse events across all medication classes compared to placebo (RR 0.98; 95% CI 0.91 to 1.08; participants; 37 trials, 4212 participants; see Analysis 18.2); which is confirmed by the low dropout rate in the medication (10%) and placebo arm (7%). For this analysis, we excluded data from one study reporting on sertraline, because this study also reported data for the SNRI venlafaxine (Davidson 2006b).
18.2. Analysis.

Comparison 18: Total effect of medication versus placebo, Outcome 2: Drop‐out rate due to treatment emergent adverse effects
We noted evidence of reductions in symptom severity for participants who received medication in 34 short‐term trials from which it was possible to retrieve data for this outcome (MD ‐4.79, 95% CI −6.64 to −2.94; 34 trials, 4639 participants; see Analysis 18.3). For this analysis, data from one study reporting on sertraline were excluded, because this study also reported data for the SNRI venlafaxine (Davidson 2006b).
18.3. Analysis.

Comparison 18: Total effect of medication versus placebo, Outcome 3: Clinically Administered PTSD Scale (CAPS): CAPS Total
The proportion of dropouts due to any cause across both the medication and placebo groups was similar (32% and 29%, respectively) and was comparable across subgroups (Chi2 = 23.93, df = 14 (P = 0.05), I2 = 41.5%; see Analysis 18.4). For this analysis data from one study were excluded, with an additional arm of phenelzine to give preference to imipramine (Kosten 1991).
18.4. Analysis.

Comparison 18: Total effect of medication versus placebo, Outcome 4: Drop‐out rate due to any cause
Comparison 19: Head‐to‐head comparisons
More participants dropped out of the reboxetine group (45%) compared to the fluvoxamine group (15%) due to treatment‐emergent side effects (see Analysis 19.1; Spivak 2006).
19.1. Analysis.

Comparison 19: Head‐to‐head comparisons, Outcome 1: Drop‐out rate due to treatment emergent adverse effects
It is unclear if there was evidence of a benefit of the reduction of symptom severity in the two head‐to‐head comparisons of nefazodone and sertraline (SMD −0.19 points, 95% CI −0.63 to 0.25; 2 trials, 80 participants; see Analysis 19.2; McRae 2004; Saygin 2002), nor in the single‐trial comparison of venlafaxine and sertraline (SMD −0.01 points, 95% CI −0.22 to 0.20; 1 trial, 352 participants; see Analysis 19.2; Davidson 2006b), and the single trial comparing reboxetine and fluvoxamine (SMD 0.67 points, 95% CI −0.11 to 1.45; 1 trial, 28 participants; see Analysis 19.2; Spivak 2006).
19.2. Analysis.

Comparison 19: Head‐to‐head comparisons, Outcome 2: Clinician administered scales: Symptom severity
There was no evidence of a benefit of nefazodone compared to sertraline for the reduction of depression symptoms on the MADRS (SMD −0.09 points, 95% CI −0.86 to 0.68; 1 trial, 26 participants; see Analysis 19.3; McRae 2004) or of reboxetine and fluvoxamine on the HAM‐D (SMD 0.67 points, 95% CI −0.11 to 1.45; 1 trial, 28 participants; see Analysis 19.3; Spivak 2006). We also found no evidence of a benefit of nefazodone compared to sertraline on the reduction in anxiety symptoms on the HAM‐A (MD −3.23 points, 95% CI −10.90 to 4.44, 1 trial, 26 participants; see Analysis 19.4; McRae 2004), or for reboxetine and fluvoxamine on the HAM‐A (MD 1.80 points, 95% CI −1.30 to 4.90; 1 trial, 28 participants; see Analysis 19.4; Spivak 2006).
19.3. Analysis.

Comparison 19: Head‐to‐head comparisons, Outcome 3: Comorbid symptoms: Depression
19.4. Analysis.

Comparison 19: Head‐to‐head comparisons, Outcome 4: Comorbid symptoms: Anxiety (Hamilton Anxiety Scale)
Comparison 20: Subgroup analyses ‐ Methodological criteria
We found evidence of a benefit in the analysis of treatment response for single‐centre trials and for multi‐centre trials (Chi2 = 0.77, df = 1 (P = 0.38); I2 = 0%; see Analysis 20.1). Symptom severity in the 22 trials which took place across multiple centres for which the CAPS total score was available was reduced to a greater extent than in the eight trials conducted within single centres (Chi2 = 1.24, df = 1 (P = 0.27); I2 = 19.1%; see Analysis 20.2).
20.1. Analysis.

Comparison 20: Subgroup analyses ‐ Methodological criteria, Outcome 1: Single versus multi‐centre trials (CGI‐I: No. of responders)
20.2. Analysis.

Comparison 20: Subgroup analyses ‐ Methodological criteria, Outcome 2: Single versus multi‐centre trials (CAPS: Total score)
There was evidence of a benefit in treatment response based on source of funding and to a lesser extent for non‐industry‐funded trials (Chi2 = 0.42, df = 1 (P = 0.52); I2 = 0%; see Analysis 20.3) on the CGI‐I. This was also observed in the analysis of the reduction of PTSD symptoms for industry‐funded trials versus non‐industry‐funded trials (Chi2 = 3.15, df = 1 (P = 0.08), I2 = 68.2%; see Analysis 20.4).
20.3. Analysis.

Comparison 20: Subgroup analyses ‐ Methodological criteria, Outcome 3: Industry versus non‐industry funded trials (CGI‐I: No. of responders)
20.4. Analysis.

Comparison 20: Subgroup analyses ‐ Methodological criteria, Outcome 4: Industry versus non‐industry funded trials (CAPS: Total score)
Comparison 21: Subgroup analyses ‐ Clinical criteria
There was evidence of a benefit of treatment response for trials which included depressed participants compared to those trials which did not (Chi2 = 2.57, df = 1 (P = 0.11), I2 = 61.1%; see Analysis 21.1). For the outcome of symptom severity, there was evidence of a benefit of the reduction of PTSD symptoms in trials which included depressed participants, as in those trials which did not (Chi2 = 0.94, df = 1 (P = 0.33), I2 = 0%; see Analysis 21.2).
21.1. Analysis.

Comparison 21: Subgroup analyses ‐ Clinical criteria, Outcome 1: Inclusion of major depression vs. non‐inclusion: (CGI‐I: No. of responders)
21.2. Analysis.

Comparison 21: Subgroup analyses ‐ Clinical criteria, Outcome 2: Inclusion of major depression vs. non‐inclusion: (CAPS: Total score)
We found evidence of a benefit between groups for trials including war trauma compared to trials without war trauma (Chi2 = 0.06, df = 1 (P = 0.81); I2 = 0%; see Analysis 21.3). RCTs which contained participants without war trauma demonstrated evidence of a reduction in symptom severity following medication treatment compared with trials containing participants with war trauma exposure (Chi2 = 4.21, df = 1 (P = 0.04), I2 = 76.3%; see Analysis 21.4). The finding of a beneficial effect in the reduction of symptom severity between trials with few war veterans versus those with many was not surprising, given the general characterisation of the war trauma subgroup of PTSD sufferers as more treatment‐resistant than other subgroups.
21.3. Analysis.

Comparison 21: Subgroup analyses ‐ Clinical criteria, Outcome 3: Inclusion of war veterans versus non‐inclusion: (CGI‐I: No. of responders)
21.4. Analysis.

Comparison 21: Subgroup analyses ‐ Clinical criteria, Outcome 4: Inclusion of war veterans versus non‐inclusion: (CAPS: Total score)
Comparison 22: Sensitivity analyses
The comparison of the analysis of treatment efficacy for non‐response as opposed to response on the CGI‐I (or equivalent) revealed similar outcomes for both the overall short‐term efficacy of medication (RR 0.72; 95% CI 0.62 to 0.83; 20 trials, 1847 participants; see Analysis 22.1), as well as for the efficacy of the NaSSA mirtazapine (RR 0.45; 95% CI 0.22 to 0.94; 1 trial, 26 participants; see Analysis 22.1), the SSRIs (RR 0.66; 95% CI 0.59 to 0.74; 8 trials, 1078 participants; see Analysis 22.1), and the TCA amitriptyline in treating PTSD (RR 0.60, 95% CI 0.38 to 0.96; 1 trial, 40 participants; see Analysis 22.1). However, the use of treatment non‐response as a summary statistic indicates that the anticonvulsants, antipsychotics, experimental medications and MAOIs were no more effective than placebo.
22.1. Analysis.

Comparison 22: Sensitivity analyses, Outcome 1: Clinical Global Impressions Scale Improvement Item (CGI‐I): non‐response
The number of participants responding to medication was significantly higher compared to placebo in both the worst‐case scenario (RR 0.83, 95% CI 0.69 to 1.00; 13 trials, 1555 participants; see Analysis 22.2), where those participants from the medication group who were not included in the analysis were regarded as non‐responders, and in the best‐case scenario (RR 1.41, 95% CI 1.12 to 1.78; 13 trials, 1498 participants; see Analysis 22.3), in which those participants excluded from the placebo control were regarded as non‐responders. The overlap in the confidence intervals for these two outcomes indicates that loss to follow‐up is unlikely to have influenced assumptions about the overall efficacy of medication.
22.2. Analysis.

Comparison 22: Sensitivity analyses, Outcome 2: "Worst case" loss to follow up analysis
22.3. Analysis.

Comparison 22: Sensitivity analyses, Outcome 3: "Best case" loss to follow up analysis
Comparison 23: Publication bias
The distribution of trials on a funnel plot for treatment response (see Figure 4) and symptom severity (see Figure 5) demonstrate no evidence for substantial publication bias.
4.

Funnel plot of comparison: 13 Comparison 13: SSRIs versus placebo, outcome: 13.1 Clinical Global Impressions scale improvement item (CGI‐I). Note: The left side favours the SSRI arm and the right side favours the placebo arm.
5.

Funnel plot of comparison: 15 Comparison 13: SSRIs versus placebo, outcome: 15.5 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms. Note: The left side favours the SSRI arm and the right side favours the placebo arm.
Discussion
Summary of main results
For the primary outcome measure of PTSD response to treatment, this review found evidence of beneficial effect for the SSRIs (i.e. citalopram, fluoxetine, paroxetine, and sertraline), based on moderate‐certainty evidence. We also found evidence of beneficial effect for the NaSSA mirtazapine and the TCA amitriptyline. The remaining medication classes (i.e. anticonvulsant, antipsychotic, GR205171, GSK561679, and RIMA groups), did not, however, provide evidence of a benefit.
Individuals diagnosed with PTSD who were treated with individual SSRI agents, and in particular the SSRI paroxetine, were more likely to withdraw from treatment due to treatment‐related adverse events. Nevertheless, the absolute proportion of individuals dropping out from treatment due to treatment‐related adverse events was low (9%), with the difference in dropouts from the control group of two percentage points unlikely to be clinically significant, based on moderate‐certainty evidence.
For the secondary outcome measure of PTSD symptom reduction on the CAPS there was also evidence of a benefit of efficacy for the SSRIs, the antipsychotics, and the SNRI venlafaxine. Medication was significantly more effective than placebo across the re‐experiencing/intrusion, avoidance/numbing, and hyperarousal cluster assessed by the CAPS for the SSRIs and the SNRIs. We also found evidence of a benefit for reducing PTSD symptoms on the CGI‐S for the SSRIs, the SNRI venlafaxine and for the antipsychotics. This was also evidence for the SSRIs, the SNRI venlafaxine, and the MAOIs on the self‐rated IES or the DTS. Direct comparisons of sertraline and nefazodone, of venlafaxine and sertraline, and of reboxetine and fluvoxamine indicated that these medications were not effective in reducing PTSD symptom severity. Moreover, the reduction of PTSD was not observed in the long‐term trials included in the review.
We found evidence of efficacy for reducing depression symptoms only observed for the antipsychotic group. For the reduction of anxiety symptoms, evidence of a benefit was observed for the MAOI, NaSSA and TCA groups. In addition, the SSRIs and the SNRI venlafaxine were the only medications showing a treatment effect on the SDS.
The review also found evidence of a difference of dropouts due to any cause for the SARI nefazodone and the TCAs, although dropout rates were low (24%, 28%, respectively). Subgroup analysis of trials where depression was and was not an inclusion criterion found differences in efficacy (i.e. for the number of participants who responded to treatment). We also found evidence of a benefit in response or reduction or both for single versus multi‐site studies and for industry‐funded versus non‐industry‐funded studies. We also found that war veterans are more resistant to pharmacotherapy than other patient groups, in terms of treatment response and reduction of symptom severity.
When studies were combined, evidence of a benefit was found on the CGI‐I and CAPS across all medications. Further, no evidence of a harm was found across studies that reported this, and dropouts due to any cause were similar across the medication classes.Also, no publication bias was observed for the SSRI group versus the placebo group. We could not assess the remaining medication classes due to lack of sufficient trials.
Overall completeness and applicability of evidence
Completeness of evidence
Although it has been suggested that the SSRIs are more effective than older antidepressants (Dow 1997; Penava 1996), class membership did not contribute significantly to the variation observed in symptom severity outcomes between trials, and the confidence intervals for the summary statistic of responder status on the SSRI trials overlapped with the NaSSA mirtazapine and the TCA amitriptyline, probably reflecting insufficient power for this comparison to reach statistical significance. Similarly, direct comparisons of sertraline and nefazodone, of venlafaxine and sertraline, and of reboxetine and fluvoxamine, did not demonstrate differences between these treatments in reducing PTSD symptoms. Although the SSRIs have been argued and have shown superior tolerability in comparison to older medication classes in this review, it is important to be aware of the need for careful monitoring after initiation of individual SSRI agents (CSM 2005). Nevertheless, the SSRI trials constitute the bulk of the evidence for the efficacy of medication in treating PTSD, both in the number of studies and their size. It is therefore reasonable to support the expert consensus (Ballenger 2000; Ballenger 2004; Foa 1999; NICE 2018) that SSRIs constitute the first‐line medication choice for PTSD.
In addition, industry‐funded trials found a larger reduction in CAPS total score than in non‐industry‐funded trials, which could be a potential bias because published trials sponsored by pharmaceutical companies appear more likely to report positive findings than trials that are not supported by for‐profit companies (Als‐Nielsen 2003; Baker 2003).
Applicability of evidence
Studies included men and women across a wide age range. Furthermore, the existing evidence base of RCTs includes a heterogeneous sample of participants with a range of different traumas, trauma duration and severity, and comorbidity. Nevertheless, people with PTSD who are recruited to clinical trials may not be representative of those with PTSD in the community (Franco 2016). Given the high prevalence and enormous personal and societal costs of PTSD, there are still relatively few RCTs of pharmacotherapy for PTSD, with some notable gaps. With a few exceptions (Baker 1995; Braun 1990; Davidson 1990; Hertzberg 1999; Hertzberg 2000; Reist 1989), trials have excluded people with comorbid substance use. Although outside the scope of the current review, there is also a need for additional work on the pharmacotherapy for the prevention of PTSD (Amos 2014).
Quality of the evidence
Sixteen RCTs had a high risk of bias related to at least one aspect of study design, with the most commonly‐observed weakness relating to attrition bias (Butterfield 2001; Chung 2004; Connor 2006; Davis 2008; Feder 2014; Hertzberg 1999; Kosten 1991; Martenyi 2002b; NCT01681849; Reich 2004; Saygin 2002; Smajkic 2001; Spivak 2006; Van der Kolk 1994; Villarreal 2016). Lack of blinding for outcome assessors may also have influenced the detection of treatment effects in three trials (Chung 2004; Saygin 2002; Smajkic 2001). There was also evidence for selective reporting bias in Davis 2008, Feder 2014 and Villarreal 2016. Even though the SSRIs were the best‐studied medication class, we rated most of these trials as unclear for randomisation procedures.
Judgements of the primary outcome measure of response to treatment with the SSRIs were based on evidence rated as being of moderate certainty. Additional research may therefore impact effect size and confidence intervals (see Table 6). Findings of treatment efficacy for the NaSSA mirtazapine and for the TCA amitriptyline were based on evidence rated as being of low‐certainty (see Table 4; Table 7), indicating that data from additional studies may also change the effect size and confidence intervals for these medication classes. The most common reasons for downgrading the quality of studies were imprecise effect estimates, low response rates, small sample size, a rating of high or unclear bias for study design (based on 50% attrition rates across groups, detection, and selective reporting bias), and heterogeneity between the medication and placebo groups.
Although a recent guideline noted that, with few exceptions, the overall effect size for medication trials for PTSD failed to exceed the limit of 0.5 defined as indicative of clinical efficacy (NICE 2018), we would caution that there is no direct translation between the effect size statistic and assessments of clinical efficacy. The findings here of efficacy for the primary outcome measure of CGI‐I response rate support the clinical consensus that medication does have an important role in the treatment of PTSD.
Trials tested in this review where depression was or was not an inclusion criterion were able to account for the substantial proportion of participants who do not appear to respond to medication. The finding that symptom severity is reduced to a greater extent in multi‐centre than in the single‐centre trials should be interpreted with caution, due to the marginal significance of this finding. The finding of an association between the presence of participants with comorbid major depression and treatment efficacy suggests that medications are likely to exert their effects on PTSD via a reduction in depressive symptoms. This review also found evidence that war veterans are more resistant to pharmacotherapy than other patient groups, for response and the reduction of symptom severity. However, several RCTs with war veteran samples were excluded from this review (Bartzokis 2005; Hamner 1997; Hamner 2003; Monnelly 2003; Raskind 2003; Stein 2002) (see Characteristics of excluded studies), and it was not possible to classify certain large‐scale unpublished trials according to trauma type (SKB627; SKB650). Given the heterogeneous phenomenology of PTSD, it remains crucial to determine the factors which do predict response to medication, and to delineate whether certain medications are more effective for particular symptom sets (including symptoms such as psychosis (Hamner 1996), dissociation (Fichtner 1990; Marshall 1998b) and vulnerability to stress (Connor 1999b)). Future RCTs and meta‐analyses should attempt to address such questions in greater detail.
Potential biases in the review process
We conducted an extensive search for studies meeting rigorous inclusion criteria and made repeated efforts to obtain missing data from the trial investigators. Nevertheless, there were insufficient data available to assess the extent to which selective publication may have introduced bias into the findings for medication classes other than the SSRIs, where no publication bias was observed (see Figure 4; Figure 5).
Furthermore, the inherent problems of meta‐analysis should be borne in mind (Bailar 1997); certainly, these are by no means a substitute for primary clinical research. This is especially the case when there is evidence that trials with negative outcomes are not being published. Also, the context of clinical practice differs from that of controlled trials in many respects, not least being the inclusion of more complex participants (including those with possible 'secondary gain' from symptoms, a group that has been specifically excluded from some RCTs) (Brady 2000).
Agreements and disagreements with other studies or reviews
The findings of this review are consistent with findings from other systematic reviews and meta‐analyses in identifying the SSRIs as first‐line agents for the treatment of PTSD (Hoskins 2015; Ipser 2012). This review classified medications based on their putative mechanisms of action (taken from CCMD antidepressant classification map) (Davies 2015), and these do not necessarily map onto the drug classification schemes used in other reviews.
Authors' conclusions
Implications for practice.
Medication treatments can be effective in PTSD, acting to increase treatment response and to reduce symptoms, and should be considered as part of the treatment of this disorder. The existing evidence base of RCTs includes a heterogeneous sample of participants with a range of different traumas, trauma duration and severity, and comorbidity. The greatest number of trials showing efficacy so far, as well as the largest, have been with the SSRIs.
Implications for research.
Given the prevalence and costs of PTSD, there is a need for improved study design and controlled clinical trials for the treatment of this disorder. The varying efficacy and tolerability of different classes of medication, including a range of newer agents potentially useful in this disorder, requires further study. Questions for future research also include the precise effects of medication on quality‐of‐life measures, appropriate dose and duration of medication, and determining factors which predict response to medication. Further research on the value of medication in PTSD in different trauma groups, in paediatric and geriatric recipients, in patients with comorbid substance use, and in treatment of refractory patients is needed. Clinical trials to determine the possible benefits of early (prophylactic), combined (with psychotherapy), and long‐term (maintenance) interventions in PTSD may also be valuable.
Feedback
Comments submitted by Danielle Stacey, 8 December 2015
Summary
In 2009, Stein DJ et al. published a Cochrane review which provided us with a very thorough explanation of the literature surrounding pharmacotherapy for post‐traumatic stress disorder (PTSD).1 After a thorough review of the publication, we have a few enquiries regarding the patient population represented in the meta‐analysis and the statistical analysis conducted. In addition, we have provided some potential recommendations for future updates.
Clinical heterogeneity
Within a systematic review we test for statistical heterogeneity, as did Stein DJ et al, however we often over look clinical heterogeneity or diversity.2 We agree with the authors’ conclusions that “the existing evidence base of RCTs includes a heterogeneous sample of participants with a range of different traumas, trauma duration and severity, and comorbidity.” As an example we can look at analysis 1.1 which includes the following patient populations: Brady 2004, patients suffering from alcoholism; Brady 2000 and Davidson 2001, patients with anxiety and MDD; Pfizer 589, predominantly war veterans; and some trials that have a combination of these various characteristics. The significantly greater reduction in symptom severity in trials with less veteran participants highlights how important clinical diversity can be when interpreting results and when analyzing comparisons for treatment within a meta‐analysis. Given this, one could argue whether meta‐analysis of trials with high clinical variability is even appropriate. At the very least it warrants a section to inform clinicians which patient populations are encompassed in each analysis. We also suggest completing an additional subgroup analysis for trials including participants with concurrent anxiety disorders. After a random selection of three trials (Brady 2000, Davidson 2001, Marshall 2004) from analysis 1.1, we found that all three included participants with concurrent anxiety disorders at various rates. Together, these three trials comprise a weight of 16.2% of the random effects model. This additional subgroup analysis will aid in further defining the treatment populations within the review and the potential impact on the pooled efficacy results.
Inclusion of studies in meta‐analysis 1.3
Focusing again on the statistical analysis, at first glance, analysis 1.3 for clinical global impression scale improvement item (CGI‐I) seemed to include a low number of RCTs for SSRIs. After scanning the included trial characteristics tables, we noticed 12 trials that had CGI‐I (or similar scale) as a primary or secondary outcome (Davidson, Davidson 2001, Brady 2000, Pfizer588, Pfizer589, Zohar 2002, Marshall 2004, Tucker 2001, Marshall 2001, Herzberg 2000, Eli Lilly, and Connor 1999). Analysis 1.3 included only 7 trials in the forest plot (Brady 2000, Connor 1999, Hertzberg 2000, Marshall 2001, Marshall 2004, Tucker 2001, and Zohar 2002). It was mentioned in the description of studies section that Davidson would not be used for statistical analysis, however this does not explain why the remainder of the trials were not included. In addition, a similar statement claimed that data from Tucker 2003 would be excluded from statistical analysis, however this trial was included in other forest plots (eg. Analysis 1.1). A brief explanation to help understand the reasoning for inclusion of trials into each analysis would help in interpreting the results.
References
As previously mentioned, we took the time to randomly pull some of the trials that were cited in the review. Our first choice was Davidson 2001. From the included studies citations, we chose the second of four trials under Davidson 2001 given the lead author and year of publication.3 It was very apparent that this trial was in fact a maintenance trial, and answering a very different question than the review. This particular Davidson publication cited another Davidson publication, which was in fact the acute treatment trial. Upon further inspection, data from the original trial was included in the analysis, which is great. However, it is confusing for readers because wherever Davidson 2001 is cited in the review, may lead them to look at the maintenance trial and not the main trial of interest. We suggest, for trials that have the same lead author and publication year (eg. Davidson 2001), adding a distinguishing mark for each will help readers.
Additional points
The combination of clinical variability, unclear trial methodology and very thorough inclusion of information can lead to confusion and misinterpretation without a clear, concise and easy to analyze presentation. To make it easier for readers to gain a quick and accurate interpretation of the included data and results, we suggest the following changes:
1. Include a risk of bias graph or a risk of bias summary figure for the included trials.2 It is apparent that a bias assessment was completed, however each risk of bias section did not include all pertinent design factors to make an accurate assessment. Furthermore, placing these factors in the characteristic tables does not help viewers get an overall idea of the included trial quality, nor is it efficient. In addition, although it seems Cochrane is not a full supporter of quality scales, this information could also be displayed with or immediately after the risk of bias tool(s).
2. Conduct a subgroup analysis for trials including patients with concurrent anxiety disorders as previously suggested in an effort to address clinical heterogeneity.
3. Include reasoning for including or excluding trials in each analysis where applicable, as previously suggested.
4. For different trials that have the same lead author and publication year (eg. Davidson 2001), add a distinguishing mark for each.
5. On page 14, the following statement contains an error, “the conclusion that a short‐term course of treatment with SSRIs may be inadequate is supported by increased relapse rates in trials of both fluoxetine (Davidson 2001) and sertraline (Martenyi 2002).” Davidson 2001 and Martenyi 2002 should be switched.
References:
1. Stein DJ, Ipser JC, Seedat S. Pharmacotherapy for post traumatic stress disorder (PTSD). Cochrane Database of Systematic Reviews 2006, Issue 1. Art. No.: CD002795. DOI: 10.1002/14651858.CD002795.pub2
2. Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org
3. Davidson J, Pearlstein T, Londborg P, Brady KT, Rothbaum B, Bell J, et al.Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: results of a 28‐week double‐blind, placebo‐ controlled study. American Journal of Psychiatry 2001;158(12): 1974–81.
Reply
We would like to thank the reviewers for their close reading of our Cochrane review ‘Pharmacotherapy for posttraumatic stress disorder’. We would note that there have been considerable advances in methodological and reporting standards since this review was published, a decade ago in 2006, and that adherence to these standards in the imminent update of the review should help to address many of the reviewer’s concerns. Specifically, new Cochrane standards require a more thorough description of important aspects of studies in the description of included studies section, under the following subheadings – design, participant characteristics, diagnosis, co‐morbidities, setting, interventions, and outcomes. The updated review will incorporate these headings and this information will make it easier for the reader to assess important aspects of studies included in the review. Nevertheless, we have responded as best we can to the individual comments kindly provided by the reviewers below.
Clinical heterogeneity
The reviewers are correct in highlighting the importance of taking clinical variability into account when interpreting the results of this meta‐analysis. It was in recognition of this fact that we originally decided to investigate whether effect sizes differed in trials of civilian trauma versus war veterans. That said, trials of very homogenous clinical populations may have limited generalizability. We have opted, in response to the reviewer’s comments, to present a general descriptive overview of the patients included in the eligible studies in the Description of studies section of the review. This was done instead of providing the suggested breakdown of the clinical background of trial participants for each analysis, in the interests of readability. As evident in this paragraph, the majority (18/22 or 82%) of RCTs included in the meta‐analysis that described comorbid diagnoses in their patients documented comorbid anxiety disorders in addition to PTSD. The exceptions in this regards were Katz 1995, Kosten 1991, Chung 2004, and Reist 1989, with only Katz 1995 providing data for inclusion in the analyses of the review’s primary outcomes. Accordingly, we feel that there is not not much utility in conducting additional subgroup analyses to assess the effects of comorbid anxiety disorders in this review.
Inclusion of studies in meta‐analysis 1.3
With regards to the observation that the analysis of the effect of medication on the CGI‐I outcome (Analysis 1.3) does not appear to contain all included studies, when considered across all medication classes, Analysis 1.3 contains a total of 13 trials, including the majority of the trials that the reviewers listed as including the CGI‐I as a primary or secondary outcome. The discrepancy noted by the reviewers is due to the fact that only those trials for which it was possible to extract data on the particular outcomes of interest were included in the analyses (after requesting data from the authors, where these were not reported in the study publication). In order to pre‐empt confusion on this point, we have altered the text describing the primary outcomes in the Effects of interventions section, to make this observation explicit.
As far as the inclusion of data from Tucker 2003 in analyses conducted within the review is concerned, it was noted in paragraph 5 (now paragraph 6) of the Description of included studies section that “Summary statistics for the sertraline arm of the Tucker 2003 trial were excluded from the analysis, in favour of including the data from the less well represented citalopram arm.” Nevertheless, we determined that data from the sertraline arm of Tucker 2003 had inadvertently been included in the review in place of data from the citalopram arm from this trial (Analysis 1.2 and 2.2). This error has now been corrected, and the results of relevant analyses updated accordingly. This correction has not changed the findings of the review.
References
Additional entries have been added under the Included Studies category in RevMan (version 5.2) to accommodate the maintenance phases of both the Davidson 2001 and Martenyi 2002 short‐term trials. Study IDs have been distinguished by appending them with alphabetical characters.
Additional points
1. We thank the reviewers for this suggestion. Cochrane methodologies have improved in the 10 years since this review was published. Inclusion of two risk of bias summary figures ‐ one that presents judgements about each risk of bias item as percentages across all included studies; and another that presents judgements about each risk of bias item for each included study is now a standard requirement for Cochrane reviews, and accordingly both will be included in the update to this review. Additionally Cochrane now supports the assessment of quality of included studies using the GRADE approach and this methodology will also be applied and incorporated in the interpretation of results in the update of the review.
2. This is not a viable option, given the fact that the vast majority of included trials that provide information on comorbid diagnoses include patients diagnosed with other anxiety disorders (please refer to the response to the first comment above).
3. We trust that we have sufficiently addressed the reviewer’s concerns regarding the rational for including or excluding RCTs in the review in our response to this particular query above.
4. We have distinguished between the acute and maintenance components of trials included in the review by assigning different labels to these study components, as described above. We have also updated the Characteristics of included studies section of the review accordingly.
5. We have made the suggested switch, and linked the text to the appropriate references for the maintenance components of the sertraline (Davidson 2001b) and fluoxetine (Martenyi 2002b) trials.
Contributors
Feedback submitted by Danielle Stacey, BSc(Pharm), PharmD Student Aaron Tejani, BSc(Pharm), PharmD
Response submitted by Jonathan Ipser
What's new
| Date | Event | Description |
|---|---|---|
| 28 February 2022 | New citation required and conclusions have changed | Review has been updated. |
| 28 February 2022 | New search has been performed | We updated the searches and identified 31 new trials. |
History
Protocol first published: Issue 1, 1998 Review first published: Issue 4, 2000
| Date | Event | Description |
|---|---|---|
| 12 April 2016 | Feedback has been incorporated | Feedback incorporated and small amendments made to the description of studies and effects of intervention sections. |
| 4 November 2008 | Amended | Converted to new review format. |
| 14 October 2005 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
The authors were supported by the SAMRC Research Unit on Risk & Resilience in Mental Disorders (Cape Town, South Africa).
The authors thank S Seedat, C Sager, N Zungu‐Dirwayi and G van der Linden for important and substantive contributions to earlier versions of this review as coauthors (Stein 2000; Stein 2006).
We thank Dianne O'Connell who provided statistical advice for the first version of this review. We would also like to thank Dr George Bartzokis, Dr Susan Brady, Dr Phebe Tucker, Prof Rothbaum, Prof Killeen, Dr Lindley, Dr Saygin and Ms Suliman for responding to requests for additional data for the update to the review, and Dr Bessel van der Kolk and Dr Randall Marshall for letting us have access to unpublished trials. We would also like to thank Prof Davis, Prof Baker, Prof Zohar, Dr Davidson, Dr Reich and Dr Tucker for responding to our request even though they were unable to provide us with missing data (most of these studies were published more than 15 years ago). Joy Oliver of the South African Cochrane Centre helped retrieve a number of trial reports in updating the review.
We are especially indebted to Cochrane UK for providing funding to update an earlier version of this review, and Elizabeth Waters and Nandi Siegfried in particular for their part in organising the funding. Nandi Siegfried also arranged for two of the review authors (TA and JI) to attend a 5‐day Cochrane workshop in preparation for updating this review. We are also grateful to Dr Tamara Kredo for continuous support.
We thank the editorial team of the Cochrane Common Mental Disorders (CCMD) Group, including current and previous Managing Editors, Jessica Sharp and Jess Hendon. Hugh McGuire and Sarah Dawson provided help with literature searches for this review and earlier versions.
The review authors and the CCMD Editorial Team are grateful to the following peer reviewers for their time and comments: Theodoros A Filippou, Nuala Livingstone, Nick Meader and Balwinder Singh. They would also like to thank Cochrane Copy‐Edit Support for the team's help.
The National Institute for Health Research (NIHR) is the largest single funder of the CCMD Group. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Appendices
Appendix 1. Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR)
Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR)
Cochrane Common Mental Disorders (CCMD) maintains two archived clinical trials registers at its editorial base in York, UK: a references register and a studies‐based register. The CCMDCTR‐References Register contains over 40,000 reports of RCTs in depression, anxiety and neurosis. Approximately 50% of these references have been tagged to individual, coded trials. The coded trials are held in the CCMDCTR‐Studies Register and records are linked between the two registers through the use of unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual, using a controlled vocabulary; (please contact the CCMD Information Specialists for further details). Reports of trials for inclusion in the Group's registers are collated from routine (weekly), generic searches of MEDLINE (1950 to 2016), Embase (1974 to 2016) and PsycINFO (1967 to 2016); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. Reports of trials are also sourced from international trial registers via the World Health Organization's trials portal (the International Clinical Trials Registry Platform (ICTRP)), pharmaceutical companies, the handsearching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCMD's generic search strategies (used to identify RCTs) can be found on the Group's website, (cmd.cochrane.org/specialised-register), with an example of the core MEDLINE search (used to inform the register) listed below. The Group’s Specialised Register fell out of date with the Editorial Group’s move from Bristol to York in the summer of 2016.
Core search strategy used to inform the Cochrane Common Mental Disorders Group's Specialised Register: OVID MEDLINE (to June 2016)
A weekly search alert based on condition + RCT filter only 1. [MeSH Headings]: eating disorders/ or anorexia nervosa/ or binge‐eating disorder/ or bulimia nervosa/ or female athlete triad syndrome/ or pica/ or hyperphagia/ or bulimia/ or self‐injurious behavior/ or self mutilation/ or suicide/ or suicidal ideation/ or suicide, attempted/ or mood disorders/ or affective disorders, psychotic/ or bipolar disorder/ or cyclothymic disorder/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ or seasonal affective disorder/ or neurotic disorders/ or depression/ or adjustment disorders/ or exp antidepressive agents/ or anxiety disorders/ or agoraphobia/ or neurocirculatory asthenia/ or obsessive‐compulsive disorder/ or obsessive hoarding/ or panic disorder/ or phobic disorders/ or stress disorders, traumatic/ or combat disorders/ or stress disorders, post‐traumatic/ or stress disorders, traumatic, acute/ or anxiety/ or anxiety, castration/ or koro/ or anxiety, separation/ or panic/ or exp anti‐anxiety agents/ or somatoform disorders/ or body dysmorphic disorders/ or conversion disorder/ or hypochondriasis/ or neurasthenia/ or hysteria/ or munchausen syndrome by proxy/ or munchausen syndrome/ or fatigue syndrome, chronic/ or obsessive behavior/ or compulsive behavior/ or behavior, addictive/ or impulse control disorders/ or firesetting behavior/ or gambling/ or trichotillomania/ or stress, psychological/ or burnout, professional/ or sexual dysfunctions, psychological/ or vaginismus/ or Anhedonia/ or Affective Symptoms/ or *Mental Disorders/ 2. [Title/ Author Keywords]: (eating disorder* or anorexia nervosa or bulimi* or binge eat* or (self adj (injur* or mutilat*)) or suicide* or suicidal or parasuicid* or mood disorder* or affective disorder* or bipolar i or bipolar ii or (bipolar and (affective or disorder*)) or mania or manic or cyclothymic* or depression or depressive or dysthymi* or neurotic or neurosis or adjustment disorder* or antidepress* or anxiety disorder* or agoraphobia or obsess* or compulsi* or panic or phobi* or ptsd or posttrauma* or post trauma* or combat or somatoform or somati#ation or medical* unexplained or body dysmorphi* or conversion disorder or hypochondria* or neurastheni* or hysteria or munchausen or chronic fatigue* or gambling or trichotillomania or vaginismus or anhedoni* or affective symptoms or mental disorder* or mental health).ti,kf. 3. [RCT filter]: (controlled clinical trial.pt. or randomized controlled trial.pt. or (randomi#ed or randomi#ation).ab,ti. or randomly.ab. or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or distribut* or expose* or fashion or number* or place* or recruit* or subsitut* or treat*)).ab. or placebo*.ab,ti. or drug therapy.fs. or trial.ab,ti. or groups.ab. or (control* adj3 (trial* or study or studies)).ab,ti. or ((singl* or doubl* or tripl* or trebl*) adj3 (blind* or mask* or dummy*)).mp. or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or randomized controlled trial/ or pragmatic clinical trial/ or (quasi adj (experimental or random*)).ti,ab. or ((waitlist* or wait* list* or treatment as usual or TAU) adj3 (control or group)).ab.) 4. (1 and 2 and 3) Records are screened for reports of RCTs within the scope of the Cochrane Common Mental Disorders Group. Secondary reports of RCTs are tagged to the appropriate study record. Similar weekly search alerts are also conducted on OVID Embase and PsycINFO, using relevant subject headings (controlled vocabularies) and search syntax, appropriate to each resource.
CCMDCTR search strategies for this review
Search‐1 (initial search for population + intervention)
Diagnosis = Post‐Traumatic Stress Disorders AND Intervention = "Antidepressive Agents" OR "Monoamine Oxidase Inhibitors" OR "Selective Serotonin Reuptake Inhibitors" OR "Tricyclic Drugs" OR Acetylcarnitine OR Alaproclate OR Amersergide OR Amiflamine OR Amineptine OR Amitriptyline OR Amoxapine OR Befloxatone OR Benactyzine OR Brofaromine OR Bupropion OR Butriptyline OR Caroxazone OR Chlorpoxiten OR Cilosamine OR Cimoxatone OR Citalopram OR Clomipramine OR Clorgyline OR Clorimipramine OR Clovoxamine OR Deanol OR Demexiptiline OR Deprenyl OR Desipramine OR Dibenzipin OR Diclofensine OR Dothiepin OR Doxepin OR Duloxetine OR Escitalopram OR Etoperidone OR Femoxetine OR Fluotracen OR Fluoxetine OR Fluparoxan OR Fluvoxamine OR Idazoxan OR Imipramine OR Iprindole OR Iproniazid OR Isocarboxazid OR Litoxetine OR Lofepramine OR Maprotiline OR Medifoxamine OR Melitracen OR Metapramine OR Mianserin OR Milnacipran OR Minaprine OR Mirtazapine OR Moclobemide OR Nefazodone OR Nialamide OR Nomifensine OR Nortriptyline OR Noxiptiline OR Opipramol OR Oxaflozane OR Oxaprotiline OR Pargyline OR Paroxetine OR Phenelzine OR Piribedil OR Pirlindole OR Pivagabine OR Prosulpride OR Protriptyline OR Quinupramine OR Reboxetine OR Rolipram OR Sertraline OR Setiptiline OR Teniloxine OR Tetrindole OR Thiazesim OR Thozalinone OR Tianeptine OR Toloxatone OR Tomoxetine OR Tranylcypromine OR Trazodone OR Trimipramine OR Venlafaxine OR Viloxazine OR Viqualine OR Zimeldine.
Search‐2 (population only)
The CCMDCTR was searched for this review using the following terms(all available years <1968 to 14 June 2016>):
CCMDCTR‐Studies Register: (PTSD or posttrauma* or post‐trauma* or "post trauma*" or "combat disorder*" or "stress disorder*"):sco,stc
CCDMDCTR‐References Register: (PTSD or posttrauma* or post‐trauma* or "post trauma*" or "combat disorder*" or "stress disorder*"):ti,ab,kw,ky,emt,mh,mc
[Key to field tags. ti:title; ab:abstract; kw:keywords; ky:other keywords; mh:MeSH headings; mc:MeSH check words; emt:EMTREE headings; sco:healthcare condition; stc:target condition]
*************************************************************
Interim searches conducted by CCMD (2004‐2011), directly on MEDLINE & Embase (when the CCMDCTR was in flux).
OVID MEDLINE
1. ((serotonin or norepinephrine or noradrenaline or dopamine or neurotransmitter) adj (uptake or reuptake or re‐uptake)).mp. 2. (antiadrenergic or anti‐adrenergic).mp. 3. (5‐hydroxytrypotophan or Acetylcarnitine or Alaproclate or alprazolam or Amersergide or Amiflamine or Amineptine or Amitriptyline or Amoxapine or anticonvulsant* or Antidepress* or antipsychotic* or anxiolytic* or Aripiprazole).mp. 4. (Befloxatone or Benactyzine or Benzodiazepine* or Brofaromine or Bupropion or Butriptyline).mp. 5. (Carbazepine or Caroxazone or cck‐4 or Chlorimipramine or Chlorphenamidine or Chlorpoxiten or Cilosamine or Cimoxatone or Citalopram or Clomipramine or clonidine or Clorgyline or Clovoxamine or Cyproheptadine or d‐Cycloserine).mp. 6. (Deanol or Demexiptiline or Deprenyl or Desipramine or Desvenlafaxine or Dibenzipin or Diclofensine or divalproex or dopamin* or Dosulepin or Dothiepin or Doxazosin or Doxepin or Duloxetine).mp. 7. (Escitalopram or Etoperidone or Femoxetine or Fenfluramine or flumazenil or Fluotracen or fluoxetine or Fluparoxan or fluphenazine or Fluvoxamine or Furazolidone or Guanfacine).mp. 8. (haloperidol or Harmaline or Harmine or hydrocortisone or Idazoxan or Imipramine or inositol or Iprindole or Iproniazid or Isocarboxazid or lamotrigine).mp. 9. (Lithium carbonate or Lithium compounds or Litoxetine or Lofepramine).mp. 10. (MAOI* or Maprotiline or medicat* or Medifoxamine or Melitracen or Metapramine or Metyrapone or Mianserin or Milnacipran or Minaprine or Mirtazapine or Moclobemide or Monoamine Oxidase Inhibitor*).mp. 11. (Naloxone or Naltrexone or Nefazodone or Nialamide or Nomifensine or noradrenerg* or Norfenfluramine or Nortriptyline or Noxiptiline or Olanzapine or Opipramol or Oxaflozane or Oxaprotiline or Oxcarbazepine).mp. 12. N‐Methyl‐3,4‐methylenedioxyamphetamine/ 13. (Pargyline or Paroxetine or pharmacother* or Phenelzine or Pheniprazine or Piribedil or Pirlindole or Pivagabine or Pizotyline or Prazosin or Pregabalin or Procaine or Propranolol or Prosulpride or Protriptyline or psychotropic*).mp. 14. (Quetiapine or Quinupramine or Quipazine or Reboxetine or Risperidone or Ritanserin or Rolipram).mp. 15. (Selegiline or seroto* or Sertraline or Setiptiline or SNRI* or SSRI* or Sulpiride).mp. 16. (Teniloxine or Tetrindole or Thiazesim or Thozalinone or Tiagabine or Tianeptine or Toloxatone or Tomoxetine or Topiramate or Tranylcypromine or Trazodone or tricyclic* or Trimipramine or Tryptophan).mp. 17. (Venlafaxine or Viloxazine or Viqualine or Yohimbine or Zimeldine).mp. 18. exp Stress Disorders, Traumatic/dt 19. or/1‐18 20. exp Stress Disorders, Traumatic/ 21. ((post‐traumatic or post traumatic or posttraumatic) and disorder*).tw. 22. PTSD.tw. 23. or/20‐22 24. randomized controlled trial/ 25. controlled clinical trial/ 26. randomi#ed.ti,ab. 27. randomly.ab. 28. placebo$.tw. 29. trial.ab. 30. drug therapy.fs. 31. ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$ or dummy)).mp. 32. (control$ adj3 (trial$ or study or studies$ or group$)).tw. 33. (animals not (humans and animals)).sh. 34. or/24‐32 35. 34 not 33 36. 19 and 23 and 35
OVID Embase
1. ((seroton* or norepinephrine or noradrenaline or dopamin* or neurotransmitter) adj (uptake or reuptake or re‐uptake)).mp. 2. (5‐hydroxytrypotophan or acetylcarnitine or alaproclate or alprazolam or amersergide or amiflamine or amineptine or amitriptyline or amoxapine or anticonvulsant* or antidepress* or antipsychotic* or anxiolytic*).mp. 3. (befloxatone or benactyzine or benzodiazepine* or brofaromine or bupropion or butriptyline).mp. 4. (caroxazone or cck‐4 or chlorimipramine or chlorphenamidine or chlorpoxiten or cilosamine or cimoxatone or citalopram or clomipramine or clonidine or clorgyline or clovoxamine or cyproheptadine or d‐cycloserine).mp. 5. (deanol or demexiptiline or deprenyl or desipramine or desvenlafaxine or dibenzipin or diclofensine or divalproex or dopamin* or dosulepin or dothiepin or doxepin or duloxetine).mp. 6. (escitalopram or etoperidone or femoxetine or fenfluramine or flumazenil or fluotracen or fluoxetine or fluparoxan or fluphenazine or fluvoxamine or furazolidone).mp. 7. (haloperidol or harmaline or harmine or hydrocortisone or idazoxan or imipramine or inositol or iprindole or iproniazid or isocarboxazid or lamotrigine).mp. 8. (lithium carbonate or lithium compounds or litoxetine or lofepramine).mp. 9. (maoi* or maprotiline or medicat* or medifoxamine or melitracen or metapramine or metyrapone or mianserin or milnacipran or minaprine or mirtazapine or moclobemide or monoamine oxidase inhibitor*).mp. 10. (naloxone or naltrexone or nefazodone or nialamide or nomifensine or noradrenerg* or norfenfluramine or nortriptyline or noxiptiline or olanzapine or opipramol or oxaflozane or oxaprotiline).mp. 11. (pargyline or paroxetine or pharmacother* or phenelzine or pheniprazine or piribedil or pirlindole or pivagabine or pizotyline or prazosin or procaine or propranolol or prosulpride or protriptyline or psychotropic*).mp. 12. (quetiapine or quinupramine or quipazine or reboxetine or risperidone or ritanserin or rolipram).mp. 13. (selegiline or seroto* or sertraline or setiptiline or snri* or ssri* or sulpiride).mp. 14. (teniloxine or tetrindole or thiazesim or thozalinone or tiagabine or tianeptine or toloxatone or tomoxetine or topiramate or tranylcypromine or trazodone or tricyclic* or trimipramine or tryptophan).mp. 15. (venlafaxine or viloxazine or viqualine or yohimbine or zimeldine).mp. 16. or/1‐15 17. posttraumatic stress disorder/ 18. ((post‐traumatic or post traumatic or posttraumatic) and disorder*).tw. 19. ptsd.tw. 20. or/17‐19 21. major clinical study/ 22. randomized controlled trial/ 23. randomization/ 24. placebo/ 25. randomi#ed.ti,ab. 26. placebo$.tw. 27. trial$.ti,ab. 28. randomly.ab. 29. ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$ or dummy)).mp. 30. (control$ adj3 (trial$ or study or studies$)).tw. 31. ((animal or nonhuman) not (human and (animal or nonhuman))).de. 32. or/21‐30 33. 32 not 31 34. 16 and 20 and 33
Appendix 2. Other database search strategies ‐ 1
Other bibliographic database searches (population only) (searched 2014 to 2020)
Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library
Date of latest search: Issue 11 of 12, 2020
ID Search Hits
#1 MeSH descriptor: [Stress Disorders, Post‐Traumatic] this term only
#2 (PTSD or ((posttrauma* or post‐trauma* or "post trauma*") near/3 (stress* or disorder* or psych* or symptom*)) or "acute stress disorder*" or "combat disorder*" or "war neuros*"):ti,ab,kw
#3 (((acute or traumatic) near/1 stress*) and (expos* or psyc*)):ti,ab,kw
#4 (traumatised near/1 (victim* or survivor*)):ti,ab,kw
#5 (traumatized near/1 (victim* or survivor*)):ti,ab,kw
#6 (trauma* near/2 (event* or memor* or flashback* or nightmare*)):ti,ab,kw
#7 ((trauma* or posttrauma* or post‐trauma* or victim* or survivor*) and (exposure near/3 (therap* or psychotherap* or training or counsel*))):ti,ab,kw
#8 MeSH descriptor: [Crisis Intervention] this term only
#9 ("critical incident" near/1 (stress or debrief* or de‐brief*)):ti,ab,kw
#10 (debriefing or de‐briefing):ti,ab,kw
#11 ("crisis intervention*" or CISD):ti,ab,kw
#12 ((stress or group* or psychological or crisis) near/3 (debrief* or de‐brief*)):ti,ab,kw
#13 (trauma* near/2 (event* or memor* or flashback* or nightmare*)):ti,ab,kw
#14 (EMDR or ("eye movement desensitization" and reprocessing)):ti,ab,kw
#15 (EMDR or ("eye movement desensitisation" and reprocessing)):ti,ab,kw
#16 (#1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15)
*************************************************************
Ovid MEDLINE(R) and Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations and Daily <2014 to November 13, 2020>
Search Strategy:
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
1 "trauma and stressor related disorders"/ or stress disorders, post‐traumatic/
2 (PTSD or ((posttrauma* or post‐trauma* or post trauma*) adj3 (stress* or disorder* or psych* or symptom?)) or acute stress disorder* or combat disorder* or war neuros*).ti,ab,kf.
3 (((acute or traumatic) adj stress*) and (expos* or psyc*)).ti,ab,kf.
4 (traumati#ed adj (victim? or survivor?)).ti,ab,kf.
5 (trauma* adj2 (event? or memor* or flashback* or nightmare?)).ti,ab,kf.
6 ((trauma* or posttrauma* or post‐trauma* or victim* or survivor?) and (exposure adj3 (therap* or psychotherap* or training or counsel*))).ti,ab,kf,hw.
7 Crisis Intervention/
8 (critical incident adj (stress or debrief* or de‐brief*)).ti,ab,kf.
9 (debriefing or de‐briefing).ti,kf.
10 (crisis intervention? or CISD).ti,ab,kf.
11 ((stress or group? or psychological or crisis) adj3 (debrief* or de‐brief*)).ti,ab,kf.
12 (trauma* adj2 (event? or memor* or flashback* or nightmare?)).ti,kf.
13 (EMDR or (eye movement desensiti#ation and reprocessing)).ti,ab,kf,sh.
14 or/1‐13
15 randomized controlled trial.pt.
16 controlled clinical trial.pt.
17 placebo.ab.
18 clinical trials as topic.sh.
19 (randomized or randomised).ab.
20 randomly.ab.
21 trial.ti.
22 or/15‐21
23 (14 and 22)
*************************************************************
Ovid Embase <2014 to 2020 Week 46>
Search Strategy:
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
1 posttraumatic stress disorder/ or "trauma and stressor related disorders"/ or combat disorders/ or psychological trauma/ or stress disorders, post‐traumatic/ or stress disorders, traumatic, acute/
2 (PTSD or ((posttrauma* or post‐trauma* or post trauma*) adj3 (stress* or disorder* or psych* or symptom?)) or acute stress disorder* or combat disorder* or war neuros*).ti,ab,kw.
3 (((acute or traumatic) adj stress*) and (expos* or psyc*)).ti,ab,kw.
4 (traumati#ed adj (victim? or survivor?)).ti,ab,kw.
5 (trauma* adj2 (event? or memor* or flashback* or nightmare?)).ti,ab,kw.
6 ((trauma* or posttrauma* or post‐trauma* or victim* or survivor?) and (exposure adj3 (therap* or psychotherap* or training or counsel*))).ti,ab,kw,hw.
7 (critical incident adj (stress or debrief* or de‐brief*)).ti,ab,kw.
8 (debriefing or de‐briefing).ti,kw.
9 (crisis intervention? or CISD).ti,ab,kw.
10 ((stress or group? or psychological or crisis) adj3 (debrief* or de‐brief*)).ti,ab,kw.
11 (trauma* adj2 (event? or memor* or flashback* or nightmare?)).ti,kw.
12 (EMDR or (eye movement desensiti#ation and reprocessing)).ti,ab,kw,sh.
13 or/1‐12
14 crossover‐procedure/ or double‐blind procedure/ or randomized controlled trial/ or single‐blind procedure/ or (random* or factorial* or crossover* or cross over* or placebo* or (doubl* adj blind*) or (singl* adj blind*) or assign* or allocat* or volunteer*).tw.
15 (13 and 14)
*************************************************************
Ovid PsycINFO <2014 to November Week 46 2020>
1 posttraumatic stress disorder/ or complex ptsd/ or desnos/ or acute stress disorder/ or combat experience/ or "debriefing (psychological)"/ or emotional trauma/ or post‐traumatic stress/ or exp stress reactions/ or traumatic neurosis/
2 exp disasters/
3 (PTSD or ((posttrauma* or post‐trauma* or post trauma*) adj3 (stress* or disorder* or psych* or symptom?)) or acute stress disorder* or combat disorder* or war neuros*).ti,ab.
4 (((acute or traumatic) adj stress*) and (expos* or psyc*)).ti,ab,id.
5 (traumati#ed adj (victim? or survivor?)).ti,ab,id.
6 (trauma* adj2 (event? or memor* or flashback* or nightmare?)).ti,ab,id.
7 (EMDR or (eye movement desensiti#ation and reprocessing)).ti,ab,id.
8 ((trauma* or posttrauma* or post‐trauma* or victim* or survivor?) and (exposure adj3 (therap* or psychotherap* or training or counsel*))).ti,ab,id.
9 crisis intervention/
10 (critical incident adj (stress or debrief* or de‐brief*)).ti,ab,id.
11 (debriefing or de‐briefing).ti,ab,id.
12 (crisis intervention? or CISD).ti,ab,id.
13 ((stress or group? or psychological or crisis) adj3 (debrief* or de‐brief*)).ti,ab,id.
14 (trauma* adj2 (event? or memor* or flashback* or nightmare?)).ti,ab,id.
15 (1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14)
16 clinical trials.sh.
17 (randomi#ed or randomi#ation or randomi#ing).ti,ab,id.
18 (RCT or at random or (random* adj3 (assign* or allocat* or control* or crossover or cross‐over or design* or divide* or division or number))).ti,ab,id.
19 (control* and (trial or study or group) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,id,hw.
20 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,id.
21 trial.ti.
22 placebo.ti,ab,id,hw.
23 treatment outcome.md.
24 treatment effectiveness evaluation.sh.
25 mental health program evaluation.sh.
26 (16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25)
27 (15 and 26)
*************************************************************
ProQuest PTSDPubs (formerly PILOTS: Published International Literature On Traumatic Stress)
Date Searched: 16 November 2020
Search Strategy
Set#: S1 Searched for: ti((posttrauma* near/4 (stress* or disorder* or psych* or symptom*))) OR ab((posttrauma* near/4 (stress* or disorder* or psych* or symptom*)))
Set#: S2 Searched for: ti((post‐trauma* near/4 (stress* or disorder* or psych* or symptom*))) OR ab((post‐trauma* near/4 (stress* or disorder* or psych* or symptom*)))
Set#: S3 Searched for: ti((post trauma* near/4 (stress* or disorder* or psych* or symptom*))) OR ab((post trauma* near/4 (stress* or disorder* or psych* or symptom*)))
Set#: S4 Searched for: ti((PTSD or acute stress disorder* or combat disorder* or war neuros*) ) OR ab((PTSD or acute stress disorder* or combat disorder* or war neuros*) )
Set#: S5 Searched for: ti((((acute or traumatic) near/2 stress*) and (expos* or psyc*)) ) OR ab((((acute or traumatic) near/2 stress*) and (expos* or psyc*)) )
Set#: S6 Searched for: ti((traumatised near/2 (victim* or survivor*)) ) OR ab((traumatised near/2 (victim* or survivor*)) )
Set#: S7 Searched for: ti((trauma* near/3 (event* or memor* or flashback* or nightmare*)) ) OR ab((trauma* near/3 (event* or memor* or flashback* or nightmare*)) )
Set#: S8 Searched for: ti(((trauma* or posttrauma* or post‐trauma* or victim* or survivor*) and (exposure near/4 (therap* or psychotherap* or training or counsel*))) ) OR ab(((trauma* or posttrauma* or post‐trauma* or victim* or survivor*) and (exposure near/4 (therap* or psychotherap* or training or counsel*))) )
Set#: S9 Searched for: ti((critical incident near/2 (stress or debrief* or de‐brief*)) ) OR ab((critical incident near/2 (stress or debrief* or de‐brief*)) )
Set#: S10 Searched for: ti((debriefing or de‐briefing)) OR ab((debriefing or de‐briefing))
Set#: S11 Searched for: ti((crisis intervention* or CISD)) OR ab((crisis intervention* or CISD))
Set#: S12 Searched for: ti(((stress or group* or psychological or crisis) near/4 (debrief* or de‐brief*)) ) OR ab(((stress or group* or psychological or crisis) near/4 (debrief* or de‐brief*)) )
Set#: S13 Searched for: ti((trauma* near/3 (event* or memor* or flashback* or nightmare*)) ) OR ab((trauma* near/3 (event* or memor* or flashback* or nightmare*)) )
Set#: S14 Searched for: ti((EMDR or (eye movement desensitisation and reprocessing))) OR ab((EMDR or (eye movement desensitisation and reprocessing)))
Set#: S15 Searched for: ti((EMDR or (eye movement desensitiZation and reprocessing))) OR ab((EMDR or (eye movement desensitiZation and reprocessing)))
Set#: S16 Searched for: (s1 or s2 or s3 or s4 or s5 or s6 or s7 or s8 or s9 or s10 or s11 or s12 or s13 or s14 or s15)
Set#: S17 Searched for: MAINSUBJECT.EXACT("Randomized Clinical Trial")
Set#: S18 Searched for: ab((randomized or randomised or placebo or randomly))
Set#: S19 Searched for: ti(trial)
Set#: S20 Searched for: (S17 or S18 or S19)
Set#: S21 Searched for: S16 and s20
Limited 2014 onwards
**************************************************
Appendix 3. Other database search strategies ‐ 2
PubMed (randomized controlled trial [pt] OR controlled clinical trial [pt] OR randomized controlled trials [mh] OR random allocation [mh] OR double‐blind method [mh] OR single‐blind method [mh] OR clinical trial [pt] OR clinical trials [mh] OR ("clinical trial" [tw]) OR ((singl* [tw] OR doubl* [tw] OR trebl* [tw] OR tripl* [tw]) AND (mask* [tw] OR blind* [tw])) OR ("latin square" [tw]) OR placebos [mh] OR placebo* [tw] OR random* [tw] OR research design [mh:noexp] OR comparative study [mh] OR evaluation studies [mh] OR follow‐up studies [mh] OR prospective studies [mh] OR cross‐over studies [mh] OR control* [tw] OR prospectiv* [tw] OR volunteer* [tw]) NOT (animal [mh] NOT human [mh]) AND (Stress Disorders, Post‐Traumatic [mh:noexp] OR "posttraumatic stress disorder" [tw] OR "post traumatic stress disorder" [tw] OR PTSD [tw]) AND (pharmacother* [tw] OR medicat* [tw] OR drug* [tw] OR Drug Therapy [mh])
PsycINFO ("randomisation" OR "randomization") OR "controlled" AND ("post‐traumatic" OR posttraumatic) AND (medication OR pharmacotherapy OR treatment) PsycINFO includes the Dissertation Abstracts International database ‐ a database of unpublished dissertations.
PTSDPubs The National PTSD Center database PTSDPubs (previously PILOTS) contains published and unpublished articles on PTSD. We searched it using the following search query: (randomisation or randomization) or controlled AND (post‐traumatic OR posttraumatic) AND (medication OR pharmacotherapy).
**************************************************
Data and analyses
Comparison 1. Alpha‐blockers versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Dropout rate due to treatment‐emergent adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1.1 Dropout rate | 1 | 304 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.91, 1.08] |
| 1.2 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.2.1 CAPS: Total score | 1 | 271 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐5.13, 5.33] |
| 1.3 Self‐rated scales | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.3.1 Total score | 1 | 271 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐0.41, 0.06] |
| 1.4 Depression scale (typically Hamilton Depression) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.4.1 Reduction of depression symptoms | 1 | 271 | Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐2.11, 0.91] |
| 1.5 Dropout rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.5.1 Dropout rate | 1 | 303 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.65, 1.62] |
Comparison 2. Anticonvulsants versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Clinical Global Impressions Scale Improvement Item (CGI‐I) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1.1 No. of responders | 4 | 315 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.63, 1.21] |
| 2.2 Dropout rate due to treatment emergent adverse effects | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.2.1 Dropout rate | 5 | 348 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.92, 1.05] |
| 2.3 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.3.1 CAPS: Total score | 5 | 411 | Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐6.54, 5.84] |
| 2.3.2 CAPS subscale: Re‐experiencing/intrusion | 3 | 141 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐3.74, 4.13] |
| 2.3.3 CAPS subscale: Avoidance/numbing | 3 | 141 | Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐8.65, 7.91] |
| 2.3.4 CAPS subscale: Hyperarousal | 3 | 141 | Mean Difference (IV, Random, 95% CI) | 0.21 [‐2.57, 2.99] |
| 2.4 Symptom severity: Other measures | 5 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.4.1 Reduction of PTSD symptoms | 5 | 180 | Std. Mean Difference (IV, Random, 95% CI) | 0.15 [‐0.31, 0.60] |
| 2.5 Self‐rated scales | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.5.1 Reduction of PTSD symptoms | 3 | 84 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.75, 0.64] |
| 2.6 Depression scale (typically Hamilton Depression) | 5 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.6.1 Reduction of depression symptoms | 3 | 131 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.57, 0.12] |
| 2.6.2 Depression Scale ‐ Change scores | 2 | 66 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.32 [‐0.81, 0.16] |
| 2.7 Anxiety ‐ Hamilton Anxiety Scale | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.7.1 Reduction of anxiety symptoms | 3 | 138 | Mean Difference (IV, Random, 95% CI) | ‐0.66 [‐3.66, 2.35] |
| 2.8 Anxiety ‐ Other scales | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.8.1 Reduction of anxiety symptoms | 1 | 28 | Std. Mean Difference (IV, Random, 95% CI) | 0.55 [‐0.21, 1.31] |
| 2.9 Sheehan Disability Scale | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.9.1 Reduction of functional disability | 2 | 270 | Mean Difference (IV, Random, 95% CI) | 0.41 [‐1.48, 2.30] |
| 2.10 Dropout rate due to any cause | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.10.1 Dropout rate | 7 | 447 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.61, 1.06] |
2.1. Analysis.

Comparison 2: Anticonvulsants versus placebo, Outcome 1: Clinical Global Impressions Scale Improvement Item (CGI‐I)
Comparison 3. Antipsychotics versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Clinical Global Impressions Scale Improvement Item (CGI‐I) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1.1 No. of responders | 2 | 43 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.16, 1.67] |
| 3.2 Drop‐out rate due to treatment emergent adverse effects | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.2.1 Dropout rate | 5 | 174 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.85, 1.03] |
| 3.3 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.3.1 CAPS: Total score | 3 | 129 | Mean Difference (IV, Random, 95% CI) | ‐14.10 [‐22.57, ‐5.62] |
| 3.3.2 CAPS subscale: Re‐experiencing/intrusion | 3 | 129 | Mean Difference (IV, Random, 95% CI) | ‐4.73 [‐7.55, ‐1.90] |
| 3.3.3 CAPS subscale: Avoidance/numbing | 3 | 129 | Mean Difference (IV, Random, 95% CI) | ‐6.01 [‐13.72, 1.70] |
| 3.3.4 CAPS subscale: Hyperarousal | 3 | 129 | Mean Difference (IV, Random, 95% CI) | ‐5.14 [‐8.16, ‐2.11] |
| 3.4 Symptom severity: Other measures | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.4.1 Reduction of PTSD symptoms | 2 | 108 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.55 [‐0.94, ‐0.17] |
| 3.5 Self‐rated scales | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.5.1 Total score | 3 | 119 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.47 [‐1.06, 0.11] |
| 3.5.2 Self‐rated subscale: Re‐experiencing/Intrusion | 1 | 15 | Std. Mean Difference (IV, Random, 95% CI) | 0.51 [‐0.58, 1.61] |
| 3.5.3 Self‐rated subscale: Avoidance/numbing | 1 | 15 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐1.38, 0.78] |
| 3.5.4 Self‐rated subscale: Hyperarousal | 1 | 15 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐1.13, 1.01] |
| 3.6 Depression Scale (typically Hamilton Depression) | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.6.1 Reduction of depression symptoms | 2 | 108 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.62 [‐1.01, ‐0.23] |
| 3.7 Anxiety ‐ Hamilton Anxiety Scale | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.7.1 Reduction of anxiety symptoms | 1 | 80 | Mean Difference (IV, Random, 95% CI) | ‐3.03 [‐6.21, 0.15] |
| 3.8 Sheehan Disability Scale | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.8.1 Reduction of functional disability | 2 | 43 | Mean Difference (IV, Random, 95% CI) | ‐6.57 [‐14.74, 1.61] |
| 3.9 Drop‐out rate due to any cause | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.9.1 Dropout rate | 4 | 133 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.54, 1.14] |
Comparison 4. Benzodiazepines versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Drop‐out rate due to treatment emergent adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1.1 Dropout rate | 1 | 32 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 4.2 Drop‐out rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.2.1 Dropout rate | 1 | 16 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.39, 1.89] |
Comparison 5. Dopamine beta‐hydroxylase inhibitors versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Drop‐out rate due to treatment emergent adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1.1 Dropout rate | 1 | 91 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.77, 1.58] |
| 5.2 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.2.1 CAPS: Total score | 1 | 86 | Mean Difference (IV, Random, 95% CI) | ‐5.27 [‐16.72, 6.18] |
| 5.2.2 CAPS subscale: Re‐experiencing/intrusion | 1 | 86 | Mean Difference (IV, Random, 95% CI) | 2.63 [‐1.67, 6.93] |
| 5.2.3 CAPS subscale: Avoidance/numbing | 1 | 86 | Mean Difference (IV, Random, 95% CI) | 0.58 [‐4.57, 5.73] |
| 5.2.4 CAPS subscale: Hyperarousal | 1 | 86 | Mean Difference (IV, Random, 95% CI) | 2.05 [‐1.82, 5.92] |
| 5.3 Drop‐out rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.3.1 Dropout rate | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [0.64, 3.00] |
Comparison 6. Ganaxolone versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Drop‐out rate due to treatment emergent adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1.1 Dropout rate | 1 | 112 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.10, 3.45] |
| 6.2 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.2.1 CAPS: Total score | 1 | 99 | Mean Difference (IV, Random, 95% CI) | ‐2.50 [‐10.92, 5.92] |
| 6.2.2 CAPS subscale: Re‐experiencing/intrusion | 1 | 99 | Mean Difference (IV, Random, 95% CI) | ‐0.80 [‐4.09, 2.49] |
| 6.2.3 CAPS subscale: Avoidance/numbing | 1 | 99 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐5.74, 2.54] |
| 6.2.4 CAPS subscale: Hyperarousal | 1 | 99 | Mean Difference (IV, Random, 95% CI) | ‐1.10 [‐3.96, 1.76] |
| 6.3 Symptom severity: Other measures | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.3.1 Reduction of PTSD symptoms | 1 | 86 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.59, 0.25] |
| 6.4 Self‐rated scales | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.4.1 Total score | 1 | 86 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.59, 0.25] |
| 6.5 Drop‐out rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.5.1 Dropout rate | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.93 [0.94, 3.92] |
Comparison 7. GR205171 versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Clinical Global Impressions Scale Improvement Item (CGI‐I) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1.1 No. of responders | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.42, 1.44] |
| 7.2 Drop‐out rate due to treatment emergent adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.2.1 Dropout rate | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 7.3 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.3.1 CAPS: Total score | 1 | 39 | Mean Difference (IV, Random, 95% CI) | ‐7.60 [‐22.59, 7.39] |
| 7.4 Drop‐out rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.4.1 Dropout rate | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [1.03, 2.41] |
Comparison 8. GSK561679 versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Clinical Global Impressions Scale Improvement Item (CGI‐I) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1.1 No. of responders | 1 | 128 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.88, 1.31] |
Comparison 9. Hypnotics versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.1.1 CAPS Total Score | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐1.00 [‐19.57, 17.57] |
| 9.2 Symptom severity: Other measures | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.2.1 Reduction of PTSD symptoms | 1 | 16 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.09 [‐1.08, 0.90] |
| 9.3 Depression Scale (typically Hamilton Depression) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.3.1 Reduction of depression symptoms | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐2.67 [‐12.68, 7.34] |
| 9.4 Drop‐out rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.4.1 Dropout rate | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 1.85 [0.59, 5.79] |
Comparison 10. MAOIs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 10.1 Drop‐out rate due to treatment emergent adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1.1 Dropout rate | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.90, 1.43] |
| 10.2 Self‐rated scales | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10.2.1 Total score | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.06 [‐1.75, ‐0.36] |
| 10.2.2 Self‐rated subscale: Re‐experiencing/Intrusion | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.07 [‐1.77, ‐0.38] |
| 10.2.3 Self‐rated subscale: Avoidance/numbing | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐1.49, ‐0.14] |
| 10.3 Depression Scale (typically Hamilton Depression) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10.3.1 Reduction of depression symptoms | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐2.80 [‐7.15, 1.55] |
| 10.4 Anxiety ‐ Other scales | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10.4.1 Reduction of anxiety symptoms | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.34, ‐0.01] |
| 10.5 Drop‐out rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.5.1 Dropout rate | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.67, 1.18] |
Comparison 11. NaSSAs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 11.1 Clinical Global Impressions Scale Improvement Item (CGI‐I) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1.1 No. of responders | 1 | 26 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.22, 0.94] |
| 11.2 Drop‐out rate due to treatment emergent adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.2.1 Dropout rate | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.68, 1.11] |
| 11.3 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.3.1 CAPS Total Score | 1 | 61 | Mean Difference (IV, Random, 95% CI) | ‐3.20 [‐14.74, 8.34] |
| 11.3.2 CAPS subscale: Re‐experiencing/intrusion | 1 | 61 | Mean Difference (IV, Random, 95% CI) | ‐2.50 [‐6.32, 1.32] |
| 11.3.3 CAPS subscale: Avoidance/numbing | 1 | 61 | Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐6.22, 5.22] |
| 11.3.4 CAPS subscale: Hyperarousal | 1 | 61 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐3.51, 3.11] |
| 11.4 Symptom severity: Other measures | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.4.1 Reduction of PTSD symptoms | 2 | 87 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐0.87, ‐0.00] |
| 11.5 Self‐rated scales | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.5.1 Total score | 2 | 87 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐1.02, 0.41] |
| 11.6 Depression Scale (typically Hamilton Depression) | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.6.1 Reduction of depression symptoms | 2 | 86 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐1.39, 0.88] |
| 11.7 Anxiety ‐ Other scales | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.7.1 Reduction of anxiety symptoms | 1 | 25 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.88 [‐1.77, ‐0.00] |
| 11.8 Drop‐out rate due to any cause | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.8.1 Dropout rate | 3 | 200 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.48, 1.52] |
Comparison 12. NK‐1 receptor antagonists versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 12.1 Drop‐out rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1.1 Dropout rate | 1 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.32, 1.04] |
Comparison 13. RIMAs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 13.1 Clinical Global Impressions Scale Improvement Item (CGI‐I) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1.1 No. of responders | 2 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.44, 1.44] |
| 13.2 Drop‐out rate due to treatment emergent adverse effects | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.2.1 Dropout rate | 2 | 159 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.91, 1.04] |
| 13.3 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 13.3.1 CAPS Total Score | 2 | 178 | Mean Difference (IV, Random, 95% CI) | ‐5.06 [‐15.93, 5.81] |
| 13.4 Drop‐out rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.4.1 Dropout rate | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.49, 2.22] |
Comparison 14. SARIs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 14.1 Drop‐out rate due to treatment emergent adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1.1 Dropout rate | 1 | 42 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.70, 1.09] |
| 14.2 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 14.2.1 CAPS: Total score | 1 | 41 | Mean Difference (IV, Random, 95% CI) | ‐5.60 [‐21.26, 10.06] |
| 14.2.2 CAPS subscale: Re‐experiencing/intrusion | 1 | 41 | Mean Difference (IV, Random, 95% CI) | ‐1.30 [‐6.80, 4.20] |
| 14.2.3 CAPS subscale: Avoidance/numbing | 1 | 41 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐9.23, 6.03] |
| 14.2.4 CAPS subscale: Hyperarousal | 1 | 41 | Mean Difference (IV, Random, 95% CI) | ‐2.90 [‐8.40, 2.60] |
| 14.3 Drop‐out rate due to any cause | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.3.1 Dropout rate | 2 | 101 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.68, 0.97] |
Comparison 15. SNRIs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 15.1 Drop‐out rate due to treatment emergent adverse effects | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1.1 Dropout rate | 2 | 687 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.88, 1.10] |
| 15.2 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 15.2.1 CAPS: Total score | 2 | 687 | Mean Difference (IV, Random, 95% CI) | ‐8.11 [‐12.29, ‐3.92] |
| 15.2.2 CAPS subscale: Re‐experiencing/intrusion | 2 | 687 | Mean Difference (IV, Random, 95% CI) | ‐1.95 [‐3.36, ‐0.54] |
| 15.2.3 CAPS subscale: Avoidance/numbing | 2 | 687 | Mean Difference (IV, Random, 95% CI) | ‐3.42 [‐5.23, ‐1.61] |
| 15.2.4 CAPS subscale: Hyperarousal | 2 | 687 | Mean Difference (IV, Random, 95% CI) | ‐2.29 [‐3.67, ‐0.91] |
| 15.3 Symptom severity: Other measures | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 15.3.1 Reduction of PTSD symptoms | 2 | 687 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.46, ‐0.16] |
| 15.4 Self‐rated scales | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 15.4.1 Total score | 1 | 358 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐0.47, ‐0.05] |
| 15.4.2 Self‐rated subscale: Re‐experiencing/Intrusion | 1 | 358 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐0.44, ‐0.02] |
| 15.4.3 Self‐rated subscale: Avoidance/numbing | 1 | 358 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐0.44, ‐0.02] |
| 15.4.4 Self‐rated subscale: Hyperarousal | 1 | 358 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐0.45, ‐0.03] |
| 15.5 Depression Scale (typically Hamilton Depression) | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 15.5.1 Reduction of depression symptoms | 1 | 329 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.42, 0.02] |
| 15.5.2 Depression Scale ‐ Change scores | 1 | 358 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.43, ‐0.01] |
| 15.6 Sheehan Disability Scale | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 15.6.1 Reduction of functional disability | 2 | 687 | Mean Difference (IV, Random, 95% CI) | ‐2.01 [‐3.30, ‐0.72] |
| 15.7 Drop‐out rate due to any cause | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.7.1 Dropout rate | 1 | 329 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.90, 1.21] |
Comparison 16. SSRIs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 16.1 Clinical Global Impressions Scale Improvement Item (CGI‐I) | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1.1 No. of responders | 8 | 1078 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.59, 0.74] |
| 16.2 CGI: Individual SSRI agents | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.2.1 Fluoxetine versus placebo | 2 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.19, 2.82] |
| 16.2.2 Paroxetine versus placebo | 3 | 728 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.55, 0.74] |
| 16.2.3 Sertraline versus placebo | 3 | 285 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.56, 0.81] |
| 16.3 Drop‐out rate due to treatment emergent adverse effects | 14 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.3.1 Dropout rate | 14 | 2399 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.96, 1.00] |
| 16.4 Drop‐out rate due to treatment emergent adverse effects: Individual SSRI agents | 14 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.4.1 Fluoxetine | 3 | 367 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.25, 2.96] |
| 16.4.2 Paroxetine | 5 | 1101 | Risk Ratio (M‐H, Random, 95% CI) | 1.55 [1.05, 2.29] |
| 16.4.3 Sertraline | 6 | 931 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.87, 2.05] |
| 16.5 Clinically Administered PTSD Scale (CAPS): reduction of PTSD symptoms | 15 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 16.5.1 CAPS: Total score | 15 | 2615 | Mean Difference (IV, Random, 95% CI) | ‐4.91 [‐7.48, ‐2.34] |
| 16.5.2 CAPS subscale: Re‐experiencing/intrusion | 8 | 1813 | Mean Difference (IV, Random, 95% CI) | ‐1.86 [‐2.82, ‐0.90] |
| 16.5.3 CAPS subscale: Avoidance/numbing | 8 | 1813 | Mean Difference (IV, Random, 95% CI) | ‐3.68 [‐4.82, ‐2.54] |
| 16.5.4 CAPS subscale: Hyperarousal | 8 | 1813 | Mean Difference (IV, Random, 95% CI) | ‐2.45 [‐3.30, ‐1.59] |
| 16.6 CAPS: Individual SSRI agents | 14 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 16.6.1 Citalopram versus placebo | 1 | 35 | Mean Difference (IV, Random, 95% CI) | 4.78 [‐15.95, 25.51] |
| 16.6.2 Fluoxetine versus placebo | 1 | 59 | Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐12.31, 10.51] |
| 16.6.3 Paroxetine versus placebo | 6 | 1375 | Mean Difference (IV, Random, 95% CI) | ‐6.32 [‐11.49, ‐1.14] |
| 16.6.4 Sertraline versus placebo | 6 | 1133 | Mean Difference (IV, Random, 95% CI) | ‐4.45 [‐7.17, ‐1.73] |
| 16.7 Symptom severity: Other measures | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 16.7.1 Reduction of PTSD symptoms | 6 | 862 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.32 [‐0.55, ‐0.09] |
| 16.8 Self‐rated scales | 10 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 16.8.1 Total score | 10 | 1698 | Std. Mean Difference (IV, Random, 95% CI) | 0.11 [‐0.50, 0.72] |
| 16.8.2 Self‐rated subscale: Re‐experiencing/Intrusion | 5 | 986 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.58, ‐0.03] |
| 16.8.3 Self‐rated subscale: Avoidance/numbing | 5 | 985 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.58, ‐0.04] |
| 16.8.4 Self‐rated subscale: Hyperarousal | 4 | 805 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.48 [‐0.98, 0.01] |
| 16.9 Depression Scale (typically Hamilton Depression) | 8 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 16.9.1 Reduction of depression symptoms | 3 | 270 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.44, 0.04] |
| 16.9.2 Depression Scale ‐ Change scores | 5 | 1551 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐0.43, ‐0.06] |
| 16.10 Anxiety ‐ Hamilton Anxiety Scale | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 16.10.1 Reduction of anxiety symptoms | 3 | 606 | Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐1.84, 1.41] |
| 16.11 Sheehan Disability Scale | 6 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 16.11.1 Reduction of functional disability | 6 | 1336 | Mean Difference (IV, Random, 95% CI) | ‐2.10 [‐3.00, ‐1.21] |
| 16.12 Drop‐out rate due to any cause | 21 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.12.1 Dropout rate | 21 | 3206 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.87, 1.03] |
| 16.13 Continuation trials | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 16.13.1 Symptom severity on the CAPS | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 9.62 [‐3.53, 22.77] |
Comparison 17. TCAs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 17.1 Clinical Global Impressions Scale Improvement Item (CGI‐I) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1.1 No. of responders | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.38, 0.96] |
| 17.2 Drop‐out rate due to treatment emergent adverse effects | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.2.1 Dropout rate | 3 | 141 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.81, 1.05] |
| 17.3 Self‐rated scales | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 17.3.1 Total score | 2 | 74 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.54 [‐1.18, 0.10] |
| 17.3.2 Self‐rated subscale: Re‐experiencing/Intrusion | 2 | 74 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.44 [‐0.99, 0.11] |
| 17.3.3 Self‐rated subscale: Avoidance/numbing | 2 | 74 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐1.51, 1.04] |
| 17.4 Depression Scale (typically Hamilton Depression) | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 17.4.1 Reduction of depression symptoms | 2 | 74 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.58, 0.25] |
| 17.5 Anxiety ‐ Hamilton Anxiety Scale | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 17.5.1 Reduction of anxiety symptoms | 1 | 33 | Mean Difference (IV, Random, 95% CI) | ‐7.60 [‐12.74, ‐2.46] |
| 17.6 Anxiety ‐ Other scales | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 17.6.1 Reduction of anxiety symptoms | 1 | 41 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.46 [‐1.08, 0.17] |
| 17.7 Drop‐out rate due to any cause | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.7.1 Dropout rate | 2 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.62, 0.99] |
Comparison 18. Total effect of medication versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 18.1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar) scale: no. of responders | 19 | 1822 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.64, 0.85] |
| 18.1.1 Anticonvulsants | 4 | 315 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.63, 1.21] |
| 18.1.2 Antipsychotics | 2 | 43 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.16, 1.67] |
| 18.1.3 MAOIs | 2 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.44, 1.44] |
| 18.1.4 GR205171 | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.42, 1.44] |
| 18.1.5 GSK561679 | 1 | 128 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.88, 1.31] |
| 18.1.6 SSRIs | 8 | 1078 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.59, 0.74] |
| 18.1.7 TCAs | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.37, 0.94] |
| 18.2 Drop‐out rate due to treatment emergent adverse effects | 37 | 4212 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.96, 1.00] |
| 18.2.1 Alpha‐blockers | 1 | 304 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.91, 1.08] |
| 18.2.2 Anticonvulsants | 5 | 348 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.92, 1.05] |
| 18.2.3 Antipsychotics | 5 | 174 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.85, 1.03] |
| 18.2.4 Benzodiazepines | 1 | 32 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.89, 1.12] |
| 18.2.5 Dopamine beta‐hydroxylase inhibitors | 1 | 91 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.54, 1.41] |
| 18.2.6 Ganaxolone | 1 | 112 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.94, 1.11] |
| 18.2.7 GR205171 | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.91, 1.10] |
| 18.2.8 MAOIs | 2 | 159 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.91, 1.04] |
| 18.2.9 NaSSAs | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.68, 1.11] |
| 18.2.10 SARIs | 1 | 42 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.70, 1.09] |
| 18.2.11 SNRIs | 2 | 687 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.88, 1.10] |
| 18.2.12 SSRIs | 13 | 2047 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.96, 1.00] |
| 18.2.13 TCAs | 3 | 141 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.81, 1.05] |
| 18.3 Clinically Administered PTSD Scale (CAPS): CAPS Total | 34 | 4639 | Mean Difference (IV, Random, 95% CI) | ‐4.79 [‐6.64, ‐2.94] |
| 18.3.1 Alpha‐blockers | 1 | 271 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐5.13, 5.33] |
| 18.3.2 Anticonvulsants | 5 | 411 | Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐6.54, 5.84] |
| 18.3.3 Antipsychotics | 3 | 129 | Mean Difference (IV, Random, 95% CI) | ‐14.10 [‐22.57, ‐5.62] |
| 18.3.4 Dopamine beta‐hydroxylase inhibitors | 1 | 86 | Mean Difference (IV, Random, 95% CI) | ‐5.27 [‐16.72, 6.18] |
| 18.3.5 Ganaxolone | 1 | 99 | Mean Difference (IV, Random, 95% CI) | ‐2.50 [‐10.92, 5.92] |
| 18.3.6 GR205171 | 1 | 39 | Mean Difference (IV, Random, 95% CI) | ‐7.60 [‐22.59, 7.39] |
| 18.3.7 Hypnotics | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐1.00 [‐19.57, 17.57] |
| 18.3.8 MAOIs | 2 | 178 | Mean Difference (IV, Random, 95% CI) | ‐5.06 [‐15.93, 5.81] |
| 18.3.9 NaSSAs | 2 | 419 | Mean Difference (IV, Random, 95% CI) | ‐4.38 [‐10.32, 1.56] |
| 18.3.10 SARIs | 1 | 41 | Mean Difference (IV, Random, 95% CI) | ‐5.60 [‐21.26, 10.06] |
| 18.3.11 SNRIs | 2 | 687 | Mean Difference (IV, Random, 95% CI) | ‐8.11 [‐12.29, ‐3.92] |
| 18.3.12 SSRIs | 14 | 2263 | Mean Difference (IV, Random, 95% CI) | ‐4.84 [‐7.64, ‐2.05] |
| 18.4 Drop‐out rate due to any cause | 48 | 5302 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.90, 1.01] |
| 18.4.1 Alpha‐blockers | 1 | 303 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.89, 1.11] |
| 18.4.2 Anticonvulsants | 7 | 447 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.90, 1.16] |
| 18.4.3 Antipsychotics | 4 | 133 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.93, 1.66] |
| 18.4.4 Benzodiazepines | 1 | 16 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.39, 1.89] |
| 18.4.5 Dopamine beta‐hydroxylase inhibitors | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.75, 1.12] |
| 18.4.6 Ganaxolone | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.65, 1.02] |
| 18.4.7 GR205171 | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [1.03, 2.41] |
| 18.4.8 Hypnotics | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.39, 1.31] |
| 18.4.9 MAOIs | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.71, 1.35] |
| 18.4.10 NaSSAs | 3 | 200 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.88, 1.12] |
| 18.4.11 NK‐1 receptor antagonists | 1 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.32, 1.04] |
| 18.4.12 SARIs | 2 | 101 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.68, 0.97] |
| 18.4.13 SNRIs | 1 | 329 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.90, 1.21] |
| 18.4.14 SSRIs | 21 | 3206 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.87, 1.03] |
| 18.4.15 TCAs | 2 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.62, 0.99] |
Comparison 19. Head‐to‐head comparisons.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 19.1 Drop‐out rate due to treatment emergent adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 19.1.1 Reboxetine versus fluvoxamine | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.42, 1.00] |
| 19.2 Clinician administered scales: Symptom severity | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 19.2.1 Nefazodone versus sertraline | 2 | 80 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.19 [‐0.63, 0.25] |
| 19.2.2 Venlafaxine versus sertraline | 1 | 352 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.22, 0.20] |
| 19.2.3 Reboxetine versus fluvoxamine | 1 | 28 | Std. Mean Difference (IV, Random, 95% CI) | 0.67 [‐0.11, 1.45] |
| 19.3 Comorbid symptoms: Depression | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 19.3.1 Nefazodone versus sertraline | 1 | 26 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.09 [‐0.86, 0.68] |
| 19.3.2 Reboxetine versus fluvoxamine | 1 | 28 | Std. Mean Difference (IV, Random, 95% CI) | 0.67 [‐0.11, 1.45] |
| 19.4 Comorbid symptoms: Anxiety (Hamilton Anxiety Scale) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 19.4.1 Nefazodone versus sertraline | 1 | 26 | Mean Difference (IV, Random, 95% CI) | ‐3.23 [‐10.90, 4.44] |
| 19.4.2 Reboxetine versus fluvoxamine | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 1.80 [‐1.30, 4.90] |
Comparison 20. Subgroup analyses ‐ Methodological criteria.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 20.1 Single versus multi‐centre trials (CGI‐I: No. of responders) | 20 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 20.1.1 Single centre trials | 8 | 284 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.55, 0.81] |
| 20.1.2 Multi‐centre trials | 12 | 1563 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.63, 0.91] |
| 20.2 Single versus multi‐centre trials (CAPS: Total score) | 30 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 20.2.1 Single centre trials | 8 | 269 | Mean Difference (IV, Random, 95% CI) | ‐2.04 [‐7.91, 3.83] |
| 20.2.2 Multi‐centre trials | 22 | 3636 | Mean Difference (IV, Random, 95% CI) | ‐5.60 [‐7.84, ‐3.37] |
| 20.3 Industry versus non‐industry funded trials (CGI‐I: No. of responders) | 19 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 20.3.1 Industry funded trials | 12 | 1360 | Risk Ratio (M‐H, Random, 95% CI) | 1.54 [1.25, 1.90] |
| 20.3.2 Non‐industry funded trials | 7 | 373 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [1.00, 1.86] |
| 20.4 Industry versus non‐industry funded trials (CAPS: Total score) | 29 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 20.4.1 Industry funded trials | 20 | 3015 | Mean Difference (IV, Random, 95% CI) | ‐6.29 [‐9.09, ‐3.49] |
| 20.4.2 Non‐industry funded trials | 9 | 675 | Mean Difference (IV, Random, 95% CI) | ‐2.43 [‐5.64, 0.79] |
Comparison 21. Subgroup analyses ‐ Clinical criteria.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 21.1 Inclusion of major depression vs. non‐inclusion: (CGI‐I: No. of responders) | 13 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 21.1.1 Trials including patients with major depressive disorder | 12 | 1127 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.60, 0.86] |
| 21.1.2 Trials excluding patients with major depressive disorder | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.10, 0.86] |
| 21.2 Inclusion of major depression vs. non‐inclusion: (CAPS: Total score) | 23 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 21.2.1 Trials including patients with major depressive disorder | 18 | 1959 | Mean Difference (IV, Random, 95% CI) | ‐6.40 [‐9.36, ‐3.44] |
| 21.2.2 Trials excluding patients with major depressive disorder | 5 | 866 | Mean Difference (IV, Random, 95% CI) | ‐9.34 [‐14.51, ‐4.18] |
| 21.3 Inclusion of war veterans versus non‐inclusion: (CGI‐I: No. of responders) | 18 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 21.3.1 Trials including war veterans | 8 | 323 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.59, 0.89] |
| 21.3.2 Trials without war veterans | 10 | 1264 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.58, 0.84] |
| 21.4 Inclusion of war veterans versus non‐inclusion: (CAPS: Total score) | 22 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 21.4.1 Trials including war veterans | 8 | 1186 | Mean Difference (IV, Random, 95% CI) | ‐3.65 [‐7.27, ‐0.02] |
| 21.4.2 Trials without war veterans | 14 | 1653 | Mean Difference (IV, Random, 95% CI) | ‐8.26 [‐10.76, ‐5.76] |
Comparison 22. Sensitivity analyses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 22.1 Clinical Global Impressions Scale Improvement Item (CGI‐I): non‐response | 20 | 1847 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.62, 0.83] |
| 22.1.1 Anticonvulsants | 4 | 315 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.46, 1.21] |
| 22.1.2 Antipsychotics | 2 | 43 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.16, 1.67] |
| 22.1.3 MAOIs | 2 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.44, 1.44] |
| 22.1.4 GR205171 | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.42, 1.44] |
| 22.1.5 GSK561679 | 1 | 128 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.88, 1.31] |
| 22.1.6 NASSAs | 1 | 26 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.22, 0.94] |
| 22.1.7 SSRIs | 8 | 1078 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.59, 0.74] |
| 22.1.8 TCAs | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.38, 0.96] |
| 22.2 "Worst case" loss to follow up analysis | 13 | 1555 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.69, 1.00] |
| 22.2.1 Anticonvulsants | 2 | 284 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.84, 1.27] |
| 22.2.2 Antipsychotics | 1 | 15 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.27, 3.72] |
| 22.2.3 MAOIs | 2 | 175 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [1.07, 1.63] |
| 22.2.4 SSRIs | 7 | 1038 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.62, 0.78] |
| 22.2.5 TCAs | 1 | 43 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.45, 1.01] |
| 22.3 "Best case" loss to follow up analysis | 13 | 1498 | Risk Ratio (M‐H, Random, 95% CI) | 1.41 [1.12, 1.78] |
| 22.3.1 Anticonvulsants | 2 | 246 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.72, 1.18] |
| 22.3.2 Antipsychotics | 1 | 15 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.42, 2.40] |
| 22.3.3 MAOIs | 2 | 172 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.06, 3.86] |
| 22.3.4 SSRIs | 7 | 1022 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [1.43, 1.86] |
| 22.3.5 TCAs | 1 | 43 | Risk Ratio (M‐H, Random, 95% CI) | 3.50 [1.13, 10.81] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ahmadpanah 2014.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre trial, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Attending the psychology section or the infirmary of Farshchian Hospital in Hamadan" Sample size: 100 randomised to prazosin, hydroxyzine and placebo Mean age: 35.50 years (SD, 6.40). Sex: 72 men and 28 women, car accident (37%); Persian Gulf war (51%); disaster (4%); other (7%). Diagnostic measure: DSM‐IV TR Inclusion criteria: Quote: “Inclusion criteria were as follows: (1) a diagnosis of PTSD according to the diagnostic criteria of the DSM‐IV TR, (2) severe sleep disorders and (3) age between 18 and 45 years" Exclusion criteria: Quote: “Exclusion criteria were the following: (1) further psychiatric comorbidities such as major depressive disorder, anxiety disorders, substance abuse (alcohol, drugs), psychosis and personality disorders, (2) women who were pregnant or intending to get pregnant or who were breast‐feeding, (3) known physical illness such as heart disease and (4) adverse experience with prazosin (hypotension caused by prazosin injection) or hydroxyzine (2 patients were subsequently excluded from the study because of symptoms of hypotension due to prazosin). Moreover, a sudden dramatic drop of the repeatedly and routinely measured blood pressure was a further criterion to exclude a patient from the study" Comorbidity: Not specified Dropout rates: Insufficient evidence to determine dropouts by groups |
|
| Interventions | Pharmacological intervention: Quote: “For the prazosin group, prazosin was administered at 1 mg before going to sleep; over the first 10 days, the dosage was increased to 15 mg and remained at this level for the remaining duration of the 8‐week study. Hydroxyzine dosage was started at 10 mg on the first evening and continually increased over the first 10 days up to 100 mg, remaining thereafter at this level until the end of the study" | |
| Outcomes | Outcomes: PSQI, MINI Time points: Not specified | |
| Notes | Dates of trial: Not stated Industry‐funded: Not specified Medication provided by industry: Not specified Any of the authors work for industry? No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "First, a ballot box containing 3 different colored chips was prepared (35 blue chips for placebo, 35 red chips for prazosin and 35 white chips for hydroxyzine). Patients drew a chip consecutively and received the corresponding compounds" |
| Allocation concealment (selection bias) | Low risk | Quote: "The pharmacy provided the compounds with the same appearance and with no recognizable labels or signs on them, so that the patient, their psychologist and even the nurses could not know the contents" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The pharmacy provided the compounds with the same appearance and with no recognizable labels or signs on them, so that the patient, their psychologist and even the nurses could not know the contents" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient evidence to determine dropouts by groups Quote: "There were no significant differences between the three groups with respect to age, gender distribution, type of experienced trauma, educational level, duration of illness or, most importantly, main medication" ITT sample was used at baseline and endpoint |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified |
Baker 1995.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, comparative, parallel, flexible dose, double‐blind
Duration of intervention: 12 weeks
Placebo run‐in: single‐blind placebo run‐in excluded 28 placebo‐responders Post‐treatment: Not specified |
|
| Participants | Setting: United States of America Sample size: 118 randomised to brofaromine and placebo Mean age: 44 years (23 ‐ 73). Sex: 92 men and 22 women (for 114 participants), MDD not present, 60% combat‐related Diagnostic measure: DSM‐III‐R Inclusion criteria: Quote: “One hundred and forty‐six outpatients of both sexes, civilians and veterans, ages 23 73, all with PTSD, were enrolled in the study. In addition to meeting DSM‐III‐R criteria for PTSD, patients were required to have a minimum Clinician Administered PTSD Scale (CAPS) score of 45, a maximum Montgomery‐Asberg Depression Scale (MADRS) score of 22 and to be symptomatic for at least 6 months. The types of trauma reported were varied” Exclusion criteria: Quote: “Women of child‐bearing potential were excluded, as were those with comorbid medical or psychiatric conditions, at immediate risk of suicide, in active pursuit of compensation, receiving other forms of active treatment such as psychotherapy, or with a known sensitivity to MAOIs. Participating patients could not receive psychotropic medication except for low‐dose choral hydrate, diphenhydramine, hydroxyzine, and benzodiazepines under specified conditions. Placebo responders, i.e., patients who showed a 30% or more improvement in the CAPS score between the screening and baseline visits, were excluded, leaving a total of 118 patients entered into the active treatment phase of the study” Comorbidity: MADRS Dropouts: 35/118 (insufficient information to determine dropout rates for the 2 groups separately) |
|
| Interventions | Pharmacological intervention: Quote: “The 12‐week trial, conducted at 12 centers throughout the country, utilized a randomized, double‐blind, flexible dose, comparative design with two parallel groups. Eligible patients were randomized to receive either brofaromine, titrated up to 150 mg, or placebo" | |
| Outcomes | Primary outcome: CAPS Secondary outcomes: IES, DTS, CGI Time points: Quote: “Safety and efficacy assessments were performed during the screening visit, at baseline, weekly at the end of weeks 1 through 4, and every other week at the end of weeks 6 through 12" | |
| Notes | Dates of trial: Not stated Industry‐funded: Not mentioned Medication provided by industry: No Any of the authors work for industry? Yes Obtained additional HAM‐D and CAPS‐2 summary statistics from Sudie Back |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the patients were randomised, but no mention is made of the method of randomisation Quote: "The 12‐week trial, conducted at 12 centers throughout the country, utilized a randomized, double‐blind, flexible dose, comparative design with two parallel groups" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Large multi‐center, double‐blind, parallel trial to assess the efficacy of brofaromine in the treatment of post traumatic stress disorder (PTSD) failed to show a significant difference between the brofaromine and placebo treatment groups" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to determine dropout rates for the 2 groups separately. Overall 29% of the participants dropped out of the study. Reasons for treatment withdrawal were provided for only 12 participants in the treatment group and only reasons given for 6 participants in the placebo group. The characteristics of the sample did not differ at baseline. All analyses were intention‐to‐treat (ITT) Quote: “Treatment groups were well matched on demographic and baseline variables, although patients in the placebo group were somewhat younger. Of the 35 patients who discontinued the study prematurely, ten brofaromine and six placebo patients did so because of adverse experiences with the medication. One patient in the brofaromine study was discontinued because of abnormal laboratory values” |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified |
Becker 2007.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "Outpatient clinics at the Durham Veteran’s Administration Medical Center and the community through approved flyers advertising the study" Sample size: 30 randomised to bupropion SR and placebo Mean age: 50.39 years (SD, 7.46) Sex: 24 men and 6 women; war trauma (n = 14), domestic violence (n = 2), rape (n = 1), motor vehicle accident (n = 2), homicide (n = 2), medical illness (n = 3), death/suicide of a loved one (n = 2), and childhood sexual or physical abuse (n = 2) Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Enrolled patients were medically stable, fulfilled the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM‐IV) criteria for PTSD, spoke English, and completed written informed consent. Participants already prescribed other psychiatric medications were included if they were considered medically stable on the dosage” Exclusion criteria: Quote: “Exclusion criteria included ulcers, seizure disorder, psychosis, bipolar disorder, eating disorders, pregnancy or lactation, and current drug/alcohol abuse or dependence. In addition, women of childbearing potential had a blood human chorionic gonadotropin pregnancy test to ensure that no one who was pregnant was enrolled" Comorbidity: Not specified Dropouts: 2 dropped out (not specified by groups) |
|
| Interventions | Pharmacological intervention: Quote: “Study medication was dispensed as follows: bupropion SR was begun at 100 mg every morning for the first 2 weeks. If indicated, dosing was then increased to 100 mg twice daily. At Week 4, if no significant improvement was shown, all patients were then prescribed a maximum dosage of 150 mg twice daily (300 mg/d). Patients were allowed to be maintained on a previously prescribed antidepressant" | |
| Outcomes | Outcomes: CAPS, DTS, PSQI, BDI, PANAS,7‐item Short PTSD Rating Interview, CGI‐I, Severity of Symptoms Scale (not clear whether primary or secondary) Time points: Quote: “Patients were evaluated at screening, baseline, and at weeks 2, 4, 6, and 8". Severity of Symptoms Scale: Quote: “Patients completed this checklist at baseline and at all subsequent visits" | |
| Notes | Dates of trial: Quote: "Enrollment began in June 2003, with completion in July 2005" Industry‐funded: Yes. Quote: "Preparation of this article was supported by an investigator‐initiated award from GlaxoSmithKline and Veterans Affairs Merit Awards MH‐0018, R01CA81595, R01MH062482, and K24DA016388" Medication provided by industry: Yes. Quote: "GlaxoSmithKline provided bupropion SR and placebo pills" Any of the authors work for industry? Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Patients were randomized to a double‐blind treatment with bupropion SR or placebo in a 2:1 ratio" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "The current study is a double‐blind placebo‐controlled trial of men and women with PTSD due to a range of trauma exposures using PTSD, depression, and sleep measures" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was insufficient information to determine dropout rates for the 2 groups separately. However, dropout rates were low (n = 2; 7%) Quote: “Initially, data analysis was conducted on the 23 participants (77%) who completed Week 8. Because only participants with a baseline and at least 1 postbaseline visit were included in end point analyses, intent‐to‐treat analysis was then conducted on 28 study participants. This group consisted of the original 23 study participants and 5 additional patients: 2 patients who completed Week 2, 2 who completed Week 4, and 1 who completed Week 6. Reasons for discontinuing with the study include allergic reaction (1), transportation difficulties (1), no further interest (1), and lost to contact (2). Accordingly, an intent‐to‐treat statistical analysis was used to include those participants with different end points. There were no significant statistical differences between the groups for age, sex, race, employment, education, or marital status" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding and medication was provided by industry Quote: "Preparation of this article was supported by an investigator‐initiated award from GlaxoSmithKline and Veterans Affairs Merit Awards MH‐0018, R01CA81595, R01MH062482, and K24DA016388. GlaxoSmithKline provided bupropion SR and placebo pills" |
Brady 2000.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel, flexible dose, double‐blind
Duration of intervention: 12 weeks
Placebo run‐in: single‐blind 2‐week placebo run‐in Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "The research was conducted at outpatient psychiatric clinics in 8 academic medical centers and 6 clinical research centers" Sample size: 187 randomised to sertraline and placebo Mean age: 39.9 years (18 ‐ 69) Sex: 50 men and 137 women, 6% combat‐related, with current major depression Diagnostic measure: DSM‐III‐R Inclusion criteria: Quote: “The subjects were male and female outpatients aged 18 years and older who met DSM‐III‐R criteria for a principal diagnosis of PTSD as determined by part 1 of the CAPS. A minimum 6‐month duration of PTSD illness was required, as well as a total severity score of at least 50 on the CAPS, part 2 at the end of a 2‐week placebo run‐in period; subjects were at least moderately ill. All patients were required to be free of psychotropic medication for at least 2 weeks prior to beginning treatment. Women’s participation was contingent on negative results of a beta‐human chorionic gonadotrophin pregnancy test and stable use of a medically accepted form of contraception” Exclusion criteria: Quote: “Exclusion criteria: (1) current or past history of bipolar, schizophrenic, or other psychotic disorder; (2) current organic mental disorder, factitious disorder or malingering, or primary diagnosis of major depression, obsessive compulsive disorder, or other anxiety disorders; (3) alcohol or substance dependence or abuse in the past 6 months; (4) evidence of clinically significant hepatic or renal disease or any other acute or unstable medical condition that might interfere with the safe conduct of the study; (5) intolerance or hypersensitivity to sertraline or nonresponse to previous adequate trial; (6) current use of any medication with clinically significant psychotropic activity within 2 weeks of randomisation; (7) cognitive behavioural therapy during the trial; and (8) psychotherapy that was initiated or that ended during the trial” Comorbidity: No information Dropouts: 35/187 (29/94 in the sertraline and 25/93 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Treatment was initiated at 25mg/d for 1 week, with flexible daily dosing and 50‐200 mg/d thereafter, based on clinical response and tolerability. Dosing changes were made in 50‐mg increments at no less than weekly intervals unless required for safety'' | |
| Outcomes | Primary outcome: CGI‐I, CGI‐S, CAPS‐2, IES. Responders defined as 30% or greater decrease on CAPS‐2 and CGI‐I of 1 or 2 Secondary outcomes: DTS, HAM‐D, Q‐LES‐Q Time points: Quote: “Primary outcome assessments were performed at baseline and at study treatment weeks, 1, 2, 3, 4, 6, 8, 10, 12. Secondary outcome assessments were performed at baseline and at the end of study treatment weeks 2, 4, 6, 8, 10, and 12" | |
| Notes | Dates of trial: Not stated. Industry‐funded: Yes, quote: "This study was funded by Pfizer Inc" Medication provided by industry? Yes. Any of the authors work for industry? Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Patients were randomized to acute treatment with sertraline hydrochloride in flexible daily dosages of 50 to 200 mg/d, following 1 week at 25 mg/d (n=94); or placebo (n=93)" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Following a 2‐week, single‐blind placebo run‐in period, patients were randomized to 12 weeks of double‐blind parallel treatment with either sertraline or matched placebo" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Twelve‐week, double‐blind, placebo‐controlled trial preceded by a 2‐week, single‐blind placebo lead‐in period, conducted between May 1996 and June 1997" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the sertraline group (29/94, 31%) compared to the placebo group (25/93; 27%) Quote: "A total of 187 patients were randomly assigned to sertraline (n=94) or placebo (n=93), of whom 93 (99%) sertraline‐treated patients and 90 (97%) placebo‐treated patients were available for at least 1 postrandomization efficacy assessment. Efficacy analyses were performed on the latter group, omitting the 1 patient taking sertraline and the 3 patients taking placebo who were unavailable for postrandomization assessment. For the total randomized sample there were no significant differences between the treatment groups in any of the baseline demographic and clinical characteristics. ITT sample was used at baseline and endpoint with LOCF" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This study was funded by Pfizer Inc" |
Braun 1990.
| Study characteristics | ||
| Methods | Design: single‐centre, randomised, placebo‐controlled, cross‐over, flexible dose, double‐blind
Duration of intervention: 5 weeks
Placebo run‐in: 2 week titrated placebo washout; 4‐day tapering of medication at end of trial Post‐treatment: Not specified |
|
| Participants | Setting: Jerusalem Health Center Ezrath Nashim Hospital Outpatient Clinic Sample size: 16 randomised to alprazolam and placebo Mean age: 37.7 years (19 ‐ 56), 25% combat‐veterans, 13% (2/16) MDD Sex: not specified Diagnostic measure: DSM‐III Inclusion criteria: Quote: “Patients referred to the Jerusalem Health Centre – Ezrath Nashim Hospital Outpatient Clinic who fulfilled DSM‐III criteria for PTSD were eligible for the study, provided that they were physically healthy, had not suffered significant head injury, and were able and willing to give written informed consent. All participants were free of psychotropic medication for at least 2 weeks before commencing treatment. During that period, the diagnosis was confirmed by semi‐structured interview, and baseline psychiatric ratings” Exclusion criteria: Not specified Comorbidity: None Dropouts: 6/16 (3/7 in the alprazolam and 3/9 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “The patients were randomly assigned to treatment with alprazolam or placebo. The initial alprazolam dose of 1.5 mg/day in divided doses was increased to the maximum tolerated dose (up to 6 mg/day) within 2 weeks. The maximum dose was maintained for a further 3 weeks. The alprazolam dose was then tapered and discontinued over the next 2 weeks with substitution of placebo. Those patients which received placebo in the first leg of the trial then commenced alprazolam treatment, while those who has received alprazolam commenced placebo. The second leg of the trial lasted 5 weeks. The complete trials thus extended 12 weeks, 5 weeks for each experimental leg separated by a 2‐week interim phase" | |
| Outcomes | Outcomes: DSM‐based PTSD scale, IES, HAM‐D, HAM‐A, Visual Analogue (not clear which outcomes are secondary or primary) Time points: Baseline and at 5 weeks Data estimation: Observed cases for participants who completed 5 weeks of both phases |
|
| Notes | Dates of trial: Not stated. Industry‐funded: Unclear Medication provided by industry: Unclear Any of the authors work for industry? Unclear |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "The authors report a random‐assignment, double‐blind, cross crossover trial comparing alprazolam and placebo in posttraumatic stress disorder (PTSD)" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "The authors report a random‐assignment, double‐blind, cross crossover trial comparing alprazolam and placebo in posttraumatic stress disorder (PTSD)" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Clinical response was monitored by means of rating scales administered at baseline and then weekly throughout the trial by a rater who was blind to treatment assignment (P.B.)" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of participants withdrew from the alprazolam group (3; 43%) compared to the placebo group (3; 33%). Sample characteristics were provided in a table for 10 participants at baseline only. Reasons for withdrawals were not provided. Observed cases for patients who completed 5 weeks of both phases were provided Quote: "Six subjects dropped out during this phase: 3 alprazolam‐treated patients withdrew after 1, 4, and 5 weeks, respectively, and 3 placebo‐treated patients withdrew after 1, 3, and 4 weeks. None of the dropout complained of adverse effect" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified |
Butterfield 2001.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 10 weeks Placebo run‐in: not specified Post‐treatment: Not specified |
|
| Participants | Setting: Duke University Medical Center Outpatient Psychiatry Service and the Durham Women Veteran’s Comprehensive Health Center Sample size: 15 randomised to olanzapine and placebo Mean age: 43.2 years (26 ‐ 73) Sex: 1 men and 15 women, 53.3% (8/15) MDD Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Patients aged 18‐70 years were recruited from Duke University Medical Center Outpatient Psychiatry Service and the Durham Women Veteran’s Comprehensive Health Center. Enrolled patients were medically stable, fulfilled the DSM‐IV criteria for PTSD, understood English, and were able to give their written informed consent". Exclusion criteria: Quote: “Exclusion criteria included a history of bipolar disorder, a psychotic disorder, mental retardation, a recent history of alcohol substance use disorder and serious suicidal or violence risk. The study began in April 1998 and was completed in February 1999” Comorbidity: MINI: MDD,GAD, PD Dropouts: 4/12 (3/7 in the olanzapine and 1/5 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Olanzapine was started at a daily dosage of 5 mg and subsequently increased to 10 mg, 15 mg and, finally, to a potential maximum daily dosage of 20 mg at weeks 1, 4 and 6, respectively. Tolerance was assessed by evaluating compliance with the dose, the percentage of patients reporting treatment‐related events and assessment with the Barnes Akathisia Scale and the Abnormal Involuntary Movement Scale at weeks 6 and 10 of the study. Weight gain was measured and side effects were recorded at baseline and at each subsequent visit" | |
| Outcomes | Primary outcome: TOP‐8, SPRINT
Secondary outcomes: IES, DTS, CGI‐I, SDS, BAS, AIMS
Time points: Quote: “Psychometric measures were administered at baseline and weekly for one month and then at weeks 4, 6, 8 and 10" Data estimation: ITT (General linear model) |
|
| Notes | Dates of trial: Quote: "The study began in April 1998 and was completed in February 1999" Industry‐funded: Yes, Quote: "This study was supported in part by a grant from Eli Lilly, Inc" Medication provided by industry: No Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Because the atypical antipsychotic olanzapine may be efficacious in treating post‐traumatic stress disorder (PTSD) symptoms, we conducted a 10‐week, double‐blind, placebo‐controlled evaluation in which 15 patients were randomized 2: 1 to either olanzapine or placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Because the atypical antipsychotic olanzapine may be efficacious in treating post‐traumatic stress disorder (PTSD) symptoms, we conducted a 10‐week, double‐blind, placebo‐controlled evaluation in which 15 patients were randomized 2: 1 to either olanzapine or placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More participants withdrew from the olanzapine group (3/7; 43%) compared to the placebo (1/5; 20%) group. Reasons for withdrawal were not explained. No information was provided on sample characteristics at endpoint. ITT population Quote: "Four patients dropped out during the trial. Our study sample comprised primarily women, many of whom had rape‐related PTSD and psychiatric comorbidities (major depression, panic disorder or generalized anxiety disorder. Intent‐to‐treat efficacy analyses were conducted as a general linear model (GLM). analysis of variance, with treatment as a grouping variable and time as a repeated measure" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This study was supported in part by a grant from Eli Lilly, Inc" |
Carey 2012.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: 1 week of single‐blind placebo run‐in Post‐treatment: Not specified |
|
| Participants | Setting: Primary care clinics, South Africa. Sample size: 28 randomised to olanzapine and placebo Mean age: 40.75 years Sex: 11 men and 17 women Diagnostic measure: DSM‐IV and MINI Inclusion criteria: Quote: “Men and women aged 18 years and over with DSM‐IV, non‐combat, chronic PTSD (PTSD symptoms of at least 3 months) were eligible to participate in the study if they (i) had a minimum score of 50 on the Clinician Administered PTSD Scale (CAPS), (ii) were willing to provide written informed consent to their participation, and (iii) were free of disallowed psychotropic medication for a washout period of 5 days for all except fluoxetine (5 weeks) prior to randomization. Trauma types reflect the profile of trauma in South Africa (i.e. domestic violence and criminal violence". Exclusion criteria: Quote: “Exclusion criteria included current major depressive disorder, a Montgomery Äsberg Depression Rating Scale (MADRS) score ≥20 at baseline, and the presence of a significant suicide risk according to the clinical judgement of the investigator. Additional exclusion criteria included a substance use disorder within 6 months of randomization, a positive urine drug screen for illicit substances, a history of severe personality disorder (based on clinician judgement), a lifetime history of schizophrenia or other psychotic disorder, pregnant or breastfeeding women, women of child‐bearing potential not willing to use contraception, an unstable medical condition, and unresolved clinically significant laboratory or electrocardiogram findings. Previous failure to respond to or intolerance of a second generation antipsychotic (SGA), failure of two or more trials of an SSRI or an SNRI given in adequate doses for an adequate duration, and initiation or change in psychotherapy within 8 weeks of screening receipt of electroconvulsive therapy in the 3 months before screening, participation in a clinical trial in the 6 months before screening, and finally an improvement of 2 or more points on the Clinical Global Impressions (CGI) severity score from screening to randomization were also criteria for exclusion from participation”. Comorbidity: MADRS (but participants with comorbid severe depression were excluded), DTS, SDS Dropouts: 10/34 (insufficient information to determine dropout rates for the 2 groups separately) |
|
| Interventions | Pharmacological intervention: Quote: “Following randomization, participants received 5mg olanzapine or matching placebo for 1week. This was followed by 7.5mg for 1week and then 10 mg for 2 weeks. If the response was satisfactory (CG I≤2) and treatment was well tolerated at the end of week four, participants were maintained at that dose. However, if response was not satisfactory (CGI ≥3) but adequately tolerated, olanzapine/placebo was increased in 2.5mg increments two weekly to a maximum dose of 15 mg. In cases of poor tolerability following a dose increase, a single dose reduction of 2.5 mg was permitted". | |
| Outcomes | Outcomes: CAPS, CGI, MADRS, DTS, SDS (not clear which outcomes are secondary or primary)
Time points: Quote: “Severity of PTSD symptoms was assessed at baseline, week 4, and week 8 using the CAPS. Participants were assessed by clinicians at baseline, then weekly for the first 4weeks and every 2 weeks thereafter, with these symptom measures, and with open‐ended questions regarding tolerability". Data estimation: ITT and LOCF |
|
| Notes | Dates of trial: Not stated. Industry‐funded: Yes. Quote: "Funding for this study was received from Eli Lilly" Medication provided by industry: Yes. Quote: "Matching olanzapine/placebo was supplied by Eli Lilly and then packaged for individual participants by an independent contract pharmacist". Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization codes were kept only by the contracted pharmacist. Following successful screening, eligible participants were randomly assigned to receive either olanzapine or placebo using a computer generated 10‐number block randomization schedule". |
| Allocation concealment (selection bias) | Low risk | Quote: "Matching olanzapine/placebo was supplied by Eli Lilly and then packaged for individual participants by an independent contract pharmacist". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information on the blinding of participants was provided; nothing on whether personnel were blinded, however. Quote: "There was no instance of unblinding until after study closure and after data entry had been completed". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to determine dropout rates for the 2 groups separately. Reasons for treatment withdrawal were provided. No information was provided on sample characteristics at endpoint. Data estimation was based on ITT and LOCF values. Quote: "Of the 10 early withdrawals, four were lost to follow‐up and one participant was withdrawn because of poor/erratic compliance at week 2. Of the remaining five participants, four were withdrawn because of lack of treatment efficacy or withdrawal of consent, and one was withdrawn at week 1 because of severe sedation (olanzapine group). No participants were withdrawn because of a serious adverse event. By using an intent‐to‐treat sample, defined as all participants with at least one post‐baseline CAPS assessment and a last observation carried forward (LOCF) approach, efficacy for olanzapine versus placebo was tested using repeated measures analysis of variance (RMANOVA)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry. Quote: "Funding for this study was received from Eli Lilly" |
Chung 2004.
| Study characteristics | ||
| Methods | Design: single‐centre trial, randomised, open‐label controlled trial, parallel, flexible dose, non‐blinded Duration of intervention: 6 weeks Placebo run‐in: 7‐day washout prior to entering trial Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Inpatients and outpatients of the Seoul Veterans Hospital". Sample size: 113 randomised to mirtazapine and sertraline Mean age: 59.8 years (SD: 6.35) Sex: 113 men, Korean war veterans, 17% (17/100) MDD Diagnostic measure: DSM‐IV, CAPS‐1 Inclusion criteria: Quote: “Male veterans of the Korean or Vietnam war with PTSD as a primary diagnosis according to DSM‐IV and CAPS‐1 criteria and possible co‐morbid major depression or dysthymia, were included in the study if symptoms of depression had been present for more than 3 months". Exclusion criteria: Quote: “Patients were excluded from the study if they had any of the following: (1) schizophrenia, bipolar disorder, organic mental disorder, factitious disorder, malingering, obsessive compulsive disorder, or other mental disorders; (2) alcohol or other substance dependency or abuse within previous 6 months; (3) use of neuroleptic drugs within previous 2 weeks; (4) non‐response to previous treatment with mirtazapine for at least 8 weeks; (5) electroshock therapy in the past; (6) clinically significant laboratory or EKG abnormalities; (7) a history of convulsive disorder or treatment with anticonvulsants because of such symptoms; (8) a baseline score below 50 on the CAPS‐2” Comorbidity: MDD, dysthymia, MDD and dysthymia Dropouts: 13/113 (7/58 in the mirtazapine and 6/55 in the sertraline group) |
|
| Interventions | Pharmacological intervention: Quote: “Treatment with mirtazapine and sertraline, respectively, started at 15 mg and 50 mg daily. After 1, 2 and 6 weeks of therapy the drug dosage could be adjusted based on clinical evaluation by psychiatrists" | |
| Outcomes | Outcomes: CAPS‐2, HAM‐D‐17, CGI‐I, CGI‐S (no distinction made between primary and secondary outcomes)
Time points: Quote: “All other assessments were performed at baseline and at weeks 1, 2 and 6" Data estimation: Completer values |
|
| Notes | Dates of trial: Started in October 2001 and ended in August 2002. Industry‐funded: Yes. Quote: "Contract/grant sponsor: Organon and Janssen; contract/grant number: GL063". Medication provided by industry: No Any of the authors work for industry? No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation. Quote: "A total of 95 outpatients and 18 inpatients were randomly assigned to treatment with mirtazapine or sertraline". |
| Allocation concealment (selection bias) | High risk | Not used, open‐label trial |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding of participants or personnel |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of participants withdrew from the mirtazapine group (7; 12%) compared to the placebo group (6; 11%). Reasons for withdrawals were provided. No information was provided on sample characteristics at endpoint. Data estimation was based on completer values. Quote: "The mean age and mean duration of illness in the mirtazapine and sertraline groups were highly comparable. As for the mean age and duration of illness, no statistically significant difference was found between the two groups for marriage status, education level, career, or monthly income. Thirteen subjects dropped out during the study, seven from the mirtazapine group (13.7%), and six from the sertraline group (12.2%). The reasons for dropping out were lack of efficacy (n=3), personal reasons (n=3), side effects (n=1) in the mirtazapine group and lack of efficacy (n=5) and for personal reasons (n=1) in the sertraline group". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry. Quote: "Contract/grant sponsor: Organon and Janssen; contract/grant number: GL063" |
Connor 1999a.
| Study characteristics | ||
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: not specified |
|
| Participants | Setting: Veterans Administration Medical Center Sample size: 54 randomised to fluoxetine and placebo Mean age: 37 (32.44) (median age in years (quartiles)) Sex: 5 men and 49 women, none combat‐related Diagnostic measure: DSM‐III and SCID Inclusion criteria: Quote: “Individuals aged 18‐55 were included if they met DSM‐III‐R criteria for PTSD according to the Structured Clinical Interview for DSM‐III‐R and were civilians" Exclusion criteria: Quote: “Exclusion criteria, also determined by SCID, comprised a history of psychosis, bipolar disorder, antisocial personality disorder, current or recurrent or recent risk of suicide, homicide, and drug or alcohol abuse disorder within the previous six months” Comorbidity: None Dropouts: 16/54 (6/27 in the fluoxetine and 11/27 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Drug or placebo was initiated at a daily dose of 10 mg or its equivalent and increased at a rate of 10 mg per week to a maximum of 60 mg/day as needed and tolerated" | |
| Outcomes | Primary outcome: DGRP (change, severity)
Secondary outcomes: SIP, DTS, SDS, VS
Time points: Quote: “Assessments were conducted at pre‐treatment baseline (week 0) and at weeks 1, 2, 3, 4, 6, 8, 10 and 12" Data estimation: ITT and LOCF |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes Medication provided by industry: Yes. Quote: "Fluoxetine and matching placebo capsules were provided by Lilly Research Laboratories, and packaging was performed by the Duke University Medical Centre Pharmacy" Any of the authors work for industry? No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was achieved by means of computer‐generated group assignment, whose results were available only to the hospital pharmacy" |
| Allocation concealment (selection bias) | Low risk | Quote: "Fluoxetine and matching placebo capsules were provided by Lilly Research Laboratories, and packaging was performed by the Duke University Medical Centre Pharmacy" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Our threefold approach to measurement – with the physician, interviewer and patient rating ‐ minimised the likelihood of halo effaces between scales yet still resulted in differences favouring drug for all three approaches. This strategy may have also helped to offset any possible breaches in the double‐blind – which may occur in any trial – although study blinding was not rigorously assessed" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The Duke was assessed by the treating physician, who was blind to the other outcome measures. The SIP was rated by the study coordinator, while the DTS, SDS, and VS measures were completed by the patient" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More participants withdrew from the placebo group (11/27; 41%) compared to the fluoxetine group (6/27; 22%). Reasons for withdrawal were explained. No information was provided on sample characteristics at endpoint. Data estimation was based on ITT and LOCF values. Quote: "The results indicate broad comparability between the treatment groups with respect to demographic features, age at onset of PTSD, and number of traumatic events. Of the 17 early dropouts, 11 (65%) came from the placebo group, whereas six (36%) came from the fluoxetine group., a non‐significant difference. Reasons for drop‐out in the drug group included lack of efficacy (n=1), loss to follow‐up (n=3), relocation (n=1) and protocol violation (n=1: reported non‐attendance). Subjects in the placebo group dropped out owing to lack of efficacy (n=5), loss to follow‐up (n=1), relocation or job concerns (n=3), and protocol violation (n=2: noncompliance; initiation of treatment with prohibited non‐protocol antidepressant). For all analyses, missing values were filled in by carrying forward the score from the previous week. Outcome analyses are presented here as an intention‐to‐treat analysis, in which the last observation was carried forward to Week 12. However, we conducted an analysis of completer patients at Week 12, with essentially similar results" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified. Medication was provided by industry, but no authors work for industry. |
Connor 2006.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks, 24 weeks Post‐treatment: participants returned for follow‐up assessments at weeks 13, 14, 16, 20, and 24 Placebo run‐in: Quote: "Subjects randomized to placebo were tapered off of tiagabine in decrements of 2 mg every 3 days as they were switched to placebo. Those randomized to tiagabine continued on the same doses of the drug as had been taken during the end of the open‐label phase" |
|
| Participants | Setting: Quote: “Outpatients were recruited through advertisement and clinical referral" Sample size: 18 randomised to tiagabine and placebo Mean age: Not specified Sex: Not specified, excluded MDD Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Inclusion criteria were as follows: age between 18 and 65 years; current diagnosis of PTSD, according to DSM‐IV, as assessed by a structured interview and clinical exam; and CGI severity (CGI‐S) score of at least 4 (i.e., moderate severity) at baseline" Exclusion criteria: Quote: “Subjects who met any of the following criteria were excluded: a primary psychiatric disorder other than PTSD, substance use disorder within the last year, clinically significant unstable medical condition or laboratory abnormality, concurrent use of psychotropic medications or cognitive behavioral therapy, failure to respond to two or more previous trials of pharmacotherapy for PTSD, prior treatment with tiagabine exceeding 8 mg/day, and pregnancy or lactation” Comorbidity: Unclear Dropouts: 5/18 (4/10 in the tiagabine and 1/8 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “The drug was initiated at 2 mg BID for 1 week and titrated upwards in 4‐mg increments (2 mg BID) per week. The dose was titrated as tolerated or until response was attained, to a maximum daily dose of 16 mg/day, and the dose remained fixed after week 8" | |
| Outcomes | Primary outcome: SPRINT, CGI‐S
Secondary outcomes: SIP, DTS, HADS, CD‐RISC, SDI, CGI‐I, CGI‐S, PSQI
Time points: Quote: “In the double‐blind phase, subjects returned for follow up assessments at weeks 13, 14, 16, 20, and 24" Data estimation: Data estimation: ITT and LOCF |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "This study was supported by a grant from Cephalon, Inc. to Dr. Davidson" Medication provided by industry: No Any of the authors work for industry? No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Subjects who demonstrated at least minimal response on the Clinical Global Impressions of Improvement scale (CGI‐I) were randomized 1:1 to 12 weeks of double‐blind treatment, either continuing tiagabine or switching to placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved. Quote: "Subjects who demonstrated at least minimal response on the Clinical Global Impressions of Improvement scale (CGI‐I) were randomized 1:1 to 12 weeks of double‐blind treatment, either continuing tiagabine or switching to placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More participants withdrew from the tiagabine group (4/10; 40%) compared to the placebo (1/8; 13%) group. Reasons for withdrawal were explained. No information was provided on sample characteristics at endpoint. ITT and LOCF estimation. Quote: "Efficacy analyses included all subjects who completed at least one postbaseline visit, who comprised the intent‐to‐treat (ITT) sample, and data reported used the last observation carried forward. Five subjects withdrew during this phase; two were lost to follow‐up (n=1, each for tiagabine and placebo) and three due to lack of further efficacy or clinical deterioration (n=3, tiagabine). In addition, 46% of subjects dropped out of the study before completing the double‐blind phase of treatment. However, when one considers the dropout rate by treatment phase over a total period of 6 months (open label 27% and double‐blind 22%, respectively), our figures are consistent with rates reported in other published trials" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This study was supported by a grant from Cephalon, Inc. to Dr. Davidson" |
Davidson 1990.
| Study characteristics | ||
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Patients were referred to the study either from the inpatient service or from the Mental Hygiene Clinic at the Durham (NC) Veterans Administration Medical Center" Sample size: 46 randomised to amitriptyline and placebo Mean age: Not specified Sex: Not specified, combat veterans, 20% (9/46) MDD Diagnostic measure: DSM‐III Criteria for PTSD (SI‐PTSD) Inclusion criteria: Quote: “To be eligible in the study, patients were required to meet DSM‐III criteria for PTSD, as well as to be free of schizophrenia, bipolar disorder, a history of serious violence within the past 5 years, and any medical condition that would contraindicate the use of amitriptyline" Exclusion criteria: See inclusion criteria Comorbidity: MD, history of PD, GAD and alcohol use Dropouts: 13/46 (8/25 in the amitriptyline and 5/21 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Patients were treated with amitriptyline hydrochloride or placebo at doses that varied between 50‐80 mg/d. The initial starting dose of 50 mg was increased to 75 mg and then to 100 mg within the first week. The dose was then increased to 150 mg then 200 mg before the end of 2 weeks. It could then be increased further if necessary and tolerated" | |
| Outcomes | Primary outcomes: CGI‐I, SI‐PTSD, CGI‐S
Secondary outcomes: HAM‐D, HAM‐A, IES, NI, EPI Time points: Quote: “The duration of treatment was 8 weeks, with the assessments bring made at baseline and weeks 1 (side effects only), 2, 4, 6 and 8" Data estimation: completer values |
|
| Notes | Dates of trial: Not stated Industry‐funded: No Medication provided by industry: No Any of the authors work for industry? No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "The clinical trial employed a balanced randomisation to amitriptyline or placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Medication was administered double‐blind, and no other psychotropic agents were permitted during the trial with the exception of chloral hydrate as necessary" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "The key ratings was performed by a rater uninvolved in prescribing and monitoring medication dose, but who was performing PTSD ratings" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the amitriptyline group (8/25, 32%) compared to the placebo group (5/21; 24%). Reasons for treatment withdrawal were provided. No information was provided on sample characteristics at endpoint. Data estimation was based on completer values. Quote: "Forty (87%) of 46 patients completed the minimum 4 weeks of treatment that was required for inclusion in the efficacy analysis. Thirty‐five patients (76%) completed 6 weeks of treatment and 33 (71%) completed 8 weeks of treatment. Three patients receiving amitriptyline dropped out of the study earlier than 4 weeks: 2 from intolerable side effects and 1 from alcohol intoxication. Three patients receiving placebo also dropped out prior to week 4: 1 due to myocardial infarction, 1 who failed to return at week 2, and 1 from worsening nightmares. In the patients who dropped out at week 4 and 6, their last observations were carried forward to week 8" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified. The trial was supported by a veterans grant, but no other disclosures were made about medication or industry |
Davidson 2001a.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 1 week single‐blind placebo run‐in Post‐treatment: Not specified |
|
| Participants | Setting: Outpatients from 12 collaborating centers Sample size: 208 randomised to sertraline and placebo Mean age: 37.1 years (18 ‐ 69) Sex: 46 men and 162 women, 5% war trauma, 40% (83/208) MDD Diagnostic measure: DSM‐III‐R Inclusion criteria: Quote: “The subjects were male and female outpatients 18 years and older who met DSM‐III‐R criteria for a primary diagnosis pf PTSD as determined by part 1 of the CAPS‐1. A minimum 6‐month duration of PTSD illness was required, as well as a total score of 50 or higher on part 2 of the CAPS‐2 at end of a 1‐week placebo run‐in period. All subjects were required to be free of psychotropic medication for at least 2 weeks before beginning treatment, or 5 weeks for fluoxetine. Female participation was contingent on a negative beta‐human chorionic gonadotropin pregnancy test and stable use of a medically accepted form of contraception" Exclusion criteria: Quote: "Exclusion criteria included (1) current or past history of bipolar, schizophrenic, or other psychotic disorder; (2) current organic mental disorder, factitious disorder, or malingering, or primary diagnosis of major depression; (3) alcohol or substance abuse or dependence in the past 6 months; (4) evidence of clinically significant hepatic or renal disease, or any other acute or unstable medical condition that might interfere with the safe conduct of the study; (5) intolerance or hypersensitivity to sertraline, or nonresponse to a previous adequate trial; and (6) current use of any medication with clinically significant psychotropic properties. Subjects were not permitted to participate in cognitive‐behavioural therapy during the trial, but were permitted to attend on‐going psychotherapy or counselling that was initiated at least three months before randomization" Comorbidity: MD, anxiety disorder, alcohol and substance dependence Dropouts: 67 (32 in the sertraline and 35 in the placebo group; insufficient information to determine 'n' in each group) |
|
| Interventions | Pharmacological intervention: Quote: “Sertraline treatment was initiated at 25 mg per day for 1 week, with flexible daily dosing thereafter in the range of 50 to 200 mg, based on clinical response and tolerability. Study visits took place at baseline and at the end of study treatment weeks 1, 2, 3, 4, 6, 8, 10, and 12, or at the time of discontinuation if before week 12" | |
| Outcomes | Primary outcomes: CAPS‐2, IES, CGI‐S, CGI‐I
Secondary outcomes: DTS, HAM‐D, HAM‐A, PSQI Time points: Quote: “Assessment of primary outcome measures was performed at baseline (except the CGI‐I) and at every study visit. The CAPS‐2 and DTS were administered every study visit, while the HAM‐D, HAM‐A, and the Pittsburgh Sleep Quality Index were administered at baseline and study end point only. Safety assessments took place at week 6 and 12" Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Financial support for this study was provided by Pfizer Inc, New York, NY" Medication provided by industry: No Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "After a 1‐week, single‐blind, placebo run‐in period, subjects were randomized, using computer‐generated random numbers, to 12 weeks of double‐blind, parallel treatment with either matched placebo or sertraline" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Treatment with either matched placebo or sertraline", although it is not clear how they were matched" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Outpatients with a DSM‐III‐R diagnosis of moderate‐to‐severe PTSD were randomized to 12 weeks of double‐blind treatment with either sertraline (N = 100) in flexible daily doses in the range of 50 to 200 mg or placebo (N = 108)" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the sertraline group (32; 30%) compared to the placebo group (35; 27%). The two groups did not differ by sample characteristics at baseline. Quote: "Two hundred eight subjects were randomly assigned to sertraline or placebo, of whom 98 sertraline‐treated subjects and 104 placebo‐treated subjects were available for at least 1 postbaseline efficacy assessment, and so constituted the intent‐to‐treat sample. Thirty‐two subjects discontinued sertraline treatment (30%) compared with 35 subjects who discontinued placebo treatment (27%). The primary reason cited for discontinuation of sertraline and placebo, respectively, was as follows: adverse events (9.1% vs 4.7%); withdrawal of consent (6.1% vs 3.8%); lost to follow‐up (5.1% vs 10.4%); protocol violation (2.0% vs 4.7%); laboratory abnormality (2.0% vs 0%); insufficient therapeutic response (0% vs 4.7%); and miscellaneous other reasons (2.0% vs 1.9%). Two sertraline subjects discontinued prematurely because of mild elevations in aspartate aminotransferase and alanine aminotransferase values. There were no serious electrocardiographic or laboratory abnormalities during the course of the study, nor were there any significant differences between the 2 treatment groups in the incidence of electrocardiographic or laboratory abnormalities. Demographic and clinical characteristics for the subject sample are shown in Table 1. The only significant difference between the 2 treatment groups in any of the baseline variables was the smaller percentage of males in the sertraline group. Women constituted the majority of the sample" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Financial support for this study was provided by Pfizer Inc, New York, NY" |
Davidson 2001b.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, double‐blind, maintenance treatment study, relapse prevention study for participants undergoing acute and continuation treatment in Brady 2000 and Davidson 2001a Duration of intervention: 28 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: At 24 centers in the United States Sample size: 96 randomised to sertraline and placebo Mean age: 43.4 years (21 ‐ 69) Sex: 29 men and 67 women Diagnostic measure: DSM‐III‐R Inclusion criteria: Quote: “The enrollees in the current 28‐week double‐blind study were male and female outpatients at least 18 years of age who had completed the previous 24 weeks of open‐label continuation treatment with sertraline and who met responder criteria at the final two visits. The responder criteria were a Clinical Global Impression improvement score ≤2 (much or very much improved) and ≥30% improvement in the total severity score in part 2 of the Clinician‐Administered PTSD Scale, both indexed against the pretreatment baseline of the original double‐blind acute treatment study. Additional entry criteria required that the patients have no clinically significant abnormalities identified in a physical examination and laboratory testing conducted at the end of week 24 of continuation treatment study and that female patients use medically acceptable birth control throughout the study. The original eligibility criteria that defined the study group have been presented in detail in a previous publication. Briefly, at baseline of the two acute treatment studies, the patients met DSM‐III‐R criteria for a principal diagnosis of PTSD as determined by part 1 of the Clinician‐Administered PTSD Scale. A minimum 6‐month duration of PTSD symptoms was required, as well as a total severity score ≥50 on part 2 of the Clinician Administered PTSD Scale at the end of a 2‐week placebo run‐in period; subjects were thus at least moderately ill" Exclusion criteria: Quote: "Subjects were excluded if they had a current or past history of bipolar disorder, schizophrenia, or organic mental disorder; a primary diagnosis of major depression, OCD, or other anxiety disorders; or alcohol or other substance dependence or abuse in the past 6 months, or if they met other exclusion criteria summarized in a previous report. Concomitant psychotropic therapy, with the exception of chloral hydrate for sleep (not more than two nights per week), was prohibited during all studies in the series. Cognitive behavior therapy was not permitted during any of the trials, and other forms of psychotherapy could not be initiated or ended during a trial" Comorbidity: Not specified Dropouts: Insufficient information to determine 'n' in each group |
|
| Interventions | Pharmacological intervention: Quote: “To preserve the double blind from the acute treatment study, all patients were discontinued from their acute‐phase study medication (sertraline or placebo) before initiating open‐label study treatment with a daily dose of 25 mg of sertraline. At the end of the first week, the daily dose of sertraline was increased to 50 mg. Dosing throughout the open‐label phase was flexible, in the range of 50 mg to 200 mg, and was based on clinical response and tolerability. The results of the study of continuation treatment with sertraline have previously been reported" | |
| Outcomes | Primary outcomes: Rates and times to event for 1) relapse, 2) relapse or discontinuation because of clinical deterioration (self‐rated), and 3) acute exacerbation of PTSD
Secondary outcomes: CAPS, HAM‐D, QLES Time points: Quote: “Patients were evaluated and rated biweekly during the 28 weeks of double‐blind treatment". Data estimation: Not applicable for primary outcomes (Kaplan Meier survival analysis) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Supported by a grant from Pfizer" Medication provided by industry: No Any of the authors work for industry? Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Ninety‐six patients were randomly assigned, in a double‐blind design, to 28 weeks of maintenance treatment with sertraline (50–200 mg, N=46; 78% were women) or placebo (N=50; 62% were women)" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved Quote: "The study examined the efficacy of sertraline, compared with placebo, in sustaining improvement and preventing relapse over 28 weeks in patients with posttraumatic stress disorder (PTSD) who had completed a 12‐week double‐blind, placebo‐controlled acute treatment study and a subsequent 24‐week open‐label study of continuation treatment with sertraline" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to determine 'n' in each group. Reasons for treatment withdrawal were provided. No information was provided on sample characteristics at endpoint Quote: "Overall, 60.9% of the patients who received sertraline (N=28) and 40.0% of the patients who received placebo (N=20) completed the study. Reasons for study discontinuation for the patients who received sertraline and for those who received placebo, respectively, were: met relapse criteria, 6.5% (N=3) versus 28.0% (N=14); clinical deterioration, 10.9% (N=5) versus 20.0% (N=10); adverse events, 8.7% (N=4) versus 6.0% (N=3); withdrew consent, 6.5% (N=3) versus 6.0% (N=3); and miscellaneous other reasons, 6.5% (N=3) versus 0% (N=0). Six patients who received sertraline and four patients who received placebo at one study site were excluded from the efficacy analyses because of the institutional review board’s concerns about a study investigator’s lack of participation in performing the required efficacy assessments" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Supported by a grant from Pfizer" |
Davidson 2003.
| Study characteristics | ||
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: 1 week single‐blind placebo run‐in Post‐treatment: Not specified |
|
| Participants | Setting: North Carolina. Quote: “Subjects were recruited by advertisements or from within the clinical practice of the investigators" Sample size: 29 randomised to mirtazapine and placebo Mean age: 46.5 years Sex: 7 men and 13 women (for 20 participants), 15% (4/26) war trauma, 73% (19/26) MDD Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Subjects were required to be 18 years of age or older, to carry a primary diagnosis of chronic PTSD by DSM‐IV criteria, with a minimum score of 20 on the Structured Interview for PTSD (SIP), to show fluency in English, and to provide written informed consent. Included in the informed consent process was an explanation that participants were free to withdraw from the trial without penalty and that the study medicine (and other treatments) were available outside of the protocol if desired. Consent was obtained from the study coordinator and treating physician" Exclusion criteria: Quote: "Subjects were excluded on the basis of the following: 1) lifetime schizophrenia, bipolar disorder, or cognitive dysfunction owing to medical condition; 2) history of alcohol or other substance use disorder within 3 months; 3) mental retardation; 4) significant risk of violence or suicide; 5) unstable medical condition; 6) clinically significant laboratory or EKG abnormality or positive urine screen for illicit drugs; 7) nonresponse to mirtazapine 30 mg/day for 8 weeks; 8) concomitant therapy with other psychotropic medications or herbs; 9) need for concurrent psychotherapy; and 10) pregnancy or lactation. Comorbidity was assessed by means of the Mini–International Neuropsychiatric Interview (MINI)" Comorbidity: MINI, MD, agoraphobia, generalised anxiety, panic disorder, social anxiety Dropouts: 6/26 (3/17 in the mirtazapine and 3/9 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Subjects were randomized to mirtazapine or placebo, at an initial dose of 15 mg daily, which was increased at weekly intervals, if tolerated, to 45 mg per day of mirtazapine or matching dose of placebo" | |
| Outcomes | Primary outcomes: SPRINT(Clinical Global Improvement & Total Scores)
Secondary outcomes: DTS, HADS, SIP, ASEX Time points: Quote: “At baseline, subjects were randomly assigned to treatment, and returned at weeks 1, 2, 3, 4, 6, and 8" Data estimation: ITT, LOCF (1 post‐baseline assessment) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Acknowledgment of financial support for this study from Organon to Dr. Davidson, as well as honoraria to him for consultation" Medication provided by industry: No Any of the authors work for industry? Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Twenty‐nine patients were randomized to receive drug up to 45 mg/day or placebo double‐blind on a 2:1 ratio for 8 weeks, with data being available for analysis in 26" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Subjects were randomized to mirtazapine or placebo, at an initial dose of 15 mg daily, which was increased at weekly intervals, if tolerated, to 45 mg per day of mirtazapine or matching dose of placebo" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Twenty‐nine patients were randomized to receive drug up to 45 mg/day or placebo double‐blind on a 2:1 ratio for 8 weeks, with data being available for analysis in 26" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More participants withdrew from the placebo group (3/9; 33%) compared to the mirtazapine (3/17; 18%) group, however the two groups did not differ with regards to sample characteristics at baseline. Reasons for treatment withdrawal were provided. Data estimation was based on intent to treat last observation carried forward efficacy analyses Quote: "Twenty‐nine subjects were randomized, three failing to return. Twenty (69%) completed the 8‐week period. Early dropouts occurred for three subjects on mirtazapine and three on placebo. Reasons for dropout in the mirtazapine group were: sedation, panic attacks, increased anxiety and irritability (n = 1 each). Placebo dropouts occurred because of lack of efficacy (n = 2) and pain (n = 1). There were no differences between drug and placebo groups in mean (SD) age (48.4 13.6 vs. 42.9 11.7), gender (n = 13 women [81%] and n = 7 men [78%]), marital status (n = 7 [40%] and n = 4 [44%] married) or ethnicity (n = 9 [47%], n = 8 [89%]). Intent to treat last observation carried forward efficacy analyses were conducted by means of analysis of covariance (ANCOVA), with baseline as covariate and treatment as a grouping variable" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Acknowledgment of financial support for this study from Organon to Dr. Davidson, as well as honoraria to him for consultation" |
Davidson 2005.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind, discontinuation study Duration of intervention: 24 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Durham. Quote: “1 of 3 clinics: Duke University Medical Center, Durham Veterans Administration Medical Center, and in private practice" Sample size: 62 randomised to fluoxetine and placebo Mean age: 44.05 (11.85) years Sex: 28 men and 29 women (for 57 participants) Diagnostic measure: criteria for PTSD Inclusion criteria: Quote: “Patients were required to fulfill criteria for PTSD. A trauma history was also obtained. All patients were required to undergo discontinuation of any psychotropic medication, and to be free thereof, for at least 2 weeks before entering open‐label treatment. Patients could be man or woman, between ages 18 and 70, and if female, be of nonpregnant status" Exclusion criteria: Quote: "Patients were excluded from the study in the presence of a history of schizophrenia, bipolar disorder, organic brain disease, alcohol or drug abuse/dependence (within the previous 6 months), mental retardation, the need for ongoing psychotropic medication, significant risks of suicide or history of suicide attempt within in the previous 6 months, history of significant violence within the previous year, medically unstable state, prior nonresponse to adequate treatment with FLU (i.e. 40 mg/d or greater for at least 8 weeks), the need for trauma‐focused psychotherapy, positive urine drug screen for illicit substances, and clinically significant abnormal laboratory tests" Comorbidity: Not specified Dropouts: 5/57 (3/27 in the fluoxetine and 2/30 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Open‐label FLU was commenced at 10 mg/d for 1 week, increasing to 20 mg at the second week, by 20‐mg increments every fortnight, up to 60 mg maximum. Dose was not increased if patients experienced troublesome side effects or if they achieved a score of 2 (much improvement) on the Clinical Global Improvement Scale (CGI‐I). Thereafter, the dose remained steady unless side effects required further lowering. At randomization, patients who had shown at least minimal improvement were assigned to continue on FLU or to receive PBO double‐blind on the basis of a preassigned randomly generated set of study numbers" | |
| Outcomes | Primary outcomes: CGI‐I
Secondary outcomes: CGI‐S, SPRINT, DTS. Medication side effects were assessed by means of the self‐rated Severity of Symptoms Scale Time points: Quote: “Subjects were seen at baseline (i.e. week 24, before randomisation), weeks 1, 2, and 4, and then monthly thereafter" Data estimation: LOCF (after randomisation, with week 24 as the covariate) |
|
| Notes | Dates of trial: Quote: "Initiated on August 27, 1997, and completed on May 30, 2001" Industry‐funded: No. Quote: "This study was supported by grant #R01 MH 56656 to the principal author" Medication provided by industry: Yes. Quote: "The authors thank Eli Lilly for providing medication and placebo" Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "At randomization, patients who had shown at least minimal improvement were assigned to continue on FLU or to receive PBO double‐blind on the basis of a preassigned randomly generated set of study numbers" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved. Quote: "Subjects received open label treatment for 6 months, followed by double‐blind randomized treatment with FLU or placebo (PBO) for 6 months" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of participants withdrew from the fluoxetine group (3; 11%) compared to the placebo group (2; 7%). Sample characteristics were provided for participants at baseline only. Reasons for withdrawals were provided. Data were estimated for LOCF scores after randomisation Quote: "Sixty‐three completed 24 weeks of treatment and showed clinical improvement which would qualify them for the next study phase. One subject decided not to continue beyond week 24, leaving 62 randomized to either remain on FLU (n = 30) or to be switched double‐blind to PBO (n = 32). Five failed to return (n = 3 on FLU and n = 2 on PBO), leaving 57 evaluable cases (n = 27 for FLU and n = 30 for PBO). Reasons for dropout (n = 51) during the first 6 months were as follows: loss to follow‐up (n = 18), nonresponse or deterioration (n = 12), adverse event (n = 13), withdrawal, noncompliance (n = 5), moved or job factors (n = 2), and other (n = 1). There were no significant differences between subjects who were randomized (n = 62) and those who were not (n = 52), other than a higher number of African Americans (n = 23) and lower number of Caucasians (n = 27) in the nonrandomized group, as compared with rates (n = 15 and n = 46) in the randomized group (x2 = 6.13, df 2, P = 0.04). Similarly, there were no differences, apart from ethnicity, when the analyzed group (n = 57) was compared with the nonrandomized (n = 52). Similarly, the 2 groups were comparable on all clinical ratings or type of trauma" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified. Medication was provided by industry, but no authors work for industry. Self‐reported measures were used |
Davidson 2006a.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind Duration of intervention: 24 weeks, followed by a taper period of up to 2 weeks and post‐study evaluation (4 ‐ 10 days after taper) Placebo run‐in: washout period of at least 7 days Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “International study at 56 outpatient psychiatric clinic sites in Argentina, Chile, Colombia, Denmark, Finland, Mexico, Norway, Portugal, South Africa, Spain, Sweden, and the United Kingdom" Sample size: 329 randomised to venlafaxine ER and placebo Mean age: 41.35 (18.60) years Sex: 151 men and 178 women, 40 combat Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Patients were enrolled in the study if they were at least 18 years of age, could provide legal consent, and were not currently hospitalized; met the DSM‐IV criteria for a primary diagnosis of PTSD; had a score of at least 60 on the Clinician‐Administered PTSD Scale, abbreviated 1‐Week Symptom Status Version (CAPS‐SX17); and had PTSD symptoms for at least the previous 6 months. In addition, they must have had a negative serum pregnancy test at screening (for women of childbearing potential); been in generally good health as determined by the investigator on the basis of medical history, physical examination, and screening laboratory results; been willing and able to return for all protocol‐defined visits; been fluent in written and spoken forms of English, Spanish, or Portuguese; and been willing and able to provide written informed consent prior to admission" Exclusion criteria: Quote: "Subjects were excluded if they had intolerance, hypersensitivity, or nonresponse to a previous adequate trial of venlafaxine; had inability to tolerate or respond to adequate trials of 3 antidepressants; had current primary major depression or panic disorder; had a current mental disorder due to a general medical condition or history of bipolar disorder, schizophrenia, or other psychotic disorder; abused or were dependent on alcohol or other drugs within 6 months of randomization or had a positive urine drug screen; showed a high risk of suicide or violence; used any investigational drug, antipsychotic, or monoamine oxidase inhibitor within 30 days of randomization; had electroconvulsive therapy within 3 months of randomization or likelihood of requiring electroconvulsive therapy during the study; used triptans or any other psychoactive drug, including fluoxetine, or herbal preparation within 7 days of randomization; had current involvement in criminal proceedings or compensation claims related to trauma; and, for women, were nursing, pregnant, or sexually active without acceptable birth control. Subjects who had initiated or changed psychotherapy of any kind within 3 months of study enrollment were also excluded" Comorbidity: Not specified Dropouts: 105/329 (49/161 in the venlafaxine ER and 56/168 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Investigators followed the protocol specified medication dosing guidelines for venlafaxine ER (75‐300 mg/d with a lead‐in dose at baseline of 37.5 mg/d) and increased the dose to the next higher level based on tolerability for patients who did not achieve remission, which was operationally defined as a score of 20 or lower on the CAPS‐SX17, a cut off that has been used elsewhere.18,19 Venlafaxine ER dosing was increased to a maximum dose of 75 mg/d at day 5, 150 mg/d at day 14, 225 mg/d at day 28, and 300 mg/d at day 42. Study visits took place at baseline and at the end of study treatment weeks 1, 2, 4, 6, 8, 12, 18, and 24 and follow‐up or at the time of discontinuation if before week 24" | |
| Outcomes | Primary outcomes: CAPS‐SX‐17
Secondary outcomes: CGI‐S, GAF, HDRS‐17, CD‐RISC, SVS, a 1‐item scale that rates the magnitude of setbacks to stressful events or personal problems experienced by participnts from the time of their last visits, Q‐LES‐Q‐SF, a 16‐item scale that assesses the degree of enjoyment and satisfaction in various areas of daily life, SDS, a 3‐item scale that evaluates the extent to which PTSD symptoms have disrupted patients’ work/school schedules, social life, and family life/home responsibilities Time points: Quote: “The primary outcome was measured at baseline and weeks 2, 4, 6, 8, 12,18, and 24. The CGI‐S, HDRS‐17 and the SDS were measured at baseline and weeks 2, 4, 6, 8, 12, 18, and 24; the Q‐LES‐Q‐SF, CD‐RISC and the SVS, at baseline and weeks 4, 12, and 24; and the GAF, at baseline and weeks 12 and 24" Data estimation: LOCF, completer analysis |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "This study was supported in part by Wyeth Pharmaceuticals" Medication provided by industry: No Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The sponsor sent each site a table of computer‐generated numbers randomized in blocks of 4, and patients were assigned packages linked to the randomization numbers in numerical order by the site investigator using the randomization tables" |
| Allocation concealment (selection bias) | Low risk | Quote: "The sponsor sent each site a table of computer‐generated numbers randomized in blocks of 4, and patients were assigned packages linked to the randomization numbers in numerical order by the site investigator using the randomization tables" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved. Quote: "Design: 6‐month, double‐blind, placebo‐controlled trial" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participaents withdrew from the venlafaxine ER group (49/161, 30%) compared to the placebo group (56/168; 33%). The 2 groups did not differ by sample characteristics at baseline. The ITT population was used at baseline and LOCF at endpoint Quote: "As shown in Figure 1, 442 patients were screened, 329 were randomized, and 224 (68%) completed the study. There were no clinically important differences in the proportion of patients in each group who completed the study. There were no unexpected differences between the venlafaxine ER and placebo groups in the reasons given for early withdrawal (Figure 1) or clinically important differences in baseline demographic or clinical characteristics (Table 1). Withdrawal rates were 30.4% for venlafaxine ER and 33.3% for placebo with no significant difference in dropouts attributable to adverse events. The venlafaxine ER group had 15 discontinuations attributed to adverse events compared with 9 discontinuations for the placebo group; 5 patients in the venlafaxine ER group withdrew because of unsatisfactory response compared with 18 patients in the placebo group. The types of trauma experienced by all randomized patients are outlined in Table 2. For patients who discontinued participation before study completion, the last post dose observed value was used for end point analysis (LOCF). In addition to the LOCF value, the observed changes at each visit were computed" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This study was supported in part by Wyeth Pharmaceuticals" |
Davidson 2006b.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: washout period of at least 7 days Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “59 outpatient centers in the United States" Sample size: 538 randomised to sertraline, venlafaxine and placebo Mean age: 32 years Sex: Not specified Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “The subjects were male and female outpatients aged 18 years or older who met DSM‐IV criteria for a primary diagnosis of PTSD based on the Structured Clinical Interview for DSM‐IV. Additional inclusion criteria included a score of at least 40 on the Davidson Trauma Scale (DTS); a score of at least 60 on the 17‐item Clinician‐administered PTSD Scale (CAPS‐SX17), 1‐week symptom status version; PTSD symptoms for at least the previous 6 months; a negative serum pregnancy test at screening (for women of childbearing potential); generally good health based on medical history, physical examination, and screening laboratory results; and likelihood of complying with protocol" Exclusion criteria: Quote: "Excluded were subjects with a decrease of more than 25% on the DTS between screening and baseline; intolerance, hypersensitivity, or nonresponse to a previous adequate trial of venlafaxine or sertraline; inability to tolerate or respond to adequate trials of 3 or more antidepressants; current primary major depression or panic disorder (determined using the structured Mini‐International Neuropsychiatric Interview); a current mental disorder due to a general medical condition or history of bipolar disorder, schizophrenia, or other psychotic disorder; alcohol or drug abuse or dependence within 6 months of randomization or a positive urine drug screen; and a high risk of suicide or violence. Additional exclusion criteria included use of any investigational drug, antipsychotic, or monoamine oxidase inhibitor within 30 days of randomization; electroconvulsive therapy within 3 months of randomization or likelihood of requiring electroconvulsive therapy during the study; triptans or any other psychoactive drug (including SSRIs or tricyclic antidepressants) or herbal preparation within 7 days of randomization; initiation of or change in psychotherapy within 3 months of randomization; current involvement in criminal proceedings or compensation claims related to trauma; and for women, nursing, pregnancy, or sexual activity without acceptable birth control. Patients who began or received any of the prohibited treatments described above during the study were considered protocol deviators. These patients were evaluated by the sponsor on a case‐by‐case basis for possible exclusion from the study. Permitted medications included zaleplon or zolpidem, 1 dose nightly as needed for insomnia, for up to 6 nights, during the 14 days after the baseline evaluation only. The use of any alternative hypnotics required prior approval of the sponsor. Short‐term treatments for allergies, colds, or flu were permitted, provided the medications used had minimal psychotropic effects" Comorbidity: Not specified Drop‐out rates: The proportion of dropouts per treatment group could not be determined due to the lack of information provided by the study |
|
| Interventions | Pharmacological intervention: Quote: “Investigators followed the protocol‐specific medication dosing guidelines for venlafaxine ER (75–300 mg/d, with a lead‐in dose at baseline of 37.5 mg/d) and the within‐label dosing guidelines for sertraline for PTSD (50–200 mg/d, with a lead‐in dose at baseline of 25 mg/d) and increased the dose to the next higher level, based on efficacy and tolerability, for patients who did not achieve remission, which was operationally defined as a score of 20 or less on the CAPS‐SX17. Venlafaxine ER dosing was increased to a maximum dose of 75 mg/d at day 5, 150 mg/d at day 14, 225 mg/d at day 28, and 300 mg/d at day 42. Sertraline dosing increased to a maximum dose of 50 mg/d at day 5, 100 mg/d at day 14, 150 mg/d at day 28, and 200 mg/d at day 42. Study visits took place at baseline and at the end of study treatment weeks 1, 2, 4, 6, 8, and 12 or at the time of discontinuation, if before week 12" | |
| Outcomes | Primary outcomes: CAPS‐SX17
Secondary outcomes: CGI‐S, GAF, HAM‐D17, DTS, a 17‐item scale that evaluates the frequency and severity of PTSD symptoms over the past week, SVS, a 1‐item scale that evaluates how much subjects were set back by stressful events or personal problems since their last visit, Q‐LES‐Q‐SF, a 16‐item scale that evaluates the degree of enjoyment and satisfaction in various areas of daily life; SDS, a 3‐item scale that evaluates how PTSD symptoms have disrupted patients’ work/school schedule, social life, and family life/home responsibilities Time points: Quote: “Primary outcomes were measured at baseline and weeks 2, 4, 6, 8, and 12.T he CGI‐S, HAM‐D, DTS, and SDS were measured at baseline and weeks 2, 4, 6, 8, and 12; the Q‐LES‐Q‐SF and SVS, at baseline and weeks 4 and 12; and the GAF, at baseline and week 12" Data estimation: Baseline–to–end point (week 12 or the time of discontinuation) and LOCF |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Funding was provided by Wyeth Pharmaceuticals" Medication provided by industry: No. Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the patients were randomised but no mention is made of the method of randomisation Quote: "Patients were randomly assigned to receive placebo or flexible doses of venlafaxine ER (37.5–300 mg/d) or sertraline (25–200 mg/d) for 12 weeks or less" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "This 12‐week, double‐blind, multicenter trial evaluated the efficacy of venlafaxine extended release (ER), sertraline, and placebo in adult outpatients (N = 538) with a primary diagnosis of posttraumatic stress disorder (PTSD), as defined in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, symptoms for 6 months or more and 17‐item Clinician‐administered PTSD Scale (CAPS‐SX17) score of 60 or more" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The proportion of dropouts per treatment group could not be determined due to the lack of information provided by the study. No information was provided on sample characteristics at end point Quote: " The CAPS‐SX17 total score was the primary outcome measure, and the primary analysis compared venlafaxine ER with placebo on the mean change in CAPS‐SX17 total score from baseline to week 12 (LOCF). Last‐observation‐carried‐forward values at week 12 were used for analysis of CAPS‐SX17 symptom cluster scores, and LOCF values at the final visit, defined as the last nonmissing postbaseline assessment (which, in a few cases, occurred after week 12), were used for the analysis of remission rates and other secondary outcomes. In addition to the analyses based on LOCF values, the observed changes (ie, the changes observed only for those patients who had nonmissing values at each visit) were summarized. A total of 538 patients of the 889 patients screened for participation met study entry criteria and were randomized to treatment (7 of whom discontinued before receiving the study drug). Of the 531 patients who received treatment, 350 (66%) completed the study. There were no clinically important differences between the 3 treatment groups in the proportion of patients in each group who completed the study. Of the 181 treated patients (34%) who withdrew from the trial, primary reasons for early withdrawal included failure to return (70 patients [13%]) and AEs (58 patients [11%]). There were no clinically important differences between the 3 treatment groups in reasons for early withdrawal. The 3 groups also had no important differences in baseline demographic or baseline clinical characteristics (details available on request)" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Funding was provided by Wyeth Pharmaceuticals" |
Davidson 2007.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind Duration of intervention: 12 weeks. Placebo run‐in: not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “38 centers across the United States" Sample size: 232 randomised to tiagabine and placebo Mean age: 42.6 (18 ‐ 64). Sex: 79 men and 153 women Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Adult patients aged 18 to 64 years who met the DSM‐IV criteria for PTSD, as determined by the Clinician‐Administered PTSD Scale (CAPS), were enrolled from 38 centers across the United States. Patients were required to have both CAPS and Davidson Trauma Scale (DTS) scores of 50 or more at screening and baseline visits" Exclusion criteria: Quote: "Patients were excluded from the study for the following reasons: (1) other psychiatric Axis I disorders (DSM‐IV) as a principal diagnosis (except PTSD), an eating disorder within 6 months of screening, or any history of obsessive‐compulsive disorder, psychotic disorder, bipolar disorder, mental retardation, or antisocial personality disorder; (2) a decrease of 50% or more in CAPS or DTS score between the screening and baseline visits; (3) a medical condition that could affect the pharmacokinetics of tiagabine; (4) unresponsiveness (by history) to 2 or more previously documented pharmacological treatments of PTSD; (5) drug or alcohol abuse (within the last 3 months) or dependence (within the last 6 months, except nicotine and caffeine dependence); (6) prior use of tiagabine; (7) a history of seizures; or (8) an involvement in PTSD‐related litigation or disability payments" Comorbidity: MADRS, MDD Dropout rates: 91/232 (39/116 in the tiagabine and 52/116 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Tiagabine was initiated at 4 mg/d at week 1, and then individually titrated through week 6 in weekly increments of up to 4 mg/d to a maximum dose of 16 mg/d. Doses were titrated upward unless, in the investigator’s judgment, an increase in dose was: (1) inadvisable because of recurrent or persistent adverse events; (2) not expected to increase efficacy; or (3) not desired by the patient. The dose established at week 6 could not be increased during weeks 7 to 12. After week 12, the study drug was tapered by 2 mg every other day. Tiagabine was administered in equal divided doses, 1 dose in the morning with breakfast and one in the evening (approximately 9:00 PM) with a snack. When downward titration was required (because of adverse events), this was applied to the morning dose when possible" | |
| Outcomes | Outcomes: CAPS, DTS, TOP‐8, CGI‐C, SDS and a participant‐rated evaluation of sleep (sleep questionnaire) (not clear which outcomes are secondary or primary) Time points: Not specified Data estimation: Completer analysis |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "This study was sponsored by Cephalon, Inc, Frazer, Pa" Medication provided by industry: No Any of the authors work for industry? No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "This 12‐week, multi center, double‐blind study randomized patients to receive either tiagabine or placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealedal |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "This 12‐week, multi center, double‐blind study randomized patients to receive either tiagabine or placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the tiagabine group (39/116, 37%) compared to the placebo group (52/116; 49%) Quote: "Of the 232 patients randomized (tiagabine, n = 116; placebo, n = 116), 227 had at least 1 dose of study medication (tiagabine, n = 114; placebo, n = 113), and 202 had at least 1 postbaseline CAPS assessment (tiagabine, n = 105; placebo, n = 97). Sixty‐six percent (77/116) of patients in the tiagabine group and 55% (64/116) in the placebo group completed the study. Main reasons for discontinuation in the tiagabine and placebo groups were lost to follow‐up (15% and 17%, respectively), adverse events (8% and 8%), withdrawal of consent (4% and 10%), lack of efficacy (2% and 5%), and ‘‘other’’ (3% and 0%). There were no significant differences between patient groups in proportion of patients who discontinued medications or reasons for medication discontinuation. The tiagabine and placebo groups were well matched with respect to baseline demographics and characteristics" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This study was sponsored by Cephalon, Inc, Frazer, Pa" |
Davis 2004.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Birmingham and Tuscaloosa VA Medical Centers and the Department of Psychiatry Clinical Research Clinic, University of Alabama at Birmingham" Sample size: 41 randomised to nefazodone and placebo Mean age: 53.8 (32 ‐ 73) Sex: 40 men and 1 woman, 98% (40/41) combat veterans, 39% (16/41) MD Diagnostic measure: DSM‐IV, SCI Inclusion criteria: Quote: “Criteria for entry included a diagnosis of chronic PTSD, aged 19 to 75 years, stable physical health, negative urine drugs‐of‐abuse screen, ability to take oral medication, free of all psychotropic medication in the previous 2 weeks (except 6 weeks for fluoxetine), and a signed informed consent form. Women of childbearing potential had to be using at least one medically approved method of birth control" Exclusion criteria: Quote: "Exclusionary criteria included a lifetime history of bipolar, psychotic, or cognitive disorder, acute suicidality or homicidality, active substance abuse/dependence within the previous 4 months (except nicotine and caffeine), unstable medical condition, history of sensitivity to nefazodone, and inability to attend follow‐up visits. Women who were pregnant, planning to become pregnant, or breastfeeding were excluded from the study" Comorbidity: MD, Dysthymia, Panic with/without agoraphobia, Lifetime alcohol and polysubstance use disorder. Drop‐out rates: 18/41 (12/26 in the nefazodone and 6/15 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Study medication was started at 1 capsule (100 mg nefazodone vs. look‐a‐like placebo) twice daily. Based on symptoms and occurrence of side effects, the investigator increased the medication in 1 capsule increment every 4 days, as tolerated, until a maximum therapeutic benefit was achieved, not to exceed 6 capsules/day (600 mg/d nefazodone). Dosing was twice daily, with the higher dose generally at bedtime. Compliance was assessed by patient report at each study visit" | |
| Outcomes | Primary outcomes: CAPS (i.e. subscales: CAPS‐B, CAPS‐C, CAPS‐D)
Secondary outcomes: HAM‐A, HAM‐D, PTSD Checklist, CADSS, GAF, CGI Time points: Quote: "Patients returned to the clinic every 2 weeks for symptom ratings, adverse effects documentation, and assessment of clinical status. The HAM‐A, HAM‐D, and CGI were administered at each study visit (every 2 weeks), while the CAPS, GAF, PTSD Checklist, and CADSS were administered at baseline and at weeks 4, 8, and 12" Data estimation: Repeated‐measures analyses of variance with the LOCF |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Partial funding for this project was provided through an unrestricted grant from Bristol‐Myers Squibb" Medication provided by industry: No Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "In this study, the pharmacist maintained the randomization log and administered the placebo or nefazodone (100 mg/capsule) in look‐a‐like capsules" |
| Allocation concealment (selection bias) | Low risk | Quote: "In this study, the pharmacist maintained the randomization log and administered the placebo or nefazodone (100 mg/capsule) in look‐a‐like capsules" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Both the investigators and the patients were kept blind to the contents of the capsules until the end of the study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of paarticipants withdrew from the nefazodone group (12/26, 46%) compared to the placebo group (6/15; 40%) Quote: "Table 2 shows the dropout occurrences during the study. Of those patients randomized to nefazodone, 12 (46%) exited before week 12. Of these, 4 were lost to follow‐up (3 after baseline and 1 after week 10), 5 withdrew due to adverse effects (4 after baseline with combined side effects of drowsiness, dizziness, agitation, gastrointestinal distress, and fatigue; 1 after week 8 with dizziness, orthostatic hypotension, and headaches), 2 withdrew due to medication being ineffective (1 after week 4 and 1 after week 8), and 1 was exited due to noncompliance after week 4. In the placebo group, 6 (40%) patients exited before week 12. Of these, 1 was lost to follow‐up after baseline, 1 withdrew consent after baseline due to adverse effects of insomnia and irritability, 2 withdrew consent with no reason given (1 after week 2 and 1 after week 4), and 2 withdrew due to medication being ineffective (1 after week 4 and 1 after week 6). Overall, 19 patients on nefazodone and 12 patients on placebo completed at least 4 weeks of therapy, and 14 patients on nefazodone and 9 patients on placebo completed the entire 12‐week study. Nefazodone therapy was generally well tolerated. Five (19%) patients on nefazodone therapy discontinued due to side effects. The most common side effects were dizziness, gastrointestinal symptoms, and headache. The average final dose of study medication was 435 mg/d for the nefazodone group" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Quote: "In addition, generalizability is limited due to this study population’s rather homogeneous composition (predominantly male combat veterans). Partial funding for this project was provided through an unrestricted grant from Bristol‐Myers Squibb" |
Davis 2008.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Tuscaloosa Veterans Affairs Medical Center (TVAMC) and Birmingham VA Medical Center" Sample size: 85 randomised to divalproex and placebo Mean age: 55.2 years (SD, 6.8) Sex: 83 men and 2 women, 4 combat‐related trauma (5%) Diagnostic measure: DSM‐IV, CAPS Inclusion criteria: Quote: “Eligibility criteria included the following: age 19 to 70 years; stable medical condition; no substance use disorder (other than caffeine and nicotine) in the previous 2 months; no use of psychotropic medications for the previous 2 weeks (6 weeks for fluoxetine); no lifetime history of bipolar, psychotic, or cognitive disorders; no history of seizure disorder; no history of sensitivity to divalproex; and no current suicidal ideation, homicidal ideation, or psychotic symptoms that might interfere with the patient’s ability to give informed consent or preclude safe maintenance on divalproex monotherapy for the duration of the study. Women of childbearing potential had to use a medically approved method of contraception and could not be pregnant or breastfeeding during the course of the study" Exclusion criteria: See inclusion criteria Comorbidity: Not specified Dropout rates: 5/82 (2/41 in the divalproex and 3/41 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Divalproex was started at a 500‐mg tablet twice daily and increased in 500‐mg increments, as tolerated, until maximum therapeutic benefit was achieved, but not to exceed 3000 mg/d. No other psychotropic medications were allowed during the study, except for low‐dose trazodone (50 mg/d), if needed, for insomnia" | |
| Outcomes | Primary outcomes: CAPS (i.e. subscale: CAPS‐D)
Secondary outcomes: CAPS (i.e. subscales: CAPS‐B, CAPS‐C), HAM‐A, TOP‐8, MADRS, CGI‐S, CGI‐I, DTS Time points: Quote: "Patients returned for assessment of symptoms and side effects at weeks 2, 4, 6, and 8 by a trained assessor" Data estimation: Quote: "Analyses of differences between groups in terms of response (≥30% improvement on CAPS and CAPS subscales) and the completers‐only groups were conducted (i.e. LOCF)" |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "This study was supported by a VA Research and Development Merit Award and an investigator‐initiated grant from Abbott Laboratories, Chicago, IL" Medication provided by industry: Yes Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was determined by a random‐number generator with allocation sequence that was concealed in the pharmacy until the entire study was concluded" |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients meeting the entrance criteria were randomized 1:1 to divalproex sodium (enteric‐coated, delayed‐release; Depakote) or visually identical placebo tablets, which were both supplied by Abbott Laboratories. Randomization was determined by a random‐number generator with allocation sequence that was concealed in the pharmacy until the entire study was concluded. Patients were assigned their randomization number at the time the study medication prescription was written" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "This 8‐week, randomized, double‐blind, placebo‐controlled study was conducted at Tuscaloosa Veterans Affairs Medical Center (TVAMC) and Birmingham VA Medical Center, approved by the TVAMC and Birmingham VA Medical Center institutional review boards, and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Trained assessor who was blind to medication assignment" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the divalproex group (2/41, 5%) compared to the placebo group (3/42; 7%) Quote: "One hundred one (N = 101) US military veterans signed informed consent, and 85 were randomized. Of those not randomized (n = 16), 9 subjects were screen failures (4 for positive urine drug screen, 3 for unstable medical condition, 1 for CAPS <45, and 1 for current alcohol abuse), 4 subjects withdrew consent, and 3 were lost to follow‐up. Of the 85 randomized patients, 2 subjects failed to return, and 1 subject had an adverse event before receiving 1 postbaseline assessment, and they were considered nonevaluable. Thus, 82 evaluable subjects were considered in the efficacy analysis (n = 41 placebo, and n = 41 divalproex). Sixty‐eight subjects completed the entire 8‐week study (9 were lost to follow‐up or withdrew consent; 3 were withdrawn for adverse events [2 divalproex, 1 placebo]; one for a serious adverse event unrelated to study medication or procedures, and one was withdrawn for lack of efficacy [placebo]). All but 2 of the 82 randomized evaluable subjects were male (98%), and all but 4 had a combat‐related trauma (95%). There were no significant intergroup differences in age, sex, trauma type, or duration of illness. All randomized subjects who took at least 1 dose of study medication and were assessed for at least 1 postbaseline visit were included in the intent‐to‐treat population for efficacy (a priori definition). The primary method of analysis for efficacy comparisons between divalproex and placebo groups was a repeated‐measures analysis of variance model using the last observation carried forward, with treatment as qualitative factor" |
| Selective reporting (reporting bias) | High risk | Information was not provided for the GAF (Global Assessment of Functioning) outcome |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This study was supported by a VA Research and Development Merit Award and an investigator‐initiated grant from Abbott Laboratories, Chicago, IL" Unequal distribution of genders |
Davis 2020.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Tuscaloosa and Birmingham Veterans Affairs Medical Centers in Alabama" Sample size: 78 randomised to mirtazapine and placebo Mean age: 38.0 years (SD, 9.9) Sex: 73 men and 5 women, 3 combat‐related trauma (4%) Diagnostic measure: DSM‐IV, MINI, DSM‐IV (CAPS) Inclusion criteria: Quote: “Male and female US military veterans of any race or ethnicity were eligible for inclusion, if all of the following criteria were met: 19‐65 years of age, current PTSD diagnosis (based on DSM‐IV criteria confirmed by the Mini‐ International Neuropsychiatric Interview (MINI) and the Clinician Administered PTSD Scale‐Diagnosis for DSM‐IV (CAPS), baseline total CAPS score of ≥45, no substance abuse or dependence for the previous 4 weeks (other than nicotine and caffeine), free of psychotropic medications for the previous 2 weeks (4 weeks for fluoxetine), and clinically stable medical examination and laboratory tests" Exclusion criteria: Quote: “Participants were excluded if 1 of the following criteria was met: lifetime history of bipolar I, psychotic ideation; history of sensitivity to mirtazapine; unstable general medical conditions that would contraindicate the use of mirtazapine; or score of ≥ 6 on question 10 (suicidal thoughts) on the Montgomery‐Asberg Depression Rating Scale (MADRS). Women of childbearing potential had to use a medically approved method of birth control during the study and were excluded if pregnant, breastfeeding, or planning to become pregnant or breastfeed during the study" Comorbidity: MDD, panic, agoraphobia, social anxiety, alcohol dependence (past year), alcohol abuse (past year), substance dependence (past year), substance abuse (past year) Dropout rates: 17/78 (8/39 in the mirtazapine and 9/39 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Participants were initiated on one capsule of 15mg mirtazapine or placebo at bedtime, which was increased by one 15mg mirtazapine or placebo capsule each week as tolerated to a maximum of 45mg mirtazapine or placebo (i.e. 3 capsules) per day. The minimum target dose was 30mg per day. Compliance was assessed by pill counts at the follow‐up visits" | |
| Outcomes | Primary outcomes: SIP
Secondary outcomes: CAPS (SX), DTS, MADRS, CGI‐S, CGI‐I, Q‐LES‐Q, PSQI, SDS Time points: Quote: "Data on most outcome measures were collected every 2 weeks. In consideration of minimizing participant burden, the CAPS‐SX was scheduled monthly. Adverse events (AEs) were recorded as they were spontaneously reported to clinical questions" Data estimation: ITT and completers |
|
| Notes | Dates of trial: Quote: "Between April 2006 and November 2010" Industry‐funded: No. Quote: "This study was funded by the Veterans Administration Clinical Science Research and Development (VA CSR&D)" Medication provided by industry: No Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Participants meeting eligibility criteria were randomised in a 1:1 double‐blind fashion to receive wither overencapsulated mirtazapine or a look‐alike placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Participants meeting eligibility criteria were randomised in a 1:1 double‐blind fashion to receive wither overencapsulated mirtazapine or a look‐alike placebo" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The participants and investigators remined blinded as to the allocation status during the placebo‐controlled phase" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors Quote: "As much as possible, each participant was evaluated by the same clinical rater throughout the study" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the mirtazapine group (8/39, 21%) compared to the placebo group (9/39; 23%) Quote: "Sixty‐one participants completed the 8‐week placebo‐controlled phase of the study (78.2% retention). 5 participants did not return after baseline, 1 relocated, 2 were lost to follow‐up weeks 2, 4, and 1 participant was pregnant at week 4 for the mirtazapine group compared to 2 participants that did not return after baseline, 1 who missed week 8, 4 who were lost to follow‐up weeks 2, 4, 6 and 1 who experienced an unrelated adverse event at week 2" ITT was used at baseline followed by number of completers at end point |
| Selective reporting (reporting bias) | Low risk | Primary outcomes were reported as specified in the protocol |
| Other bias | Low risk | No other sources of bias reported |
Dowd 2020.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre trial, randomised, placebo‐controlled, parallel arm, fixed dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Not stated Sample size: 25 randomised to eszopiclone and placebo Mean age: 43 years (SD, 12.2) Sex: 15 men and 10 women, 1 combat‐related trauma (4%) Diagnostic measure: DSM‐IV confirmed with CAPS Inclusion criteria: Quote: “Subjects included male and female outpatients, aged 18‐65 years of age with a primary DSM‐IV diagnosis of PTSD and confirmed with CAPS score of > 45. Associated sleep disturbance was operationalized as (1) positive score on diagnostic criterion item D1: “difficulty falling or staying asleep”; (2) sleep latency > 30 min; and (3) total sleep time < 6.5 h at least three times per week over the previous month" Exclusion criteria: Quote: “A lifetime history of psychotic disorders was exclusionary, as was a history of alcohol/substance abuse in the last 3 months. Concurrent use of antidepressants or benzodiazepines (on a stable dose for at least 6 wk) was allowed. Patients receiving current psychotherapy initiated within 3 months of randomization or ongoing psychotherapy of any duration directed specifically toward treatment of PTSD and/or sleep disturbance (e.g., CBT) were excluded, but general supportive individual, couple/family therapy > 3 months duration was permitted. Patients who had current legal actions related to trauma or an ongoing relationship with their assailant were also excluded" Dropout rates: 9/25 (6/13 in the eszopiclone and 3/12 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “The study was a 12‐wk, double blind, randomized controlled trial with 3 mg of ESZ or PBO at bedtime with a one‐month follow‐up visit" | |
| Outcomes | Primary outcomes: change in CAPS, PSQI
Secondary outcomes: SPRINT, Self‐report sleep diary, Actigraphy to assess for change in sleep, including total sleep time, initial insomnia and number of awakenings, MADRS, CGI‐S, LIFE‐RIFT Time points: Quote: "CAPS was assessed six times throughout the study; PSQI, SPRINT and Self‐report sleep diary was administered weekly; Actigraphy data was collected daily for the week between baseline and week 1 as well as between week 11 and week 12" Data estimation: ITT and completers |
|
| Notes | Dates of trial: Quote: "Recruitment began in September of 2012 and completed in 2015" Industry‐funded: No. Quote: "Supported by the National Institute of Mental Health, No. 5R34MH91338‐03 and No. K23 MH103394 (to Zalta AK)" Medication provided by industry: No Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the patients were randomised however no mention is made of the method of randomisation Quote: "The study was a 12‐wk, double blind, randomized controlled trial with 3 mg of ESZ or PBO at bedtime with a one‐month follow‐up visit" |
| Allocation concealment (selection bias) | Low risk | Quote: "Investigators were blind to the randomization sequence, which was designed by the Medical Center’s research pharmacy" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information on the blinding of personal was provided; nothing on whether participants were blinded, however Quote: "Investigators were blind to the randomization sequence, which was designed by the Medical Center’s research pharmacy" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors Quote: "The study was a 12‐wk, double blind, randomized controlled trial with 3 mg of ESZ or PBO at bedtime with a one‐month follow‐up visit" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More participants withdrew from the eszopiclone group (6/13; 46%) compared to the placebo (2/12; 25%) group. No information was provided on sample characteristics at endpoint. ITT and LOCF population Quote: "Of the 25 randomized subjects (13 ESZ, 12 PBO) 9 did not complete the study (ESZ = 6, PBO = 3). Four experienced worsening symptoms (2 ESZ, 2 PBO), 1 had memory disturbance (ESZ) and 4 were lost to follow up (3 ESZ, 1 PBO) ... The primary analysis used last observation carried forward approach in a modified intent to treat population that excluded subject who did not receive at least 1 dose of study drug" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was available for this study. Memory recall bias and inflammatory markers were not assessed in the final paper |
| Other bias | Low risk | No other sources of bias reported |
Dunlop 2017.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, fixed‐dose, double‐blind Duration of intervention: 6 weeks Placebo run‐in: Not specified Post‐treatment: 1‐month off‐drug follow‐up phase to monitor safety and durability of any clinical changes |
|
| Participants | Setting: Quote: “Four academic sites conducted the study: Emory University, Icahn School of Medicine at Mount Sinai, Baylor College of Medicine, and the University of California San Francisco. Recruitment was conducted by advertising and clinic referral" Sample size: 128 randomised to GSK561679 and placebo Mean age: 40.5 years (SD, 12.1) Sex: 128 women, 3 participants identified combat as their index trauma; MDD 88 (69%) Diagnostic measure: DSM‐IV‐TR, SCID Inclusion criteria: Quote: “Eligible participants were women 18 to 65 years of age who met DSM‐IV‐TR criteria for chronic PTSD, determined using the Structured Clinical Interview for DSM‐IV and confirmed through a clinical interview with a study psychiatrist. For patients with multiple DSM‐IV–qualifying traumas, we defined the “index” trauma as the trauma currently causing the greatest distress or impairment to the patient, identified from parts one and two of the Posttraumatic Diagnostic Scale (PDS). PTSD had to be at least moderately severe at the screening and baseline visits, defined as Clinician Administered PTSD Scale for DSM‐IV (CAPS) past‐month and past‐week total scores $50" Exclusion criteria: Quote: “Important exclusion criteria included the following: any current or past diagnosis of schizophrenia or other psychotic disorder, bipolar disorder, or obsessive compulsive disorder; current substance abuse or dependence; use of a psychotropic agent, other than a nonbenzodiazepine hypnotic; use of a systemic steroid medication; significant uncontrolled medical conditions, or current clinically significant suicidal or homicidal ideation; current participation in a structured psychotherapy targeting PTSD symptoms; and any current or planned litigation regarding the traumatic event" Comorbidity: MD Dropout rates: Insufficient evidence to determine dropouts by groups, number of completers 91 |
|
| Interventions | Pharmacological intervention: Quote: “The selected dose of 350 mg/day of GSK561679 was based on the tolerability and biological activity observed during phase 1 testing. Study medication was dispensed in two bottles containing 100 mg or 50 mg white tablets of GSK561679 or matching placebo. Patients took three 100‐mg tabs and one 50‐mg tab each evening between 6:00 PM and 8:00 PM and recorded the time in a dosing diary" | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: CAPS, MADRS, Safety, Measured by the number of participants that experienced an adverse event Time points: Quote: "Ratings visits occurred at baseline and weeks 1, 2, 4, and 6 postrandomization, with administration of past‐week CAPS, MADRS, and the self‐report symptom measures" Safety, measured by the number of participants that experienced an adverse event: week 6 Data estimation: Intent‐to‐treat and completer estimation |
|
| Notes | Dates of trial: Quote: "Enrolled patients between January 2010 and June 2014" Industry‐funded: No. Quote: "This work was supported by National Institute of Mental Health (NIMH) Grant Nos. U19 MH069056 (to BWD, HM) and K23 MH086690 (to BWD) and Veterans Affairs Clinical Science Research and Development project 09SNIMH‐002 (to TCN)" Medication provided by industry: Yes Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization to GSK561679 or placebo was 1:1 with permuted blocks generated separately for each site by a statistician who was not involved in the analysis of the data" |
| Allocation concealment (selection bias) | Low risk | Quote: "The investigational pharmacist assigned the eligible patient to the treatment indicated by the randomization list at the baseline visit" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. Quote: "We conducted a double‐blind, placebo‐controlled, randomized, fixed‐dose clinical trial evaluating the efficacy of GSK561679, a corticotropin‐releasing factor receptor 1 (CRF1 receptor) antagonist in adult women with PTSD" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient evidence to determine dropouts by groups.They do not describe which they used, LOCF or ITT Quote: "Kaplan‐Meier survival curves failed to demonstrate differential attrition as a function of treatment group. Among individuals (n = 91) who completed treatment and who demonstrated compliance with the medication regimen (verified via serum levels at the week 6 or early termination visit only in the GSK561679 condition), retention did not differ as a function of treatment group" |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol |
| Other bias | Low risk | No other sources of bias reported Quote: "GlaxoSmithKline contributed the study medication, the matching placebo, and the funds to support subject recruitment and laboratory testing, and Neurocrine Biosciences conducted the pharmacokinetic analyses. GlaxoSmithKline and Neurocrine Biosciences were not involved in the data collection, data analysis, or interpretation of findings" |
Feder 2014.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre trial, randomised, placebo‐controlled, parallel arm, fixed‐dose, double‐blind, cross‐over study Duration of intervention: 13 days Placebo run‐in: 2 weeks prior to randomization Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Conducted at a single site (Icahn School of Medicine at Mount Sinai, New York, New York)" Sample size: 41 randomised to ketamine and midazolam Mean age: 36.05 years (SD: 10.4) Sex: 22 men and 19 women, Combat exposure, n = 2 Diagnostic measure: DSM‐IV‐TR Inclusion criteria: Quote: “Eligible participants were between 18 and 55 years of age, had a primary diagnosis of PTSD assessed with the Structured Clinical Interview for DSM‐IV‐TR Axis I Disorders–Patient Version and a score of at least 50 on the CAPS" Exclusion criteria: Quote: “Exclusion criteria included a lifetime history of psychotic or bipolar disorder, current bulimia or anorexia nervosa, alcohol abuse or dependence in the previous 3 months, serious unstable medical illness or sleep apnea, active suicidal or homicidal ideation on presentation, or current use of any psychotropic medications. All patients underwent a physical examination and laboratory screening, including routine hematologic, biochemical, and urine toxicology testing, as well as undergoing electrocardiography to rule out unstable medical illness and active substance use" Comorbidity: No information Dropout rates: 4/41 (3/22 in the ketamine and 1/19 in the midazolam group) |
|
| Interventions | Pharmacological intervention: Quote: “For each procedure day, patients were assigned to receive a single IV infusion of ketamine hydrochloride (0.5 mg/kg) or midazolam (0.045 mg/kg), administered over 40minutes. The order of infusions (ketamine then midazolam or midazolam then ketamine) was randomly assigned, and administrations were 2 weeks apart. Midazolam was chosen as the active placebo control because its pharmacokinetic parameters and nonspecific behavioral effects are similar to those of ketamine. Ratings were administered by a trained rater during infusions and 40, 120, and 240 minutes after infusion. A different trained rater, blinded to the ratings conducted during and after infusion on infusion days, administered ratings at preinfusion baseline and 24 hours (day 1) after infusion (before patients were discharged from the hospital), 48 hours (day 2) after infusion, 72 hours (day 3) after infusion, and 7days (day 7) after infusion. Ratings were also administered 10 and 13 days after infusion, although data analyses focused on the first week after infusion owing to the expected duration of ketamine action" | |
| Outcomes | Primary outcomes: IES‐R
Secondary outcomes: MADRS, CGI‐S, CGI‐I, CADSS, BPRS, YMRS Time points: Quote: "The primary outcome was PTSD symptom severity 24 hours after infusion, assessed with the IES‐R. Twenty‐four hours after infusion was selected as the primary end point because acute sedating and other side effects were expected to have resolved, while potential symptom improvement was expected to persist at 24 hours. Secondary outcome measures were administered by a study clinician 24 hours, 48 hours, 72 hours, and 7 days after infusion. The IES‐R was also administered 48 hours, 72 hours, and 7days after infusion. The CAPS was administered at baseline and 7 days after infusion" Data estimation: Intent‐to‐treat and completer estimation (mixed model) |
|
| Notes | Dates of trial: Quote: "Patients with chronic PTSD were enrolled between May 2009 and December 2012" Industry‐funded: No. Quote: "This study was funded by grant W81XWH‐08‐1‐0602 from the Department of the Army–US Army Medical Research Acquisition Activity (Dr Charney, principal investigator)" Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "41 Randomly assigned to ketamine or midazolam" |
| Allocation concealment (selection bias) | Low risk | Quote: "Only the research pharmacy was aware of drug identity" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All study personnel, including investigators, anesthesiologists, raters, patients, and data analysts, were blinded to randomization order" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All study personnel, including investigators, anesthesiologists, raters, patients, and data analysts, were blinded to randomization order" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More participants withdrew from the ketamine group (3/22, 14%) compared to the midazolam (1/19, 5%) group. However overall dropout rates were low (10%). Reasons for treatment withdrawal include: Found a job; Temporarily lost to follow‐up; Removed from study due to delayed‐onset sedation (ketamine); Removed from study due to low baseline PTSD symptom levels. There was no information to determine whether the two groups differed by sample characteristics at baseline and endpoint. Data estimation was based on intent to treat and completer values Quote: " All 41 patients received study medication and completed 24‐hour ratings; 29 of them completed both infusions and ratings following each infusion. Of the remaining 12 participants, 6 (all of whom had been randomly assigned to receive ketamine first) completed the study at 2 weeks, following only their first infusion and ratings, because their CAPS scores were less than 50 at 2 weeks, precluding the second infusion. Two additional participants also had a CAPS score of less than 50 at 2 weeks, one who received ketamine first and the other who received midazolam first, but they both received their second infusion a week later" |
| Selective reporting (reporting bias) | High risk | Additional outcomes were used in comparison to the protocol. Hopkins Verbal Learning Test (HVLT) and the Quick Inventory of Depressive Symptomatology ‐ Self Report (QIDS‐SR) was not assessed in the published article |
| Other bias | Low risk | No other sources of bias were identified |
Friedman 2007.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 1‐week placebo run‐in period Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “The study was at outpatient psychiatric clinics at 10 VA medical centers in the United States" Sample size: 169 randomised to sertraline and placebo Mean age: 42.4 years (SD, 9.5) Sex: 135 men and 34 women Diagnostic measure: DSM‐III‐R, CAPS‐1, CAPS‐2 Inclusion criteria: Quote: “Subjects include literate male and female subjects 18 years of age and older who had a negative urine drug screen at screening on day 1 of the placebo run‐in, who at study entry had a complete medical and psychiatric history and physical examination with no significant medial problems, and who had discontinued all other psychotropic medication (except chloral hydrate for sleep) prior to entry into the study. Subjects were also included if they were judged reliable for medication compliance and, if female, were practicing acceptable method of contraception and had a negative serum beta‐human chorionic gonadotrophin pregnancy test" Exclusion criteria: Quote: “Excluded from the study were subjects with an organic mental disorder or who had a primary current diagnosis meeting DSM‐III‐R criteria for major depression single episode, dysthymic disorder, personality disorder from clusters other than cluster C, obsessive‐compulsive disorder, generalised anxiety disorder, panic disorder, simple or social phobia, agoraphobia, anxiety disorder, or bipolar disorder. Subjects who had any current psychotic features or had a history of schizophrenia, delusional disorder, or psychotic disorder were excluded. Meeting criteria for any substance use disorder in the past 6 months was also an exclusion. Also excluded were subjects who were receiving concomitant psychotropic therapy of any type, who had therapy with any depot neuroleptic within 6 months, or who would be receiving behaviour therapy during the study, as well as subjects with a history of nonresponse to adequate treatment and subjects who were taking drugs with a psychotropic component, neuroleptics, MAOIs, antidepressants, or hypnotics or anxiolytics in the previous 2 weeks (5 weeks for fluoxetine). Other exclusion criteria included history or evidence of malignancy or significant hematologic, endocrine, cardiovascular, renal, hepatic, neurologic, or gastrointestinal disease; a liver function test result greater than twice the upper limit of the normal range at screening; current impulse control problems; and current involvement in litigation for disability benefits or for damages related to the subjects disorder" Comorbidity: MD, DSM‐III‐R anxiety disorders, history of alcohol or substance abuse Dropout rates: 40/169 (26/86 in the sertraline and 14/83 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Dosage of sertraline was flexible. Subjects who were assigned to sertraline received 25 mg/day for 1 week. Subjects who did not experience dose‐limiting adverse events due to medication were increased to 50 mg/day in week 2. Subjects who failed to respond satisfactorily to 50 mg/day could, in the absence of dose‐limiting adverse events, have their dose titrated in weekly 50 mg increments to a maximum of 200 mg/day depending on the subjects response to the drug" | |
| Outcomes | Primary outcomes: CAPS‐2, IES, CGI‐S, CGI
Secondary outcomes: DTS, HAM‐D, HAM‐A, Mississippi Rating Scale for Combat‐Related PTSD‐Civilian Trauma Version, DESNOS, PSQI Time points: Quote: "Subjects program was evaluated with a series of efficacy measures that were administered at double‐blind treatment weeks 1, 2, 3, 4, 6, 8, 10, and 12. All secondary efficacy scales were administered at baseline and week 12 (or endpoint), with the exception of the DTS, which was completed by the subject at screening, at baseline, and at the end of weeks, 1, 2, 3, 4, 6, 8, 10, and 12" Data estimation: ITT, completers: LOCF |
|
| Notes | Dates of trial: Quote: "Between May 1994 and September 1996". Industry‐funded: Yes. Quote: "Supported by funding from Pfizer Inc." Medication provided by industry: No Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed centrally using a computer‐generated randomization scheme" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Following the placebo run‐in period, subjects we randomly assigned to either sertraline or matching placebo for 12 weeks of double‐blind treatment" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved Quote: "This was a 12‐week, double‐blind, randomized comparison of flexibly dosed sertraline and placebo in the treatment of PTSD. Subjects’ progress was evaluated with a series of efficacy measures that were administered at double‐blind treatment weeks 1, 2, 3, 4, 6, 8, 10, and 12" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More participants withdrew from the sertraline group (26/86, 30%) compared to the placebo group (14/83; 17%). No information was provided on sample characteristics at end point Quote: "Intent‐to‐treat efficacy analyses were conducted on endpoint data (week 12 for completers and at the last available visit for patients who did not complete the study) using all patients with at least 1 post‐baseline assessment. One hundred sixty‐nine subjects (86 sertraline; 83 placebo) were randomly assigned to treatment and dispensed double‐blind medication. All 169 subjects had follow‐up safety data. The intent‐to‐treat population included 166 subjects, 84 who were treated with sertraline and 82 who were treated with placebo (2 sertraline and 1 placebo subject had no postbaseline efficacy data). One hundred twenty—nine subjects completed the study: 60 (70%) in the sertraline group and 69 (83%) in the placebo group. Among women, there were no significant differences between sertraline and placebo groups on any of the baseline demographic or clinical characteristics. There were also no significant differences in these characteristics among men for sertraline versus placebo" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry. Quote: "Supported by funding from Pfizer Inc" |
GSK 29060 627.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 1‐week single‐blind PBO run‐in phase Post‐treatment: 14 days after the last dose of study drug (at the end of the taper phase or after premature withdrawal) |
|
| Participants | Setting: Quote: “44 centres in Austria, Belgium, Canada, France, Germany, Ireland, Netherlands, South Africa, UK, Italy, Israel, and Switzerland" Sample size: 322 randomised to paroxetine and placebo Mean age: 39.2 years (SD, 11.65) Sex: 149 men and 173 women Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “To be eligible to enter the study patients had to fulfill the following: Diagnosis of PTSD according to DSM‐IV criteria with a duration of 3 months; Male or female subjects ≥18 years of age; Signed informed consent" Exclusion criteria: Quote: “Key exclusion criteria: Patients who scored <50 on the CAPS‐2 at baseline; Patients who had a placebo run‐in medication compliance of less that 80% at the baseline visit; Patients who had recently taken tricyclic antidepressants, noradrenergic serotonin reuptake inhibitors (NSRIs), selective serotonin reuptake inhibitors (SSRIs) (other than fluoxetine), lithium and oral neuroleptics, Fluoxetine, or monoamine oxidase inhibitors (MAOIs); Patients who used investigational drug within 1 month prior to screening; Patients who had electroconvulsive therapy (ECT) in the three months prior to screening; Patients who received formal psychotherapy; Patients who met DSM‐IV criteria for substance abuse or dependence within 6 months prior to screening; Patients who posed a current suicidal or homicidal risk; Women of child‐bearing potential who were not practicing a clinically accepted method of contraception; Women who had a positive pregnancy test or who were lactating; Patients who had a current Major Depressive Episode that preceded their diagnosis of PTS" Comorbidity: Not specified Dropout rates: 105/322 (49/160 in the paroxetine and 56/162 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “The dose of paroxetine ranged from 20 to 50mg/day. All subjects randomized to paroxetine remained on 20mg/day for the first 2 weeks of active treatment, and could then be up titrated to a maximum dose of 50mg/day at intervals no more often than every 14 days (maximum of 10mg/day at least every 14 days) at the discretion of the investigator, according to clinical response and tolerability. Dose reduction during the active treatment period was allowed, if necessary, due to an adverse event (AE). If the AE abated, the dose could be returned to the original level. Such a reduction was permitted only once during the study. A gradual reduction of study drug dose over a 3‐week period was performed for subjects who either completed the 12‐week treatment period or who withdrew prematurely" | |
| Outcomes | Primary outcomes: CAPS‐2, CGI‐I
Secondary outcomes: CAPS‐2 (Cluster B, Cluster C, Cluster D), DTS, TOP‐8, MADRS, SDS, CGI‐S, EQ‐5D Time points: Not specified Data estimation: ITT, LOCF |
|
| Notes | Dates of trial: Quote: "Study Period: 8 July 1998 to 21 January 2000" Industry‐funded: Not specified Medication provided by industry: Not specified Any of the authors work for industry: Not specified |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Subjects who scored ≥50 on part 2 of the Clinician Administered Posttraumatic Stress Disorder Scale (CAPS‐2) at baseline (Day 0, the day before the start of study drug administration) were randomized (1:1) to receive either paroxetine or PBO for a 12‐week treatment period, followed by a taper phase of up to 3 weeks" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "A 12‐Week, Double‐Blind, Placebo‐Controlled, Parallel Group Study to Assess the Efficacy and Tolerability of Paroxetine in Patients Suffering from Posttraumatic Stress Disorder (PTSD)" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the paroxetine (49/160, 31%) compared to the placebo group (56/162; 35%). No information was provided on sample characteristics at endpoint. Data estimation ITT and LOCF. Participants withdrew from the study because of reported AEs, lack of efficacy, and other reasons |
| Selective reporting (reporting bias) | Unclear risk | Registered on the GSK clinical trials registry. Insufficient information is provided to determine selective reporting |
| Other bias | Unclear risk | Insufficient information is provided to determine other sources of bias |
Hamner 2008.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 10 weeks Placebo run‐in: 1‐week single‐blind placebo lead‐in Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Ralph H. Johnson VA Medical Center in Charleston, SC" Sample size: 29 randomised to divalproex and placebo Mean age: 52.35 years (SD, 6.8) Sex: 28 men and 1 women, MDD = 20 (69%) Diagnostic measure: DSM‐IV, SCID and CAPS‐1 Inclusion criteria: Quote: “Patients were eligible for the study if they were between age 18 and 65, were competent to give informed consent, and had a score ≥ 50 on the CAPS‐1" Exclusion criteria: Quote: “Exclusion criteria included: history of hypersensitivity to divalproex; unstable medical conditions or medical contraindications to the safe administration of divalproex alcohol or substance use disorders within 1 month of study entry; diagnoses of schizophrenia, schizoaffective disorder, or bipolar disorder; or change in psychotropic medication within 4 weeks of randomization" Comorbidity: Comorbid Axis I: MD, Other anxiety disorder, History of substance use disorder Dropout rates: 13/28 (6/15 in the divalproex and 7/13 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Divalproex was initiated at 750 mg/d in divided doses and increased by 250 mg every 3 to 4 days, if tolerated, until positive clinical response was achieved or desired trough levels were reached" | |
| Outcomes | Primary outcomes: CAPS‐2
Secondary outcomes: IES, CGI‐I, HAM‐D, LSAS, PSQI, PSQI‐A Time points: Quote: "Clinical evaluation and tolerability assessment were performed weekly for the first 2 weeks, then biweekly until the end of the study" Data estimation: ITT |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "The study was funded by Abbott Laboratories" Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Patients were randomized to receive placebo or divalproex" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "A 10‐week randomized, double‐blind, placebo‐control trial was conducted with patients from the Ralph H. Johnson VA Medical Centre in Charleston, SC" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of participants withdrew from the divalproex group (6/15, 40%) compared to the placebo group (7/13; 54%). ITT sample was used throughout the analysis Quote: "The study enrolled 29 patients; 13 were randomized to placebo and 16 to divalproex. One patient in the latter group did not have at least one post randomization visit for efficacy assessments, thus the analysis sample consisted of 15 in the divalproex and 13 in the placebo groups. The baseline characteristics of the subject participants were similar in both groups. The proportion of patients that completed the study was slightly higher in the divalproex group (n = 9 [56.3%]) vs placebo (n = 6 [46.2%]), although the difference was not statistically significant. Early protocol terminations occurred for 7 patients taking divalproex (2 for lack of efficacy; 1 for increased blood pressure; 1 for unsteady gait; 1 for nausea, diarrhoea, and weakness; 1 for noncompliance with protocol; and 1 was lost to follow‐up) and for 7 patients taking placebo (3 for lack of efficacy; 1 for lethargy; 1 due to an unstable medication condition; 1 for lack of time; and 1 was lost to follow‐up). Study completers and study dropouts did not statistically differ on any background characteristic, suggesting an absence of bias among completers compared to those with attrition" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry. Quote: "The study was funded by Abbott Laboratories". "All were male except for one female" |
Hertzberg 1999.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "Duke University Medical Center and the PTSD Outpatient Treatment and the Women’s Veteran Health Programs at the Durham Veterans Affairs Medical Center" Sample size: 15 randomised to lamotrigine and placebo (14 returned for assessment) Mean age: 43.4 years (29 ‐ 53) Sex: 9 men and 5 women, 71% (10/14) war combat Diagnostic measure: DSM‐IV, SIP Inclusion criteria: Quote: “All patients provided informed consent. All patients met DSM‐IV criteria for a primary diagnosis of PTSD based on clinical interview and the Structured Interview for PTSD (SIP). Comorbid Axis I diagnoses were determined by clinical interview and MINI (Sheehan et al 1994). A comprehensive trauma history was obtained using the Kilpatrick’s Potential Stressful Events Interview. All patients were evaluated with a medical and psychiatric history, laboratory studies, and ECG" Exclusion criteria: Quote: “No concomitant psychotropic medications were permitted" Comorbidity: MINI Dropout rates: 4/14 (2/10 in the lamotrigine and 2/4 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “After an initial drug‐free period, patients received study medication as follows: 25 mg/day for 2 weeks, 50 mg/day for 2 weeks, and 100 mg/day in divided dosages for 1 week. Dosages were then increased by 100 mg every 1 to 2 weeks until achieving a maximal response, reaching a maximum dosage of 500 mg/day or upon developing dose‐limiting side effects. A 2‐week medication taper was begun at week 10, with medication being reduced by 50% per week. Mean lamotrigine dose at endpoint (week 10) was 380 mg/day, with a range of 50 mg to 500 mg" | |
| Outcomes | Outcomes: SIP, DGRP‐S, DGRP‐I, Somatic symptoms during treatment were assessed using a 34‐item checklist rated on a 4‐point Likert scale (not clear which outcomes are secondary or primary) Time points: Quote: "After random allocation to treatment, patients were seen at 2‐week intervals throughout the 12‐week study" Data estimation: ITT (LOCF) values provided; LOCF (excluded 1 participant) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "This study was supported in part by Glaxo Wellcome Inc. and a R29 MH51752‐01 awarded by NIMH to JCB" Medication provided by industry: No Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Patients were randomized to a double‐blind treatment with lamotrigine or placebo in a ratio of 2 to 1, respectively" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Subjects enrolled in a 12‐week double‐blind evaluation of lamotrigine and placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More participants withdrew from the placebo group (2/4, 50%) compared to the lamotrigine group (2/10; 20%). No information was provided on sample characteristics at baseline and end point. Reasons for withdrawal were provided. ITT(LOCF) values were used Quote: "Eleven patients received lamotrigine and four patients received placebo. One lamotrigine patient moved away from the area and did not return after the first visit and was not included in any analyses. Two of the 10 lamotrigine patients (20%) developed a rash leading to discontinuation from the study were non responders. In contrast, two of the four patients (50%) developed a rash and did not complete the study" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This study was supported in part by Glaxo Wellcome Inc. and a R29 MH51752‐01 awarded by NIMH to JCB" |
Hertzberg 2000.
| Study characteristics | ||
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "PTSD outpatient treatment program at the Durham Veterans Affairs Medical Center" Sample size: 12 randomised to fluoxetine and placebo Mean age: 46 years (44 ‐ 48) Sex: 12 men, Vietnam combat veterans, 66% (8/12) MDD Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “All patients met DSM‐IV criteria for a primary diagnosis of PTSD based on clinical interview and the Structured Interview for PTSD (SIP)" Exclusion criteria: Quote: “No concomitant psychotropic medications were permitted" Comorbidity: SCID ‐ DSM‐III‐R: MD, SP, OCD, alcohol and marijuana dependence Dropout rates: 1/12 (1/6 in the fluoxetine and 0/6 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “After an initial drug‐free period, patients received study medication as follows: 10 mg/day for 1 week, then 20 mg/day for 2 weeks, then dosages were increased 10 mg/day every week until achieving a maximal response, reaching a maximum dosage of 60 mg/day or upon developing dose‐limiting side effects. Mean fluoxetine dose at endpoint (week 12) was 48 mg/day with a range of 10 mg to 60 mg" | |
| Outcomes | Outcomes: DTS, SDS, SIP, DGRP, Somatic symptoms during treatment were assessed using a 34‐item checklist rated on a 4‐point Likert scale. Patients completed this checklist at baseline and each subsequent visit (no distinction between primary and secondary outcomes) Time points: Quote: "After random allocation to treatment, patients were seen at end of weeks 1, 2, 3, 4, 6, 8, 10, and 12 after receiving study medication" Data estimation: Presumably LOCF (not clear in text) |
|
| Notes | Dates of trial: Not stated Industry‐funded: No. Quote: "This study was supported in part by an NIMH Grant MH44740‐01" Medication provided by industry: Yes Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Patients were randomized to a double‐blind treatment with fluoxetine or placebo in a 1:1 ratio" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Placebo and fluoxetine were provided by Eli Lilly Co" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved Quote: "Twelve male veterans with PTSD were enrolled in a 12 week double‐blind evaluation of fluoxetine and placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More participants withdrew from the fluoxetine group (1/6, 17%) compared to the placebo group (0/6; 0%). However, the dropouts across the two groups were low (i.e. 8%). No information was provided on sample characteristics at end point. Reasons for withdrawal were provided. They do not describe which they used, LOCF or ITT Quote: "There were no significant differences between the groups for compensation status, age, or ethnic minority status, type, chronicity or severity of trauma. One of the six fluoxetine patients did not complete the study because of medication side effects. He experienced activation symptoms and dropped out at the end of week two on a dose of 10 mg/day. All the other patients completed the full 12 weeks of the study" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified. Medication was provided by industry, but no authors work for industry |
Kaplan 1996.
| Study characteristics | ||
| Methods | Design: multi‐centre, randomised, placebo‐controlled, parallel arm, crossover, flexible‐dose, double‐blind Duration of intervention: 4 weeks Placebo run‐in: 2 week washout period Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "Patients were offered inositol therapy in two different outpatient clinics, the Beer‐Sheva Mental Health
Center and Abarbanel Mental Health Center" Sample size: 17 randomised to inositol and placebo Mean age: 39.7 years (25 ‐ 56) Sex: 8 men 5 women, 3% (3/13) combat‐related, no comorbid major depression Diagnostic measure: DSM‐III‐R Inclusion criteria: Quote: “Patients met DSM‐III‐R criteria for PTSD" Exclusion criteria: Quote: “Patients with any history of alcohol or drug abuse as well as patients with organometal comorbidity were excluded" Comorbidity: No information Dropout rates: 4/17 (insufficient evidence to determine dropouts by groups) |
|
| Interventions | Pharmacological intervention: Quote: “The trial was double‐blind and all patients were given inositol or placebo (glucose) in identical containers according to a prearranged random code, 12 grams a day of each substance. The substances were in powder form of very similar taste and texture and patients were instructed to take two teaspoons twice a day in juice or tea. The design was crossover after a washout period of 2 weeks. Patients received four weeks of inositol or placebo followed by the other substance" | |
| Outcomes | Outcomes: IES, SCL‐90 (1 centre), HAM‐A, HAM‐D (other centre) (no distinction between primary and secondary outcomes) Time points: Quote: "The patients were assessed very two weeks with the IES. At baseline all patients were assessed with the SCL‐90. At the Beer‐Sheva Clinic every two weeks and at the Abarbanel clinic biweekly with the HAM‐D and HAM‐A" Data estimation: Completer values |
|
| Notes | Dates of trial: Not stated. Industry‐funded: No information. Medication provided by industry: No Any of the authors work for industry: No information |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The trial was double‐blind and all patients were given inositol or placebo (glucose) in identical containers according to a prearranged random code, 12 grams a day of each substance" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "The trial was double‐blind and all patients were given inositol or placebo (glucose) in identical containers according to a prearranged random code, 12 grams a day of each substance. The substances were in powder form of very similar taste and texture and patients were instructed to take two teaspoons twice a day in juice or tea" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "The trial was double‐blind and all patients were given inositol or placebo (glucose) in identical containers according to a prearranged random code, 12 grams a day of each substance" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient evidence to determine dropouts by groups. No information was provided on sample characteristics at baseline and end point. Reasons for withdrawal were not provided. Completer values were used Quote: "Seventeen patients agreed to participate and were randomized, but only thirteen completed the study. Two dropped out within one week of starting the trial, and the other two at the third week of the trial" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | No information on if the trial was funded by industry Quote: "Throughout the trial patients received supportive psychotherapy only" |
Katz 1994.
| Study characteristics | ||
| Methods | Design: multi‐centre, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 14 weeks Placebo run‐in: 2 week single‐blind placebo run‐in Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "Outpatients in three countries" Sample size: 64 randomised to brofaromine and placebo Mean age: 39 years (25 ‐ 56) Sex: 49 men 15 women, 7% (8/45) combat‐related Diagnostic measure: DSM‐III‐R (confirmed by CAPS) Inclusion criteria: Quote: “This trial was restricted to male and female outpatients 18‐65 years old fulfilling DSM‐III‐R definitions; full satisfaction of CAPS criteria, therefore, provided a further objective check on diagnostic acceptability. Patients were obliged to fully satisfy B, C and D criteria and have a minimum total score of 36 for entry. In principle, a CAPS score as low as 18 can technically satisfy a PTSD diagnosis; a score of two times this level was employed to further assure the validity of the diagnosis and adequate severity" Exclusion criteria: Quote: “Patients were excluded from trial entry if they had a comorbid primary depression, based upon psychiatric interview and a Hamilton Depression Rating Scale (HAM‐D) score of > 21, with > 2 on item 1 (depressed mood). Patients were also excluded if they had a placebo response (≥ 20% decrease on the CAPS during their placebo run‐in). Patients were required to meet standard criteria of medical eligibility. Patients known to abuse alcohol or drugs were excluded from trial entry. With the exception of chloral hydrate and short acting benzodiazepines hypnotics for sleep, other medications with central nervous system (CNS) effects could be not employed in this trial" Comorbidity: HAM‐D > 21 Dropout rates: 19/64 (10/33 in the brofaromine and 9/31 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “A one‐ to four‐week drug‐free phase preceded a two‐week single‐blind placebo phase. Following a fixed titration, a flexible dosage schedule was utilized. Dosing started at 50 mg/day or 1 identical placebo tablet/day and was titrated up to a maximum of 150mg/day or 3 tablets. Final dosage was determined by the investigator, and represented the treating physicians judgment of a therapeutic best dose. Patients were studied for up to 14 weeks in the double‐blind phase. Twice daily dosing was routinely employed, with drugs taken with accompanying food. The modal dose was 2 tablets" | |
| Outcomes | Primary outcome: CAPS
Secondary outcome: CGI Time points: Quote: "Weekly CAPS ratings" Data estimation: LOCF |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "This study was supported in part by Ciba‐Geigy Corporation" Medication provided by industry: Unclear Any of the authors work for industry: Yes CGI‐C values obtained from Davidson et al, 1997 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the prticipants were randomised, but no mention is made of the method of randomisation Quote: "This was a multicentre, randomized, double‐blind, parallel trial conducted in outpatients in three countries" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Dosing started at 50 mg/day or 1 identical placebo tablet/day and was titrated up to a maximum of 150mg/day or 3 tablets" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved Quote: "Patients were studied for up to 14 weeks in the double‐blind phase" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the brofaromine group (10/33, 30%) compared to the placebo group (9/31; 29%). LOCF values were used Quote: "Patients appeared well‐mated on standard demographics criteria, and entered with similar severities of illness based upon the CAPS. Safety and tolerability were assessed via an examination of serious events, hospitalizations, and discontinuations due to medical events. One patient receiving brofaromine elected to discontinue due insomnia and agitation" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Quote: "This study was supported in part by Ciba‐Geigy Corporation; It is evident that while a majority of patients were male, a substantial proportion were female" |
Kosten 1991.
| Study characteristics | ||
| Methods | Design: multi‐centre, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "The subjects in this study were outpatient male Vietnam combat veterans seeking treatment at three Connecticut Veterans Readjustment Counselling Centers" Sample size: 60 randomised to imipramine, phenelzine and placebo Mean age: 39 years (SD: 2.6) Sex: 60 men, all combat veterans, no‐one with comorbid major depression Diagnostic measure: DSM‐III Inclusion criteria: Quote: “The subjects in this study were outpatient male Vietnam combat veterans seeking treatment at three Connecticut Veterans Readjustment Counselling Centers. We screened 102 male Vietnam veterans meeting DSM‐III criteria for PTSD, and 42 refused or were excluded due to current substance abuse" Exclusion criteria: Quote: “We excluded subjects with schizophrenia, bipolar disorder, and substance abuse within the month previous to the study, but our subjects did have substantial rates (57%) of past substance abuse, mainly alcoholism" Comorbidity: SADS, RDC: minor depression and alcoholism Dropout rates: 28/60 (12/23 in the imipramine, 4/19 phenelzine and 12/18 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “The initial dose of imipramine was 50 mg daily, adjusted up to 300 mg/day, with a mean (SD) maximal dose of 225 (SD: 55) mg (4.5 tablets) and a mean blood level of imipramine plus desipramine of 184 (SD: 80) ng/ml after a week of stable dosing (weeks 2 to 4). The starting dose of phenelzine was 15 mg daily and was adjusted up to 75 mg/daily with a maximal dose of 68 (SD: 20) mg (4.5 tablets) and mean platelet MAO activity inhibition of 94% (SD: 7%). The placebo group took a mean maximal dose of 4.4 (SD: 1.4) tablets per day. This was not significantly different from the number of tablets taken daily by the other two groups, supporting the maintenance of the double‐blind during the trial" | |
| Outcomes | Primary outcomes: IES
Secondary outcomes: Combat Scale, CAS, HAM‐D, HAM‐A, RSD Time points: Quote: "Subjects were rated weekly on the IES, Hamilton, Covi and Raskin scales and a 5‐point CGI scale was used to rate symptom change at the end of the trial" Data estimation: LOCF (completers of 3 or more weeks) |
|
| Notes | Dates of trial: Not stated Industry‐funded: No Medication provided by industry: Yes Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Subjects in this study were randomly assigned to active medication or placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Subjects in this study were randomly assigned to active medication or placebo, and evaluated under double‐blind conditions. In order to maintain the blind in this study, the nonblind physician also changed the number of placebo pills taken by “yoked” placebo patients" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More participants withdrew from the imipramine (12/23, 52%) and placebo group (12/18, 67%) compared to the phenelzine group (4/19; 21%). No information was provided on sample characteristics at end point. Reasons for withdrawal were provided. LOCF values were used Quote: "The three treatment groups did not differ in demography, rates of comorbid psychopathology, or baseline symptoms. Reasons for dropout differed somewhat among the three medication groups. The most common reason was simply failure to return without explanation, and this occurred in 50% of the placebo (N = 6) and imipramine (N = 6) dropouts and 25% of the phenelzine (N = 1) dropouts. The next common reason was “medication” side effects in 25% of the placebo (N = 3), 33% of the imipramine (N = 4), and 25% of the phenelzine (N = 1) dropouts. Clinical deterioration leading to discontinuing the medication occurred in 17% of the imipramine (N = 4) and 50% of the phenelzine (N = 2) dropouts. Finally, 25% of the placebo patients (N = 3) were administratively terminated after 7 weeks due to transfer to more intensive inpatient treatments" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Medication provided by industry and some authors work for industry |
Li 2017.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre, randomised, placebo‐controlled, parallel arm, fixed‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: Not specified Post‐treatment: Not specified |
|
| Participants | Setting: China. Sample size: 72 randomised to sertraline and placebo Mean age: 46 years (SD: 5.95) Sex: 63 men and 9 women, mood or anxiety disorders 7 (10%), military‐related trauma 27 (38%) Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “The following inclusion criteria were applied: (1) age of at least 18 years, (2) fulfilment of the DSM PTSD criteria, (3) illness duration of at least 6 months and (4) CGI‐S score of ≥ 4 at the baseline visit" Exclusion criteria: Quote: “The following exclusion criteria were applied: (1) current or past history of bipolar disorder, schizophrenia, or other psychotic disorder; (2) alcohol or substance abuse or dependence within the preceding 6 months; (3) intolerance of or hypersensitivity to sertraline; (4) any acute or unstable medical condition that could interfere with the safe conduct of the study; (5) pregnancy, nursing or failure to practice a medically accepted form of birth control and (6) receipt of cognitive‐behavioural therapy during the trial" Comorbidity: History of mood and anxiety disorders, and mental illness Dropout rates: 7/72 (4/36 in the sertraline and 3/36 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Subjects assigned to the sertraline and placebo groups received sertraline and a placebo (both 135 mg daily), respectively, for 12 weeks. All medication was given as three pills (50, 50, and 35 mg) each day at a fixed dose" | |
| Outcomes | Primary outcomes: IES‐R
Secondary outcomes: CGI‐S Time points: not specified Data estimation: completer values |
|
| Notes | Dates of trial: Not stated Industry‐funded: No. This study was funded by grants Medication provided by industry: Yes Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Seventy‐two patients were randomly assigned to 12 weeks of a double‐blind trial of either sertraline (n = 36) or a matched placebo (n = 36), using computer‐generated random numbers. Patients who met all of the inclusion and exclusion criteria were assigned to either the sertraline or placebo group using a computerised number generator and the Statistical Analysis Software stratified block randomisation method" |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomisation was performed by a professional statistician, who was blind to treatment allocation. Randomisation assignments were concealed in opaque sequentially numbered sealed envelopes. In addition, all medication was provided and prepared in a double‐blind‐form by the Company Limited of Guangzhou Guanhua Pharmaceutical Industry" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The subjects, researchers, outcome assessors and data analyst were also blind to treatment allocation" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The subjects, researchers, outcome assessors and data analyst were also blind to treatment allocation" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the sertraline group (4/36, 11%) compared to the placebo group (3/36; 8%). The 2 groups did not differ by sample characteristics at baseline. Data estimation for completer values. Quote: "Seven patients withdrew from the study; the major reasons for withdrawal included AEs, consent withdrawal, loss to follow up and other reasons. Age, sex, ethnicity, disease duration, educational background, employment, mood and anxiety disorder prevalence, and baseline IES‐R and CGI‐S scores did not differ significantly between groups". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified Medication was provided by industry, butr no authors work for industry |
Marshall 2001.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, fixed‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 1‐week placebo run‐in Post‐treatment: Not specified |
|
| Participants | Setting: Outpatients. Sample size: 551 randomised to paroxetine and placebo Mean age: 41.8 years Sex: 236 men and 315 women, approximately 45% (248/551) comorbid MDD Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Study subjects were male and female outpatients 18 years or older who met DSM‐IV criteria for chronic PTSD as determined by the diagnostic version of the Clinician‐Administered PTSD Scale, part 1, and the Mini International Neuropsychiatric Interview. The Clinician‐Administered PTSD Scale is a validated clinical interview designed to assess the frequency and severity of each of the 17 DSM‐IV‐defined PTSD symptoms as well as criterion F (social and occupational impairment). The Clinician‐ Administered PTSD Scale, part 1, is used to assess a patient’s current and lifetime DSM‐IV diagnosis of PTSD, while the Clinician‐Administered PTSD Scale, part 2, is used to evaluate symptom severity and change. Concurrent affective and anxiety disorders were allowed in study patients, provided that PTSD was considered the principal diagnosis (i.e., the main focus of attention or need for treatment) and the onset of PTSD preceded that of concurrent disorders. Furthermore, patients could not have had another axis I disorder as a principal diagnosis within 6 months of screening. A Clinician‐Administered PTSD Scale, part 2, total score of 50 points or higher was required for study entry. For female patients of childbearing potential, participation was contingent on a negative serum pregnancy test and a medically accepted method of contraception" Exclusion criteria: Quote: “Other exclusion criteria were 1) receiving disability payments or involvement in litigation related to PTSD or any other psychiatric illness, 2) alcohol or substance abuse or dependence within 6 months of screening, 3) taking psychotropic medications within 2 weeks of the first dose of study medication (or 4 weeks for fluoxetine), 4) having psychotherapy or ECT within 12 weeks of screening, 5) being a homicidal or suicidal risk, 6) intolerance to paroxetine or any other SSRI, or 7) having a serious medical condition" Comorbidity: MDD Drop‐out rates: 208/563 (140/375 in the paroxetine and 68/188 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Eligible patients entered a 1‐week placebo run‐in period to evaluate compliance with study procedures, followed by random assignment to one of the three treatments. All paroxetine‐treated patients started therapy at 20 mg/day; patients assigned to the 40‐mg/day group then received 30 mg/day during week 2 and 40 mg/day from the beginning of week 3. Chloral hydrate was permitted at doses up to 1000 mg per night for sleep disturbance only during the placebo run‐in period and week 1" | |
| Outcomes | Primary outcomes: CAPS‐2, CGI‐I
Secondary outcomes: DTS, TOP‐8, CAPS‐2, SDS, MADRS Time points: Quote: "Assessments were conducted at weeks 1, 2, 4, 6, 8, and 12. Safety assessments conducted at screening and endpoint or at study discontinuation included a complete physical examination, a laboratory evaluation (including clinical chemistry and hematology tests and a urinalysis), and an ECG. At each scheduled visit, the patient’s sitting heart rate and blood pressure were also documented" Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Supported by GlaxoSmithKline and NIMH grant MH‐01412 (to Dr. Marshall)" Medication provided by industry: No information Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the patients were randomised however no mention is made of the method of randomisation Quote: "Outpatients with chronic PTSD according to DSM‐IV criteria and a score of 50 or more on the Clinician‐Administered PTSD Scale, part 2, were randomly assigned to take placebo (N=186), 20 mg/day of paroxetine (N=183), or 40 mg/day of paroxetine (N=182) for 12 weeks" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "This study was a 12‐week, double‐blind comparison of 20 mg/ day and 40 mg/day of paroxetine and of placebo in adults with chronic PTSD" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of participants withdrew from the paroxetine groups (20mg: 61/188, 32%; 40mg: 69/187, 37%) compared to the placebo group (66/188; 36%). The two groups did not differ by sample characteristics at baseline. Reasons for withdrawal were not provided Quote: "Statistical analyses were based on the data set with the last observation carried forward for the patients who were randomly assigned to groups and who had at least one treatment assessment (intent‐to‐treat group). A total of 840 patients entered the screening/placebo run‐in phase of the study. Of these, 277 patients were not randomly assigned to treatment groups at baseline for the reasons shown in Figure 1. Of the 563 patients randomly assigned to receive treatment, 12 patients (two receiving placebo and five in each paroxetine dose group) were lost to follow‐up before the first postbaseline assessment. The remaining 551 patients had at least one treatment assessment and comprised the intent‐to‐treat group for all analyses (186 receiving placebo, 183 receiving 20 mg/day of paroxetine, and 182 receiving 40 mg/day of paroxetine). Figure 1 summarizes patient disposition throughout the study. Patient demographic characteristics did not differ across the three randomly assigned groups" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry. Quote: "Supported by GlaxoSmithKline and NIMH grant MH‐01412 (to Dr. Marshall)" |
Marshall 2007.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind and maintenance study Duration of intervention: 10 weeks acute, 12 weeks maintenance Placebo run‐in: 1 week of single‐blind placebo Post‐treatment: Not specified |
|
| Participants | Setting: New York Sample size: 52 randomised to paroxetine and placebo Mean age: 39.8 years (SD: 11.2) Sex: 17 men and 35 women, no combat‐related trauma, 62.5% (30/48) MDD Diagnostic measure: DSM‐IV, SCID Inclusion criteria: Quote: “Subjects were adults between 18 and 65 years of age, who met DSM‐IV criteria for chronic PTSD (duration at least 3 months) according to the Structured Clinical Interview for DSM‐IV and who provided written informed consent after the study procedures were explained. Comorbid affective, anxiety, and personality disorders were allowed provided that PTSD was the primary diagnosis in need of clinical intervention. Maintenance phase. We entered subjects rated as much improved or very much improved, on the Clinical Global Impression Improvement scale (CGI‐I score of 2 or 1, respectively) at week 10 into a 12‐week maintenance phase with continuation of double‐blind treatment and independent evaluations every 4 weeks, to examine whether clinical gains were maintained and/or continued to accrue" Exclusion criteria: Quote: “Concomitant psychoactive medications and concomitant trauma‐focused psychotherapy were not allowed. Patients were excluded if they had previously been treated with paroxetine 20 mg or more for at least 4 weeks; if they manifested psychotic symptoms or serious suicidal ideation; if they met criteria for substance abuse disorders within the past 3 months or lifetime criteria for schizophrenia, schizoaffective disorder, or bipolar disorder; if they had been successfully treated in the past with an antidepressant for major depressive disorder; or if they had unstable medical illness or significant abnormalities on laboratory or electrocardiogram (ECG) examinations. Prior to entering the trial, patients were medication‐free for 2 weeks if previously taking any psychoactive medication, or for 4 weeks if previously taking fluoxetine or a monoamine oxidase inhibitor" Comorbidity: SCID: social phobia, MDD, panic disorder, personality disorder Dropout rates: 12/52 (8/25 in the paroxetine and 4/27 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Non responders to placebo were randomized to paroxetine–placebo initiated at 10 mg daily. If patients remained symptomatic, dosage was increased to 20 mg for weeks 2 and 3, 40mg daily for weeks 4–6, and 60 mg daily for weeks 7–10 as tolerated" | |
| Outcomes | Primary outcomes: CGI‐I, CAPS
Secondary outcomes: CAPS‐2, IIP, DES, HAM‐A, HAM‐D‐24, and items 22 (social functioning) and 23 (occupational or important role functioning) of the CAPS‐2 Time points: Quote: "Primary outcome was assessed using x2 analysis of the CGI‐I scale at weeks 0, 5, and 10. The treating physician assessed adverse effects at each visit using an adverse effects checklist" Data estimation: ITT and observed cases (i.e. mixed effects model) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "National Institutes of Mental Health; Contract grant number: MH‐01412; Contract grant sponsor: Glaxo SmithKline (investigator‐initiated grant)" Medication provided by industry: No information Any of the authors work for industry: No information |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the patients were randomised however no mention is made of the method of randomisation Quote: "Those not rated as significantly improved were then randomly assigned to placebo (N527) or paroxetine (N525) for 10 weeks, with a flexible dosage design (maximum 60mg by week 7)" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | It is not clear whether the participants were blinded, only the personnel Quote: "Treating physicians and IE staff were kept blind to randomized assignment throughout the course of the study" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Outcome was assessed by independent evaluators blind to treatment status or side‐effect profile. Maintenance phase. We entered subjects rated as much improved or very much improved, on the Clinical Global Impression ‐ Improvement scale (CGI‐I score of 2 or 1, respectively) at week 10 into a 12‐week maintenance phase with continuation of double‐blind treatment and independent evaluations every 4 weeks, to examine whether clinical gains were maintained and/or continued to accrue" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More participants withdrew from the paroxetine group (8/25, 32%) compared to the placebo group (4/27; 15%). Reasons for withdrawal were not provided. ITT and observed cases were used for the estimation of data. Quote: "Patient demographics (age, gender, ethnicity, language status, education, and income) did not differ significantly between the paroxetine and placebo groups" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Quote: "Contract grant sponsor: Glaxo SmithKline (investigator‐initiated grant)" |
Martenyi 2002a.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind, 24 weeks relapse prevention phase (i.e. Martenyi 2002b) Duration of intervention: 12 weeks Placebo run‐in: 1‐ to 2‐week diagnostic evaluation period during which no study drug was given Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "18 study centers in Belgium, Bosnia, Croatia, Israel, South Africa and Yugoslavia" Sample size: 301 randomised to fluoxetine and placebo Mean age: 37.9 years (SD: 13.9) Sex: 244 men and 57 women, 6% combat‐related Diagnostic measure: DSM‐IV, CAPS Inclusion criteria: Quote: “Participants in the trial were men and women aged 18 to 65 years who met DSM‐IV criteria for PTSD according to the Structured Clinical Interview for DSM‐IV Axis I Disorders, Investigator Version and the CAPS, Current Diagnostic Version (CAPS‐DX), To enrol, patients must have had a total score ≥ 50 on the CAPS‐DX and a score ≥ 4 on the Clinical Global‐Impression‐Severity of Illness scale (CGI‐S) at baseline (Visit 2). Patient with a Montgomery‐Asberg Depression Rating Scale (MADRS) score > 20 at baseline were ineligible for the study" Exclusion criteria: Quote: “Exclusion criteria included a serious comorbid illness, serious suicidal risk or hetero‐ aggressivity, or a diagnosis of an Axis I psychiatric disorder as defined by DSM‐IV criteria within the 5 years prior to the primary traumatic episode. Patients diagnosed with bipolar disorder, OCD, or schizophrenia at any time were excluded. Patients with a diagnosis of any Axis I psychiatric disorder or comorbidity following the primary traumatic episode, with the exception of generalized anxiety disorder, depression, panic disorder, or social phobia, were excluded. Patients with substance abuse following the traumatic episode were allowed to enrol, provided the abuse had resolved at least 6 months prior to study entry" Comorbidity: MADRS > 20 Dropout rates: 139/301 (95/226 in the fluoxetine and 4/75 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Fluoxetine‐treated patients initially received a dosage of 20 mg/day. This dose could have been increased in 20‐mg increments at each of the 3 titration points based on predefined response criteria to a maximum dosage of 80 mg/day. Patients were seen at 3‐week intervals throughout the 12‐week acute treatment period" Description: fluoxetine 20 mg/d ‐ 80 mg/d, (avg dose: 57.8 mg/d) versus placebo x 12 weeks |
|
| Outcomes | Primary outcomes: TOPS‐8
Secondary outcomes: CAPS‐2, CGI‐S, CGI‐I, DTS, MADRS, HAM‐A, SCL‐90‐R, DES Time points: No information Data estimation: Baseline, Endpoint, LOCF (repeated measures) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "This work was sponsored by Eli Lilly and Company" Medication provided by industry: No information Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A computer generated randomization sequence was used to determine each patients treatment group assignment. Emergency codes, generated by a computer drug‐labelling system, were available to the investigator. These codes, which revealed the patients treatment group, were opened during the study only if the choice of follow‐up treatment depended on the patients treatment assignment" |
| Allocation concealment (selection bias) | Low risk | Quote: "A computer generated randomization sequence was used to determine each patients treatment group assignment. Emergency codes, generated by a computer drug‐labelling system, were available to the investigator. These codes, which revealed the patients treatment group, were opened during the study only if the choice of follow‐up treatment depended on the patients treatment assignment" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved Quote: "This was a double‐blind randomized, placebo‐controlled study conducted in Europe, Israel, and South Africa, primarily in war‐torn countries" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More participants withdrew from the fluoxetine group (95/226, 42%) compared to the placebo group (4/75, 5%). Reasons for withdrawal were not provided, only for dropout due to adverse events, although not specific by group. The two groups did not differ by sample characteristics at baseline. LOCF values were used for the estimation of data Quote: "Demographic data and baseline clinical characteristics were similar for both groups, with the exception of a statistically significant difference in CGI‐S score" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This work was sponsored by Eli Lilly and Company" Reliability and validity of instruments used |
Martenyi 2002b.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind; relapse prevention component of Martenyi 2002a Duration of intervention: 24 weeks Placebo run‐in: 1‐ to 2‐week diagnostic evaluation period during which no study drug was given at acute phase (i.e. 12 weeks) Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "18 study centres in Belgium, Bosnia, Croatia, Yugoslavia, Israel and South Africa" Sample size: 131 randomised to fluoxetine and placebo Mean age: 38.2 years Sex: 106 men and 25 women, 47% trauma combat‐related Diagnostic measure: DSM‐IV, SCID–I, CAPS–DX Inclusion criteria: Quote: “Participants were men and women aged 18–65 years who met DSM–IV criteria for PTSD according to the SCID–I and the CAPS–DX. To enrol, patients had to have a total score ≥ 50 on the CAPS–DX and a score ≥ 4 on the CGI–S scale at baseline (visit 2). Individuals with MADRS scores > 20 at baseline were ineligible for the study" Exclusion criteria: Quote: “Exclusion criteria included serious comorbid illness, concomitant psychotherapy, serious suicidal risk or risk to others, and diagnosis of an Axis I psychiatric disorder (defined by DSM–IV criteria) 5 years before the primary traumatic episode. Patients with lifetime diagnoses of bipolar disorder, obsessive–compulsive disorder (OCD) or schizophrenia were excluded. Those with a diagnosis of any Axis I psychiatric disorder or comorbidity following the primary traumatic episode, except generalised anxiety disorder, depression, panic disorder or social phobia, were also excluded. Patients with a history of alcohol or substance misuse following the primary traumatic episode were allowed to enrol if the misuse had resolved at least 6 months before study entry" Comorbidity: MADRS > 20 Dropout rates: 33/131 (12/69 in the fluoxetine and 21/62 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Fluoxetine‐treated patients initially received 20mg/day. This dose could be increased by 20‐mg increments at each of three titration points based on predefined response criteria (CGI–S ≥ 3) to a maximum dosage of 80mg/day. Acute response to fluoxetine v. placebo has been described elsewhere. After 12 weeks of acute treatment with fluoxetine or placebo, participants who responded to treatment by a 50% decrease in the eight‐item TOP–8 score from baseline, a CGI–S score ≤ 2, and failing DSM–IV diagnostic criteria for PTSD continued in a 24‐week relapse prevention phase. Those patients who had received fluoxetine were randomised either tocontinue without variation from final dosage in the acute phase or to placebo treatment. In order to maintain blinding, patients switching to placebo did not undergo a tapering regimen" | |
| Outcomes | Primary outcomes: Time to relapse on TOPS‐8 (≥ 40% increase for 12 week acute baseline) and CGI‐S (≥ 2 on CGI‐S from 12 week acute baseline)
Secondary outcomes: CAPS‐2, CGI‐I, DTS, MADRS, HAM‐A, SCL‐90‐R Time points: No information Data estimation: Baseline, endpoint, LOCF |
|
| Notes | Dates of trial: Quote: "The study was conducted from June 1998 to August 2000" Industry‐funded: Yes. Quote: "This work was sponsored by Eli Lilly & Co." Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "After a 1‐ to 2‐week evaluation period, participants were randomised to 12 weeks’ double‐blind acute treatment with fluoxetine or placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "This was a double‐blind, randomised, placebo‐controlled study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More participants withdrew from the placebo group (21/62, 34%) compared to the fluoxetine group (12/69, 17%). Reasons for withdrawal include: adverse events, clinical relapse, patient decision, lack of efficacy, noncompliance, lost to follow‐up. The 2 groups did not differ by sample characteristics at baseline. Baseline, endpoint and LOCF values were used for the estimation of data. Quote: "Demographic as well as disease characteristics following 12 weeks of acute fluoxetine treatment were similar in both groups" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This work was sponsored by Eli Lilly & Co." |
Martenyi 2007.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, fixed‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 1‐ to 2‐week, open‐label, washout period Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "Forty‐three investigational sites participated in the study, all from the United States" Sample size: 411 randomised to fluoxetine and placebo Mean age: 40.7 years (SD: 11.8) Sex: 117 men and 294 women Diagnostic measure: DSM‐IV, SCID, CAPS Inclusion criteria: Quote: “Participants in the trial were men and women aged 18 to 75 who met Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM‐IV) criteria for PTSD according to the Structured Clinical Interview for DSM‐IV Axis I Disorders, Investigator Version and CAPS Current Diagnostic Version. To enroll, patients must have had a score of 50 or more on the CAPS Current Diagnostic Version and a score of 4 or more on the Clinical Global Impression of Severity (CGI Severity) scale at baseline" Exclusion criteria: Quote: “Patients with severe (comorbid) depression were ineligible to enter the study as defined by Montgomery‐Asberg Depression Rating Scale (MADRS) score greater than 20 at baseline" Comorbidity: Not specified Dropout rates: 259 (63%) dropouts in total ‐ Insufficient information to determine drop‐outs by group |
|
| Interventions | Pharmacological intervention: Quote: “This study was a 12‐week double‐blind comparison of 20 and 40 mg/d fluoxetine and placebo in adults diagnosed with PTSD. Eligible patients entered a 1‐ to 2‐week, open‐label, washout period followed by random assignment to 1 of the 3 treatment groups in a 2:2:1 manner: 20 mg/d fluoxetine, 40 mg/d fluoxetine, and placebo, respectively. All fluoxetinetreated patients initially received 20 mg/d, and patients randomized to the 40‐mg/d treatment group were titrated up to 40 mg/d after 2 weeks" | |
| Outcomes | Primary outcomes: TOP‐8
Secondary outcomes: CAPS, DTS, MADRS, HAS, DES Time points: No information Data estimation: ITT, LOCF (mixed model) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Support for this study was provided by Eli Lilly and Company" Medication provided by industry: Yes Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Eligible patients entered a 1‐ to 2‐week, open‐label, washout period followed by random assignment to 1 of the 3 treatment groups in a 2:2:1 manner: 20 mg/d fluoxetine, 40 mg/d fluoxetine, and placebo, respectively" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "A multicenter, double‐blind, 12‐week, placebo‐controlled trial of 411 randomized patients, predominantly women diagnosed with posttraumatic stress disorder, failed to show a difference between either dose of fluoxetine treatment and placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More participants withdrew from the 20 mg fluoxetine and placebo group. However the proportion of dropouts is low. The 2 groups did not differ by sample characteristics at baseline Quote: "A total number of 411 patients were randomized into the study (163, 160, and 88 in the 20‐mg/d fluoxetine, 40‐mg/d fluoxetine, and placebo treatment groups, respectively). Of these patients, 71.5% were women and 76.9% were white, with a mean age of 40.7 years (SD = 11.8 years). Other baseline demographic and psychometric data are given in Table 1. Significantly more patients discontinued the study due to an adverse event in the 40‐mg/d fluoxetine treatment group (13.1%) than in the 20‐mg/d fluoxetine treatment group (4.3%; P = 0.005); however, neither of these rates differed significantly from the placebo treatment group (8.0%, both P > 0.20). There were no other significant differences between treatment groups for other reasons of discontinuation, including discontinuation due to lack of efficacy (20 mg fluoxetine, 6.7%; 40 mg fluoxetine, 3.8%; placebo, 6.8%; P = 0.416) or discontinuation due to patient decision (20 mg fluoxetine, 11.0%; 40 mg fluoxetine, 6.9%; placebo, 13.6%; P = 0.182)" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Support for this study was provided by Eli Lilly and Company" |
Mathew 2011.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, fixed‐dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: 2‐week placebo lead‐in Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "Conducted at 2 U.S. sites" Sample size: 47 randomised to GR205171 and placebo Mean age: 40.85 years (SD: 11.9) Sex: 16 men and 23 women (for 39 participants), 25 MDD (64%), 3 combat trauma (8%) Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Patients (aged 18–65) were recruited from media advertisement (86%) or clinician referral (14%). Diagnoses were made with the Structured Diagnostic Interview for DSM‐IV performed by an experienced research clinician, along with an independent interview by a psychiatrist. A primary diagnosis of chronic PTSD, signifying an illness duration ≥3 months, was required. A score ≥50 on the Clinician Administered PTSD Scale (CAPS), and ≥ 4 on the Clinical Global Impression (CGI) Scale severity item was required at screening and baseline. The Life Events Checklist was used to identify traumatic stressors; the event identified as causing the most distress or generating the PTSD symptoms was the focus for the CAPS ratings. Comorbid DSM‐IV depressive disorders (specifically, major depressive disorder, dysthymic disorder, and depressive disorder NOS) and anxiety disorders (except obsessive–compulsive disorder) were permitted if the onset of illness post‐dated the traumatic event and was not the primary focus of clinical attention. Patients were required to be free of all psychotropic medications for at least 1 week before starting placebo lead‐in (5 weeks for fluoxetine)" Exclusion criteria: Quote: “Exclusionary criteria included bipolar disorder, schizophrenia, schizoaffective disorder, or psychotic symptoms; current anorexia or bulimia nervosa; alcohol or drug abuse or dependence within the past 3 months (except nicotine); unstable medical or neurological illness; and for females of child‐bearing potential, pregnancy. Physical and neurological examinations, vital signs, weight, ECG, standard blood tests, urinalysis, and urine toxicology confirmed absence of unstable medical illnesses and recent illicit substance use. No psychotherapy was permitted during the trial" Comorbidity: DTS, MADRS, CGI‐S, or SDS: depressive disorder, anxiety disorder, history of alcohol dependence/abuse, substance dependence/abuse, and nicotine use) Dropout rates: 36/131 (4/22 in the GR205171 and 12/25 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Patients completing the 2‐week placebo lead‐in period and who continued to meet all eligibility criteria (including CAPS ≥50) entered the 8‐week double‐blind treatment protocol and were randomly assigned to GR205171 (5 mg capsule) or identical‐ appearing placebo capsule. This dose was selected because of repeated dose safety data in healthy volunteers and patients with social phobia. Each week, participants received a pill bottle containing 10 capsules. At the following visit, patients returned the pill bottle, with the number of remaining capsules documented for adherence testing. Patients were assigned to study medication for 8 successive weeks or until drop‐out. Participants were discontinued from the study if there was an increase of greater than 30% in the CAPS scores from baseline at any assessment. In addition to weekly ratings, participants were evaluated by a study psychiatrist, who conducted medication management focusing on symptoms, adherence, and side effects" | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: "CAPS (≥ 50% reduction in the CAPS total score from baseline), remission (CAPS ≤ 20), CGI‐I, DTS, MADRS, CGI‐S, SDS. Safety evaluations included vital signs and weight, physical and neurological examinations, 12‐lead ECG, and laboratory tests, including weekly liver function tests. Side effects were elicited at each visit by a blinded rater who administered a checklist of 87 symptoms on a scale of 0 (none) to 3 (severe)" Time points: No information Data estimation: baseline. ITT, LOCF |
|
| Notes | Dates of trial: Quote: "Enrollment began in September 2005 and ended in December 2008" Industry‐funded: Yes. Quote: "The study was funded by the National Institutes of Health, and designed in collaboration with the drug manufacturer, GlaxoSmithKline" Medication provided by industry: Yes Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomization scheme was generated by a Mount Sinai School of Medicine (MSSM) pharmacist who assigned individuals across both sites using a permuted‐block 1:1 randomization list" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Patients completing the 2‐week placebo lead‐in period and who continued to meet all eligibility criteria (including CAPS ≥50) entered the 8‐week double‐blind treatment protocol and were randomly assigned to GR205171 (5 mg capsule) or identical‐appearing placebo capsule" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All study personnel, investigators, and patients were blinded to treatment assignment until completion of the entire study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More participants withdrew from the placebo (12/25, 48%) compared to the GR205171 group (4/22, 18%). Reasons for withdrawal include: protocol violation, withdrew consent, and medical condition. Data represent ITT and LOCF values. The 2 groups did not differ by sample characteristics at baseline. Quote: "Of 47 randomized patients, eight patients (two receiving GR205171 and six receiving placebo) did not receive ratings at the first post‐baseline assessment at week 1, and were discontinued from the study. The remaining 39 patients (20 receiving GR205171, and 19 receiving placebo) were included in primary efficacy and safety analyses detailed below. There were no significant differences between treatment groups in baseline characteristics, except for higher rates of past substance abuse or dependence in the GR205171 group. There was a trend for higher retention in the active treatment arm, with a 82% completion rate for patients in the GR205171 group versus a 52% completion rate in the placebo group (p=0.09, Fisher's Exact Test)" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "The study was funded by the National Institutes of Health, and designed in collaboration with the drug manufacturer, GlaxoSmithKline" |
McCall 2018.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: Not specified Post‐treatment: 5‐ to 8‐week follow‐up |
|
| Participants | Setting: Georgia Sample size: 20 randomised to prazosin and placebo Mean age: 39.8 years (SD: 14.5) Sex: 3 men and 17 women; major depressive (50%), bipolar (20%), military combatants (n = 2), civilians (n = 18) Diagnostic measure: CAPS Inclusion criteria: Quote: “Posttraumatic stress disorder was confirmed using the Clinician‐Administered PTSD Scale. Other psychiatric diagnoses were assessed using the Structured Clinical Interview for Diagnosis for Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Major depression, bipolar depression, generalized anxiety, panic disorder, social phobia, and obsessive‐compulsive disorder were permitted. Participants reported at least moderate nightmare severity, with a Disturbing Dreams and Nightmare Severity Index (DDNSI) score of at least 10. Mild‐moderate intensity of suicidal ideation was confirmed with a Scale for Suicide Ideation (SSI) score of at least 3 but without imminent intent to commit suicide, that is, a Columbia Suicide Severity Rating Scale (C‐SSRS) suicidal ideation level of ≤4" Exclusion criteria: Quote: “Exclusions were an active diagnosis of alcohol or substance abuse, active episode of bipolar mania or hypomania, lifetime diagnosis of schizophrenia according to the Structured Clinical Interview for Diagnosis, major neurocognitive disorder, or a Mini Mental State Exam score of 24 or lower. Additional exclusion criteria were a history of fainting or syncopal episodes in the last 6 months, a history of hypotension, or in‐clinic automatic blood pressure (BP) readings with systolic BP of less than 90 mm Hg or diastolic BP of less than 50 mm Hg" Comorbidity: DSM‐IV‐TR: major depressive, bipolar Drop‐out rates: Not specified |
|
| Interventions | Pharmacological intervention: Quote: “Participants received double‐blind, randomized treatment for 8 weeks, with weekly visits to receive the next week's supply of study medication. The dosing schedule was adapted from Raskind et al, modified to allow for only bedtime dosing, and adhered to the published sex‐specific dosing by Raskind et al (i.e. men 1‐20 mg/d and women 1‐10 mg/d)" | |
| Outcomes | Outcomes: ISI, PTSD Checklist–specific version, HRSD, CGI‐S (not clear whether primary or secondary) Time points: No information Data estimation: ITT |
|
| Notes | Dates of trial: Not stated Industry‐funded: No. Quote: "American Foundation for Suicide Prevention DIG‐0‐087‐13" Medication provided by industry: Not specified Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Blocked randomization sequence was used for allocation between prazosin and identical matching placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Blocked randomization sequence was used for allocation between prazosin and identical matching placebo" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved Quote: "Twenty adult, suicidal PTSD patients with nightmares were blindly and randomly assigned 1:1 to escalating doses of prazosin versus placebo at bedtime only for 8 weeks" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Insomnia symptoms were assessed using the Insomnia Severity Index (ISI). The severity of PTSD severity was measured using the PTSD Checklist–specific version. Depression severity was tracked using the 24‐item Hamilton Rating Scale for Depression (HRSD) by a blind rater. Overall clinical status was assessed using the Clinical Global Impression–Severity (CGI‐S) rated along a 7‐point dimension. The CGI‐S was completed by a study psychiatrist blind to treatment condition" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not specified. ITT. Quote: "Characteristics were similar between groups" |
| Selective reporting (reporting bias) | Unclear risk | Additional outcomes measured and included in the published article |
| Other bias | Low risk | None identified, but more women than men recruited; and small sample size |
McRae 2004.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 1 week single‐blind placebo run‐in Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “The research was conducted at outpatient psychiatric clinics in two academic medical centers (Medical University of South Carolina and Dartmouth Medical School)" Sample size: 37 randomised to nefazodone and sertraline Mean age: 40.3 years (18 ‐ 65) Sex: 6 men and 20 women (for 26 participants) Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Subjects were male and female outpatients 18–65 years of age who met DSM‐IV criteria for a principle diagnosis of PTSD as determined by Part 1 of the CAPS‐1. To meet inclusion criteria, subjects had to have a minimum 3‐month duration of PTSD symptoms and a total severity score of at least 50 on the CAPS‐2 at the end of a 1‐week placebo wash‐out period" Exclusion criteria: Quote: “Additional exclusion criteria were: 1) any clinically significant medical condition or laboratory abnormality that could be expected to progress, recur, or change such that it might bias the assessment of the clinical and mental status of the subject; 2) history of seizure disorder or organic brain disease; 3) pregnancy or breast‐feeding; 4) current diagnosis of a psychotic disorder, bulimia or anorexia, bipolar disorder, or obsessive compulsive disorder; 5) current substance abuse or dependence (defined as not having a documented recovery of at least 3 months duration); 6) current diagnosis of major depression, panic disorder, or agoraphobia if these conditions were not deemed secondary to PTSD; 7) current use of any psychotropic medication or other medication that would interfere with assessment of effectiveness or compromise safety of study participants, including medications that are substrates of the cytochrome P450 system; 8) hypersensitivity to nefazodone or sertraline; 9) history of nonresponse to nefazodone or sertraline; and 10) treatment refractory patients (defined as patients who had three trials of psychotropic treatment of adequate dose and duration for treatment of PTSD). In addition, subjects could not be receiving PTSD‐specific psychotherapy" Comorbidity: MINI Dropout rates: 14 dropouts in total (insufficient information to determine dropout rates for the 2 groups separately) |
|
| Interventions | Pharmacological intervention: Quote: “Treatment was initiated at 25 mg twice daily for sertraline and 50 mg twice daily for nefazodone, and titrated up based on response and tolerability to a target dose of 100 mg twice daily for sertraline and 300 mg twice daily for nefazodone" | |
| Outcomes | Primary outcomes: CAPS‐2, CGI‐I
Secondary outcomes: DTS, MADRS, HAM‐A, TOP‐8, PSQI, SDS Time points: Quote: "The CAPS‐2 was carried out at the end of the placebo wash‐out period and at Weeks 4, 8, and 12. The CAPS‐2 carried out at the end of the placebo wash‐out was used as the baseline CAPS rating. The CGI‐I was rated weekly. The DTS was completed weekly. The TOP‐8 was completed at the end of the placebo wash‐out period and Weeks 2, 4, 6, 8, 10, and 12. The MADRS, HAM‐A, PSQI, and SDS were completed at the end of the placebo wash‐out period and Weeks 4, 8, and 12" Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Contract grant sponsor: Bristol‐Myers Squibb" Medication provided by industry: No Any of the authors work for industry: Yes 3 patients excluded during run‐in |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Thirty‐seven male and female outpatients meeting DSM‐IV criteria for PTSD were randomly assigned to receive nefazodone (maximum dose 600 mg /day; average dose 463 mg/day) or sertraline (maximum dose 200/day; average dose 153 mg/day). Treatment randomization information was kept by a research pharmacist" |
| Allocation concealment (selection bias) | Low risk | Quote: "All medication was packaged in identical opaque gelatin capsules with cornstarch filler. Treatment randomization information was kept by a research pharmacist" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "This 12‐week, randomized, double‐blind study compares the effectiveness, safety, and tolerability of nefazodone and sertraline for the treatment of PTSD" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There is insufficient information to determine dropout rates for the 2 groups separately. Overall 54% of the participants dropped out of the study. Did not use ITT or LOCF Quote: "For the analyzed sample there were no significant differences between groups in any of the baseline demographic and clinical characteristics. Twenty‐three subjects completed the 12‐week trial. Four subjects withdrew early because of adverse effects (two subjects from each treatment group). Five subjects did not return for scheduled visits and could not be contacted, three subjects withdrew for personal reasons, one subject was discontinued from the study after being diagnosed with sleep apnea, and one subject was discontinued by the investigator for displaying volatile behavior. There were no between group differences in rate of or reason for early termination" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Contract grant sponsor: Bristol‐Myers Squibb" Quote: "The major limitations of this study are the small sample size, high attrition rate, and lack of a placebo control group" |
NCT00659230.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 6 weeks Placebo run‐in: No information Post‐treatment: 8 weeks |
|
| Participants | Setting: United States, California, New York Sample size: 100 randomised to nepicastat and placebo Mean age: 37.96 years (10.88) Sex: 86 men and 5 women (for 91 participants), OIF/OEF Veterans, included MDD Diagnostic measure: MINI, CAPS‐DX Inclusion criteria: Quote: “Signed informed consent; Patient understands the risks and benefits and agrees to visit frequency and procedures; Male or female; Any race or ethnic origin; Served in OIF/OEF or Afghanistan conflicts or other Southwest Asia conditions; Currently Active Duty, National Guard, Reservist, Veteran, and/or Retired Military; Diagnosis of PTSD (by MINI (Mini International Neuropsychiatric Interview) and CAPS‐DX (Clinician Administered PTSD scale‐ Diagnostic Form) using Rule of Fours and total CAPS‐DX score of 45); No substance use disorders in the previous 2 weeks and no substance dependence disorders in the past 4 weeks (except for nicotine and caffeine); Free of psychotropic medication for 2 weeks prior to randomization; Physical and laboratory panel are within normal limits or not clinically significant; Women of childbearing potential must be using medically‐approved methods of birth control; ≥19 to 65 years of age" Exclusion criteria: Quote: “Lifetime history of bipolar I, schizophrenia, schizoaffective or cognitive disorders; Actively considering plans of suicide or homicide; Psychotic symptoms that in the investigator's opinion impair the patient's ability to give informed consent or make it unsafe for patient to be maintained without a neuroleptic; Unstable general medical conditions or a contraindication to the use of nepicastat; Women planning to become pregnant or breastfeed during the study; Current or pending incarceration; Terminal Illness" Comorbidity: No information Dropout rates: 21/100 (12/49 in the nepicastat and 9/51 in the placebo group) |
|
| Interventions | Pharmacological intervention: Drug: Placebo, 100 ‐ 800 mg placebo; Drug: Nepicastat, 100 ‐ 800 mg | |
| Outcomes | Primary outcomes: CAPS, subscale D (CAPS‐D)
Secondary outcomes: CAPS total, CAPS‐B, CAPS‐C Time points: CAPS‐D, CAPS‐C and CAPS Total Score: Baseline, week 2, 4 and 6; CAPS‐B: week 6 Data estimation: Completer analysis |
|
| Notes | Dates of trial: July 2009 to August 2012 Industry‐funded: No. Quote: "Tuscaloosa Research & Education Advancement Corporation" Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "This study proposes a multi‐site, randomized, double‐blind, placebo‐controlled clinical trial of the dopamine‐ß‐ hydroxylase (DBH) inhibitor, nepicastat, for the treatment of posttraumatic stress disorder (PTSD) in outpatients who have previously served in a combat zone during Operation Iraqi Freedom and Operation Enduring Freedom (OIF/OEF) or other Southwest conditions since 19800. A DBH inhibitor's mechanism of action is to decrease neuronal noradrenaline (NA) release by inhibiting DBH conversion of dopamine (DA) to NA" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "This study proposes a multi‐site, randomized, double‐blind, placebo‐controlled clinical trial of the dopamine‐ß‐ hydroxylase (DBH) inhibitor, nepicastat, for the treatment of posttraumatic stress disorder (PTSD) in outpatients who have previously served in a combat zone during Operation Iraqi Freedom and Operation Enduring Freedom (OIF/OEF) or other Southwest conditions since 19800. A DBH inhibitor's mechanism of action is to decrease neuronal noradrenaline (NA) release by inhibiting DBH conversion of dopamine (DA) to NA" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the nepicastat group (12/49, 24%) compared to the placebo group (9/51, 18%). No information was provided on if the 2 groups did differ by sample characteristics at baseline and endpoint. Data estimation for completer values Quote: “2 did not return post‐randomization, 5 lost source documents, thus 7 excluded from results analysis; 3 did not return post‐randomization, 4 lost source documents, thus 7 excluded from results analysis” |
| Selective reporting (reporting bias) | Low risk | All outcomes were assessed as specified by the protocol |
| Other bias | Low risk | No other sources of bias identified |
NCT01000493.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, fixed‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: No information Post‐treatment: follow‐up visit 14, 28 and 42 days after the last dose of study medication |
|
| Participants | Setting: United States Sample size: 129 randomised to orvepitant and placebo Mean age: 36.7 years (11.59) Sex: 30 men and 99 women, non‐combat‐related Diagnostic measure: No information Inclusion criteria: Quote: “Aged 18‐64 years, inclusive; A primary diagnosis of noncombat‐related Post traumatic Stress Disorder (PTSD); Subjects with symptom severity considered to be at least moderate to severe" Exclusion criteria: Quote: “Subjects whose symptoms are better accounted for by a diagnosis other than Post traumatic Stress Disorder (PTSD), subjects diagnosed with dementia; subjects diagnosed with a current/recent eating disorder such as anorexia nervosa or bulimia; subjects with a diagnosed history of schizophrenia, schizoaffective disorder, or Bipolar Disorder; Subjects who have a history of failing to respond to adequate treatment for PTSD with an antidepressant/anti‐anxiety drug, i..e, failure to improve following administration of at least two other antidepressants/anti‐anxiety drugs, each given for at least 4 weeks" Comorbidity: No information Dropout rates: 94/129 (55/69 in the orvepitant and 39/60 in the placebo group) |
|
| Interventions | Pharmacological intervention: Experimental: Orvepitant 60 mg/day; Placebo comparator: Placebo | |
| Outcomes | Primary outcomes:CAPS‐17
Secondary outcomes: CAPS‐17, CAPS‐A, CAPS‐B, CAPS‐C, CAPS‐D, CGI‐I, CGI‐S, SPRINT, DTS, PSQI, PSQI‐A, CAPS‐B2, HAM‐D, CPFQ, MSFQ Total Score in men and women, C‐SSRS Time points: CAPS: Baseline (Day 1 pre‐dose) and Week 1, 4, 8, 12; CGI‐I, CGI‐S, HAM‐D, CPFQ, MSFQ and C‐SSRS: Baseline, Week 1, 2, 4, 6, 8, 10 and 12; DTS, PSQI and PSQI‐A: Baseline (Day 1 pre‐dose) and up to Week 12 Data estimation: ITT, completer analysis |
|
| Notes | Dates of trial: November 2009 to June 2010 Industry‐funded: Yes. GlaxoSmithKline Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the patients were randomised however no mention is made of the method of randomisation Quote: "This is a 12‐week, randomized, multicenter, double‐blind, placebo controlled, fixed‐dose parallel group study to assess the efficacy and safety of orvepitant (60 mg/day) versus placebo in subjects with a diagnosis of noncombat‐related Posttraumatic Stress Disorder (PTSD), whose symptoms are considered moderate or severe" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Those subjects randomised to receive placebo will receive study medication identical in appearance to that received by subjects assigned to receive orvepitant" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "This is a 12‐week, randomized, multicenter, double‐blind, placebo controlled, fixed‐dose parallel group study to assess the efficacy and safety of orvepitant (60 mg/day) versus placebo in subjects with a diagnosis of noncombat‐related Posttraumatic Stress Disorder (PTSD), whose symptoms are considered moderate or severe" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A high dropout rate was reported for the orvepitant group (55/69, 78%) and the placebo group (39/60, 65%). No information was provided on whether the 2 groups differed by sample characteristics at baseline and endpoint. Data estimation for ITT and LOCF values. Reasons for withdrawal include: adverse event, lack of efficacy; protocol violation; study closed/terminated; lost to follow‐up; physician decision; withdrawal by subject |
| Selective reporting (reporting bias) | Low risk | All outcomes were assessed as specified by the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry, i.e. GlaxoSmithKline |
NCT01681849.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: No information Post‐treatment: 10 weeks |
|
| Participants | Setting: United States, Georgia Sample size: 28 randomised to paroxetine and placebo Mean age: No information Sex: 28 women Diagnostic measure: DSM‐IV and CAPS Inclusion criteria: Quote: “18 Years to 75 Years (Adult, Older Adult); Female; Healthy Volunteers; Subjects meet criteria for current PTSD as determined by the SCID interview for PTSD and the CAPS and have a score of greater than 60 on the CAPS; history of penetrative sexual abuse which occurred once a month or more, for a period of greater than a year at some time between the ages of 4‐13, as assessed by the Early Trauma Inventory (ETI); are free of psychotropic medication for four weeks before the study (subjects will not be taken off of medication for the purpose of the study); Non‐PTSD subjects will be included based on the same criteria with the exception that they do not meet criteria for PTSD" Exclusion criteria: Quote: “a history of shrapnel or other foreign bodies which would preclude MRI scanning; meningitis; traumatic brain injury; neurological disorder or organic mental disorder; history of loss of consciousness; alcohol abuse or substance abuse or dependence based on the SCID within the past 24 months; positive pregnancy test as measured by a serum beta‐HCG or urine pregnancy test on the morning of the PET scan. Women will be counseled about the risks of pregnancy during the course of the study; current or lifetime history of schizophrenia, schizoaffective disorder, or bulimia, based on the SCID; a history of serious medical or neurological illness, such as cardiovascular, gastrointestinal, hepatic, renal, neurologic or other systemic illness; evidence of a major medical or neurological illness on physical examination or as a result of laboratory studies (CBC, BUN, creatinine, blood sugar, electrolytes, liver and thyroid function tests, urinalysis, and EKG); positive urine toxicology screen; history of ongoing violence such as domestic abuse as measured by the ETI‐lifetime; post‐menopausal status as measured by menstrual history; Non‐PTSD subjects will additionally be excluded with current major depression or other major psychiatric disorder based on the SCID" Comorbidity: SCID Dropout rates: 15/28 (8/15 in the paroxetine and 7/13 in the placebo group) |
|
| Interventions | Pharmacological intervention: Experimental: Paroxetine, 10 ‐ 40 mg to reach individual therapeutic levels for 3 months; Placebo comparator: Placebo |
|
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: SPM Time points: CAPS: Baseline, end of study (up to 52 weeks); SPM: Baseline, 3 months post‐treatment Data estimation: ITT, completer analysis |
|
| Notes | Dates of trial: July 2009 to July 2015 Industry‐funded: No. Sponsors: Emory University and National Institute of Mental Health (NIMH) Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Participants then were treated in a randomized double‐blind fashion with paroxetine or placebo for three months, followed by a repeat of these assessments" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Participants then were treated in a randomized double‐blind fashion with paroxetine or placebo for three months, followed by a repeat of these assessments" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A high dropout rate was reported for the paroxetine group (8/15, 53%) and the placebo group (7/13, 54%). No information was provided on whether the 2 groups differed by sample characteristics at baseline and endpoint. Data estimation for ITT and LOCF values. Reasons for withdrawal include: protocol violation; withdrawal by subject; lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | All outcomes were assessed as specified by the protocol |
| Other bias | Low risk | No other sources of bias identified |
Padala 2006.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 2‐week washout period Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Creighton University Medical Center, Omaha, Nebraska, USA" Sample size: 20 randomised to risperidone and placebo Mean age: 41.3 years Sex: 20 women, non‐combat‐related Diagnostic measure: MINI Inclusion criteria: Quote: “To be eligible for the study, participants must have had symptoms of current PTSD and a demonstrated level of understanding sufficient to perform all tests and examinations required by the protocol. Comorbid depressive and other anxiety disorders were allowed provided they were chronologically secondary to PTSD" Exclusion criteria: Quote: “Exclusion criteria included individuals with a lifetime history of schizophrenia or bipolar I disorder, clinically unstable general medical condition or serious illness, significantly abnormal laboratory values, significant suicidality, prior treatment with risperidone, and/or enrollment in a drug related research study within the past 60 days. Pregnant or nursing females were excluded from the participation as were those meeting diagnostic criteria for substance abuse/dependence within the previous 2 months. Although history of schizophrenia was an exclusion criterion, patients with psychotic symptoms in conjunction with depression were allowed to participate. Only one patient had concomitant diagnosis of major depressive disorder with psychotic features. Concomitant psychotropic medications were not allowed. Ongoing psychotherapy was permitted; however, participants were instructed to avoid initiation of new psychotherapy during the trial" Comorbidity: MINI Drop‐out rates: 5/20 (2/11 in the risperidone and 3/9 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Those receiving risperidone began at a dosage of a 0.5 mg twice daily, which was increased weekly by up to 1mg per day, as tolerated, to a target dose of 4–6 mg per day in divided doses. Participants had the option of treating extrapyramidal effects with diphenhydramine 25–50 mg per day" | |
| Outcomes | Primary outcomes: TOP‐8
Secondary outcomes: CAPS, HAM‐A Time points: TOP‐8: at each study visit; CAPS and HAM‐A: visits 7 and 11; Safety and efficacy procedures performed at baseline were repeated at the final visit at week 13 Data estimation: No information |
|
| Notes | Dates of trial: Quote: "Data were collected from October 24, 2001 to August 4, 2004" Industry‐funded: Yes. Quote: "Sponsorship: Janssen Pharmaceuticals" Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "After washout from other psychotropic medications, 20 participants were randomized to either risperidone or placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "The study was a 12‐week, monotherapy, double‐blind, comparison of flexible‐dose risperidone with placebo in adult females aged 19–65 years with chronic PTSD as a result of domestic violence or sexual assault" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More participants withdrew from the placebo (3/9, 33%) compared to the risperidone group (2/11, 18%). Data estimation points were not clear. The 2 groups did not differ by sample characteristics at baseline Quote: "No significant differences were found in the baseline demographics between groups. Among the participants randomized to the treatment group who discontinued during the 10‐week acute phase, only one withdrew owing to an adverse event, a rash" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Sponsorship: Janssen Pharmaceuticals" |
Panahi 2011.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 10 weeks Placebo run‐in: no information Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Out‐patients who had been referred to the neuropsychiatric clinic of Baqiyatallah Hospital (Tehran,
Iran)" Sample size: 70 randomised to sertraline and placebo Mean age: 45.55 years (SD: 5.25) Sex: 70 men, 48 military (69%), 7 mood and anxiety (10%) Diagnostic measure: DSM‐IV‐TR Inclusion criteria: Quote: “The subjects were male Iranian Iran–Iraq war veteran out‐patients who had been referred to the neuropsychiatric clinic of Baqiyatallah Hospital and met DSM‐IV‐TR criteria for a primary diagnosis of PTSD. A duration of at least 6 months of illness and a Clinical Global Impression scale – Severity (CGI‐S) score of ≥ 4 were also required at the baseline visit for inclusion into the trial" Exclusion criteria: Quote: “(1) the presence of any axis I disorder other than PTSD (subjects with concurrent depression were included provided that their depression was secondary to PTSD and initiated after PTSD); (2) evidence of clinically significant hepatic or renal disorder or any other medical condition (in acute or unstable form) that might confound the procedure or the results of the trial ; (3) alcohol or substance abuse or dependency within the preceding 6 months; (4) intolerance or hypersensitivity to sertraline ; (5) concurrent use of any psychotropic medication (except for chloral hydrate or diazepam, taken as needed) with clinically significant psychotropic activity within 2 weeks of randomization (or 5 weeks for fluoxetine); (6) any cognitive‐behavioral therapy during the trial ; and (7) psychotherapy that was initiated or ended during the trial" Comorbidity: history of mood and anxiety disorders Dropout rates: 8/70 (3/35 in the sertraline and 5/35 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Sertraline treatment was initiated at a daily dose of 50 mg. Patients were visited every 2 weeks by a psychiatrist and, based on the examinations, clinical response and adverse effects, drug dosage could be flexibly adjusted to a maximum dose of 200 mg/day" | |
| Outcomes | Outcomes: IES‐R, CGI‐S, CGI‐I (not clear whether primary or secondary) Time points: No information Data estimation: ITT, LVCF, completer analysis. |
|
| Notes | Dates of trial: Not stated Industry‐funded: No. Quote: "We gratefully acknowledge the financial support for this work that was provided by the Baqiyatallah University of Medical Sciences" Medication provided by industry: Yes Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Seventy subjects were randomized, using computer‐generated random numbers, to 10 weeks of double‐blind and parallel treatment with either sertraline (n=35) or matched placebo (n=35)" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Sertraline and placebo were obtained from Razak Laboratories Co., Iran" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "The present study aimed to evaluate the clinical efficacy of sertraline against combat‐related PTSD in a randomized, double‐blind, placebo‐controlled trial" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "CGI‐S and CGI‐I data were collected by a single rater (blinded to the allocations) who was different from the psychiatrist who performed the patients’ visits and from the statistician who analyzed the data" However, no information was provided for the IES‐R scale |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the sertraline group (3/35, 9%) compared to the placebo group (5/35, 14%). Reasons for withdrawals include: adverse events; protocol violation; other reasons. The 2 groups did not differ by sample characteristics at baseline. Data estimation for ITT, LVCF and completer analysis Quote: "Three patients discontinued study treatment in the sertraline group (two because of adverse events and one because of protocol violation) and five in the placebo group (two on their own accord, two because of protocol violation, and one because of adverse events). The difference in the drop‐out rate was not significant between the groups. The groups were not significantly different in terms of age, occupation, education and the presence of chemical injury (p>0.05), nor was there any significant difference in the prevalence of mood or anxiety disorders between the groups at baseline (p>0.05)" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | Quote: "Sertraline and placebo were obtained from Razak Laboratories Co., Iran" However, no authors work for industry |
Pfizer588.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: No information Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “16 study sites in USA" Sample size: 190 randomised to sertraline and placebo Mean age: 37 years Sex: 49 men and 144 women, physical/sexual assault Diagnostic measure: DSM‐III‐R Inclusion criteria: Quote: “100% PTSD by DSM‐III‐R" Exclusion criteria: Quote: “CAPS‐2 score of <50 at baseline (one exception to this)" Comorbidity: No information Dropout rates: 49/190 (24/95 in the sertraline and 25/95 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Sertraline ‐ Mean dose for completers 156 mg/day" | |
| Outcomes | Outcomes: IES, DTS, CGI‐S, CGI‐I, CAPS‐2 (not clear whether primary or secondary) Time points: No information Data estimation: ITT, completer analysis |
|
| Notes | Dates of trial: Not stated Industry‐funded: No information Medication provided by industry: No information Any of the authors work for industry: No information |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the patients were randomised but no mention is made of the method of randomisation Quote: "Study Type: RCT" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Blindness: Double blind" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of participants withdrew from the sertraline group (24/95, 25%) compared to the placebo group (25/95, 26%). Reasons for withdrawals were not provided. No information was provided on whether the 2 groups differed by sample characteristics at baseline and endpoint. Data estimation for ITT and completer analysis |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | No information provided to determine this |
Pfizer589.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: No information Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Outpatient, Veterans Administration Medical Centres" Sample size: 169 randomised to sertraline and placebo Mean age: 45 years Sex: 135 men and 34 women, predominantly war‐trauma sample (71%) Diagnostic measure: DSM‐III‐R Inclusion criteria: Quote: “100% PTSD by DSM‐III‐R" Exclusion criteria: No criteria reported. Comorbidity: No information Dropout rates: 43/169 (28/86 in the sertraline and 15/83 in the placebo group) |
|
| Interventions | Pharmacological intervention: sertraline (mean completer dose: 156 mg/d) versus placebo x mean duration of 72 days | |
| Outcomes | Outcomes: CAPS‐2, CGI‐I, CGI‐S, IES, DTS (not clear whether primary or secondary) Time points: No information Data estimation: ITT, completer analysis |
|
| Notes | Dates of trial: Not stated Industry‐funded: No information Medication provided by industry: No information Any of the authors work for industry: No information |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Study Type: RCT" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Blindness: Double blind" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More participants withdrew from the sertraline (28/86, 33%) compared to the placebo group (15/83, 18%). Reasons for withdrawals were not provided. No information was provided on whether the 2 groups differed by sample characteristics at baseline and endpoint. Data estimation for ITT and completer analysis |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Prevalence of participants with drug abuse history higher at baseline in medication than in placebo group. No other information provided to determine this |
Raskind 2018.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 16 weeks (double‐blind phase, 24 weeks in total) Placebo run‐in: No information Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “13 Department of Veterans Affairs medical centers" Sample size: 304 randomised to prazosin and placebo Mean age: 51.85 years (SD: 13.8) Sex: 297 men and 7 women, military veterans, 115 MDD (38%) Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Participants were eligible if they met the criteria for PTSD according to the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM‐IV), and had a total score of at least 50 on the 17‐item Clinician‐Administered PTSD Scale (CAPS; scores range from 0 to 136, with higher scores indicating greater frequency and intensity of symptoms)16; if they had been exposed to one or more traumatic, life‐threatening events in a war zone that preceded the onset of recurrent nightmares; if they reported and could recall combat‐related nightmares; if they had a frequency score of at least 2 and a cumulative score of at least 5 on CAPS item B2 (“recurrent distressing dreams”); cumulative scores range from 0 to 8 [with a range of 0 to 4 for frequency and 0 to 4 for intensity], with higher scores indicating more frequent and more distressing dreams); and if, for at least 4 weeks before randomization, they were receiving a stable dose of nonexcluded medications or had been receiving supportive psychotherapy" Exclusion criteria: Quote: “Exclusion criteria were acute or unstable medical illness; a systolic blood pressure of less than 110 mm Hg while the patient was in the supine position, a decrease in systolic blood pressure of more than 20 mm Hg after 2 minutes of standing, or any decrease in blood pressure accompanied by dizziness; the presence of a psychotic or cognitive disorder; substance dependence within the previous 3 months; current cocaine or stimulant use; active suicidal or homicidal ideation with plan or intent; and psychosocial instability (defined as a short‐term or long‐term situational life crisis). Sleep apnea was not a criterion for exclusion, nor was it screened for except by means of history taking and chart review. However, veterans with a diagnosis of sleep apnea on their medical record who did not adhere to treatment for the condition were excluded. Participants were also excluded if they were receiving prazosin or another α1‐adrenergic antagonist at the time of recruitment or if they had participated in a previous trial of prazosin for PTSD; if they had received prolonged exposure therapy, eye‐movement desensitization and reprogramming, or cognitive processing therapy within 4 weeks before randomization; or if they had taken trazodone within 2 weeks before randomization. Women were excluded if they were pregnant or nursing or if they declined to use an effective birth‐control method" Comorbidity: MD, Anxiety disorders other than PTSD, Alcohol abuse and cannabis abuse Dropout rates: 59/304 (30/152 in the prazosin and 29/151 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Eligible participants were randomly assigned, in 1:1 ratio, to receive prazosin or placebo (in identical 1‐mg, 2‐mg, and 5‐mg capsules) in escalating doses. Flexible‐dose adjustment was used to achieve the maximal alleviation of nightmares with acceptable adverse effects. The maximum allowed dose was 5 mg at midmorning and 15 mg at bedtime for men and 2 mg at midmorning and 10 mg at bedtime for women. The maximum achieved daily dose of prazosin was continued as the maintenance dose" | |
| Outcomes | Primary outcomes: CAPS‐B, PSQI, CGIC
Secondary outcomes: CAPS‐B, PSQI, CGIC, PCL‐M, PHQ‐9, QOLI, SF‐12, AUDIT‐C Time points: Quote: "Interview‐based assessments were performed by trained clinician raters at weeks 14, 18, 22, and 26" Data estimation: ITT |
|
| Notes | Dates of trial: Not stated Industry‐funded: No. Quote: "There was no industry support of or involvement in the trial" Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Eligible participants were randomly assigned, in 1:1 ratio, to receive prazosin or placebo (in identical 1‐mg, 2‐mg, and 5‐mg capsules) in escalating doses. Adaptive randomization was used to achieve balance in the following stratification factors within each site: current antidepressant use (yes vs. no) and a traumatic experience before versus on or after October 7, 2001 (the date of the U.S. invasion of Afghanistan)" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Eligible participants were randomly assigned, in 1:1 ratio, to receive prazosin or placebo (in identical 1‐mg, 2‐mg, and 5‐mg capsules) in escalating doses" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Trial office personnel, site personnel, and participants were unaware of the group assignments" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Clinician raters were unaware of assignments as well as participants’ vital signs, adverse events, and dose of the trial drug or placebo" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of patients withdrew from the prazosin group (30/152, 18%) compared to the placebo group (29/151, 19%). The 2 groups did not differ by sample characteristics at baseline. ITT values were used in the analysis Quote: "A total of 271 participants (90% in the prazosin group and 89% in the placebo group) completed the 10‐week primary outcome assessments, and 284 (94% in the prazosin group and 93% in the placebo group) completed one or more of the 10‐week primary outcome assessments; 59 participants (20% in the prazosin group and 19% in the placebo group) withdrew from the trial before the 26‐week visit. The pattern of missing data did not differ significantly between the groups at either 10 weeks or 26 weeks. The two groups did not differ significantly at baseline with respect to age; race; the experience of war‐zone trauma before versus on or after October 7, 2001; behavioral ratings; coexisting psychiatric disorders; or disability status" |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported on as per the study protocol |
| Other bias | Low risk | No other sources of bias were identified |
Rasmusson 2017.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind, cross‐over study Duration of intervention: 6 weeks (6‐week, open‐label extension phase; in total 15 weeks) Placebo run‐in: No information Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “At eight Department of Veterans Affairs Medical Centers" Sample size: 112 randomised to ganaxolone and placebo Mean age: 38.3 years (SD: 10.7) Sex: 88 men and 24 women, 37 MDD (33%), 98 military (88%) Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “To be included in the trial, veteran or civilian outpatients, aged 18–65 years,1 had to be exposed to DSM‐IV PTSD A1 and A2 criteria trauma at least 6 months prior to evaluation, and meet DSM‐IV criteria for current PTSD. Exposure to potentially traumatic events was assessed using the CTQ, the LEC modified to ascertain whether endorsed items met the DSM‐IV A2 criterion, and the DRRI. Participants also had to score at least a "1" for frequency and "2" for intensity on at least one Criterion B reexperiencing symptom, three Criterion C avoidance symptoms, and two Criterion D hyperarousal symptoms over the past month. A 1‐month CAPS total score ≥ 50 and a past‐week CAPS score ≥ 50 were required at the screening and baseline visits, respectively. Substance abuse and psychiatric diagnoses other than PTSD were assessed using the MINI. Participants had to be otherwise in general good health as confirmed by medical history, physical examination, electrocardiogram, and screening laboratory tests, have a negative urine drug screen for benzodiazepines, opiates, barbiturates, phencyclidine, cocaine, amphetamines, and tetrahydrocannabinol [THC], and test negative for pregnancy and refrain from breastfeeding, if female. Participants with childbearing potential had to agree to use effective contraception" Exclusion criteria: Quote: “Participants with clinically unstable medical conditions, a history of seizures (except childhood febrile seizures), and moderate to severe traumatic brain injury (TBI) were excluded. Also exclusionary were DSM‐IV diagnoses of current or past schizophrenia or other psychotic disorders (except psychosis NOS due to the presence of sensory hallucinations clearly related to trauma), bipolar type I disorder, dementia, substance abuse or dependence on drugs or alcohol within 6 months of study entry, unwillingness to abstain from alcohol during the study, suicidal or homicidal ideation necessitating clinical intervention, history of suicide attempt in the past 10 years, alanine or aspartate transferase levels greater than two times the upper limits of normal, and unwillingness to abstain from grapefruit products that inhibit the enzyme CYP3A4, which metabolizes ganaxolone. Participants taking psychotropic medications other than approved insomnia medications (zolpidem, zaleplon, eszopiclone, or trazodone up to 150 mg at a frequency of four times a week or less) were excluded, as were subjects in evidence‐based trauma‐focused treatment for PTSD within 6 weeks of the trial. Participation in other psychotherapeutic modalities maintained for 3 months before and during the trial was permitted" Comorbidity: MINI Dropout rates: 26/112 (17/152 in the ganaxolone and 9/53 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “The trial consisted of a 6‐week, randomized, double‐ blind, placebo‐controlled phase, followed by a 6‐week, open‐label extension phase in which subjects initially randomized to ganaxolone continued ganaxolone (GNX‐GNX), while placebo‐treated subjects crossed over to ganaxolone (PLC‐GNX). A 1‐week taper of ganaxolone followed with safety assessments extending to week 15 (Fig. 1). The GNX‐GNX group received biweekly escalating ganaxolone doses of 200, 400, and 600 mg twice a day; ganaxolone was continued at 600 mg twice a day during the 6‐week extension phase" | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: CGI‐I. Ancillary symptom measures: PCL, DSM‐IV, CGI‐I, POMS, a 65‐item self‐report with 5‐point measures of five factor analytically derived dimensions of mood (vigor/activity, anger/irritability, anxiety/tension, depression/dejection, confusion/bewilderment, and fatigue/inertia), PHQ‐9, a 9‐item depression subscale of the Patient Health Questionnaire based on the nine DSM‐IV diagnostic criteria for major depressive disorder; ISI, a 5‐item 2‐week assessment of self‐perceived difficulty with sleep onset, middle of the night awakening, early morning awakening, and impairment in daily function attributed to insomnia, a 25‐item scale that typically correlates with treatment response. Safety assessments: CSSRS, a mental status examination by the study psychiatrist, vital signs including orthostatic blood pressure changes, weight, clinical blood tests, urinalysis, a urine toxicology screen, measurement of urine cotinine (a long‐acting metabolite of nicotine), a pregnancy test, and an electrocardiogram Time points: Quote: "CAPS was administered at baseline and biweekly through week 12. CGI‐I was administered biweekly beginning at week 2. The PCL was administered biweekly. The other ancillary ratings were administered at baseline, week 6, and week 12. Physical and neurological examinations were conducted at screening, week 6, week 12, and additional visits if clinically indicated" Data estimation: mITT |
|
| Notes | Dates of trial: Quote: "The trial was conducted between April 19, 2011 and January 9, 2014" Industry‐funded: No. Quote: "The study as submitted for funding and support to the INTRuST Clinical Consortium Coordinating Center" Medication provided by industry: No information Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible participants were randomized in a 1:1 ratio based on a randomly permuted block design stratified by site and sex" |
| Allocation concealment (selection bias) | Low risk | Quote: "Each gelatin capsule of ganaxolone contained 200 mg ganaxolone (3α‐hydroxy‐3β‐methyl‐5α‐pregnan‐20‐one). The placebo formulation was comprised of sucrose spheres of comparable size to the ganaxolone spray‐layered spheres encapsulated in a gelatin capsule of identical (00) size, weight, and appearance. Randomization and study drug dispensing were managed through an Interactive Web Response System (IWRS). The contents of each drug bottle were blinded; labels contained a unique bottle number corresponding to either ganaxolone or placebo. Upon completion of each participant’s baseline evaluation, the investigator or appropriate designee logged onto the IWRS to randomize the subject to treatment type and receive corresponding bottle numbers. Designated personnel at the clinical site matched the assigned bottle numbers with the correct bottles of study drug and distributed the bottles to the investigator or designee. Only the investigational drug supplier and an unblinded inventory manager had access to the bottle numbers and treatment assignment codes" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved. Quote: "We investigated ganaxolone (a synthetic 3β‐methylated derivative of allopregnanolone, a GABAergic neuroactive steroid) for treatment of PTSD in a proof‐of‐concept, multisite, double‐blind, placebo‐controlled trial" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Only the investigational drug supplier and an unblinded inventory manager had access to the bottle numbers and treatment assignment codes" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of participants withdrew from the ganaxolone group (17/152, 11%) compared to the placebo group (9/53, 17%). Reasons for withdrawal include: lost to follow‐up; withdrew consent; adverse events; non‐compliance; multiple reasons; prohibited meds; positive urine drug screen. The 2 groups did not differ by sample characteristics at baseline. mITT. values were used in the analysis. Quote: "One hundred twelve (112) participants with PTSD were randomized into the study (GNX‐GNX group: n = 59; PLC‐GNX group: n = 53); 86 completed the first 6‐week, blinded, placebo‐controlled phase of the trial (GNX: n = 42; PLC: n = 44); 53 participants remained in the study through the subsequent 6‐week open‐label phase, the 1‐week taper, and the 2‐week, post‐taper follow‐up period (GNX‐GNX: n = 27; PLC‐GNX: n = 26). There were no significant differences between participants randomized to the GNX‐GNX and PLC‐GNX groups with regard to demographic or baseline clinical characteristics, including age, sex, ethnicity, combat veteran status, smoking status, comorbid major depression, or presence of mild TBI" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified |
Reich 2004.
| Study characteristics | ||
| Methods | Design: single‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: No information Post‐treatment: Not specified |
|
| Participants | Setting: McLean Hospital, Belmont, Mass Sample size: 21 randomised to risperidone and placebo Mean age: 27.9 years (18 ‐ 56) Sex: 21 women, childhood physical, sexual, emotion or verbal abuse Diagnostic measure: DSM‐III‐R (SCID, CAPS‐1) Inclusion criteria: Quote: “To be eligible for the study patients must have had a score of ≥ 50 on the CAPS‐1. All subjects needed to report PTSD related to childhood physical, sexual, emotional, or verbal abuse. All subjects needed to be able to provide informed consent, speak fluent English, and have understanding sufficient to perform all tests and examinations required by the protocol" Exclusion criteria: Quote: “Exclusion criteria included diagnosis of organic mental disorder or psychotic disorder (schizophrenia, schizoaffective disorder, or mood disorder with psychotic features) in the last 6 months; diagnoses of substance dependence (active within the last 60 days); unstable general medical condition; previous treatment with risperidone for 1 week or more; simultaneous treatment with another antipsychotic or mood stabilizer; enrolment in a drug study within the last 60 days; significant risk of suicide or homicide; entering individual psychotherapy within 3 months of the study; entering group therapy within 1 month of the study; and pregnancy or nursing". Comorbidity: MINI, DSM‐III‐R: MD, dysthymia, PD, agoraphobia with/without PD, GAD, simple phobia, social phobia, eating disorder NOS, OCD, somatization disorder Dropout rates: 16/21 (9/12 in the risperidone and 7/9 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Those receiving risperidone began at a dosage of 0.5 mg and were instructed to increase the dosage to 1 mg after 3 days. Risperidone was then increased weekly by up to 1 mg per day per week, as tolerated, to a targeted dosage of 4 mg per day or until subjects reported predominant relief of symptoms at targeted dosage by week 5, the dosage could be increased to a maximum of 8 mg per day. Risperidone could be divided into 2 or 3 daily doses" | |
| Outcomes | Primary outcomes: CAPS‐1, CAPS‐2
Secondary outcomes: None Time points: Quote: "Subjects were subsequently administered the CAPS‐2 at weeks 1, 2, 4, and 8. The CAPS‐1 was readministered at week 8" Data estimation: LOCF and random effects time series modelling methods |
|
| Notes | Dates of trial: Quote: "Data were collected from November 18, 2001, to June 7, 2003" Industry‐funded: Yes. Quote: "Supported by a grant from Janssen Pharmaceutica" Medication provided by industry: Unclear Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Subjects were randomly assigned to receive risperidone (N=12) in flexible daily dosages in the range of 0.5 to 8 mg or placebo (N=9)" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "The study was an 8‐week, double‐blind comparison of flexible‐dosage risperidone 0.5 to 8 mg/day and placebo in female adults with chronic PTSD from childhood physical, sexual, verbal, or emotional abuse" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A substantial amount of participants withdrew from the risperidone (19/12,75%) and from the placebo group (7/9, 76%). Reasons for withdrawal were not provided. Data represent LOCF and random effects time series modelling methods. The 2 groups did not differ by sample characteristics at baseline. Quote: "Nine subjects in the risperidone group and 7 subjects in the placebo group completed 8 weeks of the study. There were no significant differences between the risperidone and placebo with respect to any of the baseline measures" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Supported by a grant from Janssen Pharmaceutica" |
Reist 1989.
| Study characteristics | ||
| Methods | Design: multi‐centre trial, randomised, placebo‐controlled, flexible‐dose, double‐blind, 4 day switched cross‐over Duration of intervention: 8 weeks Placebo run‐in: 1 week Post‐treatment: Not specified |
|
| Participants | Setting: USA (outpatient psychiatry service and local Vietnam veterans outreach centers) Sample size: 27 randomised to desipramine and placebo Mean age: 38.4 years (28 ‐ 64) Sex: 27 men, combat veterans, 33% (6/18) major depression, most prevalent comorbidity: 50% (9/18) dysthymic disorder Diagnostic measure: DSM‐III Inclusion criteria: Quote: “The patient sample originally comprised of 27 hospitalised men aged 28‐64 years who were diagnosed as having PTSD according to DSM‐III criteria" Exclusion criteria: medication, drugs and alcohol Comorbidity: SCID, SCID ‐ DSM‐III‐R for personality disorders: MD, dysthymic disorder Dropout rates: 33% (9/27). There was insufficient information to determine dropouts by group separately |
|
| Interventions | Pharmacological intervention: Quote: “Patients were treated with oral desipramine at the beginning dose of 50 mg/day. This was increased daily by 50‐mg increments to a maximum dose of 200 mg/day" | |
| Outcomes | Outcomes: HAM‐D, HAM‐A, BDI, IES (no distinction between primary and secondary outcomes) Time points: Not specified Data estimation: Completer values |
|
| Notes | Dates of trial: Quote: “Between September 1986 and February 1987” Industry‐funded: No information Medication provided by industry: Unclear Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Following this workup during the first week of hospitalisation, patients were randomly assigned to treatment with desipramine or placebo (double‐blind)" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Following this workup during the first week of hospitalisation, patients were randomly assigned to treatment with desipramine or placebo (double‐blind)" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to determine dropouts by group separately. Thirty‐three percent of the patients across both groups withdrew from the trial. No information was provided on whether the two groups differed by sample characteristics at baseline and endpoint. Completer values were used in the analysis Quote: "Of the original patients, 21 completed the cross‐over period. Six patients dropped out for the following reasons: one patient left the hospital against medical advice; one patient was excluded from continued substance abuse; two patients developed irritability and dysphoria while taking desipramine, and, during the workup period, one patient developed mania and one became intolerant of the ward structure and asked to be withdrawn" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | No other sources of bias were identified except for ongoing recreational and group therapies |
Saygin 2002.
| Study characteristics | ||
| Methods | Design: single‐centre trial, randomised, placebo‐controlled, parallel arm, flexible‐dose Duration of intervention: 24 weeks Placebo run‐in: no information Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "Out‐patient trauma clinic based in Izmit, Izmit Rehabilitation Center, (IREM)" Sample size: 60 randomised to nefazadone and sertraline Mean age: 41.5 years (SD: 9.85) Sex: 13 men and 41 women (for 54 participants), earthquake survivors, 9% (5/54) MDD Diagnostic measure: DSM‐IV (SCID‐1) and nonstructured psychiatric interview Inclusion criteria: Quote: “The patient population of IREM mainly consisted of PTSD patients" Exclusion criteria: Quote: “Subjects who had a history of alcohol or drug abuse, neurological disorder, current organic mental disorder and who are under psychiatric medication less than 2 weeks before the study were all excluded" Comorbidity: Not mentioned Dropout rates: 6/60 (6/30 in the nefazadone and 0/30 in the sertraline group) |
|
| Interventions | Pharmacological intervention: Quote: “The mean dose of sertraline was 68.33±21.70 mg/day, with a range between 50‐100 mg/day. The mean dose of nefazodone was 332.35±SD 63.5 mg/day, range being 200‐400 mg/day" | |
| Outcomes | Primary outcomes: PDS, TOP‐8, CGI Secondary outcomes: Non specified Time points: Quote: "Measurements have been made every month for a five‐months period with six assessment points including the baseline" Data estimation: Completer values |
|
| Notes | Dates of trial: Not stated Industry‐funded: No Medication provided by industry: No Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Subjects were randomly assigned to one of the two treatment groups, nefazodone or sertraline" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Major limitation of the current study stems from its design with no double‐blinded comparison with placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Major limitation of the current study stems from its design with no double‐blinded comparison with placebo". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More participants withdrew from the nefazodone (6/30, 20%) compared to the placebo group (0/30, 0%). Reasons for withdrawals were not provided. Data estimation points were for completer values. No information was provided on whether the 2 groups differed by sample characteristics at baseline and endpoint Quote: "There were 30 patients in each treatment group. As 6 patients dropped out of the nefazodone group, sample size was 54, with 30 subjects in sertraline group and 24 subjects in nefazodone group" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias identified |
Shestatzky 1988.
| Study characteristics | ||
| Methods | Design: single‐centre, randomised, placebo‐controlled, flexible‐dose, double‐blind, cross‐over study Duration of intervention: 5 weeks (plus a cross‐over of 5 weeks, 10 weeks in total) Placebo run‐in: 2 week placebo washout Post‐treatment: Not specified |
|
| Participants | Setting: Quote: "Jerusalem Mental Health Center Outpatient Clinic" Sample size: 13 randomised to phenelzine and placebo Mean age: 38.5 years (31 ‐ 50) Sex: No information Diagnostic measure: DSM‐III Inclusion criteria: Quote: “Patients referred to the Jerusalem Mental Health Center Outpatient Clinic were considered eligible for the study provided that they met DSM‐III criteria for PTSD, were medically healthy, and had not suffered significant head injury or other physical trauma" Exclusion criteria: No information Comorbidity: IES: MDD, GAD Dropout rates: 54%; Insufficient information to determine dropouts |
|
| Interventions | Pharmacological intervention: Quote: “The subjects were then randomly assigned to treatment with phenelzine or placebo. Phenelzine was begun at 30 mg/day and was increased to the maximum tolerated dosage. After 5 weeks of treatment, all subjects were switched to placebo for a 2‐week period and were then, under double‐blind conditions, crossed over to the alternate treatment (phenelzine or placebo) for a further 5 weeks" | |
| Outcomes | Primary outcomes: CAPS‐2, CGI‐I, CGI‐S
Secondary outcomes: MADRS Time points: Quote: "Clinical response was monitored by the following observer‐rated scales, administered at baseline and then weekly throughout the trial by a rater" Data estimation: ITT(LOCF) values provided |
|
| Notes | Dates of trial: Not stated Industry‐funded: No Medication provided by industry: No Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Thirteen patients meeting DSM‐III criteria for posttraumatic stress disorder participated in a random‐assignment, double‐blind crossover trial comparing phenelzine (45‐75 mg/day) and placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "The authors thank Dr. Y. Ciivant for preparing the trial medications" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved Quote: "Thirteen patients meeting DSM‐III criteria for posttraumatic stress disorder participated in a random‐assignment, double‐blind crossover trial comparing phenelzine (45‐75 mg/day) and placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Clinical response was monitored by the following observer‐rated scales, administered at baseline and then weekly throughout the trial by a rater (MS) without knowledge of treatment assignment" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There as insufficient information to determine dropout rates for the 2 groups separately. Overall 54% of the participants dropped out of the study. Reasons for treatment withdrawal were provided for only subjects in the treatment group before crossover. No information was provided on whether the 2 groups differed at baseline and endpoint. Analyses were ITT and LOCF Quote: "Seven subjects were randomly assigned to phenelzine treatment during the first leg of the trial, and six subjects received placebo. Three subjects dropped out during this phase, after 1, 2, and 5 weeks, respectively. All were receiving phenelzine and complained of lassitude, dizziness, and lack of therapeutic benefit. Following crossover, four subjects (all receiving phenelzine) failed to complete week 5 of treatment. One developed urticaria which was attributed to phenelzine and three refused further treatment on the grounds that it was ineffective. Ten subjects completed at least 4 weeks of treatment" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Much of descriptive data for 4‐week completers in both phases; supportive psychotherapy provided to participants |
SKB627.
| Study characteristics | ||
| Methods | Design: randomised, placebo‐controlled, parallel, flexible‐dose, double‐blind Duration of intervention: 84 days (mean) Placebo run‐in: Yes Post‐treatment: Not specified |
|
| Participants | Setting: Not stated Sample size: 322 randomised to paroxetine and placebo Mean age: 18 ‐ 75 years Sex: 149 men and 173 women Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “100% PTSD by DSM‐IV" Exclusion criteria: Quote: “Likelyhood of P to exaggerate symptoms, placebo run‐in mediaction compliance of less than 80% at baseline, unresolved clinically sig abnormal lab/ECG findings, taken psychotropic drugs which had not been discontinued within set periods, used investigational drug within 1 month prior to screening, had ECT, intolerance to paroxetine, received psychotherapy in last 12 months, substance abuse, suicidal/homicidal risk, of child‐bearing potential & not using contraception, pregnancy, medical disorder that precludes the administration of Paroxetine, major depressive episode preceeding PTSD diagnosis" Comorbidity: Unclear Dropout rates: 105/322 (49/160 in the paroxetine and 56/162 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Paroxetine ‐ 20‐50mg flexible dose" | |
| Outcomes | Outcomes: CAPS‐2, CGI‐I (no distinction made between primary and secondary outcomes) Time points: No information Data estimation: ITT |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes Medication provided by industry: Yes Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Study Type: RCT" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Blindness: Double blind" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of participants withdrew from the paroxetine group (49/160, 31%) compared to the placebo group (56/162, 35%). Reasons for withdrawals were not provided. No information was provided on whether the 2 groups differed by sample characteristics at baseline and endpoint. Data estimation for ITT analysis |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Insufficient information to determine other sources of bias |
SKB650.
| Study characteristics | ||
| Methods | Design: randomised, placebo‐controlled, parallel, fixed‐dose, double‐blind, relapse prevention Duration of intervention: 252 days (mean) Placebo run‐in: Yes Post‐treatment: Not specified |
|
| Participants | Setting: Not stated Sample size: 176 randomised to paroxetine and placebo Mean age: 43 (18 ‐ 82 years) Sex: 59 men and 114 women Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “100% PTSD by DSM‐IV" Exclusion criteria: Quote: “<50 CAPS at baseline, Axis I disorder diagnosis, depressive episode preceeding PTSD diagnosis, CGI decreasing by 2 or more points between screening & baseline, likely to exaggerate symptoms, placebo run‐in compliance of < 80% or >120% at baseline visit, abnormal ECG not resolved by baseline, psychotropic drugs not discontinued according to cut‐offs, herbal treatments, investigational drugs within 3 months prior to screening, ECT within 3 months prior, intolerance to any SSRI, psychotherapy 12 wks prior, substance abuse/dependence within 6 months, suicidal risk, pregnancy, not practising contraception, serious medical disorder, previous participation in similar studies, judged as unable to comply to instructions" Comorbidity: Unclear Dropout rates: 46/176 (21/88 in the paroxetine and 25/88 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Paroxetine ‐ Maximum dose 50mg/day" | |
| Outcomes | Outcomes: CAPS‐2, DTS, MADRS, SDS (no distinction made between primary and secondary outcomes) Time points: No information Data estimation: ITT and LOCF |
|
| Notes | Dates of trial: Not stated Industry‐funded: No information Medication provided by industry: No information Any of the authors work for industry: No information |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the patients were randomised but no mention is made of the method of randomisation Quote: "Study Type: RCT" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Blindness: Double blind". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A similar proportion of participants withdrew from the paroxetine group (21/88, 24%) compared to the placebo group (25/88, 28%). Reasons for withdrawals were not provided. No information was provided on whether the 2 groups differed by sample characteristics at baseline and endpoint. Data estimation for ITT and LOCF analysis |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Insufficient information to determine other sources of bias |
Smajkic 2001.
| Study characteristics | ||
| Methods | Design: single‐centre, randomised, open‐label controlled trial, parallel, flexible‐dose Duration of intervention: 6 weeks Placebo run‐in: No information Post‐treatment: Not specified |
|
| Participants | Setting: Single‐centre, Mental Health Clinic Sample size: 32 randomised to sertraline, venlafaxine and paroxetine Mean age: 51.34 years (24 ‐ 63) Sex: 14 men and 18 women, Bosnian refugees, no combat‐related trauma Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “To meet diagnostic criteria for PTSD, a subject must have a requisite number of symptoms in each of the three symptom clusters corresponding with DSM‐III‐R criteria for PTSD; Participants were either referred to us by refugee resettlement organizations in Chicago, by family members, or by themselves. The participants reported in this study were all those who started treatment for PTSD at the Refugee and Bosnian Mental Health Programs at the Chicago Health Outreach over a 3‐month period in 1995. All participants received concurrent case management and supportive counseling but no other psychosocial treatments during the period of study" Exclusion criteria: Quote: “We excluded any refugees with prior history of major mental illness (e.g., schizophrenia, bipolar disorder). No refugee who met criteria declined the treatment with medications" Comorbidity: SCID, BDI Dropout rates: 8/40 (0/15 in the sertraline, 8/13 in the venlafaxine and 0/12 in the paroxetine group) |
|
| Interventions | Pharmacological intervention: Quote: “The dosages were as follows: Venlafaxine, 37.5 mg twice daily for 14 days, then if tolerated at 2 weeks, 75 mg twice daily; Sertraline, 50 mg once daily for 14 days, then if tolerated at 2 weeks, 100 mg once daily; Paroxetine, 20 mg once daily for 14 days, then if tolerated at 2 weeks, the dosage was continued. Patients were instructed to take the prescribed capsule daily unless side effects emerged" | |
| Outcomes | Primary outcome: PSS
Secondary outcomes: GAF, BDI Time points: Quote: "Assessments were performed just before initialization of therapy and 6 weeks after initiating therapy" Data estimation: Completer values |
|
| Notes | Dates of trial: Not stated Industry‐funded: No Medication provided by industry: No Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Participants were all randomly assigned to Sertraline, Paroxetine, and Venlafaxine conditions" |
| Allocation concealment (selection bias) | High risk | Not used, open‐label trial |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding of participants and personnel |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A substantial amount of participants withdrew from the venlafaxine group (8/13, 62%) compared to the sertraline (0/15, 0%) and paroxetine groups (0/12, 0%). Reasons for withdrawal were provided. Data represent completer values. No information was provided on whether the 2 groups differed by sample characteristics at baseline and endpoint Quote: "There were no dropouts from the Sertraline and Paroxetine groups. However, Venlafaxine was discontinued in eight patients because of adverse effects. Most commonly reported adverse effects were abdominal pain, agitation, dizziness, diaphoresis, headache, nausea, and palpitations" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Use of a nonblinded, open‐trial design with no placebo control group; Scales translated into Bosnian; Participants received concurrent case management and supportive therapy |
Spivak 2006.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre, randomised, controlled, parallel, fixed‐dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: 7 to 14 days Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Local community mental health outpatient clinic near central Israel" Sample size: 40 randomised to reboxetine and fluvoxamine Mean age: 40.08 (SD: 9.2) Sex: 21 men and 19 women, motor vehicle accidents Diagnostic measure: DSM‐IV and CAPS Inclusion criteria: Quote: “Inclusion criteria for study participation required that subjects met a primary current diagnosis of PTSD according to the Structured Clinical Interview for Axis I DSM‐IV Disorders–Patient Version and the Clinician‐Administered PTSD Scale for DSM‐IV (CAPS), Part 1. All subjects should have experienced PTSD symptoms for at least 1 month before study recruitment and had a total score of at least 60 on the first 17 items of the CAPS, Part 2 (CAPS‐2) at baseline" Exclusion criteria: Quote: “Subjects with a diagnosis of any DSM‐IV Axis I psychiatric disorder (except any mood or anxiety disorder considered to be comorbid with the primary diagnosis of PTSD), past or current traumatic brain injury and loss of consciousness, past or current medical or neurological illness, past or current alcohol or any other substance abuse, or current major routine laboratory abnormality were excluded from study participation. Also excluded from the study were subjects who were involved in any current litigation or who had been treated with any psychotropic medication for a period of 2 months before study enrolment" Comorbidity: No information Dropout rates: 12/40 (9/20 in the reboxetine and 3/20 in the fluvoxamine group) |
|
| Interventions | Pharmacological intervention: Quote: “Subjects who were randomized to receive reboxetine commenced treatment at 8 mg/d (4 mg BID) and remained at this fixed dosage for the 8‐week duration of the study. Patients randomly assigned to fluvoxamine commenced treatment at 150 mg/d (75 mg BID) and also remained at this fixed dosage for the 8‐week duration of the study" | |
| Outcomes | Outcomes: CAPS‐2, TOP‐8, HAM‐D, HAM‐A (not clear whether primary or secondary) Time points: Quote: " Patients were rated at baseline and at study end point" Data estimation: Completer values |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "This study was supported in part by a grant from the Medical Corps of the Israel Defense Force and the Agis Pharmaceutics Company (Ramat Gan, Israel)" Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A computer‐generated randomization sequence was used to determine each patient’s treatment group assignment" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "After a 7‐ to 14‐day screening and diagnostic assessment period, during which time no medication was administered, subjects who met the study inclusion criteria were randomly assigned under double‐blind conditions to 8 weeks of treatment with either reboxetine (n = 20) or fluvoxamine (n = 20), which were prepared in identically looking capsules" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. Quote: "Forty patients with MVA‐related PTSD attending a local community mental health outpatient clinic were randomized to receive a fixed dose of either reboxetine (8 mg/d) or fluvoxamine (150 mg/d) in a double‐blind fashion for a period of 8 weeks" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All rating scales were administered by a trained research psychiatrist (ES) blinded to the medication condition" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More participants withdrew from the reboxetine (9/20, 45%) compared to the fluvoxamine group (3/20, 15%). Reasons for withdrawal were provided. include: Data represent completer values. The 2 groups did not differ by sample characteristics at baseline and endpoint Quote: "No significant differences between the 2 subgroups were noted with respect to age, length of education, number of traumatic events encountered, or sex distribution. Nine reboxetine‐treated subjects and 3 fluvoxamine treated subjects did not complete the study. Reasons for early termination included the respective side effects of reboxetine (tension, palpitations, insomnia, headache, nausea, and increased sweating) and fluvoxamine (asthenia, headache, constipation, sedation, nausea, and dizziness). Thus, 28 patients (15 men and 13 women) who completed the 8‐week protocol were included in the final study analysis. Of the 28 subjects, 11 received reboxetine and 17 received fluvoxamine. No significant differences between the 2 subgroups were noted with respect to age or sex distribution" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This study was supported in part by a grant from the Medical Corps of the Israel Defense Force and the Agis Pharmaceutics Company (Ramat Gan, Israel)" |
Tucker 2001.
| Study characteristics | ||
| Methods | Design: multi‐centre, randomised, placebo‐controlled, parallel, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 1 week placebo run‐in Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “37 centers in the United States and Canada" Sample size: 323 randomised to paroxetine and placebo Mean age: 40.8 years Sex: 105 men and 202 women (for 307 participants), 7% (21/307) combat‐related, 35% (108/307) with comorbid major depression Diagnostic measure: DSM‐IV (i.e. MINI and CAPS‐1) Inclusion criteria: Quote: “Male and female patients at least 18 years of age were included in the study at the initial (screening) assessment if they satisfied the DSM‐IV criteria for chronic PTSD as determined by the MINI and CAPS‐1". Exclusion criteria: Quote: “Subjects with comorbid bipolar disorder, dissociative disorder, or any psychotic disorder were not eligible for entry into this study. Following a 1‐week placebo run‐in phase, patients were reassessed at the baseline visit and excluded from the study if they scored less than 50 on the first 17 items of the CAPS‐2. Also excluded were patients who were involved in litigation or were receiving disability payments because of any psychiatric disorder, who had received formal psychotherapy or electroconvulsive therapy in the 12 weeks prior to the initial assessment, or who met DSM‐IV criteria for alcohol/drug dependence or abuse within the preceding 6 months. Women of childbearing potential practicing a clinically accepted method of contraception were included, while women who had a positive pregnancy test at screening or who were lactating were excluded. Psychoactive herbal medication were not allowed during the study nor any other antidepressants, hypnotics and sedatives, and 12 weeks depot neuroleptics" Comorbidity: MDD Dropout rates: 136/307 (70/151 in the paroxetine and 66/156 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Patients randomly assigned to paroxetine started treatment at 20 mg daily and remained at this dosage for the first 2 weeks. After week 2, the paroxetine dosage could be increased according to the judgment of the investigating physician every 2 weeks by 10 mg/day up to 50 mg/day. During the treatment period, a single dosage reduction (because of physical illness or an adverse event) was allowed for paroxetine patients taking at least 30 mg/day" | |
| Outcomes | Primary outcomes: CAPS‐2 (score < 20 = 'remission'), CGI‐I
Secondary outcomes: DTS, TOP‐8, SDS, MADRS Time points: Quote: "Following the baseline assessment, the CGI‐I was administered at weeks 1 and 2 and biweekly thereafter; the CAPS‐2 and DTS, at weeks 4, 8, and 12; the SDS, at weeks 6 and 12; and the TOP‐8 and MADRS, at week 12" Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Supported by SmithKline Beecham pharmaceuticals, Collegeville, Pa" Medication provided by industry: No information Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised but no mention is made of the method of randomisation Quote: "Male and female outpatients 18 years and older who met DSM‐IV criteria on the Clinician Administered PTSD Scale (CAPS‐2) were randomly assigned to treatment with paroxetine (20‐50mg/day) or placebo for 12 weeks" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "This was a randomized double‐blind parallel‐group, placebo‐controlled flexible‐dose study of outpatients with chronic PTSD" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the paroxetine group (70/151, 46%) compared to the placebo group (66/156, 42%). The 2 groups did not differ by sample characteristics at baseline. Quote: "A total of 323 patients were randomly assigned to double‐blind study medication; 12 paroxetine patients and 4 placebo patients were lost to follow up after the baseline visit. The demographic and mean bassline clinical characteristics of the 307 patients who comprised the ITT population are presented in Table 1. The treatment groups were comparable with respect to the distribution of gender and race and were also very similar with regard to the time since index trauma and baseline clinical measures" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Supported by SmithKline Beecham Pharmaceuticals, Collegeville, Pa" |
Tucker 2003.
| Study characteristics | ||
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible‐dose, double‐blind Duration of intervention: 10 weeks Placebo run‐in: 1 ‐ 2 week taper at end Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “University hospital outpatient setting" Sample size: 59 randomised to citalopram, sertraline and placebo Mean age: 38.5 years Sex: 15 men and 43 women (for 58 participants), 3% (2/58) combat‐related, 78% (45/58) major depression Diagnostic measure: DSM‐IV, SCID, CAPS‐1 Inclusion criteria: Quote: “Eighty‐six subjects with PTSD, according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Structured Clinical Interview (DSM‐IV, SCID‐IV) and the CAPS‐I, with CAPS scores ≥ 50 (moderate to severe PTSD), were enrolled in the trial. Several studies have used a score of 50 on the CAPS as the cutoff for including PTSD subjects in clinical trials. The CAPS total score is composed of 3 subscales corresponding to the 3 PTSD symptom clusters—subscale B (CAPSB) for re‐experiencing symptoms, subscale C (CAPSC) for avoidance and numbing, and subscale D (CAPSD) for physiologic arousal. PTSD subjects had no other primary Axis‐I condition, although individuals with depression, panic disorder, or dysthymia were included if these were assessed as secondary to PTSD. All participants were free of significant medical illness and were not on any medication affecting autonomic functioning. They were not on any psychotropic medications for at least 2 weeks at baseline, and not on fluoxetine for at least 4 weeks. Occasional diphenhydramine for sleep was allowed throughout the ten week medication trial" Exclusion criteria: Quote: "Subjects were excluded if their participation in the study was potentially detrimental, that is, if a medical condition precluded use of an SSRI, if they had previously not tolerated or responded to an adequate trial of citalopram or sertraline, if the trauma script procedure was judged to be too stressful, if their psychiatric condition was such that possible placebo treatment was unsafe, or if psychotherapy was indicated. Subjects were further excluded if they had alcohol or substance abuse or dependence within 6 months of enrollment, if they were actively suicidal, or if they were assessed as not likely to comply with protocol requirements" Comorbidity: MD Dropout rates: 14/58 (5/25 in the citalopram, 6/23 in the sertraline and 3/10 in the placebo group) (for 58 participants) |
|
| Interventions | Pharmacological intervention: Quote: “Flexible dosing started at 20 mg/day for citalopram and at 50 mg/day for sertraline, titrated up or down each week by 10 mg/day for citalopram and by 50 mg/day for sertraline, according to side effects and efficacy. Minimum dose/day was 20 mg/day for citalopram and 50 mg/day for sertraline, and maximum dose was 50 mg/day for citalopram and 200 mg/day for sertraline. Placebo treated patients started with 1 tablet/day for the first week, titrated up or down by 1 tablet per week to a minimum of 1 tablet and a maximum of 4 tablets/day. Visits were scheduled weekly for the first 4 weeks of medication, and biweekly after that, with side effects and vital signs monitored at each visit" | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: IES (revised), BDI (revised) Time points: Quote: "The CAPS and IES were administered only at screening and at weeks 1, 6, and 10, in order to prevent possible therapeutic desensitization by these scales’ repeated exploration of PTSD symptoms. The BDI was administered at weeks 1, 2, 3, 4, 6, 8, and 10" Data estimation: LOCF (2 post‐baseline assessment) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Financial support for this study was provided by Forest Pharmaceuticals, Inc" Medication provided by industry: Unclear Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Subjects with PTSD who consented to drug treatment were randomly assigned in a double‐blind fashion to citalopram, sertraline, or placebo in the ratio of approximately 2:2:1" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Subjects in all 3 treatment groups were given identical white tablets" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved Quote: "Subjects with PTSD who consented to drug treatment were randomly assigned in a double‐blind fashion to citalopram, sertraline, or placebo in the ratio of approximately 2:2:1" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the citalopram group (5/25, 20%) compared to the sertraline (6/23, 26%) and placebo groups (3/10; 30%). Reasons for withdrawal were provided. The 2 groups did not differ by sample characteristics at baseline Quote: "A second incidence rate was completed for each group by dividing the number reporting symptom by the number who dropped out. Of the total 86 subjects screened, 27 were eliminated from the trial due to: lack of sufficient PTSD symptoms (n=4), severe medical conditions or abnormal laboratory values (n=8), current alcohol or substance abuse problems (n=5), suicidal ideation requiring clinical treatment (n=2), and failure to return for the baseline visit (n=8). These subjects were given appropriate referral for clinical treatment if desired. In total, 59 subjects were randomized to medication. Of these, 13 who withdrew early had at least 2 sets of data and were included in the intent‐to‐treat data analysis, with 1 randomized subject terminating early and with only one set of data not included in the analysis. Thus, data for 58 subjects were included in the final data analysis. Table 1 summarizes demographic variables of age, gender, and race, as well as type of trauma leading to PTSD for the 58 subjects included in the data analysis. Subjects were comparable across treatment groups. Of the 14 randomized subjects who withdrew before completing the 10‐week trial, 5 received citalopram, 6 received sertraline, and 3 were on placebo. Only 2 early terminators indicated their reason for withdrawing was due to side effects.Others stated that they changed their minds about participation or simply did not return for their next visit. Thus, it is possible that more of the early dropouts found side effects to be unacceptable" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Financial support for this study was provided by Forest Pharmaceuticals, Inc" |
Tucker 2007.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre, randomised, placebo‐controlled, parallel, flexible‐dose, double‐blind Duration of intervention: 8 weeks Placebo run‐in: washout/screening (up to 30 days before randomisation) Post‐treatment: 4 weeks maintenance |
|
| Participants | Setting: Quote: “Outpatient mental health setting associated with the University of Oklahoma Health Sciences Center, Oklahoma City" Sample size: 40 randomised to topiramate and placebo Mean age: 38.5 years (SD: 10.5) (18 ‐ 64) Sex: 8 men and 30 women (for 38 participants), non‐combat, 23 MDD (61%) Diagnostic measure: DSM‐IV (SCID‐IV, CAPS) Inclusion criteria: Quote: “Selection criteria included men and nonpregnant women 18 to 64 years of age with a diagnosis of civilian, non‐combat‐related Axis I PTSD for greater than 6 months according to DSM‐IV criteria as measured by Structured Clinical Interview for DSM‐IV (SCID‐IV) and with a CAPS score ≥50. Women had to be postmenopausal or practicing reliable contraception" Exclusion criteria: Quote: "Patients with major organic psychiatric diseases, current substance dependence or abuse (excluding nicotine or caffeine), serious or unstable concurrent illness, medical conditions potentially affecting drug absorption, history of nephrolithiasis or seizures, reduced renal clearance, elevated serum liver enzyme levels, current enrolment in cognitive behavioural therapy program, a history of primary major depressive disorder or primary major anxiety disorder, or known hypersensitivity to or a prior adverse event with topiramate were excluded from the study. Pregnant or lactating women were also excluded" Comorbidity: MD, MD + panic, MD + dysthymia Dropout rates: 8/38 (5/19 in the topiramate and 3/19 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Study medication was started at 25 mg/day and was titrated by 25‐50 mg/week during an 8‐week period following a designated washout period for protocol‐prohibited medications, which included all psychotropic medications. Patients were titrated to their maximum tolerated dose until complete or nearly complete efficacy was achieved or until the maximum dosage allowed (400 mg/day) was reached. Topiramate was given twice daily" | |
| Outcomes | Primary outcomes: CAPS‐17
Secondary outcomes: HRSA, HRSD, TOP‐8, CGI, DTS, BIS, SDS, CD‐RISC, SFQ Time points: No information Data estimation: ITT with LOCF |
|
| Notes | Dates of trial: Quote: “Patients participated in this study between April 26, 2002, and February 4, 2004" Industry‐funded: Yes. Quote: "This study was supported by Ortho‐McNeil Neurologics, Inc., Titusville, NJ" Medication provided by industry: No information Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to receive either placebo (N=20) or topiramate (N=20) in a 1:1 ratio using computer generated codes; randomization was balanced using permuted blocks” |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More participants withdrew from the placebo group (3/20; 15%) compared to the topiramate (5/20; 25%) group. Common withdrawals include adverse events and patient choice. However, the 2 groups did not differ by sample characteristics at baseline and the ITT population was used at baseline and endpoint Quote: "The efficacy and safety population comprised 38 patients (topiramate, N=19; placebo, N=19). Demographic characteristics, comorbid Axis I condition secondary to PTSD, and trauma type were similar between groups .... Efficacy was analysed in the intention‐to‐treat population that included all randomised patients who had taken ≥1 dose of study medication and had ≥1 post baseline efficacy evaluation" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "This study was supported by Ortho‐McNeil Neurologics, Inc., Titusville, NJ" |
Van der Kolk 1994.
| Study characteristics | ||
| Methods | Design: multi‐centre, randomised, placebo‐controlled, parallel, flexible‐dose, double‐blind Duration of intervention: 5 weeks Placebo run‐in: 2 weeks Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Two sites: the Massachusetts General Hospital Trauma Clinic and the Boston Veterans Administration Outpatient Clinic" Sample size: 64 randomised to fluoxetine and placebo Mean age: 40.4 years (22 ‐ 55) Sex: 42 men and 22 women, 44% (28/64) combat‐related trauma, 54.8% (34/64) MDD Diagnostic measure: DSM‐II‐R Inclusion criteria: Quote: “Subjects were outpatients who met DSM‐III‐R primary Axis I diagnosis of PTSD, including a score of 45 or above on the CAPS" Exclusion criteria: Quote: "Subjects were excluded if they reported diagnoses of schizophrenia, bipolar I disorder, organic mental disorder, or drug or alcohol addiction within 6 months of the initial interview or met criteria for any of these disorders in the psychiatric evaluation. Patients were requested to abstain from illicit drug and alcohol use and to report any medications taken during the study. Also, patients with clinically significant cardiovascular, renal, hepatic, endocrine, or neurologic disease and pregnant or nursing mothers were excluded. Supportive psychotherapy was permitted" Comorbidity: MDD Dropout rates: 16/64 (12/33 in the fluoxetine and 4/31 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Subjects were instructed to take one capsule (20 mg of fluoxetine or placebo) the first day, none the second day, and one daily for the remainder of the first week unless troublesome side effects emerged. During each weekly visit, the dosage was adjusted by, at most, one additional capsule every other day. Dosage was increased until a significant clinical effect was obtained (reduction in symptoms), with a maximum possible dose of three capsules (60 mg) per day" | |
| Outcomes | Outcomes: CAPS, BDHI, HAM‐D, DES, DESI, acoustic startle response, Rorschach Inkblot Test (no distinction between primary and secondary outcomes) Time points: Quote: "At Visit 1 (baseline) and again at Visit 6 (endpoint)". Data estimation: Completer values |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Funded, in part, by a grant from Eli Lilly and Company, Indianapolis" Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the patients were randomised however no mention is made of the method of randomisation Quote: "64 subjects with PTSD entered a 5‐week randomized double‐blind trial comparing fluoxetine and placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Medication was randomized and administered in double‐blind fashion" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "64 subjects with PTSD entered a 5‐week randomized double‐blind trial comparing fluoxetine and placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More participants withdrew from the fluoxetine (12/33, 36%) compared to the placebo group (4/31, 13%). Reasons for withdrawal were not provided. Data estimation points were based on completer values. The 2 groups did not differ by sample characteristics at baseline Quote: "Sixty ‐four subjects entered the study, of whom 47 completed the study. There was no significant difference between groups on marital status, nor on education. There were no correlations between severity at intake and dropout. Age and current diagnostic criteria for major depression was not statistically significant" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Funded, in part, by a grant from Eli Lilly and Company, Indianapolis" |
Van der Kolk 2007.
| Study characteristics | ||
| Methods | Design: multi‐centre, randomised, placebo‐controlled, parallel, flexible‐dose, double‐blind, long‐term maintenance Duration of intervention: 5 weeks Placebo run‐in: No information Post‐treatment: 6 month follow‐up |
|
| Participants | Setting: Quote: “Multi‐centre, recruited via newspaper ads, the Internet, and solicitation from medical and mental health professionals" Sample size: 59 randomised to fluoxetine and placebo Mean age: 34.9 years (18 ‐ 65). Sex: 8 men and 51 women Diagnostic measure: DSM‐IV Inclusion criteria: Quote: “Individuals 18‐65 years old with current PTSD and mixed trauma exposure at least one year prior to intake were recruited via newspaper ads, the Internet, and solicitation from medical and mental health professionals" Exclusion criteria: Quote: "Exclusion criteria were unstable medical condition; contraindications to either treatment (i.e., pregnancy; glaucoma or detached retina; history of severe allergies or multiple adverse drug reactions); inability to be weaned off current psychotropic medications; psychotic or bipolar disorder; current alcohol or substance abuse/dependence; severe dissociation; active suicidality or life‐threatening mutilation; prior exposure to active study interventions; concurrent trauma‐focused treatment; unstable living situation; GAF<40; and disability compensation for PTSD or pending trauma‐related lawsuit. Initial telephone screening was used to assess likely presence/absence of inclusion and exclusion criteria; potential participants were invited for in‐person assessment" Comorbidity: Axis I and II disorders at baseline Dropout rates: 7/59 (4/30 in the fluoxetine and 3/29 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Starting dosage was 10 mg/day of fluoxetine (or pill placebo equivalent). Dosage was increased in 10 mg increments per week to a maximum of 60 mg/day or until symptom remission was achieved. Increases or decreases in dosage were based on physician judgment of clinical response and presence/absence of dose‐limiting side effects" | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: BDI Time points: CAPS: Rating was based on 1‐week interval for immediate pre‐ and posttreatment assessment; 1‐month interval was used at baseline and six‐month follow‐up Data estimation: LOCF |
|
| Notes | Dates of trial: Quote: “The study ran from July 2000 through July 2003” Industry‐funded: No. Quote: "This study was supported by grant R01MH58363 from the National Institutes of Mental Health" Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Participants were randomly assigned to 1 of 3 treatment conditions: EMDR, fluoxetine, or pill placebo. Randomization was stratified by presence/absence of concurrent supportive psychotherapy. In order to ensure approximately equal numbers in each treatment condition, random assignment was blocked in groups of 12 consecutive participants, so that in each block, 4 participants were assigned to each condition. Participants in all 3 conditions received a total of 8 weekly treatment sessions. Following cessation of treatment, participants in the 2 active treatment conditions were asked to refrain from initiating new treatment during the 6‐month follow‐up period" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Starting dosage was 10 mg/day of fluoxetine (or pill placebo equivalent)" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved (re participants) Quote: "The fluoxetine and placebo interventions were administered in a double‐blind, fixed flexible‐dose design according to standard protocol for double‐blind pharmacologic interventions for PTSD. All raters were blind to treatment condition and were never assigned the same participant for both pretreatment and posttreatment evaluation. All EMDR sessions were videotaped, and an independent evaluator assessed treatment fidelity through videotape review from randomly sampled sessions" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All raters were blind to treatment condition, and were never assigned the same participant for both pre‐ and post‐treatment evaluation" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the fluoxetine group (4/30, 13%) compared to the placebo group (3/29; 10%). The 2 groups did not differ by sample characteristics at baseline. Quote: "Significantly more patients discontinued the study due to an adverse event in the 40‐mg/d fluoxetine treatment group (13.1%) than in the 20‐mg/d fluoxetine treatment group (4.3%; P = 0.005); however, neither of these rates differed significantly from the placebo treatment group (8.0%, both P > 0.20). There were no other significant differences between treatment groups for other reasons of discontinuation, including discontinuation due to lack of efficacy (20 mg fluoxetine, 6.7%; 40 mg fluoxetine, 3.8%; placebo, 6.8%; P = 0.416) or discontinuation due to patient decision (20 mg fluoxetine, 11.0%; 40 mg fluoxetine, 6.9%; placebo, 13.6%; P = 0.182)". Twelve people dropped out during the 8‐week treatment phase, leaving 76 treatment completers. There were no significant differences in dropout rates on any baseline measure of psychopathology or across treatment conditions. Completers in each group were 24 of 29 (83%) for EMDR, 26 of 30 (87%) for fluoxetine, and 26 of 29 (90%) for pill placebo. Dropouts were significantly younger than completers (dropout mean age = 27.1 years, completer mean age = 37.6 years; F = 6.785, df = 1,86; p < .05). In addition, dropouts were more likely to have had child‐ versus adult‐onset trauma (χ2 = 6.175, df = 1,88; p = .013)Study hypotheses were tested on both treatment completer and intent‐to‐treat (ITT) samples. Intent‐to‐treat analyses were conducted using an early termination assessment, when available, or a last‐observation‐carried forward (LOCF) procedure to impute missing data. Long term effects of active‐treatment completers were assessed at 6 months posttreatment for both follow‐up completer and intent‐to‐follow (ITF) samples. For the ITF sample, missing follow‐up data were also estimated using LOCF. Participants in the 3 treatment conditions did not differ significantly on any demographic variable or on any baseline measure of psychopathology" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified |
Villarreal 2016.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: multi‐centre, randomised, placebo‐controlled, parallel, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 1‐week, single‐blind placebo lead‐in Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Two sites: the Ralph H. JohnsonVAMedical Center in Charleston, S.C., and the Raymond G. Murphy VA Medical Center in Albuquerque, N.M" Sample size: 80 randomised to quetiapine and placebo Mean age: 53 years (SD: 11) Sex: 75 men and 5 women, mostly combat veterans Diagnostic measure: DSM‐IV (i.e. CAPS‐DX, SCID‐I/P) Inclusion criteria: Quote: “Patients were included in the study if they were 18 to 65 years of age and met DSM‐IV criteria for PTSD, as established with the CAPS‐DX. Patients were also assessed with the SCID‐I/P. Veterans of both genders and any ethnic background were recruited. Participants had to have a score of at least 50 on the CAPS at baseline and be capable of giving informed consent. Female patients were required to use a medically approved contraceptive or otherwise not be of child‐bearing potential. Patients did not take any psychotropic medications or herbal remedies within 1 week prior to randomization (2 weeks for fluoxetine) and during the course of the study except for rescue medications. Medications for medical indications were held constant for 1 month prior to study entry" Exclusion criteria: Quote: "Exclusion criteria included history of sensitivity to quetiapine, use of psychotropic medications within 1week prior to randomization and throughout the duration of the study (except for short‐term use as rescue medication as specified in the Concomitant Medications section), and medical disorders that may preclude safe administration of quetiapine or exacerbate anxiety symptoms. Diabetes mellitus was not an exclusion criterion. Additional reasons for exclusion were alcohol or substance abuse or dependence within 1 month of study entry as defined by DSM‐IV criteria; schizophrenia, schizoaffective disorder, bipolar disorder, or dementia; suicidal or homicidal ideation; current pursuit of compensation or an increase in compensation for the effects of trauma; and initiation or change in psychotherapy within 3 months of randomization" Comorbidity: No information Dropout rates: 33/80 (13/42 in the quetiapine and 20/38 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “The study medication was initiated at a dose of 25 mg at bedtime and gradually titrated to 400 mg daily by the end of week 2 as tolerated. Further dose increases or decreases were allowed up to a maximum of 800 mg daily of study medication and a minimum of 50 mg daily" | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: CAPS subscales, DTS, PANSS, CGI, CGI‐S HAM‐D, HAM‐A, PSQI. Safety measures: AIMS, SAS, BARS, ASEX. Time points: The participants were evaluated at weeks 1, 2, 4, 8, and 12 Data estimation: ITT with LOCF |
|
| Notes | Dates of trial: Between 2004 and 2008 Industry‐funded: Yes. Quote: "Funded by an investigator‐initiated grant from Astra Zeneca to Dr.Hamner" Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The subjects were randomly assigned to quetiapine or placebo in a 1:1 ratio by using a computer program for random number assignment" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved (re participants) Quote: "Placebo nonresponders (participants who had less than a 30% reduction in CAPS total score) were then randomly assigned to receive double‐blind quetiapine or placebo tablets for 12 weeks" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More participaents withdrew from the placebo group (20/38, 53%) compared to the quetiapine (13/42, 31%) group. Common withdrawals include adverse events, lack of efficacy and other reasons. The 2 groups did not differ by sample characteristics at baseline and the ITT and LOCF sample was used at baseline and/or endpoint Quote: "Thirteen patients (31%) dropped out of the quetiapine group, and 20 patients (53%) dropped out of the placebo group. The mean age of the participants was 52 years, with no significant difference between the quetiapine and placebo groups. The patients in the quetiapine group had slightly more education than those in the placebo group. The majority of the patients were male combat veterans. There was no difference in the percentage of males between groups. Race distributions were also similar in the two groups" |
| Selective reporting (reporting bias) | High risk | No information on the SDS was reported |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Funded by an investigator‐initiated grant from Astra Zeneca to Dr. Hamner" |
Yeh 2011.
| Study characteristics | ||
| Methods |
NEW TRIAL ADDED TO UPDATED REVIEW Design: single‐centre, randomised, placebo‐controlled, parallel, flexible‐dose, double‐blind Duration of intervention: 12 weeks Placebo run‐in: 1‐week period of washout Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Outpatient clinic of the violence program of Federal University of São Paulo Hospital (Prove‐UNIFESP),
SãoPaulo City" Sample size: 35 randomised to topiramate and placebo Mean age: 40.1 (SD: 10.71) Sex: 10 men and 21 women (for 31 participants), Civilian Sample Diagnostic measure: DSM‐IV, SCID‐I and SCID‐II Inclusion criteria: Quote: “The inclusion criteria comprised men and women aged 18–62 years with a diagnosis of PTSD according to Diagnostic and Statistical Manual of Mental Disorders Fourth Edition (DSM‐IV), confirmed by the use of the Structured Clinical Interview for DSMIV Axis I and Axis II (SCID‐I and SCID II, respectively)" Exclusion criteria: Quote: "Women of childbearing potential had to be practicing reliable contraception and were advised not be pregnant or breastfeeding during the study. Participants with lifetime history of bipolar, psychotic, borderline personality disorder, substance dependence or abuse (excluding nicotine and caffeine) in the previous 6 months, serious or unstable concurrent illness, history of nephrolithiasis, use of psychotropic medications for the previous 2 weeks (6 weeks for fluoxetine), body mass index below 20, current suicidal ideation or psychotic symptoms, were excluded from the study" Comorbidity: No information Dropout rates: 5/35 (3/17 in the topiramate and 2/18 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Study medication started at 25 mg/day once daily, at night, and increased in 25 mg weekly, as tolerated, until complete or nearly complete efficacy was achieved or until maximum dose allowed was reached (200 mg/day)" | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: CGI, BDI Time points: Quote: "Trained interviewers administered the CAPS, and the patients completed the BDI at baseline and 12th week. Study follow‐up visits were conducted at baseline, and weeks 1, 2, 3, 4, 6, 8, and 12" Data estimation: ITT with LOCF |
|
| Notes | Dates of trial: Between April 2006 and December 2009 Industry‐funded: No Medication provided by industry: No information Any of the authors work for industry: No |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Participants were then randomly assigned to receive either placebo or topiramate, in a 1:1 ratio using computer‐generated code" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The participants, interviewers, and the practitioners in charge of the cases were all blind to the study experimental and control groups" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the topiramate group (3/17, 18%) compared to the placebo group (2/18, 11%). The 2 groups did not differ by sample characteristics at baseline and the ITT and LOCF sample was used Quote: "Thirty‐six civilian cases with a confirmed diagnosis of PTSD agreed to participate in the study and provided a signed informed consent. One patient showed remarkable improvement in the washout phase, being excluded from the study. Thus, 35 patients were randomized, but 4 subjects failed to return to the first visit and were not considered in the evaluation. Thirty‐one eligible cases were then enrolled in the study, and were considered for the efficacy analysis (n = 17 topiramate, and n = 14 placebo).Twenty‐six subjects completed the entire 12‐week study: 2 patients were withdrawn for worsening of symptoms characterized by 1‐point decrease at CGI and 3 discontinued treatment (1 due to worsening of symptoms, 1 due to an adverse event, and 1 dropout treatment). There were no significant differences between groups according to age, gender, marital status, duration of illness, and baseline levels of CAPS and BDI" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Low risk | No other sources of bias were identified |
Zohar 2002.
| Study characteristics | ||
| Methods | Design: mutli‐centre, randomised, placebo‐controlled, parallel, flexible‐dose, double‐blind Duration of intervention: 10 weeks Placebo run‐in: 1 week single‐blind placebo run‐in Post‐treatment: Not specified |
|
| Participants | Setting: Quote: “Three collaborating sites, all of which were in Israel" Sample size: 42 randomised to sertraline and placebo Mean age: 40 (SD: 6) (for 42 participants) Sex: 35 men and 7 women (for 42 participants), 76% (32/42) combat‐related trauma Diagnostic measure: DSM‐III‐R (CAPS‐1) Inclusion criteria: Quote: “The subjects were male and female outpatients aged 18 years and older who met DSM‐III‐R criteria for a primary diagnosis of PTSD as determined by part 1 of the CAPS‐1. A minimum 6‐month duration of PTSD illness was required (exceeding the 1‐month minimum required by the DSMIII‐R) as well as a CGI‐S score of 4 or higher and a total severity score of 50 of higher on the CAPS‐2 at the baseline visit. Female participation was contingent on a negative beta human chorionic gonadotropin pregnancy test and use of a medically accepted form of contraception for at least 3 months" Exclusion criteria: Quote: "Exclusion criteria included (1) presence of any other primary axis I disorder (concurrent depression was permitted only if its onset was judged to be secondary to PTSD and with a later onset of illness); (2) alcohol or substance abuse or dependence in the past 6 months; (3) evidence of clinically significant hepatic or renal disease or any other acute or unstable medical condition that might interfere with the safe conduct of the study; (4) intolerance or hypersensitivity to sertraline; (5) nonresponse to a previous adequate trial of any SSRI in the treatment of any axis I disorder; (6) current use of psychotropic medication (except infrequent chloral hydrate or temazepam on an as‐needed basis); and (7) 20% or greater reduction in the CAPS‐2 total severity score during the placebo lead‐in period" Comorbidity: Unclear. Dropout rates: 11/42 (6/23 in the sertraline and 5/19 in the placebo group) |
|
| Interventions | Pharmacological intervention: Quote: “Sertraline treatment was initiated at a daily dose of 50 mg, with flexible titration in 50‐mg increments permitted every 2 weeks (based on clinical response) to a maximum dose of 200 mg" | |
| Outcomes | Primary outcomes: CAPS‐2, CGI‐I, CGI‐S
Secondary outcome: MADRS Time points: Quote: "Assessments took place at baseline and at the end of study treatment weeks 2, 4, 6, 8, and 10" Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | Dates of trial: Not stated Industry‐funded: Yes. Quote: "Supported by Pfizer Inc." Medication provided by industry: No information Any of the authors work for industry: Yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was reported that the participants were randomised, but no mention is made of the method of randomisation Quote: "Outpatient Israeli military veterans with a DSM‐III‐R diagnosis of PTSD were randomized to 10 weeks of double blind treatment with sertraline or placebo" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on which parties were blinded and how blinding was achieved Quote: "Outpatient Israeli military veterans with a DSM‐III‐R diagnosis of PTSD were randomized to 10 weeks of double blind treatment with sertraline or placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", although no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The same amount of participants withdrew from the sertraline (6/23, 26%) and placebo group (5/19, 26%). Reasons for withdrawal were not provided. No information was provided on whether the 2 groups differed by sample characteristics at baseline. Data estimation values were based on LOCF. Quote: Fifty‐one participants were screened and signed informed consent forms. The intent‐to‐treat group consisted of 42 patients" |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study |
| Other bias | Unclear risk | Funding for study provided by industry Quote: "Supported by Pfizer Inc." |
AEs: Adverse Events; AIMS: Abnormal Involuntary Movement Scale; ANCOVA: analysis of covariance; ASEX: Arizona Sexual Experience Scale; ASI: Addiction Severity Index; AUDIT‐C: Alcohol Use Disorders Identification Test–Consumption; BAS: Barnes Akathisia Scale; BDHI: Buss‐Durkee Hostility Inventory; BDI: Beck Depression Inventory; BDI‐R: Beck Depression Inventory, Revised; BID: means twice (two times) a day; BPRS: The Brief Psychiatric Rating Scale; BUN: blood urea nitrogen; CADSS: Clinician Administered Dissociative States Scale; CAPS/CAPS‐SX17: Clinician Administered PTSD Scale, 17 items; CAPS‐1: Clinician Administered PTSD Scale‐version 1; CAPS‐2: Clinician Administered PTSD Scale ‐ part 2; CAPS‐A: Clinician Administered PTSD Scale, Criterion A: Avoidance/Numbing; symptoms; CAPS‐B: Clinician Administered PTSD Scale, Criterion B: Re‐experiencing symptoms; CAPS‐B2: Clinician Administered PTSD Scale, Criterion B2: Recurrent distressing dreams symptoms; CAPS‐C: Clinician Administered PTSD Scale, Criterion C: Avoidance symptoms; CAPS‐D: Clinician Administered PTSD Scale, Criterion D: Negative alterations in cognitions and mood; CAPS‐DX: Clinician Administered PTSD Scale, Current Diagnostic Version; CAS: Covi Anxiety Scale; CBC: complete blood count; CD‐RISC: Connor‐Davidson Resilience Scale; CES: Combat Exposure Scale; CES‐D: Center for Epidemiologic Studies Depression Scale (self‐rated); CGI‐C: Clinical Impression ‐ Improvement Scale or Change; CGI‐I: Clinical Impression ‐ Improvement Scale; CGI‐S: Clinical Global Impression – Severity scale; Co.: Company; CPFQ: Cognitive and Physical Function Questionnaire; CS: Columbia Scale; C‐SSRS: Columbia Sucidie Severity Rating Scale; CTQ: Childhood Trauma Questionnaire; DBH: dopamine‐ß‐ hydroxylase; DESI: Disorders of Extreme Stress Inventory; DES: Dissociative Experiences Scale; DGRP: Duke Global Rating for PTSD; DGRP‐I: Duke Global Rating for PTSD Improvement Scale; DGRP‐S: Duke Global Rating for PTSD Severity; scale; Dr.: Doctor; DRRI: Deployment Risk and Resilience Inventory; DSM‐III: Diagnostic and Statistical Manual of Mental Disorders, Third Edition; DSM‐III‐R: Diagnostic and Statistical Manual of Mental Disorders, Third Edition, Revised; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition; DSM‐IV A2: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Criterion A2: that exposure to a potentially traumatic experience (PTE; PTSD criterion A1) is accompanied by intense fear, helplessness, or horror; DSM‐IV‐R: Diagnostic and Statistical Manual of Mental Disorders, Revised; DTS: Davidson Trauma Scale; ECT: Electroconvulsive therapy; e.g.: example; EKG: electrocardiogram; EMDR: Eye Movement Desensitization and Reprocessing; EPI: Eyesenck Personality Inventory; ER: Extended release; ESZ: eszopiclone; EQ‐5D: EuroQol Visual Analog Scale; FLU: Fluoxetine; GAD: Generalized Anxiety Disorder; GAF: Global Assessment of Functioning; GLM: general linear model; HADS: Hospital Anxiety and Depression Scale; HAM‐A: Hamilton Anxiety Scale; HAM‐D: Hamilton Depression Rating Scale; HAS: Hamilton Anxiety Scale; HDRS‐17: Hamilton Depression Rating Scale, 17 items; HRSA: IE: Independent evaluator; IES: Impact of Events Scale; IES‐R: Impact of Events Scale Revised; IL: Illinois; Inc.: Incorporation; IPP: Inventory of Personal Problems; ITT: Intention to treat; IV: Intravenous therapy; kg: kilogram; LEC: Life Events Checklist; LIFE‐RIFT: Range of Impaired Functioning Tool; LOCF: Last Observation Carried Forward; LVCF: Last Value Carried Forward; MD: major depression; MDD: major depressive disorder; MADRS: Montgomery‐Asberg Depression Rating Scale; MAOIs: Monoamine oxidase inhibitors; mg/d: Milligrams per day; MINI: Mini International Neuropsychiatric Interview; MISS: Mississippi Scale for Combat‐Related PTSD; MSFQ: Massachusetts Sexual Function Questionnaire; n/N: number of participants in the study; NI: Newcastle Index; NA: neuronal noradrenaline; NFQ: Nightmare Frequency Questionnaire‐Revised; NIMH: National Institute of Mental Health; NJ: New Jersey; NOS: Not Otherwise Specified; NY: New York; OCD: Obsessive Compulsive Disorder; OCDS: Obsessive Compulsive Drinking Scale; OIF/OEF: Operation Iraqi Freedom and Operation Enduring Freedom; PANSS: Positive and Negative Syndrome Scale; PBO: Placebo; PCL: PTSD Check‐List; PCL‐M: PTSD Checklist–Military Version; PD: Panic disorder; PDS: Posttraumatic Stress Disorder Scale; PDRS: PTSD Dream Rating Scale; PET: Positron Emission Tomography; PHQ‐9: Patient Health Questionnaire 9‐item depression scale; PM: post meridiem; POMS: Profile of Mood States; PSQI: Pittsburgh Sleep Quality Index; PSS: PTSD Checklist: Post‐traumatic stress disorder (PTSD) Checklist; PTSD Symptoms Scale; PTSD: Post‐traumatic stress disorder; PTS: Post‐traumatic stress; Q‐LES‐Q: Quality of Life Enjoyment and Satisfaction Questionnaire; QOLI: Quality of Life Inventory; RCT: Randomised Control Trial; RDC: RMANOVA: repeated measures analysis of variance; RSD: Raskin Scale for Depression; SADS: Schedule for Affective Disorders and Schizophrenia; SAS: Simpson‐Angus Scale; SC: South Carolina; SCID: Structured Clinical Interview for DSM; SCID‐P: Structured Clinical Interview for DSM, Psychotic screen; SCL‐90‐R: Hopkins 90‐item Symptom Checklist; SCL‐90‐R: Hopkins 90‐item Symptom Checklist‐Revised; SD: Standard deviation; SDI: Sheehan Disability Inventory; SDS: Sheehan Disability Scale; SF‐12: Veterans 12‐Item Short‐Form General Health Survey; SGA: second generation antipsychotic; SI‐PTSD: DSM based PTSD scale; SIP: Structured Interview for PTSD; SNRI: Serotonin and norepinephrine reuptake inhibitor; SP: Social Phobia; SPM: Statistical Parametric Mapping; SPRINT: Short PTSD Rating Interview scale; SSRI: Selective serotonin reuptake inhibitor; SVS: Sheehan Vulnerability to the Effects of Stress Scale; TLFB: Time‐Line Follow‐Back; TOP‐8: Treatment Outcome PTSD Scale; UKUSERS: UKU Side Effect Rating Scale; US: United States; VA: Veteran affairs; VAS: Visual Analog Scale; VS: Vulnerability to the effects of stress scale; vs.: versus; YMRS: Young Mania Rating Scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aerni 2004 | All 3 participants received concurrent medication. In addition, 1 of the participants was treated with concurrent psychotherapy |
| Angelidis 2019 | Not primary diagnostic. Specific to stress disorders in general |
| Back 2016 | Augmentation study |
| Back 2018 | Not primary diagnostic. Dual diagnosis |
| Bartzokis 2005 | Augmentation trial of adjunctive risperidone for chronic, combat‐related PTSD. To be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Batki 2014 | Augmentation trial |
| Boroughs 2018 | Secondary publication |
| Borrelli 2019 | Prevention study |
| Bountress 2018 | Augmentation study. Treatment with Prolonged Exposure Therapy |
| Brady 2005 | PTSD and co‐occurring alcoholism |
| Bremner 1997 | Pharmacotherapy impact assessment trial |
| Brunet 2018 | Augmentation study. Treatment with brief memory reactivation |
| Buhmann 2018 | Augmentation study. Cognitive behavioural therapy and antidepressants |
| Coupland 1997 | Pharmacotherapy impact assessment trial |
| Eftekhari 2004 | Comparison of treatment of chronic PTSD with prolonged exposure or sertraline. No placebo or other medication used as control |
| Feder 2021 | Concurrent psychotherapy |
| Flanagan 2018 | Imaging study |
| Flanagan 2019 | Not primary diagnosis. Dual diagnosis |
| Frank 1988 | An extension of this database was subsequently published (Kosten 1991) |
| Frijling 2017 | Prevention study |
| Frommberger 1998 | Open‐label comparison of paroxetine and CBT. No placebo or alternative medication control |
| Gelpin 1996 | Trial of prophylactic treatment of recent trauma survivors with benzodiazepines. To be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Graham 2018 | No control arm |
| Hamner 1997 | Buspirone potentiation of antidepressants for combat‐veteran PTSD sufferers. May be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Hamner 1999 | Pharmacotherapy impact assessment trial |
| Hamner 2003 | Risperidone augmentation of medication for war‐veteran PTSD sample with psychotic symptoms. May be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Heresco‐Levy 2002 | Augmentation trial of d‐cycloserine for PTSD. May be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Inslicht 2018 | No control arm |
| Jacobs‐Rebhun 2000 | Trial of treatment for sleep disorders associated with PTSD |
| Kanter 2001 | Pharmacotherapy impact assessment trial |
| Kellner 2000 | Pharmacotherapy impact assessment trial. Not a treatment study |
| Le 2018 | No control arm |
| Mello 2009 | Protocol |
| Monnelly 2003 | Augmentation of risperidone for irritable aggression symptoms in combat‐veteran PTSD sufferers. May be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Morgan 1995 | Pharmacotherapy impact assessment trial |
| Naylor 2013 | Subthreshold PTSD |
| NCT00557622 | Terminated (difficulty in achieving target enrolment numbers) |
| NCT00648375 | Terminated (inadequate recruitment) |
| NCT00706173 | Withdrawn (unable to recruit eligible participants for the trial) |
| NCT01325168 | Augmentation study |
| NCT01449955 | Augmentation study |
| NCT01477762 | Augmentation study |
| NCT01664260 | Withdrawn (the research project was cancelled before any participants were enrolled) |
| Neylan 2006 | Concurrent psychotherapy |
| Otto 2003 | Augmentation trial and comparison group received medication and cognitive‐behaviour therapy |
| Pape 2018 | Genetic study |
| Petrakis 2006 | PTSD not primary diagnosis |
| Pitman 1990 | Pharmacotherapy impact assessment trial |
| Pitman 2002 | Prevention study |
| Pollack 2011 | No control arm |
| Ramaswamy 2016 | No control arm |
| Ramaswamy 2017 | Concurrent psychotherapy |
| Randall 1995 | Pharmacotherapy impact assessment trial |
| Raskind 2003 | Trial classified as an augmentation trial for treatment‐resistant PTSD. To be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Raskind 2007 | Concurrent psychotherapy |
| Raskind 2013 | Secondary publication. |
| Reist 1995 | Pharmacotherapy impact assessment trial |
| Reist 2001 | Pharmacotherapy impact assessment trial |
| Roache 2017 | PTSD not primary diagnosis. Dual diagnosis |
| Schelling 2004 | Trial of hydrocortisone for post‐operative complications in cardiac surgery. The trial is concerned with the development of PTSD following surgery, and diagnosis with PTSD does not fall within its inclusion criteria; this is better considered a prophylaxis trial, and will be included in an upcoming protocol |
| Southwick 1997 | Pharmacotherapy impact assessment trial |
| Stein 2002 | Augmentation trial of adjunctive olanzapine for SSRI resistant combated‐related PTSD. To be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Suris 2017 | Augmentation study |
| Vaiva 2003 | Pharmacotherapy impact assessment trial |
| Van der Kolk 1989 | Pharmacotherapy impact assessment trial |
| Verplaetse 2019 | PTSD not primary diagnosis |
CBT: cognitive behavioural therapy; PTSD: posttraumatic stress disorder.
Characteristics of studies awaiting classification [ordered by study ID]
Davis 2003.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Reference: Davis L, Petty F. GABAergic modulation in PTSD and treatment with anticonvulsants. In: 19th Annual Meeting, International Society for Traumatic Stress Studies, October 29 ‐ November 1, Chicago, IL. 2003. "Study ID: CRSREF: 15593141" |
Fluoxetine, Davidson.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Study ID: CRSSTD: 15593122 |
Fluoxetine, Lilly.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Study ID: CRSSTD: 15593123 |
Nagy 1996.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Reference: Nagy LM, Southwick SM, Charney DS. Placebo‐controlled trial of fluoxetine in PTSD. In: International Society for Traumatic Stress Studies 12th Annual Meeting, San Francisco, CA, November 11. 1996 |
NCT00413296.
| Methods | Study type: Interventional (clinical trial) Actual enrolment: 16 participants Allocation: Randomized Intervention Model: Parallel Assignment Masking: Double (Participant, Investigator) |
| Participants | Inclusion criteria: Quote: "ages 18‐65; primary diagnosis of PTSD based on DSM‐IV criteria and assessed by the MINI International Neuropsychiatric Interview (MINI); Davidson Trauma Scale (DTS) score of at least 40 on screening; ability to provide written informed consent" Exclusion criteria: Quote: "any primary DSM‐IV Axis I disorder other than PTSD; substance abuse during the last 6 months; a clinically unstable medical condition or clinically significant laboratory abnormalities; suicide risk or serious suicide attempt during the last year; concurrent use of psychotropic medications including benzodiazepines, barbiturates, antiepileptic drugs, antidepressants, buspirone, dietary supplements or herbal or homeopathic remedies with psychotropic effects; recent (within the last 3 months) initiation of cognitive behavioral therapy; failure of a previous trial of levetiracetam at 2000 mg/day; pregnancy or lactation; women of childbearing potential who are unwilling to practice an acceptable method of contraception" |
| Interventions | Placebo Comparator: 1 Tablets, no active ingredient, 1 ‐ 6 tablets/day for 12 weeks in the 2nd phase of the trial Drug: Placebo Active Comparator: 2 Levetiracetam Other name: Keppra Drug: Levetriracetam tablets, 500 mg each (1 ‐ 6 tablets/day) for 8 weeks during the open‐label phase and for 12 weeks during the 2nd phase of the study |
| Outcomes | Primary outcome measures: Clinical Global Impressions ‐ Improvement (CGI‐I) (Time frame: 20 weeks) Secondary outcome measures:
|
| Notes | First Posted: December 19, 2006 Last Update Posted: July 21, 2014 Study start date: November 2005 Actual primary completion date: September 2007 Actual study completion date: March 2008 Sponsors and Collaborators: Duke University, UCB Pharma Principal Investigator: Jonathan Davidson, M.D., Duke University |
NCT00672776.
| Methods | Study type: Interventional (clinical trial) Actual enrolment: 40 participants Allocation: Randomized Intervention Model: Parallel Assignment Masking: Quadruple (participant, care provider, investigator, outcomes assessor) |
| Participants | Inclusion criteria: Quote: "Male and female patients meeting Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM IV) criteria for PTSD assessed by the Structured Clinical Interview for DSM IV (SCID); All patients with PTSD will be greater than 18 years of age, and will be required to give informed consent; Patients will be recruited from newspaper advertisements and fliers; All patients must be free of major medical illness on the basis of history and physical examination, lab testing, and electrocardiogram, and must not be actively abusing substances or alcohol; Patients should be free of psychotropic medications for four weeks before the study" Exclusion Criteria: Quote: "Pregnant and breast‐feeding women will not be studied. Female subjects will be required to have a negative pregnancy test before the study. Female subjects of childbearing age will be advised to use barrier contraception for the duration of the study, in addition to other forms of contraception that they may be using; Serious medical or neurological illness or a hypersensitivity to paroxetine; Past or present steroid use; Electroconvulsive therapy (ECT) within the 6 months prior to study entry; Organic mental disorders or epilepsy; History of head trauma; Cerebral infectious disease or dyslexi; History of psychosis, schizophrenia, or eating disorders; Active suicidality or homicidality" |
| Interventions | Experimental: 1, paroxetine Placebo comparator: 2, placebo |
| Outcomes | Primary outcome measures: brain function with traumatic reminders (Time frame: 3 months), PET measurement of brain activation before and after paroxetine or placebo treatment |
| Notes | Study start date: June 2008 Actual primary completion date: September 2007 Actual study completion date: September 2007 Principal investigator: J. Douglas Bremner, M.D. |
NCT01517711.
| Methods | Study type: Interventional (clinical trial) Actual enrolment: 40 participants Allocation: randomized Intervention model: Parallel assignment Masking: quadruple (participant, care provider, investigator, outcomes assessor) |
| Participants | Inclusion criteria: Quote: "Men and women, military veterans and non‐veterans, aged 21‐55 years; Active PTSD as determined by diagnostic evaluation and standardized interview (Structured Clinical Interview for the DSM (SCID)); Literacy and ability to give informed consent; In women of child‐conceiving potential, a negative pregnancy test and use of an approved birth control method; Glasgow Coma Scale (GCS) score of 15, Extension of GCS with 7‐point Amnesia Scale score of 6 (amnesia for traumatic event of 30 min or fewer) or 7 (no amnesia for impact of head); Clinically judged to be at low risk for adverse sequelae from taking tramadol; Concomitant medications must be approved by the PI" Exclusion criteria: Quote: "Pregnant or nursing women; Homeless persons; Suicidal or homicidal ideation with plans or intent". History of opioid dependence or abuse; Psychosis or history thereof, substance dependence or abuse (other than tobacco dependence; lifetime opioid abuse is exclusionary) within the past 60 days, anorexia nervosa, antisocial personality disorder, or other psychiatric disorder judged by the investigator to be more clinically significant than PTSD; Serious or unstable illness, endocrinopathy, or metabolic instability, including renal insufficiency, liver disease, hydrocephalus, history of stroke, history of seizures, history of brain tumour; Use of non‐study medications except those approved by the PI; Newly started in psychotherapy (< 3months); History of hypersensitivity, allergy, or other significant adverse effects from tramadol" |
| Interventions | Drug: Tramadol, Tramadol hydrochloride Extended Release (ER) will be supplied as tablets of Ultram® ER 100 mg. Tramadol ER (100 ‐ 300 mg) or matching placebo will be self‐administered by oral route every morning (with or without food) for 6 weeks. Participants will be instructed to take it the same way (either with food or without food) each time they take their dose. Each participant will be provided with 1 week supply of Tramadol ER or matching placebo on visits 2 (week 0) and 3 (week 1) and 2 weeks supply on visits 4 (week 2) and 5 (week 4) Other name: Ultram® ER Drug: Placebo, Matching placebo will be self‐administered by oral route every morning (with or without food) for 6 weeks. Participants will be instructed to take it the same way (either with food or without food) each time they take their dose. Each participant will be provided with 1 week supply of Tramadol ER or matching placebo on visits 2 (week 0) and 3 (week 1) and 2 weeks supply on visits 4 (week 2) and 5 (week 4) |
| Outcomes | Primary outcome measures: Efficacy as measured by a reduction in PTSD symptoms. (Time frame: Baseline and weeks 1, 2, 4, and 6) Efficacy will be determined by change in PTSD symptoms as measured by the Clinician‐Administered PTSD Scale (CAPS) and Clinicians Global Impressions scale. Secondary outcome measures:
|
| Notes | Study start date: September 2011 Actual primary completion date: September 2014 Actual study completion date: July 2015 Sponsors and collaborators: INTRuST, Post‐Traumatic Stress Disorder ‐ Traumatic Brain Injury Clinical Consortium; U.S. Army Medical Research and Materiel Command Principal Investigator: Thomas Geracioti, MD., University of Cincinnati |
NCT02637895.
| Methods | Vortioxetine for posttraumatic stress disorder Study type: Interventional Study phase: Phase 4 Allocation: Randomised Intervention model: Parallel assignment Masking: quadruple (participant, care provider, investigator, outcomes assessor) Primary purpose: Treatment |
| Participants | Inclusion: Men and women between the ages of 18 and 65; Fulfill Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM‐5) criteria for primary diagnosis of PTSD; Able to give consent; Willingness to sign the treatment contract; a negative urine toxicology; for women of reproductive age, use of an effective birth control method* for the duration of the study or abstinence; Duration of illness of PTSD for at least 3 months; An initial score at screening, and visit 3 (randomisation) of ≥ 50 on the CAPS for PTSD Studies Exclusion: Lifetime or current diagnosis of schizophrenia or other psychotic disorder, dementia, bipolar disorder; person is currently participating in another clinical trial in which s/he is or will be exposed to an investigational or non‐investigational drug or device, or has done so within the preceding month; current evidence or history of significant unstable medical illness or organic brain impairment, including stroke, Central Nervous System (CNS) tumour, demyelinating disease, cardiac, pulmonary, gastrointestinal, renal or hepatic impairment that would likely interfere with the action, absorption, distribution, metabolism, or excretion of Vortioxetine. History of moderate or more severe Traumatic Brain Injury (TBHI) will also be exclusionary; People who in the investigator's judgment pose a current suicidal or homicidal risk; DSM‐5 substance abuse or dependence within the past 90 days. Person has a positive urine toxicology test for illegal substances; diagnosis of anorexia nervosa, bulimia, or Obsessive Compulsive Disorder (OCD) in the past year; person has a documented history of hepato‐biliary disease including a history of, or positive laboratory results for hepatitis (hepatitis B surface antigen and/or hepatitis C antibody), and clinically significant hepatic enzyme elevation, including any one of the following enzymes greater than 3 times the upper limit of normal (ULN) value (ALT, AST, ALP), or total or direct bil > 1.5 x ULN, unless consistent with presumed or diagnosed Gilbert's disease; person has taken systemic corticosteroids within 2 weeks of the randomisation visit; treatment with any other psychoactive medication within 2 weeks of visit 1, including all antidepressants, psychoactive herbal or nutritional treatments (St Johns Wort, SAM‐e), lithium, other mood stabilisers, oral antipsychotics, depot antipsychotics within 12 weeks, beta blockers, thioridazine, pimozide, opiates, anxiolytics, and sedatives (with the exception of zolpidem, eszopiclone, and zaleplon). Also any treatment with any medication that the PI judges not acceptable for this study; Pregnancy or lactation*; people who, in the opinion of the investigator, would be noncompliant with the visit schedule or study procedures (e.g. illiteracy, planned vacations, or planned hospitalisations during the study); any laboratory abnormality that in the investigator's judgement is considered to be clinically significant; People who are receiving exposure‐based psychotherapy that targets PTSD symptoms; Current or planned litigation or other actions related to secondary gain regarding the traumatic event; person has clinical evidence of, or ElectroCardiogram (ECG) results indicating any of the following at either screen or randomisation visit unless repeat ECG shows that the parameter had returned to within normal range by the randomisation visit: QTc > 450 msec for men, or > 475 msec for women;any cardiac condition or ECG evidence that the investigator feels may pose a potential safety concern |
| Interventions | Placebo comparator: Placebo; placebo pill once daily for 12 weeks of active treatment Intervention: drug: placebo Active comparator: Vortioxetine Vortioxetine pill 10 mg once daily up to 4 weeks followed by 20 mg once daily if tolerated for the rest of the study. Patients unable to tolerate the 20 mg/day dose may be reduced to 10 mg/day between weeks 4 and 8. The dose of study medication should remain stable for weeks 8 ‐ 12 Intervention: Drug: Vortioxetine |
| Outcomes | Primary outcome measures: An improved PTSD scale score (Time frame: from baseline to week 12). Mean changes in the Clinician‐Administered PTSD Scale (CAPS) Secondary outcome measures:
|
| Notes | Starting date: December 2016 Contact information: Principal investigator: Philip Harvey, PhD., University of Miami Responsible parties: University of Miami, Takeda, Emory University Estimated primary completion date: May 2019 Estimated study completion date: July 2019 |
NCT02709018.
| Methods | A controlled trial of Losartan in posttraumatic stress disorder (LOSe‐PTSD) Study type: Interventional Study phase: Not applicable Allocation: Randomised Intervention model: Parallel assignment Masking: quadruple (participant, care provider, investigator, outcomes assessor) Primary purpose: Treatment |
| Participants | Inclusion criteria; Person must be a man or woman between 18 and 70 years of age, inclusive; must have a primary DSM‐5 diagnosis of Posttraumatic Stress Disorder; must have a Clinical Administered PTSD Scale for PTSD (CAPS‐5) ≥ 25 persistent at screening for at least 3 months duration; person must be willing and able to adhere to the prohibitions and restrictions specified in this protocol; must be willing and able to fill out self‐administered questionnaires; Subject must be able to be compliant with self‐administration of medication; Subject must be able to swallow the study medication whole with aid of water; must sign an informed consent document indicating that they understand the purpose of and procedures required for the study and are willing to participate in the study Exclusion criteria: Persons who have current or imminent risk of suicide as assessed by the Columbia‐Suicide Severity Rating Scale (C‐SSRS) at each study visit; person with active psychosis; has a history of moderate or severe drug or alcohol use disorder according to DSM‐5 criteria within 3 months before screening; has a history of allergy to losartan or other angiotensin receptor blockers (ARBs); has a medical illness likely to result in imminent hospitalisation or for which treatments are contraindicated based on lab results, medical history and physical exam; has serious cognitive impairment felt likely to interfere with the ability to participate meaningfully in the study. Participants with mild to moderate traumatic brain injury (TBI) will not be excluded from the study. Only those with evidence of significant cognitive impairment at Screening (as evidenced by confusion, inability to track discussion or answer questions, or other clear and significant indicators of cognitive impairment) will be excluded; Concurrent ACE Inhibitors or Angiotensin Receptor Blockers or Prazosin; patients on other antihypertensives may be enrolled if, after consultation with their prescribing physician, it is determined that the addition of losartan would not be contraindicated; concurrent antidepressants or antipsychotics. People who have elected, in consultation with their healthcare provider, to discontinue any antidepressants or antipsychotics, must be off the medications for a minimum of 2 weeks prior to study randomisation. Stable bedtime doses of sleep agents (e.g., trazodone ≤ 200 mg; eszopiclone; zolpidem; lorazepam) will be allowed as long as the dose has been stable for at least 2 weeks prior to study randomisation. Benzodiazepines taken for other than sleep are not permitted; Person is a woman who is pregnant, or breast‐feeding, or planning to become pregnant; is unable to comply with the study‐specific requirements; people with abnormal liver, renal or EKG findings as determined by physician; person exhibits clinically‐significant hypertension as determined by medical evaluation and/or BP > 190/100; systolic blood pressure (SBP) < 90 mmHg; liver function tests (LFT's) > 2 times the upper limit of normal; People with chronic kidney disease 4, as determined by history, baseline labs (including eGFR < 45 ml/minute) and evaluation by a physician will be excluded |
| Interventions | Experimental: losartan Losartan flexibly‐dosed from 25 ‐ 100 mg per day over 10 weeks Intervention: Drug: losartan Placebo comparator: placebo Placebo flexibly dosed from 25 ‐ 100 mg per day over 10 weeks Intervention: drug: Placebo |
| Outcomes | Primary outcome measures: The primary outcome for this study is mean change in Clinician‐Administered PTSD Scale for DSM‐5 (CAPS‐5) over the treatment period of 10 weeks between the losartan arm and the placebo arm. (Time frame: 10 weeks ). Clinician‐Administered PTSD Scale for DSM‐5, also known as CAPS 5, is the gold standard in PTSD assessment. The CAPS‐5 is a 30‐item structured interview that can be used to make current (past month) diagnosis of PTSD, make a lifetime diagnosis of PTSD and assess PTSD symptoms over the past week Secondary outcome measures: Change in CAPS‐5 associated with CC homozygosity for rs4311 SNP in the angiotensin converting enzyme gene (ACE) compared to T carriers, among participants randomised to losartan. (Time frame: 10 weeks) |
| Notes | Starting date: July 2016 Contact information: Investigators: Murray B Stein, MD, MPH., University of California, San Diego; Kerry J Ressler, MD., McLean Hospital and Harvard Medical School Responsible parties: University of California, San Diego, Mclean Hospital, Massachusetts General Hospital, Henry M. Jackson Foundation, Walter Reed National Military Medical Center, Atlanta Research and Education Foundation New York University School of Medicine, George Washington University Estimated primary completion date: September 2019 Estimated study completion date: September 2019 |
NCT02933606.
| Methods | Phase II Study of BNC210 in PTSD (RESTORE) Study type: Interventional Study phase: Phase 2 Allocation: Randomised Intervention model: Parallel assignment Masking description: Double‐blind Primary purpose: Treatment |
| Participants | Key inclusion criteria: Signed and dated informed consent; Men or women between 18 and 70 years of age, inclusive; Diagnosed with current PTSD as defined by the CAPS‐5 for DSM‐5; Currently not using any psychiatric medications except for: no more than 1 selective serotonin reuptake inhibitor (SSRI) (fluvoxamine is excluded) or serotonin noradrenaline reuptake inhibitor (SNRI) within the licensed prescribing dose range. Persons must have been on a stable dose for at least 3 months prior and through screening, with the intent to remain on the same dose through to week 16. As needed (PRN) use of benzodiazepines (BZD) at a frequency not exceeding 2 days per week in the 3 months prior to screening. The total dose must not exceed 30 mg/day in diazepam equivalents; Persons not currently receiving psychotherapy except long‐term supportive counselling or those that have received intensive regular psychotherapy for a minimum of 3 months prior to screening; women of childbearing potential must have a negative serum pregnancy. Women not of childbearing potential must be postmenopausal. Sterilised men must be at least 1 year post‐vasectomy to be considered of non‐child bearing potential. Men and women of childbearing potential must agree to use 2 effective methods of contraception Key exclusion criteria: Current and ongoing exposure to the trauma that caused the PTSD; Failed more than 3 trials of antidepressant medication(s) prescribed for the treatment of PTSD. Each trial must have lasted at least 6 weeks to be considered a failed attempt. A trial that was terminated due to intolerability or side effects does not constitute a failed attempt; The use of psychiatric medications within 2 weeks of screening except for SSRIs, SNRIs or limited PRN BZD use as per inclusion criterion 4. Restricted psychiatric medications include (but are not limited to) antidepressants not allowed by inclusion criterion 4, anti‐anxiety drugs (except limited BZD use per inclusion criterion 4), mood stabilisers, stimulants, antipsychotics, hypnotics and acetylcholinesterase inhibitors; history of significant traumatic brain injury; depression as measured by Montgomery‐Asberg depression scale (MADRS) rating > 23; bipolar and psychotic disorders as identified at screening using the MINI International Neuropsychiatry Interview (V7.0) (M.I.N.I); A score ≥ 7 on the McLean Screening Instrument for Borderline Personality Disorder (MSI‐BPD) at screening; history of seizure disorders, uncontrolled sleep apnoea or severe neurologic disease; increased risk of suicide, defined as: any previous suicide attempt disclosed by the participant at screening using the Columbia Suicide Severity Rating Scale (C‐SSRS). Any suicidal ideation with intent (yes to item 4 and/or 5) or suicidal behaviour in the past year, as captured at screening using the C‐SSRS. A score > 4 on item 10 of the MADRS at Screening; the use of alprazolam or flunitrazepam within 3 months of screening; any clinically significant abnormalities in laboratory test results, vitals signs, or ECG at screening; positive result for human immunodeficiency virus (HIV), hepatitis B surface antigen (HBsAg), or hepatitis C (HCV) at screening; Any moderate‐to‐severe substance use disorder (any type) in the 12 months prior to screening as identified by the DSM‐5 using the M.I.N.I (V7.0); Current Australian serving Defense personnel or any member of the US military currently serving on active duty; people involved with ongoing insurance or workplace claims that in the opinion of the Investigator are likely to have an impact on the mental health, presentation or capacity of the patient to engage in the study |
| Interventions | Experimental: BNC210 600 mg twice a day; Suspension administered orally for 12 weeks Intervention: Drug: BNC210 Experimental: BNC210 300 mg twice a day; .Suspension administered orally for 12 weeks Intervention: Drug: BNC210 Experimental: BNC210 150 mg twice a day; Suspension administered orally for 12 weeks Intervention: Drug: BNC210. Placebo comparator: Placebo twice a day; Suspension administered orally for 12 weeks Intervention: Drug: Placebo |
| Outcomes | Primary outcome measures: Clinician‐Administered PTSD Scale for the Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM‐5) (CAPS‐5). (Time frame: 12 weeks). Investigator‐rated PTSD symptom severity Secondary outcome measures:
|
| Notes | Starting date: 30 June 2016 Contact information: Responsible party: Bionomics Limited Actual primary completion date: 05 July 2018 Actual study completion date: 25 July 2018 |
Nefazodone 1, BMS.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Study ID: CRSSTD: 15593132 |
Nefazodone 2, BMS.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | “Study ID: CRSSTD: 15593133” |
Olanzapine, Davidson.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | “Study ID: CRSSTD: 15593134” |
Paroxetine 2 ‐ SB.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | “Study ID: CRSSTD: 15593135” |
Paroxetine 3 ‐ SB.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | “Study ID: CRSSTD: 15593136” |
Sertraline 2, Pfizer.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | “Study ID: CRSSTD: 15593137” |
Sertraline 3, Pfizer.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | “Study ID: CRSSTD: 15593138” |
Sertraline 4, Pfizer.
| Methods | There was insufficient evidence from the previous update to complete the table |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | “Study ID: CRSSTD: 15593139” |
CAPS‐D: Clinician‐Administered PTSD Scale, Criterion D; ID: identifier (i.e. CRSSTD, CRSREF); Inc.: inclusive; MD: medical doctor; mg: milligram; PI.: investigator; PTSD: posttraumatic stress disorder; UCB: Union Chimique Belge; wks: weeks
Characteristics of ongoing studies [ordered by study ID]
NCT01221792.
| Study name | Carvedilol versus placebo for treatment in post traumatic stress disorder (PTSD) |
| Methods | Study type: Interventional Study phase: Phase 2 Allocation: Randomized Intervention model: Parallel assignment Masking: quadruple (participant, care provider, investigator, outcomes assessor) Primary purpose: Treatment |
| Participants | Inclusion criteria: Diagnosis of Post Traumatic Stress Disorder according to DSM‐IV; must be able to speak, read and understand the English language and be able to provide written informed consent Exclusion criteria: Current, unstable and significant medical condition/illness; bronchial asthma or related bronchospastic condition; AV block; Sick Sinus Syndrome; Bradycardia; Peripheral hear disease; Unstable thyroid disorder; History of seizure disorder; Women who are pregnant, lactating or planning to become pregnant; bipolar; schizophrenia; dementia; intolerance or hypersensitivity to alpha or beta blockers |
| Interventions | Drug: Carvedilol: oral, twice‐daily dosing using 3.125 mg tablets.1 week titration (6.25 mg/day) prior to a 3‐week flexible dosing option ranging from 6.25 mg/day to 15.625 mg/day followed by a 1 week taper (6.25 mg/day) Other name: Coreg® Drug: Placebo Non‐active comparator |
| Outcomes | Primary outcome mMeasures: Davidson Trauma Scale (DTS) ( Time frame: 5 weeks). The DTS is a 17‐item self report measure to assess the 17 DMS‐IV symptoms of PTSD. Respondents are asked to identify the trauma that is most disturbing to them and to rate, in the past week, how much trouble they have had with each symptom. The DTS can be used to make a preliminary determination about whether the symptoms meet DSM criteria for PTSD, or scores can be calculated for each of the 3 PTSD symptom clusters. The DTS will be assessed at each study visit (visit 1, 2, 3, 4, 5, 6 and 7). The primary efficacy outcome will be change from baseline (visit 2) to week 5 (visit 7) Secondary outcome measures:
|
| Starting date | October 2010 |
| Contact information | Study Director: Arifulla Khan, MD., Columbia Northwest Pharmaceuticals, LLC |
| Notes | Responsible party: Arifulla Khan, MD, Columbia Northwest Pharmaceuticals Estimated primary completion date: June 2011 Estimated study completion date: August 2011 |
ACE: angiotensin converting enzyme; ALT, AST, ALP: higher than normal; AV: Atrioventricular block; bil: total bilirubin; BZD: benzodiazepines; CAPS‐5: Clinically‐Administered PTSD Scale (CAPS) for DSM‐5; C‐SSRS: Columbia Suicide Severity Rating Scale; DSM‐5: Diagnostic and Statistical Manual of Mental Disorders, 5th Edition; ECG/EKG: ElectroCardiogram; eGFR: Estimated Glomerular Filtration Rate; MADRS: Montgomery‐Asberg Depression Rating Scale; MD: medical doctor; mg: milligrams; M.I.N.I: MINI International Neuropsychiatry Interview; MPH: Master of Public Health; msec: minutes per second; PI: principal investigator; PRN: (pro re nata) as needed; PTSD: posttraumatic stress disorder; QTc: Torsades de Pointes; SAM‐e: s‐adenosyl methionine; SNP: single nucleotide polymorphisms; SNRIs: serotonin and norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; ULN: upper limit of normal;
Differences between protocol and review
In recognition of the possibility of different effects for different types of medication, all of the comparisons were stratified by medication class. This review classified medications based on their putative mechanisms of action (taken from CCMD antidepressant classification map) (Davies 2015), and they do not necessarily map onto the drug classification schemes used in other reviews.
We added an additional acceptability outcome: 'dropouts due to any cause'.
To prevent unit‐of‐analysis issues for trials comparing multiple dosages of the same agent to placebo, we averaged the mean and standard deviation of the outcome of interest across dosage groups. We included outcome data from multiple treatment arms in the same comparison if the agents tested were from different medication classes. We turned off the subtotals of the outcome if the placebo group appeared twice in the analysis to accommodate the second medication. In the case of trials testing multiple agents from the same classes, and in calculating the total effect across all medication classes, we restricted data from multi‐arm RCTs to the agent that was least represented in the database.
Heterogeneity of treatment response and symptom severity were not assessed by means of Deeks’s stratified test of heterogeneity (Deeks 2001), as planned in the original protocol, nor was this assessed visually from the forest plot of relative risk, given the small number of studies. The number needed to treat for an additional beneficial outcome (NNTB) was also not calculated, due to a lack of data provided by studies.
This review incorporates the GRADE approach, with summary of findings tables.
Contributions of authors
Taryn Williams, Dan Stein and Jonathan Ipser co‐ordinated the work on the update of this review. Taryn Williams and Jonathan Ipser compiled the updated version of the review, including rechecking all studies for eligibility and risk of bias, completing all GRADE tables, analysing the data, and updating the abstract, results, discussion and authors' conclusion sections of the review. Nicole Phillips also assisted with the extraction of data and the assessment of risk of bias and provided feedback on the final draft.
Sources of support
Internal sources
No sources of support provided
External sources
SA MRC Unit on Risk and Resilience in Mental Disorders, Cape Town, South Africa
Declarations of interest
Taryn Williams: declares no conflicts of interest outside of her funding from the Medical Research Council of South Africa, SAMRC Unit on Risk & Resilience in Mental Disorders.
Nicole Phillips: receives funding from the Neuroscience Institute’s Fellowship Programme 2021.
Dan J Stein: has received research grants and/or consultancy honoraria from Discovery Vitality, Johnson & Johnson, Lundbeck, Sanofi, Servier, Takeda, and Vistagen.
Jonathan Ipser: none known.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Ahmadpanah 2014 {published data only}
- Ahmadpanah M, Sabzeiee P, Mohammad Hosseini S, Torabian S, Haghighi M, Jahangard L, et al.Comparing the effect of prazosin and hydroxyzine on sleep quality in patients suffering from posttraumatic stress disorder. Neuropsychobiology 2014;69(4):235–42. [10.1159/000362243] [DOI] [PubMed] [Google Scholar]
Baker 1995 {published data only}
- Baker DG, Diamond BI, Gillette G, Hamner M, Katzelnick D, Keller T, et al.A double-blind randomized placebo-controlled multi-center study of brofaromine in the treatment of post-traumatic stress disorder. Psychopharmacology 1995;122(4):386-9. [DOI] [PubMed] [Google Scholar]
- Connor KM, Hidalgo RB, Crockett B, Malik M, Katz RJ, Davidson JR.Predictors of treatment response in patients with posttraumatic stress disorder. Progress in Neuro-Psychopharmacology & Biological Psychiatry 2001;25(2):337-45. [DOI] [PubMed] [Google Scholar]
Becker 2007 {published data only}
- Becker ME, Hertzberg MA, Moore SD, Dennis MF, Bukenya DS, Beckham JC.A placebo-controlled trial of bupropion SR in the treatment of chronic posttraumatic stress disorder. Journal of Clinical Psychopharmacology 2007;27(2):193-7. [DOI: 10.1097/JCP.0b013e318032eaed] [DOI] [PubMed] [Google Scholar]
Brady 2000 {published data only}
- Brady K, Pearlstein T, Asnis GM, Baker D, Rothbaum B, Sikes CR, et al.Efficacy and safety of sertraline treatment of posttraumatic stress disorder. Archives of General Psychiatry 2000;283(14):1837-44. [DOI] [PubMed] [Google Scholar]
- Davidson J, Pearlstein T, Londborg P, Brady KT, Rothbaum B, Bell J, et al.Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: results of a 28-week double-blind, placebo-controlled study. American Journal of Psychiatry 2001;158(12):1974-81. [DOI] [PubMed] [Google Scholar]
- Davidson JR, Landerman LR, Farfel GM, Clary CM.Characterizing the effects of sertraline in post-traumatic stress disorder. Psychological Medicine 2002;32(4):661-70. [DOI] [PubMed] [Google Scholar]
- Gaffney M.Factor analysis of treatment response in posttraumatic stress disorder. Journal of Traumatic Stress 2003;16(1):77-80. [DOI] [PubMed] [Google Scholar]
- Rapaport MH, Endicott J, Clary CM.Posttraumatic stress disorder and quality of life: results across 64 weeks of sertraline treatment. Journal of Clinical Psychiatry 2002;63(1):59-65. [DOI] [PubMed] [Google Scholar]
- Rapaport MH, Farfel G, Clary CM.Quality of life improvement in PTSD with sertraline treatment: results of a multicenter, placebo-controlled trial. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):294. [Google Scholar]
Braun 1990 {published data only}
- Braun P, Greenberg D, Dasberg H, Lerer B.Core symptoms of posttraumatic stress disorder unimproved by alprazolam treatment. Journal of Clinical Psychiatry 1990;51(6):236-8. [PubMed] [Google Scholar]
Butterfield 2001 {published data only}
- Butterfield MI, Becker ME, Connor KM, Sutherland S, Churchill LE, Davidson JR.Olanzapine in the treatment of post-traumatic stress disorder: a pilot study. International Clinical Psychopharmacology 2001;16(4):197-203. [DOI] [PubMed] [Google Scholar]
Carey 2012 {published data only}
- Carey P, Suliman S, Ganesan K, Seedat S, Stein DJ.Olanzapine monotherapy in posttraumatic stress disorder: efficacy in a randomized, double-blind, placebo-controlled study. Human Psychopharmacology Clinical and Experimental 2012;27:386–91. [DOI: 10.1002/hup.2238] [DOI] [PubMed] [Google Scholar]
Chung 2004 {published data only}
- Chung MY, Min KH, Jun YJ, Kim SS, Kim WC, Jun EM.Efficacy and tolerability of mirtazapine and sertraline in Korean veterans with posttraumatic stress disorder: a randomized open label trial. Human Psychopharmacology 2004;19(7):489–94. [DOI: 10.1002/hup.615] [DOI] [PubMed] [Google Scholar]
Connor 1999a {published data only}
- Barnett SD, Tharwani HM, Hertzberg MA, Sutherland SM, Connor KM, Davidson JR.Tolerability of fluoxetine in posttraumatic stress disorder. Progress in Neuro-Psychopharmacology and Biological Psychiatry 2002;26(2):363-7. [DOI] [PubMed] [Google Scholar]
- Connor KM, Sutherland SM, Tupler LA, Malik ML, Davidson JR.Fluoxetine in post-traumatic stress disorder: randomized, double-blind study. British Journal of Psychiatry 1999;175:17-22. [DOI] [PubMed] [Google Scholar]
- Malik ML, Connor KM, Sutherland SM, Smith RD, Davison RM, Davidson JR.Quality-of-life and posttraumatic stress disorder: a pilot study assessing changes in SF-36 scores before and after treatment in a placebo- controlled trial of fluoxetine. Journal of Traumatic Stress 1999;12(2):387-93. [DOI] [PubMed] [Google Scholar]
Connor 2006 {published data only}
- Connor KM, Davidson JR, Weisler RH, Zhang W, Abraham K.Tiagabine for posttraumatic stress disorder: effects of open-label and double-blind discontinuation treatment. Psychopharmacology 2006;184:21–5. [DOI: 10.1007/s00213-005-0265-3] [DOI] [PubMed] [Google Scholar]
Davidson 1990 {published data only}
- Davidson JR, Kudler H, Smith R, Mahorney SL, Lipper S, Hammett E, et al.Treatment of posttraumatic stress disorder with amitriptyline and placebo. Archives of General Psychiatry 1990;47(3):259-66. [DOI] [PubMed] [Google Scholar]
- Davidson JR, Kudler HS, Saunders WB, Erickson L, Smith RD, Stein RM, et al.Predicting response to amitriptyline in posttraumatic stress disorder. American Journal of Psychiatry 1993;150(7):1024-9. [DOI] [PubMed] [Google Scholar]
Davidson 2001a {published data only}
- Davidson J, Londborg P, Pearlstein T, Rothbaum B, Brady K, Farfel G.Double-blind, randomized, 28-week continuation study of sertraline and placebo in posttraumatic stress disorder. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):292. [Google Scholar]
- Davidson J, Pearlstein T, Londborg P, Brady KT, Rothbaum B, Bell J, et al.Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: results of a 28-week double-blind, placebo controlled study. American Journal of Psychiatry 2001;158(12):1974–81. [DOI] [PubMed] [Google Scholar]
- Davidson JR, Landerman LR, Farfel GM, Clary CM.Characterizing the effects of sertraline in post-traumatic stress disorder. Psychological Medicine 2002;32(4):661-70. [DOI] [PubMed] [Google Scholar]
- Davidson JR, Rothbaum BO, Van der Kolk BA, Sikes CR, Farfel GM.Multicenter, double-blind comparison of sertraline and placebo in the treatment of posttraumatic stress disorder. Archives of General Psychiatry 2001;58(5):485-92. [DOI] [PubMed] [Google Scholar]
- Rapaport MH, Endicott J, Clary CM.Posttraumatic stress disorder and quality of life: results across 64 weeks of sertraline treatment. Journal of Clinical Psychiatry 2002;63(1):59–65. [DOI] [PubMed] [Google Scholar]
Davidson 2001b {published data only}
- Davidson J, Londborg P, Pearlstein T, Rothbaum B, Brady K, Farfel G.Double-blind, randomized, 28-week continuation study of sertraline and placebo in posttraumatic stress disorder. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):292. [Google Scholar]
- Davidson J, Pearlstein T, Londborg P, Brady KT, Rothbaum B, Bell J, et al.Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: results of a 28-week double-blind, placebo-controlled study. American Journal of Psychiatry 2001;158(12):1974-81. [DOI] [PubMed] [Google Scholar]
- Rapaport MH, Endicott J, Clary CM.Posttraumatic stress disorder and quality of life: results across 64 weeks of sertraline treatment. Journal of Clinical Psychiatry 2002;63(1):59-65. [DOI] [PubMed] [Google Scholar]
Davidson 2003 {published data only}
- Davidson JR, Weisler RH, Butterfield MI, Casat CD, Connor KM, Barnett S, et al.Mirtazapine vs. placebo in posttraumatic stress disorder: a pilot trial. Biological Psychiatry 2003;53(2):188-91. [DOI] [PubMed] [Google Scholar]
Davidson 2005 {published data only}
- Davidson JT, Connor KM, Hertzberg MA, Weisler RH, Wilson WH, Payne VM.Maintenance therapy with fluoxetine in posttraumatic stress disorder: a placebo-controlled discontinuation study. Journal of Clinical Psychopharmacology 2005;25(2):166–9. [DOI: 10.1097/01.jcp.0000155817.21467.6c] [DOI] [PubMed] [Google Scholar]
Davidson 2006a {published data only}
- Davidson J, Baldwin D, Stein DJ, Kuper E, Benattia I, Ahmed S, et al.Treatment of posttraumatic stress disorder with venlafaxine extended release: a 6-month randomized controlled trial. Archives of General Psychiatry 2006;63(10):1158-65. [DOI] [PubMed] [Google Scholar]
Davidson 2006b {published data only}
- Davidson J, Rothbaum BO, Tucker P, Asnis G, Benattia I, Musgnung JJ.Venlafaxine extended release in posttraumatic stress disorder: a sertraline- and placebo-controlled study. Journal of Clinical Psychopharmacology 2006;26(3):259-67. [DOI] [PubMed] [Google Scholar]
Davidson 2007 {published data only}
- Davidson JR, Brady K, Mellman TA, Stein MB, Pollack MH.The efficacy and tolerability of tiagabine in adult patients with post-traumatic stress disorder. Journal of Clinical Psychopharmacology 2007;27(1):85–8. [DOI: 10.1097/JCP.0b013e31802e5115] [DOI] [PubMed] [Google Scholar]
Davis 2004 {published data only}
- Davis LL, Ambrose S, English B, Dafore ME, Farley J, Bartolucci Al, et al.A placebo-controlled study of nefazodone for the treatment of chronic posttraumatic stress disorder. In: 20th Annual Meeting, International Society for Traumatic Stress Studies, December 6 - 9, New Orleans, LA. 2001.
- Davis LL, Jewell ME, Ambrose S, Farley J, English B, Bartolucci A, et al.A placebo-controlled study of nefazodone for the treatment of chronic posttraumatic stress disorder: a preliminary study. Journal of Clinical Psychopharmacology 2004;24(3):291-7. [DOI: 10.1097/01.jcp.0000125685.82219.1a] [DOI] [PubMed] [Google Scholar]
Davis 2008 {published data only}
- Davis LL, Davidson JR, Ward C, Bartolucci A, Bowden CL, Petty F.Divalproex in the treatment of posttraumatic tress disorder: a randomized, double-blind, placebo-controlled trial in a veteran population. Journal of Clinical Psychopharmacology 2008;28(21):84–8. [DOI: 10.1097/jcp.0b013e318160f83b] [DOI] [PubMed] [Google Scholar]
Davis 2020 {published data only}
- Davis LL, Pilkinton P, Lin C, Parker P, Estes S, Bartolucci A.A randomized, placebo-controlled trial of mirtazapine for the treatment of posttraumatic stress disorder in v eterans. Journal of Clinical Psychiatry 2020;81(6):e1-8. [DOI: 10.4088/JCP.20m13267] [DOI] [PubMed] [Google Scholar]
Dowd 2020 {published data only}
- Dowd SM, Zalta AK, Burgess HJ, Adkins EC, Valdespino-Hayden Z, Pollack MH.Double-blind randomized controlled study of the efficacy, safety and tolerability of eszopiclone vs placebo for the treatment of patients with post-traumatic stress disorder and insomnia. World Journal of Psychiatry 2020;10(3):21-8. [DOI: 10.5498/wjp.v10.i3.21] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dunlop 2017 {published data only}
- Dunlop BW, Binder EB, Iosifescu D, Mathew SJ, Neylan TC, Pape JC, et al.Corticotropin-releasing factor receptor 1 antagonism Is ineffective for women with posttraumatic stress disorder. Biological Psychiatry 2017;82(12):866–74. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Feder 2014 {published data only}
- Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, et al.Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder. A randomized clinical trial. JAMA Psychiatry 2014;71(6):681-8. [DOI: 10.1001/jamapsychiatry.2014.62] [DOI] [PubMed] [Google Scholar]
Friedman 2007 {published data only}
- Eli Lilly.Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
- Friedman MJ, Marmar CR, Baker DG, Sikes CR, Farfel GM.Randomised, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. Journal of Clinical Psychiatry 2007;68(5):711-20. [DOI] [PubMed] [Google Scholar]
GSK 29060 627 {published data only}
- GSK 29060 627.A 12-week, double-blind, placebo-controlled, parallel group study to assess the efficacy and tolerability of paroxetine in patients suffering from posttraumatic stress disorder (PTSD). https://www.gsk-studyregister.com/en/trial-details/?id=29060/648.
Hamner 2008 {published data only}
- Hamner MB, Faldowski RA, Robert S, Ulmer HG, Horner MD, Lorberbaum JP.A preliminary controlled trial of divalproex in posttraumatic stress disorder. Annals of Clinical Psychiatry 2008;21(2):89-94. [PubMed] [Google Scholar]
Hertzberg 1999 {published data only}
- Hertzberg MA, Butterfield MI, Feldman ME, Beckham JC, Sutherland SM, Connor KM, et al.A preliminary study of lamotrigine for the treatment of posttraumatic stress disorder. Biological Psychiatry 1999;45(9):1226-9. [DOI] [PubMed] [Google Scholar]
Hertzberg 2000 {published data only}
- Hertzberg MA, Feldman ME, Beckham JC, Kudler HS, Davidson JR.Lack of efficacy for fluoxetine in PTSD: a placebo controlled trial in combat veterans. Annals of Clinical Psychiatry 2000;12(2):101-5. [DOI] [PubMed] [Google Scholar]
Kaplan 1996 {published data only}
- Kaplan Z, Amir M, Swartz M, Levine J.Inositol treatment of posttraumatic stress disorder. Anxiety 1996;2(1):51–2. [DOI: ] [DOI] [PubMed] [Google Scholar]
Katz 1994 {published data only}
- Connor KM, Hidalgo RB, Crockett B, Malik M, Katz RJ, Davidson JR.Predictors of treatment response in patients with posttraumatic stress disorder. Progress in Neuro-Psychopharmacology and Biological Psychiatry 2001;25(2):337–45. [DOI] [PubMed] [Google Scholar]
- Katz RJ, Lott MH, Arbus P, Crocq L, Herlobsen P, Lingjaerde O, et al.Pharmacotherapy of post-traumatic stress disorder with a novel psychotropic. Anxiety 1994;1(4):169–74. [DOI: https://doi.org/10.1002/anxi.3070010404 ] [DOI] [PubMed] [Google Scholar]
Kosten 1991 {published data only}
- Kosten TR, Frank JB, Dan E, McDougle CJ, Giller EL Jr.Pharmacotherapy for posttraumatic stress disorder using phenelzine or imipramine. Journal of Nervous and Mental Disease 1991;179(6):366–70. [DOI] [PubMed] [Google Scholar]
Li 2017 {published data only}
- Li W, Ma Y-B, Yang Q, Li BL, Meng Q-G, Zhang Y.Effect and safety of sertraline for treat posttraumatic stress disorder: a multicenter randomised controlled study. International Journal of Psychiatry in Clinical Practice 2017;21(2):151-5. [DOI: 10.1080/13651501.2017.1291838] [DOI] [PubMed] [Google Scholar]
Marshall 2001 {published data only}
- Food and Drugs Administration (FDA).Paxil (paroxetine) [NDA 20-031/S-029 Paxil (Paroxetine hydrochloride) tablets; GlaxoSmithKline; Post-traumatic Stress Disorder]. www.accessdata.fda.gov/drugsatfda_docs/nda/2001/20-031S029.pdf (accessed 26 November 2021).
- GlaxoSmithKline.A 12 week, double-blind, fixed dose comparison of 20 and 40 mg daily of paroxetine and placebo in Patients suffering from Posttraumatic Stress Disorder (PTSD). GSK - clinical study register [www.gsk-clinicalstudyregister.com]:Study ID: PAR 29060 651 (BRL-02960).
- Marshall RD, Beebe KL, Oldham M, Zaninelli R.Efficacy and safety of paroxetine treatment for chronic PTSD: A fixed-dose, placebo-controlled study. American Journal of Psychiatry 2001;158(12):1982-8. [DOI] [PubMed] [Google Scholar]
Marshall 2007 {published data only}
- Marshall R, Blanco C, Lewis-Fernandez R, Simpson B, Lin SH, Garcia W, et al.Randomized controlled trial of paroxetine in adults with chronic PTSD. In: 18th Annual Meeting, International Society for Traumatic Stress Studies, November 7 - 10, Baltimore, MD. 2002.
- Marshall RD, Lewis-Fernandez R, Blanco C, Simpson HB, Lin S, Garcia W, et al.A controlled trial of paroxetine for chronic PTSD, dissociation and interpersonal problems in mostly minority adults. Draft manuscript 2004. [DOI] [PubMed]
- Marshall RD, Lewis-Fernandez R, Blanco C, Simpson HB, Lin S-H, Vermes D, et al.A controlled trial of paroxetine for chronic PTSD, dissociation, and interpersonal problems in mostly minority adults. Depression and Anxiety 2007;24(2):77–84. [DOI: 10.1002/da] [DOI] [PubMed] [Google Scholar]
Martenyi 2002a {published data only}
- Martenyi F, Brown EB, Zhang H, Prakash A, Koke SC.Fluoxetine versus placebo in posttraumatic stress disorder. Journal of Clinical Psychiatry 2002;63(3):199-206. [DOI] [PubMed] [Google Scholar]
Martenyi 2002b {published data only}
- Martenyi F, Brown EB, Zhang H, Koke SC, Prakash A.Fluoxetine v. placebo in prevention of relapse in post-traumatic stress disorder. British Journal of Psychiatry 2002;181:315-20. [DOI] [PubMed] [Google Scholar]
Martenyi 2007 {published data only}
- Martenyi F, Brown EB, Caldwell CB.Failed efficacy of fluoxetine in the treatment of posttraumatic stress disorder: results of a fixed-dose, placebo-controlled study. Journal of Clinical Psychopharmacology 2007;27(2):166–70. [DOI: 10.1097/JCP.0b013e31803308ce] [DOI] [PubMed] [Google Scholar]
Mathew 2011 {published data only}
- Mathew SJ, Vythilingam M, Murrougha JW, Zarate CA, Federa A, Luckenbaugh DA, et al.A selective neurokinin-1 receptor antagonist in chronic PTSD: a randomized, double-blind, placebo-controlled, proof-of-concept trial. European Neuropsychopharmacology 2011;21(3):221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
McCall 2018 {published data only}
- McCall WV, Youssef N, Branch F, Nolla T, McCloud L, Moraczewski J, et al.A randomized controlled trial (RCT) of prazosin versus placebo for suicidal posttraumatic stress disorder (PTSD) patients with nightmares-a pilot study. Sleep 2018;41(Supplement 1):A351-2. [DOI: 10.1097/JCP.0000000000000968] [DOI] [PubMed] [Google Scholar]
McRae 2004 {published data only}
- McRae AL, Brady KT, Mellman TA, Sonn SC, Killeen TK, Timmerman MA, et al.Comparison of nefazodone and sertraline for the treatment of posttraumatic stress disorder. Depression and Anxiety 2004;19(3):190–6. [DOI: 10.1002/da.20008] [DOI] [PubMed] [Google Scholar]
NCT00659230 {published data only}
- NCT00659230.Nepicastat for Posttraumatic Stress Disorder (PTSD) in OIF/OEF Veterans (Nepicastat). clinicaltrials.gov/ct2/show/NCT00659230 (first received 06 May 2008).
NCT01000493 {published data only}
- NCT01000493.Orvepitant (GW823296) in adult Post Traumatic Stress Disorder. clinicaltrials.gov/ct2/show/NCT01000493 (first received 23 October 2009).
NCT01681849 {published data only}
- NCT01681849.Neural circuits in women with abuse and Posttraumatic Stress Disorder. clinicaltrials.gov/ct2/show/NCT01681849 (first received 10 September 2012).
Padala 2006 {published data only}
- Padala PR, Madison J, Monnahan M, Marcil W, Price P, Ramaswamy S, et al.Risperidone monotherapy for post-traumatic stress disorder related to sexual assault and domestic abuse in women. International Clinical Psychopharmacology 2006;21(5):275–80. [DOI] [PubMed] [Google Scholar]
Panahi 2011 {published data only}
- Panahi Y, Moghaddam BR, Sahebkar A, Nazari MA, Beiraghdar F, Karami G, et al.A randomized, double-blind, placebo-controlled trial on the efficacy and tolerability of sertraline in Iranian veterans with post-traumatic stress disorder. Psychological Medicine 2011;41(10):2159–66. [DOI: 10.1017/S0033291711000201] [DOI] [PubMed] [Google Scholar]
Pfizer588 {unpublished data only}
- Food and Drug Administration (FDA).Review and evaluation of clinical data [Pfizer NDA application: Sertraline (Zoloft) for PTSD]. www.accessdata.fda.gov/drugsatfda_docs/nda/99/19-839S026_Zoloft_Clinr_P1.pdf 1999;(accessed 28 November 2021).
- Pfizer.Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
Pfizer589 {unpublished data only}
- Food and Drug Adminstration (FDA).Review and evaluation of clinical data [Pfizer NDA application: Sertraline (Zoloft) for PTSD]. www.accessdata.fda.gov/drugsatfda_docs/nda/99/19-839S026_Zoloft_Clinr_P1.pdf 1999;(accessed 28 November 2021).
- Friedman MJ, Marmar CR, Baker DG, Sikes CR, Farfel GM.Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. Journal of Clinical Psychiatry 2007;68(5):711-20. [DOI] [PubMed] [Google Scholar]
- Pfizer.Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
Raskind 2018 {published data only}
- Raskind MA, Peskind ER, Chow B, Harris C, Davis-Karim A, Holmes HA, et al.Trial of prazosin for post-traumatic stress disorder in military veterans. New England Journal of Medicine 2018;378(6):507-17. [DOI: 10.1056/NEJMoa1507598] [DOI] [PubMed] [Google Scholar]
Rasmusson 2017 {published data only}
- Rasmusson AM, Marx CE, Jain S, Farfel GM, Tsai J, Sun X, et al.A randomized controlled trial of ganaxolone in posttraumatic stress disorder. Psychopharmacology 2017;234(15):2245–57. [DOI: 10.1007/s00213-017-4649-y] [DOI] [PubMed] [Google Scholar]
Reich 2004 {published data only}
- Reich DB, Winternitz S, Hennen J, Watts T, Stanculescu C.A preliminary study of risperidone in the treatment of posttraumatic stress disorder related to childhood abuse in women. Journal of Clinical Psychiatry 2004;65(12):1601-5. [DOI] [PubMed] [Google Scholar]
Reist 1989 {published data only}
- Reist C, Kauffmann CD, Chicz Demet A, Chen CC, Demet EM.REM latency, dexamethasone suppression test, and thyroid releasing hormone stimulation test in posttraumatic stress disorder. Progress in Neuropsychopharmacology and Biological Psychiatry 1995;19(3):433-43. [DOI] [PubMed] [Google Scholar]
- Reist C, Kauffmann CD, Haier RJ, Sangdahl C, DeMet EM, Chicz-DeMet A, et al.A controlled trial of desipramine in 18 men with posttraumatic stress disorder. American Journal of Psychiatry 1989;146(4):513-6. [DOI] [PubMed] [Google Scholar]
Saygin 2002 {published data only}
- Saygin MZ, Sungur MZ, Sabol EU, Cetinkaya P.Nefazodone versus sertraline in treatment of posttraumatic stress disorder. Bulletin of Clinical Psychopharmacology 2002;12(1):1-5. [Google Scholar]
Shestatzky 1988 {published data only}
- Shestatzky M, Greenberg D, Lerer B.A controlled trial of phenelzine in posttraumatic stress disorder. Psychiatric Research 1988;24(2):149-55. [DOI] [PubMed] [Google Scholar]
SKB627 {unpublished data only}
- Food and Drug Administration (FDA).NDA 20-031/S-029 Paxil (paroxetine hydrochloride) tablets; GlaxoSmithKline; Post-traumatic Stress Disorder. www.accessdata.fda.gov/drugsatfda_docs/nda/2001/20-031S029.pdf;(accessed 28 November 2021).
- GlaxoSmithKline.A 12-week, double-blind, placebo-controlled, parallel group study to assess the efficacy and tolerability of paroxetine in patients suffering from posttraumatic stress disorder (PTSD). GSK - clinical study register [www.gsk-clinicalstudyregister.com]:Study ID: 29060/627.
- SmithKline Beecham.Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
- Stein DJ, Davidson J, Seedat S, Beebe K.Paroxetine in the treatment of post-traumatic stress disorder: pooled analysis of placebo-controlled studies. Expert Opinion on Pharmacotherapy 2003;4(10):1829-38. [DOI] [PubMed] [Google Scholar]
SKB650 {published data only}
- SmithKline Beecham.Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
Smajkic 2001 {published data only}
- Smajkic A, Weine S, Djuric-Bijedic Z, Boskailo E, Lewis J, Pavkovic I.Sertraline, paroxetine, and venlafaxine in refugee posttraumatic stress disorder with depression symptoms. Journal of Traumatic Stress 2001;14(3):445-52. [DOI] [PubMed] [Google Scholar]
Spivak 2006 {published data only}
- Spivak B, Strous RD, Shaked G, Shabash E, Kotler M, Weizman A.Reboxetine versus fluvoxamine in the treatment of motor vehicle accident-related posttraumatic stress disorder: a double-blind, fixed-dosage, controlled trial. Journal of Clinical Psychopharmacology 2006;26(2):152–6. [DOI: 10.1097/01.jcp.0000203195.65710.f0] [DOI] [PubMed] [Google Scholar]
Tucker 2001 {published data only}
- GlaxoSmithKline.A 12-week, double-blind, placebo-controlled, parallel group study to assess the efficacy and tolerability of paroxetine in patients suffering from posttraumatic stress disorder (PTSD). GSK - Clinical Study Register [www.gsk-clinicalstudyregister.com] 2000:Study ID:GSK 29060/648.
- Tucker P, Zaninelli R, Yehuda R, Ruggiero L, Dillingham K, Pitts CD.Paroxetine in the treatment of chronic posttraumatic stress disorder: results of a placebo-controlled, flexible-dosage trial. Journal of Clinical Psychiatry 2001;62(11):860-8. [DOI] [PubMed] [Google Scholar]
Tucker 2003 {published data only}
- Tucker P, Potter-Kimball R, Wyatt DB, Parker DE, Burgin C, Jones DE, et al.Can physiologic assessment and side effects tease out differences in PTSD trials? A double-blind comparison of citalopram, sertraline, and placebo. General Psychopharmacology 2003;37(3):135-49. [PubMed] [Google Scholar]
Tucker 2007 {published data only}
- Tucker P, Trautman RP, Wyatt DB, Thompson J, Wu SC, Capece JA, et al.Efficacy and safety of topiramate monotherapy in civilian posttraumatic stress disorder: a randomized, double-blind, placebo-controlled study. Journal of Clinical Psychiatry 2007;68(2):201-6. [DOI] [PubMed] [Google Scholar]
Van der Kolk 1994 {published data only}
- Van der Kolk BA, Dreyfuss D, Michaels M, Shera D, Berkowitz R, Fisler R, et al.Fluoxetine treatment in posttraumatic stress disorder. Journal of Clinical Psychiatry 1994;55(12):517-22. [PubMed] [Google Scholar]
Van der Kolk 2007 {published and unpublished data}
- Van der Kolk, Spinazzola J, Blaustein M, Hopper J, Hopper E, Korn D, et al.A randomized clinical trial of EMDR, fluoxetine and pill placebo in the treatment of PTSD: treatment effects and long-term maintenance. Draft manuscript 2004. [DOI] [PubMed]
- Van der Kolk BA, Spinazzola J, Blaustein ME, Hopper JW, Hopper EK, Korn DL, et al.A randomized clinical trial of eye movement desensitization and reprocessing (EMDR), fluoxetine, and pill placebo in the treatment of posttraumatic stress disorder: treatment effects and long-term maintenance. Journal of Clinical Psychiatry 2007;68(1):37-46. [DOI] [PubMed] [Google Scholar]
Villarreal 2016 {published data only}
- Villarreal G, Hamner MB, Cañive JM, Robert S, Calais LA, Durklaski V, et al.Efficacy of quetiapine monotherapy in posttraumatic stress disorder: a randomized, placebo-controlled trial. American Journal of Psychiatry 2016;173(12):1205-12. [DOI: 10.1176/appi.ajp.2016.15070967] [DOI] [PubMed] [Google Scholar]
Yeh 2011 {published data only}
- Yeh MS, Mari JJ, Costa MC, Andreoli SB, Bressan RA, Mello MF.A double-blind randomized controlled trial to study the efficacy of topiramate in a civilian sample of PTSD. CNS Neuroscience and Therapeutics 2011;17(5):305–10. [DOI: 10.1111/j.1755-5949.2010.00188.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zohar 2002 {published data only}
- Amital D, Zohar J, Kotler M, Bleich A, Miodovnik H, Nevo A, et al.A placebo-controlled study of sertraline in posttraumatic stress disorder. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):269. [Google Scholar]
- Amital D, Zohar J, Kotler M, Bleich A, Miodovnik H, Nevo A, et al.A placebo-controlled trial pilot study of sertraline in PTSD. In: Annual Meeting of the American Psychiatric Association. American Psychiatric Association, 1999:157.
- Zohar J, Amital D, Miodownik C, Kotler M, Bleich A, Lane RM.Double-blind placebo-controlled pilot study of sertraline in military veterans with posttraumatic stress disorder. Journal of Clinical Psychopharmacology 2002;22(2):190-5. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Aerni 2004 {published data only}
- Aerni A, Traber R, Hock C, Roozendaal B, Schelling G, Papassotiropoulos A, et al.Low-dose cortisol for symptoms of posttraumatic stress disorder. American Journal of Psychiatry 2004;161(8):1488-90. [DOI] [PubMed] [Google Scholar]
Angelidis 2019 {published data only}
- Angelidis A, Kyrgiou A, Van der Wee N, Van der Does W, Putman P.P.3.26 Effects of hydrocortisone on cognitive performance and threat-interference under acute stress in highly anxious females. European Neuropsychopharmacology 2019;29(Supplement 2):S698-9. [DOI: ] [Google Scholar]
Back 2016 {published data only}
- Back SE, McCauley JL, Korte KJ, Gros DF, Leavitt V, Gray KM, et al.A double-blind, randomized, controlled pilot trial of N-Acetylcysteine in veterans with posttraumatic stress disorder and substance use disorders. Journal of Clinical Psychiatry 2016;77(11):e1439-46. [DOI: 10.4088/JCP.15m10239] [DOI] [PMC free article] [PubMed] [Google Scholar]
Back 2018 {published data only}
- Back SE, Flanagan JC, Jones JL, Augur I, Peterson AL, Young-McCaughan S, et al.Doxazosin for the treatment of co-occurring PTSD and alcohol use disorder: design and methodology of a randomized controlled trial in military veterans. Contemporary Clinical Trials 2018;73:8-15. [DOI: 10.1016/j.cct.2018.08.009] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bartzokis 2005 {published data only}
- Bartzokis G, Lu PH, Turner J, Mintz J, Saunders CS.Adjunctive risperidone in the treatment of chronic combat-related posttraumatic stress disorder. Biological Psychiatry 2005;57(5):474-9. [DOI] [PubMed] [Google Scholar]
Batki 2014 {published data only}
- Batki SL, Pennington DL, Lasher B, Neylan TC, Metzler T, Waldrop A, et al.Topiramate treatment of alcohol use disorder in veterans with PTSD: a randomized controlled pilot trial. Alcoholism: Clinical and Experimental Research 2014;38(8):2169–77. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Boroughs 2018 {published data only}
- Boroughs MS, Ehlinger PP, Batchelder AW, Safren SA, O'Cleirigh C.Posttraumatic stress symptoms and emerging adult sexual minority men: implications for assessment and treatment of childhood sexual abuse. Journal of Traumatic Stress 2018;31(5):665-75. [DOI: 10.1002/jts.22335] [DOI] [PMC free article] [PubMed] [Google Scholar]
Borrelli 2019 {published data only}
- Borrelli J Jr, Starr A, Downs DL, North CS.Prospective study of the effectiveness of paroxetine on the onset of posttraumatic stress disorder, depression, and health and functional outcomes after trauma. Journal of Orthopaedic Trauma 2019;33(2):e58-e63. [DOI: ] [DOI] [PubMed] [Google Scholar]
Bountress 2018 {published data only}
- Bountress KE, Badour C, Flanagan J, Gilmore AK, Back SE.Treatment of co-occurring posttraumatic stress disorder and substance use: does order of onset influence outcomes? Psychological Trauma: Theory, Research, Practice and Policy 2018;10(6):662-5. [DOI: 10.1037/tra0000309] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brady 2005 {published data only}
- Brady KT, Sonne S, Anton RF, Randall CL, Back SE, Simpson K.Sertraline in the treatment of co-occurring alcohol dependence and posttraumatic stress disorder. Alcoholism: Clinical and Experimental Research 2005;29(3):395–401. [DOI: 10.1097/01.ALC.0000156129.98265.57] [DOI] [PubMed] [Google Scholar]
Bremner 1997 {published data only}
- Bremner JD, Innis RB, Ng CK, Staib LH, Salomon RM, Bronen RA, et al.Positron emission tomography measurement of cerebral metabolic correlates of yohimbine administration in combat-related posttraumatic stress disorder. Archives of General Psychiatry 1997;54(3):246-54. [DOI] [PubMed] [Google Scholar]
Brunet 2018 {published data only}
- Brunet A, Saumier D, Liu A, Streiner DL, Tremblay J, Pitman RK.Reduction of PTSD symptoms with pre-reactivation propranolol therapy: a randomized controlled trial. American Journal of Psychiatry 2018;175(5):427-33. [DOI: 10.1176/appi.ajp.2017.17050481] [DOI] [PubMed] [Google Scholar]
Buhmann 2018 {published data only}
- Buhmann CB, Nordentoft M, Ekstroem M, Carlsson J, Mortensen EL.Long-term treatment effect of trauma-affected refugees with flexible cognitive behavioural therapy and antidepressants. Psychiatry Research 2018;264:217-23. [DOI: 10.1016/j.psychres.2018.03.069] [DOI] [PubMed] [Google Scholar]
Coupland 1997 {published data only}
- Coupland NJ, Lillywhite A, Bell CE, Potokar JP, Nutt DJ.A pilot controlled study of the effects of flumazenil in posttraumatic stress disorder. Biological Psychiatry 1997;41(9):988-90. [DOI] [PubMed] [Google Scholar]
Eftekhari 2004 {unpublished data only}
- Eftekhari A, Miller H, Zoellner L, Feeny N.Prolonged exposure vs. sertraline: Secondary outcomes in PTSD treatment. In: 20th Annual Meeting, International Society for Traumatic Stress Studies, November 14 - November 18, New Orleans, LA. 2004.
Feder 2021 {published data only}
- Feder A, Costi S, Rutter SB, Collins AB, Govindarajulu U, Jha MK, et al.A randomized controlled trial of repeated ketamine administration for chronic posttraumatic stress disorder. American Journal of Psychiatry 2021;178(2):193–202. [DOI: 10.1176/appi.ajp.2020.20050596] [DOI] [PubMed] [Google Scholar]
Flanagan 2018 {published data only}
- Flanagan JC, Hand A, Jarnecke AM, Moran-Santa Maria MM, Brady KT, Joseph JE.Effects of oxytocin on working memory and executive control system connectivity in posttraumatic stress disorder. Experimental and Clinical Psychopharmacology 2018;26(4):391-402. [DOI: 10.1037/pha0000197] [DOI] [PMC free article] [PubMed] [Google Scholar]
Flanagan 2019 {published data only}
- Flanagan JC, Allan NP, Calhoun CD, Badour CL, Moran-Santa Maria M, Brady KT, et al.Effects of oxytocin on stress reactivity and craving in veterans with co-occurring PTSD and alcohol use disorder. Experimental and Clinical Psychopharmacology 2019;27(1):45-54. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Frank 1988 {published data only}
- Frank JB, Kosten TR, Giller EL Jr, Dan E.A randomized clinical trial of phenelzine and imipramine for post-traumatic stress disorder. American Journal of Psychiatry 1988;145(10):1289-91. [DOI] [PubMed] [Google Scholar]
Frijling 2017 {published data only}
- Frijling JL.Preventing PTSD with oxytocin: effects of oxytocin administration on fear neurocircuitry and PTSD symptom development in recently trauma-exposed individuals. European Journal of Psychotraumatology 2017;8(1):1302652. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Frommberger 1998 {published data only}
- Frommberger U, Nyberg E, Richter H, Stieglitz RD, Berger M.Paroxetine vs. cognitive-behavioral therapy in the treatment of depression in PTSD patients. In: XXIst Collegium Internationale Neuro psychopharmacologicum, Glasgow, Scotland. 12th 16th July. 1998.
Gelpin 1996 {published data only}
- Gelpin E, Bonne O, Peri T, Brandes D, Shalev AY.Treatment of recent trauma survivors with benzodiazepines: a prospective study. Journal of Clinical Psychiatry 1996;57(9):390-4. [PubMed] [Google Scholar]
Graham 2018 {published data only}
- Graham B, Garcia NM, Burton MS, Cooper AA, Roy-Byrne PP, Mavissakalian MR.High expectancy and early response produce optimal effects in sertraline treatment for post-traumatic stress disorder. British Journal of Psychiatry 2018;213(6):704-8. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamner 1997 {published data only}
- Hamner M, Ulmer H, Horne D.Buspirone potentiation of antidepressants in the treatment of PTSD. Depression and Anxiety 1997;5(3):137-9. [PubMed] [Google Scholar]
Hamner 1999 {published data only}
- Hamner MB, Ulmer HG, Horne DF, George MS, Arana GW.Procaine administration and behavioral responsivity in post-traumatic stress disorder: a pilot study of tolerability. Human Psychopharmacology 1999;14(2):105-11. [Google Scholar]
Hamner 2003 {published data only}
- Hamner MB, Faldowski RA, Ulmer HG, Frueh BC, Huber MG, Arana GW.Adjunctive risperidone treatment in post-traumatic stress disorder: a preliminary controlled trial of effects on comorbid psychotic symptoms. International Clinical Psychopharmacology 2003;18(1):1-8. [DOI] [PubMed] [Google Scholar]
Heresco‐Levy 2002 {published data only}
- Heresco-Levy U, Kremer I, Javitt DC, Goichman R, Reshef A, Blanaru M, et al.Pilot-controlled trial of D-cycloserine for the treatment of post-traumatic stress disorder. International Journal of Neuropsychopharmacology 2002;5(4):301-7. [DOI] [PubMed] [Google Scholar]
Inslicht 2018 {published data only}
- Inslicht S, Niles A, Metzler T, Milad M, Orr SP, Marmar C, et al.Randomized controlled trial of hydrocortisone and D-cycloserine on fear extinction in PTSD. Biological Psychiatry 2018;83(9 Supplement 1):S352. [Google Scholar]
Jacobs‐Rebhun 2000 {published data only}
- Jacobs-Rebhun S, Schnurr PP, Friedman MJ, Peck R, Brophy M, Fuller D.Posttraumatic stress disorder and sleep difficulty. American Journal of Psychiatry 2000;157(9):1525-6. [DOI] [PubMed] [Google Scholar]
Kanter 2001 {published data only}
- Kanter ED, Wilkinson CW, Radant AD, Petrie EC, Dobie DJ, McFall ME, et al.Glucocorticoid feedback sensitivity and adrenocortical responsiveness in posttraumatic stress disorder. Biological Psychiatry 2001;50(4):238-45. [DOI] [PubMed] [Google Scholar]
Kellner 2000 {published data only}
- Kellner M, Wiedemann K, Yassouridis A, Levengood R, Guo LS, Holsboer F, et al.Behavioral and endocrine response to cholecystokinin tetrapeptide in patients with posttraumatic stress disorder. Biological Psychiatry 2000;47(2):107-11. [DOI] [PubMed] [Google Scholar]
Le 2018 {published data only}
- Le QA, Doctor JN, Zoellner LA, Feeny NC.Effects of treatment, choice, and preference on health-related quality-of-life outcomes in patients with posttraumatic stress disorder (PTSD). Quality of Life Research 2018;27(6):1555-62. [DOI: ] [DOI] [PubMed] [Google Scholar]
Mello 2009 {published data only}
- Mello MF, Yeh MS, Neto JB, Braga LL, Fiks JP, Mendes DD, et al.A randomized, double-blind, placebo-controlled trial to assess the efficacy of topiramate in the treatment of post-traumatic stress disorder. BMC Psychiatry 2009;9(28):1-20. [DOI: 10.1186/1471-244X-9-28] [DOI] [PMC free article] [PubMed] [Google Scholar]
Monnelly 2003 {published data only}
- Monnelly EP, Ciraulo DA, Knapp C, Keane T.Low-dose risperidone as adjunctive therapy for irritable aggression in posttraumatic stress disorder. Journal of Clinical Psychopharmacology 2003;23(2):193-6. [DOI] [PubMed] [Google Scholar]
Morgan 1995 {published data only}
- Morgan CA 3rd, Grillon C, Southwick SM, Nagy LM, Davis M, Krystal JH, et al.Yohimbine facilitated acoustic startle in combat veterans with post-traumatic stress disorder. Psychopharmacology 1995;117(4):466-71. [DOI] [PubMed] [Google Scholar]
Naylor 2013 {published data only}
- Naylor JC, Dolber TR, Strauss JL, Kilts JD, Strauman TJ, Bradford DW, et al.A pilot randomized controlled trial with paroxetine for subthreshold PTSD in operation enduring freedom/operation Iraqi freedom era veterans. Psychiatry Research Journal 2013;206(0):318–20. [DOI: 10.1016/j.psychres.2012.11.008] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00557622 {published data only}
- NCT00557622.Clinical evaluation of BRL29060A (paroxetine hydrochloride hydrate) in posttraumatic stress disorder (PTSD). clinicaltrials.gov/ct2/show/record/ NCT00557622 (first received 14 November 2007).
NCT00648375 {published data only}
- NCT00648375.Effectiveness of propranolol for treating people with post-traumatic stress disorder. clinicaltrials.gov/ct2/show/record/NCT00648375 (first received 01 April 2008).
NCT00706173 {published data only}
- NCT00706173.Trial of hydrocortisone for post-traumatic stress disorder (PTSD). clinicaltrials.gov/ct2/show/record/NCT00706173 (first received 27 June 2008).
NCT01325168 {published data only}
- NCT01325168.The effect of intranasal oxytocin on emphatic abilities in patients with post traumatic stress disorder (PTSD). clinicaltrials.gov/ct2/show/record/NCT01325168 (first received 29 March 2011).
NCT01449955 {published data only}
- NCT01449955.Rapamycin as a means of interference with reconsolidation of posttraumatic stress disorder-related traumatic memory. clinicaltrials.gov/ct2/show/record/NCT01449955 (first received 10 October 2011).
NCT01477762 {published data only}
- NCT01477762.Cortisol suppression and startle responses in posttraumatic stress disorder (PTSD) (CSS). clinicaltrials.gov/ct2/show/record/NCT01477762 (first received 18 April 2017).
NCT01664260 {published data only}
- NCT01664260.Efficacy mechanism of N-acetylcysteine in patients with posttraumatic stress disorder. clinicaltrials.gov/ct2/show/record/NCT01664260 (first received 14 August 2012).
Neylan 2006 {published data only}
- Neylan TC, Lenoci M, Samuelson KW, Metzler TJ, Henn-Haase C, Hierholzer RW, et al.No improvement of posttraumatic stress disorder symptoms with guanfacine treatment. American Journal of Psychiatry 2006;163(12):2186–8. [DOI] [PubMed] [Google Scholar]
Otto 2003 {published data only}
- Otto MW, Hinton D, Korbly NB, Chea A, Ba P, Gershuny BS, et al.Treatment of pharmacotherapy-refractory posttraumatic stress disorder among Cambodian refugees: a pilot study of combination treatment with cognitive-behavior therapy vs sertraline alone. Behaviour Research and Therapy 2003;41(11):1271-6. [DOI] [PubMed] [Google Scholar]
Pape 2018 {published data only}
- Pape JC, Carrillo-Roa T, Rothbaum BO, Nemeroff CB, Czamara D, Zannas AS, et al.DNA methylation levels are associated with CRF1 receptor antagonist treatment outcome in women with post-traumatic stress disorder. Clinical Epigenetics 2018;10(1):136. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Petrakis 2006 {published data only}
- Petrakis IL, Poling J, Levinson C, Nich C, Carroll K, Ralevski E, et al.Naltrexone and disulfiram in patients with alcohol dependence and comorbid post-traumatic stress disorder. Biological Psychiatry 2006;60(7):777-83. [DOI: 10.1016/j.biopsych.2006.03.074] [DOI] [PubMed] [Google Scholar]
Pitman 1990 {published data only}
- Pitman RK, Van der Kolk BA, Orr SP, Greenberg MS.Naloxone-reversible analgesic response to combat-related stimuli in posttraumatic stress disorder. Archives of General Psychiatry 1990;47(6):541-4. [DOI] [PubMed] [Google Scholar]
Pitman 2002 {published data only}
- Pitman RK, Sanders KM, Zusman RM, Healy AR, Cheema F, Lasko NB, et al.Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biological Psychiatry 2002;51(2):189-92. [DOI] [PubMed] [Google Scholar]
Pollack 2011 {published data only}
- Pollack MH, Hoge EA, Worthington JJ, Moshier SJ, Wechsler RS, Brandes M, et al.Eszopiclone for the treatment of posttraumatic stress disorder and associated insomnia: a randomized, double-blind, placebo-controlled trial. Journal of Clinical Psychiatry 2011;72(7):892-7. [DOI: 10.4088/JCP.09m05607gry] [DOI] [PubMed] [Google Scholar]
Ramaswamy 2016 {published data only}
- Ramaswamy S, Driscoll D, Smith LM, Bhatia SC, Petty F.Failed efficacy of ziprasidone in the treatment of post-traumatic stress disorder. Contemporary Clinical Trials Communications 2016;2:1-5. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ramaswamy 2017 {published data only}
- Ramaswamy S, Driscoll D, Reist C, Smith LM, Albers LJ, Rose JR.A double-blind, placebo-controlled randomized trial of vilazodone in the treatment of posttraumatic stress disorder and comorbid depression. Primary Care Companion for CNS Disorders 2017;19(4):17m02138. [DOI: ] [DOI] [PubMed] [Google Scholar]
Randall 1995 {published data only}
- Randall PK, Bremner JD, Krystal JH, Nagy LM, Heninger GR, Nicolaou AL, et al.Effects of the benzodiazepine antagonist flumazenil in PTSD. Biological Psychiatry 1995;38(5):319-24. [DOI] [PubMed] [Google Scholar]
Raskind 2003 {published data only}
- Raskind MA, Peskind ER, Kanter ED, Petrie EC, Radant A, Thompson CE, et al.Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. American Journal of Psychiatry 2003;160(2):371-3. [DOI] [PubMed] [Google Scholar]
Raskind 2007 {published data only}
- Raskind MA, Peskind ER, Hoff DJ, Hart KL, Holmes HA, Warren D.A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Society of Biological Psychiatry 2007;61:928–34. [DOI: 10.1016/j.biopsych.2006.06.032] [DOI] [PubMed] [Google Scholar]
Raskind 2013 {published data only}
- Raskind MA, Peterson K, Williams T, Hoff DJ, Hart K, Holmes H, et al.A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. American Journal of Psychiatry 2013;170(9):1003-10. [DOI: 10.1176/appi.ajp.2013.12081133] [DOI] [PubMed] [Google Scholar]
Reist 1995 {published data only}
- Reist C, Kauffmann CD, Chicz Demet A, Chen CC, Demet EM.REM latency, dexamethasone suppression test, and thyroid releasing hormone stimulation test in posttraumatic stress disorder. Progress in Neuropsychopharmacology and Biological Psychiatry 1995;19(3):433-43. [DOI] [PubMed] [Google Scholar]
Reist 2001 {published data only}
- Reist C, Duffy JG, Fujimoto K, Cahill L.Beta-adrenergic blockade and emotional memory in PTSD. International Journal of Neuropsychopharmacology 2001;4(4):377-83. [DOI] [PubMed] [Google Scholar]
Roache 2017 {published data only}
- Roache JD, Raj JJ, Blount T, Hale W, Mintz, J, Murff WR.SSRI treatment of dual diagnosis PTSD and alcohol dependence In veterans: opposite effects of sertraline In EOA and LOA subtypes. In: UT Health Science Center at San Antonio and South Texas Veterans Health Care System, San Antonio, TX, USA. 2017.
Schelling 2004 {published data only}
- Schelling G, Kilger E, Roozendaal B, Quervain DJ, Briegel J, Dagge A, et al.Stress doses of hydrocortisone, traumatic memories, and symptoms of posttraumatic stress disorder in patients after cardiac surgery: a randomized study. Biological Psychiatry 2004;55(6):627-33. [DOI] [PubMed] [Google Scholar]
Southwick 1997 {published data only}
- Southwick SM, Krystal JH, Bremner JD, Morgan CA 3rd, Nicolaou AL, Nagy LM, et al.Noradrenergic and serotonergic function in posttraumatic stress disorder. Archives of General Psychiatry 1997;54(8):749-58. [DOI] [PubMed] [Google Scholar]
Stein 2002 {published data only}
- Stein MB, Kline NA, Matloff JL.Adjunctive olanzapine for SSRI-resistant combat-related PTSD: a double-blind, placebo-controlled study. American Journal of Psychiatry 2002;159(10):1777-9. [DOI] [PubMed] [Google Scholar]
Suris 2017 {published data only}
- Suris A, Holliday R, Adinoff B, Holder N, North CS.Facilitating fear-based memory extinction with dexamethasone: a randomized controlled trial in male veterans with combat-related PTSD. Psychiatry 2017;80(4):399-410. [DOI: 10.1080/00332747.2017.1286892] [DOI] [PubMed] [Google Scholar]
Vaiva 2003 {published data only}
- Vaiva G, Ducrocq F, Jezequel K, Averland B, Lestavel P, Brunet A, et al.Immediate treatment with propranolol decreases posttraumatic stress disorder two months after trauma. Biological Psychiatry 2003;54(9):947-9. [DOI] [PubMed] [Google Scholar]
Van der Kolk 1989 {published data only}
- Van der Kolk BA, Greenberg MS, Orr SP, Pitman RK.Endogenous opioids, stress induced analgesia, and posttraumatic stress disorder. Psychopharmacology Bulletin 1989;25(3):417-21. [PubMed] [Google Scholar]
Verplaetse 2019 {published data only}
- Verplaetse TL, Ralevski E, Roberts W, Gueorguieva R, McKee SA, Petrakis IL.Alcohol abstainer status and prazosin treatment in association with changes in posttraumatic stress disorder symptoms in veterans with comorbid alcohol use disorder and posttraumatic stress disorder. Alcoholism: Clinical and Experimental Research 2019;43(4):741-6. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
Davis 2003 {published data only}
- Davis L, Petty F.GABAergic modulation in PTSD and treatment with anticonvulsants. In: In: 19th Annual Meeting, International Society for Traumatic Stress Studies, October 29 - November 1, Chicago, IL. 2003.
Fluoxetine, Davidson {published data only (unpublished sought but not used)}
Fluoxetine, Lilly {published data only (unpublished sought but not used)}
Nagy 1996 {unpublished data only}
- Nagy LM, Southwick SM, Charney DS.Placebo-controlled trial of fluoxetine in PTSD. In: International Society for Traumatic Stress Studies 12th Annual Meeting, San Francisco, CA, November 11. 1996.
NCT00413296 {published data only}
- NCT00413296.Levetiracetam in post-traumatic stress disorder (PTSD). clinicaltrials.gov/ct2/show/record/NCT00413296 (first received 19 December 2006).
NCT00672776 {published data only}
- NCT00672776.Effects of Paxil CR on neural circuits in posttraumatic stress disorder (PTSD). clinicaltrials.gov/ct2/show/record/NCT00672776 (first received 06 May 2008).
NCT01517711 {published data only}
- NCT01517711.Tramadol Extended-Release (ER) for posttraumatic stress disorder (PTSD). clinicaltrials.gov/ct2/show/record/NCT01517711 (first received 25 January 2012).
NCT02637895 {published data only}
- NCT02637895.Vortioxetine for posttraumatic stress disorder. clinicaltrials.gov/ct2/show/record/NCT02637895 (first received 22 December 2015).
NCT02709018 {published data only}
- NCT02709018.A controlled trial of losartan in posttraumatic stress disorder (LOSe-PTSD). clinicaltrials.gov/ct2/show/record/NCT02709018 (first received 15 March 2016).
NCT02933606 {published data only}
- NCT02933606.Phase II study of BNC210 in PTSD (RESTORE). clinicaltrials.gov/ct2/show/record/NCT02933606 (first received 14 October 2016).
Nefazodone 1, BMS {published data only (unpublished sought but not used)}
Nefazodone 2, BMS {published data only (unpublished sought but not used)}
Olanzapine, Davidson {published data only}
Paroxetine 2 ‐ SB {published data only (unpublished sought but not used)}
Paroxetine 3 ‐ SB {published data only (unpublished sought but not used)}
Sertraline 2, Pfizer {published data only (unpublished sought but not used)}
Sertraline 3, Pfizer {unpublished data only}
Sertraline 4, Pfizer {published data only}
References to ongoing studies
NCT01221792 {published data only}
- NCT01221792.Carvedilol versus placebo for treatment in post traumatic stress disorder (PTSD). clinicaltrials.gov/ct2/show/record/NCT01221792 (first received 15 October 2010).
Additional references
Albucher 2002
- Albucher RC, Liberzon I.Psychopharmacological treatment in PTSD: a critical review. Journal of Psychiatric Research 2002;36(6):355-67. [DOI] [PubMed] [Google Scholar]
Als‐Nielsen 2003
- Als-Nielsen B, Chen W, Gluud C, Kjaergard LL.Association of funding and conclusions in randomized drug trials - A reflection of treatment effect or adverse events? JAMA 2003;290(7):921-8. [DOI] [PubMed] [Google Scholar]
Amos 2014
- Amos T, Stein DJ, Ipser JC.Pharmacological interventions for preventing post-traumatic stress disorder (PTSD). Cochrane Database of Systematic Reviews 2014, Issue Issue 7. Art. No.: CD006239. Art. No: CD006239. [DOI: 10.1002/14651858.CD006239.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association.Diagnostic and statistical manual of mental disorders (DSM-III). 3rd edition. Washington, DC: American Psychiatric Association, 1980. [Google Scholar]
APA 1994
- American Psychiatric Association.Diagnostic and statistical manual of mental disorders (DSM-IV). 4th edition. Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
APA 2013
- American Psychiatric Association.Diagnostic and statistical manual of mental Disorders, fifth edition. Arlington, VA: American Psychiatric Publishing, 2013. [Google Scholar]
Asnis 2004
- Asnis GM, Kohn SR, Henderson M, Brown NL.SSRIs versus non-SSRIs in post-traumatic stress disorder: an update with recommendations. Drugs 2004;64(4):383-404. [DOI] [PubMed] [Google Scholar]
Bailar 1997
- Bailar JC 3rd.The promise and problems of meta-analysis. New England Journal of Medicine 1999;337(8):559-61. [DOI] [PubMed] [Google Scholar]
Baker 2003
- Baker CB, Johnsrud MT, Crismon ML, Rosenheck RA, Woods SW.Quantitative analysis of sponsorship bias in economic studies of antidepressants. British Journal of Psychiatry 2003;183:498-506. [DOI] [PubMed] [Google Scholar]
Baldwin 2014
- Baldwin DS, Anderson IM, Nutt DJ, Allgulander C, Bandelow B, Den Boer JA.Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: A revision of the 2005 guidelines from the British Association for Psychopharmacology. Journal of Psychopharmacology 2014;28(5):403-39. [DOI: 10.1177/0269881114525674] [DOI] [PubMed] [Google Scholar]
Ballenger 2000
- Ballenger JC, Davidson JR, Lecrubier Y, Nutt DJ, Foa EB, Kessler RC, et al.Consensus statement on posttraumatic stress disorder from the International Consensus Group on Depression and Anxiety. Journal of Clinical Psychiatry 2000;61(Suppl 5):60-6. [PubMed] [Google Scholar]
Ballenger 2004
- Ballenger JC, Davidson JR, Lecrubier Y, Nutt DJ, Marshall RD, Nemeroff CB, et al.Consensus statement update on posttraumatic stress disorder from the international consensus group on depression and anxiety. Journal of Clinical Psychiatry 2004;65(Suppl 1):55-62. [PubMed] [Google Scholar]
Bandelow 2008
- Bandelow B, Zohar J, Hollander E, Kasper S, Möller H-J, WFSBP Task Force on Treatment Guidelines for Anxiety Obsessive-compulsive Post-traumatic Stress Disorders, et al.World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-raumatic tress disorders –first revision. World Journal of Biological Psychiatry 2008;9(4):248-312. [DOI: ] [DOI] [PubMed] [Google Scholar]
Basoglu 1992
- Basoglu M, Marks IM, Senguen S.Amitriptyline for PTSD in a torture survivor: a case study. Journal of Traumatic Stress 1992;5:77-83. [Google Scholar]
Beck 1961
- Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J.An inventory for measuring depression. Archives of General Psychiatry 1961;4:561-71. [DOI] [PubMed] [Google Scholar]
Bernardy 2017
- Bernardy NC, Friedman MJ.Pharmacological management of posttraumatic stress disorder. Current Opinion in Psychology 2017;14:116-21. [DOI: ] [DOI] [PubMed] [Google Scholar]
Bisson 2007
- Bisson JI, Andrew M.Psychological treatment of post-traumatic stress disorder (PTSD). Cochrane Database of Systematic Reviews 2007, Issue 12. Art. No: CD003388. [DOI: 10.1002/14651858.CD003388.pub3] [DOI] [PubMed] [Google Scholar]
Bisson 2013
- Bisson JI, Roberts NP, Andrew M, Cooper R, Lewis C.Psychological therapies for chronic post‐traumatic stress disorder (PTSD) in adults. Cochrane Database of Systematic Reviews 2013, Issue 12. Art. No: CD003388. [DOI: 10.1002/14651858.CD003388.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Blake 1990
- Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, et al.A clinician rating scale for assessing current and lifetime PTSD: The CAPS-1. Behavior Therapy 1990;21:187-8. [Google Scholar]
Bleich 1986
- Bleich A, Siegel B, Garb R, Lerer B.Post-traumatic stress disorder following combat exposure: clinical features and psychopharmacological treatment. British Journal of Psychiatry 1986;149:365-9. [DOI] [PubMed] [Google Scholar]
Bradley 2005
- Bradley R, Greene J, Russ E, Dutra L, Westen D.A multidimensional meta-analysis of psychotherapy for PTSD. American Journal of Psychiatry 2005;162(2):214-27. [DOI] [PubMed] [Google Scholar]
Bremner 2004
- Bremner JD, Vermetten E.Neuroanatomical changes associated with pharmacotherapy in posttraumatic stress disorder. Annals of the New York Academy of Science 2004;1032:154-7. [DOI] [PubMed] [Google Scholar]
Breslau 1991
- Breslau N, Davis GC, Andreski P.Traumatic events and post traumatic stress disorder in an urban population of young adults. Archives of General Psychiatry 1991;48(3):216-22. [DOI] [PubMed] [Google Scholar]
Brom 1989
- Brom D, Kleber RJ, Defares PB.Brief psychotherapy for posttraumatic stress disorders. Journal of Consulting and Clinical Psychology 1989;57(5):607-12. [DOI] [PubMed] [Google Scholar]
Brophy 1991
- Brophy MH.Cyproheptadine for combat nightmares in post-traumatic stress disorder and dream anxiety disorder. Military Medicine 1991;156(2):100-1. [PubMed] [Google Scholar]
Brunello 2001
- Brunello N, Davidson JR, Deahl M, Kessler RC, Mendlewicz J, Racagni G, et al.Posttraumatic stress disorder: diagnosis and epidemiology, comorbidity and social consequences, biology and treatment. Neuropsychobiology 2001;43(3):150-62. [DOI] [PubMed] [Google Scholar]
Burstein 1984
- Burstein A.Treatment of posttraumatic stress disorder with imipramine. Psychosomatics 1984;25(9):681-7. [DOI] [PubMed] [Google Scholar]
Burton 1999
- Burton JK, Marshall RD.Categorizing fear: the role of trauma in a clinical formulation. American Journal of Psychiatry 1999;156(5):761-6. [DOI] [PubMed] [Google Scholar]
Cahill 1994
- Cahill L, Prins B, Weber M, McGaugh JL.β-adrenergic activation and memory for emotional events. Nature 1994;371(6499):702–4. [DOI: 10.1038/371702a0] [DOI] [PubMed] [Google Scholar]
Canive 1997
- Canive JM, Lewine JD, Orrison WW Jr, Edgar CJ, Provencal SL, Davis JT, et al.MRI reveals gross structural abnormalities in PTSD. Annals of the New York Academy of Sciences 1997;821:512-5. [DOI] [PubMed] [Google Scholar]
Canive 1998
- Canive JM, Clark RD, Calais LA, Qualls C, Tuason VB.Bupropion treatment in veterans with posttraumatic stress disorder: an open study. Journal of Clinical Psychopharmacology 1998;18(5):379-83. [DOI] [PubMed] [Google Scholar]
Charney 1993
- Charney DS, Deutch AY, Krystal JH, Southwick SM, Davis M.Psychobiologic mechanisms of posttraumatic stress disorder. Archives of General Psychiatry 1993;50(4):294-306. [DOI] [PubMed] [Google Scholar]
Charney 2004
- Charney D.Psychobiological mechanisms of resilience and vulnerability: Implications for successful adaptation to extreme stress. American Journal of Psychiatry 2004;161(2):195-216. [DOI] [PubMed] [Google Scholar]
Chen 1991
- Chen C.The obsessive quality and clomipramine treatment of PTSD. American Journal of Psychiatry 1991;148(8):1087-8. [DOI] [PubMed] [Google Scholar]
Connor 1998
- Connor KM, Davidson JR.The role of serotonin in posttraumatic stress disorder: neurobiology and pharmacotherapy. CNS Spectrums 1998;3(7S2):43-51. [Google Scholar]
Connor 1999b
- Connor KM, Davidson JR, Weisler RH, Ahearn E.A pilot study of mirtazapine in post-traumatic stress disorder. International Clinical Psychopharmacology 1999;14(1):29-31. [DOI] [PubMed] [Google Scholar]
Covi 1984
- Covi L, Lipman RS.Primary depression or primary anxiety: a possible psychometric approach to a diagnostic dilemma. Clinical Neuropharmacology 1984;7:S502-3. [Google Scholar]
Creamer 2003
- Creamer M, Bell R, Failla S.Psychometric properties of the Impact of Event Scale—Revised. Behaviour Research and Therapy 2003;41(12):1489–96. [DOI] [PubMed] [Google Scholar]
CSM 2005
- Committee on Safety of Medicines.Report of the CSM expert working group on the safety of selective serotonin reuptake inhibitor antidepressants. https://www.neuroscience.ox.ac.uk/publications/474047 2005.
Cyr 2000
- Cyr M, Farrar MK.Treatment for posttraumatic stress disorder. Annals of Pharmacotherapy 2000;34(3):366-76. [DOI] [PubMed] [Google Scholar]
Davidson 1987
- Davidson J, Walker I, Kilts C.A pilot study of phenelzine in the treatment of post-traumatic stress disorder. British Journal of Psychiatry 1987;150:252-5. [DOI] [PubMed] [Google Scholar]
Davidson 1989
- Davidson JR, Nemeroff CB.Pharmacotherapy in posttraumatic stress disorder: historical and clinical considerations and future directions. Psychopharmacology Bulletin 1989;25(3):422-5. [PubMed] [Google Scholar]
Davidson 1993
- Davidson JR, Kudler HS, Saunders WB, Erickson L, Smith RD, Stein RM, et al.Predicting response to amitriptyline in posttraumatic stress disorder. American Journal of Psychiatry 1993;150(7):1024-9. [DOI] [PubMed] [Google Scholar]
Davidson 1997a
- Davidson JR, Malik ML, Sutherland SN.Response characteristics to antidepressants and placebo in post-traumatic stress disorder. International Clinical Psychopharmacology 1997;12(6):291-6. [DOI] [PubMed] [Google Scholar]
Davidson 1997b
- Davidson JR.Biological therapies for posttraumatic stress disorder: an overview. Journal of Clinical Psychiatry 1997;58(Suppl 9):33-6. [PubMed] [Google Scholar]
Davidson 1997c
- Davidson JR, Book SW, Colket L, Tupler LA, Roth S, David D, et al.Assessment of a new self-rating scale for posttraumatic stress disorder. Psychological Medicine 1997;27(1):153-60. [DOI] [PubMed] [Google Scholar]
Davidson 1998
- Davidson JR, Weisler RH, Malik M, Tupler LA.Fluvoxamine in civilians with posttraumatic stress disorder. Journal of Clinical Psychopharmacology 1998;18(1):93-5. [DOI] [PubMed] [Google Scholar]
Davidson 2000
- Davidson JR.Pharmacotherapy of posttraumatic stress disorder: treatment options, long term follow-up, and predictors of outcome. Journal of Clinical Psychiatry 2000;61(Suppl 5):52-6. [PubMed] [Google Scholar]
Davies 2015
- Davies SJ, Champion C, Dawson S, Sharp J, Churchill R.The Subway Map - a new way to visualize antidepressant classification (a Cochrane CCDAN initiative). In: 14th ICGP and 19th JSNP Joint Congress; 3 October. Tsukuba, Japan, 2014: 78. [Abstract P14]. 2015.
Davis 2000
- Davis LL, Nugent AL, Murray J, Kramer GL, Petty F.Nefazodone treatment for chronic posttraumatic stress disorder: an open trial. Journal of Clinical Psychopharmacology 2000;20(2):159-64. [DOI] [PubMed] [Google Scholar]
Deeks 2001
- Deeks JJ, Altman DG, Bradburn MJ.Statistical methods for examining heterogeneity and combining results from several studies in meta-analysis. In: Egger M, Davey SG, Altman DG, editors(s). Systematic Reviews in Health Care: Meta-Analysis in Context. 2nd edition. London: BMJ Publication Group, 2001. [Google Scholar]
Deeks 2002
- Deeks JJ.Issues in the selection of a summary statistic for meta-analysis of clinical trials with binary outcomes. Statistics in Medicine 2002;21(11):1575-600. [DOI] [PubMed] [Google Scholar]
Deeks 2003
- Deeks J, Higgins J, Altman D.Cochrane Reviewers’ Handbook 4.2.1 (updated December 2003). In: Alderson P, Green S, Higgins J, editors(s). The Cochrane Library. Chichester, UK: John Wiley & Sons, Ltd., 2003. [Google Scholar]
Deeks 2011
- Deeks JJ, Altman DG, Bradburn MJ.Statistical methods for examining heterogeneity and combining results from several studies in meta-analysis. In: Egger M, Davey SG, Altman DG editor(s). Systematic Reviews in Health Care: Meta-Analysis in Context. 2nd Edition. BMJ Publication Group, 2008:285–12. [Google Scholar]
De Jong 2001
- De Jong J, Komproe TV, Ivan H, Von Ommeren M, El Masri M, Araya M, et al.Lifetime events and posttraumatic stress disorder in 4 postconflict settings. JAMA 2001;286(5):555-62. [DOI: 10.1001/jama.286.5.555] [DOI] [PubMed] [Google Scholar]
Dillard 1993
- Dillard ML, Bendfeldt F, Jernigan P.Use of thioridazine in post-traumatic stress disorder. Southern Medical Journal 1993;86(11):1276-8. [DOI] [PubMed] [Google Scholar]
Dow 1997
- Dow B, Kline N.Antidepressant treatment of posttraumatic stress disorder and major depression in veterans. Annals of Clinical Psychiatry 1997;9(1):1-5. [DOI] [PubMed] [Google Scholar]
Duffy 1992
- Duffy JD.Rapid response to buspirone in a case of post-traumatic stress disorder. Annals of Clinical Psychiatry 1992;4:193-6. [Google Scholar]
Duffy 1994
- Duffy JD, Malloy PF.Efficacy of buspirone in the treatment of posttraumatic stress disorder: An open trial. Annals of Clinical Psychiatry 1994;6(1):33-7. [DOI] [PubMed] [Google Scholar]
Dunner 1985
- Dunner FJ, Edwards WP, Copeland PC.Clinical efficacy of alprazolam in PTSD patients. In: 138th Annual Meeting of the American Psychiatric Association, Los Angeles, CA. 1985.
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A.Meta-analyses involving cross-over trials: Methodological issues. International Journal of Epidemiology 2002;31(1):140-9. [DOI] [PubMed] [Google Scholar]
Falcon 1985
- Falcon S, Ryan C, Chamberlain K, Curtis G.Tricyclics: possible treatment for post-traumatic stress disorder. Journal of Clinical Psychiatry 1985;46(9):385-9. [PubMed] [Google Scholar]
Famularo 1988
- Famularo R, Kinscherff R, Fenton T.Propranolol treatment for childhood posttraumatic stress disorder, acute type. A pilot study. American Journal of Diseases of Children 1988;142(11):1244-7. [DOI] [PubMed] [Google Scholar]
Fesler 1991
- Fesler FA.Valproate in combat-related posttraumatic stress disorder. Journal of Clinical Psychiatry 1991;52(9):361-4. [PubMed] [Google Scholar]
Fichtner 1990
- Fichtner CG, Kuhlman DT, Gruenfeld MJ, Hughes JR.Decreased episodic violence and increased control of dissociation in a carbamazepine-treated case of multiple personality. Biological Psychiatry 1990;27(9):1045-52. [DOI] [PubMed] [Google Scholar]
Fichtner 1994
- Fichtner CG, Arora RC, O'Connor FL, Crayton JW.Platelet paroxetine binding and fluoxetine pharmacotherapy in posttraumatic stress disorder: preliminary observations on a possible predictor of clinical treatment response. Life Sciences 1994;54(3):39-44. [DOI] [PubMed] [Google Scholar]
Foa 1999
- Foa EB, Davidson JR, Frances A.The expert consensus guideline series: treatment of posttraumatic stress disorder: the expert consensus panel for PTSD. Journal of Clinical Psychiatry 1999;60(Suppl. 16):1-75. [PubMed] [Google Scholar]
Ford 1996
- Ford N.The use of anticonvulsants in posttraumatic stress disorder: case study and overview. Journal of Traumatic Stress 1996;9(4):857-63. [DOI] [PubMed] [Google Scholar]
Forster 1994
- Forster PL, Schoenfield FB, Marmar CR, Lang AJ.Lithium for irritability in post-traumatic stress disorder. Journal of Traumatic Stress 1994;8(1):143-9. [DOI] [PubMed] [Google Scholar]
Franco 2016
- Franco S, Hoertel N, McMahon K, Wang S, Rodríguez-Fernández JM, Peyre H, et al.Generalizability of pharmacologic and psychotherapy clinical trial results for posttraumatic stress disorder to community samples. Journal of Clinical Psychiatry 2016;77(8):975-81. [DOI: doi: 10.4088/JCP.15m10060] [DOI] [PubMed] [Google Scholar]
Friedman 2016
- Friedman MJ.PTSD: National Center for PTSD. U.S Department of Veterans Affairs 2016.
Gersons 2000
- Gersons BP, Carlier IV, Lamberts RD, Van der Kolk BA.Randomized clinical trial of brief eclectic psychotherapy for police officers with posttraumatic stress disorder. Journal of Trauma Stress 2000;13(2):333-47. [DOI] [PubMed] [Google Scholar]
Gilboa‐Schechtman 2010
- Gilboa-Schechtman E, Foa EB, Shafran N, Aderka IM, Powers MB, Rachamim L, et al.Prolonged exposure versus dynamic therapy for adolescent PTSD: a pilot randomized controlled trial. Journal of the American Academy of Child and Adolescent Psychiatry 2010;49(10):1034-42. [DOI: 10.1016/j.jaac.2010.07.014] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gist 2015
- Gist R.Psychological debriefing. In: Encyclopaedia of Clinical Psychology. Chichester: John Wiley & Sons, Inc, 2015. [Google Scholar]
Glover 1993
- Glover H.A preliminary trial of nalmefene for the treatment of emotional numbing in combat veterans with post-traumatic stress disorder. Israel Journal of Psychiatry and Related Sciences 1993;30(4):255-63. [PubMed] [Google Scholar]
Gupta 1998
- Gupta S, Popli A, Bathurst E, Hennig L, Droney T, Keller P.Efficacy of cyproheptadine for nightmares associated with posttraumatic stress disorder. Comprehensive Psychiatry 1998;39(3):160-4. [DOI] [PubMed] [Google Scholar]
Gury 1999
- Gury C, Cousin F.Pharmacokinetics of SSRI antidepressants: half-life and clinical applicability. Encephale 1999;25(5):470-6. [PubMed] [Google Scholar]
Guy 1976
- Guy W.ECDEU assessment manual for psychopharmacology. Washington, DC: U.S. National Institute of Health, 1976. [Google Scholar]
Hamilton 1959
- Hamilton M.The assessment of anxiety states by rating. British Journal of Medical Psychology 1959;32(1):50-5. [DOI] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M.A rating scale for depression. Journal of Neurology, Neurosurgery & Psychiatry 1960;23:56-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamner 1996
- Hamner MB.Clozapine treatment for a veteran with comorbid psychosis and PTSD (letter). American Journal of Psychiatry 1996;153(6):841. [DOI] [PubMed] [Google Scholar]
Hamner 1998
- Hamner MB, Frueh BC.Response to venlafaxine in a previously antidepressant treatment-resistant combat veteran with post-traumatic stress disorder. International Clinical Psychopharmacology 1998;13(5):233-4. [DOI] [PubMed] [Google Scholar]
Harmon 1996
- Harmon RJ, Riggs PD.Clonidine for posttraumatic stress disorder in preschool children. Journal of the American Academy of Child & Adolescent Psychiatry 1996;35(9):1247-9. [DOI] [PubMed] [Google Scholar]
Harvey 2003
- Harvey AG, Bryant RA, Tarrier N.Cognitive behaviour therapy for posttraumatic stress disorder. Clinical Psychology Review 2003;23(3):501-22. [DOI] [PubMed] [Google Scholar]
Hertzberg 1996
- Hertzberg MA, Feldman ME, Beckham JC, Davidson JR.Trial of trazodone for posttraumatic stress disorder using a multiple baseline group design. Journal of Clinical Psychopharmacology 1996;16(4):294-8. [DOI] [PubMed] [Google Scholar]
Hertzberg 1998
- Hertzberg MA, Feldman ME, Beckham JC, Moore SD, Davidson JR.Open trial of nefazodone for combat-related posttraumatic stress disorder. Journal of Clinical Psychiatry 1998;59(9):460-4. [DOI] [PubMed] [Google Scholar]
Hidalgo 1999
- Hidalgo R.Nefazodone in posttraumatic stress: results from six open-label trials. International Clinical Psychopharmacology 1999;14(2):61-8. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JT, Thompson SG, Deeks JJ, Altman DG.Measuring inconsistency in meta-analyses. BMJ 2003;327(6):557-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Greens S, editor(s).Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1/.
Hogben 1981
- Hogben GL, Cornfield RB.Treatment of traumatic war neurosis with phenelzine. Archives of General Psychiatry 1981;38(4):440-5. [DOI] [PubMed] [Google Scholar]
Hopewell 2002
- Hopewell S, Clarke M, Lusher A, Lefebvre C, Westby M.A comparison of handsearching versus MEDLINE searching to identify reports of randomized controlled trials. Statistics in Medicine 2002;21(11):1625-34. [DOI] [PubMed] [Google Scholar]
Horowitz 1979
- Horowitz M, Wilner N, Alvarez W.Impact of Event Scale: a measure of subjective stress. Psychosomatic Medicine 1979;41(3):209-18. [DOI] [PubMed] [Google Scholar]
Horrigan 1996
- Horrigan JP.Guanfacine for PTSD nightmares. Journal of the American Academy of Child & Adolescent Psychiatry 1996;35(8):975-6. [DOI] [PubMed] [Google Scholar]
Hoskins 2015
- Hoskins M, Pearce J, Bethell A, Dankova L, Barbui C, Tol WA, et al.Pharmacotherapy for post-traumatic stress disorder: systematic review and meta-analysis. British Journal of Psychiatry 2015;206(2):93–100. [DOI: 10.1192/bjp.bp.114.148551] [DOI] [PubMed] [Google Scholar]
Ipser 2012
- Ipser JC, Stein DJ.Evidence-based pharmacotherapy of post-traumatic stress disorder (PTSD). International Journal of Neuropsychopharmacology 2012;15(6):825–40. [DOI: ] [DOI] [PubMed] [Google Scholar]
Irwin 1989
- Irwin M, Van-Putten T, Guze B, Marder SR.Pharmacologic treatment of veterans with posttraumatic stress disorder and concomitant affective disorder. Annals of Clinical Psychiatry 1989;1(2):127-30. [Google Scholar]
Izrayelit 1998
- Izrayelit L.Schizoaffective disorder and PTSD successfully treated with olanzapine and supportive psychotherapy. Psychiatric Annals 1998;28(8):424-6. [Google Scholar]
Keck 1992
- Keck PE, McElroy SL, Friedman LM.Valproate and carbamazepine in the treatment of panic and posttraumatic stress disorders. Journal of Clinical Psychopharmacology 1992;12(1S):36-41. [DOI] [PubMed] [Google Scholar]
Kessler 1995
- Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB.Posttraumatic stress disorder in the National Comorbidity Survey. Archives of General Psychiatry 1995;52(12):1048-60. [DOI] [PubMed] [Google Scholar]
Kessler 2005
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE.Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry 2005;62:617-27. [DOI: 10.1001/archpsyc.62.6.617] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kinzie 1989
- Kinzie JD, Leung P.Clonidine in Cambodian patients with post traumatic stress disorder. Journal of Nervous and Mental Disease 1989;177(9):546-50. [DOI] [PubMed] [Google Scholar]
Koenen 2017
- Koenen KC, Ratanatharathorn A, Ng L, McLaughlin KA, Bromet EJ, Stein DJ, et al.Posttraumatic stress disorder in the World Mental Health Surveys. Psychological Medicine 2017;47(13):2260–74. [DOI: 10.1017/S0033291717000708] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kolb 1984
- Kolb LC, Burris BC, Griffiths S.Propranolol and clonidine in the treatment of the chronic post-traumatic stress disorders of war. In: Van der Kolk BA, editors(s). Post-traumatic Stress Disorder: Psychological and Biological Sequelae. Washington, DC: American Psychiatric Press, 1984:98-105. [Google Scholar]
LaPorta 1992
- LaPorta LD, Ware MR.Buspirone in the treatment of posttraumatic stress disorder. Journal of Clinical Psychopharmacology 1992;12(2):133-4. [DOI] [PubMed] [Google Scholar]
Lerer 1987
- Lerer B, Bleich A, Kotler M, Garb R, Hertzberg M, Levin B.Posttraumatic stress disorder in Israeli combat veterans: effect of phenelzine treatment. Archives of General Psychiatry 1987;44(11):976-81. [DOI] [PubMed] [Google Scholar]
Leyba 1998
- Leyba CM, Wampler TP.Risperidone in PTSD. Psychiatric Services 1998;49(2):245-6. [DOI] [PubMed] [Google Scholar]
Liebowitz 1989
- Liebowitz NR, El Mallakh RS.Trazodone for the treatment of anxiety symptoms in substance abusers. Journal of Clinical Psychopharmacology 1989;9(8):449-51. [PubMed] [Google Scholar]
Lipper 1986
- Lipper S, Davidson JR, Grady TA, Edinger JD, Hammett EB, Mahorney SL, et al.Preliminary study of carbamazepine in post traumatic stress disorder. Psychosomatics 1986;27(12):849-54. [DOI] [PubMed] [Google Scholar]
Looff 1995
- Looff D, Grimley P, Kuller F, Martin A, Shonfield L.Carbamazepine for PTSD. Journal of the American Academy of Child & Adolescent Psychiatry 1995;34(6):703-4. [DOI] [PubMed] [Google Scholar]
Lowenstein 1988
- Lowenstein RJ, Hornstein N, Farber B.Open trial of clonazepam in the treatment of post traumatic stress symptoms in multiple personality disorder. Dissociation 1988;1:3-12. [Google Scholar]
Marshall 1996
- Marshall RD, Stein DJ, Liebowitz MR, Yehuda R.Psychopharmacology of posttraumatic stress disorder. Psychiatric Annals 1996;26:217-26. [Google Scholar]
Marshall 1998a
- Marshall RD, Davidson JR, Yehuda R.Pharmacotherapy in the treatment of posttraumatic stress disorder and other trauma-related disorders. In: Yehuda R, editors(s). Psychological Trauma. Washington, DC: American Psychiatric Press, 1998:133-77. [Google Scholar]
Marshall 1998b
- Marshall RD, Schneier FR, Fallon BA, Knight CB, Abbate LA, Goetz D, et al.An open trial of paroxetine in patients with noncombat-related posttraumatic stress disorder. Journal of Clinical Psychopharmacology 1998;18(1):10-8. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall RD, Pierce D.Implications of recent findings in posttraumatic stress disorder and the role of pharmacotherapy. Harvard Review of Psychiatry 2000;7(5):247-56. [PubMed] [Google Scholar]
Milanes 1984
- Milanes F, Mack C.Phenelzine treatment of post-vietnam stress syndrome. VA Practitioner 1984;1:40-9. [Google Scholar]
Moher 1999
- Moher D, Cook DJ, Eastwood S, Olkin I, Rennie D, Stroup DF.Improving the quality of reports of meta-analyses of randomized controlled trials: the QUOROM statement. Lancet 1999;354(9193):1896-900. [DOI] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M.A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382-9. [DOI] [PubMed] [Google Scholar]
Mulrow 1997
- Mulrow CD, Oxman AD.Cochrane Collaboration Handbook. Oxford: Update Software, 1997. [Google Scholar]
Neal 1997
- Neal LA, Shapland W, Fox C.An open trial of moclobemide in the treatment of post-traumatic stress disorder. International Clinical Psychopharmacology 1997;12(4):231-7. [DOI] [PubMed] [Google Scholar]
NICE 2018
- NICE.Post-traumatic stress disorder: management (NG116). Clinical guideline Published: December 2018. National Institute for Health and Care Excellence 2018.
Olivera 1990
- Olivera AT, Fero D.Affective disorders, DST, and treatment in PTSD patients: clinical observations. Journal of Traumatic Stress 1990;3:407-14. [Google Scholar]
Penava 1996
- Penava SJ, Otto MW, Pollack MH, Rosenbaum JF.Current status of pharmacotherapy for PTSD: an effect size analysis of controlled studies. Depression and Anxiety 1996;4(5):240-2. [DOI] [PubMed] [Google Scholar]
Ravindran 2009
- Ravindran LN, Stein MB.Pharmacotherapy of PTSD: premises, principles, and priorities. Brain Research 2009;1293:24–39. [DOI: 10.1016/j.brainres.2009.03.037] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ressler 2018
- Ressler KJ.Alpha-adrenergic receptors in PTSD -failure or time for precision medicine? New England Journal of Medicine 2018;378:575-6. [DOI: 10.1056/NEJMe1716724] [DOI] [PubMed] [Google Scholar]
RevMan 2020 [Computer program]
- Review Manager 5 (RevMan 5).Version 5.4. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2020.
Rose 1998
- Rose S, Bisson J.Brief early psychological interventions following trauma: a systematic review of the literature. Journal of Traumatic Stress 1998;11(4):697-710. [DOI] [PubMed] [Google Scholar]
Rose 2002
- Rose S, Bisson J, Wessely S.Psychological debriefing for preventing post traumatic stress disorder (PTSD). Cochrane Database of Systematic Reviews 2002, Issue 2. Art. No: CD000560. [DOI: 10.1002/14651858.CD000560] [DOI] [PubMed] [Google Scholar]
Rubin 1993
- Rubin MR, Solt V, Chen C-J, et al.Anafranil's effect on the obsessions and sleep in PTSD. In: Proceedings and Abstracts of the 146th Annual Meeting of the American Psychiatric Association, San Francisco, CA. 1993:102.
Sargent 1940
- Sargent WW, Slater E.Acute war neuroses. Lancet 1940;140:1-2. [Google Scholar]
Shalev 1996
- Shalev A, Bonne E, Eth S.Treatment of posttraumatic stress disorder: a review. Psychosomatic Medicine 1996;58(2):162-82. [DOI] [PubMed] [Google Scholar]
Sheehan 1996
- Sheehan DV, Harnett-Sheehan K, Raj BA.The measurement of disability. International Clinical Psychopharmacology 1996;11 Suppl 3:89-95. [DOI] [PubMed] [Google Scholar]
Sherin 2011
- Sherin JE, Nemeroff CB.Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues in Clinical Neuroscience 2011;13(3):263-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Simpson 1991
- Simpson MA.Buspirone, benzodiazepines, and post-traumatic stress disorder. Journal of Traumatic Stress 1991;4:305-8. [Google Scholar]
Solomon 1997
- Solomon SD, Davidson JR.Trauma: prevalence, impairment, service use, and cost. Journal of Clinical Psychiatry 1997;58(Suppl 5):1-11. [PubMed] [Google Scholar]
Spitzer 1996
- Spitzer R, Williams J, Gibbons M.Instructional manual for the structured clinical interview for DSM-IV. New York. NY: New York State Psychiatric Institute, 1996. [Google Scholar]
Stahl 2013
- Stahl SM.Stahl’s essential psychopharmacology. Neuroscientific Basis and Practical Application. Fourth Edition. Cambridge, UK: Cambridge University Press, 2013. [Google Scholar]
Szymanski 1991
- Szymanski HV, Olympia J.Divalproex in posttraumatic stress disorder. American Journal of Psychiatry 1991;148(8):1086-7. [DOI] [PubMed] [Google Scholar]
Thomson 1994
- Thomson SG.Why sources of heterogeneity in meta-analysis should be investigated. BMJ 1994;309(6965):1351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ursano 2004
- Ursano RJ, Bell C, Eth S, Friedman M, Norwood A, Pfefferbaum B, et al.Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. American Journal of Psychiatry 2004;161(11 Suppl):3-31. [PubMed] [Google Scholar]
Verbeke 2000
- Verbeke M, Molenberghs G.Linear mixed models for longitudinal data. New York: Springer 2000.
Watkins 2018
- Watkins LE, Sprang KR, Rothbaum BO.Treating PTSD: a review of evidence-based psychotherapy interventions. Frontiers in Behavioral Neuroscience 2018;12:258. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weathers 1999
- Weathers FW, Ruscio AM, Keane TM.Psychometric properties of nine scoring rules for the Clinician-Administered Posttraumatic Stress Disorder Scale. Psychological Assessment 1999;11:124-33. [Google Scholar]
Weiss 1997
- Weiss DS, Marmar CR.The Impact of Event Scale—Revised. In: Wilson JP, Keane TM, editors(s). Assessing psychological trauma and PTSD: a handbook for practitioners. New York, NY: Guilford Press, 1997:399-411. [Google Scholar]
Wells 1991
- Wells GB, Chu C, Johnson R, Nasdahl C, Ayubi MA, Sewell E, et al.Buspirone in the treatment of post-traumatic stress disorder and dream anxiety disorder. Military Medicine 1991;11(4):340-3. [PubMed] [Google Scholar]
White 1983
- White NS.Post-traumatic stress disorder. Hospital and Community Psychiatry 1983;34(11):1061-2. [DOI] [PubMed] [Google Scholar]
Yehuda 1995
- Yehuda R, McFarlane AC.Conflict between current knowledge about posttraumatic stress disorder and its original conceptual basis. American Journal of Psychiatry 1995;152(12):1705-13. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Stein 2000
- Stein DJ, Zungu‐Dirwayi N, Linden G, Seedat S.Pharmacotherapy for post traumatic stress disorder (PTSD). Cochrane Database of Systematic Reviews 2000, Issue 4. Art. No: CD002795. [DOI: 10.1002/14651858.CD002795] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stein 2006
- Stein DJ, Ipser JC, Seedat S, Sager C, Amos T.Pharmacotherapy for post traumatic stress disorder (PTSD). Cochrane Database of Systematic Reviews 2006, Issue 1. Art. No: CD002795. [DOI: 10.1002/14651858.CD002795.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]