

ABSTRACT
In vivo modeling combined with CRISPR/Cas9-mediated somatic genome editing has contributed to elucidating the functional importance of specific genetic alterations in human tumors. Our recent work uncovered tumor suppressor pathways that affect EGFR-driven lung tumor growth and sensitivity to tyrosine kinase inhibitors and reflect the mutational landscape and treatment outcomes in the human disease.
KEYWORDS: Lung cancer, EGFR, targeted therapy, tumor suppressor genes, multiplexed in vivo genome editing
Tyrosine kinase inhibitors (TKIs) are the standard of care treatment for oncogenic epidermal growth factor receptor (EGFR)-driven lung adenocarcinomas.1 Despite the efficacy of TKIs, responses are heterogeneous and drug resistance inevitably emerges, underscoring the need to identify determinants of therapeutic sensitivity.1 Understanding how genotypes influence responses to drugs could help advance treatment strategies for different subsets of patients with EGFR-driven lung cancer and delay or prevent the emergence of resistance. Sequencing data of human tumors show that EGFR alterations co-occur with alterations in many putative tumor suppressor genes.2 Whether these alterations have biological implications and whether their relative alteration frequency reflects their functional importance remains largely unknown. Understanding the extent to which tumor suppressor gene alterations contribute to TKI sensitivity and resistance could improve treatment approaches (Figure 1). Moreover, the identification of combinations of genetic alterations that alter tumor fitness could also lead to the discovery of novel vulnerabilities of genomic subsets of EGFR mutant tumors.2
Figure 1.
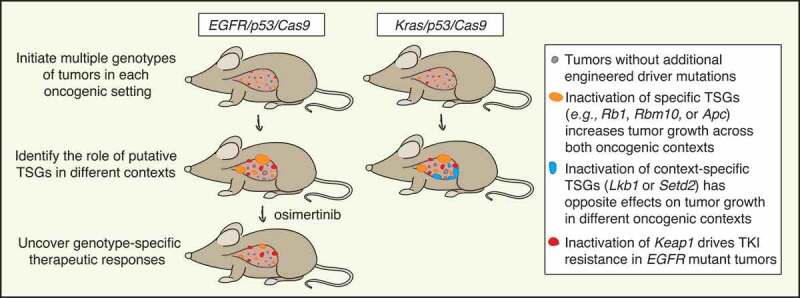
Dissecting the role of tumor suppressor genes using multiplexed genome editing across oncogenic contexts. Schematic of multiplexed genome editing in a mouse model of Epidermal growth factor receptor with activating point mutation L858R (EGFRL858R) mutant and Transformation related protein 53 (Trp53, best known as p53)-deficient lung adenocarcinoma (expressing Cas9; EGFR/p53/Cas9). Tumors are initiated by intratracheal administration of a lentiviral pool of vectors that lead to simultaneous tumor suppressor gene (TSG) inactivation mediated by CRISPR/Cas9 system. We modeled EGFR/p53 mutant tumors and 10 tumor genotypes via inactivation of putative tumor suppressor genes and quantified their impact on tumor growth compared to a different oncogenic context (Kirsten rat sarcoma viral oncogene homologue with activating point mutation G12D, KrasG12D mutant and p53-deficient model; Kras/p53/Cas9) and on sensitivity to treatment with osimertinib.
Prior to our study, there were almost no in vivo studies on the function of tumor suppressor genes in oncogenic EGFR-driven lung cancer. This was at least partially due to the absence of suitable autochthonous EGFR-driven lung cancer models with which to investigate the biological consequences of gene inactivation.3 In our recent work, we leveraged tumor barcoding with high-throughput barcode sequencing (Tuba-seq)4, 5 and applied it to a novel model of EGFR mutant and Transformation related protein 53 (Trp53, best known as p53)-deficient lung adenocarcinoma to study the functional importance of tumor suppressor genes that are frequently altered in human lung tumors.3 –6 Our findings revealed tumor suppressor genes that when inactivated promoted tumor growth, most notably RNA binding motif protein 10 (Rbm10), Retinoblastoma 1 (Rb1), and Adenomatous polyposis coli (Apc).3 Importantly, these tumor suppressor genes were also some of the most frequently altered genes in EGFR/P53 mutant human lung tumors, supporting the importance of these tumor suppressor pathways in driving EGFR mutant lung tumors.3 AT-rich interaction domain 1A (Arid1a) and Cyclin-dependent kinase inhibitor 2A (Cdkn2a) inactivation appeared to increase tumor growth only later during tumor progression, suggesting that specific tumor suppressor genes can have different effects at different stages of tumor development. Inactivation of other putative tumor suppressor genes that we investigated did not promote tumor growth, indicating that the function of some tumor suppressor genes may be highly context-dependent (Figure 1).3
Mutual exclusivity between genomic alterations in human tumors can imply redundancy of biological processes or synthetic lethality. Given the complexity of tumorigenesis, multiple factors (e.g., tumor subtype, mutational processes and load, environment) may play a role in determining genetic epistasis, suggesting that experimental approaches are particularly important for understanding why this is observed.7 Oncogenic EGFR alterations (mainly affecting EGFR exons 18 through 21) and oncogenic Kirsten rat sarcoma viral oncogene homologue (KRAS) missense mutations (mainly at codons 12, 13, and 61) represent major drivers of lung adenocarcinoma.2 Given that both these oncogenes are components of the same pathway, we anticipated that tumor suppressor genes would have similar impacts on in vivo growth of EGFR and KRAS mutant lung tumors. Although inactivation of Rbm10, Rb1, or Apc did have similar effects on EGFR and KRAS mutant tumor growth, Serine/threonine kinase 11 (Stk11, also known as Lkb1) and SET domain containing 2, histone lysine methyltransferase (Setd2) inactivation had opposite effects in the two oncogenic settings. Lkb1 and Setd2 inactivation are two of the strongest drivers of KRAS mutant tumor growth; however, their inactivation reduced EGFR mutant tumor growth.8 These results correlate with the relative frequency of LKB1 and SETD2 alterations in human EGFR and KRAS mutant lung tumors, suggesting the existence of a synthetic lethal relationship between Lkb1/Setd2 inactivation and oncogenic EGFR. These findings underscore the importance of quantitative modeling of genetic alterations in vivo. More broadly, the observation that inactivation of certain genes can have different effects depending on the specific oncogenic alteration present (e.g., KRAS vs. EGFR mutation) revealed surprising context specificity of the role of these genes in cancer (Figure 1).3
Osimertinib, a third generation TKI that leads to better overall survival compared to other TKIs, has been approved as first-line therapy for patients with metastatic EGFR-driven lung cancer.9 Recently, osimertinib was also approved as adjuvant therapy for early-stage EGFR mutant tumors.10 Outcomes upon osimertinib treatment are variable; thus, the importance of uncovering how co-incident genomic alterations contribute to sensitivity and resistance is of great clinical relevance. We quantified the impact of inactivating 10 putative tumor suppressor genes on the response to osimertinib within our mouse model and found that Kelch-like ECH associated protein 1 (Keap1) inactivation reduced sensitivity to this therapy.3 Our in vivo data mirror clinical data suggesting that KEAP1 pathway alterations predict poor clinical responses to TKIs.3 Thus, we established a causal link between this tumor suppressor pathway and TKI sensitivity in EGFR mutant lung adenocarcinoma.
The multiplexed in vivo CRISPR/Cas9 screening approach that we used allows us to interrogate multiple genes simultaneously and assess their contributions to tumor phenotypes.3,4,5,6, 8,11 Thus, it provides quantitative cause-and-effect information and avoids confounding factors that are inevitable in human tumors (e.g., high mutation burden). However, our model systems do not yet recreate the extent of genomic complexity and intratumoral heterogeneity found in human lung tumors. Future studies will also be required to uncover the molecular mechanisms by which these tumor suppressors normally constrain tumor growth and by which their inactivation sensitizes tumors to therapy. Our study represents an initial step in defining the role of tumor suppressor genes in oncogenic EGFR-driven lung tumors. We anticipate that assessing broader panels of putative tumor suppressors will further elucidate the functional genomic landscape of this disease and allow genotypes to be related to diverse cancer phenotypes, including their response to different therapies. Despite the efficacy of single agent osimertinib, quantifying the impact of rational combination therapies on defined genotypes of lung tumors will drive further gains in the treatment of specific subsets of tumors. We envision that these types of multiplexed in vivo studies will ultimately contribute to the development of tailored treatments for patients with EGFR mutant lung cancer.
Funding
G.F. is supported by a European Respiratory Society (ERS) long-term research fellowship [ERS LTRF202101-00866] and was previously supported by an American Italian Cancer Foundation (AICF) postdoctoral fellowship; C.L. was the Connie and Bob Lurie Fellow of the Damon Runyon Cancer Research Foundation [DRG-2331]; H.C. was supported by a Tobacco-Related Disease Research Program Postdoctoral Fellowship [28FT-0019]; This work was supported by the National Institutes of Health [CA231253 (to M.M.W. and D.A.P.), CA234349 (to M.M.W and D.A.P.), CA263715 (to K.P. and M.M.W.), CA120247 and CA196530 (to K.P.)], the Ginny and Kenneth Grunley Fund for Lung Cancer Research, and the Yale Cancer Center Class of ’61 Research Award (to K.P.).
Disclosure statement
K.P. is a co-inventor on a patent licensed to Molecular MD for EGFR T790M mutation testing (through MSKCC). K.P. has received Honoraria/Consulting fees from Takeda, NCCN, Novartis, Merck, AstraZeneca, Tocagen, Maverick Therapeutics and Dynamo Therapeutics and research support from AstraZeneca, D2G Oncology, Kolltan, Roche, Symphogen and Boehringer-Ingelheim. M.M.W. has received honoraria from Genentech, Merck, and Amgen. M.M.W. and D.A.P. have ownership interest in, and are founders, consultants, and advisory board members for, D2G Oncology Inc.
References
- 1.Ramalingam SS, Yang JC, Lee CK, Kurata T, Kim DW, John T, Nogami N, Ohe Y, Mann H, Rukazenkov Y, et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J Clin Oncol. 2018;36:841–3. [DOI] [PubMed] [Google Scholar]
- 2.Skoulidis F, Heymach JV.. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19:495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foggetti G, Li C, Cai H, Hellyer JA, Lin WY, Ayeni D, Hastings K, Choi J, Wurtz A, Andrejka L, et al. Genetic determinants of EGFR-driven lung cancer growth and therapeutic response In vivo. Cancer Discov. 2021;11(7):1736–1753. doi: 10.1158/2159-8290.CD-20-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers ZN, McFarland CD, Winters IP, Naranjo S, Chuang CH, Petrov D, Winslow MM . A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat Methods. 2017;14:737–742 doi: 10.1038/nmeth.4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogers ZN, McFarland CD, Winters IP, Seoane JA, Brady JJ, Yoon S, et al. Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat Genet. 2018;50:483–486 doi: 10.1038/s41588-018-0083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winters IP, Murray CW, Winslow MM. Towards quantitative and multiplexed in vivo functional cancer genomics. Nat Rev Genet. 2018;19(12):741–755. doi: 10.1038/s41576-018-0053-7. [DOI] [PubMed] [Google Scholar]
- 7.van de Haar J, Canisius S, Yu MK, Voest EE, Wessels LFA, Ideker T. Identifying epistasis in cancer genomes: a delicate affair. Cell. 2019;177:1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai H, Chew SK, Li C, Tsai MK, Andrejka L, Murray CW, et al. A functional taxonomy of tumor suppression in oncogenic KRAS-driven lung cancer. Cancer Discov. 2021;11(7):1754–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-Mutated advanced NSCLC. N Engl J Med. 2020;382:41–50. [DOI] [PubMed] [Google Scholar]
- 10.Wu YL, Tsuboi M, He J, John T, Grohe C, Majem M, et al. Osimertinib in resected EGFR-Mutated non-small-cell lung cancer. N Engl J Med. 2020;383:1711–1723. [DOI] [PubMed] [Google Scholar]
- 11.Li C, Lin WY, Rizvi H, Cai H, McFarland CD, Rogers ZN, et al. Quantitative in vivo analyses reveal a complex pharmacogenomic landscape in lung adenocarcinoma. Cancer Res. 2021;81(17):4570–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]