

Abstract
Background and Objectives:
Portopulmonary hypertension (PoPH) is a rare complication of portal hypertension associated with poor survival. Scarce data is available on predictors of survival in PoPH with conflicting results. We sought to characterize the outcomes and variables associated with survival in a large cohort of patients with PoPH in an American population of patients.
Study design and Methods:
We identified PoPH patients from the Cleveland Clinic Pulmonary Hypertension Registry between 1998–2019. We collected prespecified data, particularly focusing on hepatic and cardiopulmonary assessments and tested their effect on long-term survival.
Results:
Eighty patients with PoPH with a mean ± standard deviation age of 54 ±10 years, (54% females) were included in the analysis. The median Model for End Stage Liver disease with sodium (MELD-Na) score was 13.0 (10.0–18.0) at PoPH diagnosis. World Health Association functional class III-IV was noted in 57%. Mean pulmonary arterial pressure was 47 ±10 mmHg and pulmonary vascular resistance 6.0 ±2.8 Woods units. A total of 63 (78.5%) patients were started on pulmonary arterial hypertension (PAH)-specific treatment during the first 6 months of diagnosis. Survival rates at 1-, 3- and 5-year were 77%, 52% and 34%, respectively. Cardiopulmonary hemodynamics as well as PAH-specific treatment did not affect survival. In multivariable model, MELD-Na, resting heart rate and the presence of hepatic encephalopathy were independent predictors of survival.
Conclusions:
PoPH patients have poor 5-year survival which is strongly associated to the severity of underlying liver disease and not to the hemodynamic severity of PoPH; therefore efforts should be focused in facilitating liver transplantation for these patients.
Keywords: Pulmonary arterial hypertension, Portal hypertension, Cirrhosis, Outcome, Mortality, MELD score
Introduction:
The presence of pulmonary arterial hypertension (PAH) in the context of portal hypertension is termed portopulmonary hypertension (PoPH). Approximately 2–6% of patients with portal hypertension have PoPH[1, 2] and PoPH accounts for 5.3–10% of all cases of PAH [3]. The survival of patients with PoPH is marekedly reduced and most patients die of complications related to their liver and PAH [4, 5]. In fact, a retrospective study from the US based REVEAL registry reported worse survival in PoPH (5-year survival: 40%) as compared to idiopathic PAH (5-year survival: 64%), even in the presence of a better hemodynamic profile at diagnosis[3].
Limited data are available regarding predictors of survival in PoPH with conflicting results. On one hand the earlier French (2008)[6] experience reported cardiac index (CI) to be an important prognostic factor, the more recent UK[7] and French (2008–18)[5] experiences did not find pulmonary hemodynamics at diagnosis to be associated with survival. Data from American population are limited to a study from the REVEAL registry which carried limitations due to a registry based design. The REVEAL registry lacked specific details on the etiology and severity of the underlying cause of portal hypertension, precluding identification of liver related prognostic factors [3]. Studies from the United Kingdom (UK)[7], France[5] and Spain[4] contributed valuable information, but with limitations, in part due to a) the lack of patient specific data related to type and severity of the liver disease, and b) heterogeneous inclusion, with the recruitment of patients with various levels of liver disease severity.
It remains unanswered whether PAH specific therapies alter survival in PoPH, since PoPH patients have been excluded from most PAH treatment trials. In fact, current recommendations regarding the management of PoPH patients are extrapolated from other PAH conditions.[8, 9] Swanson et al[10] reported that the 5-year survival was significantly higher in PoPH patients treated with PAH-specific therapies as compared to no PAH treatment (45% vs 14%). In contrary, both UK[7] and French[5] studies found no difference in survival when treating PoPH patients with PAH-specific agents. In the present study we sought to identify prognostic factors that drive survival at the time of POPH diagnosis in a American population of POPH patients. We hypothesize that variables that reflect the type and severity of the liver disease would have a greater impact in predicting survival than variables that indicate the severity of the pulmonary vascular disease at the time of PoPH diagnosis.
Methods
a). Study subjects and design:
This retrospective study was approved by the Cleveland Clinic institutional review board (study number: 19–1469). Written informed consent was waived. Patients with PoPH were identified from the Cleveland Clinic Pulmonary Hypertension Registry. We included consecutive PoPH patients at the time of the first RHC at our institution, including both newly diagnosed (incident cases) and referred (prevalent cases) patients, between October 1998 and November 2019. All candidates for liver transplantation undergo an echocardiogram to screen for pulmonary hypertension. Patients with echocardiograms that showed an estimated right ventricular systolic pressure (RVSP) ≥ 50 mmHg or right ventricular dysfunction / dilation, underwent RHC.
All patients had end-stage cirrhosis and evidence of portal hypertension, either by distinctive clinical manifestations or elevated hepatic venous pressure gradient (≥ 6 mmHg). In addition, all patients had pre-capillary PH characterized by mean pulmonary artery pressure (mPAP) ≥ 25 mmHg and pulmonary vascular resistance (PVR) ≥ 3 Wood units[11],[12]. We included patients with PAWP ≤15 mmHg or PAWP >15 mmHg, to test the impact on survival of this hemodynamic determination, i.e. isolated or combined pre- and postcapillary PH. All patients underwent an extensive workup to identify other causes for PH, following current recommendations.[8] The diagnosis of PoPH was established after agreement between two PH experts. We collected demographic, spirometric, functional (World Heath Assocaition (WHO) class and distance walked in the six-minute walk test) and echocardiographic data closest to the initial RHC at our institution. Patients were followed until death, liver transplant or end of study (June 2020).
b). Right heart catheterization:
All subjects underwent RHC in the outpatient setting under local anesthesia. In supine position, with the transducer located in mid-thoracic line (4th intercostal space), pulmonary pressures were measured at end-expiration using waveform analysis. Cardiac output (CO) was measured by thermodilution technique, averaging at least three measurements with less than 15% variation. Cardiac index (CI= CO/body surface area) and PVR ((mPAP-PAWP)/CO) were calculated.
c). Liver disease characteristics and liver transplant evaluation:
The diagnosis of cirrhosis with portal hypertension was established by Cleveland Clinic hepatologists. The severity of the liver disease was assessed by Model for End stage Liver Disease (MELD), MELD-Na and the Child Turcotte Pugh (CTP) scoring systems. The MELD-Na[13] score uses the international normalized ratio (INR), serum creatinine, total bilirubin and sodium; meanwhile the CTP score utilizes serum bilirubin, albumin, INR, ascites and hepatic encephalopathy.[14]
d). PAH therapy:
We recorded data on the use of PAH-specific therapies throughout the first 6 months from PoPH diagnosis, using a similar strategy as Savale et al.[5] We divided the PAH treatment groups as no therapy, monotherapy, dual or triple therapy. We recorded whether patients received parenteral prostacyclin analogues during the first 6 months from PoPH diagnosis.
e). Statistical analysis
Data were summarized as mean with standard deviation for continuous variables and as count and percentage for categorical variables. Kaplan-Meir analysis with log-rank test and Cox proportional-hazard regression modeling were performed for time-to-event outcomes. Patients were censored at the time of liver transplantation or end-of-study. Harrell’s C-index was calculated to evaluate the goodness of fit. Subgroup analysis was conducted to evaluate the impact of PAWP (≤ 15 and > 15 mmHg), treatment with PAH-specific medications (yes vs no) and year of PoPH diagnosis (before 2008 and 2008 and after).[15] Multivariable regression models were constructed using a list of carefully preselected variables based on current knowledge, in order to avoid overfitting. Hazard ratios (HR) with 95% confidence intervals were reported. No imputations were performed. All analyses were performed using SAS 9.4 software (SAS Institute, Cary, NC). The level of statistical significance was set at p < 0.05 (two-tailed).
Results
a). Study population
A total of 103 patients with PoPH were identified in our registry, but only 80 patients were included in the final analysis. We excluded 23 patients as a) medical records were incomplete (n=13), with only one visit in our system with limited testing), and b) PoPH developed after liver transplant (n-10). The baseline patients’ characteristics are depicted in table 1. The mean ± SD age at the diagnosis of cirrhosis and POPH were 47 ± 13 years and 54 ± 10 years, respectively. In our cohort, 54% of the patients were women. Alcoholic cirrhosis and hepatitis C were the two most common etiologies for cirrhosis followed by NASH. The median (IQR) MELD-Na and Child-Turcotte-Pugh (CTP) scores at the time of PoPH diagnosis were 14.1 ± 2.3 and 8.1 ± 2.0, respectively. The WHO class at presentation was I-II and III-IV in 33 (42.9%) and 44 (57.1%) patients, respectively (unavailable in 3 cases). Six-minute walk distance (n=58) was 328 ± 115 m.
Table 1:
Baseline patient characteristics.
| Variables | n | Mean ± SD, n (%) |
|---|---|---|
| Age at RHC, years | 80 | 54.4±10.2 |
| Age at liver disease diagnosis, years | 70 | 47.3±13.3 |
| Female gender | 87 | 47(54.0) |
| Race | 80 | |
| White | 67(84.8) | |
| African American | 8(10.1) | |
| Other | 4(5.1) | |
| BMI, kg/m 2 | 75 | 31.6±6.4 |
| Etiology of liver disease | 80 | |
| • Alcohol related | 15(18.7) | |
| • Hepatitis C | 16(20) | |
| • Alcohol and Hepatitis C both | 10(12.5) | |
| • NAFLD | 16(20) | |
| • Others | 23(28.7) | |
| CTP score | 78 | 8.1±2.0 |
| INR | 78 | 1.2±0.20 |
| Total bilirubin, mg/dl | 78 | 2.6±1.8 |
| Serum sodium, mmol/dl | 78 | 138.6±4.1 |
| Serum Creatinine, mg/dl | 78 | 1.3±0.58 |
| MELD | 78 | 13.2±4.4 |
| MELD-NA | 78 | 14.1±5.3 |
| Serum Aspartate Aminotransferase, U/L | 78 | 56.9±39.4 |
| Serum Alanine Aminotransferase, U/L | 77 | 32.5±28.5 |
| Serum Alkaline Phosphatase, U/L | 78 | 144.2±91.6 |
| Albumin, g/dl | 78 | 3.3±0.62 |
| Ascites | 79 | |
| Absent | 32(40.5) | |
| Present | 47(59.5) | |
| Hepatic encephalopathy | 79 | |
| Absent | 39(49.4) | |
| Present | 40(50.6) | |
| TIPS | 76 | 14(18.4) |
| WHO class | 77 | |
| I | 8(10.4) | |
| II | 25(32.5) | |
| III | 40(51.9) | |
| IV | 4(5.2) | |
| Distance walked in 6MWT, m | 58 | 328±115 |
| LVEF, % | 75 | 59.7±5.2 |
| RVSP, mmHg | 71 | 67.1±20.1 |
| Right Ventricular function (echocardiography) | 75 | |
| normal | 36 (48) | |
| abnormal | 39 (52) |
BMI, Body Mass Index; CTP, Child-Turcotte-Pugh; INR, International Normalized Ratio; LVEF, Left Ventricular Ejection Fraction, Model for End stage Liver Disease; MELD- Na, Model for End stage Liver Disease with serum sodium; NAFLD, Non-alcoholic Fatty liver disease; WHO, New York Heart Association; RHC, Right Heart Catheterization; RVSP, Right Ventricle systolic Pressure; TIPS; Transjugular Intrahepatic Portosystemic Shunt.
b). Pulmonary hemodynamics at initial RHC
The pulmonary hemodynamic determinations during initial RHC are shown in table 2. The average mPAP and PVR at diagnosis were 47 ± 10 mmHg and 6.0 ± 2.8 Woods units, respectively. A total of 23 patients had combined pre- and post-capillary PH. Demographics, WHO functional class, MELD-Na or CTP scores at diagnosis were not significantly different between patients with isolated pre- or combined pre- and post-capillary PH (e-Table 1). However, hepatic encephalopathy was more common in patients with isolated pre-capillary PH. There was no difference in the PAH treatment strategy between patients with isolated pre-capillary or combined pre- and post-capillary PH.
Table 2:
Pulmonary hemodynamics at the time of POPH diagnosis.
| Pulmonary hemodynamics | Overall cohort | No PAH specific therapy | Any PAH specific therapy | P-value | |||
|---|---|---|---|---|---|---|---|
| (n=17) | (n=63) | ||||||
| n | Mean ± SD or median (IQR) | n | Mean ± SD or median (IQR) | n | Mean ± SD or median (IQR) | ||
| Heart rate, bpm | 80 | 72.6±12.2 | 18 | 74.1±13.1 | 62 | 72.2±12.0 | 0.58a |
| RA pressure, mmHg | 80 | 10.4± 5.8 | 18 | 9.8±6.8 | 62 | 10.5±5.6 | 0.64a |
| Mean PAP, mmHg | 80 | 47.4±10.0 | 17 | 45.4±11.9 | 63 | 47.9±9.5 | 0.36a |
| PAWP, mmHg | 80 | 13.9±5.9 | 17 | 14.2±6.9 | 63 | 13.8±5.6 | 0.81a |
| Cardiac index (thermo), L.min−1.m2 | 74 | 3.1±1.09 | 17 | 3.4±1.2 | 57 | 3.2±1.06 | 0.34a |
| Stroke volume, ml | 78 | 86.1±26.6 | 16 | 87.2±30.8 | 62 | 85.8±25.6 | 0.85a |
| MvO2, % | 64 | 67.8±8.7 | 14 | 67.9±12.0 | 50 | 67.8±7.6 | 0.98a |
| PVR, Wood units | 80 | 6.0±2.8 | 17 | 5.3±2.3 | 63 | 6.2±2.9 | 0.24a |
| Stroke volume index, ml/m2 | 78 | 42.5±11.9 | 16 | 45.0±14.9 | 62 | 41.9±11.1 | 0.34a |
| Transpulmonary gradient, mmHg | 80 | 33.4±9.6 | 17 | 31.1±10.3 | 63 | 34.1±9.4 | 0.27a |
Statistics presented as Mean ± SD, Median [P25, P75], Pulmonary hemodynamics between patients who were not started on Pulmonary arterial hypertension (PAH) specific therapy are compared to those who were started on PAH specific therapy in the first six months.
p values: a=ANOVA
c). Treatment
A total of 63 patients (78.5%) were started on PAH-specific therapy during the first six months of POPH diagnosis. The majority (62.5%) of treated patients received monotherapy. Phosphodiesterase-5 (PDE-5) inhibitors were the most common treatment, followed by prostacyclin analogues. Intravenous prostacyclin analogues were given to 26.3% patients over the course of their disease. There were no significant differences in the MELD-Na score, CTP score, WHO class or pulmonary hemodynamics between patients who received PAH-specific treatment and those who did not (Table 2). The major reason for patients not receiving POPH specific therapy was their transplant candidacy.
d). Survival
The median follow period was 34 (IQR: 13–65) months. The median survival time was 38 (IQR: 14–95) months in our study. The 1-, 3- and 5-year survival rate was 77.1%, 52.0% and 34.4%, respectively. The MELD (13.8±4.3 vs 11.5±4.3, p=0.03), MELD-Na (15.1±5.3 vs 11.7±4.7, p=0.01) and CTP (8.4±1.9 vs 7.3±1.9, p=0.02) scores were higher in patients that died (n=57) compared to those alive at end of study (n=23). On univariable analysis (Table 3), higher MELD, MELD-Na and CTP scores (p<0.0001 for all three) and their individual components were significantly associated with survival. The C-indexes for MELD, MELD-Na and CTP scores were similar at 0.70, 0.71 and 0.71, respectively. Remarkably, none of the traditional markers of PAH severity were associated with survival. In multivariable analysis (Table 3), MELD-Na (HR=1.13 (95% CI: 1.06–1.20), p=.0003) and absence of hepatic encephalopathy (HR=0.05 (95% CI: 0.26–0.92), p=.026) were significantly associated with survival, with a C-index of 0.75. A MELD-Na score of 11 provided the highest sensitivity (80%) and specificity (52%) for predicting survival (Figure-1)
Table 3:
Univariable and multivariable results of the Cox survival analysis using preselected baseline variables
| Univariable analysis | Multivariable analysis | ||||||
|---|---|---|---|---|---|---|---|
| Variable | n | Hazard Ratio (HR) | 95%CI | P value | HR | 95%CI | P Value |
| Age, years | 80 | 1.00 | 0.98–1.03 | 0.61 | |||
| MELD score, per unit | 78 | 1.21 | 1.12–1.30 | <0.0001 | |||
| MELD-Na, per unit | 78 | 1.16 | 1.09–1.23 | <0.0001 | 1.13 | 1.06–1.2 | 0.0003 |
| CTP score, per unit | 78 | 1.35 | 1.19–1.54 | <0.0001 | |||
| ALP, IU/L | 78 | 1.00 | 1.00–1.01 | 0.03 | |||
| Albumin, g/dl | 78 | 0.49 | 0.31–0.78 | 0.002 | |||
| Resting heart rate, bpm | 79 | 1.02 | 1.00–1.04 | 0.05 | 1.02 | 1.00–1.05 | 0.05 |
| Mean PAP, mmHg | 80 | 1.00 | 0.98–1.03 | 0.97 | |||
| CI (thermo), L.min−1.m2 | 74 | 1.03 | 0.79–1.34 | 0.81 | |||
| PVR, Wood units | 80 | 0.94 | 0.86–1.04 | 0.24 | |||
| MvO2, % | 64 | 1.00 | 0.97–1.03 | 1.00 | |||
| RV dysfunction, present | 75 | 0.97 | 0.57–1.65 | 0.92 | |||
| PAH treatment during first 6 months, yes | 80 | 1.07 | 0.57–2.02 | 0.84 | |||
| Hepatic Encephalopathy, present | 79 | 2.90 | 1.66–5.08 | 0.0002 | 2.04 | 1.09–3.85 | 0.03 |
| Ascites, present | 79 | 2.09 | 1.20–3.65 | 0.009 | |||
| WHO functional class 1–2 vs. 3–4 | 77 | 0.95 | 0.55–1.62 | 0.85 | |||
| Serum sodium, mmol/dl | 78 | 0.91 | 0.84–0.98 | 0.02 | |||
| Total bilirubin, mg/dl | 78 | 1.23 | 1.06–1.42 | 0.006 | |||
| INR | 78 | 2.65 | 0.83–8.45 | 0.10 | |||
| Serum creatinine, mg/dl | 78 | 2.44 | 1.63–3.67 | <0.0001 | |||
| Etiology of liver disease | 80 | ||||||
| Alcohol v/s others | 15 | 0.83 | 0.39–1.72 | 0.61 | |||
| NAFLD v/s others | 16 | 0.79 | 0.38–1.65 | 0.54 | |||
| Hepatitis C v/s others | 16 | 0.57 | 0.27–1.19 | 0.13 | |||
All characteristics at the time of POPH diagnosis unless specified otherwise
Abbreviations: ALP, Alkaline Phosphatase; BMI, Body Mass Index; CTP, Child-Turcotte-Pugh; INR, International Normalized Ratio; LVEF, Left Ventricular Ejection Fraction, NAFLD, Non-alcoholic Fatty Liver Disease, Model for End stage Liver Disease; MELD- Na, Model for End stage Liver Disease with serum sodium; WHO, New York Heart Association;
Figure 1:
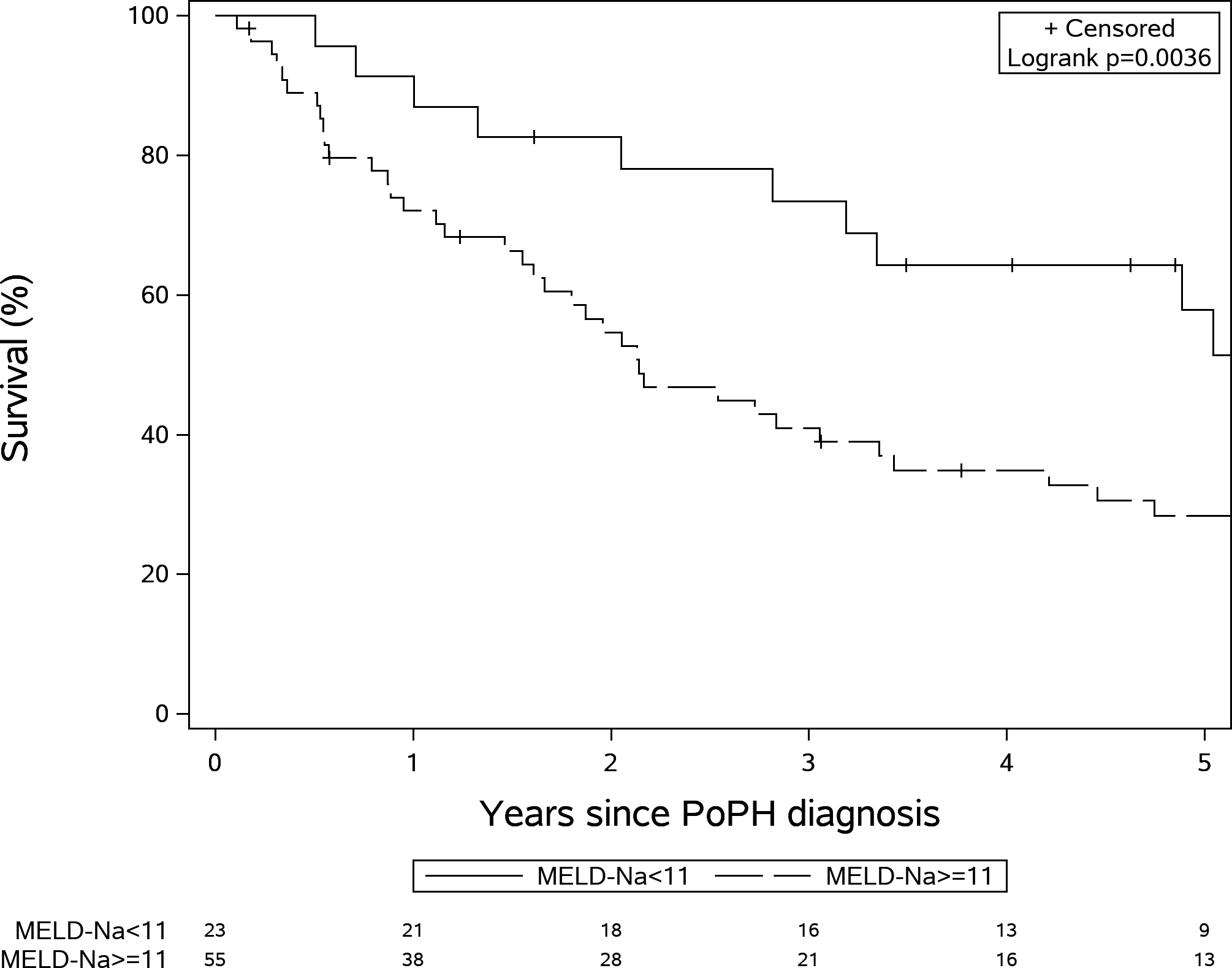
Kaplan Meir survival curves according to Model for End Stage Liver Disease- sodium (MELD-Na) <11 and ≥ 11 (Log rank test P=0.003)
e). Sensitivity analyses
1. Isolated pre- (PAWP≤15mmHg) versus combined pre- and post-capillary PH (PAWP>15mmHg).
Patients with isolated pre-capillary PH (n=57) had lower RA pressure (8.2±4.0 mmHg vs 15.8±6.3 mmHg, p<0.001), lower mPAP (45.6±9.3 vs 51.7±10.5 mmHg, p=0.001) with similar PVR (6.1±2.6 vs 5.8±3.2 Wood units, p=0.63)) and transpulmoanry gradient (34.7±9.1 vs. 30.4±10.7 mmHg, p=0.09) when compared to combined pre- and post-capillary PH (n=23) (supplementary table 1). The proportion of patients receiving PAH specific therapy (80.7% vs. 78.2%, p=1.00) and the survival (p=0.28) was similar between the 2 groups. In fact, survival rates at 1-, 3- and 5-year were 78.6%, 51.3% and 32.9% for those with PAWP ≤ 15 mmHg and 73.7%, 54.0% and 38.7%, for those with PAWP >15 mmHg (Figure 2). Patient characteristics are presented in supplementary table 1.
Figure 2:
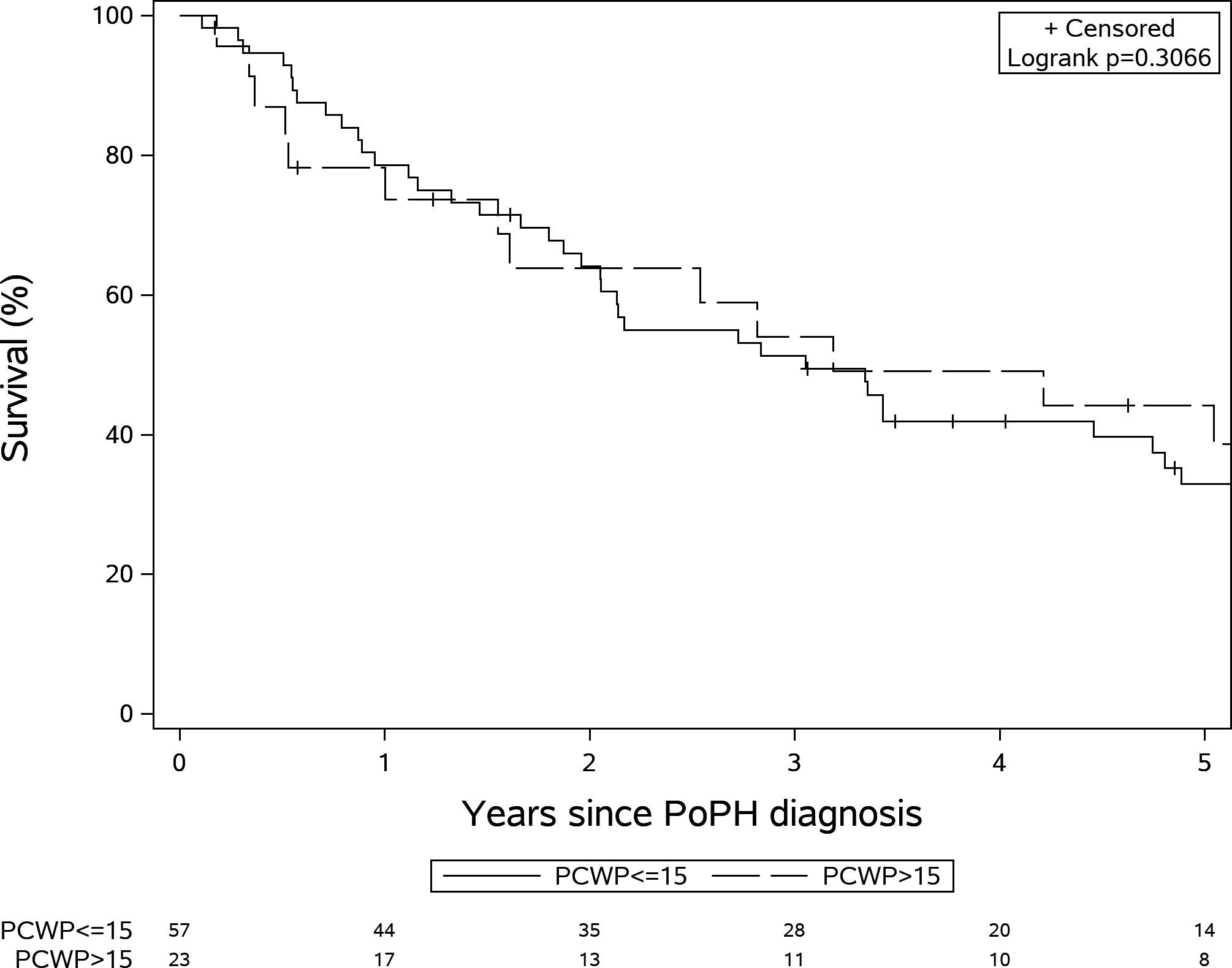
Kaplan-Meir survival stratified by pulmonary capillary wedge pressure (PCWP) >15 mm Hg and PCWP≤15 mm Hg (Log rank test P=0.31).
In the univariable analysis, MELD, MELD Na, CTP, creatinine, albumin and presence of ascites were significant predictors of survival both in PoPH patients with PAWP ≤ 15 mmHg or > 15 mmHg. In PoPH patients with PAWP ≤ 15 mmHg, MELD-Na (HR (95%CI): 1.20 (1.04–1.39), p=0.01) and presence of hepatic encephalopathy (HR (95%CI): 2.41 (1.14–5.10), p=0.02) were predictors of survival in a multivariable model. Meanhwile, in PoPH patients with PAWP > 15 mmHg only the MELD-Na remained a significant predictor of survival (HR (95%CI): 1.20 (1.04–1.39), p=0.01)
2. PAH-specific therapy versus no PAH treatment.
No variables of interest were significantly different between PoPH patients treated (n=63) or not treated (n=17) with PAH-specific therapies (MELD-Na was 14.5±5.3 in the treated vs 12.4±5.1 in the not treated group, p=0.15, PVR was 6.2±2.9 Wood units in treated vs 5.3±2.3 Wood units in the not treated group, p=0.24; PAWP was 13.8±5.6 mmHg in the treated vs 14.2±6.9 mmHg in the not treated group, p=0.81). There was no difference in survival between patients who were or were not treated with PAH-therapy during the first six months after POPH diagnosis (p=0.76) (Figure-3). In addition, the number of PAH therapies (p=0.21, Figure S1) or treatment with parenteral PAH therapy (p=0.10) Figure S2) had no significant impact on survival. A comparison of baseline characteristics and pulmonary hemodynamics are presented in Supplementary table 2 and 3.
Figure 3:
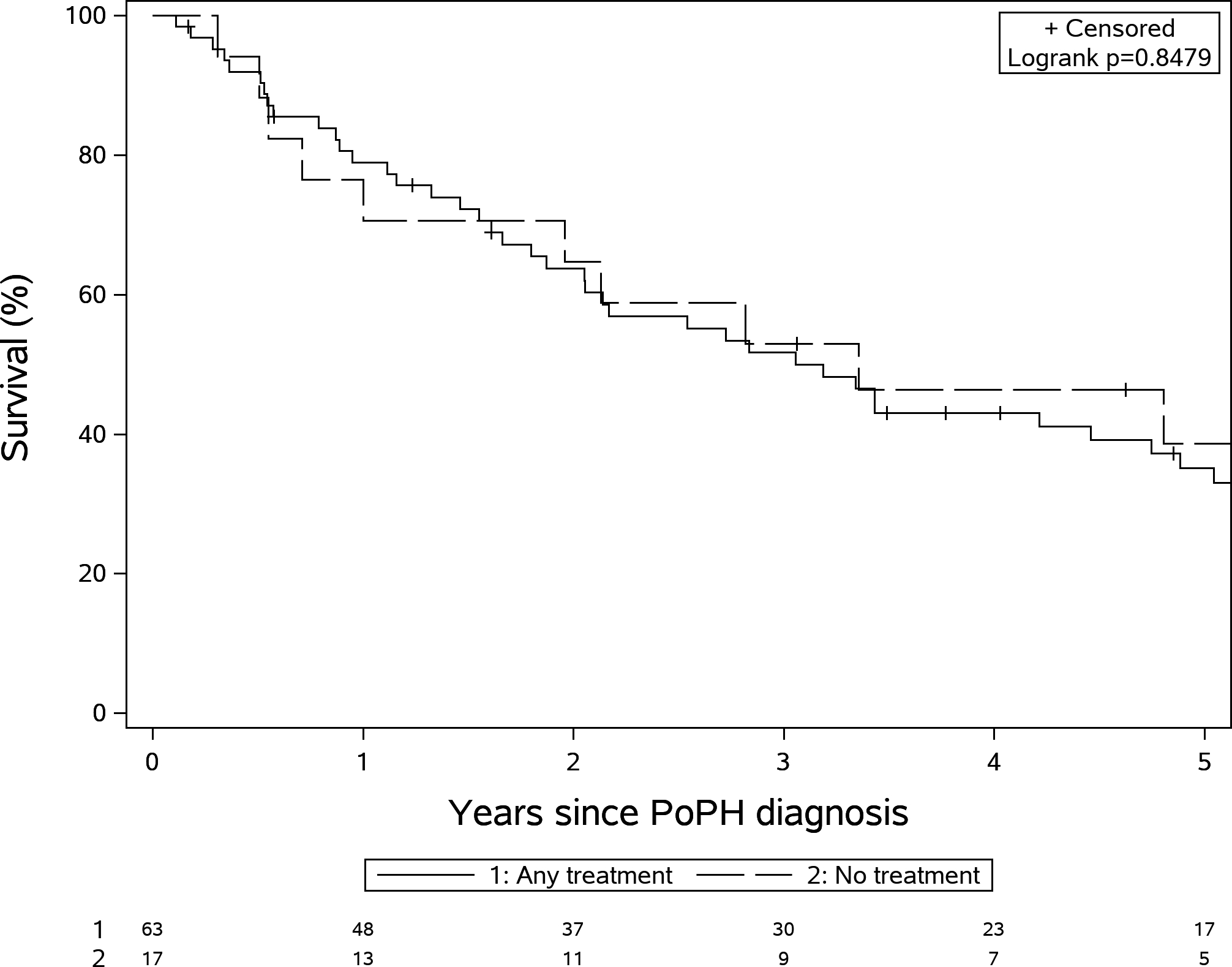
Kaplan Meir survival stratified by treatment with PAH--specific therapies (Log rank test P=0.85).
3. Year of PoPH diagnosis.
Patients that were diagnosed before the year 2008 (n=32) were younger (51±9 vs 56±11 years) but had similar MELD-Na score (13.0±4.9 vs 14.8±5.5, p=0.14) and PVR (5.8±2.2 vs 6.2±3.1, p=0.57) when compared to those diagnosed in the year 2008 or after (n=48). Patients diagnosed before 2008 were less likely to receive PDE-5 inhibitors (53.1% vs. 77.1%, p=0.02) and ETRA (29.2% vs. 3.1%, p=0.003) as treatment options (Supplementary table 4). However, the overall survival was similar between patients diagnosed before and after 2008 (median survival 37 vs. 42 months; log rank test p=0.58)
Discussion:
Patients with PoPH have poor survival, worse than other etiologies of PAH, thought to be related to the severity of the underlying hepatic condition. In the present study we found that the 5-year survival of PoPH patients was only 34.4%% (similar to the REVEAL registry), and it was predominantly affected by the severity of the underlying liver disease. Importantly, in our cohort, the severity of cardiopulmonary hemodynamics at the time of PoPH diagnosis, the presence of combined pre and postcapillary PH and treatment with PAH-specific therapies during the first 6 months after PoPH diagnosis were not predictive of survival.
Several centers have reported their experience in managing patients with PoPH[4, 5, 7]. The mean age at diagnosis of PoPH (54±10 years) in our cohort was similar to that observed in the French National Pulmonary Hypertension (55±10 years)[5] and UK national (53±12 years) registries [7]. We observed female predominance (54%) as reported by Swanson et al (57%)[10], a finding not noted in the French registry[5] (42%). Female gender has been identified as a risk factor for POPH, associated with higher PVR and lower MELD scores[16]. However, female gender was not associated with worse outcomes in our cohort. We noted that alcoholic cirrhosis (alone or combined with hepatitis C) was the most common etiology of liver disease, similarly to other studies[3, 5, 7], and consistent with the epidemiology of cirrhosis. Baseline pulmonary hemodynamics were also in agreement with data from other centers[3, 5, 7].
Although every PoPH patient included in our study had pre-capillary PH, we purposely included subjects with combined pre- and post-capillary PH since a PAWP > 15 mmHg is quite common in these patients in association with volume overload, left ventricular diastolic or renal dysfunction. We performed sensitivity analysis and found significant differences in PVR, MELD-Na, MELD or CPD scores, use of PAH-specific therapies or survival between PoPH with isolated pre- or combined pre- and post-capillary PH.
Therapies specific for PAH were administered to 78.5% of our patients during the first six months of PoPH diagnosis, which is lower than the reported in the French experience (90% [5]) likely reflecting a broader timespan of our study (1998–2019 vs 2007–2017 for Savale et al[5]), and differences in the approach to the disease, in which patients who are not considered candidates for liver transplant may not be offered PAH treatment[12]. Interestingly, we did not find a survival benefit of PAH-specific therapy, in agreement with others [7],[5],[17]. Results from the PORTICO study showed that 12-week treatment with macitentan significantly reduced PVR (35% (95% CI: 28–41) in POPH [18]. It remains unknown whether the PVR improvement would translate into better survival. A recent meta-analysis by Deroo et al. showed that survival was significantly better in PoPH patients treated with both PAH-specific therapy and liver transplant as compared to PAH-specific therapies alone [19].
It remains unclear whether earlier identification and therefore treatment of PoPH may offer a survival advantage, since the majory of these patient die of complications of their liver disease.[20] Earlier PAH treatment of PoPH may help achieve and maintain the pulmonary hemodynamic cut-offs set by liver transplant centers, facilitating liver transplantation. Liver transplantation remains the best treatment option to improve outcomes, in this is supported by our findings that the severity of liver disease drives survival. Patients are offered liver transplant at our center if they are able to satisfactorily decrease mPAP with PAH treatment and they achieve a PVR < 3 Wood units in the presence of adequate RV function. In our cohort, a total of 27 PoPH patients were listed for liver transplant, of whom 8 died on the waiting list. Of the remaining 19, 11 were removed from the list due to worsening PoPH and 3 for other reasons. Meanwhile, 5 patients were ultimately transplanted and were alive at end of study. A total of 51 PoPH patients were not offered liver transplantation, predominantly due to the presence of PoPH with insuffient response to therapy (n=32). The large number of patients not offered liver transplantation, a potentially life saving treatment modality, raises important questions. Would a) earlier recognition of PoPH, b) more aggressive PAH treatment, and c) a shift in focus from mPAP to PVR to assess response to PAH therapies, be better strategies in facilitating liver transplantation and ultimately improving the outcomes in these patients?
The low 5-year survival (34.4%) in our study was comparable to the US based REVEAL (40%) [3] and United Kingdom national registry[7] (35%) but lower than the French PH registry[5] (51%). The higher mortality in our population is likely attributed to the higher MELD-Na at PoPH diagnosis when compared to the French registry5 (14 vs 11), likely a reflection of earlier liver transplant evaluation and PoPH screening strategies. Research is needed to establish the best time to start screening cirrhotic patients for the presence of PoPH. The timing PoPH diagnosis may have an impact on the effect of the PAH-specific therapies since the vascular plasticity and response to therapy may vary over time. Treatment of PoPH in patients with advanced liver disease is unlikely to alter the natural course of the hepatic disease, unless the treatment facilitates the candidacy for liver transplantation[12].
The severity of liver disease at the time of POPH diagnosis was the predominant factor associated with mortality in our study. Our results are consistent with Savale et al[5](2007–2017) who observed a strong association between MELD-Na score and mortality. In contrast with a previous study (1984–2004) by the French National center [6] that found that cardiac index was associated with survival, none of the pulmonary hemodynamic determinations in our study predicted survival. A study from the UK national registry (2001–2010) [7] reported that neither the severity of liver disease nor the severity of PoPH predicted survival, but authors did not assess the role of MELD score, which has become a main indicator of liver cirrhosis severity [21].
Our study has a few limitations, including its single center retrospective nature and the broad study entry period. These limitations reflect the rarity of the condition. Sensitivity analysis showed no significant changes in survival when comparing patients diagnosed with PoPH before and after the year 2008. We also recognize that we studied the pulmonary hemodynamics at the time of initial RHC and we did not collect follow-up RHC data. Pulmonary hemodynamics may change overtime with potentially different prognostic implications. We also acknowledge that we only collected data regarding PAH specific therapy within the first six months of POPH diagnosis, strategy similar to Savale et al. This may have impacted the effect of treatment and hemodynamics on overall survival and needs to be studied further. In addition, 21.5% of patients in our study did not receive PAH specific therapy due to various reasons including transplant candidacy (when deemed ineligible for liver transplant the majority of our patients were not interested in PAH treatment, that is predominantly oriented to improve hemodynamics before liver transplantation), cost and availability of treatment which may effect the results. Despite these limitations our study included detailed data on a large number of PoPH patients, demonstrating a poor overall survival directly linked with the severity of liver disease.
Conclusions:
Our study showed that 34% of patients with PoPH, mostly diagnosed at the time of liver transplant evaluation, survive at 5 years. The poor survival is predominantly driven by the severity of the underlying liver disease. In our cohort of end-stage disease patients, PAH-specific therapies within first six months of POPH diagnosis did not significantly alter the course of the disease.
Supplementary Material
Acknowledgements
Financial conflict of interest statements:
Manik Aggarwal, MD; Manshi Li, MS; Abhishek Bhardwaj, MD; William D. Wallace, MD; Xiaofeng Wang, PHD; William D. Carey, MD; Raed A. Dweik MD and Adriano R. Tonelli MD have no significant conflicts of interest with any companies or organization whose products or services may be discussed in this article. Gustavo A. Heresi MD, MS: Gustavo A. Heresi received personal fees for being a member in Bayer Healthcare – Advisory Board and Speaking.
Funding sources:
A.R.T is supported by National Institute of Health (NIH) grant # R01HL130307.
Abbreviations
- ALP
Alkaline Phosphatase
- INR
International Normalized Ratio
- CO
Cardiac Output
- CTP
Child Turcotte Pugh
- MELD
Model for End stage Liver Disease
- MELD- Na
Model for End stage Liver Disease with serum sodium
- mPAP
Mean Pulmonary artery pressure
- WHO
New York Heart Association
- PAWP
Pulmonary Artery Wedge Pressure
- PAH
Pulmoanry arterial Hypertension
- PH
Pulmoanry Hypertension
- PDE-5i
Phosphodiesterase-5 inhibitors
- PoPH
Portopulmonary hypertension
- PVR
Pulmonary vascular resistance
- RHC
Right Heart Catheterization
References:
- [1].Hadengue A, Benhayoun MK, Lebrec D, et al. Pulmonary hypertension complicating portal hypertension: Prevalence and relation to splanchnic hemodynamics. Gastroenterology 1991; 100: 520–528. [DOI] [PubMed] [Google Scholar]
- [2].Castro M, Krowka MJ, Schroeder DR, et al. Frequency and Clinical Implications of Increased Pulmonary Artery Pressures in Liver Transplant Patients. Mayo Clin Proc 1996; 71: 543–551. [DOI] [PubMed] [Google Scholar]
- [3].Krowka MJ, Miller DP, Barst RJ, et al. Portopulmonary hypertension: A report from the US-based REVEAL registry. Chest 2012; 141: 906–915. [DOI] [PubMed] [Google Scholar]
- [4].Lázaro Salvador M, Quezada Loaiza C, Rodríguez Padial L, et al. Portopulmonary hypertension: prognosis and management in the current treatment era. Results from the REHAP Registry. Intern Med J. Epub ahead of print 2020. DOI: 10.1111/imj.14751. [DOI] [PubMed] [Google Scholar]
- [5].Savale L, Guimas M, Ebstein N, et al. Portopulmonary hypertension in the current era of pulmonary hypertension management. J Hepatol 2020; 73: 130–139. [DOI] [PubMed] [Google Scholar]
- [6].Le Pavec J, Souza R, Herve P, et al. Portopulmonary hypertension: Survival and prognostic factors. Am J Respir Crit Care Med 2008; 178: 637–643. [DOI] [PubMed] [Google Scholar]
- [7].Sithamparanathan S, Nair A, Thirugnanasothy L, et al. Survival in portopulmonary hypertension: Outcomes of the United Kingdom National Pulmonary Arterial Hypertension Registry. J Hear Lung Transplant 2017; 36: 770–779. [DOI] [PubMed] [Google Scholar]
- [8].Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53: 1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Galiè N, Humbert M, Vachiery J-L, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2015; 46: 903–975. [DOI] [PubMed] [Google Scholar]
- [10].Swanson KL, Wiesner RH, Nyberg SL, et al. Survival in Portopulmonary Hypertension: Mayo Clinic Experience Categorized by Treatment Subgroups. Transplantation 2008; 8: 2445–2453. [DOI] [PubMed] [Google Scholar]
- [11].Simonneau G, Gatzoulis MA, Adatia I, et al. Updated Clinical Classification of Pulmonary Hypertension. J Am Coll Cardiol 2013; 62: D34–D41. [DOI] [PubMed] [Google Scholar]
- [12].AbuHalimeh B, Krowka MJ, Tonelli AR. Treatment Barriers in Portopulmonary Hypertension. Hepatology 2019; 69: 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kim WR, Biggins SW, Kremers WK, et al. Hyponatremia and Mortality among Patients on the Liver-Transplant Waiting List. N Engl J Med 2008; 359: 1018–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Infante-Rivard C, Esnaola S, Villeneuve J-P. Clinical and statistical validity of conventional prognostic factors in predicting short-term survival among cirrhotics. Hepatology 1987; 7: 660–664. [DOI] [PubMed] [Google Scholar]
- [15].Krowka MJ, Fallon MB, Mulligan DC, et al. Model for end-stage liver disease (MELD) exception for portopulmonary hypertension. Liver Transplant 2006; 12: S114–S116. [DOI] [PubMed] [Google Scholar]
- [16].DuBrock HM, Cartin-Ceba R, Channick RN, et al. Gender Differences in Portopulmonary Hypertension. Chest. Epub ahead of print 2020. DOI: 10.1016/j.chest.2020.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Legros L, Chabanne C, Camus C, et al. Oral pulmonary vasoactive drugs achieve hemodynamic eligibility for liver transplantation in portopulmonary hypertension. Dig Liver Dis 2017; 49: 301–307. [DOI] [PubMed] [Google Scholar]
- [18].Sitbon O, Bosch J, Cottreel E, et al. Macitentan for the treatment of portopulmonary hypertension (PORTICO): a multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir Med 2019; 7: 594–604. [DOI] [PubMed] [Google Scholar]
- [19].Deroo R, Trépo E, Holvoet T, et al. Vasomodulators and liver transplantation for portopulmonary hypertension: evidence from a systematic review and meta-analysis. Hepatology. Epub ahead of print 2020. DOI: 10.1002/hep.31164. [DOI] [PubMed] [Google Scholar]
- [20].Sahay S, Al Abdi S, Melillo C, et al. Causes and Circumstances of Death in Portopulmonary Hypertension. Transplant Direct 2021; 7: e710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kamath PS, Kim WR. The Model for End-stage Liver Disease (MELD). Hepatology 2007; 45: 797–805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.