

SUMMARY
Domestic dogs (Canis lupus familiaris) are the most variable-sized mammalian species on Earth displaying a 40-fold size difference between breeds1. Although dogs of variable size are found in the archeological record2–4, the most dramatic shifts in body size are the result of selection over the last two centuries, as dog breeders selected and propagated phenotypic extremes within closed breeding populations5. Analyses of over 200 domestic breeds have identified approximately 20 body size genes regulating insulin processing, fatty acid metabolism, TGFβ signaling and skeletal formation6–10. Of these, Insulin-like Growth Factor 1 (IGF1) predominates, controlling approximately 15% of body size variation between breeds8. The identification of a functional mutation associated with IGF1 has thus far proven elusive6,10,11. Here, to identify and elucidate the role of an ancestral IGF1 allele in the propagation of modern canids, we analyzed 1,431 genome sequences from 13 species, including both ancient and modern canids, thus allowing us to define the evolutionary history of both ancestral and derived alleles at this locus. We identified a single variant in an antisense long non-coding RNA (IGF1-AS) that interacts with the insulin-like growth factor-1 (IGF1) gene creating a duplex. While the derived mutation predominates in both modern grey wolves and large domestic breeds, the ancestral allele, which predisposes to small size, was common in small-sized breeds and smaller wild canids. Our analyses demonstrate that this major regulator of canid body size nearly vanished in Pleistocene wolves, before its recent resurgence resulting from human-imposed selection for small-sized breed dogs.
Keywords: IGF1, body size, ancient DNA, domestication, canid evolution, long non-coding RNA
eTOC blurb
Plassais et al. assemble a catalog of thousands of genomes, inclusive of ancient and modern canids, in a search for genetic variants passed from ancient to modern dogs. Plassais et al. identifie an ancient mutation at the IGF1 locus, which has been under human selection, that contributes to a significant portion of body size in modern dogs.
RESULTS AND DISCUSSION
To identify functional mutation(s) at the IGF1 locus that explain body size differences in both modern and historical canids, we analyzed 1,431 genomes representing 13 species that encompassed ancient canines, modern breed dogs and wild canids. We generated a catalog of 1,297 modern dog genomes from 230 breeds (1,156 dogs), 140 indigenous and village dogs from around the world, and one dingo (Data S1A) from which we identified 64.92 million biallelic variants, including small indels. Using data from a maximum of four individuals per modern breed (two males and two females), resulting in a total of 456 individuals from 179 breeds, we calculated the association with body size at the locus surrounding the IGF1 gene on CFA15 (41.20–41.27 Mb in Canfam 3.1).
The top 10 most associated variants on CFA15 displayed a high degree of linkage disequilibrium (LD), with p-values driven largely by ten dogs from three non-European breeds12: four Chow Chows, two Afghan hounds and four Tibetan mastiffs (Table S1 and Data S1A). Among the 10 most associated variants, we identify a previously reported intronic single nucleotide polymorphism (SNP)(rs22437444)10,11, as well as a new candidate SNP (rs22397284; chr15:41219654.g.T<C, p-value=10−29). Unlike the remaining ten most associated GWAS markers and a previously reported intronic SINE element10,11, SNP rs22397284 is polymorphic in other wild canid species (Tables S1–S2), demonstrating the highest significant association with body mass in wild canids at the IGF1 locus (n=80, p-value=10−19). Of note, using a set of 19 wild canids, 255 domestic dogs and 58 village dogs, we did not detect any copy number variations associated with body size variations on IGF1 locus (Data S1B). While there exists the possibility that variants polymorphic only in dogs, such as the SINE element, play functional roles in body size regulation in domestic dogs, we focused our study on the new candidate SNP, rs22397284, as it is the only variant we identified that is associated with body size variation in both dogs and the other canid species analyzed here (Table S2).
We first observed that 75% (3rd quartile) of domestic dogs homozygous for the C allele of rs22397284 have a breed body mass average (BMA) <15 kg (herein defined as “small breeds”; Figure 1A), while 75% of dogs homozygous for the T allele have a BMA >25 kg (1st quartile; defined as “large breeds”; Data S1A). We confirmed these results via Sanger sequencing of the SNP in 144 poodle varieties (standard, miniature and toy) and in three distinct schnauzer breeds: 48 miniatures, 42 standards and 48 giants (Data S1C). The schnauzer breeds differ in body mass by up to six-fold between miniature and giant, yet were likely developed from a close lineage12,13. Miniature and toy poodles and the miniature and standard schnauzers are largely homozygous for the small size-associated C allele (C allele frequency >0.95) (Table S1 and Data S1C), and giant schnauzers are fixed for the large size-associated T allele (Figure 1B). Standard poodles, however, possess both alleles in equal frequency, perhaps reflecting a lack of strong selection for size in the large poodle variety12.
Figure 1. Insulin-like growth factor 1 (IGF1) in Canidae.
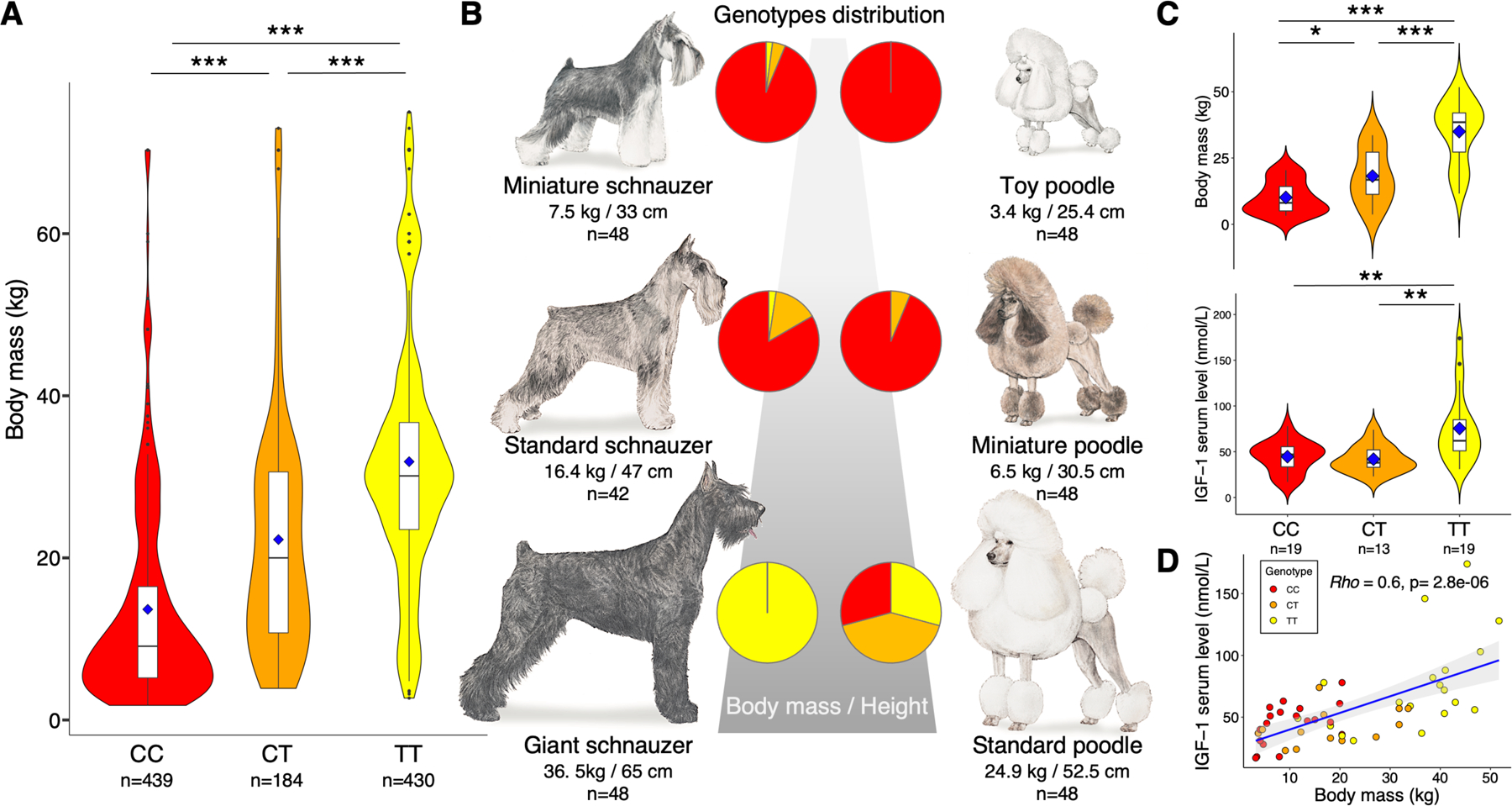
(A) IGF1-AS variant genotypes and body mass range collected from 1,162 dogs of 230 breeds. Dots represent outliers. Blue diamonds indicate breed body mass averages, boxplots represent interquartile ranges and black horizontal bars show median for each (***P < 0.0001, Mann–Whitney–Wilcoxon tests). (B) Distribution of IGF1-AS alleles in three schnauzer breeds and poodle varieties. Pie chart indicates population proportion based on genotypes. Red=CC, orange=CT, and yellow=TT. (C) Body mass and serum levels of IGF-1 protein (nmol/L) as functions of IGF1-AS genotype. IGF-1 serum protein levels were assayed in 51 dogs, including 13 mixed-breed dogs (*P<0.01, **P<0.001, ***P < 0.0001, Mann–Whitney–Wilcoxon tests); (D) Positive correlation observed between body mass and IGF-1 serum level (Rho Spearman test). Blue line shows the regression line, grey area represents confidence interval. See also Table S1 and Data S1.
Next, we Sanger sequenced the candidate variant in 51 dogs, including 13 mixed-breed dogs, for which we also measured both exact body mass and IGF-1 serum level (Figure 1C). We observed significant relationships between genotype, body mass and IGF-1 serum level (P-values <0.001, Mann–Whitney–Wilcoxon tests), and a direct correlation between body mass and IGF-1 serum level (P=10−6, ρ=0.6, Spearman test) (Figure 1D). These results confirm that this non-coding variant could impact IGF-1 production through a regulatory mechanism, particularly since this SNP is located within the last exon of a predicted 1,204 bp long non-coding RNA (lncRNA), which is itself an antisense of the IGF1 gene (herein referred to as IGF1-AS). Because of the strong association between both C and T alleles with body mass variation observed across breeds, we termed the “small allele” the small body mass-associated C allele, and the “large allele” the large body mass-associated T allele.
To better understand the origin of the IGF1-AS variant, we extended our analysis to include 33 previously published ancient dog genomes2,3,14–18 (Figure 2A and Table S3). In order to account for low coverage and expected DNA damage (i.e., as the IGF1-AS variant is a transition) that often characterizes ancient genomes, we computed the posterior probability of each genotype (PP) under different priors, with or without re-scaling base quality scores based on the likely damaged positions19 (Methods). We found the variant alleles were heterozygous in a previously described ~9,500 year old Siberian sled dog16. In addition, 50% of ancient dog genomes dating from 10,930 to 100 years before present (ybp) were homozygous for the small allele (n=13; PP>0.9), while 32% were homozygous for the large allele (n=9; PP>0.9). Surprisingly then, both small and large alleles have been segregating in dogs for at least 9,500 years.
Figure 2. Detection of IGF1-AS variants in ancient and modern genomes.
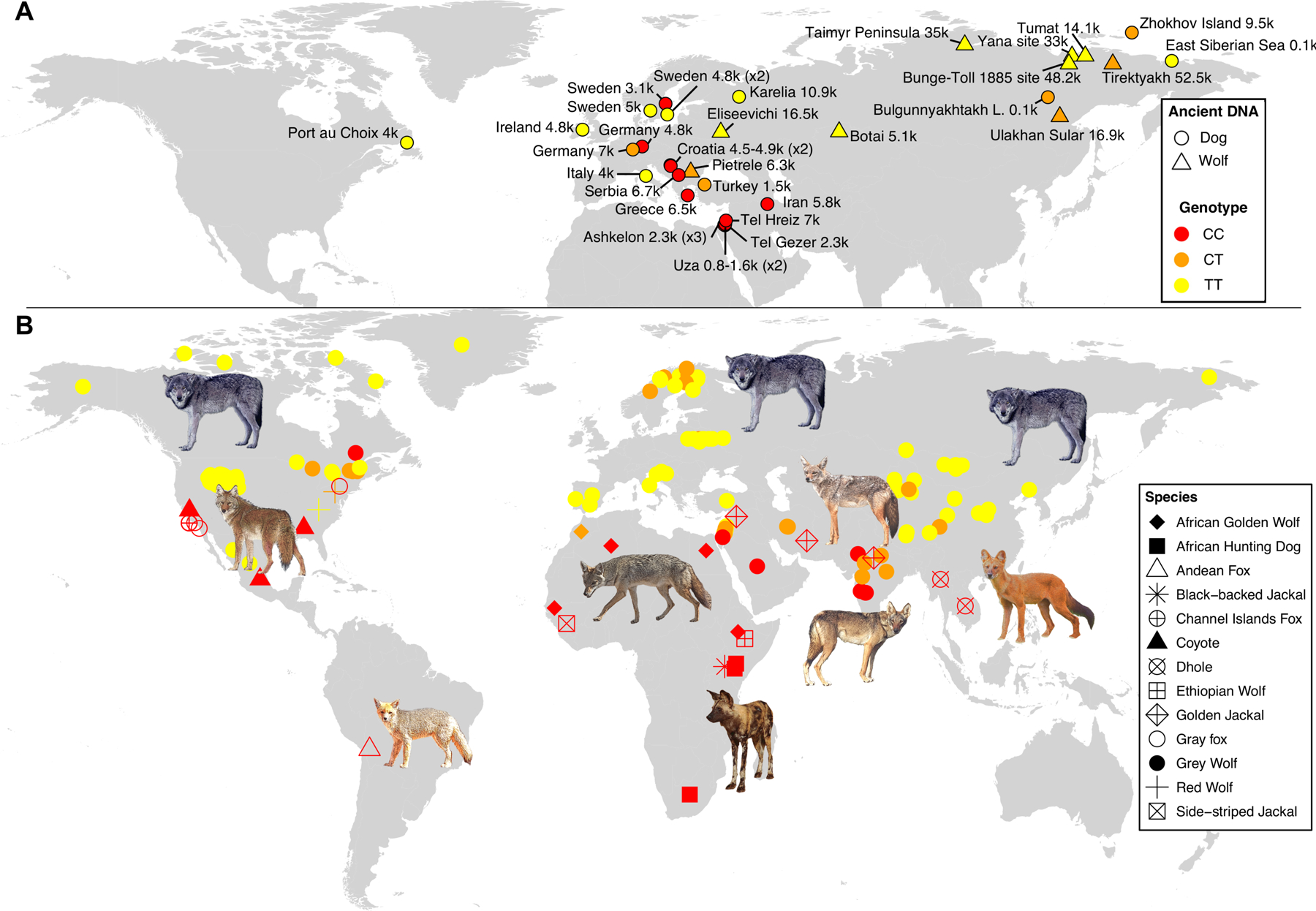
(A) Map of DNA sampling locations for 35 ancient canids, colored by their genotypes. Circles=dogs; triangles=wolves. Data were merged when several samples were collected from the same site with the same predicted age. Number of samples are indicated between brackets. Ages are given in thousand years before present (k). (B) Genotypes for the IGF1-AS variant in 13 species: 92 whole genome sequences and 58 DNA samples that were Sanger sequenced for the IGF1-AS variant. Map demonstrates a North/South geographic gradient of alleles corresponding to body size. See also Figures S1–S3 and Tables S1–S3.
The body mass of many of the archaeological dogs has been estimated2–4, and our characterization of the large and small alleles correlates with the body mass of ancient dogs. We first estimated a body mass of 24.8 kg for the ~9,500 year old heterozygous dog using direct measures of the mandible (Figure S1A–S1B and Methods)20,21. We also found that three Israeli dogs (~2,300 ybp), estimated to weigh~14.6 kg (based on the methodology described in Harcourt and Wing4(Figure S1C)), all possessed the small allele (Table S3). A pre-contact American dog sample from Newfoundland (~4,000 ybp), described as a large dog3, was homozygous for the large allele. In addition, our analyses indicated that the frequency of the large allele was higher in ancient dogs excavated from Northern latitude sites (latitude>55°N; n=8; freq(T)=0.75), while ancient dogs from Southern latitude sites (latitude<45°N, mostly from the Mediterranean region) were more likely to possess the small allele (n=12; freq(C)=0.79) (Figure 2A). This fits with what is known as Bergmann’s rule, which states that populations and species of small size live in comparatively warmer climates while larger species and populations tend to live in colder climates22,23. The antiquity of these alleles and their geographic distribution in ancient dogs suggests that each has been segregating in the ancestors of dogs, which could also explain the observed significant associations between these alleles and body mass variation observed in other canid species (Table S1).
To explore this hypothesis, we analyzed genome-wide data from nine ancient and 68 modern grey wolves from different locations (Figure 2 and Tables S2–S3). We also genotyped 46 additional modern grey wolves sampled from nine countries on three continents using Sanger sequencing (Table S2). As opposed to the previously reported variants in IGF110, and as we previously described in this work, our analyses indicate that the IGF1-AS variant identified here segregates not only in dogs, but also in both modern and ancient wolves where it is also associated with body size variation (Tables S1–S3). Indeed, we observe the small allele, albeit at low frequency, in ancient wolves (n=9, freq(C)= 0.16) and we also identify the small allele in a 53,000-year-old Pleistocene Siberian wolf (heterozygous; PP(CT)>0.9), further demonstrating the antiquity of the small allele (Figure 2A and Table S3).
We next estimated body mass for three ancient wolves (Figure S1B). We observe a Pleistocene wolf (16,500 ybp) that is homozygous for the large allele with an expected body mass of ~39.6 kg, while ancient heterozygous wolves (52,500 and 16,900 ybp) have an expected mass of 21.8 and 38.1 kg, similar to modern heterozygous canids (Figure S1C). In addition, we used generalized linear models (GLM) to test if associations exist between the distribution of the small allele, latitude, and temperature (Figures 2B). We observe that the frequency of the small allele is higher in modern smaller-size wolves (~25kg)24,25 from lower latitudes (e.g. Middle East, n=11, freq=0.47; Asia, n=28, freq=0.2), than in comparatively larger wolves (~40kg)26 from higher latitudes (e.g. North America, n=34, freq=0.09; Europe, n=28, freq=0.11; Siberia, n=3, freq=0; binomial GLM, latitude: ∆AIC=12.74, p-value<0.005, Tjur’s R2=0.23, temperature: ∆AIC=11.76, p-value<0.0005, Tjur’s R2=0.34), which matches with the predictions made by Bergman’s rule.
Although the large allele is more frequent in both modern and ancient grey wolves, the antiquity of the small allele makes it difficult to determine which allele is ancestral. To address this, we analyzed 24 additional genomes from 11 distantly related canid species including four coyotes, two red wolves, five African golden wolves, one Ethiopian wolf, three African hunting dogs, three golden jackals, one black-backed jackal, one side-striped jackal, two dholes, one gray fox and one Andean fox (Figure 2B and Table S2), representing a body mass range of five to 35 kg26. We found strong statistical support for the relationship between latitude, temperature, and the distribution of the small allele in all canid populations tested (binomial GLMM: ∆AIC (null models) =16.09 and 15.33 for latitude and temperature respectively). Small allele frequency in canid populations decreases with latitude and increases with temperatures (p-values <0.0005, Tjur’s , see: Methods). Interestingly, except for the two red wolves from North America (20–35kg), all canids (coyotes, jackals, African wolves and hunting dogs, dholes and foxes) possessed the small allele in a homozygous state, suggesting that the small allele is the ancestral state.
We next performed a comparative genomic analysis which shows that 60–70% of the genomic DNA defined by IGF1-AS exons in dogs is conserved among the most closely related mammals, as identified on an IGF1 maximum likelihood phylogenetic tree generated by the Ensembl database (http://www.ensembl.org/) (Figure 3A and Methods). Specifically, the small allele (C) was present in ferret, panda, and cat, supporting the hypothesis that the small allele represents the ancestral state.
Figure 3. Proposed ancestry for Canis lupus lineage based on IGF1-AS allele distribution.
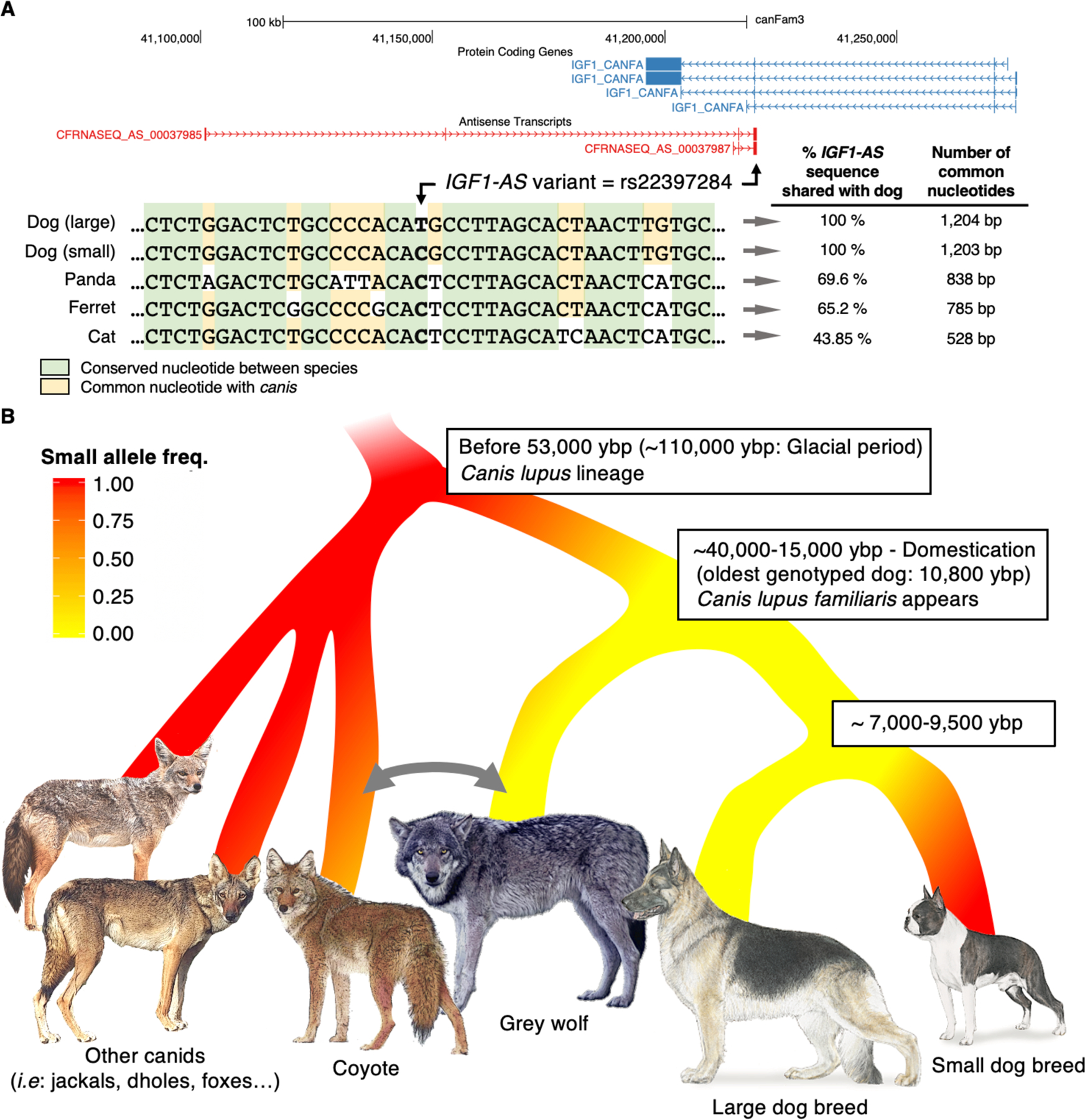
(A) IGF1 locus from UCSC browser showing four IGF1 transcripts (blue) and two IGF1-AS predicted transcripts (CFRNASEQ_AS_00037985, CFRNASEQ_AS_0003798) (red) that overlap IGF1 transcripts by 182 bp. Black arrow indicates the position of IGF1-AS variant (rs22397284). Conservation between dogs, ferret, panda and cats for 40 nucleotides surrounding the IGF1-AS variant (bold) and for the full length IGF1-AS predicted transcript (CFRNASEQ_AS_00037985). The C allele, associated with small sizes in canids, and shared by the four species corresponds to the ancestral allele. (B) Canidae ancestor was likely small and carried the C allele. The large allele arose some time before 53,000 years before present (53,000 ybp) and generated bigger animals (Canis lupus). The ancestral small allele continues to exist in the grey wolf population, albeit at a low frequency. Approximately 15,000 ybp, canine domestication likely began with large wolf-like dogs2. Shortly thereafter, human selection of small canids with the ancestral C allele led to preponderance of small modern domestic breeds. Grey arrow reflects actual hybridization observed between coyotes and wolves in East of America. See also Figures S2–S3.
To further explore this observation, we genotyped the IGF1-AS loci using Sanger sequencing in 10 Channel Island and two grey foxes (Table S2), both species weighting 1.4–5.5 kg26. We observe that all are homozygous for the small allele (freq=1, n=12). As in grey wolves, the previously described body size-associated SINE and SNP (rs22437444) originally identified in dogs10 do not show any association with size in small wild canids (Table S1). Conversely, for the IGF1-AS variant, all small canids (including small grey wolves) living in warmer regions carried the small allele, which suggests that IGF1-AS may be a major contributor to body size variation in canids other than dogs.
Finally, we obtained body mass data from 79 adult coyotes sampled from across North America (Tables S2). We observed that coyote body size is variable, following a west-to-east gradient of small (West Coast mean body mass = 9.18 ±2 kg) to large (East Coast mean body mass 16.03 ±3 kg), as previously reported27 (Figures 4A and 4C).
Figure 4. Relationship between IGF1-AS variant genotype and individual body mass measures in coyotes.
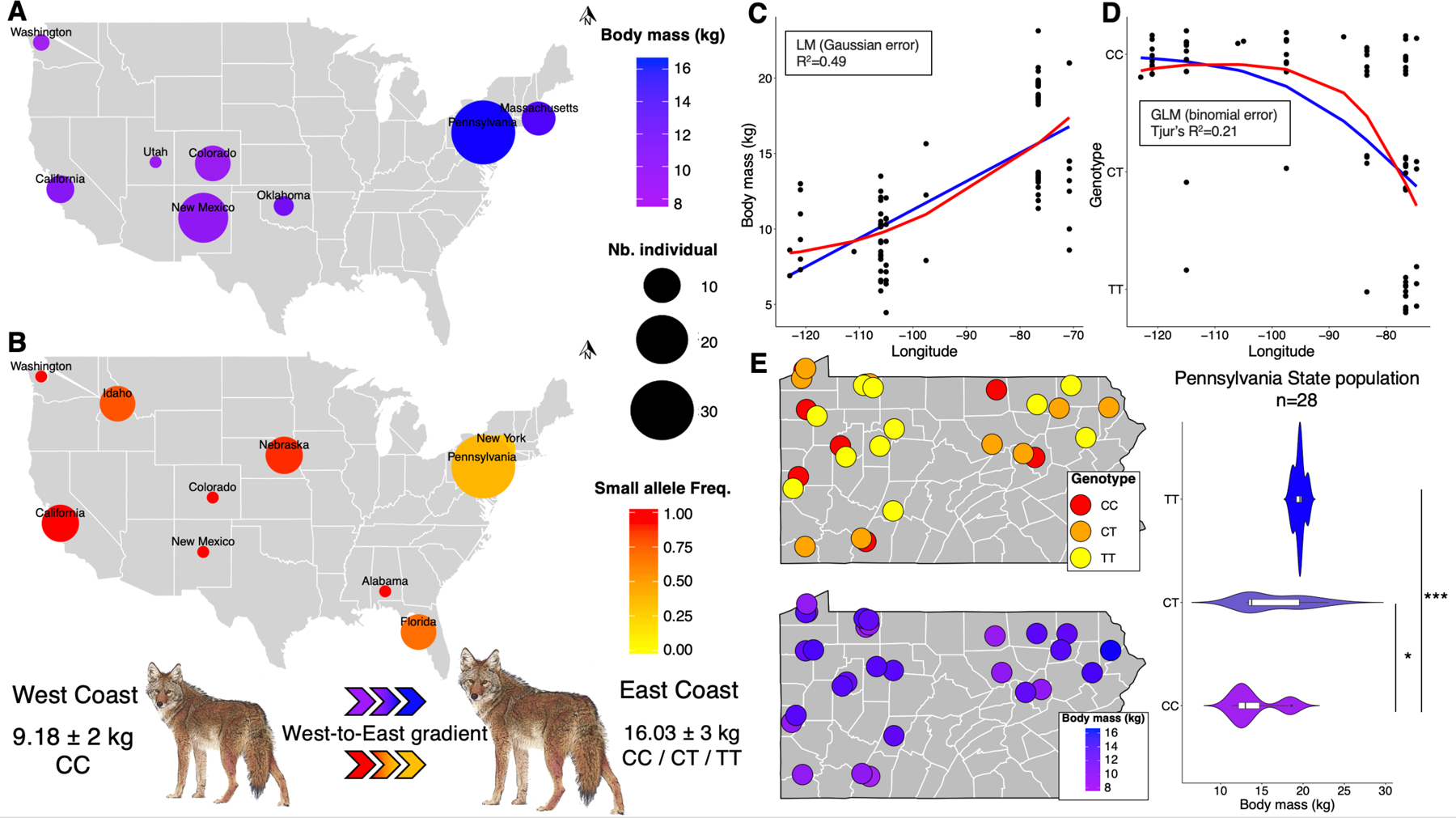
(A) Mean body mass reported by U.S. state for 79 coyotes sampled by universities and museums, as indicated in Methods. Circle size indicates number of individuals. (B) Frequency of the C allele of IGF1-AS variant in 76 samples (distinct from those in A - see Methods) drawn from eight states across the U.S. Both maps illustrate the West-to-East gradient for the coyote population supported by statistical models (C-D). Linear (blue) and quadratic (red) relationships between longitude and, body mass (C), or small allele frequency (D). Lines indicate predicted values from generalized linear models (with binomial error for small allele frequency and Gaussian error for body mass). In both cases, quadratic and linear effects received similar statistical support. West coast coyotes are primarily homozygous (C allele freq = 0.93, mean body mass = 9.18 kg ± 2 SD); East coast coyotes carry all three genotypes (Mean body mass = 16.03 kg ± 3 SD). Mid-U.S. states (Nebraska and Oklahoma) were not included in these estimations. (E) Analysis of 28 coyotes from Pennsylvania demonstrates a significant relationship between IGF1-AS allele status and body mass (*P<0.01, ***P < 0.0001, Mann–Whitney–Wilcoxon tests), but exclude the hypothesis of a local geographic effect on their distributions (at the state scale). See also Tables S1–S2.
Sanger sequencing of the IGF1-AS variant in 76 coyotes from locations spanning the U.S. revealed a high frequency of the small allele (freq=0.93; n=21) in West coast coyotes, and a significantly lower frequency (freq=0.47; n=44; binomial GLM, p<0.001; Tjur’s R² = 0.21) among coyotes from the East coast (Figures 4B and 4D), where hybridization with wolves was recently described27, suggesting that the large allele was recently introgressed from wolves into coyotes (Figure 3B). Finally, we genotyped 28 distinct coyotes from Pennsylvania state for which individual body mass data is available, demonstrating a significant relationship between IGF1-AS allele state and individual body mass (Mann–Whitney–Wilcoxon tests: p<0.0001) (Figure 4E). These data demonstrate a strong association between IGF1-AS allele state and body size gradient in coyote populations across the U.S. Thus, at the IGF1 locus, the IGF1-AS variant is likely the main canid body size mutation, such that small canids are homozygous for the small allele and large grey wolves are homozygous for the large allele, as are large dogs.
Because of its variability among canid species, and its low frequency in grey wolves, it is possible that the small allele was introgressed into dogs via gene flow from a small canid population. Using D-statistics28, however, we found no evidence of excess allele sharing between small dogs and comparatively smaller wild canids (Figure S2). This supports recent findings suggesting that gene flow from wild canids is not a significant feature of the more recent evolutionary history of dogs14. We did not detect any introgression from small wild canids (golden jackal, dhole, African golden wolf) into grey wolves living in warmer temperatures (Figure S2), according to a previous report14, which may indicate that hybridization between wolves and other small canids, except coyotes, remains rare outside of North America. Lastly, our analyses demonstrate that Middle-Eastern grey wolves (Iran, Israel, Saudi Arabia, Syria, n=4) share a closely related haplotype containing the IGF1-AS small allele with small domestic dog breeds (Figure S3), thus supporting the idea of a common origin for the variant observed in small dogs and wild canids11.
Finally, we validated the existence and the structure of the IGF1-AS long non-coding RNA using RNA-Sequencing (Figures 3A, S4 and Data S1F). We confirmed that the last exon of IGF1-AS overlaps the third coding exon of IGF1 (Figures S4A) and that the candidate SNP is thus located 200 bp downstream of the last common nucleotide shared between IGF1 and IGF1-AS. We also showed that IGF1-AS interacts with IGF1 mRNA creating a 182 bp lncRNA/IGF1 mRNA duplex (Figure S4B) and we do not detect differential expression between small and large dogs for either the lncRNA or IGF1 mRNA (Figure S4C). Knowing that antisense overlapping lncRNAs can regulate mRNA translation rate with no effect on mRNA levels29,30, it is possible that IGF1-AS could act as a regulatory mechanism associated with IGF-1 production, perhaps by affecting the affinity of a ribosomal binding motif for the C versus T allele.
Altogether, our results indicate that the selection for small dogs targeted an ancestral allele at a SNP in a lncRNA that is antisense to the established body size gene IGF131,32. Our analyses reveal that the large allele (T) likely arose in wolves more than 53,000 years ago33 (Figure 3B). The frequency of this allele then increased, likely due to natural selection in grey wolves during the Pleistocene, perhaps due to lower temperatures, and became fixed in Northern latitude wolves, while the small allele persisted in wolves from Southern latitudes.
The latitudinal association of the two alleles also exists in ancient dogs from Northern and Southern Eurasia, suggesting that dogs may have either been under similar body size selective pressures, or they experienced gene flow with local wolves. The availability of both the small and large sized associated alleles within the global dog population has also allowed dog breeders, beginning in the 19th century12, to take advantage of the morphological plasticity conferred by these alleles to produce breed dogs of highly divergent sizes. Selection has also allowed for the near fixation of these alleles in modern large and small breeds.
The human penchant for novelty has meant that domestic animal populations often possess phenotypic variability that may not have existed within the more homogeneous wild progenitors34,35. Often, these novel characteristics only appeared after domestic animals became acclimated to the human niche and experienced a commensurate reduction in natural selection. Thus, many targets of human selection have been driven to frequencies that would have been actively selected against in settings where humans had less influence over any individual’s survival. The evidence presented here demonstrates that humans have also targeted standing variation present within wild ancestors. In the case of dogs, the 40-fold size divergence has been driven in large part by selection on two divergent alleles in IGF1-AS, both segregating in wolves for over 53,000 years.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests of resources and reagents should be directed to and will be fulfilled by the Lead Contacts Elaine Ostrander (eostrand@mail.nih.gov) and Jocelyn Plassais (jocelyn.plassais@univ-rennes1.fr).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Genomes sequenced for this work, as well as all publicly available data used for alignment, are available via the Short Read Archive (ncbi.nlm.nih.gov/sra) and their associated accession numbers are listed in the Supplementary Data 1A and in the key resources table. Newly generated genomes are now available on SRA: accession number PRJNA685036. RNA-Seq raw data are registered on SRA (accession number: PRJNA690861). The three newly sequenced ancient wolves are registered on The European Nucleotide Archive (ENA) under the permanent study accession number: PRJEB42199. Raw data for tables and figures (including original gel pictures), and all raw data for statistics (including GWAS results, GLM, Wilcoxon-Mann-Whitney, Spearman) are publicly available on Dataverse. The DOI is listed in the key resources table and with the corresponding methods. Other data are contained within the article and its supplementary information.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Canis Familiaris | Various veterinary referral hospitals | N/A |
| Three Canid bone paleontological remains | This paper | Botai (sample ID: AL2350); Pietrele (sample ID: AL3185); Eliseevichi (sample ID: AL2657). |
| Chemicals, peptides, and recombinant proteins | ||
| Trizol | Life Technologies | 15596026 |
| RNAlater | Life Technologies | AM7020M |
| Critical commercial assays | ||
| TruSeq DNA nano kit | Illumina | FC-121-4001 |
| TruSeq Stranded mRNA library Prep Kit High Throughput | Illumina | RS-122-2103 |
| Illumina TruSeq Nano DNA library prep HT | Illumina | 20000903 |
| TruSeq® Stranded mRNA LT-Set A | Illumina | RS-122-2101 |
| NextSeq High Output v2.5 75 cycle kit | Illumina | 20024906 |
| Deposited data | ||
| DNA sequencing data | This paper | SRA:PRJNA685036 ncbi.nlm.nih.gov/sra |
| RNA sequencing data | This paper | SRA:PRJNA690861 ncbi.nlm.nih.gov/sra |
| Three ancient wolf DNA sequencing data | This paper | ENA: PRJEB42199 http://www.ebi.ac.uk/ena |
| Dog genome reference (CanFam3.1, ENSEMBL release-85) | ENSEMBL | http://www.ensembl.org/index.html |
| Dog genome Annotation (CanFam3.1-plus) | 66 | http://tools.genouest.org/data/tderrien/canFam3.1p/annotation/trackhub/canfam3.1ptrackhub/hub.txt |
| Oligonucleotides | ||
| gDNA targeted primer: IGF1 Forward:CACTGATCCAGAAGAATCCAACT | 9 | N/A |
| gDNA targeted primer: IGF1 Reverse: CAAAGAACCATGTAAGCCTATTTGT | 9 | N/A |
| gDNA targeted primer:IGF1-AS mutation Forward:GTGGGCTTGTCTGTGCAAAT | This paper | N/A |
| gDNA targeted primer:IGF1-AS mutation Reverse:CCTGAGCATAAAAACTAGGCAGA | This paper | N/A |
| gDNA targeted primer:IGF1-SINE Forward:CACTGATCCAGAAGAATCCAACT | This paper | N/A |
| gDNA targeted primer:IGF1-SINE Reverse:CAAAGAACCATGTAAGCCTATTTGT | This paper | N/A |
| cDNA targeted primer:IGF1-AS short isoform Forward:AGCTGGTCATCAATTTGCCCC | This paper | N/A |
| cDNA targeted primer:IGF1-AS short isoform Reverse:AAGGAAAGACTCAGTTTGGGTGT | This paper | N/A |
| cDNA targeted primer:IGF1-AS long isoform Forward:TGGAAACCACTGGATCTGAGCT | This paper | N/A |
| cDNA targeted primer:IGF1-AS long isoform Reverse:AAGGAAAGACTCAGTTTGGGTGT | This paper | N/A |
| cDNA targeted primer:IGF1-AS last exon RPA Forward:GCACCACAGAGGAAGGATGAT | This paper | N/A |
| cDNA targeted primer:IGF1-AS last exon RPA Reverse:TGGGATGTGTAGGTTGACCAG | This paper | N/A |
| cDNA targeted primer:IGF1 RPA Forward:TGCTCTCAACATCTCCCATCTCT | This paper | N/A |
| cDNA targeted primer:IGF1 RPA Reverse:ACCGTTTTGGCCAGACTCTTT | This paper | N/A |
| cDNA targeted primer:IGF1-AS/IGF1 exon 3 RPA Forward:CCTTGGGCATGTCAGTGTGG | This paper | N/A |
| cDNA targeted primer:IGF1-AS/IGF1 exon 3 RPA Reverse:GACAGGCATCGTGGATGAGTG | This paper | N/A |
| cDNA targeted primer:GAPDH qRT-PCR Forward:AAGCAGGGATGATGTTCTGG | This paper | N/A |
| cDNA targeted primer:GAPDH qRT-PCR Reverse:CCTCATGACCACCGTCCA | This paper | N/A |
| cDNA targeted primer:IGF1 qRT-PCR Forward:CCTGCACTCCCTCTACTTGC | This paper | N/A |
| cDNA targeted primer:IGF1 qRT-PCR Reverse:CTCAAGCCTGCCAAGTCTG | This paper | N/A |
| cDNA targeted primer:IGF1-AS qRT-PCR Forward:TGAAGCTTCCCCAACAATTC | This paper | N/A |
| cDNA targeted primer:IGF1-AS qRT-PCR Reverse:TGGGTGTAGACGAGATCCTTG | This paper | N/A |
| Software and algorithms | ||
| Read alignment: bwa v0.7.17 | 42 | https://sourceforge.net/projects/bio-bwa/files/ |
| Samtools 1.6 | 43 | http://samtools.sourceforge.net/ |
| Variant caller: GATK v4.1.4.0 | 44,45 | http://gatkforums.broadinstitute.org/gatk |
| PicardTools 2.9.2 | http://broadinstitute.github.io/picard | http://github.com/broadinstitute/picard |
| Vcftools v0.1.16 | 74 | http://vcftools.sourceforge.net/ |
| Linear mixed model: GEMMA v0.94.1 | 75 | http://www.xzlab.org/software.html |
| Utility: PLINK v1.9 | 76 | http://zzz.bwh.harvard.edu/plink/ |
| Graphics and data analysis: R v3.3.0 | The Comprehensive R Archive Network (CRAN) | https://cran.r-project.org |
| Phred/Phrap/Consed package | 50–52 | http://www.phrap.org/phredphrapconsed.html |
| AdapterRemoval2 | 82 | https://github.com/MikkelSchubert/adapterremoval |
| RSeQC | 69 | https://github.com/MonashBioinformaticsPlatform/RSeQC |
| MapDamage v2.58 | 19 | https://github.com/ginolhac/mapDamage |
| FilterUniqSamCons | 84 | |
| ANGSD 0.614 | 85 | https://github.com/ANGSD/angsd |
| MultiQC tool | 65 | https://multiqc.info/ |
| STAR | 67 | https://github.com/alexdobin/STAR |
| Stringtie | 68 | https://ccb.jhu.edu/software/stringtie/ |
| RSEM v1.3 | 91 | https://github.com/deweylab/RSEM |
| Data visualization: Integrative Genomics Viewer: I.G.V 2.8.2 | 70 | http://software.broadinstitute.org/software/igv/ |
| Beagle v4.1 | 88 | http://faculty.washington.edu/browning/beagle/beagle.html |
| PHYLIP | 90 | https://evolution.genetics.washington.edu/phylip.html |
| FigTree v1.4.4 | Graphical viewer of phylogenetic trees | http://tree.bio.ed.ac.uk/software/figtree/ |
| Other | ||
| KOD Xtreme HotStart Polymerase | Merck | 71975-3 |
| ExoSap-ItTM reaction | Thermo Fisher Scientific | 78201.1.ML |
| BigDye Terminator v3.1 | Thermo Fisher Scientific | 4337458 |
| RecoverAllTM Total Nucleic Acid Isolation Kit | Thermo Fisher Scientific | A26135 |
| High-Capacity cDNA Reverse Transcription kit | Thermo Fisher Scientific | 4368814 |
| TURBODNase | Thermo Fisher Scientific | AM2238 |
| RNAse A/T1 Mix | Thermo Fisher Scientific | EN0551 |
| NucleoSpin RNAclean-up kit | Macherey-Nagel | 740948.50 |
| SYBR™ Safe | Thermo Fisher Scientific | S33102 |
| Ancient DNA genotyping determination and all statistical analyses related to the paper | This paper | Dataverse :doi: 10.7910/DVN/JBXYZD https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD |
| The two VCF files built for this work (one containing 1,156 dogs and one containing 86 wild canids) are publically available on the dog genome project website | This paper | https://research.nhgri.nih.gov/dog_genome/data_release/index.shtml |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
We extracted DNA from whole blood samples collected into EDTA or ACD anticoagulant from 331 dogs and 133 wild canids. Three ancient DNA were extracted from tooth or bone samples in a dedicated ancient DNA laboratory using appropriate sterile techniques and equipment. We extracted RNA from 42 testes collected by registered veterinarians during routine sterilization procedures with consent from the dog owner. All procedures were reviewed and approved by the Animal Care and Use Committee of the National Human Genome Reseach Institute (NHGRI) at the National Institutes of Health. We provide a full description of the specimens in the Methods Details.
METHODS DETAILS
Modern canids whole genome sequencing datasets.
WGS data was gathered from the Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra;), or Genome Sequence Archive (https://bigd.big.ac.cn/gsa/)(n = 965 unique individuals)(2)8,36–40, some of which was produced via the Dog10k project41(n=371), or newly generated by the NIH Intramural Sequencing Center (n = 97) and are now available on NCBI: accession number PRJNA685036. All Biosample numbers for the initial 1,297 modern dog genomes as well as coverage levels are listed in the Data S1A. To create the vcf file containing the 1,297 modern dogs, sequence reads were aligned to the CanFam 3.1 reference genome (http://genome.ucsc.edu/cgi-bin/hgGateway?db=canFam3) using the BWA-MEM algorithm42 (current version BWA 0.7.17) and sorted with SAMtools (current version SAMtools 1.6)43.
For non-PCR-free libraries, PCR duplicates were marked as secondary reads using PicardTools (http://github.com/broadinstitute/picard; current version PicardTools 2.9.2). GATK44,45 (version GATK 4.1.4.0) was used to perform base recalibration using 2,738,537 dbSNP v131 variants. SNVs and small indels were called using GATK HaplotypeCaller, which first calls variants per-individual in gVCF mode with subsequent joint-calling utilizing all individuals46. Variant quality score recalibration was conducted with GATK best practices and default parameters for SNV and indels separately as follows: SNV recalibration: 172,254 Illumina Canine HD Chip variants (training, true, prior = 15); 2,738,537 dbSNP v131 variants (known, training, prior = 6); 3,627,539 published variants from Axelsson et al.47 (known, prior = 6). Indel recalibration: 714,278 variants as known, training and truth sets with a prior of six47 and maxGaussians set to 4. After alignment and variant calling, samples were removed if they were low quality, e.g., less than 2x average depth.
The final datasets consisted of one VCF file of 75.6 million variants and contained 1,156 modern dog genomes encompassing 230 breeds, 140 indigenous and village dogs (including 15 New Guinea singing dogs) and one dingo, sampled from around the world (Data S1A). We built a second VCF file containing only wild canid genomes (n=86) obtained using SRA data from published papers8,36–40. This file contains 68 grey wolves, four coyotes, two red wolves, five African golden wolves, one Ethiopian wolf, three African hunting dog, three golden jackals, one black-backed jackal, one side-striped jackal, two dholes, one gray fox and one Andean fox (Table S2), representing a weight range of five to 60 kg26. In order to check IGF1-AS alleles in wild canids, as well as other previously reported body size variants7–9,48, we only retained biallelic variants that were present in the domestic dog VCF file, generating a total of 64.9 million variants. Both VCF files are publically available on the NHGRI Dog Genome Project website (https://research.nhgri.nih.gov/dog_genome/data_release/index.shtml).
Additional dog samples, Sanger sequencing and IGF-1 serum level.
Whole blood samples were collected into EDTA or ACD anticoagulant and genomic DNA was extracted using a standard phenol-chloroform extraction protocol. All procedures were reviewed and approved by the Animal Care and Use Committee of the National Human Genome Reseach Institute (NHGRI) at the National Institutes of Health. We obtained blood samples from 144 poodles (48 standard, 48 miniature and 48 toy poodle variants) and 136 schnauzers (48 giant, 42 standard, and 48 miniature breeds) (Data S1C). The top 10 most associated variants with dog body size, including the IGF1-AS alleles, as well as a previously identified SINE element at the IGF1 locus10, were validated using Sanger sequencing and agarose gel migration (1%), respectively. Primers were designed using Primer3plus49 and are listed in the key resources table. Targeted regions were amplified using polymerase chain reaction (PCR) with KOD Xtreme HotStart Polymerase (Merck). PCR products were purified by ExoSap-ItTM reaction (Thermo Fisher Scientific) and sequenced using BigDye Terminator v3.1 (Thermo Fisher Scientific) on an ABI 3730 DNA analyzer (Applied Biosystems). Sequence traces were analyzed using Phred/Phrap/Consed package50–52. Serum levels of IGF-1 in 51 dogs, including 13 mixed dogs, were measured by ELISA (Veterinary diagnostic laboratory, Michigan State University) following standard methods (Data S1D).
Additional modern canid samples.
We obtained DNA samples from 133 wild canids including 75 coyotes, 10 Channel Island foxes, two grey foxes, and 46 additional grey wolves sampled from nine countries on three continents (Table S2). Following the same protocol previously described, the IGF1-AS alleles and the nine other most associated variants with dog body size, as well as the previously identified SINE element at the IGF1 locus10, were validated using Sanger sequencing and agarose gel migration (1%) respectively. Additional weight measures for 79 adult coyotes were collected directly off websites from the University of Washington Burke Museum (UWBM), Museum of Vertebrate Zoology (MVZ), California Academy of Science (CAS), The Museum of Southwestern Biology (MSB), Denver Museum of Nature and Science (DMNS), Sam Noble Oklahoma Museum of Natural History (OMNH), Museum of Comparative Zoology (MCZ) and Princeton University with permissions (Table S2).
Ancient DNA data, archaeological samples and context.
We obtained BAM files from 39 published ancient genomes representing six wolves and 33 dogs2,3,14–18. For each sample we provide detailed information (individual ID/ archeological ID, species, age, location, depth, latitude, longitude, the associated reference and IGF1-AS genotype) in Supplementary Table 3. Additional information regarding the archeological records can be found within their original associated papers referenced in Supplementary Table 3. We completed this dataset with three additional unpublished wolf samples (AL2350, AL3185 and AL2657), registered on The European Nucleotide Archive (ENA) under the permanent study accession number: PRJEB42199, with the following archeological samples and context:
Botai, Kazakhstan (sample ID: AL2350).
Botai is an Eneolithic settlement site in Northern Kazakhstan with early evidence of horse husbandry53. Both dogs and wolves have been identified from the site. This wolf specimen was recovered in 2018 from a trash pit, adjacent to a pit house, alongside the bones of horse and aurochs. The specimen consisted of a cranium, with attached mandibles, and the first three cervical vertebrae. The anterior portion of the snout had been removed from the middle of the tooth row, and the cranium displayed damage from putative projectile injury. It is radiocarbon dated to 5,169 ybp.
Pietrele, Giurgiu province, Romania (sample ID: AL3185).
The site is a c. 9m high Chalcolithic tell-settlement situated close to the Danube. The occupation phases of the tell date to the period 4,450 – 4,250 calBC54. The wolf bone comes from the uppermost layers. It is radiocarbon dated to 6,307 ybp.
Eliseevichi, W Russia (sample ID: AL2657).
The Epigravettian Eliseevichi site is located in the Russian Plain on the right bank of the Sudost’ River, a tributary of the Desna. Based on two AMS dates on canid material55,56 the calibrated age of the site is about 16,500 ybp. The faunal assemblage is dominated by woolly mammoth, reindeer, large canids and polar fox57. Remains of eight complexes made from mammoth skulls and bones were recovered58,59. Two large canids skulls and one mandible are described as from Palaeolithic dogs55,60. The analysed mandible (AL3185 – 23781 (3)) is a complete left jaw, with a broken first molar, from an adult canid (Figure S1A).
Ancient DNA sequencing.
DNA was extracted from tooth or bone samples in a dedicated ancient DNA laboratory using appropriate sterile techniques and equipment. Extraction was carried out following the Dabney extraction protocol61 but with the addition of a 30 minutes pre-digest stage62. Illumina libraries were built following Meyer and Kircher (2010)63 but with the addition of a six base-pair barcode added to the IS1_adapter.P5 and IS3_adapter.P5+P7 adapter pair. The libraries were then amplified on an Applied Biosystems StepOnePlus Real-Time PCR system to check that library building was successful, and to determine the minimum number of cycles to use during the indexing amplification PCR reaction. A six base-pair barcode was used during the indexing amplification reaction resulting in each library being double-barcoded with an “internal adapter” directly adjacent to the ancient DNA strand and forming the first bases sequenced, with a traditional external barcode sequenced during Illumina barcode sequencing. We included negative blanks (no bone powder and nuclease free water) for every batch, these were them through the entire process assess for contamination. The three samples were then sequenced on multiple Hi-Seq 2500/4000 lanes and the paired-end sequencing data were aligned to the dog canFam3.1 genome using BWA42 with permissive parameters including disabled seed64 (-I 16500 -n 0.01 -o 2). Contamination estimation process is fully-detailed by Bergstrom et al.2.
Validation of IGF1-AS transcripts.
We generated RNA-Seq data from 42 testes derived from various size breeds (Data S1F) that we registered on SRA under the accession number PRJNA690861. RNA was extracted from testes using the RecoverAll™ Total Nucleic Acid Isolation Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Quality score were measured by Agilent Bioanalyzer on a Total RNA 6000 Nano chip to obtain RIN score for RNA integrity. Illumina libraries were generated using the TruSeq® Stranded mRNA LT-Set A (Illumina Cat No RS-122–2101) for the 42 samples, with all having unique barcodes. Library quality control was performed on an Agilent Bioanalyzer. Pooled samples were run on the NextSeq 550 (Illumina) using the NextSeq High Output v2.5 75 cycle kit (Illumina Cat No 20024906). We then used the nextflow-based RNASeq pipeline from the nf-core community (version 3.1; https://nf-co.re/rnaseq/3.1) in order to uniformly process all testes RNAseq datasets. Briefly, the pipeline included QC of reads using the MultiQC tool65, the mapping of the reads on both the genome (CanFam3.1) and the transcriptome (CanFam3.1-plus)66 with the STAR program67 and the detection of new transcripts with the Stringtie program (option --stringtie_ignore_gtf and merge)68. We also double-checked the strandness of the reads using the RSeQC tool (“infer_experiment.py” program)69 which confirmed that the data were stranded single-end utlizing a protocol where the reads come from the reverse strand (historically known as -fr-firststrand).
In order to validate the structural annotation of the lncRNA IGF1-AS, we visualized all BAM files using the Integrative Genomics Viewer (I.G.V v2.8.2)70 (Figures S4A). We also manually checked the distribution of reads in the heterozygous samples. No allele specific expression were observed between T and C alleles. To complete the RNA-Seq analysis and to confirm the presence of both predicted transcripts (CFRNASEQ_AS_00037985, CFRNASEQ_AS_00037987), we also performed reverse transcription with 1 μg of total RNA from testes using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. We then Sanger sequenced cDNAs from ten dogs (five small and five large) using primer pairs specific for each transcript. Primers were designed using Primer3plus49 and listed in the key resources table. Targeted regions were amplified using polymerase chain reaction (PCR) with KOD Xtreme HotStart Polymerase (Merck). PCR products were purified by ExoSap-ItTM reaction (Thermo Fisher Scientific) and sequenced using BigDye Terminator v3.1 (Thermo Fisher Scientific) on an ABI 3730 DNA analyzer (Applied Biosystems). Sequence traces were analyzed using Phred/Phrap/Consed package50–52. At the end, we confirmed that IGF1-AS transcripts contain three exons. The first transcript corresponds to a 1,204 bp RNA(CFRNASEQ_AS_00037985; chr15:41,101,001–41,219,825) while the second corresponds to a 1,001 bp RNA (CFRNASEQ_AS_00037987; chr15:41,214,777–41,219,825). Both transcripts only differ by their first exon, with different sequences.
Ribonuclease Protection Assay (RPA).
Total RNA (approximatively 5 µg) from six dogs (three small and three large) were digested with TURBODNase (Thermo Fisher Scientific) for 30 minutes at 16°C and RNAse A/T1 Mix (Thermo Fisher Scientific) for one hour and 30 minutes at 16°C to remove all the genomic DNA contamination and single-strand RNAs. RNA was purified after each step with the NucleoSpin RNAclean-up kit (Macherey-Nagel). The cDNA from endogenous double-stranded RNAs (dsRNA) was produced using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) and the mixture of the two gene-specific primers listed in the key resources table. The double-stranded cDNA was amplified in 25 µl PCR reaction system. After 35-cycle amplification, the products were checked by electrophoresis on 2% agarose gel with SYBR™ Safe (Thermo Fisher Scientific). Note: Total RNAs used for these experiments were isolated from testes under nondenaturating conditions to preserve prospective natural RNA duplex.
QUANTIFICATION AND STATISTICAL ANALYSIS
Association analyses.
Only domestic dog samples with ≥10x sequence coverage were retained, selecting a maximum of two males and two females per breed; those with the deepest coverage were selected when more than three individuals were available. All other samples were removed (including wild canids, village dogs, unknown and mixed breed samples), generating a dataset of 456 dogs representing 179 breeds (Data S1A). For weight and height phenotypes we used the published breed standard, as has been done previously (male + female average)7–9,48. Standard breed weights (SBW) and height (SBH) were obtained from several sources: weights and height previously listed in Plassais et al8, from the American Kennel Club Book of Standards12, and the Fédération Cynologique Internationale (http://www.fci.be/en/Nomenclature/). SBW and SBH were applied to all samples from the same breed. For wild canids, we determined mean body mass for each species and wolf subspecies using several sources: body mass listed in Padilla & Hilton26, from Lopez71, Estes et al72, and the Wildlife Institute of India73. We used vcftools (--min-alleles 2 --max-alleles 2 --plink)74, retaining only biallelic variants (single nucleotide variants [SNP] and small indels<200bp), generating a dataset of 64.9 million biallelic variants.
For domestic dogs, we performed association studies using GEMMA v0.94.175 as linear-mixed model methods, removing variants with missing values > 1%, and correcting each analysis by sex and a previously calculated relatedness matrix. We used the Wald test to determine P values and Bonferroni correction was used to identify significant associations (cutoff = −log10 (0.05/number of variants) = 8.46). For wild canids, we performed association studies using PLINK v1.976 (using --assoc --adjust --geno 0.05 options). Throughout the paper, all violin plot P values were estimated by Mann–Whitney–Wilcoxon tests (*P<0.01, **P<0.001, ***P < 0.0001). The relationship between IGF-1 serum level and body mass was tested using a Spearman correlation test (P-value = 2.8e-6, rho = 0.6) and violin plots were constructed in R (https://www.r-project.org/). In addition, copy number variations were analyzed using the same dataset published in Serres Armero et al77 from which we extracted the IGF1 locus (chr15:40500000–41500000). GWAS results for CFA15, CNV analysis, the raw data for figures and all statistics results are publicly available on Dataverse (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD) (since each GWAS result file contains ~370k makers on CFA15 and sized ~49 Mb, only the top 1,000 markers are shown).
Bergmann’s Rule and geographic distribution analyses.
We tested whether the spatial distribution of ‘small allele frequency’ follows the biogeographic pattern and the underlying mechanism described in Bergmann’s rule, i.e. number of small animals (and thus here, the frequency of “small allele C”) decreases with latitude and temperature22. To ascertain worldwide map representation when the sampling geographic coordinates were missing, we used the calculated centroid position of the associated historical country of wild distribution using maps R package 3.3.0. When coyote sampling geographic coordinates were missing we used the calculated centroid position of the associated sampling U.S. state using maps R package 3.3.0. Using geographic location of each sample, we extracted local temperature data from CHELSA78, specifically the bioclimatic variable bio1 (annual mean temperature) corresponding to the 1979–2013 period. For both latitude and temperature that we tested separetely, we used generalized linear mixed models (GLMM; lme4 R package; version 1.1–2579) assuming a binomial error distribution, with small allele frequency used as the response variable (CC = 1, CT = 0.5, TT = 0) and the latitude (or temperature) used as fixed effect (herearfter, M1). We accounted for species-specific intercepts using the species identity as random effect. We also accounted for potential non-linear relationship by adding a quadratic term to the model (M2). We compared both models with a null model (M0, including the species random effect only) using the Akaike Information Criterion (AIC)80 to select the best model in terms of parcimony and data fitting. We assessed the proportion of deviance explained by latitude (or temperature) using the Tjur’s R² computed with the ‘performance’ R package version 0.7.281.
We tested whether the spatial distribution of small allele and weight followed a West-East linear gradient across U.S. for coyotes. We first used generalized linear models79, assuming a binomial error distribution with the small allele frequency used as the response variable and the longitude used as explanatory variable. We also accounted for potential non-linear relationship by adding a quadratic term to the model. We repeated the analysis to test if body mass followed the same West-East gradient using linear models with a Gaussian error distribution with body mass used as the response variable. Finally, we extended our analysis to latitude and local mean annual temperature for each coyote sample to test (and thus exclude) the Bergmann’s rule hypothesis which could explain the geographic pattern observed in coyotes across U.S. Maps and GLM figures were drawn in R (https://www.r-project.org/). All details about GLM statistics (models, AIC, parameters, SD, Z score, P-values, Tjur’s R²) are publicly available on Dataverse (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD).
IGF1-AS genotyping in ancient DNA.
For the 42 samples, raw reads were filtered, allowing one mismatch to the indices used in library preparation. Adapter sequences were removed using AdapterRemoval82. Reads were aligned using Burrows-Wheeler Aligner (BWA) version 0.7.5ar405 to canFam3.142, with default parameters apart from disabling the seed option (-I 1024)83. FilterUniqueSAMCons84 was then used to remove duplicates. BAM files from different sequencing lanes were merged using samtools v 1.3.143. Each BAM files was then re-scaled using MapDamage v219. We then produced two BAM for each sample, each containing only the reads mapping to the lncRNA SNP region using samtools: one BAM with and one without rescaling.
We then calculated likelihood of all ten possible genotypes at the position of interest by first running the following command in ANGSD v0.93385:
angsd -GL 1 -out <output_file_name> -doCounts 1 -i <input_file_name> -doGlf 4 -nThreads 2 -r chr15:41219654
For the sake of numerical representation and computational efficiency, angsd reports the likelihood ratio of each genotype compared to the ”best” one on a logarithmic scale. We thus used a custom script to rescale these values and applied a Bayesian framework to obtain genotype posterior probabilities.
Let Ω denote the set of possible genotypes at any given position in a genome. For a diploid individual we have that Ω = {AA, AC, AG, AT, CC, CG, CT, GG, GT, T T}. We denote G the event that an individual possesses a specific genotype. The events Gj constitute a partition of the sample space, i.e.
Let D denote the data we collected from sequencing, from Bayes Theorem we have:
| (Eq. 1) |
where P(Gj) is the probability associated with the event Gj i.e., our prior, while P(Gj|D) is the probability of the individual having the genotype Gj given the data, i.e. our posterior. Because the events Gk are exhaustive and mutually exclusive, we can use the law of total probability to express the denominator of Eq.1:
| (Eq. 2) |
Thus, we can rewrite our posterior as:
| (Eq. 3) |
Finally, we can explicitly express the probability of observing the data D given the event Gj being true in terms of likelihood:
| (Eq. 4) |
where k is a positive constant which reflects the proportionality relationship between likelihoods and probabilities. We then define ai as the value reported by ANGSD for the i-th genotype, with Li its likelihood and Lbest the likelihood of the best genotype. We can express ai as:
| (Eq. 5) |
Therefore,
| (Eq. 6) |
where K is equal to k Lbest.
By substituting Eq. 6 into Eq. 3 we obtain the following expression for our posterior probability:
| (Eq. 7) |
which can be simplified even further when adopting a uniform prior.
We used this framework to compute posteriors employing two different priors: a uniform prior (all ten genotypes have the same probability value of 0.1) and a more realistic prior, which takes into account our knowledge that this is a biallelic site (P(CC)=P(TT)=P(CT)=0.31 while each of the remaining seven genotypes have a probability of 0.01). At the end, we applied this method on our ancient genome dataset, and we determined the genotypes of 35 genomes (26 dogs and nine wolves). All genotype determinations are publicly available on Dataverse (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD).
Weight estimations of ancient DNA samples.
We estimated weight for four samples: three wolves (AL3185, CGG32, CGG33) and one ancient dog (CGG6). The reference material used for the osteometric comparison is composed of four groups. The recent northern wolf reference group (rNw, n=39) consists of mandibles from Palaearctic wolves from locations within Belgium, Sweden and Russia at latitudes above 50°N. The Pleistocene wolf reference group (PlW, n=18) contains mandibles from European and Siberian natural and Palaeolithic sites dating from the pre- and post-Last Glacial Maximum, located at latitudes above 44°N. The Palaeolithic dog reference set (PalD, n=18) is composed of mandibles from European and Siberian Upper Palaeolithic sites, all located above 44°N. The recent northern dog reference group (rNd, n=39) contains specimens from localities in Siberia, Sakhalin Island, and Greenland at latitudes above 50°N. For more details on the reference groups, see Germonpré et al.60,86.
To assign the Eliseevichi mandible to one of the reference groups, a biplot was used (JMP version 15.0; significance <0.05, SAS Institute Inc) (Figure S1B). For all reference groups, density ellipses (0.95) are given. These ellipses are both density contours and confidence curves that show where a given percentage (here 95%) of the data is expected to lie; they are computed from the bivariate normal distribution fit to the X and Y variables. The variables, expressed in mm, are measured on the mandibles as proposed by von den Driesch20. Following measurements are utilised: Total Length (TL): the total length from the condyle process to the Infradentale; Hp2p3: the height of the mandible between p2 and p3. Body mass estimates (BMe) are calculated based on the regression equations formulated on the base of a combined data set of modern wolves and dogs, all of known body mass at death, by Losey et al21 (Figure S1B). The equations include the measurements as defined by von Den Driesch20 for the skull (Total Skull Length, TL) and for the mandible (length from the condyle process to the border of the canine alveolus, LPcC). Finally, we used predicted weight based on the estimators of Harcourt and Wing from 15 Israeli ancient dogs published by Stager et al. in 20084. The three samples (ASHQ01, ASHQ06, ASHQ08), all homozygous for the small allele (C), came from the same expedition, the same site, and with the same estimated age. Hence they were likely of the same body size (Figure S1C and Table S3). Figures were drawn in R (https://www.r-project.org/).
Comparative approach.
The IGF1 maximum likelihood phylogenetic tree was generated using Ensembl database (http://www.ensembl.org/) and the IGF1 dog transcript ENSCAFT00000086858 which we compared to genomes of 288 species. Ensembl gene trees are generated by the Gene Orthology/Paralogy prediction method pipeline (http://www.ensembl.org/), and then generated by TreeBeST (http://treesoft.sourceforge.net/treebest.shtml). Ferret, panda and Carnivores (including cat) are the closest species to canids for which genomes are available on UCSC genome browser. We then aligned the two canine IGF1-AS transcripts (CFRNASEQ_AS_00037985, CFRNASEQ_AS_00037987) on other mammalian genomes using the BLAT tool on the UCSC genome browser (https://genome.ucsc.edu/) to identify conserved sequences between species. To draw figure 3A, we zoomed in on a 40-bp sequence centered on the IGF1-AS variant in dogs, and manually identified the conserved nucleotides between mammals. Sequence alignments and their associated statistics are publicly available on Dataverse (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD).
Detection of introgression.
To detect potential gene flow on CFA15 that may exist between canid populations36, we used the D-statistic28. We used the “genomics_general” package (https://github.com/simonhmartin/genomics_general) and first analyzed the 1.9M biallelic variants identified on CFA15, and then zoomed in on 2-Mb region (40–42Mb) spanning the IGF1 locus. The D-statistic measures the excess of shared-derived sites between a potential introgressor (P3) and a putatively admixed group (P2) over the shared-derived sites between P3 and a third group (P1) that is assumed to be unadmixed and sister to the P2 group. For example, if the D-statistic (P1=Grey wolf, P2, P3, Andean fox) deviates positively from 0, the result suggests that more gene flow exists between P2 and P3, while a negative value indicates a closer relationship between grey wolves (P1) and (P3). In absence of gene flow, D should be approximately zero. We used the Andean fox as an outgroup to define the ancestral alleles36. For domestic breeds, we defined two groups (small and large) keeping the 10 smallest CC dogs (three Chihuahuas, one Pekingese, two Pomeranian, four Yorkshire Terrier) and the 10 largest TT dogs (two English mastiffs, four Great Danes, one Irishwolfhound, one Komondor, one Leonberger, one Scottish deerhound), thus representing the extreme phenotypes for weight/height (Data S1A). We then calculated the frequency of the derived allele in all seven additional species: grey wolf, African golden wolf, red wolf, coyote, golden jackal, small and large dog breeds populations. We evaluated standard errors using a block jackknife approach with a block size of one Mb87. The D-statistic was calculated separately over all combinations of species as P1, P2 and P3. We then split grey wolves by continent and ran the same analysis testing for geographic effects (i.e: testing potential gene flow existing between small dogs and Middle East grey wolf, for example). In total, we performed all the 2,400 possible comparisons using grey wolf, African golden wolf, red wolf, coyote, golden jackal, small and large dog breeds populations. As a note, since the four WGS coyotes used in this analysis were originally sampled from the West coast, we could not investigate the hypothesis of a recent introgression with wolves on East coast27, and represent it on Figure S2. Significant values were estimated following: P= 2*pnorm(-abs (jacknife Z score)). Only results for dogs and small grey wolf populations (Middle East, Asia) were drawn using R (https://www.r-project.org/) and are represented on Figure S2. All D-statistics analyses are detailed and publicly available on Dataverse (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD).
Haplotype analysis.
We first converted both domestic dog breed and wild canid vcf files into plink format using vcftools74 (--plink option) and merged them into a single plink file using PLINK v1.976, keeping only variants with 90% of their genotypes (--merge --geno 0.9). Data were phased and haplotypes determined on CFA15 using the program Beagle v4.188, with sliding windows of 1,000 SNPs and a 50-SNP overlap. To identify which haplotype contains the IGF1-AS variant, we focused on a 2,682 base pair (bp) region centered on the mutation and corresponding to 38 polymorphic markers. We used the Andean fox (Lycalopex culpaeus) to define ancestral alleles36 (n.b: the wild canids had to be imputed and there is no reference for any of these species, which can disrupt the phasing process). Using PHASE (v2.1.1)89, we identified a total of 37 haplotypes, 23 containing the IGF1-AS small allele C, and 14 with the T allele (Figure S3 and the1,389 samples are detailed in Data S1E). To perform phylogenetic analysis, we computed a pairwise identity-by-state distance matrix using PLINK v1.976 (--distance 1-ibs option). Bootstrapped distance matrices were created by randomly resampling markers with replacement 100 times and input into PHYLIP90 using neighbor and consensus to construct neighbor-joining dendrograms. Andean fox was used to root the tree36. For domestic breeds we used the two groups (small and large) as defined previously (Data S1A). As a note, since the four WGS coyotes used in this analysis were originally sampled from the West coast, we could not investigate the hypothesis of a recent introgression with wolves on East coast27, and represent it on Figure S3. Dendrograms were visualized using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/). Raw data to construct the tree are publicly available on Dataverse (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD).
RNA-sequencing analysis and qRT-PCR.
FASTQ files were quantified to transcript per million (TPM) expression values using RSEM91 version 1.3 (options: rsem-calculate-expression–num-threads 10–paired-end–bowtie2) with CanFam 3.1 as the reference genome for alignment and CanFam 3.1-plus used to call gene counts66. We also ran the same analysis on 51 previously published RNA-seq samples obtained from the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra). To confirm the RNA-Seq results, reverse transcription was performed with 1 μg of total RNA from testes using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems) according to the manufacturer’s instructions. We then performed qPCR on diluted cDNA samples (1:20 dilutions from the 1–2 μg obtained after cDNA reverse transcription) using the Power SYBR Green PCR Master Mix kit (Applied Biosystems). qPCR reactions were run on the CFX384 TouchTM Real-Time PCR Detection System (Bio-rad) using standard procedures. For each sample, we performed three biological replicates and the experiment was performed three times. Relative normalized expressions were determined using CFX MaestroTM Analysis Software (Bio-Rad). Primers for IGF1, IGF1-AS and GAPDH (reference gene) were designed using Primer3plus49 and are listed in the key resources table. On Figure S4, violin plots were constructed in R (https://www.r-project.org/) and P values were calculated by Mann–Whitney–Wilcoxon tests (***P < 0.0001). qRT-PCR raw data are publicly available on Dataverse (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD).
Supplementary Material
A) List of the 1,297 WGS dogs. B) CNV analysis. C) Additional Sanger sequenced dogs and their genotypes. D) IGF1 serum level raw data. E) Haplotypes. F) RNA-Seq data.
Document S1. Figures S1-S4 and Tables S1-S3.
Highlights.
An ancestral variant on IGF1 locus regulates body size in ancient and modern dogs
Variant alleles are associated with body size in dogs, wolves and coyotes.
The large body size-associated allele arose more than 53,000 years ago in wolves.
Human selection for small size may have been a major force during domestication.
Acknowledgements
We thank University of Washington Burke Museum, Museum of Vertebrate Zoology, California Academy of Science, The Museum of Southwestern Biology, Denver Museum of Nature and Science, Sam Noble Oklahoma Museum of Natural History, Museum of Comparative Zoology and Princeton University for providing access to their collection of coyotes body mass measures. We thank Pontus Skoglund and Anders Bergström for facilitating access to unpublished data. We gratefully acknowledge Robert K. Wayne and Roland Kays for insightful comments on the manuscript and as well as Robert K. Wayne for sharing samples, the CaniDNA biobank which provided RNA samples for functional experiments, and the ZIN RAS (grant n° 075-15-2021-1069) for sharing ancient samples. The MoEFCC and The Maharashtra Forest Department provided necessary approvals for which we are also grateful. We also thank Luc Paillard, Agnes Mereau, Yann Audic, Pascale Quignon, Stéphane Dreano, Marion Haas, Cédric Coulouarn and Jenny Serra-Vinardell for constructive comments. Finally, we gratefully acknowledge the Dog10K Consortium for whole genome sequencing a subset of samples and the many dog owners who provided samples for this study. This work was supported by funding from the Intramural Program of the National Human Genome Research Institute (J.P., H.G.P., A.N.H., E.A.O.). J.P is also funded by Region Bretagne and Ligue contre le Cancer. B.V.H is funded by Princeton University. L.F., J.H., and G.L. were supported either by the ERC (grant ERC-2013-StG-337574-UNDEAD and ERC-2019-StG-853272-PALAEOFARM) and Natural Environmental Research Council grants (NE/K005243/1 and NE/K003259/1). L.F. and A.C were supported by the Wellcome Trust (210119/Z/18/Z). BH’s research was funded by DST, Govt. of India and Maharashtra Forest Department. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no competing interests.
Inclusion and diversity: We worked to ensure sex balance in the selection of non-human subjects. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
SUPPLEMENTAL INFORMATION
Supplemental information includes four figures, three tables and one supplementary data and can be found with this article online
Research data for this article (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD).
References
- 1.Wayne RK, and Ostrander EA (1999). Origin, genetic diversity, and genome structure of the domestic dog. Bioessays 21, 247–257. [DOI] [PubMed] [Google Scholar]
- 2.Bergström A, Frantz L, Schmidt R, Ersmark E, Lebrasseur O, Girdland-Flink L, Lin AT, Storå J, Sjögren K-G, Anthony D, et al. (2020). Origins and genetic legacy of prehistoric dogs. Science 370, 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leathlobhair MN, Perri AR, Irving-Pease EK, Witt KE, Linderholm A, Haile J, Lebrasseur O, Ameen C, Blick J, Boyko AR, et al. (2018). The evolutionary history of dogs in the Americas. Science 361, 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Master D, Schloen D, and Stager L (2008). Ashkelon 1: Introduction and Overview (1985–2006) (Eisenbrauns; ). [Google Scholar]
- 5.Worboys M, Strange J-M, and Pemberton N (2018). The Invention of the Modern Dog: Breed and Blood in Victorian Britain (Johns Hopkins University Press,). [Google Scholar]
- 6.Chase K, Carrier DR, Adler FR, Jarvik T, Ostrander EA, Lorentzen TD, and Lark KG (2002). Genetic basis for systems of skeletal quantitative traits: Principal component analysis of the canid skeleton. PNAS 99, 9930–9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayward JJ, Castelhano MG, Oliveira KC, Corey E, Balkman C, Baxter TL, Casal ML, Center SA, Fang M, Garrison SJ, et al. (2016). Complex disease and phenotype mapping in the domestic dog. Nat. Commun 7, 10460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plassais J, Kim J, Davis BW, Karyadi DM, Hogan AN, Harris AC, Decker B, Parker HG, and Ostrander EA (2019). Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun 10, 1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rimbault M, Beale HC, Schoenebeck JJ, Hoopes BC, Allen JJ, Kilroy-Glynn P, Wayne RK, Sutter NB, and Ostrander EA (2013). Derived variants at six genes explain nearly half of size reduction in dog breeds. Genome Res 23, 1985–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L, Padhukasahasram B, Karlins E, Davis S, Jones PG, et al. (2007). A single IGF1 allele is a major determinant of small size in dogs. Science 316, 112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray MM, Sutter NB, Ostrander EA, and Wayne RK (2010). The IGF1 small dog haplotype is derived from Middle Eastern grey wolves. BMC Biol 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.American Kennel Club (2006). The Complete Dog Book 20th edn (Ballantine Books; ). [Google Scholar]
- 13.Parker HG, Dreger DL, Rimbault M, Davis BW, Mullen AB, Carpintero-Ramirez G, and Ostrander EA (2017). Genomic Analyses Reveal the Influence of Geographic Origin, Migration, and Hybridization on Modern Dog Breed Development. Cell Rep 19, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramos-Madrigal J, Sinding M-HS, Carøe C, Mak SST, Niemann J, Samaniego Castruita JA, Fedorov S, Kandyba A, Germonpré M, Bocherens H, et al. (2021). Genomes of Pleistocene Siberian Wolves Uncover Multiple Extinct Wolf Lineages. Curr Biol 31, 198–206.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frantz LAF, Mullin VE, Pionnier-Capitan M, Lebrasseur O, Ollivier M, Perri A, Linderholm A, Mattiangeli V, Teasdale MD, Dimopoulos EA, et al. (2016). Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science 352, 1228–1231. [DOI] [PubMed] [Google Scholar]
- 16.Sinding M-HS, Gopalakrishnan S, Ramos-Madrigal J, Manuel M. de, Pitulko VV, Kuderna L, Feuerborn TR, Frantz LAF, Vieira FG, Niemann J, et al. (2020). Arctic-adapted dogs emerged at the Pleistocene–Holocene transition. Science 368, 1495–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skoglund P, Ersmark E, Palkopoulou E, and Dalén L (2015). Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds. Curr. Biol 25, 1515–1519. [DOI] [PubMed] [Google Scholar]
- 18.Botigué LR, Song S, Scheu A, Gopalan S, Pendleton AL, Oetjens M, Taravella AM, Seregély T, Zeeb-Lanz A, Arbogast R-M, et al. (2017). Ancient European dog genomes reveal continuity since the Early Neolithic. Nat. Commun 8, 16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jónsson H, Ginolhac A, Schubert M, Johnson PLF, and Orlando L (2013). mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von den Driesch A (1976). A Guide to the Measurement of Animal Bones from Archaeological Sites (Harvard University Press; ). [Google Scholar]
- 21.Losey RJ, Osipov B, Sivakumaran R, Nomokonova T, Kovychev EV, and Diatchina NG (2015). Estimating Body Mass in Dogs and Wolves Using Cranial and Mandibular Dimensions: Application to Siberian Canids. Int J Osteoarchaeol 25, 946–959. [Google Scholar]
- 22.Freckleton RP, Harvey PH, and Pagel M (2003). Bergmann’s Rule and Body Size in Mammals. Am Nat 161, 821–825. [DOI] [PubMed] [Google Scholar]
- 23.Gohli J, and Voje KL (2016). An interspecific assessment of Bergmann’s rule in 22 mammalian families. BMC Evol Biol 16, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ardalan A, Kluetsch CFC, Zhang A-B, Erdogan M, Uhlén M, Houshmand M, Tepeli C, Ashtiani SRM, and Savolainen P (2011). Comprehensive study of mtDNA among Southwest Asian dogs contradicts independent domestication of wolf, but implies dog-wolf hybridization. Ecol. Evol 1, 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis SJM (1981). The Effects of Temperature Change and Domestication on the Body Size of Late Pleistocene to Holocene Mammals of Israel. Paleobiology 7, 101–114. [Google Scholar]
- 26.Padilla LR, and Hilton CD (2015). Canidae. In Fowler’s Zoo and Wild Animal Medicine, Volume 8 (Saunders; ), pp. 457–467. [Google Scholar]
- 27.Hinton JW, Heppenheimer E, West KM, Caudill D, Karlin ML, Kilgo JC, Mayer JJ, Miller KV, Walch M, vonHoldt B, et al. (2019). Geographic patterns in morphometric and genetic variation for coyote populations with emphasis on southeastern coyotes. Ecol. Evol 9, 3389–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durand EY, Patterson N, Reich D, and Slatkin M (2011). Testing for ancient admixture between closely related populations. Mol. Biol. Evol 28, 2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zucchelli S, Cotella D, Takahashi H, Carrieri C, Cimatti L, Fasolo F, Jones MH, Sblattero D, Sanges R, Santoro C, et al. (2015). SINEUPs: A new class of natural and synthetic antisense long non-coding RNAs that activate translation. RNA Biol 12, 771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohyama T, Takahashi H, Sharma H, Yamazaki T, Gustincich S, Ishii Y, and Carninci P (2020). An NMR-based approach reveals the core structure of the functional domain of SINEUP lncRNAs. Nucleic Acids Res 48, 9346–9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker J, Liu J-P, Robertson EJ, and Efstratiadis A (1993). Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75, 73–82. [PubMed] [Google Scholar]
- 32.Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu J-L, Ooi GT, Setser J, Frystyk J, Boisclair YR, et al. (2002). Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest 110, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.vonHoldt BM, Cahill JA, Fan Z, Gronau I, Robinson J, Pollinger JP, Shapiro B, Wall J, and Wayne RK (2016). Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv 2, e1501714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linderholm A, and Larson G (2013). The role of humans in facilitating and sustaining coat colour variation in domestic animals. Semin. Cell Dev. Biol 24, 587–593. [DOI] [PubMed] [Google Scholar]
- 35.Bannasch DL, Kaelin CB, Letko A, Loechel R, Hug P, Jagannathan V, Henkel J, Roosje P, Hytönen MK, Lohi H, et al. (2021). Dog colour patterns explained by modular promoters of ancient canid origin. Nat Ecol Evol, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gopalakrishnan S, Sinding M-HS, Ramos-Madrigal J, Niemann J, Samaniego Castruita JA, Vieira FG, Carøe C, Montero M. de M., Kuderna L, Serres A, et al. (2018). Interspecific Gene Flow Shaped the Evolution of the Genus Canis. Curr. Biol 28, 3441–3449.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kardos M, Åkesson M, Fountain T, Flagstad Ø, Liberg O, Olason P, Sand H, Wabakken P, Wikenros C, and Ellegren H (2018). Genomic consequences of intensive inbreeding in an isolated wolf population. Nat Ecol Evol 2, 124–131. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y-H, Wang L, Xu T, Guo X, Li Y, Yin T-T, Yang H-C, Hu Y, Adeola AC, Sanke OJ, et al. (2018). Whole-Genome Sequencing of African Dogs Provides Insights into Adaptations against Tropical Parasites. Mol. Biol. Evol 35, 287–298. [DOI] [PubMed] [Google Scholar]
- 39.Robinson JA, Ortega-Del Vecchyo D, Fan Z, Kim BY, vonHoldt BM, Marsden CD, Lohmueller KE, and Wayne RK (2016). Genomic Flatlining in the Endangered Island Fox. Curr. Biol 26, 1183–1189. [DOI] [PubMed] [Google Scholar]
- 40.Sinding M-HS, Gopalakrishan S, Vieira FG, Castruita JAS, Raundrup K, Jørgensen MPH, Meldgaard M, Petersen B, Sicheritz-Ponten T, Mikkelsen JB, et al. (2018). Population genomics of grey wolves and wolf-like canids in North America. PLOS Genet 14, e1007745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ostrander EA, Wang G-D, Larson G, vonHoldt BM, Davis BW, Jagannathan V, Hitte C, Wayne RK, Zhang Y-P, and Dog10K Consortium (2019). Dog10K: an international sequencing effort to advance studies of canine domestication, phenotypes and health. Natl Sci Rev 6, 810–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Axelsson E, Ratnakumar A, Arendt M-L, Maqbool K, Webster MT, Perloski M, Liberg O, Arnemo JM, Hedhammar Å, and Lindblad-Toh K (2013). The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 495, 360–364. [DOI] [PubMed] [Google Scholar]
- 48.Plassais J, Rimbault M, Williams FJ, Davis BW, Schoenebeck JJ, and Ostrander EA (2017). Analysis of large versus small dogs reveals three genes on the canine X chromosome associated with body weight, muscling and back fat thickness. PLoS Genet 13, e1006661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, and Leunissen JAM (2007). Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35, W71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ewing B, Hillier L, Wendl MC, and Green P (1998). Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8, 175–185. [DOI] [PubMed] [Google Scholar]
- 51.Ewing B, and Green P (1998). Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8, 186–194. [PubMed] [Google Scholar]
- 52.Gordon D (2003). Viewing and editing assembled sequences using Consed. Curr Protoc Bioinformatics 11, Unit11.2. [DOI] [PubMed] [Google Scholar]
- 53.Outram AK, and Bogaard A (2019). Subsistence and Society in Prehistory: New Directions in Economic Archaeology (Cambridge University Press; ). [Google Scholar]
- 54.Benecke N, Hansen S, Nowacki D, Reingruber A, Ritchie K, and Wunderlich J (2013). Pietrele in the Lower Danube region: integrating archaeological, faunal and environmental investigations. Documenta Praehistorica 40, 175–193. [Google Scholar]
- 55.Sablin MV, and Khlopachev GA (2002). The Earliest Ice Age Dogs: Evidence from Eliseevichi 1. Current Anthropology 43, 795–799. [Google Scholar]
- 56.Germonpré M, Sablin MV, Stevens RE, Hedges REM, Hofreiter M, Stiller M, and Després VR (2009). Fossil dogs and wolves from Palaeolithic sites in Belgium, the Ukraine and Russia: osteometry, ancient DNA and stable isotopes. Journal of Archaeological Science 36, 473–490. [Google Scholar]
- 57.Demay L, Patou-Mathis M, Khlopachev GA, Sablin MV, and Vercoutère C (2019). L’exploitation de la faune par les groupes humains du Pléniglaciaire supérieur à Eliseevichi 1 (Russie). L’Anthropologie 123, 345–402. [Google Scholar]
- 58.Polikarpovich KM (1968). Palaeolithic of the Upper Dnieper. Minsk. (in Russian)
- 59.Germonpré M, and Sablin M (2017). Chapter 2. Humans and mammals in the Upper Palaeolithic of Russia. In The Oxford Handbook of Zooarchaeology Oxford Handbook of Zooarchaeology (Oxford University press; ). [Google Scholar]
- 60.Germonpré M, Lázničková-Galetová M, Losey RJ, Räikkönen J, and Sablin MV (2015). Large canids at the Gravettian Předmostí site, the Czech Republic: The mandible. Quaternary International 359–360, 261–279. [Google Scholar]
- 61.Dabney J, Knapp M, Glocke I, Gansauge M-T, Weihmann A, Nickel B, Valdiosera C, García N, Pääbo S, Arsuaga J-L, et al. (2013). Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. PNAS 110, 15758–15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Damgaard PB, Margaryan A, Schroeder H, Orlando L, Willerslev E, and Allentoft ME (2015). Improving access to endogenous DNA in ancient bones and teeth. Sci Rep 5, 11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meyer M, and Kircher M (2010). Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc 2010, pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 64.Meyer M, Kircher M, Gansauge M-T, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, Filippo C. de, et al. (2012). A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science 338, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ewels P, Magnusson M, Lundin S, and Käller M (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wucher V, Legeai F, Hédan B, Rizk G, Lagoutte L, Leeb T, Jagannathan V, Cadieu E, David A, Lohi H, et al. (2017). FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res 45, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- 70.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative Genomics Viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lopez BH (1978). Of Wolves and Men (Littlehampton Book Services Ltd; ). [Google Scholar]
- 72.Estes RD (2012). The Behavior Guide to African Mammals: Including Hoofed Mammals, Carnivores, Primates, 20th Anniversary Edition (University of California Press; ). [Google Scholar]
- 73.Wildlife Institute of India (2018). National Studbook of Indian Wolf (Canis lupus pallipes) II Edition (Wildlife Institute of India, Central Zoo Authority; ). [Google Scholar]
- 74.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou X, and Stephens M (2012). Genome-wide efficient mixed-model analysis for association studies. Nat. Genet 44, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. (2007). PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am J Hum Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Serres-Armero A, Davis BW, Povolotskaya IS, Morcillo-Suarez C, Plassais J, Juan D, Ostrander EA, and Marques-Bonet T (2021). Copy number variation underlies complex phenotypes in domestic dog breeds and other canids. Genome Res 31, 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Karger DN, Conrad O, Böhner J, Kawohl T, Kreft H, Soria-Auza RW, Zimmermann NE, Linder HP, and Kessler M (2017). Climatologies at high resolution for the earth’s land surface areas. Sci Data 4, 170122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bates D, Mächler M, Bolker B, and Walker S (2015). Fitting Linear Mixed-Effects Models Using lme4. J Stat Softw 67, 1–48. [Google Scholar]
- 80.Burnham KP, and Anderson DR (2002). Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach 2nd ed. (Springer-Verlag; ). [Google Scholar]
- 81.Lüdecke D, Ben-Shachar MS, Patil I, Waggoner P, and Makowski D (2021). performance: An R Package for Assessment, Comparison and Testing of Statistical Models. J Open Source Softw 6, 3139. [Google Scholar]
- 82.Lindgreen S (2012). AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res Notes 5, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schubert M, Ginolhac A, Lindgreen S, Thompson JF, AL-Rasheid KA, Willerslev E, Krogh A, and Orlando L (2012). Improving ancient DNA read mapping against modern reference genomes. BMC Genomics 13, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kircher M (2012). Analysis of high-throughput ancient DNA sequencing data. Methods Mol Biol 840, 197–228. [DOI] [PubMed] [Google Scholar]
- 85.Korneliussen TS, Albrechtsen A, and Nielsen R (2014). ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics 15, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Germonpré M, Fedorov S, Danilov P, Galeta P, Jimenez E-L, Sablin M, and Losey RJ (2017). Palaeolithic and prehistoric dogs and Pleistocene wolves from Yakutia: Identification of isolated skulls. J Archaeol Sci 78, 1–19. [Google Scholar]
- 87.Kunsch HR (1989). The Jackknife and the Bootstrap for General Stationary Observations. Ann. Statist 17, 1217–1241. [Google Scholar]
- 88.Browning SR, and Browning BL (2007). Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. Am J Hum Genet 81, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stephens M, and Scheet P (2005). Accounting for Decay of Linkage Disequilibrium in Haplotype Inference and Missing-Data Imputation. Am J Hum Genet 76, 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Felsenstein J (1989). PHYLIP-Phylogeny Inference Package (Ver. 3.2). Cladistics, 164–166.
- 91.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) List of the 1,297 WGS dogs. B) CNV analysis. C) Additional Sanger sequenced dogs and their genotypes. D) IGF1 serum level raw data. E) Haplotypes. F) RNA-Seq data.
Document S1. Figures S1-S4 and Tables S1-S3.
Data Availability Statement
Genomes sequenced for this work, as well as all publicly available data used for alignment, are available via the Short Read Archive (ncbi.nlm.nih.gov/sra) and their associated accession numbers are listed in the Supplementary Data 1A and in the key resources table. Newly generated genomes are now available on SRA: accession number PRJNA685036. RNA-Seq raw data are registered on SRA (accession number: PRJNA690861). The three newly sequenced ancient wolves are registered on The European Nucleotide Archive (ENA) under the permanent study accession number: PRJEB42199. Raw data for tables and figures (including original gel pictures), and all raw data for statistics (including GWAS results, GLM, Wilcoxon-Mann-Whitney, Spearman) are publicly available on Dataverse. The DOI is listed in the key resources table and with the corresponding methods. Other data are contained within the article and its supplementary information.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Canis Familiaris | Various veterinary referral hospitals | N/A |
| Three Canid bone paleontological remains | This paper | Botai (sample ID: AL2350); Pietrele (sample ID: AL3185); Eliseevichi (sample ID: AL2657). |
| Chemicals, peptides, and recombinant proteins | ||
| Trizol | Life Technologies | 15596026 |
| RNAlater | Life Technologies | AM7020M |
| Critical commercial assays | ||
| TruSeq DNA nano kit | Illumina | FC-121-4001 |
| TruSeq Stranded mRNA library Prep Kit High Throughput | Illumina | RS-122-2103 |
| Illumina TruSeq Nano DNA library prep HT | Illumina | 20000903 |
| TruSeq® Stranded mRNA LT-Set A | Illumina | RS-122-2101 |
| NextSeq High Output v2.5 75 cycle kit | Illumina | 20024906 |
| Deposited data | ||
| DNA sequencing data | This paper | SRA:PRJNA685036 ncbi.nlm.nih.gov/sra |
| RNA sequencing data | This paper | SRA:PRJNA690861 ncbi.nlm.nih.gov/sra |
| Three ancient wolf DNA sequencing data | This paper | ENA: PRJEB42199 http://www.ebi.ac.uk/ena |
| Dog genome reference (CanFam3.1, ENSEMBL release-85) | ENSEMBL | http://www.ensembl.org/index.html |
| Dog genome Annotation (CanFam3.1-plus) | 66 | http://tools.genouest.org/data/tderrien/canFam3.1p/annotation/trackhub/canfam3.1ptrackhub/hub.txt |
| Oligonucleotides | ||
| gDNA targeted primer: IGF1 Forward:CACTGATCCAGAAGAATCCAACT | 9 | N/A |
| gDNA targeted primer: IGF1 Reverse: CAAAGAACCATGTAAGCCTATTTGT | 9 | N/A |
| gDNA targeted primer:IGF1-AS mutation Forward:GTGGGCTTGTCTGTGCAAAT | This paper | N/A |
| gDNA targeted primer:IGF1-AS mutation Reverse:CCTGAGCATAAAAACTAGGCAGA | This paper | N/A |
| gDNA targeted primer:IGF1-SINE Forward:CACTGATCCAGAAGAATCCAACT | This paper | N/A |
| gDNA targeted primer:IGF1-SINE Reverse:CAAAGAACCATGTAAGCCTATTTGT | This paper | N/A |
| cDNA targeted primer:IGF1-AS short isoform Forward:AGCTGGTCATCAATTTGCCCC | This paper | N/A |
| cDNA targeted primer:IGF1-AS short isoform Reverse:AAGGAAAGACTCAGTTTGGGTGT | This paper | N/A |
| cDNA targeted primer:IGF1-AS long isoform Forward:TGGAAACCACTGGATCTGAGCT | This paper | N/A |
| cDNA targeted primer:IGF1-AS long isoform Reverse:AAGGAAAGACTCAGTTTGGGTGT | This paper | N/A |
| cDNA targeted primer:IGF1-AS last exon RPA Forward:GCACCACAGAGGAAGGATGAT | This paper | N/A |
| cDNA targeted primer:IGF1-AS last exon RPA Reverse:TGGGATGTGTAGGTTGACCAG | This paper | N/A |
| cDNA targeted primer:IGF1 RPA Forward:TGCTCTCAACATCTCCCATCTCT | This paper | N/A |
| cDNA targeted primer:IGF1 RPA Reverse:ACCGTTTTGGCCAGACTCTTT | This paper | N/A |
| cDNA targeted primer:IGF1-AS/IGF1 exon 3 RPA Forward:CCTTGGGCATGTCAGTGTGG | This paper | N/A |
| cDNA targeted primer:IGF1-AS/IGF1 exon 3 RPA Reverse:GACAGGCATCGTGGATGAGTG | This paper | N/A |
| cDNA targeted primer:GAPDH qRT-PCR Forward:AAGCAGGGATGATGTTCTGG | This paper | N/A |
| cDNA targeted primer:GAPDH qRT-PCR Reverse:CCTCATGACCACCGTCCA | This paper | N/A |
| cDNA targeted primer:IGF1 qRT-PCR Forward:CCTGCACTCCCTCTACTTGC | This paper | N/A |
| cDNA targeted primer:IGF1 qRT-PCR Reverse:CTCAAGCCTGCCAAGTCTG | This paper | N/A |
| cDNA targeted primer:IGF1-AS qRT-PCR Forward:TGAAGCTTCCCCAACAATTC | This paper | N/A |
| cDNA targeted primer:IGF1-AS qRT-PCR Reverse:TGGGTGTAGACGAGATCCTTG | This paper | N/A |
| Software and algorithms | ||
| Read alignment: bwa v0.7.17 | 42 | https://sourceforge.net/projects/bio-bwa/files/ |
| Samtools 1.6 | 43 | http://samtools.sourceforge.net/ |
| Variant caller: GATK v4.1.4.0 | 44,45 | http://gatkforums.broadinstitute.org/gatk |
| PicardTools 2.9.2 | http://broadinstitute.github.io/picard | http://github.com/broadinstitute/picard |
| Vcftools v0.1.16 | 74 | http://vcftools.sourceforge.net/ |
| Linear mixed model: GEMMA v0.94.1 | 75 | http://www.xzlab.org/software.html |
| Utility: PLINK v1.9 | 76 | http://zzz.bwh.harvard.edu/plink/ |
| Graphics and data analysis: R v3.3.0 | The Comprehensive R Archive Network (CRAN) | https://cran.r-project.org |
| Phred/Phrap/Consed package | 50–52 | http://www.phrap.org/phredphrapconsed.html |
| AdapterRemoval2 | 82 | https://github.com/MikkelSchubert/adapterremoval |
| RSeQC | 69 | https://github.com/MonashBioinformaticsPlatform/RSeQC |
| MapDamage v2.58 | 19 | https://github.com/ginolhac/mapDamage |
| FilterUniqSamCons | 84 | |
| ANGSD 0.614 | 85 | https://github.com/ANGSD/angsd |
| MultiQC tool | 65 | https://multiqc.info/ |
| STAR | 67 | https://github.com/alexdobin/STAR |
| Stringtie | 68 | https://ccb.jhu.edu/software/stringtie/ |
| RSEM v1.3 | 91 | https://github.com/deweylab/RSEM |
| Data visualization: Integrative Genomics Viewer: I.G.V 2.8.2 | 70 | http://software.broadinstitute.org/software/igv/ |
| Beagle v4.1 | 88 | http://faculty.washington.edu/browning/beagle/beagle.html |
| PHYLIP | 90 | https://evolution.genetics.washington.edu/phylip.html |
| FigTree v1.4.4 | Graphical viewer of phylogenetic trees | http://tree.bio.ed.ac.uk/software/figtree/ |
| Other | ||
| KOD Xtreme HotStart Polymerase | Merck | 71975-3 |
| ExoSap-ItTM reaction | Thermo Fisher Scientific | 78201.1.ML |
| BigDye Terminator v3.1 | Thermo Fisher Scientific | 4337458 |
| RecoverAllTM Total Nucleic Acid Isolation Kit | Thermo Fisher Scientific | A26135 |
| High-Capacity cDNA Reverse Transcription kit | Thermo Fisher Scientific | 4368814 |
| TURBODNase | Thermo Fisher Scientific | AM2238 |
| RNAse A/T1 Mix | Thermo Fisher Scientific | EN0551 |
| NucleoSpin RNAclean-up kit | Macherey-Nagel | 740948.50 |
| SYBR™ Safe | Thermo Fisher Scientific | S33102 |
| Ancient DNA genotyping determination and all statistical analyses related to the paper | This paper | Dataverse :doi: 10.7910/DVN/JBXYZD https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JBXYZD |
| The two VCF files built for this work (one containing 1,156 dogs and one containing 86 wild canids) are publically available on the dog genome project website | This paper | https://research.nhgri.nih.gov/dog_genome/data_release/index.shtml |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.