

ABSTRACT
The tumor microenvironment (TME) profoundly influences tumor progression and affects immunotherapy responses and resistance. Understanding its heterogeneity is the key for developing immunotherapy. However, the available methods can only partially portray the TME heterogeneity with a small number of cell types. Here, we developed a deep learning-based frame with a design visible, DCNet, that embeds the relationships between cells and their marker genes in the neural network, and can infer the cell landscape with more than 400 cell types based on bulk RNA-seq data. DCNet accurately recapitulated the cell landscape of multiple single cell RNA-seq datasets, which showed better robustness and stability. Based on the cell landscape of TCGA patients, which was built with DCNet, the patients were divided into two groups with significant differences in survival time and distinct cell-type populations. DCNet provides a foundation for decoding TME heterogeneity. The source code of DCNet can be found on GitHub: https://github.com/xindd/DCNet.
KEYWORDS: Tumor microenvironment, deep learning, cell landscape
Introduction
Immunotherapy has achieved remarkable success in treating advanced tumors.1 But because of the highly heterogeneous TME, only a relatively small fraction of patients obtain clinical benefit. The TME, comprising numerous and heterogeneous cell types counting fibroblats, immune cells, adipocytes, endothelial cells, becomes the hotspot of research in this field in the next few years and is being further explored by novel techniques.2,3 Single-cell transcriptome sequencing (scRNA-seq) as a novel sequencing technique can quantify transcripts in individual cells and provides a comprehensive cell landscape of the TME.4,5 But it is difficult for the generating of scRNA-seq data in a large population of patients for its high cost and technical challenges.
Bulk RNA-seq data source is a good choice for the identification of TME as a large amount of patient high-throughput sequencing data is available. Several methods have been developed for deconvolution of bulk gene expression to evaluate cell landscape in bulk RNA-seq samples, such as CIBERSORTx,6 EPIC,7 MCP-counter,8 quanTIseq,9 TIMER,10 and xCell.11 These methods expand the application direction of expression profile generated by the bulk RNA-seq and make it possible for researchers to identify the TME from bulk RNA-seq data. Bogdan A. Luca et al. have proposed the ECOTyper method, a new machine learning framework for analyzing cell states and ecosystems, which used Cibersortx and NMF methods to evaluate the 12 major cell lineages abundance of bulk samples in tumors and defined 69 cell states in TME.12 But the existing methods usually focus on a very limited number of cell types and those methods are prone to different deviations for the dependent selection of marker genes. Recently, some deep learning methods such as multi-layer perceptrons (MLP), convolutional neural networks (CNN), long and short-term memory networks (LSTM), and autoencoders (AE) have been applied in the field of bioinformatics13–17 and shown more improvement and progress. However, as a black box model, due to their multilayer nonlinear structure, the deep learning methods are often considered to be nontransparent and their predictions not traceable. Many researchers try to “light up the black box” from different angles of analysis and visualization.18,19 For example, the structural relationships of GO (Gene Ontology) terms are embedded in an ANN (artificial neural network), enabling interpretation of gene functional relationship networks.18 It provides a new angle for the application of neural networks in biology.
In this study, we present DCNet, an interpretable deep learning method, that embeds the relationships between 434 cell types and their marker genes into the structure of the ANN. It can identify the cell landscapes with better robustness and accuracy than other methods such as TIMER. Then 10,176 tumor patient cell landscapes of TCGA were obtained using the method of DCNet, and the patients was divided into two subtypes with significant differences in survival time and TME characteristics. Then four survival-related network modules were identified through the analysis of the cell co-expression network in TME. The patients were categorized into high risk and low risk group according the average expression value of genes in the survival-related network modules. Cell landscapes of non-small cell lung cancer in TCGA shows that there is significantly different in TME infiltration between the lung adenocarcinoma and squamous cell carcinoma. The significantly different cell types were identified between two subtypes, and a support vector machine (SVM) classifier was constructed using those significantly different cell types to predict lung cancer subtypes, and the precision of the SVM classifier was 0.975, as indicated in the ROC curve.
Result
Design of DCNet
Based on the AE neural network, we designed a deep learning model, called DCNet, with biologically meaning, that can identify the abundance of cell types from bulk RNA-seq data. It was designed to embed the relationships between 434 cell types and their marker genes in the ANN, which makes DCNet contain a specific biological network structure between two layers of neural networks [Detail in method]. The processing of DCNet is as follows: According to the relationships among cell types, the hierarchical structure of cell types in the patient TME was built, which bases on OBO database (Cell Ontology database) and CellMarker database [Supplementary Table 1], and 434 cells all located on the leaf node. For example, we combine the content of B cells, T cells, etc. to characterize the content of immune cells, and the relationships between cell and their marker genes were shown [Figure 1a]. Here we used marker genes expression in bulk tumors and the relationships between cells and their marker genes to infer the abundance of cells. Gene expression levels were considered as the input and output neurons in DCNet. Here expression levels of 9078 marker genes and all (21,136) genes are used as input and output neurons, respectively [Figure 1b]. Finally, the encode layer and latent layer (434 neurons) of the neural network are transferred, and the activation value of the latent layer represents the abundance of cells [Figure 1c].
Figure 1.
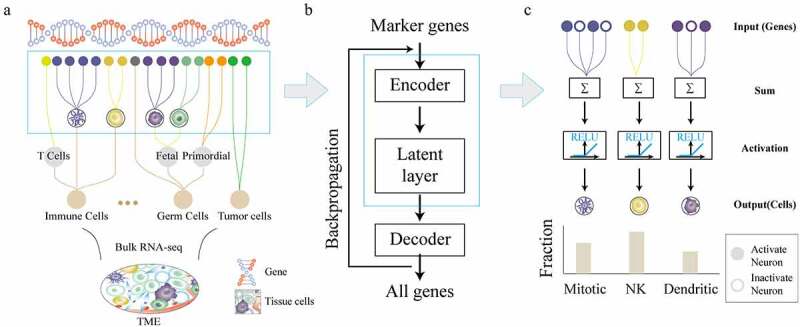
DCNet architecture and design process. (a) Different cell types in the tissue, such as stromal cells, red blood cells, macrophages, T cells, B cells, neuronal cells, etc. These cells can be further divided into different cell types, which has a corresponding marker gene. (b) The basic structure of the DCNet model. The input is the expression levels of marker genes in bulk RNA-seq, and the output is the expression levels of all genes. The middle layer artificial neural network. (c) The gray neurons are activated neurons, and the white ones are inactive neurons. The first level relationship of the DCNet model is the corresponding relationship between cells and marker genes. The middle layer of the DCNet model represents the relative content of cells.
Training and selection of DCNet
Considering the incompleteness of the input data, DCNet model was trained and tested over various dropout rates (0.00, 0.01, 0.03, 0.05, 0.07, 0.1, 0.3, 0.5) for TCGA dataset. The independent dataset (CRC) was used to evaluate the generalization performance of the DCNet model. Here root mean square error (RMSE) of neurons was used as the evaluation index to describe DCNet model fitting. The training results of the DCNet models indicate that the RMSE value of AE gradually decreases as the number of neurons increases from 64 to 4096 (power of 2), and when the number of neurons reaches 2048, its RMSE value is the lowest relative to other AE models. The RMSE value of Full-DCNet increases significantly in training and test dataset of TCGA [Figure 2a]. The RMSE value gradually increases (DCNet-D0, DCNet-D01, DCNet-D03, DCNet-D05, DCNet-D07, DCNet-D10, DCNet-D30, DCNet-D50) with the increase of dropout rate. Meanwhile, the RMSE distribution of all genes in DCNet has a smaller fluctuation range, which is superior to other models. Overall, AE2048 and DCNet models showed better performance.
Figure 2.
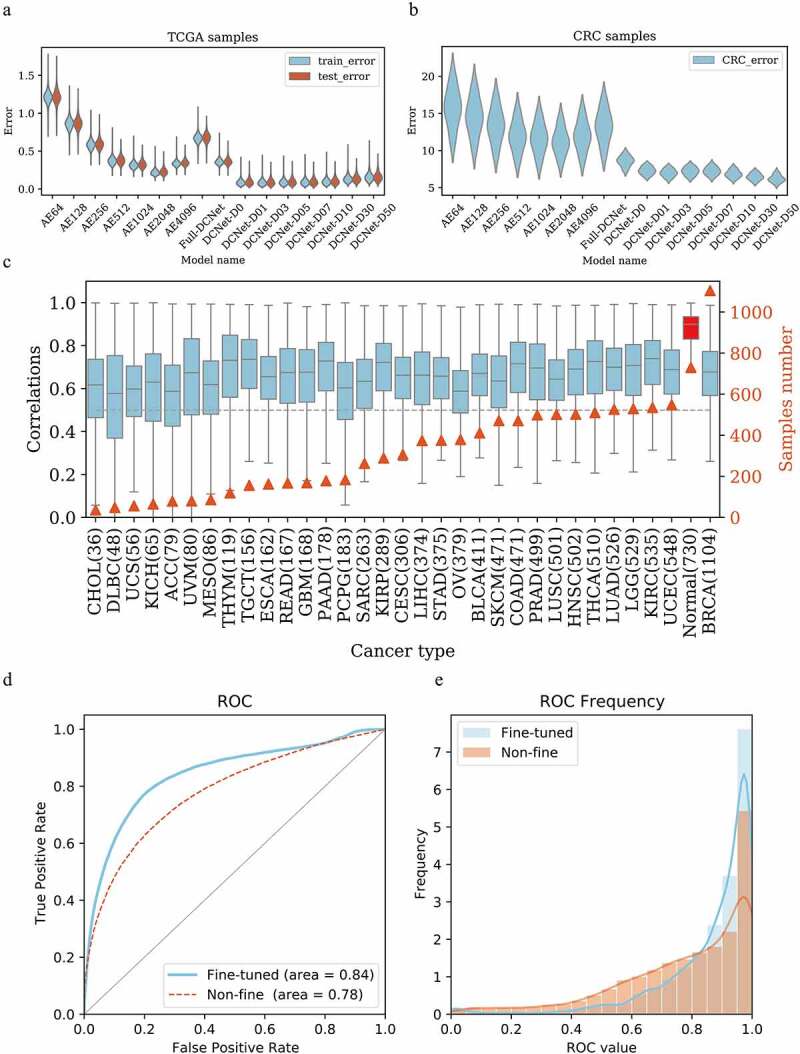
DCNet model determination and performance evaluation. (a) The root mean square error distribution of different models in the training set and test set in the TCGA sample. y is the root mean square error, and x is different neural network models. AE64~ AE4096 are three-layer autoencoders with 64 ~ 4096 neurons in each layer. Full-DCNet is similar to the DCNet architecture, but there is a fully connected neural network between neurons. DCNet-D0 means that the input data is not randomly censored, and DCNet-D01, DCNet-D03, DCNet-D05,DCNet-D07, DCNet-D10, DCNet-D30, and DCNet-D50 means that the input data is randomly censored by 1%, 3%, 5%, 7%, 10%, 30%, and 50%, respectively. Blue is the training set and red is the test set. (b) The root mean square error distribution of different models in the CRC sample. y is the root mean square error, and x is different neural network models, same as Figure A. (c) For samples of different cancer types, the DCNet predicted value is correlated with the true value. The left ordinate is the correlation distribution value, the right ordinate is the number of samples, the abscissa is the cancer type, and the gray dashed line is the correlation 0.5. (d) Draw the ROC curve of the marker gene in single-cell levels, the red is the network without fine-tuning, the blue is the network after the fine-tuning, the abscissa is the false positive rate, and the ordinate is the true positive rate. (e) Frequency distribution diagram of ROC value calculated by gene. The abscissa is the ROC value, and the ordinate is the frequency distribution of the corresponding ROC value.
In CRC independent dataset, different neural network models show great differences in the distribution of error values [Figure 2b]. The RMSE value of the Full-DCNet increases higher, and the RMSE distribution range of all genes becomes larger. DCNet shows higher prediction accuracy and better prediction effect of the samples with the increase of dropout rate. When the input sample data is missing more than 10% (dropout rate>0.1), it may cause too much missing information, such a sample may be a meaningless sample. In summary, here we recommend to choose DCNet model with dropout (dropout rate = 0.1).
Another important factor that affects the classification performance of neural networks is the distribution of the class labels in the training and test data set. If the class labels of the samples in training data set is not uniform, it will lead to the bias of the prediction. We used oversampling method to reduce the imbalance of class labels in the training dataset. We also checked the effect of sample size on classification accuracy for different types of cancer patients in training data set. The correlation coefficients between the output of DCNet and the real cell abundance were calculated for each cancer [Figure 2c]. As the sample size increases, the predicted correlation of patients does not increase or decrease significantly, which represents the sample size has little effect on the model’s predictions. Interestingly, the correlation of normal patients is the highest, with an average level of over 0.9. This is not difficult to understand based on the characteristics of neural networks. During the training process, the neural network tried to find the generalization characteristics of all samples. All cells of TCGA patient originated from normal tissue, so this leads to higher prediction performance of the network for normal patients.
We designed a fine-tuning network to update the network weights of the first and second layers with learning rates of 1e-6 and 1e-4. The ROC curve is drawn to show that the AUC level reaches up to 0.78 [Figure 2d]. After fine-tuning the model, the loss function of the network is significantly improved, and the ROC of the second-level weight is higher [Figure 2e]. The AUC value of 80% of the genes is greater than 0.5. Therefore, for different samples, fine-tuning the network can improve the performance of the model.
DCNet portrayed the cell landscapes accurately
DCNet is a neural network model with a specific biological network structure, and its input layer and the intermediate layer are connected according to the relationships between cells and their marker genes. The intermediate layer neurons are composed of 434 cell types. In the intermediate layer, the ReLU activation function is used to ensure that the activation value of the neuron is greater than zero, and the activation value of intermediate layer neurons is used to represent the abundance of the cell. It means that given the gene expression level of the sample, 434 cells abundance can be inferred from the activation value of the intermediate layer neurons.
Two single-cell sequencing datasets (GSE81861, GSE72056) were downloaded from GEO database and were used as the validation datasets to verify whether the intermediate layer neurons could infer the abundance of cells. There are six cell types including B cells, endothelial cell, epithelial cell, fibroblast cell, macrophage cell, mast cell in the GSE81861 dataset, and 6 cell types including B cells, CAFs cell, endothelial cell, macrophage cell, NK cell, and T cell in the GSE72056 dataset. The number of cells is considered as cell abundance, and the cumulative sum of gene expression levels in all single cells is used as bulk gene expression. Using a trained DCNet model, we predicted the abundance of 434 cells expressed by the activation value of the intermediate layer. We respectively calculated the cell abundance of B cell, endothelial cell, epithelial cell, fibroblast cell, macrophage cell, mast cell, and T cell with the methods of Marker_sum, CIBERSORTx, EPIC, MCP-counter, quanTIseq, Timer, xCell. We checked whether the intermediate layer activation value of DCNet can accurately characterize the abundance of the cell through the correlation between the predicted value and the true value. In the CRC dataset, the correlation values are 0.95 for T cells, while just −0.09 for macrophage cells in xCell method. The similar results were got for MCP-counter method, where the correlation values are 0.96 and 0.98 for B cells and T cells, while just 0.30 for NK cell and endothelial cell in the melanoma dataset. The correlation value of T cells and B cells can also reach 0.80 and 0.98 in the melanoma sample (Marker_sum). However, the correlation of B cells in CRC samples is −0.13, and the evaluation of other cells also fluctuates greatly. Although DCNet’s evaluation results for T cells and B cells are not the highest (0.81, 0.95 in the melanoma sample), its correlation values for all types of cells are stable. Those results show that the predicted cell abundance values of DCNet are positively correlated with the sum of the cell abundance in each sample, which is considered as actual cell abundance in all cell types (all correlation coefficient values>0.7), but the correlation results showed instability in EPIC, TIMER, CIBERSORT, MCP-counter [Figure 3a]. The reliability of those methods tends to depend on the accuracy of marker gene screening. However, DCNet portrays the relationships between marker genes and cells, which avoids the contribution of individual marker genes to cell content. The 1000 cells simulation data generated using R package(immunedeconv) was used to analyze the correlation between DCNet and other methods in T cell and B cell. In T cells, the correlation between the DCNet method and the true value is 0.97, and the correlation with other methods exceeds 0.9, except for TIMER and CIBERSORTx, which have lower correlation with other software [Figure 3b]. And in B cells, all methods show high consistency [Supplementary Figure 1].
Figure 3.
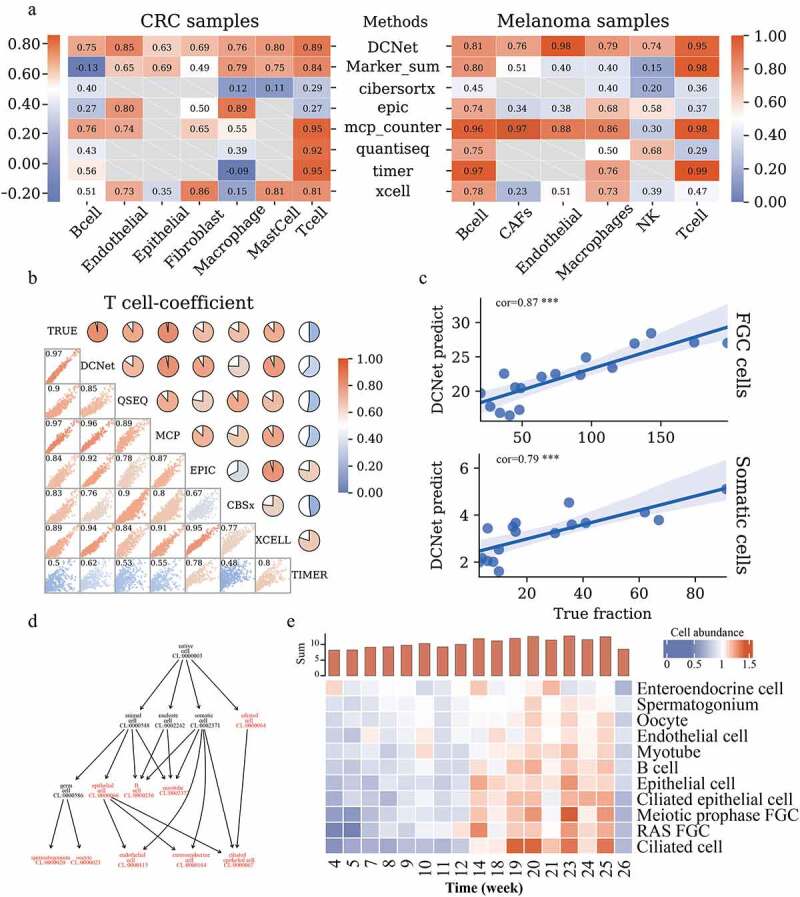
Comparison of cell content assessed by DCNet with other methods. (a) Heatmaps of the correlation between the estimated and true levels of cells on CRC samples and Melanoma samples by different methods. The left side is the CRC sample, and the right side is the Melanoma sample. The redder the color, the closer the correlation level is to 1. The methods include DCNet, Marker_sum (the expression value of marker genes screened in this topic), CIBERSORTx, EPIC, MCP-counter, quanTIseq, TIMER, xCell. The longitudinal direction indicates different cell types. Gray represents that this cell type can not be identified by the corresponding deconvolution method. (b) In the simulation data, the consistency analysis of different methods. The scatter chart shows the distribution of 1000 simulation samples among different methods, and the pie chart shows the level of correlation. The redder the color, the higher the correlation. The diagonal lines are the names of different models, and TRUE is the true level of cell content. (c) The correlation between the predicted value of the DCNet model and the true level. The abscissa is the true cell content, and the ordinate is the cell content predicted by DCNet. The upper part represents FGC cells and the lower part represents Somatic cells. (d) Cell ontology hierarchical relationships among FGC cell, somatic cell, and cell subclasses of those cells, each node represents a cell ontology identity. Highlighted red indicates the cell types recognized by DCNet. (e) enriched cell types evaluated by DCNet shows a big difference in cell abundance levels of human embryos at the developmental stage. The redder the color, the higher the level of cell abundance. The y-axis of the histogram (at the top of 3E) is the sum of the overall cell abundance level of human embryos at each week.
Next, we tested the capability of DCNet to identify the cell abundance of other cells, not just immune cells. A single cell RNA-seq data set (GSE86146) was obtained from the GEO database. It provides the cell abundance information of human embryonic germ cells (FGC) and stomatic cells, during the development for human germline cells. We obtained the FGC cell and somatic cell abundance by using DCNet and calculated the correlation coefficient between the predicted and actual cell abundance of FGC and somatic cells respectively. The values of the correlation coefficient show high consistency with 0.87 in FGC cells and 0.79 in somatic cell (p < .01), which suggests that DCNet can identify the cell abundance accurately for FGC and somatic cell accurately [Figure 3c]. The DCNet can not only estimate the abundance of FGC cell and somatic cell, but also the abundance of cell subclasses of those cells such as ciliated cells, epithelial cells, pre-meiotic embryonic germ cells, and embryonic germ cells. According to the cell ontology [Figure 3d], these cells have hierarchical relationship which come from OBO database. For example, somatic cell (CL:000237) were divided into ciliated cells, epithelial cells, B cell and so on. Germ cell (CL:0000586) were further divided into spermatogonium and oocyte. This result shows that DCNet has the ability to identify cell types that are closer to leaf nodes of the cell ontology structure.
It can be seen that the overall trend of those cells abundance increases gradually from 4 to 14 weeks of pregnancy, and the cell abundance remains relatively high level after 14 weeks [Figure 3e, Supplementary table 2]. By the second and third months of embryonic development, the primordia of almost all organs have basically been formed. This is followed by the internal cell proliferation and increase in volume. This phenomenon may be related to the cells maintaining a high level and active state after 14 weeks. These cells play an important role in the process of development and overall development. These breakthroughs span several stages of development, which is consistent with previous research.20,21 The above result shows that DCNet can not only predict the cell abundance of the common immune cells mentioned above but also quantify the cell abundance of other cell types.
The patients from TCGA cohort exhibits two significantly different types of TME
To further study the potential of DCNet to evaluate the TME, we built the cell abundance landscapes of 434 cell types for 10,176 tumor samples of TCGA using DCNet. Based on the information of the cell abundance landscapes, two TME cancer subtypes with obvious difference in the cell abundance, named immunoprotective subtype (IPS) and nonimmune protective subtype (NIPS), were identified by using the consensus clustering method.22 There are more samples in IPS, and there are more cancer types than NIPS [Figure 4a]. There is a statistical difference in survival time between two TME cancer subtype group (log-rank test, p < .0001) [Figure 4b]. Patients in the cancer IPS group have better survival than patients in the cancer NIPS group.
Figure 4.
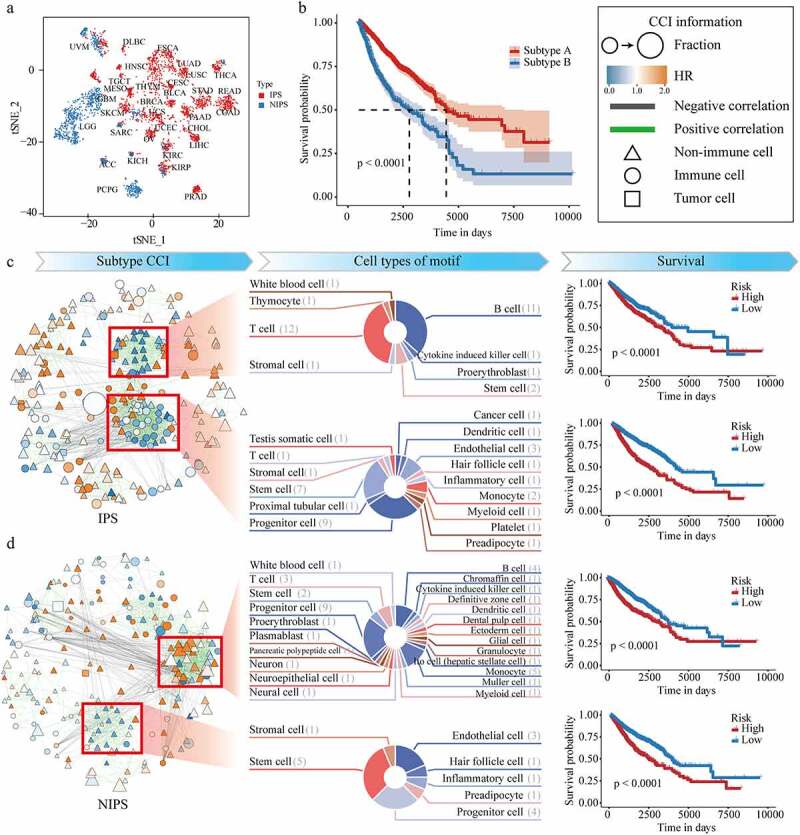
Recognition of cancer subtypes based on DCNet method and mining of cell interaction networks. (a) tSNE distribution map, x is tSNE_1, y is tSNE_2. Each dot represents a sample, and the text represents the type of cancer. Red is IPS and blue is NIPS. (b) Survival curve of IPS and NIPS. The abscissa is the survival time, and the ordinate is the survival rate. (c) and (d) respectively represent IPS and NIPS. The left side of the two graphs is the cell interaction network. The size of the dot is the expression level of the cell. The redder the color, the higher the risk (HR), and the green line is positive Correlation, the black line is negative correlation, triangles represent nonimmune cells, circles represent immune cells, and squares are tumor cells. The middle list of (c) and (d) shows the cell composition information of the four motifs, and the right list shows the survival curves of the high- and low-risk groups according to the four modules.
To investigate the cell abundance characteristics of two cancer subtypes in the TME, we analyzed the differences in cell-type composition, cell abundance, and hazard risk for each cancer subtype. For each cell, the patients were divided into two groups according to the median of this cell abundance and the hazard ratio of each cell was calculated in each cancer subtype respectively. Then the correlations between cells were calculated by using the cell abundance co-expression level (see the detail in the method section). Based on information of the hazard risk for each cell and the correlations between cells, the cell-cell related networks were constructed for each cancer subtype. The cell-cell related network describes biological interactions among cells and provides a systematic understanding of the TME [Figures 4C, D, left panel]. From a global perspective, it represents the different TME between NIPS and IPS. The patients of IPS have more immune cells and low hazard cells infiltration, and stronger connections between cells, in which the number of edges in the cell-cell related networks for cancer IPS and NIPS is 1528 and 1266. Two network modules (motifs) for the IPS were discovered by using MCODE method respectively [Figures 4C, D, middle panel]. In motif-1, there are fewer cell types, but more abundant of T cells and B cells. In motif-2, it shows there are more connections between immune cells and cancer cells, which is more likely to occur in the killing process of tumor cells. But for the patients of NIPS, they have more nonimmune cells and high-risk cells, which may contribute to the worse prognosis of those patients. Two network modules (motifs) were also discovered in NIPS, most of cells in motif-1 are nonimmune cells, and not communicate with tumor cells.
According to the mean HR of different motifs, patients are divided into high and low risk groups, all of which have a large difference in survival (log-rank p < .0001). These four motifs can be used as markers for dividing sample risks [Figures 4C, D, right panel].
It is difficult to measure the cell abundance landscapes in the process of clinical diagnosis and treatment, which causes the difficult in identifying the patients’subtype. But several gene expression signatures are easy to be detected in routine clinical practice. We further identified the TME specific genes as diagnostic markers. First, we analyzed the differentially expressed cell types based on the cell abundance of the two subtypes, and obtained top 1500 differentially expressed marker genes of these cells. Single and multiple Cox’s proportional-hazards regressions were fitted on differentially expressed marker gene expression and, and we identified 33 gene signatures whose risk score was confirmed to be an independent prognostic factor [Figure 5a]. GO function enrichment analysis of the prognostic signature was performed and these 33 genes are significantly enriched in functions linked to binding for post-transcriptional gene silencing, mRNA binding participates in post-transcriptional gene silencing, and other functions [Figure 5b].
Figure 5.
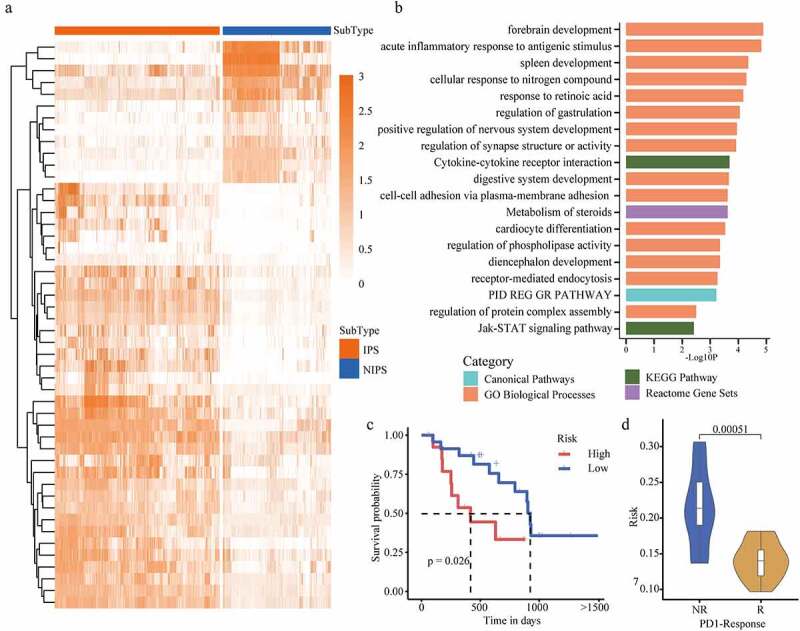
PD1 response samples verify cancer subtypes. (a) A heatmap of the expression level of differential genes. The darker the color, the higher the expression level of the gene. The upper band represents the type of cancer pressure type, red is IPS, and blue is NIPS. (b) Gene function enrichment, the abscissa is -Log10P level, and the ordinate is the function name. Blue is Canonical pathway, green is KEGG pathway, Orange is GO biological process, and purple is response group gene combination. (c) Survival curves of PD1 response and non-response samples. (d) The risk of PD1 response and non-response samples are compared. NR is the non-response group, R is the response group, and the ordinate is the risk score.
Next, those genes were gathered as a signal prognostic signature, which may be a potential biomarker for the prognosis of patients. The risk score was calculated to evaluate the predictive value of those genes. According to risk score values, we have divided the PD1 immune response patients into low risk group (risk score lower), and high risk group (risk score higher). Then survival curve analysis demonstrated that the overall survival rate of high risk patients was significantly poorer than that of low risk patients (p < .05) [Figure 5c]. We analyzed the difference in risk scores between PD1 response and non-response patients groups, and found that the risk of non-responders was significantly higher than the risk scores of patients in the response group (p = .00051) [Figure 5d]. All in all, our results suggest that the cell abundance landscapes evaluated by DCNet can be used to describe the TME accurately and to to identify the TME-specific gene markers, which is meaningful for clinical applications. There are many communications between nonimmune cells and tumor cells, which may be contributed to understand the role of immune cells in the TME.
Lung cancer subtypes show significant differences in cell landscape
We analyzed the abundance difference of cells between the two subtypes of non-small cell lung cancer (LUAD and LUSC in TCGA). The cells with a significant difference in abundance of LUAD and LUSC are identified (log2(Fold Change+1)>1, log2(Fold Change+1)<-1, p < .01) in comparison to normal patients. Further, lung-specific and immune cell types were retained. There are 15 deferentially expressed cells in LUSC and 18 deferentially expressed cells in LUAD, and 12 common deferentially expressed cell types in both cancer subtypes, which presents that LUAD and LUSC have significant differences in TME of lung cancer [Figure 6a, Supplementary table 3]. Among them, the cell abundance of Neuroepithelial cell, Myofibroblast and Basal cell were significantly higher in LUSC than in LUAD [Figure 6b]. However, the content of Granulocyte, Alveolar cell, Leukocyte, Secretory cell, Myeloid-derived suppressor cell, Ciliated cell is higher in LUAD [Figure 6c]. This reflects the difference between LUAD and LUSC in the TME. Here, we consulted the literature and found that all these cell types were specifically related to lung cancer subtypes.23–31 For example, basal cell is a candidate cell of origin for LUSC. More importantly, distinct basal cell lineage trajectories may be involved in homeostasis and injury repair.25 Isaeva, O.I. et al. found that tumor-infiltrating B-cells were significantly associated with LUAD prognosis, and the main mechanism was that the positive effect of IgG4 was related to the activation of the myeloid suppressor cell, thereby avoiding immunosuppression.30
Figure 6.
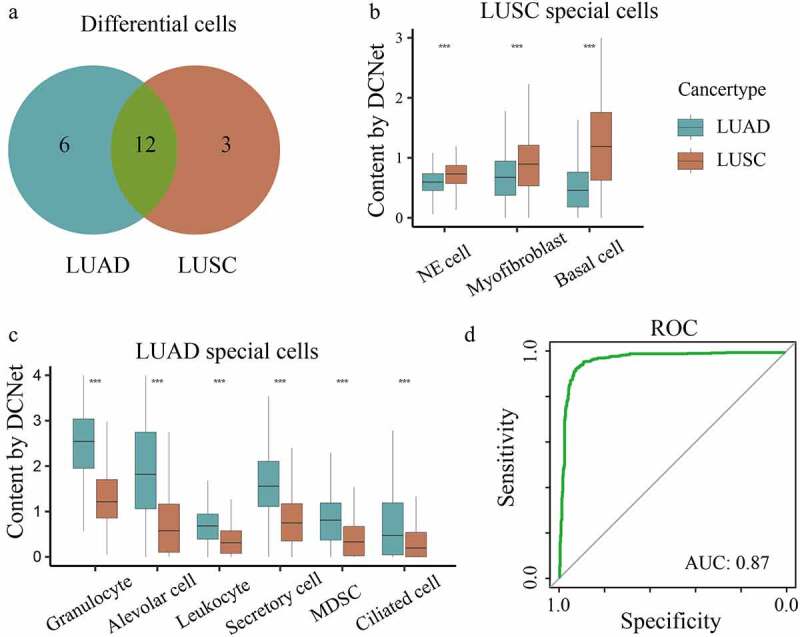
Predict cell types involved in the tumor microenvironment. (a) Differential cells of non-small cell lung cancer. Blue is the number of LUAD differential cells, Orange is the number of LUSC differential cells, green is the number of cells verified in the paper. (b) The content of LUAD specific cell types is compared with LUSC, where x is the cell type and y is the cell content evaluated by DCNet (t test ***, p < .001). (c) The content of LUSC specific cell types is compared with LUAD, where x is the cell type and y is the cell content evaluated by DCNet (t test ***, p < .001). (d) ROC curve, x is sensitivity, y is specificity.
Based on these different cells, we constructed a SVM classifier to distinguish the patients of LUAD and LUSC, with AUC = 0.87 for the ROC value [Figure 6d], which reflects the accuracy of subtype specific cell recognition and further proves that these cells are significantly related to non-small cell lung cancer subtype.
Discussion
Here, the new method DCNet is proposed for portraying the TME based on the relationships between 434 cell types and 9078 marker genes, using the deep learning method. It can evaluate the cell abundance landscapes of patients from bulk RNA-seq sequencing data. The design purpose of the DCNet method is to try to summarize the relationship between marker genes and cells, and to avoid the contribution of individual marker genes to cell types. In the assessment of the content of 434 cells, the analysis results of immune cell types are more stable and more robust than existing methods. Similarly, in the nonimmune cell types, it also shows higher accuracy and important biological significance. Furthermore, based on the DCNet method, we identified two cancer subtypes and found four modules in the cell-cell interaction network. The TME-specific biomarker genes were identified, that can divided the patients into lower risk group and higher risk group. Cell types that may participate in the microenvironment of lung cancer have been found. In actual application, we recommend choosing DCNet with a dropout of 0.1, and fine-tuning the DCNet model in the sample data set. This can improve the accuracy of DCNet’s evaluation. Because DCNet method covers all cell types currently available, it is recommended that researchers chose the appropriate or top ranked cell types to analysis according the purpose of research and the dataset used.
In general, we provide an important neural network model method DCNet, which can evaluate the cell abundance landscapes of patients from bulk RNA-seq data. The performance of DCNet is better than other existing methods, and it is a comprehensive and accurate method for analyzing the cell abundance landscapes. It has been verified in practical applications.
Data and methods
Data Available
From the GDC database (https://portal.gdc.cancer.gov/projects) downloaded 32 kinds of cancer samples and normal samples, a total of 10,906 patients’ RNA-seq expression profiles and clinical data. Two subtypes of non-small cell lung cancer data were used separately: lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC). Download five sets of data from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). GSE81861 (CRC): 11 samples of colorectal cancer patients, 7 cell types.32 GSE75688 (BC): Single-cell sequencing data of 11 breast cancer cells and lymph node metastases.33 It is including 515 single cell RNA-sequencing data, which was sequenced with Illumina HiSeq 2500. The gene expression level is represented as TPM (Transcript per million), which are already normalized and can be comparable across samples in the next analysis. GSE86146 (FGC): 2167 individual PGCs and their gonadal niche cells, covering the developmental stages of female and male human embryos from 4 to 26 weeks after pregnancy.21 GSE72056 (Melanoma): 31 melanoma samples and their 6 cell types.34 GSE78220 (PD1): Transcriptome samples and corresponding clinical data of 38 melanoma biopsy specimens before anti-PD-1 treatment.35 The cell types and corresponding marker gene are obtained from the CellMarker36 database (http://bio-bigdata.hrbmu.edu.cn/CellMarker/).
CellMarker data preprocessing
We downloaded 13,605 marker genes across 467 cell types in 158 human tissues from the CellMarker database. The data was processed as follows: As some cells and their marker genes repeatedly appear in different human tissues, tissue-specific duplications were deleted, keeping only one cell and its marker genes as representative of the duplicated group. The marker genes which not detected in TCGA gene sets were also deleted for the purpose of the DCNet model trainning [detail in TCGA data preprocessing], then 33 cell types were discarded because none of marker-genes were detected. Finally, 434 cell types including immune cells, cancer cells, stromal cells and so on, and their corresponding 9078 marker genes were kept for further analysis. The cell abundance for major cell types was calculated by integrating the category information of the cellmarker database (http://biocc.hrbmu.edu.cn/CellMarker/) and Cell Ontology database (OBO: http://www.obofoundry.org/ontology/cl.html). In the 434 cell types identified by DCNet, there were 77 cell types without Cell Ontology IDs, and 137 cell types have no owns Cell Ontology IDs but were assigned to its parent’s term IDs, and 220 cell types have owned Cell Ontology IDs in the OBO database. For the cell types without Cell Ontology IDs or only with its parents Cell Ontology IDs, the hierarchical relationship information between the cell types was obatined from the CellMarker database. For the cell types with owns Cell Ontology IDs, the ontology structure information between the cell types was downloaded from the cell OBO database. Integrating those information, the Supplementary Table 1 was created and it includes information of the parent and child cell type name, the cell ontology ids of parent and child cell type, the data source database (CellMarker or OBO).
The cell abundance of parent cell types was calculated by the accumulation of the cell abundance of its child cell types for the CellMarker database and its leaf node in the cell ontology structure for OBO database.
TCGA data preprocessing
For the expression profile data of TCGA, the genes whose expression level is 0 in more than 1/3 samples were deleted, 21,136 genes were retained, and log normalization was performed. We divided the gene expression of each sample into input data (expression level of 9078 marker genes) and output data (expression level of 21,136 genes). Due to the uneven distribution of cancer samples, we adopted an oversampling method to enlarge the number of samples while balancing the sample categories. In addition, during the experimental measurement process, some genes cannot be detected due to the low expression intensity of some genes or experimental errors, which will cause the input dimension of the model to not match the marker gene dimension. In order to solve this problem, the input data was randomly deleted with the probability of 0.1, 0.3, 0.5 (0 fill), which could not only increase the training sample, but also reduce the risk of overfitting. Finally, all samples were divided into training set and test set with a ratio of 80% and 20%.
We also obtained clinical data of 1487 patients with tumor metastasis and drug treatment information from TCGA cohorts using the TCGAbiolink37 package. If the number of patients under treatment with a certain drug is less than 48, then the information of this drugs and the patients was deleted. Finally, a total of 7 drugs and 740 patients were kept.
DCNet neural network construction and training
DCNet model trains a deep neural network, which embeds the relationships between cells and their marker genes, to predict more than 400 cell types proportion within bulk seq dataset. The depth of DCNet model is three layers including input layer, hidden layer and output layer. Each cell type is represented by a hidden neuron in the DCNet model. The number of neurons in DCNet is determined based on the corresponding relationships between cells and their marker genes [Supplementary Figure 2A]. The number of neurons in the input layer is equal to the number of total marker genes (9078), the number of neurons in the hidden layer is equal to the number of cell types (434), and the number of neurons in the output layer is equal to the number of TCGA genes (21,136). The construction and training details of the model are described as follows:
First, DCNet model is constructed based on the autoencoder neural network. For the input layer, DCNet input training data set is denoted as , and is the number of patients. For each patient , represents an expression vector for expression of total marker genes, and the dimension is the number of marker genes , and represents an expression vector for the observed expression of total TCGA genes. For the hidden layer, the neurons denotes the hidden layer and its activation value is used to characterize cell proportion in tumor microenvironment, the dimension is the number of cell types [Supplementary Figure 2B]. For the output layer, denotes the ouput vector and it represents an expression vector for the prediction expression of all TCGA genes, represents the number of all TCGA genes.
The neurons are not fully connected between the input layer and the hidden layer. The connection matrix is defined as , where is one-zero matrix with size . indicates that there is a connection relationship between the ith marker gene(the th neuron in the input layer) and the th cell type (the th neuron in the hidden layer) and indicates that there no connection relationship [Supplementary Figure 2C]. The neurons are fully connected between the hidden layer and the output layer.
In order to ensure that the output values of the hidden layer and the output layer are positive, the rectified linear activation function (RELU) is chosen as the constraint function between those layers. For the input layer and the hidden layer, RELU definition is shown as follows:
Here represents the activation value of neurons in the hidden layer, which denotes cell proportion. represents the number of corresponding marker genes of cell . The initialization weight matrix is defined as , is a random weight matrix with size . represents only those edges between cell type and their marker gens are connected and weighted. is the weight of the relationship between the neuron in the hidden layer and neuron the input layer. is input neuron , which represents expression value of marker gene . represents the bias of the th neuron in the hidden layer.
For the hidden layer and the output layer, RELU definition is shown as follows:
Here represents the output value of neurons v in the output layer, which represents the prediction expression of the th TCGA gene. is the weight of the relationship between neurons in the hidden layer and neurons in the output layer, which denotes the expression level of the v th gene in the th cell., and represents the bias of neurons in the output layer.
To train the DCNet model, its parameters are set as adam optimizer, relu activation function, L2loss loss function, learning rate 1e-4, number of iterations 600, 256 samples per batch, cpu training respectively. DCNet model were coded using the mxnet framework in python and its source code has been uploaded to GitHub (http://github.com/xindd/DCNet).
Other neural network methods for comparison
In addition, in order to verify that the performance of the structured neural network for DCNet, we built and trained other neural networks for comparison. First, we designed five deep autoencoder models (AE), the encoding layer and the decoding layer are both two layers, the number of neurons increases by the power of 2, from 64 to 1024 (named: AE64, AE128, AE256, AE512, AE1024). Second, we designed a fully connected neural network with three layers (Full-DCNet). and the number of neurons is same as DCNet model. Its neurons are fully connected, which is different from the structured neural network of DCNet. Finally, 0.00, 0.01, 0.03, 0.05, 0.07, 0.1, 0.3, 0.5 dropout ratio parameters were set to train DCNet model respectively (DCNet-D0, DCNet-D01, DCNet-D03, DCNet-D05, DCNet-D07, DCNet-D10, DCNet-D30, DCNet-D50). All neutral network were coded using the mxnet framework in python, and the parmameters are same to the training of DCNet.
Comparison with other methods
For the datasets used in this study, we evaluated their cell abundance profiles by used CIBERSORTx (Cell Frections module, https://cibersort.stanford.edu/index.php), EPIC (http://epic.gfellerlab.org/), MCP-Counter (https://github.com/ebecht/MCPcoun
ter), quanTIseq (https://icbi.i-med.ac.at/software/quantiseq/doc/index.html), TIMER (https://cistrome.shinyapps.io/timer/) and xCell (https://xcell.ucsf.edu/) respectively. Each method is set to default parameters.The immuneconv package38 of the R language, which is a comprehensive method to estimate the cell abundance by integrating methods of CIBERSORT, EPIC, MCP-Counter, quanTIseq, TIMER and xCell. We used it to construct simulation data of bulk RNA-seq expression profiles based on BC samples for comparison with other models.
Related statistical analysis
Marker genes should be cell-specific high expression in the corresponding cell type. It means that the relationships between the cell of the hidden layer and its marker gene of output layer should have higher weight. We used relationships’weight value bewteen the hidden layer and ourput layer as genes score vector, and created a vector containing the values 0, 1 based on whether the gene is a cell marker gene. We calculated the AUC value with the above two vector. The value of ROC curves is higher means that the marker genes are cell-specific higher expression.
FGC dataset was analyzed by DCNet, and the abundance of 434 cell types were obtained. Sort the cell type in descending order according to the abundance of each cell type and select the top 20 cell types as enriched cell types in FGC dataset, and then only cell types with cell ontology identity were kept for further analysis. We used an R package ontoProc (https://github.com/vjcitn/ontoProc) to visualize structure of cell ontology of those cell types.
The R package of ConsensusCluster,22 which provides a consensus clustering approach was used to classify pancancer patients into different cancer subtypes according the cell landscape identified by DCNet model. In brief, using a manhattan distance, the cluster method of partition around medoids (PAM) was resampled by 0.8% from all cell type features in 1000 iterations. The result is a co-classification matrix with the matrix element value equal to the frequency at which each pair of samples was found in the same cluster in the 1000 iterations. The consensus cluster result was obtained by a final k-mean clustering. In order to select the number of clusters K, the cophenetic correlation coefficient was calculated and the optimal number of consensus cluster was selected as K preceding the largest drop in the cophenetic correlation coefficient.
To identify differentially enrichment cell types, foldchange>2 or foldchange<0.5 and q-value<0.01 were used as standards by using limma package. Univariate and multivariate Cox regression models are used for cell type risk assessment. T test was used to identify differentially expressed genes (DEGs) between cancer subtypes. Function enrichment analyses of DEGs were performed using Metascape (http://metascape.org).
A risk model was constructed with the formula: Risk score = Σ(log10(HR)×genes expression), and the patients were divide into high-risk and low-risk groups based on the median risk score. Kaplan-Meier plots were used to compare survival curves of high risk and low risk groups.
Cell-cell interactions network was constructed and analyzed by using cytoscape software (https://cytoscape.org/). Network motifs were identified by MCODE (Molecular Complex Detection), which is a cytoscape plugin and it can detect subnetworks in an interactome.39
Supplementary Material
Funding Statement
This work was supported by the Natural Science Foundation of Hainan Province [No.821MS045, 821MS0777, 621MS041]; National Natural Science Foundation of China [No.3170115932160179]; Major Science and Technology Program of Hainan Province [No.ZDKJ202003].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Riley RS, June CH, Langer R, Mitchell MJ.. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18(3):175–12. doi: 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hui L, Chen Y.. Tumor microenvironment: sanctuary of the devil. Cancer Lett. 2015;368(1):7–13. doi: 10.1016/j.canlet.2015.07.039. [DOI] [PubMed] [Google Scholar]
- 3.Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79(18):4557–4566. doi: 10.1158/0008-5472.CAN-18-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han X, Zhou Z, Fei L, Sun H, Wang R, Chen Y, Chen H, Wang J, Tang H, Ge W. Construction of a human cell landscape at single-cell level. Nature. 2020;581(7808):303–309. doi: 10.1038/s41586-020-2157-4. [DOI] [PubMed] [Google Scholar]
- 5.Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326–341. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Racle J, de Jonge, K, Baumgaertner, P, Speiser , DE, Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017;6:e26476. doi: 10.7554/eLife.26476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becht E,Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautès-Fridman C, Fridman WH, de Reyniès A . Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17(1):218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, Krogsdam A, Loncova Z, Posch W, Wilflingseder D, Sopper S, Ijsselsteijn M, Brouwer TP, Johnson D, Xu Y, Wang Y, Sanders ME, Estrada MV, Ericsson-Gonzalez P, Charoentong P, Balko J, de Miranda NFDCC, Trajanoski Z. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11(1):1–20. doi: 10.1186/s13073-018-0611-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li B, Severson E, Pignon J-C, Zhao H, Li T, Novak J, Jiang P, Shen H, Aster JC, and Rodig S, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17(1):174. doi: 10.1186/s13059-016-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luca BA, Steen CB, Matusiak M, Azizi A, Varma S, Zhu C, Przybyl J, Espín-Pérez A, Diehn M, Alizadeh AA, et al. Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell. 2021;184(21):5482–5496 e28. doi: 10.1016/j.cell.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyö D, Moreira AL, Razavian N, Tsirigos A. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat Med. 2018;24(10):1559–1567. doi: 10.1038/s41591-018-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmauch B, Romagnoni A, Pronier E, Saillard C, Maillé P, Calderaro J, Kamoun A, Sefta M, Toldo S, Zaslavskiy M. A deep learning model to predict RNA-Seq expression of tumours from whole slide images. Nat Commun. 2020;11(1):3877. doi: 10.1038/s41467-020-17678-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saltz J, Gupta R, Hou L, Kurc T, Singh P, Nguyen V, Samaras D, Shroyer KR, Zhao T, Batiste R, et al. Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images. Cell Rep. 2018;23(1):181–193.e7. doi: 10.1016/j.celrep.2018.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dwivedi SK, Tjärnberg A, Tegnér J, Gustafsson M. Deriving disease modules from the compressed transcriptional space embedded in a deep autoencoder. Nat Commun. 2020;11(1):856. doi: 10.1038/s41467-020-14666-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen HH, Chiu Y-C, Zhang T, Zhang S, Huang Y, Chen Y. GSAE: an autoencoder with embedded gene-set nodes for genomics functional characterization. BMC Syst Biol. 2018;12(Suppl 8):142. doi: 10.1186/s12918-018-0642-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma J, Yu MK, Fong S, Ono K, Sage E, Demchak B, Sharan R, Ideker T. Using deep learning to model the hierarchical structure and function of a cell. Nat Methods. 2018;15(4):290–298. doi: 10.1038/nmeth.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azodi CB, Tang J, Shiu SH. Opening the black box: interpretable machine learning for geneticists. Trends Genet. 2020;36(6):442–455. doi: 10.1016/j.tig.2020.03.005. [DOI] [PubMed] [Google Scholar]
- 20.La H, Yoo H, Lee EJ, Thang NX, Choi HJ, Oh J, Park JH, Hong K. Insights from the applications of single-cell transcriptomic analysis in germ cell development and reproductive medicine. Int J Mol Sci. 2021;22(2):823. doi: 10.3390/ijms22020823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Dong J, Yan L, Yong J, Liu X, Hu Y, Fan X, Wu X, Guo H, Wang X, et al. Single-Cell RNA-Seq analysis maps development of human germline cells and gonadal niche interactions. Cell Stem Cell. 2017;20(6):858–873.e4. doi: 10.1016/j.stem.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 22.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rare neuroepithelial stem cells may underlie small-cell lung cancer. Cancer Discov. 2019;9(12):1641. doi: 10.1158/2159-8290.CD-RW2019-154 [DOI] [Google Scholar]
- 24.Karvonen HM, Lehtonen ST, Sormunen RT, Lappi-Blanco E, Sköld CM, Kaarteenaho RL. Lung cancer-associated myofibroblasts reveal distinctive ultrastructure and function. J Thorac Oncol. 2014;9(5):664–674. doi: 10.1097/JTO.0000000000000149. [DOI] [PubMed] [Google Scholar]
- 25.Hynds RE, Janes SM. Airway Basal Cell Heterogeneity and Lung Squamous Cell Carcinoma. Cancer Prev Res. 2017;10(9):491–493. doi: 10.1158/1940-6207.CAPR-17-0202. [DOI] [PubMed] [Google Scholar]
- 26.Qu Y, Cheng B, Shao N, Jia Y, Song Q, Tan B, Wang J. Prognostic value of immune-related genes in the tumor microenvironment of lung adenocarcinoma and lung squamous cell carcinoma. Aging. 2020;12(6):4757–4777. doi: 10.18632/aging.102871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Li Z, Zhou K, Wang C, Jiang L, Zhang L, Yang Y, Luo W, Qiao W, Wang G. Deciphering cell lineage specification of human lung adenocarcinoma with single-cell RNA sequencing. Nat Commun. 2021;12(1):6500. doi: 10.1038/s41467-021-26770-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khadirnaikar S, Chatterjee A, Shukla SK. Genetic and epigenetic landscape of leukocyte infiltration identifies an immune prognosticator in lung adenocarcinoma. Cancer Biomark. 2021;32(4):505–517. doi: 10.3233/CBM-203071. [DOI] [PubMed] [Google Scholar]
- 29.Yao E, Lin C, Wu Q, Zhang K, Song H, Chuang P-T. Notch Signaling Controls Transdifferentiation of Pulmonary Neuroendocrine Cells in Response to Lung Injury. Stem Cells. 2018;36(3):377–391. doi: 10.1002/stem.2744. [DOI] [PubMed] [Google Scholar]
- 30.Isaeva OI, Sharonov GV, Serebrovskaya EO, Turchaninova MA, Zaretsky AR, Shugay M, Chudakov DM. Intratumoral immunoglobulin isotypes predict survival in lung adenocarcinoma subtypes. J Immunother Cancer. 2019;7(1):279. doi: 10.1186/s40425-019-0747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park WY, Kim MH, Shin DH, Lee JH, Choi KU, Kim JY, Park DY, Lee CH, Sol MY. Ciliated adenocarcinomas of the lung: a tumor of non-terminal respiratory unit origin. Mod Pathol. 2012;25(9):1265–1274. doi: 10.1038/modpathol.2012.76. [DOI] [PubMed] [Google Scholar]
- 32.Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. 2017;49(5):708–718. doi: 10.1038/ng.3818. [DOI] [PubMed] [Google Scholar]
- 33.Chung W, Eum HH, Lee H-O, Lee K-M, Lee H-B, Kim K-T, Ryu HS, Kim S, Lee JE, Park YH, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8(1):15081. doi: 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165(1):35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, Luo T, Xu L, Liao G, Yan M, et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2018;47(D1):D721–D728. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sturm G, Finotello F, Petitprez F, Zhang JD, Baumbach J, Fridman WH, List M, Aneichyk T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics. 2019;35(14):i436–i445. doi: 10.1093/bioinformatics/btz363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4(1):2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.