

Abstract
Retinitis pigmentosa occurs due to mutations that cause rod photoreceptor degeneration. Once most rods are lost, gradual degeneration of cone photoreceptors occurs. Oxidative damage and abnormal glucose metabolism have been implicated as contributors to cone photoreceptor death. Herein, we show increased phosphorylation of key enzymes of glucose metabolism in the retinas of rd10 mice, a model of RP, and retinas of wild type mice with paraquat-induced oxidative stress, thereby inhibiting these key enzymes. Dietary supplementation with glucose and pyruvate failed to overcome the inhibition, but increased reducing equivalents in the retina and improved cone function and survival. Dichloroacetate reversed the increased phosphorylation of pyruvate dehydrogenase in rd10 retina and increased histone acetylation and levels of TP53-induced glycolysis and apoptosis regulator (TIGAR), which redirected glucose metabolism toward the pentose phosphate pathway. These data indicate that oxidative stress induced damage can be reversed by shifting glycolytic intermediates toward the pentose phosphate pathway which increases reducing equivalents and provides photoreceptor protection.
Keywords: Retinal degeneration, Pentose phosphate pathway, TIGAR, Metabolism
Graphical Abstract
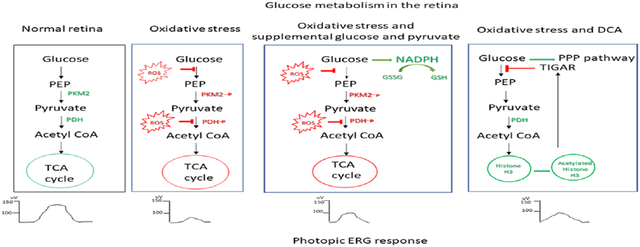
Introduction
Retinitis Pigmentosa (RP) is an inherited retinal degeneration that has a prevalence of roughly 1:4000 (1, 2). Pathogenic mutations have been identified in nearly 100 genes (http://www.sph.uth.tmc.edu/retnet/) and can result in autosomal recessive, autosomal dominant, or X-linked recessive inheritance. The key feature that links all of these different mutations with different modes of inheritance into a single disease process is that the mutations differentially affect rod photoreceptors and spare cone photoreceptors. Rod photoreceptors are responsible for vision in low light settings and therefore, the first symptom of RP is usually difficulty seeing when illumination is poor often referred to as night blindness. While this can be troubling in certain situations, it has little impact on most activities of daily life. However, after the majority of rods die, cone photoreceptors begin to degenerate resulting in gradual constriction of visual fields and eventually severe visual disability often culminating in blindness. In order to prevent severe vision loss in patients with RP, it is critical to understand the mechanism(s) by which cones die.
Rod photoreceptors constitute 95% of cells in the outer retina and after they are eliminated, oxygen consumption is markedly reduced and remaining cones are exposed to high levels of oxygen (3, 4) causing oxidative stress from excess production of superoxide radicals and other ROS (5). This results in progressive oxidative damage to cones in RP retina (6) which contributes to cone cell death, because antioxidants, including N-acetylcysteine, or expression of components of the endogenous antioxidant defense system, promote cone survival and function in multiple RP models (7-11). In patients with RP, administration of oral N-acetylcysteine for 6 months improved cone function suggesting that long-term treatment with N-acetylcysteine might reduce cone cell death and visual disability (12). Therefore, oxidative damage appears to be one contributor to cone cell death in RP.
The hostile environment that cones are exposed to after rod degeneration results in metabolic abnormalities, particularly in the transport and utilization of glucose, which may also contribute to cone cell death (13-15). Glucose enters the retina by GLUT1 transport through retinal pigmented epithelial (RPE) cells and vascular endothelial cells and this transport is perturbed in the setting of RP resulting in substrate deficiency (13). However, there may be other abnormalities as well, because glucose metabolism is complex and has several vulnerable control points. Glucose is metabolized by glycolysis in the cytoplasm to form pyruvate which enters mitochondria where it is converted to acetyl CoA by the pyruvate dehydrogenase complex (PDC) and to oxaloacetate by pyruvate carboxylase. Acetyl CoA and oxaloacetate enter the TCA cycle which drives oxidative phosphorylation through which the vast majority of ATP is generated. Cells shift their reliance on glycolysis or the TCA cycle with regard to ATP production depending on the situation, with particularly important factors being the availability of oxygen and substrates. Cancer cells extensively utilize glycolysis even when oxygen is not limiting, which is referred to as aerobic glycolysis (16, 17). Photoreceptors appear to utilize aerobic glycolysis more than many other normal non-malignant tissues, but oxidative phosphorylation is still responsible for the majority of ATP generation in normal photoreceptors (18).
A major control point for the shift between reliance on the TCA cycle and glycolysis for energy production is pyruvate kinase, the rate limiting enzyme of glycolysis. There are 4 isozymes of pyruvate kinase, PKL, PKR, PKM1, and PKM2, which differ in their regulatory properties (19, 20). The predominant isozyme in photoreceptors is PKM2 (21, 22) which is also predominant in cancer cells (23). The relative abundance of one of the precursors of pyruvate, fructose-1,6-bisphosphate (FBP), regulates PKM2, because FBP binds to PKM2 and stabilizes the active tetrameric form. When FBP is low, the lack of binding of FBP to PKM2 causes its dissociation into the inactive dimeric form and reduces utilization of the TCA cycle. PKM2 is also regulated by phosphorylation of tyrosine residue 105 (Y105) which disrupts binding of FBP to PKM2 and thus reduces PKM2 activity (24). The PDC complex provides a second major control point because its enzymatic activity is reduced when it is phosphorylated by one of four isoforms of pyruvate dehydrogenase kinase (PDK) (25, 26). PDK expression is increased by hypoxia and its activity is stimulated by the products of pyruvate dehydrogenase, acetyl CoA and NADH, shifting glucose metabolism away from the TCA cycle (27). Lactate dehydrogenase (LDH) provides a third control point; when the production of lactate is favored, the depletion of pyruvate reduces TCA cycle flux.
The shift in emphasis between glycolysis and the TCA cycle is an important adaptive response that allows cells to cope with changes in substrate and oxygen availability, but it is also vulnerable to dysregulation during disease. Abnormal inhibition of PDC resulting in energy deficiency has been demonstrated in cardiovascular (28, 29) and neurodegenerative diseases (30). In this study, we sought to determine if changes in these control points of glucose metabolism contribute to metabolic abnormalities previously reported in RP retinas.
Materials and Methods
Study approval
Mice were treated in accordance with the Association for Research in Vision and Ophthalmology and the protocol was approved by the Johns Hopkins Animal Care and Use Committee (approval number MO20M118).
Study Design
The primary objective of the study was to determine if there is reduced TCA cycle flux in rd10 mouse retina (JAX:004297) and if so, whether resultant energy deficiency contributes to cone photoreceptor dysfunction and death. A secondary objective was to determine if increasing TCA cycle flux in rd10 retina could improve cone function and survival. The primary outcome measures for TCA cycle flux was the phosphorylation status of enzymes which regulate entry of substrates into the TCA cycle. The outcome measure for cone function was mean photopic b-wave amplitude at P50 and P65, which is markedly reduced in untreated rd10 mice. The outcome measures for cone survival were the levels of M-cone and S-cone opsin relative to actin in rd10 retina at P50 and P65. Littermate rd10 mice were arbitrarily assigned to various treatment groups. No data were excluded.
Study treatments
Supplemental dietary glucose and/or pyruvate
Three independent experiments were performed. For randomization, rd10 litters were subdivided into two groups by randomly swapping pups among litters. All treatments were started at P8 and continued until P50, the predetermined endpoint of the study. Pups were weaned at P21 and continued to receive assigned supplemented or unsupplemented drinking water. The groups included, the pyruvate glucose group, the glucose group, the pyruvate group and the water treatment group. The animals in the pyruvate glucose group received 42 mM glucose (D9434, Sigma) and 10 mM pyruvate (S8636, Sigma) in drinking water, the glucose group received 42 mM glucose in drinking water, the pyruvate group received 10 mM in drinking water and the water group received only drinking water. The drug water was refreshed every alternate day.
DCA
For the DCA study group, all litters were randomly subdivided into 2 groups, the DCA group and the water group. Both treatments started at P14 and continued until P65. The DCA treatment included 200 mg of sodium dichloroacetate (347795, Sigma) in 100 ml of drinking water. The water group received only drinking water. The DCA water was refreshed every alternate day.
Electroretinogram recordings (ERG)
An investigator blind to each treatment group performed ERGs using a Diagnosys instrument (Espion) with custom made platinum electrodes as previously described (7, 48). Rd10 mice were anesthetized by intraperioneal injections with ketamine (100 mg/kg, 1049007, Henry Schein®) and xylazine (5 mg/kg, sc-362950Rx, Lloyd Laboratories Inc.). The pupils were dilated with 0.5% tropicamide and 0.5% phenylephrine hydrochloride (005728, Santen Pharmaceutical Co.). The mice were placed on a heated pad and the eye electrodes were placed on the cornea after the application of gonioscopic prism solution (17478-064-12, Alcon Labs). In addition, a reference electrode was attached subcutaneously between the eyes on the scalp and a ground electrode was attached to the tail. The Ganzfeld light illuminator was then placed above the head in a position between both eye and photopic ERGs were recorded at 3 light intensities (0.6, 1.0, 1.39 log cd-s/m2) with a background light of 30 cd/m2 For each flash intensity, 5 readings were taken and averaged.
Immunoblots
Mice were anesthetized by inhalation of Isoflurane (NDC10019-360-60, Baxter), euthanized by cervical dislocation, and eyes were removed. Retinas were isolated and homogenized in cell lysis buffer (9803, Cell Signaling) after addition of phosphatase and protease inhibitors (5872S, Cell Signaling). Lysates were electrophoresed by using NuPAGE 4-12% Bis-Tris gels (5872S, Invitrogen) and transferred to nitrocellulose membranes using the iBlot2NC system (IB23001, Invitrogen). Membranes were probed with the primary antibodies for phospho-PKM2 Tyr105 (1:1000, 3827S, Cell Signaling) PMID: 18337823, PKM2 (1:1000, 4053S, Cell Signaling) PMID: 34733164, phospho-LDHA Tyr10 (1:1000, 8176S, Cell Signaling) PMID: 21969607, LDHA (1:1000, 2012, Cell Signaling) PMID: 21969607, phospho-PDHE1-A Ser293 (1:1000, NB110-93479SS Novus Biological) PMID: 29425506, PDHE1-A (1:1000, ABS2082, EMD Millipore) PMID: 15569252, Actin (1:100, A2066, Sigma) PMID: 22864983, S opsin (1:1000. ABN1660, EMD Millipore) PMID: 33082517, M opsin (1:1000, NBP1-98471, Novus) validated by Novus on Western blots, Acetyl-Histone H3-K9 (1:1000, A7255, ABclonal) PMID: 34803511, H3 histone (1:1000, 9715S, Cell Signaling) PMID: 31586055, TIGAR (1:1000, 22136-1-AP, Proteintech) validated by Proteintech on western blots, Glut1 (1:1000, NB300-666, Novus) PMID: 30332638 and GRK1 (1:1000, PA5-13725, ThermoFischer Scientific) PMID: 32019921. Secondary antibody used was HRP conjugated donkey anti-rabbit IgG (1:10,000, NA934V, GE Healthcare) PMID: 34645815. The membranes were visualized with SuperSignal ™ West Duration Extended Duration Substrate (3407S, Thermo Fisher Scientific) and imaged using ChemiDoc™ XRS Molecular Imager (BIO-RAD). The protein bands were quantified using Image Lab Software (BIO-RAD).
Real time RT-PCR
RNA from retina samples were isolated using TRIzol® Reagent (15596026, Invitrogen). Genomic DNA was eliminated from RNA samples prior to cDNA synthesis using DNA-free™ (AM1906, Ambion®). RNA was reverse transcribed to cDNA using iScript™ cDNA Synthesis Kit (1708890, BIO-RAD). Real time PCR was performed using Rotor-Gene SYBR Green PCR Kit (204072, QIAGEN). The following primers were used.
PDK1 forward: GGACTTCGGGTCAGTGAATGC
PDK1 reverse: TCCTGAGAAGATTGTCGGGGA
PDK2 forward: AGGGGCACCCAAGTACATC
PDK2 reverse: TGCCGGAGGAAAGTGAATGAC
PDK3 forward: TCCTGGACTTCGGAAGGGATA
PDK3 reverse: GAAGGGCGGTTCAACAAGTTA
PDK4 forward: AGGGAGGTCGAGCTGTTCTC
PDK4 reverse: GGAGTGTTCACTAAGCGGTCA
BCKDK forward: ACATCAGCCACCGATACACAC
BCKDK reverse: GAGGCGAACTGAGGGCTTC
Cyclophilin A forward: TGGGGCTCTTCAAAAGCTCC
Cyclophilin A reverse: AGGAACTATCACCGGATCTTCAA
Fold Induction of Real time RT-PCR samples.
The Ct values of the gene of interest (PDKs) and house keeping gene (Cyclophilin) were initially obtained for the control and treated groups.
ΔCt Values were obtained for each group. ΔCt = Ct (of gene of interest) - Ct (house keeping gene). An average ΔCt value for the control group was calculated. Then the ΔΔCt values Calculated. ΔΔCt = ΔCt (all samples)- Average ΔCt (Control group).
Fold Induction = 2 −ΔΔα.
NADPH assay
NADPH concentration were estimated in retina samples using the Amplite™ Colorimetric NADPH Assay Kit (15272, AAT Bioquest®).
Measurement of reduced glutathione
Retinas were isolated from pyruvate/glucose treated and control rd10 mice at P50. The extracted retinas were immersed in 10% trichloroacetic acid and frozen immediately at −70°C. The reduced glutathione in the samples was measured using the reduced glutathione (GSH) assay kit (K464, BioVision). Protein measurement for normalization was done using Bradford reagent (5000006, BIO-RAD)
ATP Assay
Retinas were isolated from pyruvate/glucose treated, DCA-treated, and corresponding control rd10 mice at P50. ATP in the samples were determined using the ATP Bioluminescence Assay Kit CLS II (11699695001, Sigma) using the manufacturer’s instruction.
Immunohistochemistry
Pig eyes were removed, embedded in Optical Cutting Temperature medium (Sakura-Finetek) and 10 μm frozen sections were permeabilized with 0.2% Triton X in PBS, blocked at room temperature for 1 hour with blocking buffer (5% donkey serum, 1% BSA and 0.2% Triton X in PBS). The sections were then stained with anti-PDHE1-A antibody (1:1000, ABS2082, EMD Millipore) and peanut agglutinin (PNA, 1:100, 2315097, Vector Laboratories). The secondary antibody was goat anti-rabbit Alexa Fluor 594, (1:250, 2534095, ThermoFisher).
Paraquat treatment
C57BL/6J mice (JAX:000664) were given an intravitreous injection of 1.5 mM paraquat (856177, Millipore Sigma) in PBS. Retinas were isolated 16 hours or 7 days later used for immunoblots.
Statistical Analyses
For comparisons of photopic b wave values, amplitudes at the same flash intensities were compared between drug water and water treated controls. All comparisons in the manuscript were done by a Mann Whitney non-parametric statistical analysis and graphs were plotted using GraphPad Prism 6 (La Jolla, CA). P values below 0.05 were considered statistically significant. Asterisks shown on graphs denote: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
Results
Altered phosphorylation of glycolytic enzymes at regulatory control points for glucose metabolism favors reduced metabolic activity in RP retina
The predominant isoform of pyruvate kinase in photoreceptors is PKM2 (21, 22) and the predominant isoform of LDH is LDHA (31). Pyruvate dehydrogenase (E1-A) in the PDC complex contains the E1 active site and plays a key role in the synthesis of acetyl CoA from pyruvate, thus forming an important link between glycolysis and TCA cycle.
Immunohistochemical staining demonstrated that the PDHE1-A isoform of pyruvate dehydrogenase is present in photoreceptors (Figure 1A-C). Phosphorylation of serine residues of PDHE1-A by PDK reduces PDHE1-A activity (25, 26, 32). There was a significant increase in phosphorylation of S293 of PDHE1-A in the retinas of rd10 mice versus wild type mice (Figure 1D and E). This indicates reduction in PDHE1-A activity which was accentuated by reduction in total PDHE1-A as indicated by a decrease in PDHE1-A/actin ratio (Figure 1D and F). The mechanism of phosphorylation of PDHE1-A in rd10 retinas compared to wild type mice is due to the upregulation of pyruvate dehydrogenase kinase 2 (PDK2, Figure 1M) and branched chain ketoacid dehydrogenase kinase (BCKDK, Figure 1N), for which PDHE1-A is a substrate.
Figure 1. Altered phosphorylation indicating reduced activity of glucose metabolism regulatory enzymes in the retinas of rd10 mice.

Immunostaining demonstrates expression of the PDHE1-A isoform of pyruvate dehydrogenase (red) in the inner plexiform layer (IPL), outer plexiform layer (OPL) and photoreceptor inner segments (IS) of pig retina (A). There is staining for PDHE1-A within cone matrix sheaths labeled with FITC-labeled peanut agglutinin (green) demonstrating PDHE1-A expression in cones (B, C). Postnatal day (P) 35, wild type C57BL/6 mice and rd10 mice in a C57BL/6 background were euthanized and retinal homogenates were prepared. (D) Immunoblot of retinal homogenates from 7 C57BL/6 mice and 7 rd10 mice probed with anti-pPDHE1-A (S293), anti-PDHE1-A, and anti-Actin antibodies. Densitometry of the blot in (D) and a blot from a repeat experiment demonstrated a significant increase in pPDHE1-A/PDHE1-A ratio (p=0.0339) (E) and a significant decrease in PDHE1-A/Actin ratio (F) in rd10 vs wild type retinas (n=14). (G) Immunoblot of retinal homogenates from 7 C57BL/6 mice and 7 rd10 mice probed with anti-pPKM2 (Y105), anti-PKM2, and anti-Actin antibodies. Densitometry of the blot in (G) and a blot from a repeat experiment demonstrated a significant increase in pPKM2/PKM2 ratio (H) and a significant decrease in PKM2/Actin ratio (p=0.0236) (I) in rd10 vs wild type retinas (n=14). (J) Immunoblot of retinal homogenates from 7 C57BL/6 mice and 7 rd10 mice probed with anti-LDHA (Y10), anti-LDHA, and anti-Actin antibody. Densitometry of the blot in (J) demonstrated a significant reduction in pLDHA/LDHA ratio (K) and a significant decrease in LDHA/Actin ratio (p=0.0274) (L) in rd10 vs wild type retinas (n=7). Real-time RT-PCR showed increased expression of mRNA for pyruvate dehydrogenase kinase 2 (PDK 2, M) and branched chain ketoacid dehydrogenase kinase (p=0.0134, BCKDK, N) in the retinas of rd10 mice compared to wild type retinas.
* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 by Mann-Whitney test
PDHE1-A, Pyruvate dehydrogenase E1-A subunit; PKM2, isoform of pyruvate kinase; LDHA, isoform of lactate dehydrogenase; ONL, outer nuclear layer; INL, inner nuclear layer, IS, inner segment of photoreceptor; RPE, retinal pigmented epithelium.
The retinas of rd10 mice also showed a significant increase in phosphorylation of Y105 of PKM2 (Figure 1G and H) which reduces PKM2 activity (24). This inhibitory effect was accentuated by reduction in total PKM2 (Figure 1G and I). Contrary to PDHE1-A and PKM2 in which phosphorylation is inhibitory, phosphorylation of Y10 increases LDHA activity favoring the production of lactate from pyruvate and NAD+ from NADH (33). Phosphorylation of Y10 of LDHA was significantly reduced in rd10 retina (Figure 1J and K) suggesting decreased LDHA activity, which was accentuated by reduction in total LDHA (Figure 1J and L). The net effect of the changes in these 3 control points is reduction in glucose metabolism in rd10 retina due to the reduced activity of PKM2, PDHE1-A, and LDHA. This is consistent with a previous study demonstrating reduced levels of TCA cycle intermediates in RP retina (34).
Oxidative stress in the retina causes down-regulation of the activity of glycolytic enzymes
To investigate whether the excessive oxidative stress in rd10 retina may contribute to down-regulation of glycolytic enzymes, we utilized another model in which intraocular injection of paraquat, a superoxide radical generator, causes oxidative stress in the retina (35). Mice were given no treatment or an intraocular injection of 1.5 mM paraquat, and after 16 hours, immunoblots of retinal homogenates showed that compared with retinas from untreated eyes, those from eyes treated with paraquat had a significant increase in phosphorylation of S293 of PDHE1-A (Figure 2A and B) and no significant change in total PDHE1-A as indicated by PDHE1-A/actin ratio (Figure 2A and C). There was no significant change in the phosphorylation status of PKM2 or LDHA (Figure 2D-I). The mechanism of phosphorylation of PDHE1-A, by oxidative stress-induced by paraquat, is due to the upregulation of pyruvate dehydrogenase kinase 3 (PDK3) and branched chain ketoacid dehydrogenase kinase (BCKDK), for which PDHE1-A is a substrate (Figure 2J and K). One week after intraocular injection of 1.5 mM paraquat, there was increased phosphorylation of Y105 of PKM2 (Supplemental Figure 1A and B) and reduction of total PKM2 (Supplemental Figure 1A and C), which both reduce PKM2 activity and there was persistent increased phosphorylation of S293 in PDHE1-A (Supplemental Figure 1D and E). There was still no significant change in phosphorylation of Y10 in LDHA (Supplemental Figure 1G and H).
Figure 2. Paraquat-induced oxidative stress induces increased phosphorylation of PDHE1-A in the retina.
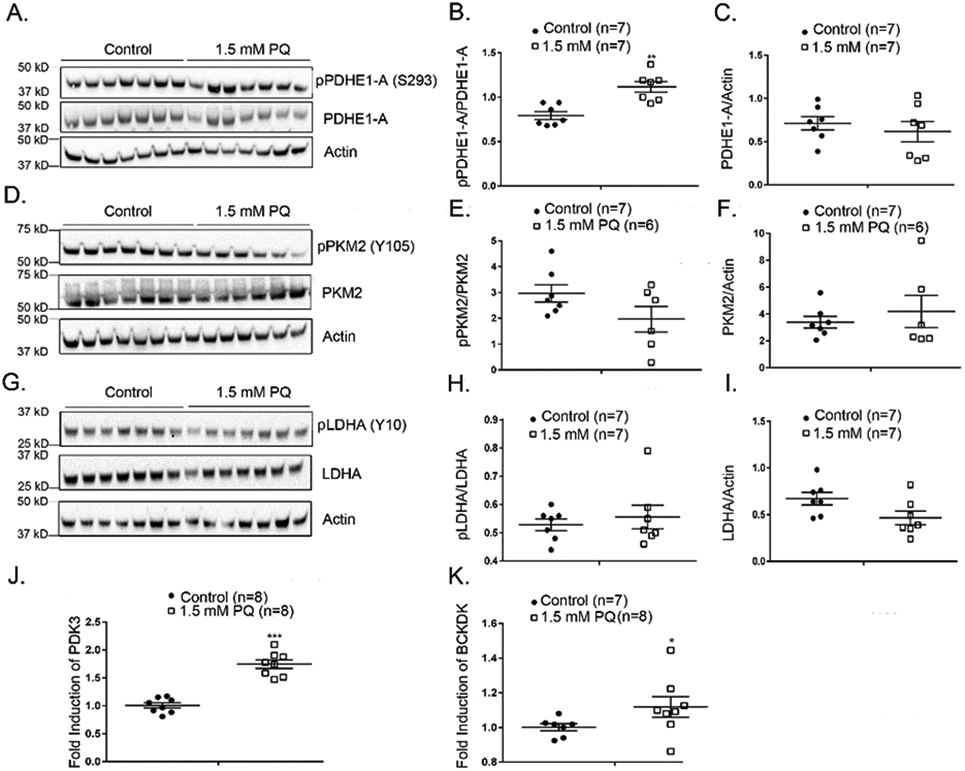
C57BL/6 mice were given no treatment (n=7) or an intraocular injection of 1.5 mM (n=7) paraquat and after 16 hours were euthanized and retinal homogenates were prepared. (A) An immunoblot of retinal homogenates from untreated or paraquat-treated mice probed with anti-pPDHE1-A (S293), anti-PDHE1-A, and anti-Actin antibodies. Densitometry of the blot in (A) demonstrated a significant increase in pPDHE1-A/PDHE1-A ratio in retinas of mice treated with 1.5 mM paraquat (B) and no significant difference in PDHE1-A/Actin ratio (C). (D) An immunoblot of retinal homogenates from untreated or paraquat-treated mice probed with anti-pPKM2 (Y105), anti-PKM2, and anti-Actin antibodies. Densitometry of the blot in (D) demonstrated no significant difference in pPKM2/PKM2 ratio in retinas of mice treated with 1.5 mM paraquat (E) and no significant difference in PKM2/Actin ratio (F). (G) Immunoblot of retinal homogenates from untreated or paraquat-treated mice probed with anti-LDHA (Y10), anti-LDHA, and anti-Actin antibody. Densitometry of the blot in (G) demonstrated no statistical difference in pLDHA/LDHA ratio (H) or LDHA/Actin ratio (I) in retinas of mice treated with 1.5 mM paraquat. Real-time RT-PCR shows increased expression of mRNA for pyruvate dehydrogenase kinase 3 (PDK 3, J) and branched chain ketoacid dehydrogenase kinase (p=0.0426, BCKDK, K) in the retina 16 hours after injection of 1.5 mM paraquat.
* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 by Mann-Whitney test.
To be certain that the phosphorylation differences were not due to injection alone, we compared phosphorylation of PDHE1-A (S293) between untreated and PBS-injected C57BL/6 retinas after 16 hours of injections (Supplemental Figure 2 A, B & C). No difference in the phosphorylation status of PDHE1-A (S293) between the two treatments proved that paraquat alone was the source of the oxidative stress and phosphorylation differences in C57BL/6 retinas. The down-regulation of these two critical enzymes, PDHE1-A and PKM2, that promote entry into the TCA cycle is the same as what was observed in retinas of rd10 mice and suggests that oxidative stress contributes to reduced flux through the TCA cycle in the setting of RP.
Dietary supplementation of rd10 mice with glucose and pyruvate improves retinal function
One strategy to try and overcome the down-regulation of glycolytic enzymes in rd10 mice and determine whether it contributes to cone dysfunction and death is to provide excess substrates in the diet. We tested the effect of excess dietary glucose which feeds into glycolysis and excess pyruvate which feeds into the TCA cycle. At P50, mean photopic b-wave amplitude was significantly higher at 2 flash intensities in eyes of mice given 42 mM glucose in drinking water compared with a corresponding control group (Figure 3A). This improvement in cone function was not accompanied by a demonstrable improvement in cone survival, because compared with control rd10 mice, those given supplemental glucose showed no difference in the amount of S opsin or M opsin in the retina (Figure 3B and C). Pyruvate (10 mM) supplementation caused a significant increase in mean photopic b-wave amplitude at the highest flash intensity (Figure 3D), and caused no difference in S opsin or M opsin levels (Figure 3E and F). Supplementation of drinking water with 42 mM glucose and 10 mM pyruvate caused a substantial, statistically significant improvement in mean photopic b-wave amplitude at all stimulus intensities (Figure 3G) and a significant increase in S opsin, but not M opsin levels in the retina (Figure 3H and I). Thus, supplemental dietary glucose and pyruvate each improve cone function in rd10 mice, and when combined also provide some improvement in cone survival. In addition to cone protection, there were significant increases in scotopic a wave and b wave amplitudes at multiple light intensities in rd10 mice treated with 10 mM pyruvate and 42 mM glucose versus control rd10 mice, indicating functional protection of rods and second order neurons (Supplemental Figure 3 A & B).
Figure 3. Dietary supplementation with glucose and pyruvate improves retinal function in rd10 mice.
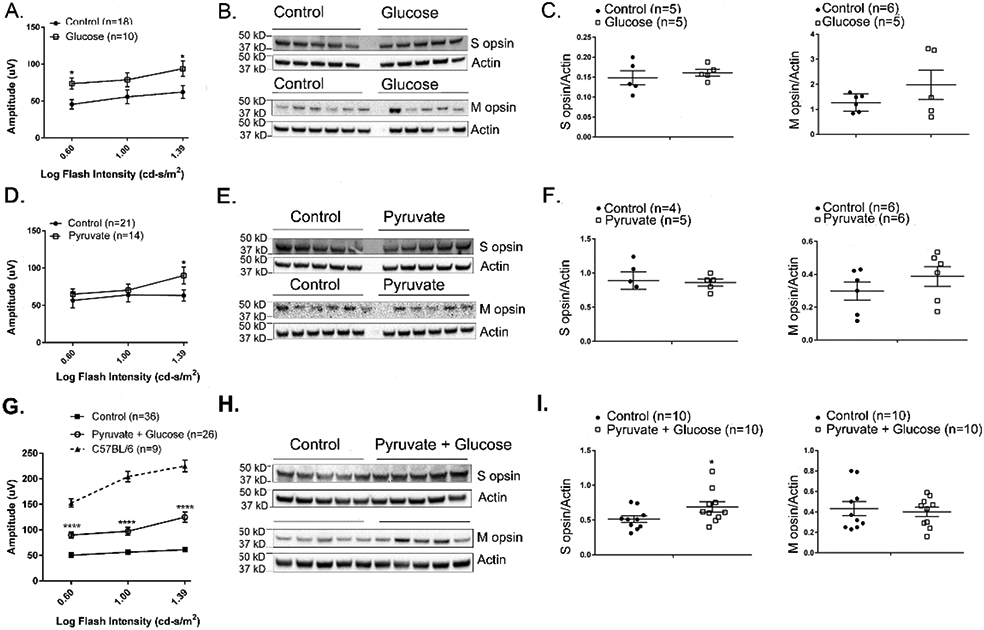
Starting at P8, rd10 mice were given normal drinking water (control) or drinking water supplemented with 42 mM glucose, 10 mM pyruvate, or both. (A) At P50, mean photopic b-wave amplitude was significantly higher at 2 flash intensities in eyes of mice treated with glucose (n=10) compared with their corresponding control group (n=18). (B) Immunoblots of retinal homogenates of P50 rd10 mice treated with glucose and their corresponding controls probed with antibodies directed against S or M cone opsin. (C) Densitometry of blots in (B) showed no difference in S opsin/Actin ratio or M opsin/Actin ratio between glucose-treated versus control rd10 mice. (D) Mean photopic b-wave amplitude was significantly higher only at the highest flash intensity in eyes of P50 rd10 mice treated with pyruvate (n=14) compared with their corresponding control group (n=21). (E) Immunoblots of retinal homogenates of P50 rd10 mice treated with pyruvate and their corresponding controls probed with antibodies directed against S or M cone opsin. (F) Densitometry of blots in (E) showed no difference in S opsin/Actin ratio or M opsin/Actin ratio between retinas from pyruvate-treated versus control rd10 mice. (G) Mean photopic b-wave amplitude was significantly higher at 3 flash intensities in eyes of P50 rd10 mice treated with 10 mM pyruvate and 42 mM glucose (n=26) compared with their corresponding control group (n=36). Mean photopic b-wave amplitude of C57BL/6 mice at P50 are also shown. (H) Immunoblots of retinal homogenates of P50 rd10 mice treated with pyruvate and glucose and their corresponding controls probed with antibodies directed against S or M cone opsin. (I) Densitometry of blots in (H) showed a significant increase in S opsin/Actin ratio (0.0399), but not M opsin/Actin ratio between retinas from rd10 mice treated with 10 mM pyruvate and 42 mM glucose versus control rd10 mice.
* p ≤ 0.05, *** p ≤ 0.001, **** p ≤ 0.0001 by Mann-Whitney test.
Supplemental glucose and pyruvate do not alter the phosphorylation status of glycolytic enzymes or increase ATP levels, but increase reducing equivalents in rd10 retina
We next sought to determine if the improvement in cone function in rd10 mice given supplemental glucose and pyruvate was due to increased flux through the TCA cycle and hence an increase in ATP levels. We first tested whether the increase in substrates reversed the blocks at control point enzymes. At P50, compared to control rd10 mice, those given supplemental glucose and pyruvate showed no significant difference in phosphorylation at key residues or total enzyme level for PDHE1-A (Figure 4A-C), PKM2 (Figure 4D-F), or LDHA (Figure 4G-I). There was also no difference in retinal ATP levels in glucose and pyruvate-supplemented P50 rd10 mice compared with control rd10 mice (Figure 4J). These data suggest that the increased levels of glucose and pyruvate did not alter the blocks in TCA cycle entry. The pentose phosphate pathway acts parallel to glycolysis to convert glucose-6-phosphate into ribulose 5-phosphate and yields two NADPH (36). Ribulose 5-phosphate is a precursor for nucleic acids and histidine and when glycolysis is down-regulated, increased flux through the pentose phosphate pathway promotes increased synthesis of nucleic acids and histidine and the accompanying increases in NADPH provides an increase in reducing equivalents. Compared with control rd10 mouse retina, the retinas of rd10 mice given supplemental glucose and pyruvate had a significant increase in NADPH (Figure 4K) as well as an increase in the reduced form of glutathione (Figure 4L). This is consistent with increased flux through the pentose phosphate pathway and suggests that the mechanism by which supplemental glucose and pyruvate improve cone function in rd10 mice is reduction of oxidative stress by increased production of glutathione, because other antioxidants have been shown to improve cone function in models of RP (5, 7, 9).
Figure 4. Supplemental glucose and pyruvate do not alter phosphorylation or levels of glucose metabolism regulatory enzymes or increase ATP levels in rd10 mouse retina, but increases reducing equivalents.
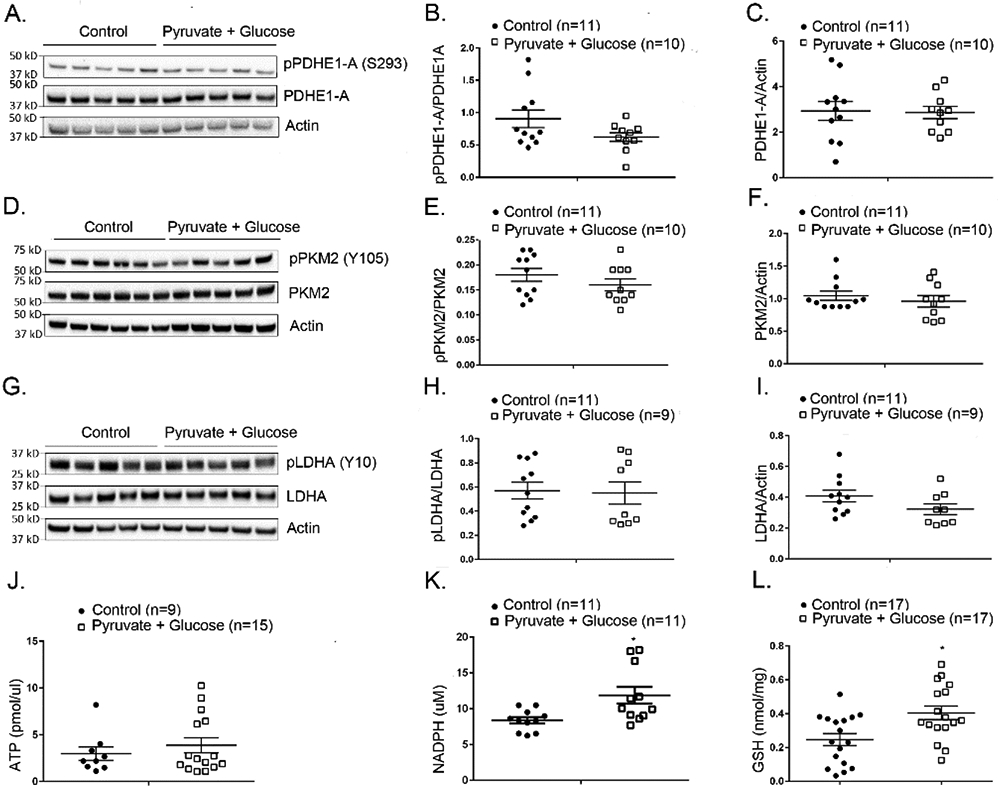
Starting at P8, rd10 mice were given normal drinking water (control) or drinking water supplemented with 42 mM glucose and 10 mM pyruvate. At P50 mice were euthanized and retinal homogenates were prepared. (A) Immunoblot of retinal homogenates from 5 rd10 mice treated with glucose + pyruvate and 5 control rd10 mice probed with anti-pPDHE1-A (S293), anti-PDHE1-A, and anti-Actin antibodies. Densitometry of the blot in (A) and a blot from a repeat experiment demonstrated no significant difference in pPDHE1-A/PDHE1-A ratio (B) or PDHE1-A/Actin ratio (C). (D) Immunoblot of retinal homogenates from 5 rd10 mice treated with glucose + pyruvate and 6 control rd10 mice probed with anti-pPKM2 (Y105), anti-PKM2, and anti-Actin antibodies. Densitometry of the blot in (D) and a blot from a repeat experiment demonstrated no significant difference in pPKM2/PKM2 ratio (E) or PKM2/Actin ratio (F) in rd10 mice treated with glucose + pyruvate versus controls. (G) Immunoblot of retinal homogenates from 5 rd10 mice treated with glucose + pyruvate and 5 control rd10 mice probed with anti-LDHA (Y10), anti-LDHA, and anti-Actin antibody. Densitometry of the blot in (G) and a blot from a repeat experiment demonstrated no significant difference in pLDHA/LDHA ratio (H) or LDHA/Actin ratio (I). (J) There were no significant differences in mean ATP level in retinas of glucose/pyruvate-treated versus control rd10 mice. (K) Measurement of NADPH in retinal homogenates demonstrated significantly more NADPH in retinas of rd10 mice treated with glucose + pyruvate versus rd10 controls (p=0.0109). (L) Measurement of reduced glutathione (GSH) in retinal homogenates showed significantly more GSH in retinas of rd10 mice treated with glucose + pyruvate versus rd10 controls (p=0.0216).
* p ≤ 0.05 by Mann-Whitney test.
Dichloroacetate improves cone function and survival in rd10 mouse retina
Since increasing glycolytic substrates did not activate the metabolic activity of the three regulated enzymes of glucose metabolism, we sought another strategy to increase metabolic flux. Dichloroacetate (DCA) is a small molecule that inactivates the PDKs which are enzymes that phosphorylate and inactivate PDHE1-A activity (37, 38). Therefore, DCA should increase TCA flux by increasing PDHE1-A activity.
Treatment of rd10 mice with DCA in drinking water starting at P14 caused a dose-dependent reduction in phosphorylation of S293 in PDHE1-A (Figure 5A and B) at P35. The effect was similar for 13 and 26 mM DCA and therefore 13 mM DCA was used for additional experiments. We also examined a longer time point and found that compared with control P65 rd10 mice, those treated with 13 mM DCA between P14 and P65 had a significant reduction in phosphorylation at S293 in PDHE1-A and no difference in total PDHE1-A (Supplemental Figure 4A-C). Compared with control rd10 mice, those given 13 mM DCA in drinking water showed a significant increase in mean photopic b-wave amplitudes at all stimulus intensities at P50 (Figure 5C) and at the 2 highest stimulus intensities at P65 (Figure 5D). There was also a significant increase in S opsin, but not M opsin, in the retinas of P65 DCA-treated rd10 mice indicating improved cone survival (Figure 5E-G). These data demonstrate that DCA significantly improves cone function and survival in rd10 mice. In addition to cone function, 13 mM DCA also increased mean scotopic a wave amplitude at P50 (Supplemental Figure 4D) and mean scotopic b wave amplitudes at P50 and P65 (Supplemental Figure 4 E & F) demonstrating protection of rod and second order neuron function.
Figure 5. Dichloroacetate (DCA) improves cone function and survival in rd10 mice.
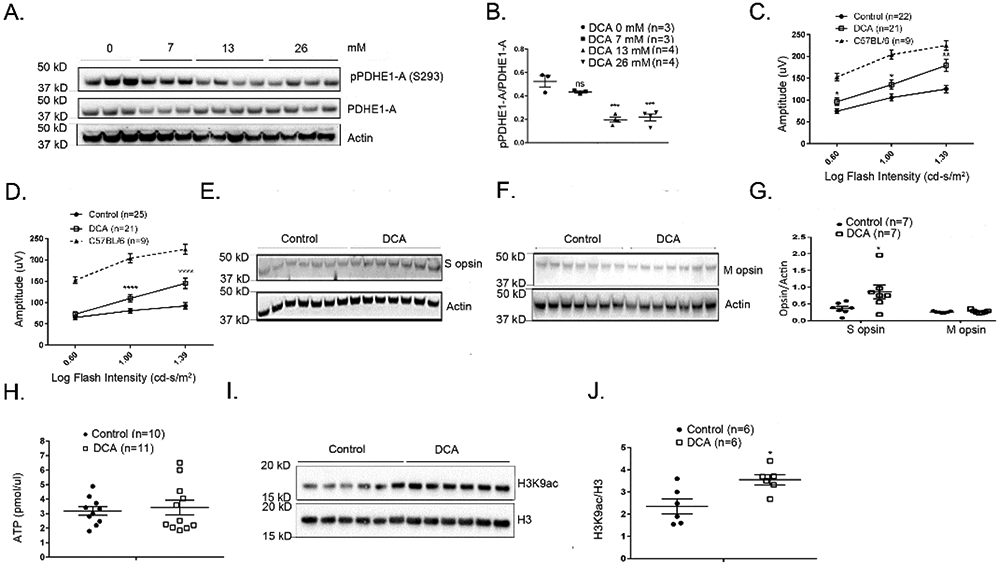
(A) Immunoblot of retinal homogenates of P35 rd10 mice treated for 3 weeks with 0, 7, 13 or 26 mM DCA probed with anti-pPDHE1-A (S293), anti-PDHE1-A, and anti-Actin antibodies. (B) Densitometry of the blot in (A) demonstrated a significant decrease in pPDHE1-A/PDHE1-A ratio in retinas of rd10 mice treated with 13 or 26 mM DCA. The concentration of DCA used for subsequent experiments was 13 mM. Starting at P14, rd10 mice were given normal drinking water (control) or drinking water supplemented with 13 mM DCA and at P50 (C) and P65 (D), mean (±SEM) photopic b-wave amplitude was significantly higher at multiple flash intensities in eyes of rd10 mice treated with DCA. Mean photopic b-wave amplitude of C57BL/6 mice at P50 are also shown. (E) Immunoblots of retinal homogenates from control or DCA-treated P65 rd10 mice probed with an antibody directed against S cone opsin or M cone opsin (F). (G) Densitometry of blots in (E) and (F) showed that compared with control rd10 mice, those treated with DCA had a significantly higher S opsin/Actin ratio (p=0.0239), but no difference in M opsin/Actin ratio. (H) Starting at P14, rd10 mice were given drinking water supplemented with 13 mM DCA or unsupplemented water and at P35 there was no difference in mean retinal ATP level in DCA-treated versus control rd10 mice. (I) Immunoblot of retinal nuclear extracts from P35 DCA-treated and control rd10 mice probed with anti-H3K9 ac (acetylated H3K9) antibody and anti-H3 antibody. (J) Densitometry of blot in (I) demonstrated a significant increase in H3K9 ac/H3 ratio in DCA-treated rd10 mice (p=0.0260).
* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 by Mann-Whitney test.
The product of DCA-induced increased activity of PDHE1-A is acetyl CoA which can be utilized in different ways. The excess acetyl CoA could enter the TCA cycle resulting in greater production of ATP, but there was no significant increase in mean retinal ATP level at P35 in DCA-treated versus control rd10 mice (Figure 5H). Acetyl CoA is also used for histone acetylation and therefore we assessed for acetylation of histone 3 (H3) at lysine 9 (K9), a common site of acetylation that alters gene expression. Immunoblots using an antibody that specifically recognizes acetylation of H3 at K9 (H3K9ac), showed a significant increase in H3K9ac in P35 DCA-treated versus control rd10 mice (Figure 5I and J).
Modulation of TP53-induced glycolysis and apoptosis regulator (TIGAR) in the retinas of rd10 and wild type mice
In a variety of tissues, H3K9ac results in increased expression of several genes including regulators of gene expression such as p53 (39, 40). TIGAR is a p53-regulated enzyme that lowers fructose-2,6-bisphosphate shifting glucose metabolism from glycolysis to the pentose phosphate pathway which reduces ROS (41, 42). TIGAR was significantly increased in retinas from DCA-treated P35 rd10 mice compared with retinas from corresponding controls (Figure 6A and B). Thus, DCA results in enhanced pentose phosphate pathway activity in rd10 retina, beyond that induced by altered phosphorylation of glycolytic enzymes. We then investigated the effect of oxidative stress on TIGAR levels in the retina and found that after intraocular injection of paraquat, there was a significant decrease in TIGAR in the retina (Figure 6 C and D). The level of TIGAR was also significantly decreased in the retinas of P35 rd10 mice compared with the retinas of P35 wild type mice (Figure 6E and F). The reduction in TIGAR in rd10 mouse retina was due to oxidative stress because it was mitigated by treatment with 7 mg/ml N-acetylcysteine (NAC) in drinking water (Figure 6G and H). Dietary supplementation of rd10 mice with 42 mM glucose and 10 mM pyruvate caused an increase in retinal TIGAR that was not statistically significant (Figure 6I and J).
Figure 6. TIGAR is down-regulated by oxidative stress and upregulated by antioxidants and DCA.
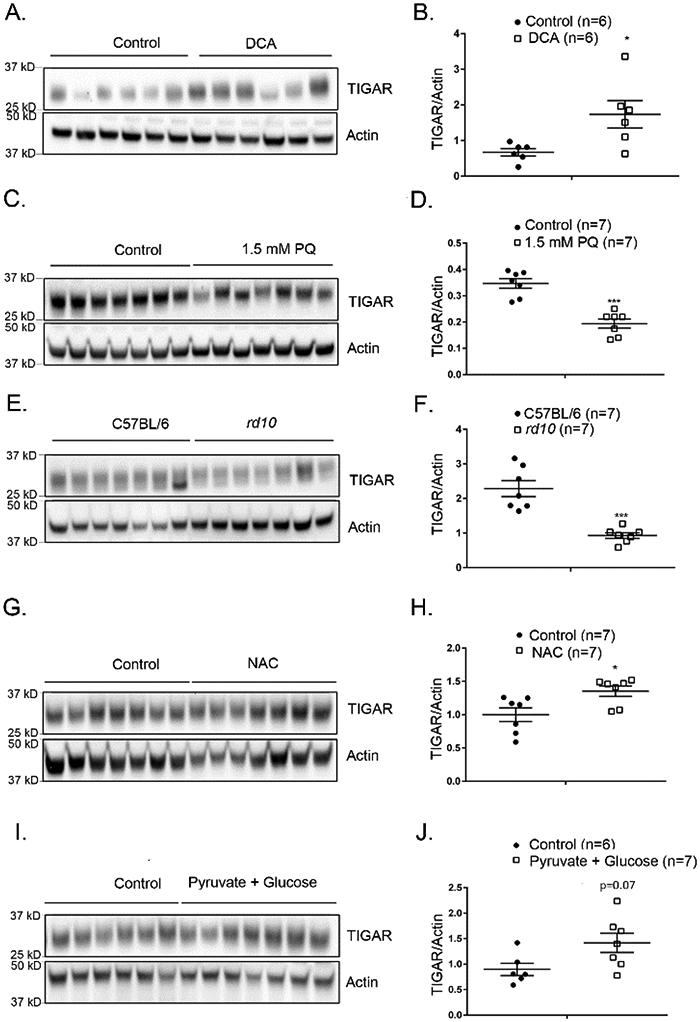
(A) Immunoblot of retinal homogenates of rd10 mice at P35 given no treatment (Control, n=6) or treated with 13 mM DCA (n=6) immunoblotted with anti-TIGAR antibody and anti-actin antibody. (B) Densitometry of blot in (A) showed a significant increase in TIGAR/Actin ratio in retinas of mice treated with DCA (p=0.0130). (C) Wild type C57BL/6 mice were given an intraocular injection of 1.5 mM paraquat (n=7) or no treatment (Control, n=7) and after 16 hours they were euthanized and retinal homogenates were immunoblotted with anti-TIGAR antibody and anti-Actin antibody. (D) Densitometry of the blot in (C) showed a significant decrease in TIGAR/actin ratio in retinas from eyes injected with 1.5 mM paraquat. (E) Retinal homogenates from C57BL/6 mice (n=7) and rd10 mice (n=7) at P35 were immunoblotted with anti-TIGAR antibody and anti-Actin antibody. (F) Densitometry of blot in (E) showed a significant decrease in TIGAR/Actin ratio in retinas of rd10 mice. (G) Starting at P8, rd10 mice were given drinking water supplemented with 7 mg/ml N-acetylcysteine (NAC, n=7) or unsupplemented water (Control, n=7) and at P50 mice were euthanized and retinal homogenates were immunoblotted with anti-TIGAR antibody and anti-Actin antibody. (H) Densitometry of the blot in (G) showed a significant increase in TIGAR/actin ratio in retinas of mice treated with NAC (p=0.0379). (I) Starting at P8, rd10 mice were given drinking water supplemented with 42 mM glucose and 10 mM pyruvate (n=7) or unsupplemented water (Control, n=7) and at P50 mice were euthanized and retinal homogenates were immunoblotted with anti-TIGAR antibody and anti-Actin antibody. (J) Densitometry of the blot in (I) showed an increase in TIGAR/Actin ratio in retinas of mice treated with 42 mM glucose and 10 mM pyruvate in drinking water.
* p ≤ 0.05, *** p ≤ 0.001 by Mann-Whitney test.
Discussion
Retinitis pigmentosa occurs from a large number of mutations that cause degeneration of rod photoreceptors and subsequently there is gradual degeneration of cone photoreceptors. It is important to elucidate the mechanism of cone photoreceptor death, because it could provide valuable therapeutic targets. Progressive oxidative damage (6-12) and abnormal glucose uptake and metabolism (13-15) have been implicated as contributors to cone cell death, but it is unknown whether they influence each other. In this study, we demonstrated increased phosphorylation of key regulatory enzymes, PDHE1-A and PKM2, which shifts glucose metabolism away from the TCA cycle in retinas of P35 rd10 mice with RP. We hypothesized that the high level of oxidative stress in RP retina contributed to the altered phosphorylation of the regulatory enzymes. To test this hypothesis, we compared the phosphorylation status of PDHE1-A and PKM2 in the retinas of normal wild type mice and wild type mice that were given an intraocular injection of paraquat which causes severe oxidative stress in all cells throughout the retina. In this oxidative stress model there was increased phosphorylation of PDHE1-A and PKM2 in the retina supporting the hypothesis that oxidative stress contributes to the altered phosphorylation of the enzymes. An alternative hypothesis for the increased phosphorylation of PDHE1-A and PKM2 in rd10 retina is that the phosphorylation status of these regulatory enzymes is normally substantially less in photoreceptors compared to inner retinal cells and the loss of photoreceptors at P35 results in an apparent increase in phosphorylation. The results in paraquat-injected eyes argues against this alternative hypothesis, because after injection there is oxidative stress in the retina without selective dropout of photoreceptors (35) and there is still increased phosphorylation of PDHE1-A and PKM2. The similar phosphorylation profiles in both models suggest that oxidative stress causes altered phosphorylation of glycolytic enzymes in retinal neurons. Since the analysis was done in whole retina, we have not proven that the changes occur in photoreceptors, but there is no reason to believe that photoreceptors would differ from other retinal neurons in this respect. Increased phosphorylation of PDHE1-A and PKM2 would be expected to reduce TCA cycle activity and this is consistent with a proteomics analysis that demonstrated reduced TCA cycle intermediates in the retinas of mice with RP compared with retinas of controls (34).
Since the TCA cycle and electron transport chain are sites of substantial generation of reactive oxygen species (ROS), which is exaggerated when there is tissue hyperoxia, the reduced flux through them may help to compensate for high oxygen levels in RP retina. However, does the reduced energy production from reduced TCA cycle flux contribute to cone dysfunction and death over time? To address this question, we sought to overcome the block in glucose metabolism by providing excess substrate. In rd10 mice, excess dietary glucose or pyruvate caused small improvements in cone cell function, and when combined there was substantial improvement in cone ERG function and cone survival. However, this improvement was not due to increased ATP production, but could be explained by an increase in NADPH and the reduced form of glutathione because other agents that neutralize ROS provide improvement in cone function and survival (7-12). It has previously been shown that supplementation with glucose, can increase glucose and insulin levels in the retina (43) and also upregulate Txnip protein in cone inner segments, thereby shunting metabolites towards glucose metabolism and cone outer segment synthesis (15). Both insulin and Txnip proteins have previously been shown to rescue cone photoreceptor cells and therefore, cone functional protection could be due to these effects as well.
We sought to overcome the block in TCA cycle entry with DCA which reversed the increase in phosphorylated PDHE1-A in rd10 retina. This also improved cone function and survival, but did not increase ATP levels. DCA increased histone acetylation and increased levels of TIGAR, a glucose regulatory enzyme that is modulated by histone acetylation through an increase in p53. This caused us to investigate the effect of RP on TIGAR levels in the retina and we found that they were reduced in rd10 mice compared with wild type mice. Retinal TIGAR levels were also reduced in wild type mice by intraocular injection of paraquat indicating that oxidative stress can cause TIGAR reduction. The excess oxidative stress in rd10 retina contributes to TIGAR reduction in that setting, because it was compensated by treatment with N-acetylcysteine. Thus, oxidative stress in the retina alters the phosphorylation of glycolytic enzymes reducing flux through the TCA cycle and increasing flux through the pentose phosphate pathway, but it also decreases TIGAR which functions to direct glucose metabolism toward the pentose phosphate pathway and reduce intracellular reactive oxygen species (41, 42). These two opposing effects may cancel each other in RP retina or at least the reduction in TIGAR may decrease the benefit of reduced TCA cycle flux, because overcoming the reduction in TIGAR by DCA results in sufficient pentose pathway activity to increase reducing equivalents and promote cone function and survival. The beneficial effect of TIGAR in RP retina is consistent with its effects in other settings. In hippocampal neurons, TIGAR protects against oxidative stress-induced damage (44). Overexpressing TIGAR in mouse brain cells with a lentiviral vector increased NADPH and reduced ischemia-induced cell death, and knockdown of TIGAR reduced NADPH and significantly increased ischemia-induced cell death (45).
While reduced TCA cycle flux may be an adaptation to oxidative stress in the retina and slow cone degeneration in RP over the short-term, it is also possible that there are long-term maladaptive consequences (46). Replenishment of TCA cycle intermediates slowed retinal degeneration in a mouse model of RP (34, 47). The mechanism of this effect has not been elucidated. There could be increased energy production which could provide benefit, but there may be other effects. Compared with the retinas of untreated RP mice, those of α-ketoglutarate-treated RP mice had higher levels of docosahexaenoic acid, which has antioxidant activity, and higher levels of aconitic acid and glutamine, which can promote production of antioxidants (48). Thus, the beneficial effects of α-ketoglutarate and other TCA cycle intermediates in RP models do not necessarily mean that down-regulation of the TCA cycle and associated ATP production through the electron transport chain ultimately contributes to cone degeneration, because these TCA intermediates feed into pathways other than the TCA cycle through which they may provide benefit. Additional studies are needed to determine if the shift away from TCA cycle flux in RP retina which provides short term benefit, has long term negative consequences from energy deficiency.
Conclusions
Oxidative stress in the retina of mice with RP causes phosphorylation and thereby inhibition of key regulatory enzymes in the glucose metabolic pathway that reduces TCA cycle flux. Supplemental glucose and pyruvate do not overcome the inhibition and increase energy production, but increase flux through the pentose phosphate pathway, increasing reducing equivalents which reduces oxidative stress and promotes cone function and survival. DCA also fails to overcome the inhibition but also increases pentose phosphate pathway activity through upregulation of TIGAR reducing oxidative stress and promoting cone function and survival. These findings demonstrate a link between oxidative stress and altered glucose metabolism in RP retina and suggest that targeting each may have therapeutic potential.
Supplementary Material
Highlights.
Oxidative stress inhibits key enzymes in glucose metabolism pathway in the retina.
Oxidative stress inhibits TIGAR production in the retina and is reversed by NAC.
Supplementation with glycolytic substrates and DCA upregulate TIGAR production.
TIGAR directs substrates into the PPP pathway
PPP pathway produces reducing equivalents, protecting retina from oxidative stress.
Acknowledgements
Funding: Supported by R01EY031041 from the National Eye Institute and grant #136302 from Fighting Blindness Canada
Abbreviations
- ATP
Adenosine triphosphate
- DCA
Dichloroacetate
- LDHA
Lactate Dehydrogenase
- NAC
N-Acetyl Cysteine
- NADPH
Reduced nicotinamide adenine dinucleotide phosphate
- PDC
Pyruvate Dehydrogenase Complex
- PDHE1-A
Pyruvate Dehydrogenase E1 Component Subunit Alpha
- PDK
Pyruvate Dehydrogenase Kinase
- PKM2
Pyruvate Kinase Muscle Isozyme
- ROS
Reactive Oxygen Species
- RP
Retinitis Pigmentosa
- RPE
Retinal Pigmented Epithelium
- TCA
Tricarboxylic acid
- TIGAR
TP53-induced Glycolysis and Apoptosis Regulator
Footnotes
Declarations of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophalmol Scand Suppl. 2002;233:1–34. [DOI] [PubMed] [Google Scholar]
- 2.Bertelsen M, Jensen H, Bregnho JF, and Rosenberg T. Prevalence of generalized retinal dystrophy in Denmark. Ophthalmic Epidemiology 2014;21(4):217–23 [DOI] [PubMed] [Google Scholar]
- 3.Yu DY, Cringle SJ, Su EN, and Yu PK. Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Invest Ophthalmol Vis Sci. 2000;41:3999–4006. [PubMed] [Google Scholar]
- 4.Yu DY, Cringle SJ, Valter K, Walsh N, Lee D, and Stone J. Photoreceptor death, trophic factor expression, retinal oxygen status, and photoreceptor function in the P23H rat. Invest Ophthalmol Vis Sci. 2004;45:2013–9. [DOI] [PubMed] [Google Scholar]
- 5.Usui S, Oveson BC, Lee SY, Jo YJ, Yoshida T, Miki A, et al. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa J Neurochem 2009;110 1028–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen J, Yan X, Dong A, Petters RM, Peng Y-W, Wong F, et al. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J Cell Physiol. 2005;203(3):457–64. [DOI] [PubMed] [Google Scholar]
- 7.Komeima K, Rogers BS, Lu L, and Campochiaro PA. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci USA. 2006;103(38):11300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komeima K, Rogers BS, and Campochiaro PA. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 2007;213(3):809–15. [DOI] [PubMed] [Google Scholar]
- 9.Lee SY, Usui S, Zafar AB, Oveson BC, Jo YJ, Lu L, et al. N-acetylcysteine promotes long term survival of cones in a model of retinitis pigmentosa. j Cell Physiol. 2011;226:1843–9 [DOI] [PubMed] [Google Scholar]
- 10.Usui S, Komeima K, Lee SY, Jo Y-J, Ueno S, Rogers BS, et al. Increased expression of catalase and superoxide dismutase 2 reduces cone cell death in retinitis pigmentosa. Molec Ther. 2009;17:778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong W, MacColl Garfinkel AE, Benowitz LI, and Cepko CL. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J Clin Invest. 2015;125(4):1433–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase 1 trial. J Clin Invest. 2019;130:1527–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ait-Ali N, Fridlich R, Millet-Puel G, Clerin E, Delalande F, Jaillard C, et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell. 2015;161:817–32. [DOI] [PubMed] [Google Scholar]
- 14.Petit L, Ma S, Cipi J, Cheng SY, Zeiger M, Hay N, et al. Aerobic glycolysis is essential for normal rod function and controls secondary cone death in retinitis pigmentosa. Cell Rep. 2018;23(9):2629–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Lee S-J, Scott PA, Lu X, Emery D, Liu Y, et al. Two-step reactivation of dormant cones in retinitis pigmentosa. Cell Rep. 2016;15(2):372–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldfeder A. The relative metabolism in vitro of analogous mammary tumors; oxygen uptake and aerobic glycolysis of mammary tumors autogenous to dba and C3H strains of mice. Cancer Res 1950;10(2):89–92. [PubMed] [Google Scholar]
- 17.Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, et al. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advnced solid tumors. Invest New Drugs. 2015;33(3):603–10. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Kondo M, and Bill A. Glucose metabolism in cat outer retina. Invest Ophthalmol Vis Sci. 1997;38:48–55. [PubMed] [Google Scholar]
- 19.Yamada K, Noguchi T, Matsuda T, Takenaka M, Monaci P, Nicosia A, et al. Identification and characterization of hepatocyte-specific regulatory regions of the rat pyruvate kinase L gene. The synergistic effect of multiple elements. J Biol Chem. 1990;265:19885–91. [PubMed] [Google Scholar]
- 20.Yamada K, and Noguchi T. Regulation of pyruvate kinase M gene expression. Biochem Biophys Res Commun. 1999;256:257–62. [DOI] [PubMed] [Google Scholar]
- 21.Lindsay KJ, Du J, Sloat SR, Contreras L, Linton JD, Turner SJ, et al. Pryruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Proc Natl Acad Sci USA. 2014;111 (43):15579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajala RVS, Rajala A, Kooker C, Wang Y, and Anderson RE. The Warburg effect mediator pyruvate kinase M2 expression and regulation in the retina. Sci Rep. 2016;6:37727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isofom of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–3. [DOI] [PubMed] [Google Scholar]
- 24.Hitosugi T, Kang S, Vander Heiden MG, Chung T-W, Elf S, Lythgoe K, et al. Tyrosine phosphorylation ihibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2(97):ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linn TC, Pettit FH, and Reed LJ. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc Natl Acad Sci USA. 1969;62(1):234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiang PK, and Sacktor B. Control of pyruvate dehydrogenase activity in intact cardiac mitochondria. Regulation of the inactivation and activation of the dehyrogenase. J Biol Chem. 1975;250(9):3399–408. [PubMed] [Google Scholar]
- 27.Pettit FH, Pelley JW, and Reed LJ. Regulation of pyruvate dehydrogenate kinase and phosphatase by acetyl-CoA/CoA and NADH/NAD ratios. Biochem Biophys Res Commun. 1975;65(2):575–82. [DOI] [PubMed] [Google Scholar]
- 28.Bersin RM, and Stacpoole PW. Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am Heart J. 1997;134(5):841–55. [DOI] [PubMed] [Google Scholar]
- 29.Deuse T, Hua X, Wang D, Maeqdefessel LH,J, Scheja L, Bolanos JP, et al. Dichloroacetate prevents restenosis in preclinical animal models of vessel injury. Nature. 2014;509(7502):641–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andreassen OA, Ferrante RJ, Huang HM, Dedeoglu A, Park L, Ferrante KL, et al. Dicholoroacetate exerts therapeutic effects in transgenic mouse models of Huntington's disease. Ann Neurol. 2001;50(1):112–7. [DOI] [PubMed] [Google Scholar]
- 31.Casson RJ, Wood JPM, Han G, Kittipassorn T, Peet DJ, and Chidlow G. M-type pyruvate kinase isoforms and lactacte dehydrogenase A in the mammalian retina: metabolic implications. Invest Ophthalmol Vis Sci. 2016;57(1):66–80. [DOI] [PubMed] [Google Scholar]
- 32.Sugden MC, and Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003;284(5):e855–e62. [DOI] [PubMed] [Google Scholar]
- 33.Fan J, Hitosugi T, Chung TW, Xie J, Ge Q, Gu T-L, et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD+ redox homeostasis in cancer cells. Mol Cell Biol. 2011;31(24):4938–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wert KJ, Velez G, Kanchustambham VL, Shankar V, Evans LP, Sengillo JD, et al. Matabolite therapy guided by liquid biopsy proteomics delays retinal neurodegeneration. EBioMedicine. 2020;52:102636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cingolani C, Rogers B, Lu L, Kachi S, Shen J, and Campochiaro PA. Retinal degeneration from oxidative damage. Free Radic Biol Med. 2006;40:660–9. [DOI] [PubMed] [Google Scholar]
- 36.Stincone A, Prigione A, Cramer T, Wamelink MMC, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev. 2015;90:927–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McAllister A, Allison SP, and Randle PJ. Effets of dichloroacetate on the metaboism of glucose, pyruvate, acetate, 3-hydroxybutyrate and palmitate in rat diaphragm and heart muscle in vitro and on extraction of glucose, lactate, pyruvate and free fatty acids by dog heart in vivo. Biochem J. 1973;134(1067–1081). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitehouse S, Cooper RH, and Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974; 141 (761–774). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 2016;17(2):366–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chatterjee B, Ghosh K, and Kanade SR. Resveratrol modulates epigenetic regulators of promoter histone methylation and acetylation that restores BRCA1, p53, p21CIP1 in human breast cancer cell lines. Biofactors. 2019;45(5):818–29. [DOI] [PubMed] [Google Scholar]
- 41.Bensaad K, Tsuruta A, Selak MA, Calvo Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006;126(1):107–20. [DOI] [PubMed] [Google Scholar]
- 42.Bensaad K, Cheung EC, and Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28(19):3015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanguesa G, Shaligram S, Akther F, Roglans N, Laguna JC, Rahimian R, et al. Type of supplemented simple sugar, not merely calorie intake, determines adverse effects on metabolism and aortic function in female rats. Am J Physiol Heart Circ Physiol. 2017;312(2):H289–H304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lei B, Liu J, Yao Z, Xiao Y, Zhang X, Zhang Y, et al. NF-κB-induced upregulation of miR-146a-5p promoted hippocampal neuronal oxidative stress and pyroptosis via TIGAR in a model of alzheimer's disease. Front Cell Neurosci. 2021;15:653881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li M, Sun M, Cao L, Gu JH, Ge J, Chen J, et al. A TIGAR-regulated metabolic pathway is critical for protection of brain ischemia. J Neurosci. 2014;34(22):7458–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, and Colman RF. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet. 2008;40(10):1230–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowe AA, Patel PD, Gordillo R, and Wert KJ. Repenishment of TCA cycle intermediates provides photoreceptor resilience against neurodegeneration during progression of retinitis pigmentosa. JCI insight. 2021;Epub ahead of print, 10.1172/jci.insight.150898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu S, He L, and Yao K. The antioxidative function of alpha-ketoglutarate and its applications. Biomed Res Int. 2018;2018:3408467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.