

Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disorder characterized by dementia. Inhibition of soluble epoxide hydrolase (sEH) regulates inflammation involving in central nervous system (CNS) diseases. However, the exactly mechanism of sEH in AD is still unclear. In this study, we evaluated the vital role of sEH in amyloid beta (Aβ)-induced AD mice, and revealed a possible molecular mechanism for inhibition of sEH in the treatment of AD. The results showed that the sEH expression and activity were remarkably increased in the hippocampus of Aβ-induced AD mice. Chemical inhibition of sEH by TPPU, a selective sEH inhibitor, alleviated spatial learning and memory deficits, and elevated levels of neurotransmitters in Aβ-induced AD mice. Furthermore, inhibition of sEH could ameliorate neuroinflammation, neuronal death, and oxidative stress via stabilizing the in vivo level of epoxyeicosatrienoic acids (EETs), especially 8,9-EET and 14,15-EET, further resulting in the anti-AD effect through the regulation of GSK3β-mediated NF-κB, p53, and Nrf2 signaling pathways. These findings revealed the underlying mechanism of sEH as a potential therapeutic target in treatment of AD.
Keywords: Alzheimer’s disease, Soluble epoxide hydrolase, TPPU, Epoxyeicosatrienoic acids, GSK3β
1. Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder characterized by cognitive dysfunction, language impairment, and emotional changes as clinical symptoms (Tarawneh and Holtzman, 2012). The number of AD patients is about 47 million worldwide in 2016, which will exceed 131 million by 2050 (Eguchi et al., 2018; Prince et al., 2016). So far, the clinicopathologic feature of AD is mainly focused on amyloid beta (Aβ) plaques and neurofibrillary tangles (Yang et al., 2018). The Aβ plaques are the extracellular deposit of Aβ produced by the cleavage of amyloid precursor protein (APP) (Tanzi et al., 1987), and the neurofibrillary tangles consist of abnormal filaments of Tau hyperphosphorylation by glycogen synthase kinase 3 beta (GSK3β) (Mandelkow and Mandelkow, 1998). Recent evidence has indicated that Aβ plaques and neurofibrillary tangles mainly result in synaptic loss or dysfunction and neuronal death, allowing in the cognitive impairment of AD (Carter and Lippa, 2001; Shankar and Walsh, 2009). Moreover, Aβ activates microglia and astrocyte to promote the production of pro-inflammatory cytokines, including necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), which induce neuronal damage (Rubio-Perez and Morillas-Ruiz, 2012; Wang et al., 2015). In addition, Aβ plays an important role in oxidative stress and affect synaptic plasticity and cognitive dysfunction in AD (Butterfield et al., 2001; Kadowaki et al., 2005). Hence, it is important to develop a new target to prevent AD by multi-pathogenic process.
Soluble epoxide hydrolase (sEH) is an α/β hydrolase fold protein with a C-terminal hydrolase and a N-terminal phosphatase function, which is mainly expressed in the cytosol of tissues, including liver, kidney, lungs, intestine, heart, and brain (Newman et al., 2005). sEH can hydrolyze epoxyeicosatrienoic acids (EETs) and other epoxy-fatty acids (EpFAs) to produce their corresponding diols, such as dihydroxyeicosatrienoic acids (DHETs) (Morisseau and Hammock, 2005), whereby decreasing and eliminating the beneficial effects of EETs, such as anti-inflammatory and hypotensive effects (Falck et al., 2003; Imig et al., 1996; Lee et al., 1999; Roman, 2002; Zeldin, 2001). Inhibition of sEH to stabilize the level of EETs is able to suppress the transcription of nuclear factor kappa B (NF-κB) responsible for cytokine-mediated inflammation and further inhibit NF-κB-mediated gene transcription, exerting the anti-inflammatory effect (Campbell, 2000; Node et al., 1999). Recently, several studies have revealed that sEH is related to hypertension, Parkinson’s disease (PD), non-alcoholic fatty liver disease (NAFLD), and diabetes (Duflot et al., 2014; Imig, 2016; Kodani and Morisseau, 2019; Lee et al., 2019). Inhibition of sEH or Ephx2 genetic deletion can protect against MPTP-induced neurotoxicity in striatum (STR) and substantia nigra (SN) and alleviate MPTP-mediated loss of TH positive cells in SN in PD (Huang et al., 2018; Qin et al., 2015; Ren et al., 2018). In addition, some of evidence demonstrate that the expression of sEH is higher in AD patients’ brains (Ghosh et al., 2020; Grinan-Ferre et al., 2020), and Ephx2 abolishment also improves the symptom of APP/presenilin 1 (PS1) transgenic (Tg) mice (Lee et al., 2019). However, it is a question worth thinking about whether inhibition of sEH alleviates neuroinflammation, neuronal death, and oxidative stress in AD, and the molecular mechanism for the treatment of AD via suppressing sEH. Therefore, we evaluated the role of sEH in Aβ-induced AD mice, and explored its underlying therapeutic mechanism via chemical inhibition of sEH by TPPU, a selective sEH inhibitor.
2. Materials and methods
2.1. Materials
TPPU was purchased from R&D Systems, Inc (Minneapolis, USA). The primary antibodies COX-2, Grp78, IKKβ, p-IKKβ, p65, p-p65, caspase 9, cleaved-caspase 9, caspase 3, cleaved-caspase 3, and p-GSK3β (Ser9) were purchased from Cell Signaling Technology (CST, Danvers, MA, USA). The primary antibodies Tau, p-Tau (Thr205), p-Tau (Ser262), p-Tau (Ser404), HO-1, GSK3β, and sEH were purchased from ABclonal (Wuhan, China). The primary antibodies GAPDH, Aβ, Bax, IL-6, and GFAP were purchased from Proteintech (Wuhan, China). The primary antibodies Bcl-2, Iba-1, Nrf2, and Keap1 were purchased from Abcam (Cambridge, United Kingdom). Aβ1–42 (Aβ) was purchased from Sigma Aldrich (St. Louis, Missouri, USA).
2.2. Animals and experimental procedure
Male C57BL/6 mice (23–25 g, 8 weeks) were afforded from the Experimental Animal Center of Dalian Medical University (Dalian, China). All animal experiments were in accordance with the Institutional Animal Care and Use Committee of Dalian Medical University. All the animals were kept under a light/dark cycle of 12 h per day at a controlled temperature of 22–24 °C and 50–60% humidity with water and food ad libitum.
Firstly, the mice model of Aβ-induced AD was applied to reveal the sEH expression level in AD mice. Mice were randomly divided into two groups (6 mice/group): the sham group (only administrated with vehicle) and Aβ group. Aβ was prepared in saline at a concentration of 400 pmol/2 μL, followed by aggregation via incubation at 37 °C for 7 days. Mice were anesthetized and placed in a stereotaxic apparatus (ZSDichuang, Beijing, China), and 2 μL of Aβ was injected for 2 min into the lateral ventricle coordinates from bregma: anteroposterior (AP) = − 0.3 mm, mediolateral (ML) = − 1.0 mm, and dorsoventral (DV) = 2.5 mm. The sham group was administrated with saline (i.p.) described as the treatment group.
Secondly, the mice model of Aβ-induced AD was also used to investigate the effect of chemical inhibition of sEH by a sEH inhibitor TPPU in the course of AD. Mice were randomly divided into four groups (12 mice/group): the sham group (only administrated with vehicle), TPPU (4 mg/kg) group, Aβ group, and Aβ + TPPU (4 mg/kg) group. The mice model of Aβ-induced AD was constructed as the abovementioned method of Aβ infusion (i.c.v.). Mice in TPPU group or Aβ + TPPU group were administrated with TPPU (4 mg/kg, dissolved in saline, i.p.) for 14 days after 2 days of i.c.v. infusion of saline or Aβ. The sham group was administrated with saline (i.p.) described as the treatment group.
2.3. Morris Water Maze Test
Morris Water Maze (MWM) test was used to evaluate the spatial learning and memory ability of mice according to previously described (Hou et al., 2019). The behavioral apparatus included a circular water pool (120 cm diameter, 40 cm height) filled with water (23 ± 2 °C, 30 cm). The pool was divided into four quadrants, and a cylindrical platform was located in the middle of a quadrant and placed about 1 cm below the surface of the water. Mice were placed in the pool facing the wall of four quadrants and given 90 s to find the platform. If they could not find the platform in this period, they were gently guided to the platform for 30 s before being removed from the platform. In the spatial navigation trial, mice were tested in two times every day, lasted for 4 days, and the experimental data from the 4th day was recorded. In the spatial probe trial, the platform was removed from the pool at 5th day, the mice were gently released into four quadrants respectively, allowed to swim freely in the pool for 60 s., and the experimental data from the 5th day was recorded. The data for escape latency, the time spent, time ratio, distances, and distance ratio in the target quadrant, and the number of platform crossings were recorded by video-tracking software (Huaibei Zhenghua Biological Instrument Equipment Co., Ltd., Suixi, China).
2.4. Determination of MDA, GSH, and T-SOD
The levels of MDA, glutathione (GSH), and total superoxide dismutase (T-SOD) in the frontal cortex were determined by corresponding kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to their instructions.
2.5. Western Blot
The hippocampal samples of mice were washed twice with PBS, added an appropriate amount of lysis RIPA buffer containing protease and phosphatase inhibitors, and then the lysates were centrifuged at 12000 g at 4 °C for 10 min. The supernatants were collected for western blot analysis after protein concentrations were determined by BCA Protein Assay Kit (Beyotime Institute of Biotechnology, Hangzhou, China). The equal amount of protein was separated by 10%−12% SDS-PAGE gel and transferred to polyvinylidenedifluoride (PVDF) membranes. After blocking via the incubation of 5% skim milk for 1 h at 37 °C, membranes were incubated with specific primary antibodies overnight at 4 °C. The membranes were conjugated with appropriate secondary antibodies at 37 °C for 2 h. The protein bands were detected by enhanced chemiluminescence (ECL). Image J software (Rawak Software Inc., Germany) was used to quantitate protein expression level.
2.6. Immunohistochemistry staining
Mouse brains were fixed with 4% paraformaldehyde, and 20% and 30% sucrose solution for 24 h, successively. Hippocampus samples were cut into 10 μm thick sections (Leica, Germany) for the immunohistochemistry and immunofluorescence assay. The sections were blocked with 10% goat serum at 37 °C for 1.5 h, and then were conjugated with primary antibodies Iba-1 (1:100) and Aβ (1:500) at 4 °C overnight. After the sections was washed with PBS for three times, they were incubated with a horseradish peroxidase-conjugated secondary antibody at 37 °C for 30 min, and then stained with DAB kit (Beyotime, Shanghai, China).
2.7. Immunofluorescence staining
The sections were blocked with 10% goat serum in 0.3% Triton-100/PBS at 37 °C for 1.5 h, and then were incubated with the primary antibody GFAP (1:100) at 4 °C overnight. After the sections was washed with PBS for three times, they were conjugated to the secondary antibody ALEX Fluor 594 at 37 °C for 1.5 h. Added a small amount of DAPI (Beyotime, China) to cover the tissue sections and leaved them at room temperature for 3–5 min. Absorbed DAPI staining solution and rinsed the slices with PBS for 5 min each time for three times.
2.8. TUNEL staining
The terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay was performed by using an in situ cell death detection kit (Roche, Mannheim, Germany) following the manufacturer’s instruction. Briefly, the brain sections were washed with 0.85% NaCl and PBS, and then fixed in 4% formaldehyde for 15 min. After being washed with PBS, the sections were treated with proteinase K solution for 15 min. After another PBS wash, the sections were covered with the TUNEL reaction mixture and incubated for 1 h. By terminating the reaction by three washes with PBS, the sections were examined and photographed using a fluorescence microscope (Leica, Wetzlar, Germany).
2.9. LC-MS/MS analysis
The levels of dopamine (DA), dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 5-hydroxytryptamine (5-HT), 5-hydroxyindole acetic acid (5-HIAA), norepinephrine (NE), 8,9-EET, 11,12-EET, 14,15-EET, 8,9-DHET, 11,12-DHET, and 14,15-DHET in the hippocampus were determined by LC-MS/MS based on the previous methods (Liu et al., 2019; Zhao et al., 2020). Hippocampus tissues were weighed, homogenized in the ice-water bath, and then centrifuged at 20000 g for 20 min. The supernatant was collected, and then analyzed using a multiple reaction monitoring (MRM) method and an AB Sciex Qtrap® 5500 LC-MS/MS system (Foster City, CA, USA). The levels of DA, DOPAC, HVA, 5-HT, 5-HIAA, NE, 8,9-EET, 11,12-EET, 14,15-EET, 8,9-DHET, 11,12-DHET, and 14,15-DHET were calculated based on themselves standard curves.
2.10. Statistical analysis
All data were analyzed using GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA, USA). The Data were presented as means ± SEM. The statistical analyses were performed by two-way analysis of variance (ANOVA) followed by Tukey’s post-hoc testing. Difference was considered significant at p < 0.05.
3. Results
3.1. The sEH expression level and activity were increased in the AD mice
Recently, some of studies demonstrated that the sEH expression level was higher than that of healthy individuals both in the brain of AD patients and in the hippocampus of SAMP8 and 5×FAD mice (Ghosh et al., 2020; Grinan-Ferre et al., 2020). However, the sEH activity has not been reported in AD mice and patients, therefore, we constructed an AD mice model induced by Aβ to investigate the sEH expression level and activity. As shown in Fig. 1A and B, the sEH expression level in the hippocampus of AD mice was nearly twofold increase of normal mice. EETs, their corresponding diols DHETs, and their ratio of DHETs/EETs are important indexes to reflect the sEH activity, therefore, we used LC-MS/MS to study the content of EETs and DHETs in hippocampus of AD mice. The contents of 8,9-EET and 14,15-EET were significantly decreased in the hippocampus of AD mice except 11,12-EET, and those of 8,9-DHET, 11,12-DHET, and 14,15-DHET in the hippocampus of AD mice was significantly increased (Fig. 1C). The ratios of DHETs and their corresponding epoxides EETs, including 8,9-DHET/8,9-EET, 11,12-DHET/11,12-EET, and 14,15-DHET/14,15-EET, were all increased in AD mice (Fig. 1D–F). This finding suggested that sEH played a role in the AD pathology.
Fig. 1.
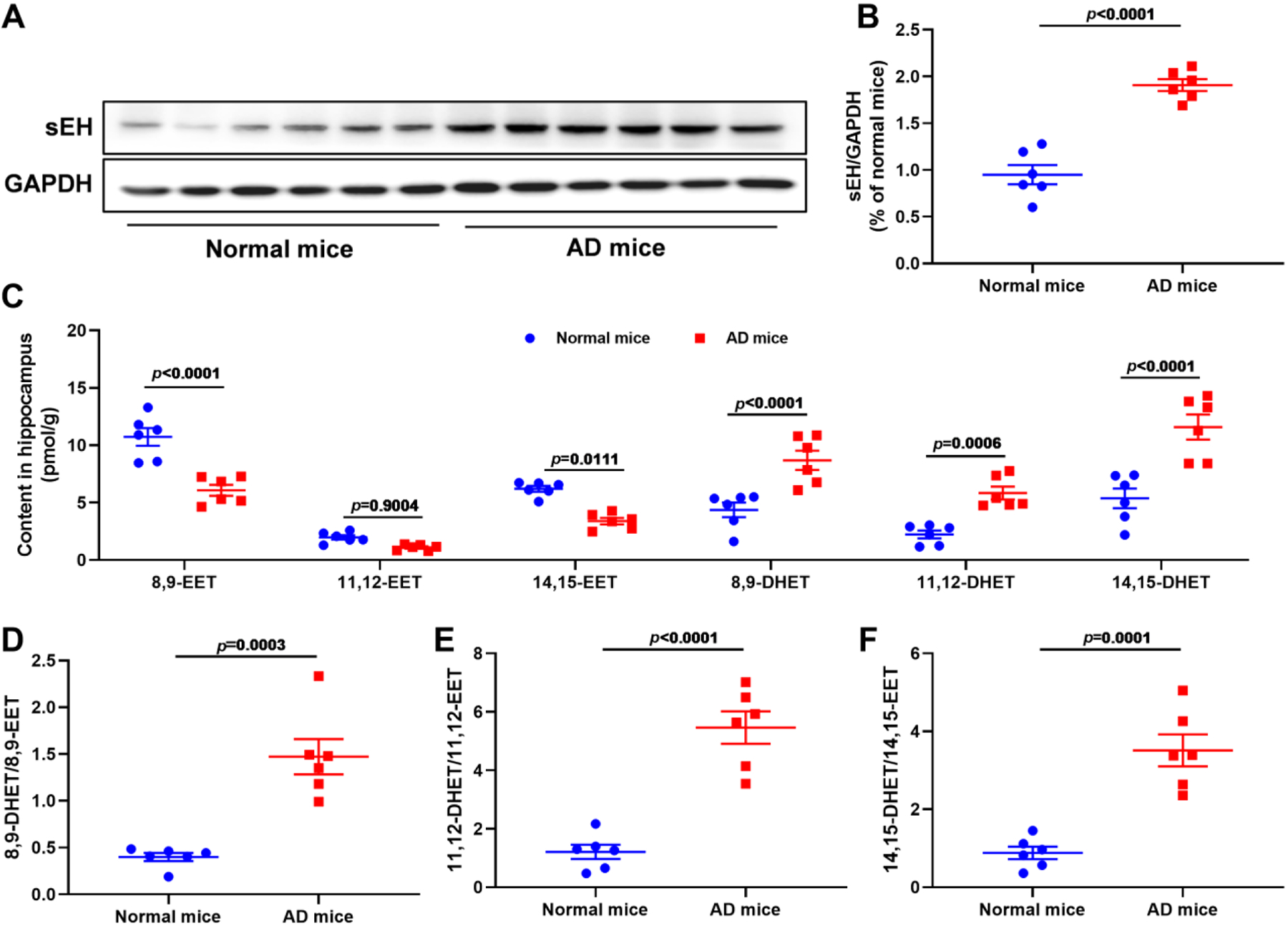
The sEH expression level and activity were increased in Aβ-induced AD mice AD mice. (A) The sEH expression levels in normal and AD mice. (B) Quantitative data of sEH. Data represent mean ± SEM, n = 6. (C) The content of 8,9-EET, 11,12-EET, 14,15-EET, and their corresponding diols 8,9-DHET, 11,12-DHET, and 14,15-DHET in the hippocampus of normal and AD mice. Data represent mean ± SEM, n = 6. (D-F) The ratio of 8,9-DHET/8,9-EET (D), 11,12-DHET/11,12-EET (E), and 14,15-DHET/14,15-EET (F) in normal and AD mice. Data represent mean ± SEM, n = 6.
3.2. Inhibition of sEH by TPPU alleviated spatial learning and memory deficits in Aβ-induced AD mice
To explore the effect of sEH against AD, TPPU, a selective sEH inhibitor, was used to investigate the influence of spatial learning and memory deficits in Aβ-induced AD mice. Compared with the sham group, AD mice took longer to reach the platform (Fig. 2B and H), while inhibition of sEH by TPPU significantly reduced the escape time of AD mice (Fig. 2B and H). After the 90 s training session with a platform, the platform was removed to evaluate the time, time ratio, distance, distance ratio, and number of platform crossings in target quadrant of AD mice during 60 s exploring session, the swimming trajectory mice was recorded and depicted in Fig. 2I. As shown in Fig. 2C–G and I, AD mice spent less time in the target quadrant compared with that of the sham group, while the result was reversed after specific inhibition of sEH by TPPU. The similar results, including time ratio, distances, distance ratio, and number of platform crossings in the target quadrant, were observed (Fig. 2D–G) These findings all suggested that inhibition of sEH by TPPU could alleviate spatial learning and memory deficits in Aβ-induced AD mice.
Fig. 2.
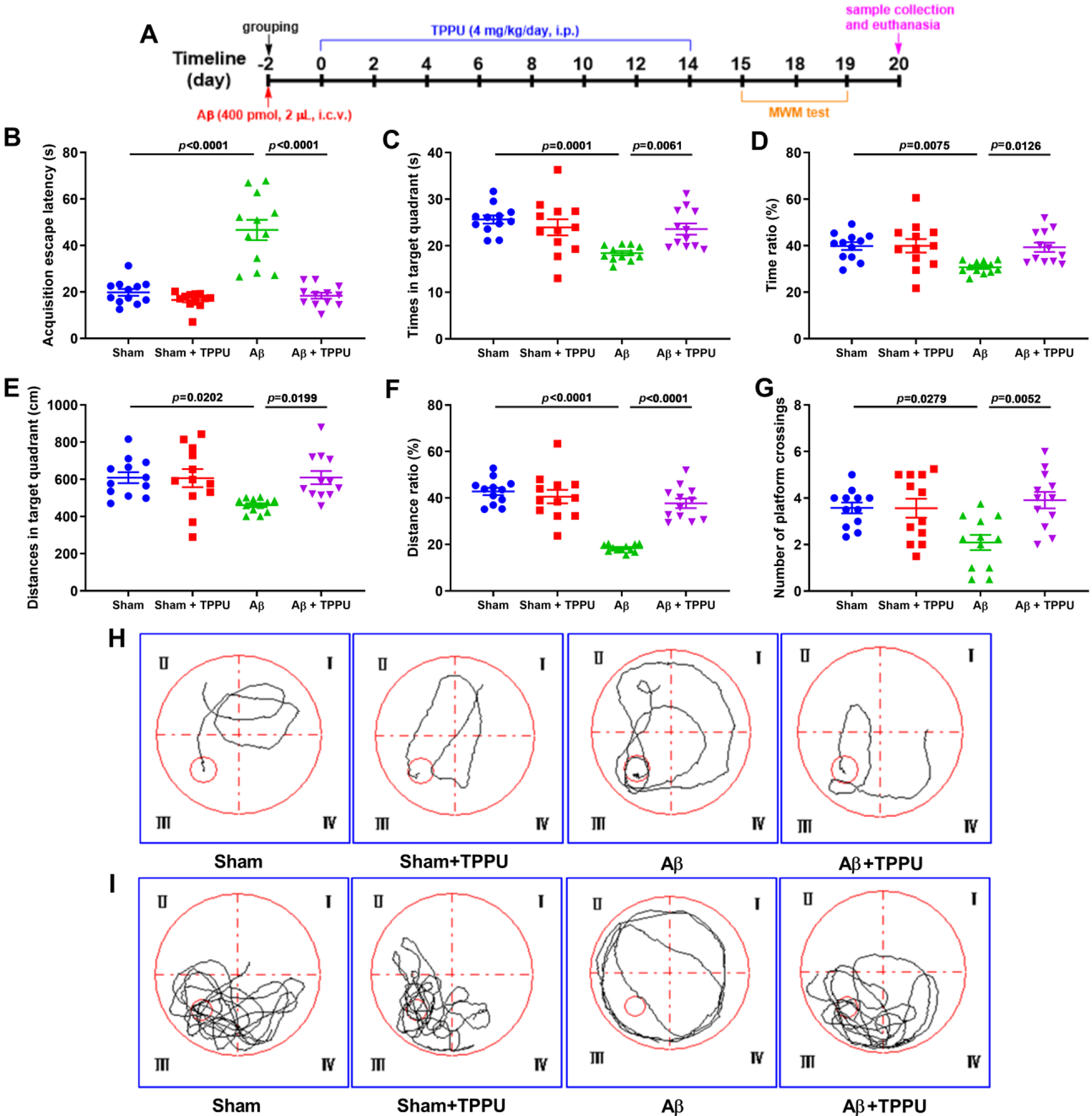
Inhibition of sEH by TPPU alleviated spatial learning and memory deficits in Aβ-induced AD mice assessed by the Morris water maze (MWM) test. (A) The flow chart of experiments. (B) The escape latency of mice in 90 s training session with a platform. (C-G) The effect of inhibition of sEH on times (C), time ratio (D), distances (E), distance ratio (F), and number of platform crossings (G) in target quadrant during 60 s probe session without a platform. (H) The representative swimming trajectory of mice during 90 s training session with a platform. (I) The representative swimming trajectory of mice during 60 s probe session without a platform. Data represent mean ± SEM, n = 12.
3.2. Inhibition of sEH by TPPU alleviated loss of neurotransmitters in Aβ-induced AD mice
The levels of three neurotransmitters with their metabolites, such as DA and its metabolites DOPAC and HVA, 5-HT and its metabolite 5-HIAA, and NE, were determined by LC-MS/MS analysis (Fig. 3). Aβ treatment led to the decrease of levels of DA (p = 0.0058), DOPAC (p = 0.0266), HVA (p = 0.0001), 5-HT (p = 0.0316), 5-HIAA (p = 0.0015), and NE (p = 0.0112) compared with the sham group, while TPPU treatment supressed the decrease of the Aβ-induced abovementioned neurotransmitters, except for 5-HT and NE. This result revealed that inhibition of sEH by TPPU could ameliorate loss of neurotransmitters.
Fig. 3.
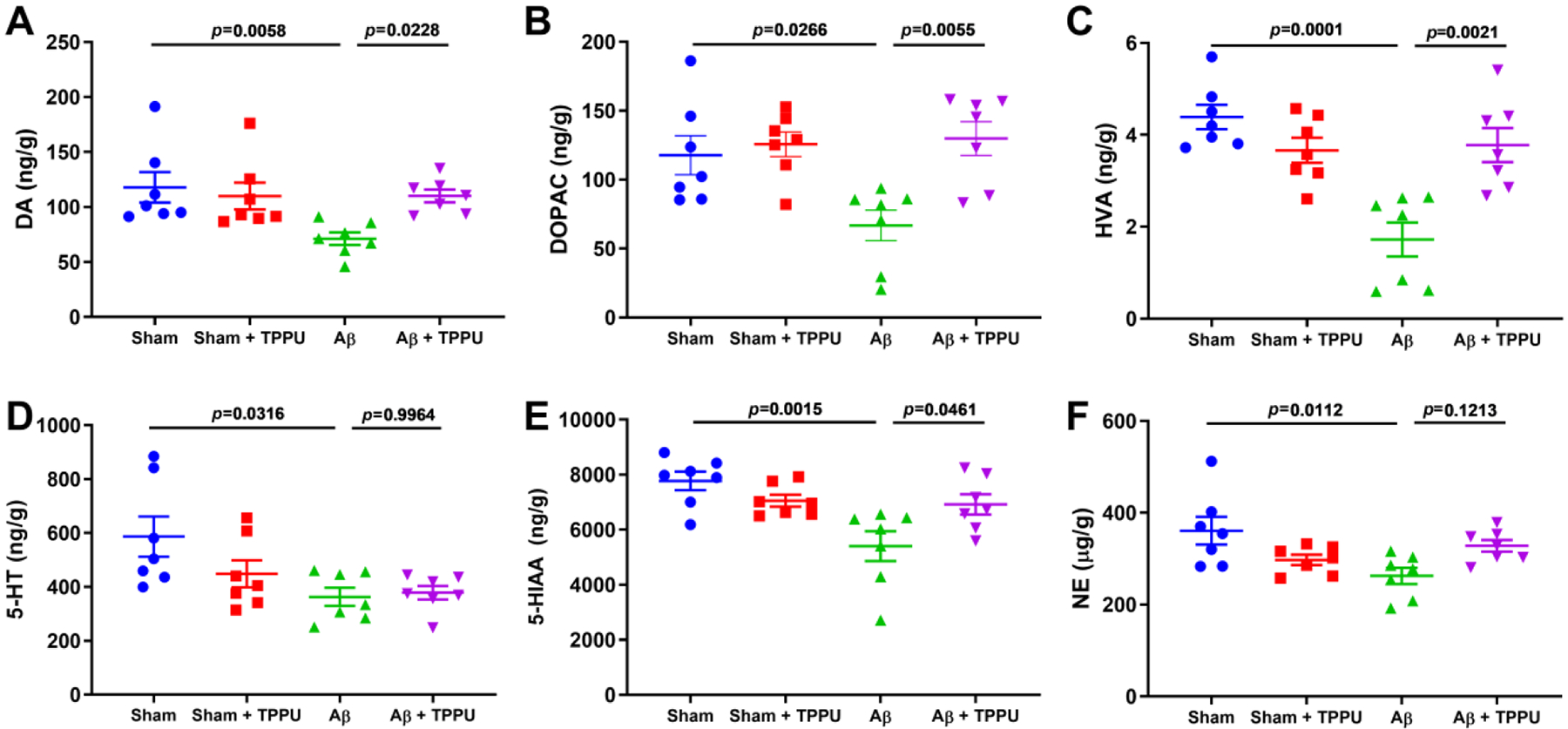
Inhibition of sEH by TPPU alleviated loss of neurotransmitters in Aβ-induced AD mice. (A-F) The effect of TPPU treatment on neurotransmitters DA (A), DOPAC (B), HVA (C), 5-HT (D), 5-HIAA (E), and NE (F) in Aβ-induced AD mice. Data represent mean ± SEM, n = 7.
3.3. Inhibition of sEH by TPPU alleviated neuroinflammation in Aβ-induced AD mice
Neuroinflammation results in the production of cytotoxic substances to aggravate Aβ aggregation and Tau pathology, allowing in the development of AD (Ahmad et al., 2019; Liu et al., 2014), therefore, we investigated the inhibition effect of sEH on microglia, astrocyte, and inflammatory NF-κB signaling pathway. Firstly, we used Iba-1 and GFAP, the markers of microglia and astrocyte, respectively, to inspect the effect of TPPU on the activation of microglia and astrocyte (Fig. 4A–E). As shown in Fig. 4A–E, Aβ treatment promoted the activation of microglia stained by Iba-1 and astrocyte stained by GFAP in the dorsal, CA1, CA3, and DG of the hippocampus compared with the sham group, whereas inhibition of sEH by TPPU significantly suppressed the activation of these two kinds of cells. Meanwhile, expression levels of Iba-1 and GFAP were both up-regulated in the hippocampus of Aβ-induced AD mice (Fig. 4F and G), and this result was reversed by inhibition of sEH with TPPU. Furthermore, Aβ treatment increased expression levels of proteins involved in NF-κB signaling pathway, including COX-2, IL-6, p-p65/p65, and p-IKKβ/IKKβ, in the hippocampus. To the contrary, administration of TPPU significantly down-regulated expression levels of the aforementioned proteins. This result fully revealed that inhibition of sEH could effectively alleviate neuroinflammation in Aβ-induced AD mice.
Fig. 4.
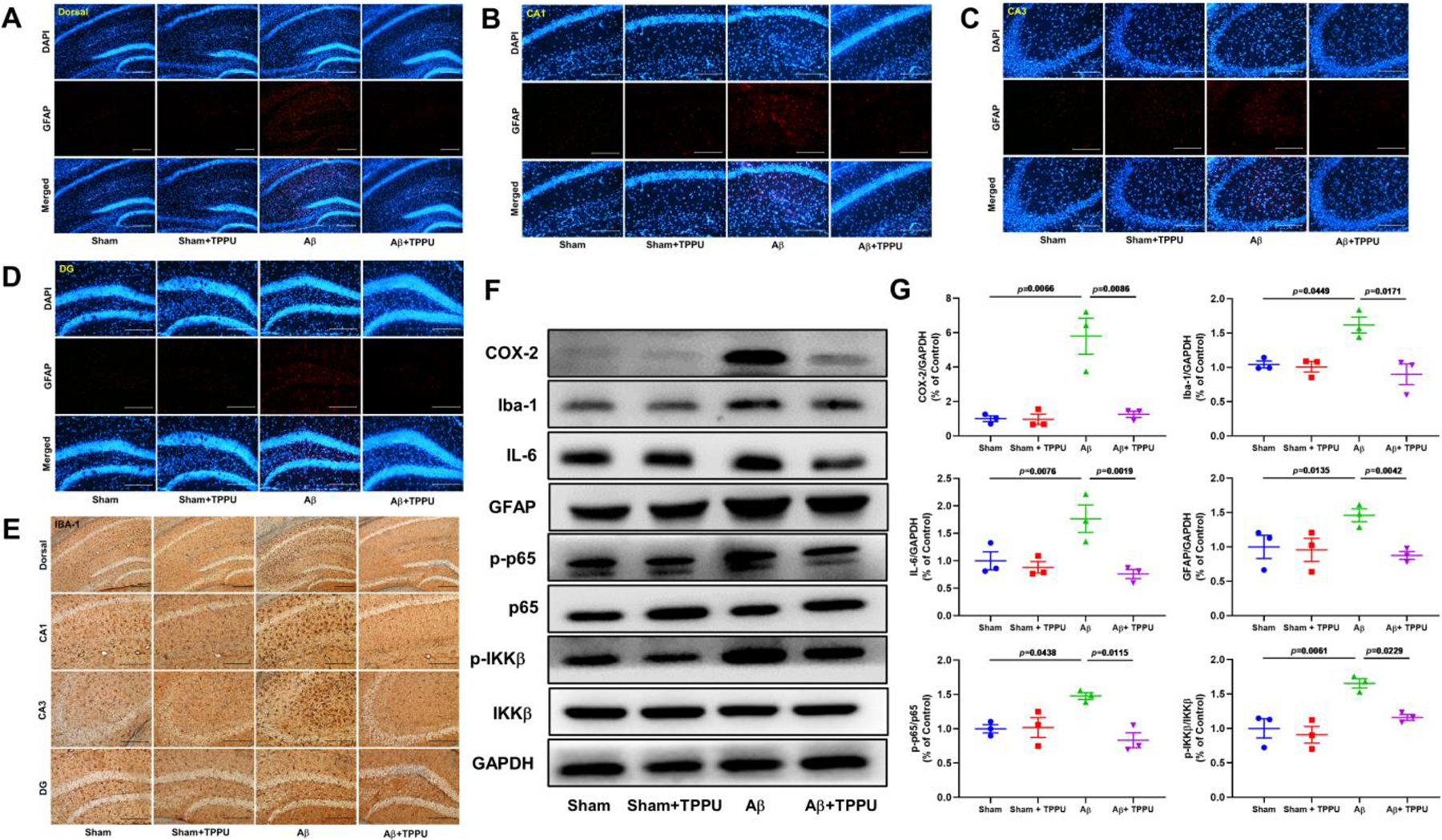
Inhibition of sEH by TPPU alleviated neuroinflammation in Aβ-induced AD mice. (A-D) Representative immunohistochemistry images for GFAP in the dorsal (A), CA1 (B), CA3 (C), and DG (D) of the hippocampus. (E) Representative immunohistochemistry images for IBA-1 in the dorsal, CA1, CA3, and DG of the hippocampus. (F) Inhibition of sEH by TPPU suppressed NF-κB signaling pathway involved in COX-2, Iba-1, IL-6, GFAP, p-p65, p65, p-IKKβ, and IKKβ. (G) Quantitative data of COX-2, Iba-1, IL-6, GFAP, p-p65/p65, and p-IKKβ/IKKβ. Data represent mean ± SEM, n = 3.
3.4. Inhibition of sEH by TPPU alleviated neuronal death via inhibition of apoptosis in Aβ-induced AD mice
Neuronal apoptosis plays an essential pathogenic role for Aβ-mediated AD. In the present study, cell apoptosis was determined by TUNEL staining. As depicted in Fig. 5A, compared with the sham group, Aβ-induced AD mice showed significant increasing in TUNEL-positive staining cells, while few apoptotic cells were observed in the hippocampus of Aβ-induced AD mice after administration of TPPU. Inhibition of sEH by TPPU might prevent apoptosis of hippocampal neuron cells caused by Aβ. Moreover, the results of western blot also indicated that administration of TPPU significantly down-regulated expression levels of Bax, cleaved-caspase 3/caspase 3, cleaved-caspase 9/caspase 9, and p53, while up-regulated Bcl-2 expression level (Fig. 5B and C). These results revealed that inhibition of sEH alleviated Aβ-induced apoptosis of hippocampal neuron cells via suppressing p53 signaling pathway.
Fig. 5.
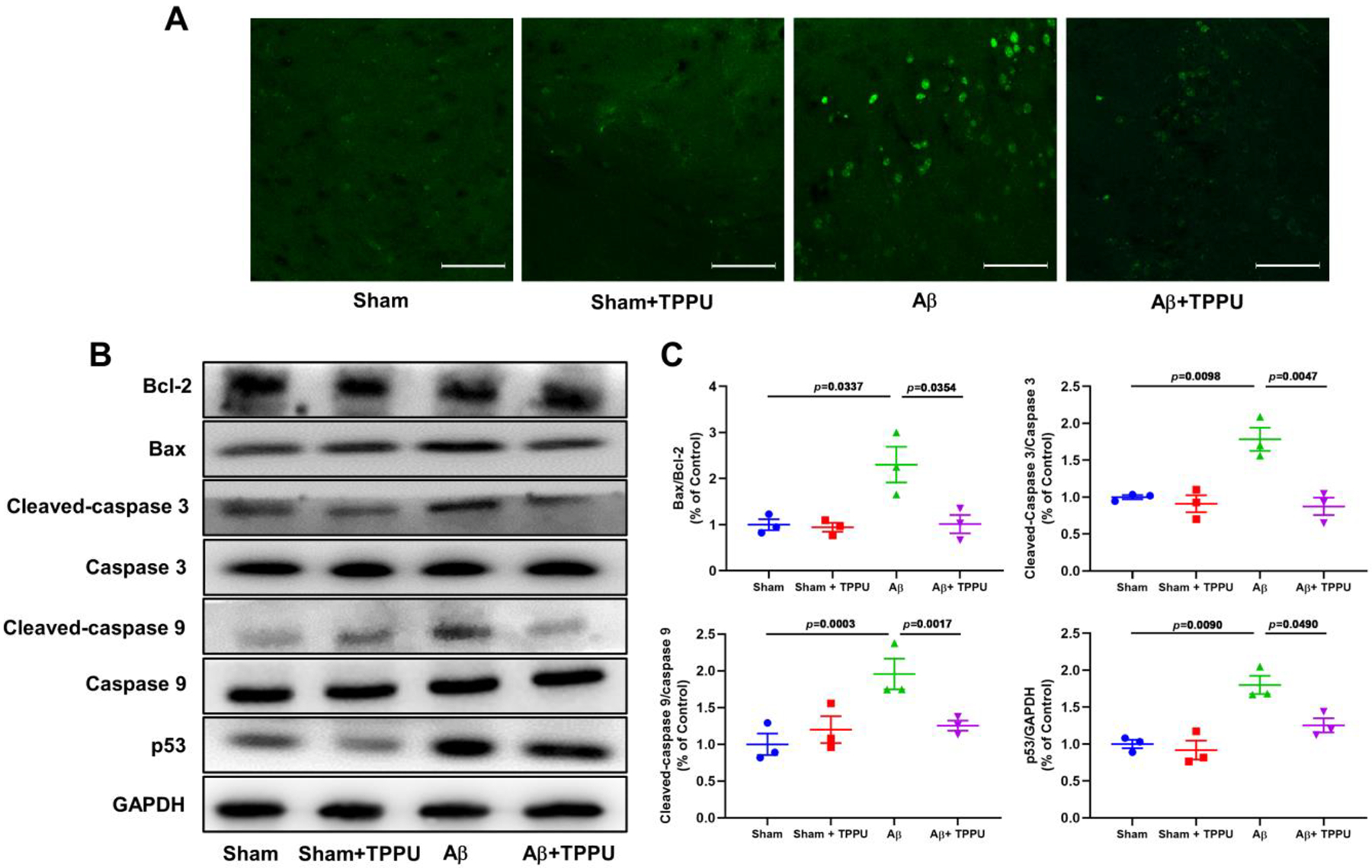
Inhibition of sEH by TPPU alleviated apoptosis of neurocyte in Aβ-induced AD mice. (A) Representative immunohistochemistry images for apoptosis of neurocyte in the hippocampus assayed by TUNEL. (B) Inhibition of sEH by TPPU suppressed p53 signaling pathway involved in Bcl-2, Bax, cleaved-caspase 3, caspase 3, cleaved-caspase 9, caspase 9, and p53. (C) Quantitative data of Bax/Bcl-2, cleaved-caspase 3/caspase 3, cleaved-caspase 9/caspase 9, and p53. Data represent mean ± SEM, n = 3.
3.5. Inhibition of sEH by TPPU alleviated oxidative and endoplasmic reticulum stresses in Aβ-induced AD mice
Oxidative and endoplasmic reticulum (ER) stresses can be caused by a variety of free oxygen radicals and have been implicated in the pathogenesis of AD, thus, we evaluated the effect of inhibition of sEH on Aβ-induced oxidative and endoplasmic reticulum stress. Aβ treatment decreased levels of GSH and T-SOD in mice, while increased MDA level (Fig. 6A–C). However, the positive results were detected in Aβ-induced AD mice after administration of TPPU (Fig. 6A–C), which demonstrated that inhibition of sEH might alleviate Aβ-induced oxidative stress. Furthermore, the results of western blot suggested that expression levels of Grp78 and Keap1 were increased (Fig. 6D), while expression levels of HO-1 and Nrf2 were decreased in Aβ-induced AD mice (Fig. 6D). Administration of TPPU resulted in the down-regulation of expression levels of Grp78 and Keap1 and up-regulation of expression levels of HO-1 and Nrf2, which indicated the protective effect on oxidative and ER stresses by sEH inhibition in Aβ-induced AD mice.
Fig. 6.
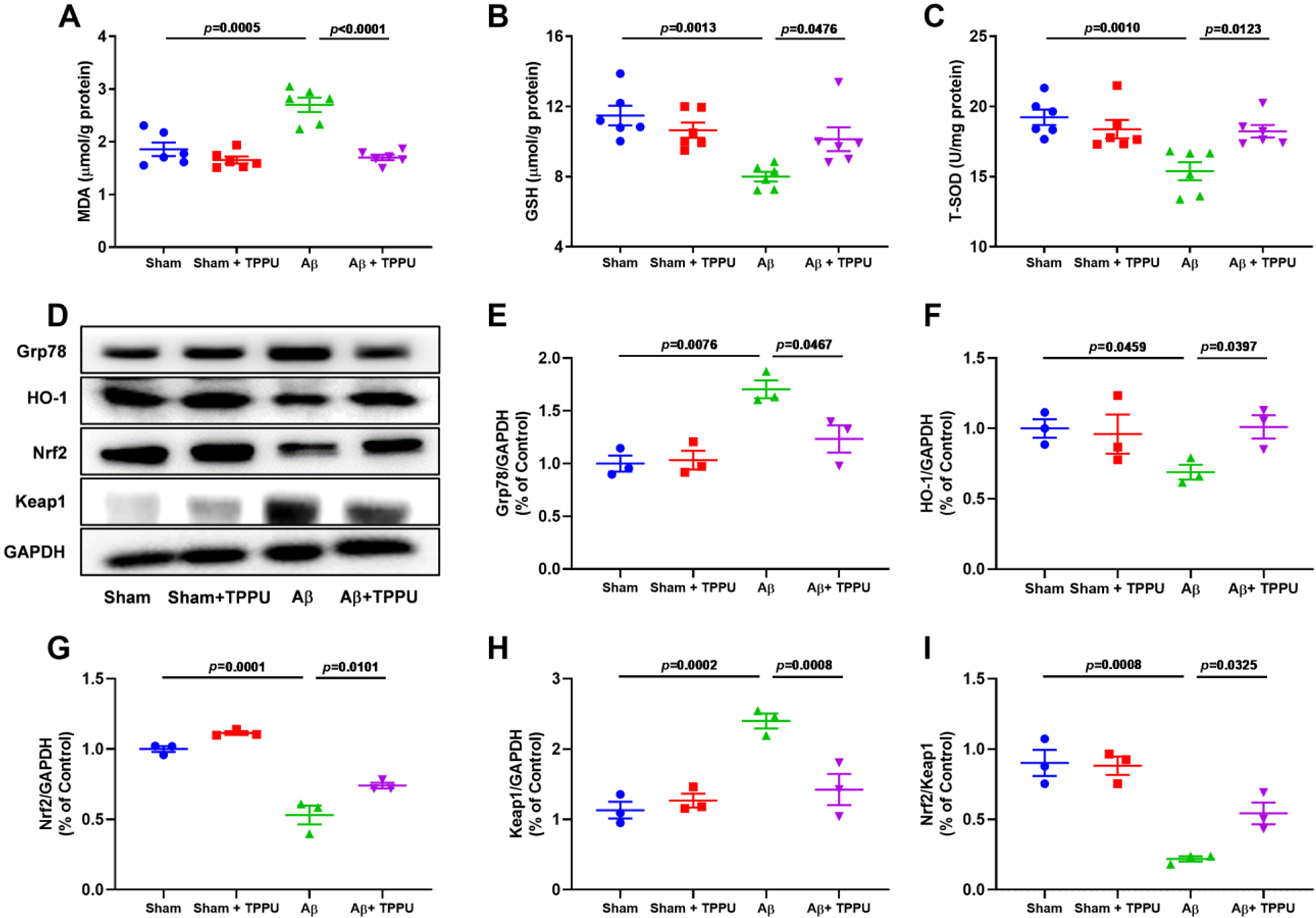
Inhibition of sEH by TPPU alleviated oxidative stress and endoplasmic reticulum stress in Aβ-induced AD mice. (A-C) TPPU treatment alleviated the increase of MDA (A) and the decrease of GSH (B) and T-SOD (C) in Aβ-induced AD mice. Data represent mean ± SEM, n = 6. (D) Inhibition of sEH by TPPU suppressed Nrf2 signaling pathway involved in Grp78, HO-1, Nrf2, and Keap1. (E-I) Quantitative data of Grp78 (E), HO-1 (F), Nrf2 (G), Keap1 (H), and Nrf2/Keap1 (I). Data represent mean ± SEM, n = 3.
3.6. Inhibition of sEH by TPPU alleviated Aβ aggregation and the hyperphosphorylation of Tau in Aβ-induced AD mice
In order to detect the effect of TPPU on Aβ aggregation and the hyperphosphorylation of Tau, immunohistochemistry and western blot were also used in present study. As shown in Fig. 7A, the formation of Aβ plaques was significantly increased in AD mice compared with the sham mice, whereas TPPU treatment could suppress the Aβ aggregation, which result was further supported by western blot (Fig. 7B and C). In addition, administration of TPPU significantly suppressed phosphorylation levels of Aβ-induced Tau protein (Fig. 7B and C), such as p-Tau (Thr205), p-Tau (Ser262), and p-Tau (Ser404). The aforementioned results demonstrated that inhibition of sEH alleviated Aβ aggregation and the hyperphosphorylation of Tau in Aβ-induced AD mice.
Fig. 7.
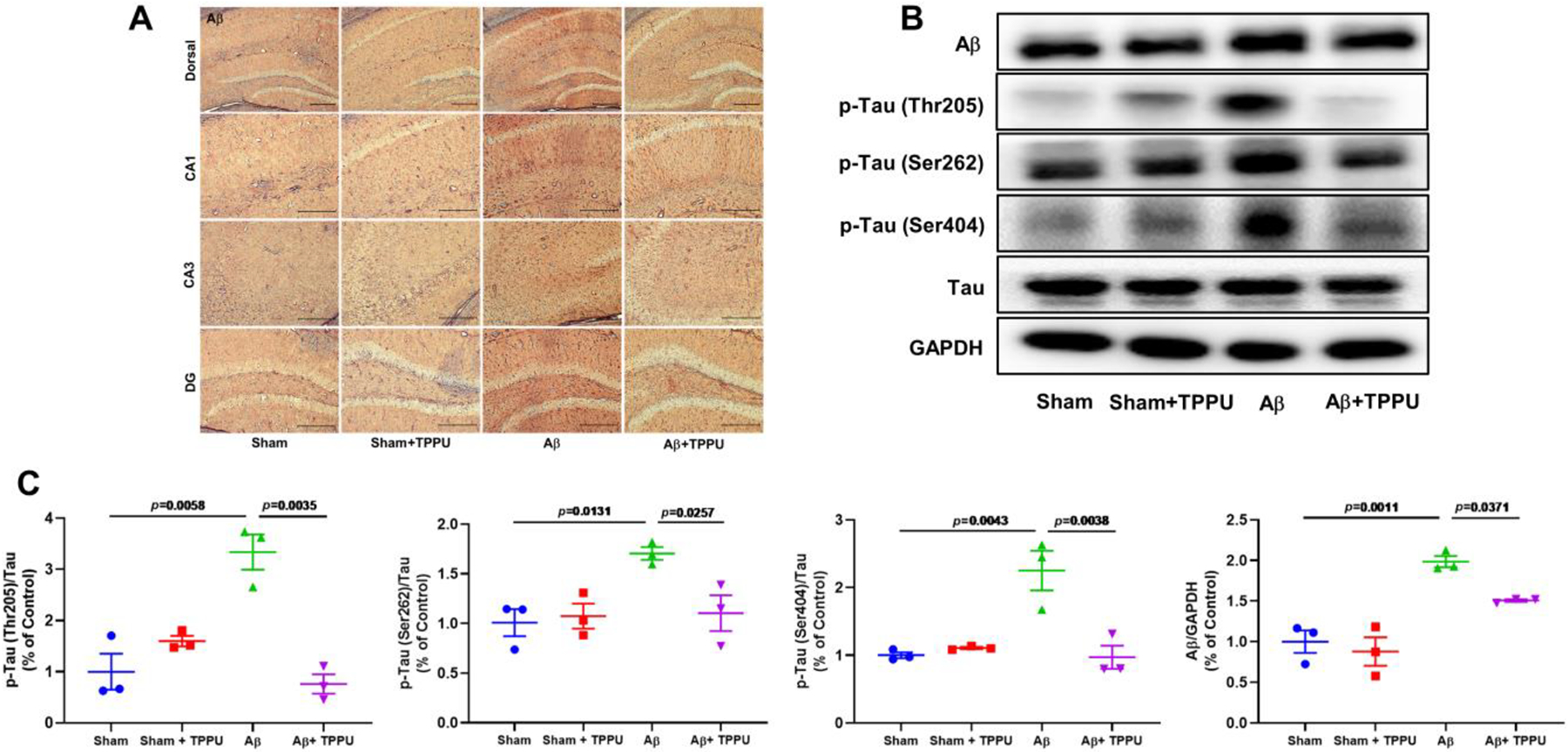
Inhibition of sEH by TPPU alleviated Aβ aggregation and the hyperphosphorylation of Tau in Aβ-induced AD mice. (A) Representative immunohistochemistry images for Aβ aggregation in the the dorsal, CA1, CA3, and DG of the hippocampus. (B) TPPU treatment down-regulated levels of Aβ and p-Tau. (C) Quantitative data of Aβ, p-Tau (Thr205)/Tau, p-Tau (Ser262)/Tau, and p-Tau (Ser404)/Tau. Data represent mean ± SEM, n = 3.
3.7. Inhibition of sEH by TPPU suppressed GSK3β signaling pathway via stabilizing the level of EETs in Aβ-induced AD mice.
LC-MS/MS was applied for the determination of EETs and DHETs. TPPU treatment significantly raised endogenous levels of 8.9-EET (8.56 ± 1.10 pmol/g) and 14,15-EET (7.97 ± 0.72 pmol/g) in Aβ-induced AD mice, and their corresponding diols 8.9-DHET and 11,12-DHET significantly decreased to 1.44 ± 0.29 and 4.26 ± 0.19 pmol/g in Aβ-induced AD mice after administration of TPPU, respectively (Fig. 8A). The ratio of DHETs and EETs can reflect the sEH activity, therefore, we analyzed the ratio of EETs and its corresponding metabolites. As shown in Fig. 8A, compared with the sham group, i.c.v. administration of Aβ significantly increased the ratios of 8,9-DHET with 8,9-EET (p < 0.0001), 11,12-DHET with 11,12-EET (p = 0.0014), and 14,15-DHET with 14,15-EET (p < 0.0001), whereas TPPU treatment significantly reversed the abovementioned results.
Fig. 8.
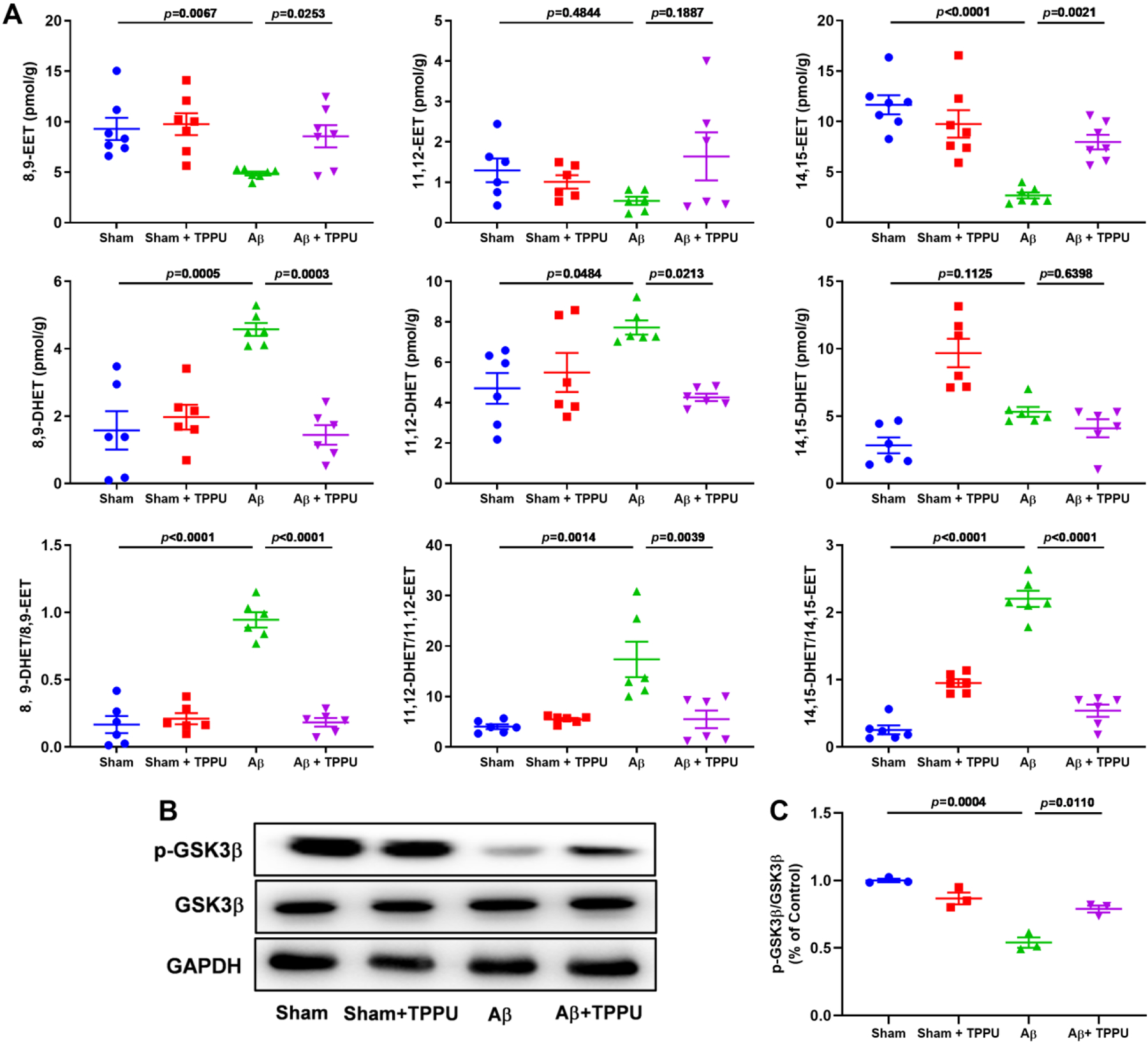
Inhibition of sEH by TPPU alleviated the increase of sEH expression level and suppressed GSK3β signaling pathway via stabilizing the level of EETs in Aβ-induced AD mice. (A) The effect of TPPU treatment on 8,9-EET, 11,12-EET, 14,15-EET, their corresponding diols 8,9-DHET, 11,12-DHET, and 14,15-DHET, 8,9-DHET/8,9-EET, 11,12-DHET/11,12-EET, and 14,15-DHET/14,15-EET in Aβ-induced AD mice. Data represent mean ± SEM, n = 6–7. (B) TPPU treatment suppressed the level of p-GSK3β in Aβ-induced AD mice. (C) Quantitative data of p-GSK3β/GSK3β. Data represent mean ± SEM, n = 3.
GSK3β is an important serine/threonine kinase involving Aβ-mediated neuron cell apoptosis, Tau phosphorylation, chronic inflammation, and oxidative stress (Hernandez et al., 2013; Kettunen et al., 2015), meanwhile EETs can significantly regulate its activity (Wang et al., 2018; Zhao et al., 2020), therefore, we also determined the inhibition effect of sEH through GSK3β signaling pathway. As shown in Fig. 8B and C, the phosphorylated level of GSK3β was significantly decreased (p = 0.0003) in Aβ-induced AD mice, while administration of TPPU significantly improved (p = 0.0088) its phosphorylated level. These results verified that inhibition of sEH suppressed GSK3β signaling pathway via stabilizing levels of EETs in Aβ-induced AD mice.
4. Discussion
In this study, Aβ (i.c.v.) was used to construct a model of AD mice to investigate the effect of inhibition of sEH with TPPU on AD mice. The sEH expression level and activity were increased in Aβ-induced AD mice. In vivo experimental results indicated that inhibition of sEH by TPPU could alleviate spatial learning and memory deficits in Aβ-induced mice in the training and probe sessions (with a platform or without a platform) of the MWM test. Furthermore, inhibition of sEH would increase levels of neurotransmitters in the hippocampus of Aβ-induced AD mice, including DA, DOPAC, HVA, 5-HT, 5-HIAA, and NE. It could also attenuate the Aβ-mediated neuroinflammation and neuronal apoptosis via suppressing the activation of microglia and astrocyte involved in NF-κB and p53 signaling pathways, respectively. In addition, chemical inhibition of sEH supressed oxidative and ER stresses through the Keap1-Nrf2 cascade, and prevented the formation of Aβ plaque and hyperphosphorylation of Tau protein. It was worth noting that endogenous levels of EETs were significantly increased in Aβ-induced AD mice after administration of TPPU, especially 8,9-EET and 14,15-EET, allowing in the inactivation of GSK3β. These findings suggested that inhibition of sEH could stabilize levels of EETs, resulting in the anti-AD effect through the regulation of GSK3β-mediated NF-κB, p53, and Nrf2 signaling pathways, which indicated that sEH might be a therapeutic target of AD.
AD is a neurodegenerative disorder mainly characterized by Aβ plaques and Tau hyperphosphorylation further causing neuronal death, neuroinflammation, and oxidative stress (Butterfield et al., 2001; Mandelkow and Mandelkow, 1998; Shankar and Walsh, 2009; Wang et al., 2015). sEH is a bifunctional enzyme with a N-terminal phosphatase region and a C-terminal hydrolase domain (Newman et al., 2005). Because its C-terminal hydrolase rapidly metabolizes or inactivates EETs to produce DHETs, sEH has become an important topic to investigate the biological role of EpFAs, especially EETs (Morisseau and Hammock, 2005). A study by Griñán-Ferré et al. demonstrated that the sEH level was higher than that of healthy individuals both in the brain of AD patients and in the hippocampus of AD mice (Grinan-Ferre et al., 2020), and Ghosh et al., also found a nearly twofold increase of sEH protein in postmortem AD brains compared to control brains (Ghosh et al., 2020). The similar result was observed in the activated astrocytes of the hippocampus of APP/PS1 mice (Hung et al., 2019; Lee et al., 2019). Consistent with previous studies, the overexpression of sEH was found in the hippocampus of Aβ-induced AD mice (Fig. 1A and B), which was further supported by the decrease of EETs and increase of DHETs in the hippocampus of AD mice (Fig. 1C). Recently, Ren et al. reported that inhibition of sEH or Ephx2 genetic deletion was able to ameliorate the pathogenesis of PD and other neurodegenerative diseases (Ren et al., 2018). Griñán-Ferré and co-workers found that two sEH inhibitors AS-2586114 and UB-EV-52 could improve cognitive impairment in two kinds of AD mice models: SAMP8 and 5×FAD (Grinan-Ferre et al., 2020), moreover, Ephx2 genetic depletion improved nesting building ability, learning, and memory of AD-like behaviors in the APP/PS1 Tg mice as well (Lee et al., 2019). In our study, we found that chemical inhibition of sEH by TPPU enhanced levels of neurotransmitters (Fig. 3), and alleviated spatial learning and memory impairments and progression of AD pathology (Fig. 2). Therefore, it is worth to further study the underlying mechanism of sEH in AD. Some of evidence has indicated that sEH is able to stabilize the level of EETs, and further suppresses the GSK3β activity, allowing in the protective effect against pulmonary fibrosis and renal injury (Deng et al., 2017; Zhao et al., 2020). In this study, the sEH activity was increased in AD mice (Fig. 1), and chemical inhibition of sEH enhanced in vivo levels of EETs in AD mice (Fig. 8), leading to the inhibition of GSK3β, which firstly revealed the vital relationship of sEH and GSK3β in AD.
GSK3β, a ubiquitously expressed and constitutively active serine-threonine kinase, involves the regulation of many key cell biology pathways (Lauretti et al., 2020; Lee et al., 2006; Ly et al., 2013), such as inflammation, neuronal death, oxidative stress, Aβ, and Tau metabolism and toxicity. GSK3β is found hyperactive in the brain of AD patients and compelling evidence supports its contribution to AD pathology (Leroy et al., 2007). Its activation can facilitate the production of TNF-α and IL-1β and 6, and activate classical inflammation pathway -- NF-κB pathway via regulating the phosphorylation of p65 (Koistinaho et al., 2011). In this study, we found that inhibition of sEH by TPPU displayed the anti-AD effect via suppressing GSK3β-mediated NF-κB activation and neuroinflammatory gene expression (Iba-1, COX-2, GFAP, and IL-6) (Fig. 4). Besides, GSK3β involves the neuronal apoptosis and oxidative stress in the pathology of AD. GSK3β can direct bond to the activation domain-1 and basic domain of p53 (Watcharasit et al., 2003), promote its phosphorylation, and activate its transcriptional activity (Turenne and Price, 2001). Furthermore, inhibition of GSK3β by LiCl, a GSK3β inhibitor, is able to regulate the transcriptional action of p53 via sharply decreasing p53-dependent target genes Mdm2, p21, and Bax mRNA levels (Watcharasit et al., 2003). Agreed with the previous report, p53-dependent apoptosis was observed in our study, represented as up-regulation expression levels of p53, Bax, and cleaved-caspases 3 and 9 after Aβ treatment (Fig. 5). However, inhibition of sEH by TPPU suppressed GSK3β-mediated p53 apoptotic signaling pathway in Aβ-induced AD mice. Xiang et al. reported that LiCl increased SOD and GSH levels, decreased the MDA level in AD mice, the mechanism of which suppressed GSK3β activity and consequently enhanced Nrf2 and HO-1 expression levels (Xiang et al., 2020). In line with the aforementioned, inhibition of sEH by TPPU decreased the MDA levels, increased GSH and T-SOD activities and HO-1 expression via activating GSK3β-Nrf2-Keap1 cascade in AD mice. In conclusion, inhibition of sEH alleviated AD involved in GSK3β signaling pathway via stabilizing in vivo levels of EETs.
5. Conclusion
In summary, we revealed the close correlation of the sEH expression level and activity with AD. We used the model of Aβ-induced AD mice to investigate the vital role and molecular mechanism of sEH in AD. Chemical inhibition of sEH by TPPU could significantly alleviate spatial learning and memory deficits, and elevate in vivo levels of neurotransmitters DA, DOPAC, HVA, 5-HT, 5-HIAA, and NE, in Aβ-induced AD mice. Furthermore, its inhibition could ameliorate neuroinflammation, neuronal apoptosis, and oxidative stress via stabilizing levels of EETs, especially 8,9-EET and 14,15-EET, further resulting in the anti-AD effect through the regulation of GSK3β-mediated NF-κB, p53, and Nrf2 signaling pathways. These findings suggested that sEH could be regarded as a potential target for effective treatment of AD.
Acknowledgments
This work is supported by Open Research Fund of the State Key Laboratory of Cognitive Neuroscience and Learning (No. CNLZD1801), Distinguished professor of Liaoning Province, Natural Science Foundation of Liaoning Province (No. 2020-MS-256), Dalian Science and Technology Leading Talents Project (2019RD15), Revolutionizing Innovative, Visionary Environmental Health Research Program of the National Institute of Environmental Health Sciences (No. R35 ES030443), Superfund Basic Research Program of the National Institutes of Environmental Health Sciences (No. P42 ES04699) and Dalian Young Star of Science and Technology (No. 2019RQ123).
Footnotes
Declaration of interest
The University of California has patents on TPPU and its use in inflammatory disease (C.M. and B.D.H.). Otherwise, the authors declare no competing interests.
References
- Ahmad MH, Fatima M, Mondal AC, 2019. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J Clin Neurosci 59, 6–11. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A, 2001. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med 7, 548–554. [DOI] [PubMed] [Google Scholar]
- Campbell WB, 2000. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol Sci 21, 125–127. [DOI] [PubMed] [Google Scholar]
- Carter J, Lippa CF, 2001. Beta-amyloid, neuronal death and Alzheimer’s disease. Curr Mol Med 1, 733–737. [DOI] [PubMed] [Google Scholar]
- Deng BQ, Luo Y, Kang X, Li CB, Morisseau C, Yang J, Lee KSS, Huang J, Hu DY, Wu MY, Peng A, Hammock BD, Liu JY, 2017. Epoxide metabolites of arachidonate and docosahexaenoate function conversely in acute kidney injury involved in GSK3beta signaling. Proc Natl Acad Sci U S A 114, 12608–12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duflot T, Roche C, Lamoureux F, Guerrot D, Bellien J, 2014. Design and discovery of soluble epoxide hydrolase inhibitors for the treatment of cardiovascular diseases. Expert Opin Drug Discov 9, 229–243. [DOI] [PubMed] [Google Scholar]
- Eguchi K, Shindo T, Ito K, Ogata T, Kurosawa R, Kagaya Y, Monma Y, Ichijo S, Kasukabe S, Miyata S, Yoshikawa T, Yanai K, Taki H, Kanai H, Osumi N, Shimokawa H, 2018. Whole-brain low-intensity pulsed ultrasound therapy markedly improves cognitive dysfunctions in mouse models of dementia - Crucial roles of endothelial nitric oxide synthase. Brain Stimul 11, 959–973. [DOI] [PubMed] [Google Scholar]
- Falck JR, Krishna UM, Reddy YK, Kumar PS, Reddy KM, Hittner SB, Deeter C, Sharma KK, Gauthier KM, Campbell WB, 2003. Comparison of vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am J Physiol Heart Circ Physiol 284, H337–349. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Comerota MM, Wan DB, Chen FD, Propson NE, Hwang SH, Hammock BD, Zheng H, 2020. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci Transl Med 12, eabb1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinan-Ferre C, Codony S, Pujol E, Yang J, Leiva R, Escolano C, Puigoriol-Illamola D, Companys-Alemany J, Corpas R, Sanfeliu C, Perez B, Loza MI, Brea J, Morisseau C, Hammock BD, Vazquez S, Pallas M, Galdeano C, 2020. Pharmacological Inhibition of Soluble Epoxide Hydrolase as a New Therapy for Alzheimer’s Disease. Neurotherapeutics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez F, Lucas JJ, Avila J, 2013. GSK3 and tau: two convergence points in Alzheimer’s disease. J Alzheimers Dis 33 Suppl 1, S141–S144. [DOI] [PubMed] [Google Scholar]
- Hou L, Sun F, Sun W, Zhang L, Wang Q, 2019. Lesion of the Locus Coeruleus Damages Learning and Memory Performance in Paraquat and Maneb-induced Mouse Parkinson’s Disease Model. Neuroscience 419, 129–140. [DOI] [PubMed] [Google Scholar]
- Huang HJ, Wang YT, Lin HC, Lee YH, Lin AM, 2018. Soluble Epoxide Hydrolase Inhibition Attenuates MPTP-Induced Neurotoxicity in the Nigrostriatal Dopaminergic System: Involvement of alpha-Synuclein Aggregation and ER Stress. Mol Neurobiol 55, 138–144. [DOI] [PubMed] [Google Scholar]
- Hung CC, Lee YH, Kuo YM, Hsu PC, Tsay HJ, Hsu YT, Lee CC, Liang JJ, Shie FS, 2019. Soluble epoxide hydrolase modulates immune responses in activated astrocytes involving regulation of STAT3 activity. J Neuroinflammation 16, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, 2016. Epoxyeicosatrienoic Acids and 20-Hydroxyeicosatetraenoic Acid on Endothelial and Vascular Function. Adv Pharmacol 77, 105–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR, 1996. Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol 7, 2364–2370. [DOI] [PubMed] [Google Scholar]
- Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, Masutani H, Yodoi J, Urano Y, Nagano T, Ichijo H, 2005. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ 12, 19–24. [DOI] [PubMed] [Google Scholar]
- Kettunen P, Larsson S, Holmgren S, Olsson S, Minthon L, Zetterberg H, Blennow K, Nilsson S, Sjolander A, 2015. Genetic variants of GSK3B are associated with biomarkers for Alzheimer’s disease and cognitive function. J Alzheimers Dis 44, 1313–1322. [DOI] [PubMed] [Google Scholar]
- Kodani SD, Morisseau C, 2019. Role of epoxy-fatty acids and epoxide hydrolases in the pathology of neuro-inflammation. Biochimie 159, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho J, Malm T, Goldsteins G, 2011. Glycogen synthase kinase-3beta: a mediator of inflammation in Alzheimer’s disease? Int J Alzheimers Dis 2011, 129753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauretti E, Dincer O, Pratico D, 2020. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys Acta Mol Cell Res 1867, 118664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Lu T, Weintraub NL, VanRollins M, Spector AA, Shibata EF, 1999. Effects of epoxyeicosatrienoic acids on the cardiac sodium channels in isolated rat ventricular myocytes. J Physiol 519 Pt 1, 153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HT, Lee KI, Chen CH, Lee TS, 2019. Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer’s disease. J Neuroinflammation 16, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Chung YH, Joo KM, Lim HC, Jeon GS, Kim D, Lee WB, Kim YS, Cha CI, 2006. Age-related changes in glycogen synthase kinase 3beta (GSK3beta) immunoreactivity in the central nervous system of rats. Neuroscience letters 409, 134–139. [DOI] [PubMed] [Google Scholar]
- Leroy K, Yilmaz Z, Brion JP, 2007. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol 33, 43–55. [DOI] [PubMed] [Google Scholar]
- Liu C, Cui G, Zhu M, Kang X, Guo H, 2014. Neuroinflammation in Alzheimer’s disease: chemokines produced by astrocytes and chemokine receptors. Int J Clin Exp Pathol 7, 8342–8355. [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu Z, Wei M, Hu M, Yue K, Bi R, Zhai S, Pi Z, Song F, Liu Z, 2019. Pharmacodynamic and urinary metabolomics studies on the mechanism of Schisandra polysaccharide in the treatment of Alzheimer’s disease. Food Funct 10, 432–447. [DOI] [PubMed] [Google Scholar]
- Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W, 2013. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest 123, 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E, 1998. Tau in Alzheimer’s disease. Trends Cell Biol 8, 425–427. [DOI] [PubMed] [Google Scholar]
- Morisseau C, Hammock BD, 2005. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol 45, 311–333. [DOI] [PubMed] [Google Scholar]
- Newman JW, Morisseau C, Hammock BD, 2005. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res 44, 1–51. [DOI] [PubMed] [Google Scholar]
- Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK, 1999. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285, 1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince A-HM, Knapp M, Guerchet M, Karagiannidou M, 2016. World Alzheimer report 2016. London: Alzheimer's Disease International 2016, 2016. [Google Scholar]
- Qin X, Wu Q, Lin L, Sun A, Liu S, Li X, Cao X, Gao T, Luo P, Zhu X, Wang X, 2015. Soluble Epoxide Hydrolase Deficiency or Inhibition Attenuates MPTP-Induced Parkinsonism. Mol Neurobiol 52, 187–195. [DOI] [PubMed] [Google Scholar]
- Ren Q, Ma M, Yang J, Nonaka R, Yamaguchi A, Ishikawa KI, Kobayashi K, Murayama S, Hwang SH, Saiki S, Akamatsu W, Hattori N, Hammock BD, Hashimoto K, 2018. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc Natl Acad Sci U S A 115, E5815–E5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman RJ, 2002. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82, 131–185. [DOI] [PubMed] [Google Scholar]
- Rubio-Perez JM, Morillas-Ruiz JM, 2012. A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal 2012, 756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Walsh DM, 2009. Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL, 1987. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 235, 880–884. [DOI] [PubMed] [Google Scholar]
- Tarawneh R, Holtzman DM, 2012. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb Perspect Med 2, a006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turenne GA, Price BD, 2001. Glycogen synthase kinase3 beta phosphorylates serine 33 of p53 and activates p53’s transcriptional activity. BMC Cell Biol 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Yang J, Zhang J, Wang Y, Hwang SH, Qi W, Wan D, Kim D, Sun J, Sanidad KZ, Yang H, Park Y, Liu JY, Zhao X, Zheng X, Liu Z, Hammock BD, Zhang G, 2018. Lipidomic profiling reveals soluble epoxide hydrolase as a therapeutic target of obesity-induced colonic inflammation. Proc Natl Acad Sci U S A 115, 5283–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Tan MS, Yu JT, Tan L, 2015. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS, 2003. Glycogen synthase kinase-3beta (GSK3beta) binds to and promotes the actions of p53. J Biol Chem 278, 48872–48879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J, Cao K, Dong YT, Xu Y, Li Y, Song H, Zeng XX, Ran LY, Hong W, Guan ZZ, 2020. Lithium chloride reduced the level of oxidative stress in brains and serums of APP/PS1 double transgenic mice via the regulation of GSK3beta/Nrf2/HO-1 pathway. Int J Neurosci 130, 564–573. [DOI] [PubMed] [Google Scholar]
- Yang EJ, Mahmood U, Kim H, Choi M, Choi Y, Lee JP, Cho JY, Hyun JW, Kim YS, Chang MJ, Kim HS, 2018. Phloroglucinol ameliorates cognitive impairments by reducing the amyloid beta peptide burden and pro-inflammatory cytokines in the hippocampus of 5XFAD mice. Free Radic Biol Med 126, 221–234. [DOI] [PubMed] [Google Scholar]
- Zeldin DC, 2001. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem 276, 36059–36062. [DOI] [PubMed] [Google Scholar]
- Zhao WY, Luan ZL, Liu TT, Ming WH, Huo XK, Huang HL, Sun CP, Zhang BJ, Ma XC, 2020. Inula japonica ameliorated bleomycin-induced pulmonary fibrosis via inhibiting soluble epoxide hydrolase. Bioorg Chem 102, 104065. [DOI] [PubMed] [Google Scholar]