

ABSTRACT
Background
Fetal-neonatal iron deficiency causes learning/memory deficits that persist after iron repletion. Simplified hippocampal neuron dendrite structure is a key mechanism underlying these long-term impairments. Early life choline supplementation, with postnatal iron repletion, improves learning/memory performance in formerly iron-deficient (ID) rats.
Objectives
To understand how choline improves iron deficiency–induced hippocampal dysfunction, we hypothesized that direct choline supplementation of ID hippocampal neurons may restore cellular energy production and dendrite structure.
Methods
Embryonic mouse hippocampal neuron cultures were made ID with 9 μM deferoxamine beginning at 3 d in vitro (DIV). At 11 DIV, iron repletion (i.e., deferoxamine removal) was performed on a subset of ID cultures. These neuron cultures and iron-sufficient (IS) control cultures were treated with 30 μM choline (or vehicle) between 11 and 18 DIV. At 18 DIV, the independent and combined effects of iron and choline treatments (2-factor ANOVA) on neuronal dendrite numbers, lengths, and overall complexity and mitochondrial respiration and glycolysis were analyzed.
Results
Choline treatment of ID neurons (ID + Cho) significantly increased overall dendrite complexity (150, 160, 180, and 210 μm from the soma) compared with untreated ID neurons to a level of complexity that was no longer significantly different from IS neurons. The average and total length of primary dendrites in ID + Cho neurons were significantly increased by ∼15% compared with ID neurons, indicating choline stimulation of dendrite growth. Measures of mitochondrial respiration, glycolysis, and ATP production rates were not significantly altered in ID + Cho neurons compared with ID neurons, remaining significantly reduced compared with IS neurons. Iron repletion significantly improved mitochondrial respiration, ATP production rates, overall dendrite complexity (100–180 μm from the soma), and dendrite and branch lengths compared with untreated ID neurons.
Conclusions
Because choline partially restores dendrite structure in ID neurons without iron repletion, it may have therapeutic potential when iron treatment is not possible or advisable. Choline's mechanism in ID neurons requires further investigation.
Keywords: iron deficiency, choline, neuron development, dendrites, mitochondria, oxidative phosphorylation, glycolysis, energy metabolism, ATP, brain development
Introduction
Iron deficiency with or without anemia is the most common micronutrient deficiency (∼2 billion people worldwide) and remains one of the top 5 leading causes of chronic disability burden (1–3). Iron deficiency disproportionately affects low- and middle-income populations, with prevalence rates reaching 50–80% of pregnant women and children in low-resource countries (1, 4). However, early life (conception to 3 y of age) iron deficiency remains a problem in high-resource countries as well, affecting an estimated 15–42% of pregnant women/children in the United States (5–7). This is important clinically because fetuses, infants, and toddlers are most susceptible to brain iron deficiency and subsequent neurobehavioral dysfunction due to the high iron needs during this period of rapid brain growth and development. Preclinical models and human epidemiology studies demonstrate incomplete neurobehavioral recovery from early life iron deficiency despite iron treatment (8–10). Documented long-term effects include poorer cognition and increased risk of depression, anxiety, schizophrenia, and autism in adolescence and adulthood. Most children with iron deficiency are diagnosed and treated late in the course (11). In addition, many young children do not get adequate iron in their diet, and iron may be contraindicated in populations at risk for common infectious diseases such as malaria (1, 12). Therefore, alternative therapeutic strategies, beyond simple iron repletion in response to anemia screening, that work by addressing the fundamental neurobiology disrupted by early life iron deficiency are needed.
Using transgenic mouse models, we have shown that the learning and memory deficits that persist after recovery from early life iron deficiency are caused by insufficient iron availability in hippocampal pyramidal neurons and not systemic effects (e.g., anemia) (13–15). Dendritic arbor complexity, one of the main determinants of proper neural circuit formation, remains blunted in these hippocampal neurons after iron repletion that occurs late in the energy-demanding critical period of dendrite formation (similar to clinical timing of iron treatment) (15). Moreover, the simplified arbor complexity appears to be due to reduced cytochrome-dependent mitochondrial energy production (16, 17). This suggests that reduced complexity of the dendritic arbor may be the iron-dependent driving force underlying the persistent learning and memory impairments and could be a target for alternative or adjunct clinical interventions.
Choline is a metabolic precursor to pathways (i.e., acetylcholine, methyl donor, and lipid synthesis) that are important for energy-dependent neuron structural development (18–20). There is an intimate relation between choline metabolism and mitochondrial function: acetylcholine synthesis requires the mitochondrial metabolite acetyl-CoA, choline conversion to the methyl donor betaine occurs in the mitochondria, and mitochondrial function depends on proper membrane construction with choline-dependent lipids (21). Choline deficiency in adult rats impairs brain mitochondrial function, including reduced oxygen consumption rates, altered mitochondrial membrane lipid composition, and increased mitochondrial production of reactive oxygen species (22).
Choline is critical for proper neuronal development with choline deprivation impairing differentiation, proliferation, migration, and survival in neural precursor cells and blunting neurite outgrowth in differentiated postmitotic neurons (23–25). Fetal or neonatal choline supplementation improves hippocampal gene expression, structure, function, and behavioral outcomes in normal rodents or in multiple rodent models of gestational neurodevelopmental pathology (26–33). We previously showed that prenatal choline supplementation in pregnant iron-deficient anemic (IDA) rats, followed by postnatal iron repletion, recovers offspring hippocampal gene expression and learning/memory behaviors without altering systemic anemia effects (26, 27). These findings suggest that choline is not acting on peripheral iron metabolism but rather on the brain (or specifically the neuron) during early life iron deficiency.
The goal of this study was to determine whether direct choline supplementation of iron-deficient (ID) hippocampal neurons restores energy production and dendrite complexity and whether choline supplementation requires concomitant iron repletion.
Methods
Animals
Timed-pregnant, wild-type CD1 mice (Charles River Laboratories) arrived on embryonic day (E) 13 (E13) and were housed onsite for 3 d until embryo harvest at E16. During this time, they were given free access to standard irradiated rodent chow (Teklad #2918; Envigo) and drinking water at constant temperature and humidity on a 12-h light/dark cycle. All animal procedures were conducted in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals. The local Institutional Animal Care and Use Committee approved these procedures.
Neuron culture experimental design
The experimental design and treatment group designations are outlined in Figure 1. Primary hippocampal cells were collected and pooled from multiple mixed-sex E16 CD1 mouse embryos and plated for gene expression, immunocytochemistry, cellular bioenergetics, or cellular ATP analyses as previously described (16, 28). Prior to use, the neuronal growth medium was “conditioned” on separate postnatal mixed glia cultures as previously described (16). At 3 d in vitro (DIV), all cultures were treated with 67.5 μM 5-fluoro-2′-deoxyuridine (F0503; Sigma-Aldrich)/136 μM uridine (U6381; Sigma-Aldrich) (5-FU), an antimitotic drug used to inhibit glia proliferation as previously described (16). Neuronal iron deficiency was created beginning at 3 DIV by treating cultures with 9 μM deferoxamine (DFO; 14595; Cayman Chemical) as described (16). Iron-sufficient cultures (0 μM DFO) were treated with the same volume of vehicle (sterile, ultrapure water in neuronal growth medium). Concentrations listed are the final concentrations in the culture medium. This creates a nearly pure neuronal culture with a degree of neuronal iron deficiency that is similar to rodent iron deficiency models and is physiologically relevant to human iron-deficient neonates as described (16). At 7 DIV, half of the medium was removed and replaced with fresh glia-conditioned neuronal growth medium containing 5-FU and 0 μM or 9 μM DFO for iron-sufficient and iron-deficient cultures, respectively. In contrast to our previous work with this model, the current cultures did not receive a medium change a week later at 14 DIV. This difference was due to a medium change at 11 DIV associated with the iron repletion procedure described below.
FIGURE 1.

Experimental design and group designations for iron repletion and choline supplementation of primary mouse hippocampal neuron culture model of iron deficiency. The general in vitro timeline of key neurodevelopmental processes is shown at the bottom. Numbers indicate the number of days of in vitro culture after harvesting and plating cells from the embryonic day 16 (E16) wild-type, mouse hippocampus. Each of the 6 treatment groups are indicated by a bar between 3 DIV and 18 DIV. Gray bars indicate the time period of DFO treatment and thus culture in iron-deficient media. White bars indicate the days without DFO treatment and thus culture in iron-sufficient media. The vehicle is sterile distilled, deionized water (same volume as DFO). Iron repletion between 11 DIV and 18 DIV was accomplished by removal of DFO and subsequent culture in iron-sufficient media. Black arrows between 11 DIV and 18 DIV indicate the period of choline treatment. The group designations for each of the 6 treatment groups are shown on the right. Cho, choline; DFO, deferoxamine; DIV, days in vitro; FID, formerly iron deficient; ID, iron deficient; IS, iron sufficient.
Iron repletion
At 11 DIV, for the formerly iron-deficient (FID) and FID + choline (Cho) groups, iron repletion was accomplished by removing all culture medium from a subset of DFO-treated cultures and immediately replacing with a half-volume of medium that was removed from iron-sufficient (IS) cultures (Figure 1). Factors secreted into the medium by growing neurons are required for neuron survival (29), necessitating the use of donor culture medium to maintain neuron health while eliminating the DFO. A half-volume of fresh glia-conditioned medium containing 5-FU and 0 μM DFO was added to bring the medium back to the full volume. The cultures for the IS, IS + Cho, ID, and ID + Cho groups were subjected to a “mock repletion” in which all culture medium was removed and immediately replaced with a half-volume of the same medium and then a half-volume of fresh glia-conditioned medium with 5-FU and 0 μM DFO (IS groups) or 9 μM DFO (ID groups).
Choline supplementation
The Neurobasal/B27 medium used in these studies contains 30 μM choline chloride to support the choline needs of developing neurons (30). At 11 DIV, IS + Cho, ID + Cho, and FID + Cho cultures were supplemented with an additional 30 μM choline chloride (Millipore-Sigma). Choline supplementation was repeated every other day until 18 DIV. IS, ID, and FID cultures that did not receive choline supplementation were treated with the same volume of vehicle (sterile, ultrapure water in neuronal growth medium).
mRNA expression analysis
Total RNA was extracted from hippocampal cultures at 18 DIV using a NucleoSpin RNA XS kit (Macherey-Nagel) as described (28). Alternatively, cells were lysed with TRIzol Reagent (Thermo-Fisher Scientific), and the manufacturer's protocol for phenol-chloroform extraction of the aqueous layer was followed. Total RNA was then recovered from the aqueous layer using the Monarch RNA Cleanup kit (New England Biolabs). RNA purity/concentration analysis, cDNA synthesis, qPCR, and calculation of relative mRNA levels were carried out as previously described (28) with the following modifications. A QuantStudio qPCR machine was used to perform qPCR on cDNA equivalent to 5 ng total RNA input with a Luna Universal Probe qPCR Master Mix (New England Biolabs) and a 10-μL final volume. TaqMan probes for transferrin receptor 1 (Tfrc) and the reference gene TATA box binding protein were previously described (28).
Plasmids and neuronal transfection
To enable visualization of individual neurons, a plasmid expressing an enchanced green fluorescent protein (eGFP)-tagged intrabody specific for PSD95 [pCAG_PSD95.FingR-eGFP-CCR5TC was a gift from Don Arnold (31); Addgene plasmid 46295] was nucleofected into hippocampal cells as previously described (16, 28). Nucleofected cells were then mixed 1:1 with nonelectroporated cells before plating.
Dendrite morphology analysis
At 18 DIV, hippocampal cultures on 12-mm coverslips were fixed and microtubule-associated protein 2 (MAP2) immunocytochemistry was performed as described (16). Aminomethylcoumarin acetate (AMCA)-conjugated donkey anti-mouse IgG (715-155-151, RRID:AB_2340807; Jackson Immunoresearch) secondary antibody was used at 1:100. Two-channel images of individual green fluorescent protein (GFP)–positive neurons were collected with a ZEISS Axiovert 200M microscope with 20× Plan-Apo objective (NA = 0.75) and MicroManager software. The GFP and MAP2 images were merged using Adobe Photoshop's “Apply Image” function with the “Darken” blending algorithm. The dendritic arbors of individual neurons were traced using FIJI and the semiautomated Simple Neurite Tracer plugin. Custom batch image-processing FIJI macros and Photoshop actions are available upon request.
Neuronal bioenergetics
In total, 8500 hippocampal cells were plated in each well of a poly-D-lysine (PDL)-laminin–coated Seahorse XFe24 cell culture microplate (Agilent Technologies). Cultures were prepared, treated, and grown as described above, except a lower DFO dose (4 μM) was used to create neuronal iron deficiency as we have described (16). At 18 DIV, the neuronal growth medium was exchanged for XF assay medium (XF DMEM, pH 7.4 plus 25 mM XF glucose, 1 mM XF sodium pyruvate, 2 mM XF glutamine) and incubated at 37°C (non-CO2 incubator) for ∼30 min prior to bioenergetic analyses. The XF assay medium for ID and ID + Cho groups contained 4 μM DFO. Similarly, the assay medium was supplemented with an additional 30 μM choline for the “+ Cho” groups. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were simultaneously measured using a Seahorse XFe24 Extracellular Flux Analyzer as described (16, 28). ECAR measurements were converted to proton efflux rate (PER) for all glycolytic calculations as described and suggested by the manufacturer. The Seahorse XF Mito Stress Test was performed, and mitochondrial respiration parameters were calculated as previously described (28) with slight modifications as follows: 1 μM oligomycin (ATP synthase inhibitor), 3 μM carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) (uncouples oxygen consumption from ATP production), and 1 μM antimycin A/rotenone (electron transport chain complex III and I inhibitors, respectively) were used. To assess the relative contribution of oxidative phosphorylation and glycolysis to ATP production, the Seahorse XF ATP Rate Assay was performed according to the manufacturer's instructions using 1.5 μM oligomycin and 0.5 μM antimycin A/rotenone. Mitochondrial respiration parameters and mitochondrial, glycolytic, and total ATP production rates were calculated from both ATP Rate Assay and Mito Stress Test data.
Cell density normalization
To normalize the neuronal bioenergetics data to cell density, cultures were treated with 1 μM Hoechst 33342 and imaged using fluorescent and transmitted light microscopy. Cell density images were captured using a ZEISS Axio Observer Z.1 (Photometrics QuantEM 512SC camera) with a 4× Achroplan objective (NA = 0.1), brightfield transmitted light and fluorescent filter set 49 (excitation: 365 nm, beamsplitter: 395 nm, emission: 445/50 nm) or a ZEISS Celldiscoverer 7 (ZEISS Axiocam 506 mono camera) with a 5× Plan-Apochromat objective (NA = 0.35), LED illumination for oblique contrast transmitted light (725 nm) and fluorescence (385 nm), and fluorescent quad bandpass filter set 90HE (beamsplitter: RQFT 405 + 493 + 575 + 653 nm, emission: QBP 425/30 + 514/30 + 592/25 + 709/100 nm). Imaging on the ZEISS Axio Observer Z.1 was supported by the resources and staff at the University of Minnesota University Imaging Centers (RRID: SCR_020997).
Statistical design and analyses
During long-term primary neuron culture, a unique, independent environment is created for each individual neuron in each dish/well by the inherent differences in cell density/distribution, neuronal subtype distribution, medium volume, and excitatory/inhibitory input strengths (29). Thus, the experimental unit is defined as the individual neuron for dendrite morphology analysis. Likewise, each dish/well has an independent environment and is defined as the experimental unit for statistical analysis of biochemical outcome measures. Sample sizes are shown in each figure legend. Data were pooled from 2 (qPCR), 3 (dendrite morphology), or 6 (cellular bioenergetics) unique culture preparations to further account for the variability created by the factors described above.
Statistical outliers were identified using the robust regression followed by outlier identification (ROUT) test with Q = 1%. For Sholl analysis, a 2-factor (treatment group and distance from soma) ANOVA with repeated measures (distance from soma for each individual neuron) and the Geisser–Greenhouse correction was used to assess the effect of treatments on overall dendrite complexity throughout the dendritic arbor. Tukey post hoc multiple comparison test was used to determine statistical differences between experimental groups at each individual distance from the soma. Supplemental Table 1 shows the complete results of the 2-factor ANOVA and multiple comparisons test for the Sholl analysis. For all other outcome measures, two-factor (iron status and choline treatment status) ANOVA with Tukey post hoc multiple comparison test was used. For these non-repeated-measures analyses, when variances of residuals were unequal across treatment groups, as determined by the Spearman test for heteroscedasticity, the data were ln-transformed prior to ANOVA. X–Y data are presented as mean ± SEM for easier visualization of group comparisons. All other data are presented as mean ± SD. Statistical analyses and data graphing were carried out using Prism version 9.1.1 (GraphPad Software) software.
Results
Neuronal iron status
At 18 DIV, DFO-induced neuronal iron deficiency caused a 2-fold increase in the mRNA levels of Tfrc (Figure 2, ID group), indicating functional neuronal iron deficiency (16, 32). Choline treatment of iron-deficient neurons (ID + Cho) did not significantly alter Tfrc mRNA levels compared to untreated ID neurons, indicating persistent iron deficiency. Iron repletion, with or without choline supplementation, restored Tfrc mRNA back to levels that were statistically lower than in ID neurons and no longer statistically different from IS neuron levels.
FIGURE 2.
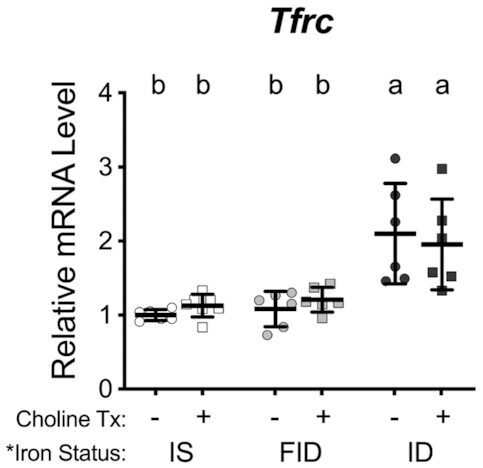
Iron repletion restores transferrin receptor 1 (Tfrc) mRNA levels of iron-deficient neurons. Hippocampal neurons cultured from embryonic day 16 (E16) mice were treated with 0 μM or 9 μM deferoxamine (DFO) beginning at 3 d in vitro (DIV). At 11 DIV, DFO was removed to begin iron repletion and 30 μM choline chloride was added to begin choline supplementation. At 18 DIV, cells were collected, total RNA was extracted, and cDNA was synthesized. qPCR was performed for Tfrc as an index of neuronal iron status. Relative mRNA levels are calculated relative to an internal control cDNA sample and a reference gene (i.e., TATA box binding protein). The data from 6–7 independent culture vessels are presented as mean ± SD. Groups that do not share a common letter are statistically different by 2-factor ANOVA and Tukey test. An asterisk (*), if present, indicates a statistically significant effect of iron or choline status. A delta symbol (∂), if present, indicates a statistically significant interaction effect. FID, formerly iron deficient; ID, iron deficient; IS, iron sufficient.
Neuronal dendrite complexity
Consistent with previous findings (16), chronic neuronal iron deficiency (ID group) from 3 DIV through 18 DIV reduced the overall complexity of the dendritic arbor (Figure 3) as quantified by Sholl analysis (at 70 μm, 120 μm, and 150–250 μm from the soma; Figure 3B and Supplemental Table 1) compared with IS neurons. FID neurons had significantly increased dendrite complexity (between 100 and 180 μm from the soma) compared with ID neurons that were statistically indistinguishable from IS neurons (Figure 3B). Choline supplementation of ID neurons (ID + Cho) significantly increased dendrite complexity at 150 μm, 160 μm, 180 μm, and 210 μm from the soma compared with untreated ID neurons (Figure 3B), despite ID + Cho neurons remaining iron deficient (Figure 1). ID + Cho neurons had a statistical trend (0.05 < P < 0.1) toward increased dendrite complexity at 120 μm, 170 μm, 190 μm, and 200 μm from the soma (Supplemental Table 1). ID + Cho dendrite complexity was not statistically different from IS neuron dendrite complexity at any distance from the soma (Supplemental Table 1). There was an overall group effect of iron status (and interaction with choline treatment) on total dendrite lengths (Figure 3C), with a significant reduction in ID neurons compared with IS neurons. Total dendrite length was significantly increased in FID neurons to a level that was no longer statistically lower than IS neurons. ID + Cho neurons exhibited total dendrite lengths that were statistically indistinguishable from IS neurons but were also not statistically different from ID neurons, suggesting partial recovery (Figure 3C).
FIGURE 3.

Iron repletion or choline supplementation restores dendrite complexity of iron-deficient neurons. Hippocampal neurons cultured from embryonic day 16 (E16) mice were nucleofected with an eGFP-tagged PSD95 intrabody at plating. Iron deficiency was induced beginning at 3 d in vitro (DIV) by treating with 9 μM deferoxamine (DFO). At 11 DIV, DFO was removed to begin iron repletion and 30 μM choline chloride was added to begin choline supplementation. (A) Representative 18 DIV images of neurons expressing the PSD95 intrabody (top row) after immunocytochemistry for MAP2 (second row). Merged images (third row) were used to trace the dendritic arbors (bottom row) for green fluorescent protein–positive neurons. (B) Sholl analysis (mean ± SEM; n = 61–100 neurons). Note: X–Y data variances are shown as SEMs rather than SDs for easier visualization. For a given distance from the soma, colored asterisks (*) indicate a statistical difference compared with the ID group by repeated-measures 2-factor ANOVA and Tukey test (P < 0.05). The full statistical comparisons are shown in Supplemental Table 1. (C) Average total length of the entire dendritic arbor (mean ± SD, n = 61–100 neurons). Groups that do not share a common letter are statistically different by 2-factor ANOVA and Tukey test (P < 0.05). An asterisk (*), if present, indicates a statistically significant effect of iron or choline status. A delta symbol (∂), if present, indicates a statistically significant interaction effect. Cho, choline; eGFP, enchanced green fluorescent protein; FID, formerly iron deficient; ID, iron deficient; IS, iron sufficient.
To determine whether choline supplementation specifically stimulated dendrite outgrowth or de novo dendrite branching, the number and length of primary dendrites and branches were quantified (Figures 4 and 5). There was an overall group effect of iron status on all measures except the average length of primary dendrites and the number of branches. There was a group effect of choline treatment only on the average and total primary dendrite length (Figure 3B and C), both having an interaction with iron status. Because all treatments were after primary dendritogenesis was complete (i.e., 3 DIV), iron deficiency and its treatments had little effect on the number of primary dendrites (Figure 4A). ID + Cho neurons had significantly increased average and total primary dendrite length compared with ID neurons such that these measures were no longer significantly different from IS neurons (Figure 4B and C). As a surrogate measure for in vivo hippocampal neuron apical dendrite length, which is reduced in the developing brain of IDA rats (33), the average length of the longest dendrite for each neuron was measured. ID neurons had significantly reduced average lengths for each neuron's longest dendrite (Figure 4D). FID and ID + Cho treatments both partially increased the average length of the longest dendrite such that it was not statistically different from either ID or IS neurons (Figure 4D). The number and length of dendrite branches were completely restored in FID neurons to a level that was significantly increased compared with ID neurons and no longer significantly different from IS neurons (Figure 5). Choline supplementation (ID + Cho) partially rescued the branching deficits to levels that remained statistically similar to ID neurons but were also statistically indistinguishable from IS neurons.
FIGURE 4.
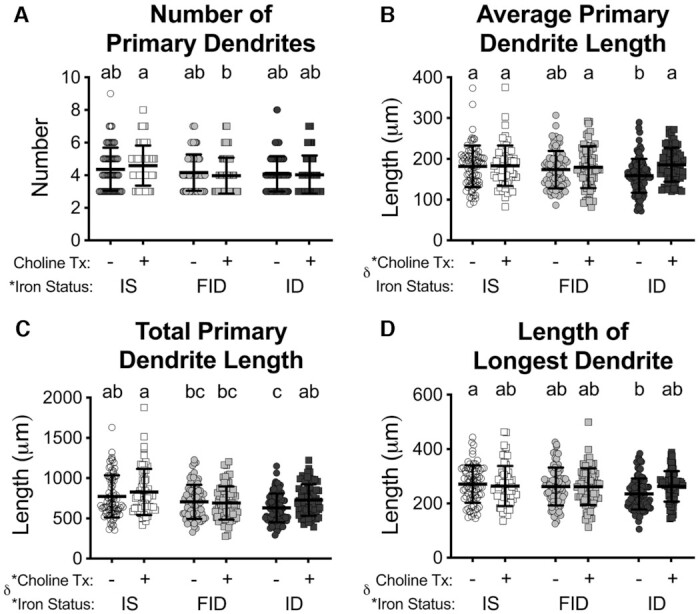
Iron repletion or choline supplementation restores primary dendrite length of iron-deficient neurons. From the tracings of 18 d in vitro neurons (see Figure 1), the average (A) number and (B) length of primary dendrites were measured and the (C) total primary dendrite length was calculated. (D) As an in vitro surrogate for the apical dendrite, the average length of the longest dendrite was calculated. Data are mean ± SD, n = 61–100 neurons. Groups that do not share a common letter are statistically different by 2-factor ANOVA and Tukey test (P < 0.05). An asterisk (*), if present, indicates a statistically significant effect of iron or choline status. A delta symbol (∂), if present, indicates a statistically significant interaction effect. FID, formerly iron deficient; ID, iron deficient; IS, iron sufficient.
FIGURE 5.
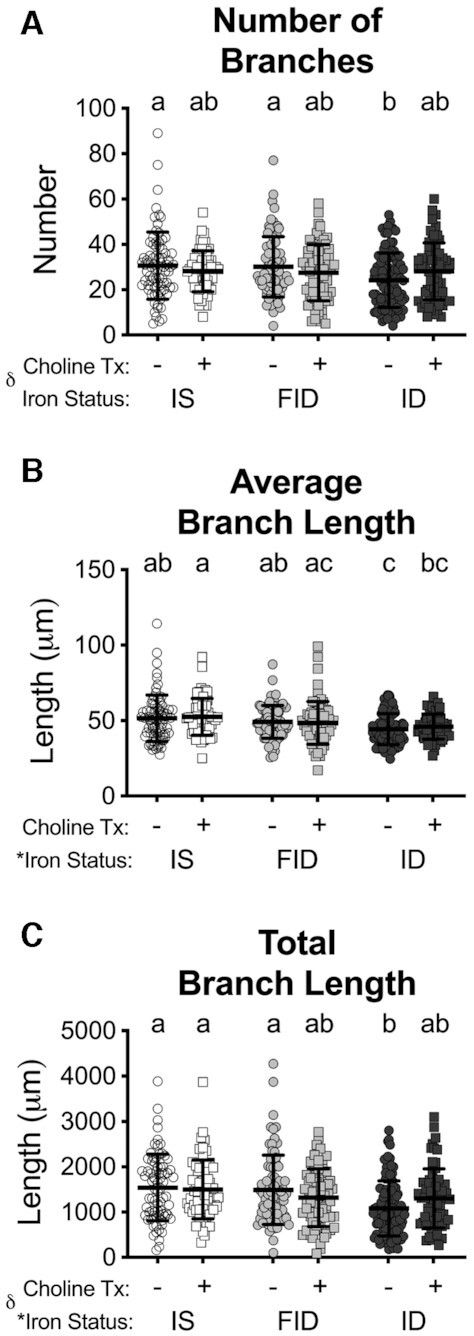
Iron repletion or choline supplementation restores dendrite branching of iron-deficient neurons. From the tracings of 18 d in vitro neurons (see Figure 1), the average (A) number and (B) length of dendrite branches were measured and the (C) total branch length was calculated. Data are mean ± SD, n = 61–100 neurons. Groups that do not share a common letter are statistically different by 2-factor ANOVA and Tukey test (P < 0.05). An asterisk (*), if present, indicates a statistically significant effect of iron or choline status. A delta symbol (∂), if present, indicates a statistically significant interaction effect. FID, formerly iron deficient; ID, iron deficient; IS, iron sufficient.
Neuronal energy metabolism
Neuronal bioenergetics measurements were normalized to cell density, which did not significantly differ between treatment groups. OCR and PER data from the ATP rate assay experiments are shown in Figure 6A–C. There was a significant overall effect of iron status, but not choline status, on all bioenergetic parameters calculated from ATP rate assay data, except for coupling efficiency and glycolytic ATP production rate (Figure 6D–K). Consistent with our previous findings in this neuron culture model (16), ID neurons had an overall impaired energetic state, with significantly reduced basal and ATP-linked respiration and mitochondrial and total ATP production rates compared with IS neurons, without significantly affecting nonmitochondrial respiration and coupling efficiency (Figure 6 and Figure 7). Iron repletion significantly increased basal and ATP-linked mitochondrial respiration and mitochondrial and total ATP production rates in FID neurons compared with ID neurons such that they were no longer significantly different from IS neurons (Figure 6E–C), suggesting that rescue of ID-induced dendrite complexity deficits may require concomitant rescue of neuronal energy status. Other than a trend (P = 0.1) toward an overall group effect of choline on nonmitochondrial and basal respiration, choline supplementation did not appear to significantly alter any of the energetic parameters calculated from the ATP rate assay data.
FIGURE 6.

Iron repletion rescues hippocampal neuron mitochondrial respiration and glycolysis. Hippocampal neurons cultured from embryonic day 16 (E16) mice were treated with 4 μM deferoxamine (DFO) to induce iron deficiency beginning at 3 d in vitro (DIV). At 11 DIV, DFO was removed to begin iron repletion and 30 μM choline chloride was added to begin choline supplementation. (A, B) At 18 DIV, real-time OCR and PER were measured at baseline and following drug treatments for the ATP Rate Assay (n = 9–10 independent wells per group). (C) Comparison of basal OCR to basal PER. X–Y data are presented as mean ± SEM (relative to initial IS OCR or PER). Note: X–Y data variances are shown as SEMs rather than SDs for easier visualization. (D) Nonmitochondrial respiration, (E) basal mitochondrial respiration, (F) ATP-linked mitochondrial respiration, and (G) coupling efficiency were calculated from OCR data. OCR and PER data were used to calculate ATP production rates due to (H) mitochondrial oxidative phosphorylation and (I) glycolysis. The sum of these 2 rates is the (J) total ATP production rate, and the ratio is the (K) ATP rate index. Bar graphs show all individual values with mean ± SD (n = 9–10). Groups that do not share a common letter are statistically different by 2-factor ANOVA and Tukey test (P < 0.05). An asterisk (*), if present, indicates a statistically significant effect of iron or choline status. A delta symbol (∂), if present, indicates a statistically significant interaction effect. Cho, choline; FID, formerly iron deficient; ID, iron deficient; IS, iron sufficient; OCR, oxygen consumption rate; PER, proton efflux rate.
FIGURE 7.

Choline supplementation increases hippocampal neuron oxygen consumption and ATP production rates. Hippocampal neurons cultured from embryonic day 16 (E16) mice were treated with 4 μM deferoxamine to induce iron deficiency beginning at 3 d in vitro (DIV). At 11 DIV, control and iron-deficient cultures were supplemented with 30 μM choline chloride. (A, B) At 18 DIV, real-time OCR and PER were measured at baseline and following drug treatments for the Mito Stress Test (n = 14–23 independent wells per group). (C) Comparison of basal OCR to basal PER. X–Y data are presented as mean ± SEM (relative to initial IS OCR or PER). Note: X–Y data variances are shown as SEMs rather than SDs for easier visualization. (D) Nonmitochondrial respiration, (E) basal mitochondrial respiration, (F) ATP-linked mitochondrial respiration, and (G) maximal mitochondrial respiration were calculated from OCR data. OCR and PER data were used to calculate ATP production rates due to (H) mitochondrial oxidative phosphorylation and (I) glycolysis. The sum of these 2 rates is the (J) total ATP production rate and the ratio is the (K) ATP rate index. Bar graphs show all individual values with mean ± SD (n = 14–23). Groups that do not share a common letter are statistically different by 2-factor ANOVA and Tukey test (P < 0.05). An asterisk (*), if present, indicates a statistically significant effect of iron or choline status. A delta symbol (∂), if present, indicates a statistically significant interaction effect. Cho, choline; FID, formerly iron deficient; ID, iron deficient; IS, iron sufficient; OCR, oxygen consumption rate; PER, proton efflux rate.
The ATP rate assay data suggest that, unlike iron repletion, choline supplementation increases dendrite complexity without significantly affecting neuronal energetics. To test this further, we performed a mitochondrial stress test experiment on choline-supplemented IS and ID neurons (Figure 7). As previously observed, there was a significant group effect for iron status on all parameters except ATP rate index (Figure 7D–K). There was also a significant group effect for choline for these same measures, except maximal respiration. Nonmitochondrial respiration was significantly increased in IS + Cho neurons compared with untreated IS neurons (Figure 7D). Glycolytic and total ATP production rates were increased in ID + Cho neurons compared with ID neurons to a level that was no longer statistically different from IS neurons (Figure 7I and J). However, choline supplementation of ID neurons (ID + Cho) did not significantly increase any of the calculated bioenergetic parameters compared with ID neurons.
Discussion
The most striking finding of this study was that choline supplementation partially restores the dendrite complexity impairments of developing ID hippocampal neurons, even without restoring neuronal iron status. This finding is consistent with in vivo studies showing that prenatal choline supplementation, followed by postnatal iron repletion, improves the hippocampal transcriptome and learning and memory behaviors in adulthood after recovery from fetal-neonatal iron deficiency anemia (26, 27). As the functional capacity of hippocampal pyramidal neurons is directly related to the complexity of their dendritic arbors (34), our current study suggests that choline supplementation may be able to improve long-term neuronal function without needing to immediately restore iron status during the iron-sensitive period of neuron development. This is a potentially important finding for developing therapeutic strategies in populations in which 1) iron treatment is not possible, 2) iron deficiency diagnosis is delayed, or 3) treatment of iron deficiency needs to be delayed (e.g., coexisting malarial infection). In these cases, choline supplementation could potentially be used to protect the brain until complete brain iron repletion is possible. This hypothesis will need to be further tested in animal models of early life iron deficiency and human clinical trials.
Previous preclinical models of gestational choline supplementation consistently show a positive effect at multiple levels of analysis of the hippocampus. When given to a mouse model of Rett syndrome, it increases dendrite complexity and synaptic function of hippocampal pyramidal neurons, ultimately resulting in better performance on learning and memory tasks in adulthood (35–38). Early life choline supplementation also improves neurobehavioral outcomes in animal models of Down and fetal alcohol syndromes and early life iron deficiency anemia (26, 38–41). However, during studies of the beneficial effects of choline on IDA rats, we also showed that prenatal choline supplementation of IS rat dams resulted in dysregulated gene expression pathways in the adult offspring hippocampus. These pathways were implicated in cognitive and psychological impairments, suggesting a possible negative effect of excess choline on brain development (26, 27). In contrast, in our current in vitro study, choline supplementation of IS neurons did not alter their dendrite complexity. This finding suggests that choline supplementation of the neuron, beyond what is considered sufficient, is not directly detrimental to neuronal structural development. Translationally, this may allay concerns of using a strategy of universal choline supplementation when there is a high risk of gestational iron deficiency, but some proportion of the population remains iron sufficient. Corroborating our finding, 2 other studies have shown that prenatal choline supplementation of normal, healthy rats increases hippocampal neuron dendrite complexity (35, 36). The fact that additional choline did not alter dendrite complexity of normal, healthy neurons in the current study suggests that the findings in the whole animal model (35, 36) may be due to additional systemic or nonneuronal effects of choline supplementation on neuronal structural development and function. Going forward in whole animal and human studies, it will be important to consider potential systemic and brain-specific effects, both positive and negative, in evaluating the therapeutic potential of choline supplementation for improving neurodevelopment.
This study also demonstrates that when iron is given back within the sensitive period of rapid neuronal development, iron repletion of the neuron is sufficient to rescue overall dendrite complexity and energy production. Translationally, this is a key finding in terms of implementing strategies to prevent the long-term neurodevelopmental deficits caused by early life iron deficiency. The data suggest that if brain iron deficiency is detected early enough, brain iron repletion alone would be sufficient to restore a key contributor to neural circuit formation and function (i.e., dendrite complexity). Yet, the increased incidence of long-term neurodevelopmental abnormalities associated with early life iron deficiency suggests that, with current diagnosis and management strategies [e.g., testing for anemia at 12 mo of age (42)], iron treatment is rarely timely (11). This “late” detection and treatment of iron deficiency, as well as the associated persistent neurobehavioral deficits, occur because iron is prioritized the red blood cells over the brain when iron demand is greater than iron supply (43–46). Thus, the developing brain becomes ID before anemia is present and also does not become iron replete with iron therapy until after anemia has resolved (47). Our data provide further support to ongoing efforts to identify peripheral biomarkers that index iron status of the developing brain prior to the onset of anemia, so that brain iron deficiency can be detected and iron treatment initiated early enough to prevent long-term neurobehavioral consequences.
Despite the apparent complete rescue of dendrite complexity by iron repletion alone, it remains to be seen whether this results in a full recovery of synaptic function. The Sholl analysis data show a distal shift in the distance at which peak branching occurs for the iron-repleted neurons (Figure 1B), indicating that there may be residual, subtle differences in dendrite structure that could alter the synaptic properties of the individual neuron and thus the circuitry. After recovery from fetal-neonatal iron deficiency anemia, FID rat hippocampal pyramidal neurons exhibit a distal shift in the distance to the first branch point but a proximal shift in peak branching distance (33, 48). These shifts in dendrite branching are associated with persistently impaired synaptic plasticity, structure, and function (33, 49). To fully understand the long-term functional implications of dendrite complexity recovery by iron repletion (or choline supplementation), future studies will need to directly assess synapse formation and the electrophysiologic properties of these neurons, both in vitro and in vivo.
The iron repletion data indicate, as we hypothesized, that neuronal energy status and dendrite complexity go hand-in-hand in developing ID neurons, suggesting rescuing the energy metabolism deficits is a prerequisite for recovery of the dendrite complexity impairments. In contrast, choline supplementation improved the dendrite complexity deficits caused by iron deficiency to near IS neuron levels, without iron repletion and without major improvements in neuronal bioenergetics. Choline supplementation did have a significant stimulatory effect on several measures of mitochondrial respiration and neuronal ATP production rates when both IS and ID neurons were considered together (Figure 7, choline group effect). However, this suggests that the amount of choline in the basal neuronal culture media may be suboptimal for these processes rather than choline having a stimulatory effect on iron-dependent mitochondrial function and ATP production. Together, these findings suggest that it may not be necessary to completely rescue the ID-induced energy metabolism deficits in order to nevertheless enhance the more functionally relevant neuron structural deficits.
The disconnect between the dendrite structure and neuronal energetics data in ID + Cho neurons indicates that choline's stimulatory effect on dendrite structure may be predominantly mediated through mechanisms that are independent of supporting iron-dependent energy metabolism [e.g., epigenetic regulation of genes critical for neuron maturation through choline's role as a metabolic precursor to the methyl donor pathway (27)]. However, dendrite outgrowth is a highly energy-dependent process (50). Thus, an alternative hypothesis is that choline's stimulatory effect on ID neuron dendrite complexity requires an increase in local dendritic ATP, despite the lack of change in global neuronal ATP production rates. This redistribution of energy could be accomplished by a stimulation of mitochondrial trafficking into the growing dendrites, a process that is impaired in ID neurons (28).
It is important to note that a technical limitation of these experiments was that the dendrite structure and neuronal bioenergetics data had to be obtained from different sets of cultures that were grown under unique conditions (dishes compared with seahorse plates). Future studies will need to obtain neuronal bioenergetics and neuron structure data from identical conditions to definitively determine whether choline's improvement of dendrite structure involves significant stimulation of energy production.
Supplementary Material
Acknowledgments
The authors’ contributions were as follows—TWB, LML, and MKG: designed the research; TWB, WCvH, and ORK: conducted the neuronal dendrite morphology studies and performed the statistical analyses; TWB: conducted and analyzed the cellular bioenergetic studies; TWB and MKG: wrote the manuscript and had responsibility for final content; and all authors: read and approved the final manuscript.
Notes
Supported by NIH (National Institute of Child Health and Human Development) grants R01 HD029421 (MKG) and F32 HD085576 (TWB).
Author disclosures: The authors report no conflicts of interest.
Supplemental Table 1 is available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/jn/.
Abbreviations used: 5-FU, 67.5 μM 5-fluoro-2′-deoxyuridine/136 μM uridine; Cho, choline; DFO, deferoxamine; DIV, days in vitro; E, embryonic day; ECAR, extracellular acidification rate; eGFP, enchanced green fluorescent protein; FID, formerly iron deficient; GFP, green fluorescent protein; ID, iron deficient; IDA, iron-deficient anemia; IS, iron sufficient; MAP2, microtubule-associated protein 2; OCR, oxygen consumption rate; PER, proton efflux rate; Tfrc, transferrin receptor 1.
Contributor Information
Thomas W Bastian, Department of Pediatrics, School of Medicine, University of Minnesota, Minneapolis, MN, USA.
William C von Hohenberg, Department of Pediatrics, School of Medicine, University of Minnesota, Minneapolis, MN, USA.
Olivia R Kaus, Department of Pediatrics, School of Medicine, University of Minnesota, Minneapolis, MN, USA.
Lorene M Lanier, Department of Neuroscience, University of Minnesota, Minneapolis, MN, USA.
Michael K Georgieff, Department of Pediatrics, School of Medicine, University of Minnesota, Minneapolis, MN, USA.
References
- 1. McLean E, Cogswell M, Egli I, Wojdyla D, de Benoist B. Worldwide prevalence of anaemia, WHO vitamin and mineral nutrition information system, 1993–2005. Public Health Nutr. 2009;12(4):444–54. [DOI] [PubMed] [Google Scholar]
- 2. Yip R. Iron deficiency: contemporary scientific issues and international programmatic approaches. J Nutr. 1994;124(Suppl 8):1479S–90S. [DOI] [PubMed] [Google Scholar]
- 3. Vos T, Abajobir AA, Abate KH, Abbafati C, Abbas KM, Abd-Allah F, Abdulkader RS, Abdulle AM, Abebo TA, Abera SFet al. . Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1211–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gupta A, Gadipudi A. Iron deficiency anaemia in pregnancy: developed versus developing countries. EMJ Hematol. 2018;6:101–9. [Google Scholar]
- 5. Cogswell ME, Looker AC, Pfeiffer CM, Cook JD, Lacher DA, Beard JL, Lynch SR, Grummer-Strawn LM. Assessment of iron deficiency in US preschool children and nonpregnant females of childbearing age: National Health and Nutrition Examination Survey 2003–2006. Am J Clin Nutr. 2009;89(5):1334–42. [DOI] [PubMed] [Google Scholar]
- 6. Mei Z, Cogswell ME, Looker AC, Pfeiffer CM, Cusick SE, Lacher DA, Grummer-Strawn LM. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999–2006. Am J Clin Nutr. 2011;93(6):1312–20. [DOI] [PubMed] [Google Scholar]
- 7. Auerbach M, Abernathy J, Juul S, Short V, Derman R. Prevalence of iron deficiency in first trimester, nonanemic pregnant women. J Matern Fetal Neonatal Med. 2021;34(6):1002–5. [DOI] [PubMed] [Google Scholar]
- 8. Georgieff MK. Long-term brain and behavioral consequences of early iron deficiency. Nutr Rev. 2011;69:S43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Georgieff MK. Iron deficiency in pregnancy. Am J Obstet Gynecol. 2020;223(4):516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barks A, Hall AM, Tran PV, Georgieff MK. Iron as a model nutrient for understanding the nutritional origins of neuropsychiatric disease. Pediatr Res. 2019;85(2):176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Georgieff MK. Iron assessment to protect the developing brain. Am J Clin Nutr. 2017;106(Suppl 6):1588S–93S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sazawal S, Black RE, Ramsan M, Chwaya HM, Stoltzfus RJ, Dutta A, Dhingra U, Kabole I, Deb S, Othman MKet al. . Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet. 2006;367(9505):133–43. [DOI] [PubMed] [Google Scholar]
- 13. Carlson ES, Tkac I, Magid R, O'Connor MB, Andrews NC, Schallert T, Gunshin H, Georgieff MK, Petryk A. Iron is essential for neuron development and memory function in mouse hippocampus. J Nutr. 2009;139(4):672–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carlson ES, Fretham SJ, Unger E, O'Connor M, Petryk A, Schallert T, Rao R, Tkac I, Georgieff MK. Hippocampus specific iron deficiency alters competition and cooperation between developing memory systems. J Neurodev Disord. 2010;2(3):133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fretham SJ, Carlson ES, Wobken J, Tran PV, Petryk A, Georgieff MK. Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus. 2012;22(8):1691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bastian TW, von Hohenberg WC, Mickelson DJ, Lanier LM, Georgieff MK. Iron deficiency impairs developing hippocampal neuron gene expression, energy metabolism, and dendrite complexity. Dev Neurosci. 2016;38(4):264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bastian TW, Rao R, Tran PV, Georgieff MK. The effects of early-life iron deficiency on brain energy metabolism. Neurosci Insights. 2020;15:263310552093510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trujillo-Gonzalez I, Zeisel SH. Present knowledge in nutrition (Eleventh Edition). Academic Press;2020. Chapter 18, Choline; p. 305–18. [Google Scholar]
- 19. Blusztajn J, Slack B, Mellott T. Neuroprotective actions of dietary choline. Nutrients. 2017;9(8):815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ueland PM. Choline and betaine in health and disease. J Inherit Metab Dis. 2011;34(1):3–15. [DOI] [PubMed] [Google Scholar]
- 21. Zeisel SH. Metabolic crosstalk between choline/1-carbon metabolism and energy homeostasis. Clin Chem Lab Med. 2013;51(3):467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pacelli C, Coluccia A, Grattagliano I, Cocco T, Petrosillo G, Paradies G, De Nitto E, Massaro A, Persichella M, Borracci Pet al. . Dietary choline deprivation impairs rat brain mitochondrial function and behavioral phenotype. J Nutr. 2010;140(6):1072–9. [DOI] [PubMed] [Google Scholar]
- 23. Posse de Chaves E, Vance DE, Campenot RB, Vance JE. Axonal synthesis of phosphatidylcholine is required for normal axonal growth in rat sympathetic neurons. J Cell Biol. 1995;128(5):913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Albright CD, Tsai AY, Friedrich CB, Mar M-H, Zeisel SH. Choline availability alters embryonic development of the hippocampus and septum in the rat. Dev Brain Res. 1999;113(1–2):13–20. [DOI] [PubMed] [Google Scholar]
- 25. Yen C-LE, Mar M-H, Meeker RB, Fernandes A, Zeisel SH. Choline deficiency induces apoptosis in primary cultures of fetal neurons. FASEB J. 2001;15(10):1704–10. [DOI] [PubMed] [Google Scholar]
- 26. Kennedy BC, Dimova JG, Siddappa AJ, Tran PV, Gewirtz JC, Georgieff MK. Prenatal choline supplementation ameliorates the long-term neurobehavioral effects of fetal-neonatal iron deficiency in rats. J Nutr. 2014;144(11):1858–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tran PV, Kennedy BC, Pisansky MT, Won K-J, Gewirtz JC, Simmons RA, Georgieff MK. Prenatal choline supplementation diminishes early-life iron deficiency–induced reprogramming of molecular networks associated with behavioral abnormalities in the adult rat hippocampus. J Nutr. 2016;146(3):484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bastian TW, von Hohenberg WC, Georgieff MK, Lanier LM. Chronic energy depletion due to iron deficiency impairs dendritic mitochondrial motility during hippocampal neuron development. J Neurosci. 2019;39(5):802–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1(5):2406–15. [DOI] [PubMed] [Google Scholar]
- 30. Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented neurobasal? A new serum-free medium combination. J Neurosci Res. 1993;35(5):567–76. [DOI] [PubMed] [Google Scholar]
- 31. Gross GG, Junge JA, Mora RJ, Kwon H-B, Olson CA, Takahashi TT, Liman ER, Ellis-Davies GCR, McGee AW, Sabatini BLet al. . Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron. 2013;78(6):971–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of mammalian iron metabolism. Cell. 2010;142(1):24–38. [DOI] [PubMed] [Google Scholar]
- 33. Brunette KE, Tran PV, Wobken JD, Carlson ES, Georgieff MK. Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev Neurosci. 2010;32(3):238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McAllister AK. Cellular and molecular mechanisms of dendrite growth. Cereb Cortex. 2000;10(10):963–73. [DOI] [PubMed] [Google Scholar]
- 35. Li Q, Guo-Ross S, Lewis DV, Turner D, White AM, Wilson WA, Swartzwelder HS. Dietary prenatal choline supplementation alters postnatal hippocampal structure and function. J Neurophysiol. 2004;91(4):1545–55. [DOI] [PubMed] [Google Scholar]
- 36. Meck WH. Developmental periods of choline sensitivity provide an ontogenetic mechanism for regulating memory capacity and age-related dementia. Front Integr Neurosci. 2008;1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meck WH, Smith RA, Williams CL. Pre- and postnatal choline supplementation produces long-term facilitation of spatial memory. Dev Psychobiol. 1988;21(4):339–53. [DOI] [PubMed] [Google Scholar]
- 38. Chin EWM, Lim WM, Ma D, Rosales FJ, Goh ELK. Choline rescues behavioural deficits in a mouse model of Rett syndrome by modulating neuronal plasticity. Mol Neurobiol. 2019;56(6):3882–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nag N, Mellott TJ, Berger-Sweeney JE. Effects of postnatal dietary choline supplementation on motor regional brain volume and growth factor expression in a mouse model of Rett syndrome. Brain Res. 2008;1237:101–9. [DOI] [PubMed] [Google Scholar]
- 40. Moon J, Chen M, Gandhy SU, Strawderman M, Levitsky DA, Maclean KN, Strupp BJ. Perinatal choline supplementation improves cognitive functioning and emotion regulation in the Ts65Dn mouse model of Down syndrome. Behav Neurosci. 2010;124(3):346–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ryan SH, Williams JK, Thomas JD. Choline supplementation attenuates learning deficits associated with neonatal alcohol exposure in the rat: effects of varying the timing of choline administration. Brain Res. 2008;1237:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baker RD, Greer FR; The Committee on Nutrition . Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0–3 years of age). Pediatrics. 2010;126(5):1040–50. [DOI] [PubMed] [Google Scholar]
- 43. Petry CD, Eaton MA, Wobken JD, Mills MM, Johnson DE, Georgieff MK. Iron deficiency of liver, heart, and brain in newborn infants of diabetic mothers. J Pediatr. 1992;121(1):109–14. [DOI] [PubMed] [Google Scholar]
- 44. Zamora TG, Guiang SF, Georgieff MK, Widness JA. Iron is prioritized to red blood cells over the brain in phlebotomized anemic newborn lambs. Pediatr Res. 2016;79(6):922–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rao R, Ennis K, Lubach GR, Lock EF, Georgieff MK, Coe CL. Metabolomic analysis of CSF indicates brain metabolic impairment precedes hematological indices of anemia in the iron-deficient infant monkey. Nutr Neurosci. 2018;21(1):40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ennis KM, Dahl LV, Rao RB, Georgieff MK. Reticulocyte hemoglobin content as an early predictive biomarker of brain iron deficiency. Pediatr Res. 2018;84(5):765–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rao R, Tkac I, Townsend EL, Gruetter R, Georgieff MK. Perinatal iron deficiency alters the neurochemical profile of the developing rat hippocampus. J Nutr. 2003;133(10):3215–21. [DOI] [PubMed] [Google Scholar]
- 48. Jorgenson LA, Wobken JD, Georgieff MK. Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev Neurosci. 2003;25(6):412–20. [DOI] [PubMed] [Google Scholar]
- 49. Jorgenson LA, Sun M, O'Connor M, Georgieff MK. Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area CA1 of the developing rat hippocampus. Hippocampus. 2005;15(8):1094–102. [DOI] [PubMed] [Google Scholar]
- 50. Rangaraju V, Lewis TL, Hirabayashi Y, Bergami M, Motori E, Cartoni R, Kwon S-K, Courchet J. Pleiotropic mitochondria: the influence of mitochondria on neuronal development and disease. J Neurosci. 2019;39(42):8200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.