

Abstract
Mutations in the exonuclease domain of POLE are associated with tumors harboring very high mutation burdens. The mechanisms linking this significant mutation accumulation and tumor development remain poorly understood. Pole+/P286R;Trp53+/– mice showed accelerated cancer mortality compared to Pole+/P286R;Trp53+/+ mice. Cells from Pole+/P286R mice showed increased p53 activation, and subsequent loss of p53 permitted rapid growth, implicating canonical p53 loss of heterozygosity in POLE mutant tumor growth. However, p53 status had no effect on tumor mutation burden or single base substitution signatures in POLE mutant tumors from mice or humans. Pten has important roles in maintaining genome stability. We find that PTEN mutations are highly enriched in human POLE mutant tumors, including many in POLE signature contexts. One such signature mutation, PTEN-F341V, was previously shown in a mouse model to specifically decrease nuclear Pten and lead to increased DNA damage. We found tumors in Pole+/P286R mice that spontaneously acquired PtenF341V mutations and were associated with significantly reduced nuclear Pten and elevated DNA damage. Re-analysis of human TCGA (The Cancer Genome Atlas) data showed that all PTEN-F341V mutations occurred in tumors with mutations in POLE. Taken together with recent published work, our results support the idea that development of POLE mutant tumors may involve disabling surveillance of nuclear DNA damage in addition to POLE-mediated hypermutagenesis.
Graphical Abstract
Graphical Abstract.
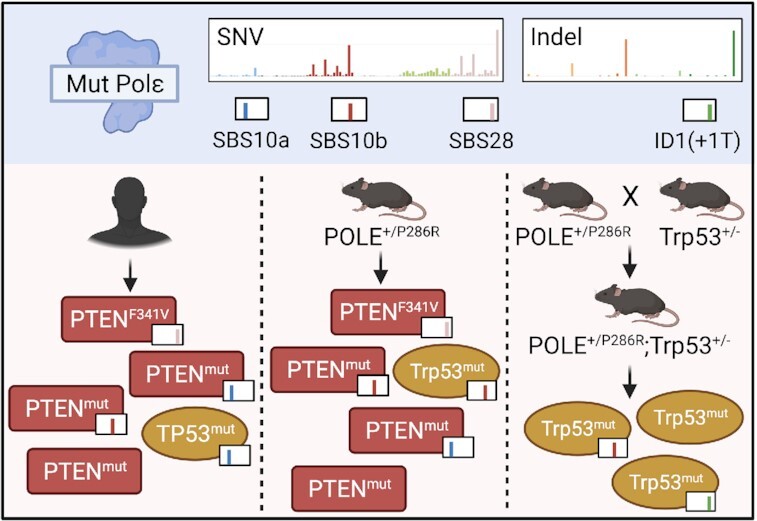
POLE signature mutations in PTEN or TP53 are increased in tumors depending on the genetic background.
INTRODUCTION
Tumors with Pol ϵ exonuclease domain mutations are associated with incredibly high mutation burdens exceeding >10 mutations per megabase (mut/Mb) and unique mutation signatures that distinguish them from other cancers. These signatures include TCT>TAT transversions, TCG>TTG transitions and TTT>TGT transversions, designated as single base substitution (SBS) signatures 10a, 10b and 28, respectively (1). These signatures are significantly elevated in POLE mutant tumors from both humans (1,2) and mice (3). An additional signature, SBS 14, is found in human POLE tumors that also show evidence of microsatellite instability and inactivation of one or more mismatch repair (MMR) factors. SBS 14 is dominated by NCT>NAT transversions, with N representing any base (1,4). However, the mechanism(s) by which mutations in POLE lead to tumorigenesis are not well understood. We used our established germline Pole-P286R mouse, which models the most recurrent human POLE mutation (2,5–7), to (i) investigate how POLE mutations drive tumorigenesis in vivo and (ii) determine the extent to which tumor development is driven by mutagenesis per se or by nonmutagenic processes.
We previously reported the curious finding that PoleS459F/S459F mice were viable and developed to adulthood, yet PoleP286R/P286R mice were not viable, even in the same background strain (3). This was somewhat surprising because the exonuclease activity of the human POLE-S459F enzyme is 100-fold lower than that of POLE-P286R in in vitro studies (3). Since mutation accumulation and mutation spectra have been shown to be influenced by the nature of the mutant POLE allele (8–10), it follows that these and other factors also likely contribute to normal embryonic development.
To further understand what factors might contribute to the high mutagenesis observed in murine POLE mutant tumors and to the PoleP286R/P286R embryonic lethality phenotype, we bred Pole+/P286R mice with Trp53+/– mice (11). The rationale guiding this cross was driven by several lines of evidence suggesting that p53 activation is a downstream target of POLE dysfunction. Bellelli et al. found that Pole4–/– mice, which lack a noncatalytic Pol ϵ holoenzyme subunit, had developmental abnormalities and increased tumorigenesis (12). Pole4–/– was embryonic lethal in a C57BL/6J background, but this lethality was rescued when p53 was inactivated. They also reported that homozygous Pole4–/– embryos had significantly increased p53 protein levels compared to wild-type and heterozygous embryos (12). We have previously reported that mRNA levels of p21 and PUMA are elevated in human cell lines engineered to express mutant POLE alleles (9). Additionally, human POLE-mutated tumors are associated with enrichment in mutations in major tumor suppressor genes, including TP53 (13,14). Therefore, we reasoned that PoleP286R/P286R and perhaps Pole+/P286R survival might be enhanced in the absence of functional p53.
While the sheer number of mutated genes in POLE-mutated tumors has made identifying tumor driving mutations difficult, a number of studies have made attempts. Enrichment of TP53 mutations in POLE mutant tumors, in particular the R231X nonsense mutation, was noted in several studies (14,15). A more careful subsequent look at p53 mutations in POLE mutant tumors found that p53-R213X was significantly elevated specifically in colorectal tumors with the POLEP286R mutant allele (13). Neither endometrial tumors nor those with the POLEV411L mutant allele showed enrichment in the p53-R213X mutation. Almost half of mouse endometrial tumors formed due to conditional knock-in of Pole+/P286R in uterine tissue stained positively for p53, though the nature of the mutant alleles was not examined further (16). Another candidate that has been reported in addition to p53 in human POLE mutant tumors is the tumor suppressor PTEN (17,18), though its association with POLE mutations was scored as indeterminate.
In the current study, we looked at how mutations in Trp53 affected growth and mutagenesis in cells and tumors from mice harboring germline PoleP286R mutations. We then used information from human tumor datasets to investigate the relationship between mutagenesis and tumorigenesis in POLE mutant tumors. We found that loss of one Trp53 allele in the mouse germline accelerated spontaneous tumorigenesis without affecting mutation burden or mutation signature. Similar to previous observations in engineered human cell lines, p53 was activated in mouse cells with mutant alleles of Pole. While TP53 driver mutations were not significantly enriched in human POLE mutant tumors, we found enriched and recurrent PTEN mutations occurring in mutant POLE signature contexts. Analyzing a human endometrial tumor proteogenomic dataset revealed that POLE-dependent mutations correlated with reduced Pten protein levels, while p53 levels were unchanged. Further, we found spontaneously occurring PtenF341V mutations in two separate PoleP286R mouse tumors that displayed loss of nuclear Pten staining. Previous studies using a germline PtenF341V mouse model have shown that this allele drives increased tumorigenesis coupled with decreased genome stability. Taken together, we propose that loss of p53 or loss of nuclear Pten can facilitate POLE mutant tumor development, likely through loss of DNA damage surveillance.
MATERIALS AND METHODS
Mice and breeding
Pole+/P286R mice were generated in C57BL/6J strain as previously described (3), and were back-crossed to C57BL/6J (The Jackson Laboratory) to minimize genetic drift. Heterozygous Trp53tm1Tyj mouse (C57BL/6J) was purchased from The Jackson Laboratory (strain #002101) for cross-breeding. The animal protocol applicable to the experiments reported here was approved by the Tulane University Institutional Animal Care and Use Committee (protocols 4445 and 921). Mice were housed and maintained at the Tulane University Health Science Center vivarium and were under the care and supervision of staff veterinarian and trained animal care and veterinary technicians. All animals were monitored closely and sacrificed by CO2 inhalation if the following criteria were observed: ulcerating tumor, visible tumor >1 cm, moribund and visible weight loss (>10%).
Pole-P286R and Trp53 genotyping strategy
Tail gDNA was extracted and PCR amplified. PCR products were directly treated with DdeI restriction enzyme (NEB) per product protocol and run on agarose gel as previously described (3). The Pole locus was PCR amplified using the following primers:
Forward: 5′-TTTGGGACTGAGGTGGTCTG-3′
Reverse: 5′-CCCCACAAAAGCATTCTAAGTTTCC-3′
The Trp53 locus was PCR amplified using the following primers:
For Trp53 wild-type allele:
exon 6: 5′-AGCGTGGTGGTACCTTATGAGC-3′;
exon 7: 5′-GGATGGTGGTATACTCAGAGCC-3′
For Trp53D neo allele:
neo19: 5′-GCTATCAGGACATAGCGTTGGC-3′;
exon 7: 5′-GGATGGTGGTATACTCAGAGCC-3′
Targeted panel and whole genome sequencing of tumors
Genomic DNA from tumor and tail of mice was extracted by phenol/chloroform extraction. Samples were sent to the Beijing Genomics Institute Americas for sequencing. Agilent SureSelectXT Mouse All Exon Kit was used for enrichment. Illumina HiSeq4000 was used for paired-end sequencing.
Variant detection from next-generation sequencing data
FASTQ files were aligned to the reference genome using BWA-MEM 0.7.12, and PicardTools 1.133 to sort the BAM file and mark duplicates. Variant calling was completed as described (3).
Mouse embryonic fibroblast generation and population doubling level experiments
Mouse embryonic fibroblasts (MEFs) were derived from timed mating of double heterozygous crosses (Pole+/P286R;Trp53+/– × Pole+/P286R;Trp53+/–) at embryonic days 12.5–13.5 from viable Pole wild-type, heterozygous and homozygous embryos. Genomic DNA from embryo heads was used for genotyping, while the remaining embryo was minced, trypsinized and plated for cell culture under aseptic conditions in 10-cm plates. After 24–48 h, cells were counted.
For population doubling level (PDL) measurement, 1.9 × 105 cells were seeded in six-well plates in triplicate and grown at 37°C in 5% CO2. After 3 days, each triplicate was harvested individually and live cells were counted using a Countess Automated Cell Counter (Invitrogen). This procedure was repeated every 3 days until the cells were immortalized as per the NIH3T3 protocol. PDL was calculated as follows:
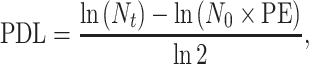 |
where Nt is the number of viable cells counted after passage, N0 is the number of cells seeded prior to passage and PE is the plating efficiency. Passage 0 was the initial seeding from the 10-cm plate into six-well plates.
Expression of p53 targets
Total RNA was extracted using Invitrogen TRIzol reagent (Cat. #15596026) and chloroform as per product protocol. RNA was precipitated in isopropanol and resuspended in DEPC water. RNA was immediately made into cDNA (First-Strand cDNA Synthesis Kit, GE). Real-time PCR was performed using QuantStudio 6 (Thermo Fisher Scientific). Expression of targets was normalized to TBP. The following primers were used:
p21: 5′-CAGGCACCATGTCCAATCC-3′;
5′-GAGACAACGGCACACTTTGCT-3′
PUMA: 5′-GCGGCGGAGACAAGAAGA-3′;
5′-AGTCCCATGAAGAGATTGTACATGAC-3′
Immunofluorescence staining
Mouse thymus tissues were harvested and fixed in 10% formalin by standard methods. Paraffin-embedded tissues were sectioned, mounted in xylene, boiled in citrate buffer and then stained with rabbit anti-Pten (Cell Signaling Technologies, Cat. #9559T), mouse anti-phospho-Histone-H2A.X (Ser139) (EMP Millipore, Cat. #05-636) and DAPI (Invitrogen, Cat. #D1306).
Driver gene mutation enrichment analysis in human cancer cohorts
Frequency of TP53 and PTEN driver mutations in POLE mutant and POLE wild-type tumors was counted, and proportions were analyzed via Fisher’s exact test to account for small sample sizes.
CPTAC endometrial carcinoma protein analysis
Individual sample protein abundance was plotted based on reported z-score per Dou et al. (19). Individual ΔZ was calculated as ΔZ = Ztumor – Zadjacent normal tissue, if adjacent normal tissue protein analysis was reported.
RESULTS
Loss of p53 does not suppress PoleP286R/P286R embryonic lethality
Loss of p53 function has previously been implicated in POLE mutant tumor development in humans (13,14) and in mice (16). We have also shown that p53 is activated in human cancer cell lines engineered to express cancer mutant alleles of POLE (9). We previously characterized the effects of the Pole-P286R mutation on tumor development and mutagenesis in a mouse model (3). We noted that PoleP286R/P286R was likely embryonic lethal due to lack of any observed viable offspring. Inactivation of p53 has been used to rescue embryonic lethality for many mouse mutant alleles, including Pole4–/– (12,20).
We set out to test the effects of p53 loss on POLE mutant tumor development by inactivating Trp53 in our Pole-P286R mice. Prior to this cross, our Pole+/P286R mice had been maintained by backcrossing to Pole+/+ C57BL/6J (Jackson Laboratories, Bar Harbor, ME) for six generations. We crossed Pole+/P286R and Trp53+/– mice with one another to generate double heterozygous, Pole+/P286R;Trp53+/– mice. Breeding double heterozygous mice (Pole+/P286R;Trp53+/– × Pole+/P286R;Trp53+/–) produced 1 PoleP286R/P286R;Trp53+/– pup out of 38 live births; however, this offspring failed to thrive and died 1 day post-utero (Supplementary Table S1). We also observed one PoleP286R/P286R embryo in utero out of one litter genotyped at approximately embryonic days 13.5–14.5 (PoleP286R/P286R;Trp53+/–, red arrow, Supplementary Figure S1A). Compared to littermates, the PoleP286R/P286R embryo was noticeably underdeveloped and likely would not have survived to live birth. In addition, we observed one Pole+/P286R;Trp53–/– embryo, which was also comparatively smaller than its littermates. The lack of expected Mendelian ratio of alleles in viable offspring suggests that loss of p53 does not suppress lethality in developing PoleP286R/P286R embryos. However, since PoleP286R/P286R offspring can be produced by other labs, though at sub-Mendelian frequencies (21), we cannot rule out the contribution of strain differences to our results.
While breeding Pole+/P286R;Trp53+/– mice, we also observed that they displayed accelerated mortality compared to Pole+/P286R;Trp53+/+ mice. In order to directly test the effect of Trp53 status on survival of Pole+/P286R, we set up a survival cohort of double heterozygous breeding pairs (Pole+/P286R;Trp53+/– × Pole+/P286R;Trp53+/–) to generate sufficient numbers of littermates of all possible genotypes. Survival of Pole+/P286R;Trp53+/– mice was decreased compared to Pole+/+;Trp53+/+ mice, with median survival decreasing from 7.7 to 4.7 months (Figure 1A, P = 0.03, log-rank). No significant difference in survival of male versus female mice within the same genotype was observed.
Figure 1.

Loss of Trp53 allele accelerates Pole-dependent cancer mortality. (A) Kaplan–Meier survival estimate of Pole+/P286R;Trp53+/– mice compared to Pole+/P286R;Trp53+/+ mice. All mice were products of Pole+/P286R;Trp53+/– × Pole+/P286R;Trp53+/– cross. Pole+/+;Trp53+/+ (n = 8); Pole+/+;Trp53+/– (n = 10); Pole+/P286R;Trp53+/+(n = 11); Pole+/P286R;Trp53+/– (n = 13). (B) Cohort mean age of death stratified by thymic and disseminated (spleen) lymphomas. Mean age: 3.6, 4.0, 7.7 and 5.6 months, respectively. Only mice with either a thymic lymphoma or a splenic lymphoma were included. For comparisons of the means between two groups, unpaired two-tailed t-test and Welch’s correction for nonequal SD were used. (C) Mean age of death for Pole+/P286R and Pole+/P286R;Trp53+/– mice from all tumors [expanded to mice beyond cohort shown in panels (A) and (B)], 7.0 and 4.1 months, respectively. (D) Mean age of death for Pole+/P286R and mice [expanded to mice beyond cohort shown in panels (A) and (B)] stratified by thymic and disseminated (spleen) lymphomas. Mean age: 2.8, 3.3, 7.9 and 5.1 months, respectively. For comparisons of the means between two groups, unpaired two-tailed t-test and Welch’s correction for nonequal SD were used.
Tumors from Pole+/P286R;Trp53+/– mice and Pole+/P286R;Trp53+/+ mice showed similar lymphoid tissue specificity (Supplementary Table S2). Full autopsies were performed on each animal and lymphoid organ tumors were the most plausible cause of death for almost all Pole+/P286R mice, regardless of p53 status. Thymic and splenic lymphomas were almost always mutually exclusive. In Pole+/P286R;Trp53+/– animals, we found 4 with enlarged thymus and normal spleen, 8 with enlarged spleen and normal thymus, and 1 with concurrent enlarged thymus and enlarged spleen from 13 animals. In Pole+/P286R;Trp53+/+ animals, we found 2 with enlarged thymus and normal spleen, 7 with enlarged spleen and normal thymus, 0 with concurrent enlarged thymus and enlarged spleen, and 2 with no obvious lymphoid organ involvement in 11 animals.
While overall cancer mortality was accelerated by loss of one allele of p53, the effect was not the same across tissue types. Pole+/P286R mice with thymic lymphomas died rapidly regardless of germline Trp53 status (Figure 1B). Occurrence of disseminated lymphomas in the spleen, lymph nodes and liver was slightly accelerated in Pole+/P286R;Trp53+/– mice compared to Pole+/P286R;Trp53+/+ mice (Figure 1B, 5.6 months versus 7.7 months, respectively), though this difference was not significant in the survival cohort. We noted that, with a single exception, all Pole+/P286R animals had either a clear visible thymic lymphoma or clear splenic lymphoma at necropsy, not both. Only those animals with cancer in either the thymus or the spleen are plotted in Figure 1B. When we looked more broadly at each of the first 63 mice [Pole+/P286R;Trp53+/+ (n = 28) and Pole+/P286R;Trp53+/– (n = 35)] generated from the beginning of this colony and compared to Pole+/P286R mice from our previous study (3), overall tumor mortality was accelerated in Pole+/P286R;Trp53+/– mice (Figure 1C). Interestingly, this difference was entirely due to more rapid development of splenic lymphomas, as thymic lymphoma development remained independent of Trp53 genotype (Figure 1D). Based on these expanded data, loss of one allele of Trp53 significantly decreased survival, likely by accelerating disseminated tumor mortality.
p53 activation in cells with mutant Pole restrains immortalization
We used MEFs to address the effects of Pole-P286R on p53 in untransformed mouse cells. We found that time to immortalization was dependent on both Pole and Trp53 status (Figure 2A). Pole+/P286R;Trp53–/– MEFs did not plateau and continued steady PDLs throughout the experiment. PDLs for Pole+/P286R;Trp53+/– MEFs slowed through 21 days, followed by rapid proliferation, indicating likely immortalization through inactivation of the remaining Trp53 allele. Consistent with NIH3T3 mouse embryo cells in culture (22), PDLs for both Pole+/P286R;Trp53+/+ and Pole+/+;Trp53+/+ MEFs plateaued after ∼9 days and remained so after >30 days of passaging.
Figure 2.

Activation of p53 target expression in Pole+/P286R MEFs and immortalized mouse cell lines. (A) PDLs of MEFs passaged according to NIH3T3 protocol double heterozygous crosses. Genotypes include Pole+/+;Trp53+/+ (n = 2, black), Pole+/+;Trp53–/– (n = 1, red), Pole+/P286R;Trp53+/+ (n = 4, green), Pole+/P286R;Trp53+/– (n = 2, purple) and Pole+/P286R;Trp53–/– (n = 2, blue). (B) qPCR expression of p21 and PUMA (encoded by Cdkn1 and Bbc3, respectively) in freshly derived Pole+/P286R MEF cell lines. Each pair of bars (p21 and PUMA) represents one cell line, measured in triplicate. 4226 cell line, created from p53 wild-type MMVT-Wnt1 tumor cells, nontreated and doxorubicin-treated (Doxo) cells, included as positive control for p53 activation. (C) qPCR expression of p21 and PUMA in immortalized Pole+/P286R and Pole+/P286R cell lines made from two independent litters. (D) The region encoding the Trp53 DNA binding domain in the Pole+/P286R MEF line from litter B [shown in panel (C)] was resequenced via Sanger sequencing. (E) Expression of p21 and PUMA was measured by qPCR in one Pole+/P286R thymic tumor compared to normal tissue determined by qPCR. Nontreated (NT) and irradiated (IR) thymus included as positive controls for p53 activation.
Consistent with the proliferation assays, Cdkn1a and Bbc3 expression was elevated in MEF cells freshly harvested and plated from Pole+/P286R;Trp53+/+ embryos (Figure 2B). As in our human cell line models (9), activation of p53 targets was also significantly elevated in immortalized Pole+/P286R mouse cell lines when compared to Pole+/+ cell lines (Figure 2C, left). However, we noted that one spontaneously immortalized Pole+/P286R cell line had significantly reduced p53 activation (Figure 2C, right). Sequencing the DNA encoding the DNA binding domain of p53 from these cells showed that they acquired a p53-R246Q mutation (Figure 2D), which has a known dominant negative function in mice (23,24). Interestingly, the R246Q mutation is due to a C>T-TCG POLE signature 10b mutation on the noncoding strand (Figure 2D), indicating that mutant Pole was likely directly responsible for p53 inactivation. Thymic tumors also had significantly reduced levels of p53 activation compared to normal thymus tissue, presumably allowing uncontrolled growth (Figure 2E). Taken together, these data establish, in two model systems, a direct link between mutant POLE-mediated mutagenesis and p53 activation and selective pressure to mutate the Trp53 to enable immortalization and transformation.
Loss of p53 has minimal impact on POLE-mediated tumor mutation accumulation
Since we had previously showed that Pole+/P286R could accelerate cancer mortality in mice and was associated with hypermutant POLE mutagenesis phenotype, we investigated the effect of p53 loss on mutagenesis. We selected four tumors from Pole+/P286R;Trp53+/– mice and performed whole exome sequencing. Each tumor had mutations in the remaining Trp53 allele, including one known loss of function mutation [R210C (25–27)], one loss of function frameshift mutation (Q97fs), one mutation that was shown to occur multiple times in human breast cancer patients [L194R (28)] and one charge reversal that was predicted to reduce function in a saturation mutagenesis screen [E14K (25)] (Figure 3A). These results indicate that tumors in Pole+/P286R;Trp53+/– mice demonstrate selective pressure for inactivating p53 in the development of tumors driven by Pole mutation. All four mutations occurred in POLE signature sequence contexts, strongly suggesting that the mutator polymerase first made the DNA synthesis error, and cells with this p53 mutation subsequently proliferated and expanded during tumor development.
Figure 3.

Mouse Pole+/P286R tumors display high mutation burden and POLE signatures independent of p53 status. (A) Spontaneous somatic Trp53 mutations arise in all tumors from germline Pole+/P286R;Trp53+/– mice. (B) Single nucleotide variants (SNVs) and insertion and deletion (indel) frameshift mutations in tumors from Pole+/P286R;Trp53+/+ and Pole+/P286R;Trp53+/– mice. For comparisons of the means between two groups, unpaired two-tailed t-test and Welch’s correction for nonequal SD were used. Mutation spectra of SNVs (C) and indels (D) from two Pole+/P286R;Trp53+/+ thymic tumors (top) and two Pole+/P286R;Trp53+/– splenic tumors (bottom) are shown.
Somewhat surprisingly, however, p53 status did not affect overall base pair substitution in tumors from Pole+/P286R mice (Figure 3B, left). We did notice that indel mutations were elevated in tumors from Pole+/P286R;Trp53+/– (Figure 3B, right), but this difference was not significant. All POLE mutation signatures (SBS 10a, 28 and 10b) were also present in tumors from Pole+/P286R;Trp53+/– mice (Figure 3C) (3), as were the more recently described +A insertions into runs of A nucleotides, particularly those ≥5 nt (29) (Supplementary Figure S2).
Using the TCGA (The Cancer Genome Atlas) endometrial and colorectal tumor datasets, we examined how tumor mutation burden and POLE signature mutations in human tumors are affected by the presence of TP53 mutations. We stratified all tumors with POLE driver mutations (9,30) into those with known TP53 driver mutations [TP53 mut, as defined by cBioPortal (31)] and those without known driver mutations (TP53 wt). The TP53 wt set may contain mutations of unknown significance. Consistent with our data in mice, incidence of SNVs and indels did not differ between POLE mutant tumors that were TP53 mutant or TP53 wild type (Figure 4A).
Figure 4.

Driver mutations in TP53 do not affect mutagenesis in human endometrial (EC) and colorectal (CRC) cancers from TCGA. (A) Mutation burden of SNVs (mut/Mb) and number of indels, categorized by POLE, POLE/MMR and MMR SBS signature status from human tumors. Comparison is stratified by TP53 driver mutation status. P-values indicated and determined by ANOVA. (B) Frequencies of TP53 driver mutations in human endometrial and colorectal tumors from the for TCGA dataset with mutations in POLE are no different than in POLE wild-type tumors. Fisher’s exact test was performed comparing all TP53 driver mutations, and P-values indicated above stacked bars. (C) The single POLE-dependent TP53 mutation hotspot observed in human tumors (R213) lacks the sequence context to be a hotspot in mouse Trp53. The threshold for declaring significance is P < 0.05.
Mutagenesis in POLE mutant tumors and cell lines has been shown to be a composite of several different signatures that depends in part on the mutant allele of POLE and whether MMR is functional or not (4,9). We asked whether p53 status differentially affected mutagenesis based on POLE and MMR status by categorizing tumors based on mutational signatures: POLE (SBS 10a, 10b or 28 inblue), POLE/MMR (SBS 14 ingreen) and MMR (32) (SBS 6, 15, 20, 21, 26 or 44 inred). TP53 driver mutation status did not affect SNVs or indels (Figure 4A) in POLE, POLE/MMR and MMR tumors. Taken together, these results suggest that the acceleration of tumor development in POLE mutant tumors that have lost p53 function is not due to simple increased or altered pattern of mutagenesis.
Lack of p53 mutation enrichment in human POLE mutant tumors
Previous analyses of cancer mutations showed enrichment for the p53 R213X mutation in POLE mutant tumors (13,14). In order to examine all TP53 driver mutations in POLE mutant tumors, we first defined POLE-mutated tumors (POLE mut) by two criteria as described: TMB >10 mut/Mb and the presence of SBS 10a, 10b and 28 (9). Tumors not satisfying both criteria were considered wild type for POLE (POLE wt). We limited this analysis to TCGA uterine and colorectal tumors (UCEC and CRC, respectively) since they account for 90% (61 of 68) of all POLE mutant tumors in the TCGA database. Samples in cBioPortal were further classified as having a driver mutation in TP53 or not (31,33) based on OncoKB and Cancer Hotspots databases of curated cancer mutations known to have functional and clinical relevance in model organisms and patients as described (34,35). One UCEC patient was not included as no specific gene mutation data were available in cBioPortal (TCGA-EY-A1G8).
Surprisingly, the overall frequency of TP53 driver mutations was no different in POLE-mutated and POLE wild-type tumors in the TCGA UCEC and CRC cohorts (Figure 4B). R213X mutations were clearly enriched in POLE mutant tumors, accounting for 33% and 80% of the TP53 driver set of mutations in UCEC and CRC tumors, respectively (Figure 4B). As others have noted, the R213 codon in human TP53 combined with the 3′ base of the immediate upstream codon results in the POLE signature hotspot, TCGA. POLE-dependent mutagenesis then generates an opal TGA nonsense mutation encoding R213X (Figure 4C). We note that the absence of p53 R213X mutations in our mouse tumor samples is easily explained by alternate arginine codon usage (CGC) in the mouse genome eliminating the POLE signature context.
Enrichment of mutations in other tumor suppressors in TCGA human tumors and mice
Since p53 driver mutations were not enriched in POLE-mutated tumors (Figure 4B), we examined the relationship between mutations in POLE and in other commonly mutated cancer genes and pathways. Driver mutations in PTEN, a negative regulator of the PI3K/AKT pathway, were significantly enriched in both UCEC and CRC (Figure 5A). This enrichment had been previously noted in two endometrial tumor studies (17,19), but not in TCGA. PTEN driver mutations are well known to be significantly enriched in UCEC tumors (36) and indeed we observed that 56% of all TCGA uterine tumors have such mutations. We found an even further increase in POLE mutant tumors, with almost all (94%) harboring a PTEN driver mutation (Figure 5A, P < 0.0001, Fisher’s exact test). While the absolute number of POLE mutant tumors is lower, this enrichment still occurred in CRC tumors (Figure 5A, P < 0.0001, Fisher’s exact test). We validated this PTEN driver mutation enrichment in POLE mutant tumors in two separate endometrial tumor cohorts: the CPTAC proteogenomic dataset (Figure 5B) and the MSK-IMPACTS endometrial dataset (Supplementary Figure S3). Mutations in POLE also occur in tumors other than uterine or colorectal, albeit quite infrequently. However, even in these disparate tumors we still found PTEN mutation enrichment in POLE mutant tumors (Figure 5B), further supporting the idea that these pathways interact.
Figure 5.

Driver mutations in PTEN are enriched in human and mouse POLE tumors. (A) Frequencies of PTEN driver mutations are significantly enriched in human POLE mutant endometrial and colorectal tumors versus tumors with wild-type POLE from the TCGA dataset. Fisher’s exact test was performed comparing all PTEN driver mutations, and P-values indicated above stacked bars. (B) The frequencies of PTEN driver mutations are also elevated in POLE mutant tumors in the CPTAC endometrial dataset as well as in tumor types in TCGA that are not otherwise enriched in POLE mutations (all other tumors). Fisher’s exact test was performed comparing all PTEN driver mutations, and P-values indicated above stacked bars. (C) Lollipop plot of PTEN mutations. Recurrent PTEN mutations (n > 2) from POLE mutant (above) and POLE wild-type tumors are plotted. Mutations occurring in POLE signature motifs are shown in blue. (D) For each of the recurrent human PTEN driver mutations observed in human POLE mutant tumors, the flanking nucleotide context is shown (above) with the mutated base pair shown in red. The same flanking nucleotides for the conserved amino acid residues in mouse Pten are also shown (below) to denote POLE mutation signature context presence (E7, F341) or absence (R130, E299).
Of all PTEN driver mutations in POLE mutant tumors, we found significant enrichment in those occurring in POLE signature contexts, including recurrent E7X, R130Q, E299X, F341C and F341V PTEN mutations (Figure 5B and C, and Supplementary Figures S4 and S5). R130 is critical for Pten catalytic activity (37,38) and F341V mutations have previously been shown to drive tumor formation in a mouse model (39). Together, these mutations accounted for 71% and 58% of all PTEN driver mutations in UCEC and CRC POLE mutant tumors, respectively, and also suggest that each was acquired due to aberrant DNA synthesis by the mutant DNA polymerase.
Based on the enrichment of PTEN mutations in human POLE-mutated tumors, we re-examined our Pole+/P286R mouse tumors [this study and (3)]. We found that Pten mutations were enriched in Pole+/P286R tumors from germline wild-type Trp53 mice (50%, Supplementary Table S3). These included multiple POLE signature mutations and several known driver mutations. We observed two occurrences of F341V Pten mutations in our mouse Pole mutant tumors. That this known PTEN driver mutation is recurrent in both human and mouse POLE mutant tumors strongly suggests a functional link between these two mutant alleles.
Analysis of protein levels in tumors can provide valuable information regarding the effects of gene mutations on protein function. We analyzed Pten protein levels in POLE mutant tumors reported in the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC) endometrial proteogenomics study (19). Pten levels were lower in all tumors relative to normal tissues (Figure 6A), consistent with PTEN mutation enrichment in endometrial tumors leading to reduced function. While PTEN nonsense mutations were weakly associated with lower Pten levels across all tumors, POLE mutant tumors with nonsense mutations had among the lowest Pten protein levels measured (Figure 6B). In the few samples that had matched normal and tumor tissues, POLE mutant tumors with nonsense mutations in PTEN were associated with the strongest decrease in Pten protein abundance (Figure 6C). Interestingly, the PTEN R130Q mutation by itself saw no change in Pten protein levels, while the F341V mutations showed lower, though not statistically significant, reductions in Pten protein levels (Figure 6B). As previously reported for all tumors in the CPTAC set, p53 levels in POLE-mutated tumors were not significantly different from those in adjacent normal tissue, even when accounting for p53 mutation status (19), tumor subtypes (Supplementary Figure S6A) or within an individual with matched adjacent normal tissue (Supplementary Figure S6B).
Figure 6.

POLE-mediated PTEN mutations predict different effects on Pten protein abundance in human tumors. (A)PTEN protein abundance (z-score) of adjacent normal tissue and endometrial tumors, categorized into the four genomic subtypes, POLE, MSI, CNV-low [endometroid-like (ENDO)] or CNV-high [serous-like (SEROUS)] (22,39). P-values indicated and determined by ANOVA. (B) Pten protein abundance (z-score) in tumors categorized by PTEN mutation status. Indicated P-values were determined by ANOVA. (C) Changes in individual tumor Pten protein abundance relative to matched normal (ΔZ) were plotted based on PTEN mutation status.
Strikingly, we found that the same PTEN-F341V recurrent mutation in human tumors also arose spontaneously in two different Pole mice. Engineered PtenF341V/– mice develop tumors with a different tissue spectra than Pten–/– mice (39). Caserta et al. made this mouse model based on observations that PTEN-F341V mutations were recurrent in human endometrial tumors. Re-analysis of the 10 967 tumors in the TCGA PanCancer studies shows that all 9 PTEN-F341V/C mutations occur in human tumors with POLE driver mutations. Leone et al. further described the Pten-F341V mutation in mice as contributing to tumor development through a noncanonical nuclear genome stability role for Pten (39). They showed evidence of reduced nuclear Pten protein coinciding with increased DNA damage in cells and tumors from these mice. We found γ-H2AX foci significantly enriched in a Pole+/P286R tumor that spontaneously acquired a Pten-F341V mutation (Figure 7), consistent with what was seen in a tumor from germline PtenF341V/– mice, and consistent with a role for Pten in maintaining genome stability. Another mutation observed in our mouse cohort, Pten-D24L, is associated with lack of cytoplasm localization (40). This is supported by lack of staining in the tumor that harbors this mutation. The repeated co-occurrence of the PTEN-F341V mutation with mutations in POLE from tumors in both humans and mice suggests that both mutations may potentially cooperate in generating and subsequently failing to protect from genome destabilizing DNA damaging events or stress that contributes to tumorigenesis.
Figure 7.

Pten mutations have different effects on Pten subcellular localization and DNA damage levels in mouse tumors. Tumors from mice with the indicated Pole and Trp53 genotypes and somatic Pten mutations were processed and stained for DAPI (blue) and Pten (green) and γ-H2AX (red). Shown are representative confocal images at 10× magnification and zoomed-in insets.
DISCUSSION
Loss of p53 function, which rescues viability of many mutant alleles, including Pole4–/–, failed to render PoleP286R/P286R mice viable. Inactivation of one Trp53 allele accelerated tumorigenesis via POLE-mediated mutation in the remaining Trp53 allele. Data collected from cells suggest that the mutant Pole-P286R polymerase activates p53, suggesting possible selective pressure toward inactivating p53 via mutation. Mutagenesis per se is unlikely to be the primary cause of p53 activation, as we observed no difference in mutation burden or signature between mouse tumors with or without p53 mutations. Instead, POLE mutant tumors are significantly biased toward mutations in PTEN. The recurrence in both human and mouse tumors of a PTEN mutation that specifically reduces nuclear Pten abundance and fails to guard against genome instability suggests involvement of mutant Pole in the replication stress that is normally surveilled by Pten.
As the most frequently mutated gene in cancer, loss of p53 function, whether through mutational inactivation or inhibition, has long been known to accelerate tumor development (41,42). Despite this vast knowledge, it was unclear until recently to what extent loss of p53 function impacted base pair substitutions and indels. A comprehensive analysis of 10 000 whole exomes from tumors in TCGA found a minimal effect (2.2-fold) of TP53 mutations on overall tumor mutation burden and no change in mutational signatures (43). The lack of unique mutational signatures in p53 mutant tumors led to the idea that loss of p53 function is unlikely to cause de novo mutagenesis, but instead permits the accumulation of mutations via other pathways (25,43). We find that this remains true in POLE mutant tumors where there is no difference in mutation burden or signature between those tumors with or without p53 driver mutations.
Several lines of evidence have pointed toward p53 inactivation being a late event in POLE mutant tumors. Li et al. showed that in mouse endometrial Pole mutant tumors, p53 staining was increased but only in subclonal fractions of the tumor indicating that p53 mutation was a late event (16). They further showed that p53 mutation was correlated with evidence of tetraploidization. Though POLE mutant tumors are characterized by base pair substitutions, tetraploidy has been seen in some human POLE mutant endometrial tumors (44). Molecular classification studies have also shown p53 mutations to be late and subclonal events in human endometrial tumors (45). It was concluded based on several factors that these p53 mutations were passenger events (46) underscoring the need for a more detailed mechanistic understanding.
Coupled with this absence of an effect on mutagenesis, the relative lack of TP53 and Trp53 mutations in human and mouse tumors led us to explore other possible cancer driver gene mutations that might play roles in driving POLE mutant tumor development. This type of analysis is complicated by the fact that since POLE mutant tumors acquire so many mutations, essentially every gene has a mutation. Thus, discerning driver mutations from passengers presents certain challenges. Our previously engineered Pole+/P286R mice with wild-type Trp53 provided an excellent opportunity to address this complication.
Potential cancer driver genes contributing to POLE mutant tumorigenesis would be predicted to share several characteristics. Mutations should be enriched in the same gene in both human and mouse POLE mutant tumors. Mutations either should be enriched in the same residue or, if homology is low, then should be enriched in functionally conserved residues or motifs. Mutations should occur repeatedly, but not necessarily exclusively, in POLE signature motifs. Finally, ideally, there should be evidence indicating that those mutations disrupt normal gene product function. Mutations satisfying all these criteria in both mouse and human POLE mutant tumors would provide corroborating evidence supporting the idea that those genes are in fact cancer driver genes.
Mutations in PTEN satisfy all these criteria and further hint at possible mechanisms of how Pten dysfunction may interact with the mutant polymerase to promote tumor development. First, PTEN mutations are significantly enriched in both mouse and human POLE mutant tumors. More than 90% of TCGA endometrial tumors with mutations in POLE also have a known driver mutation in PTEN (Figure 5A), and enrichment is also seen in colorectal cancers. POLE mutant tumors with PTEN mutations also occur sporadically in other tumor types.
Several specific PTEN mutations occur in both human and mouse POLE mutant tumors, further supporting the idea that the PTEN and POLE pathways are interacting. Once such mutation, F341V/C, occurred twice in our mouse tumors and is recurrent in human POLE mutant tumors, likely due to a combination of them occurring in a POLE signature context and its unique effects on Pten function discussed later. The PTEN-F154, Y68 and D24 mutations found in our POLE mouse tumors are also recurrent in human tumors with wild-type POLE, suggesting that these are not simply passenger mutations (Supplementary Table S4).
Mutational signatures can also explain both the occurrence of a hotspot mutation and differences between species abundance. The strongest PTEN hotspot mutation (R130Q) and highly recurrent nonsense mutation (E299X) observed in human POLE mutant tumors both have different sequence contexts in mice that preclude their mutation in mouse tumors (Figure 5C), thus explaining their absence in our mouse tumors. The E7X mutation, however, is recurrent in human tumors, yet absent in mice. The simplest explanation is that our tumor sample size is too small.
The functional effects of mutations in PTEN have been examined extensively using many different assays. With the exception of F154L, all Pten mutations we observed in mouse POLE mutant tumors either are known to cause loss of function (39,47) or are immediately adjacent to a known susceptible residue (I122L). Just looking at known PTEN driver mutations, we find that nonsense mutations significantly reduce the amount of Pten present in actual human POLE mutant tumors (Figures 5 and 6). The unchanged levels of Pten-R130Q and only modestly changed Pten-F341V/C suggest that these missense mutations may have novel functions critical to enhancing POLE mutant tumor development.
How might Pten defects help promote POLE mutant tumor development? The simplest explanation is through canonical loss of Pten and subsequent PI3K-AKT activation of downstream targets that enhance cellular growth. This is the likely situation in tumors with PTEN nonsense mutations. However, several lines of evidence suggest that the primary mechanism might be more complicated, possibly through increased replication stress tolerance. Pten-F341 is a recurrent mutation site in both mouse and human POLE mutant tumors, yet PtenF341V/F341V mice develop tumors while retaining normal Akt activation and embryonic development (39). Heterozygous mutations at F341 are sufficient to drive tumorigenesis as Pten+/F341V mice develop tumors in a number of different organs, including the thymus. Even more intriguing is the observation that Pten is depleted specifically from nuclei from PtenF341V mice, which also coincides with increased incidence of γ-H2AX foci. These results are consistent with other work showing that Pten has nuclear roles in suppressing genome instability, chromatin compaction, chromosome condensation, checkpoint control and replisome stability (48,49). Pten has both phosphatase-dependent and -independent activities that are critical in promoting both normal replisome assembly and stalled fork protection and reassembly, particularly through targets including Mcm2, Rpa1 and Rad51 (50–52).
Nonsense mutations (e.g. E7X and E299X) and other missense mutations that destabilize the Pten protein (e.g. R173C/H and F341C/V) would certainly lose the ability to regulate the PI3K-AKT pathway. However, they would also have the same loss of function effects on nuclear Pten activities. The remaining PTEN hotspot mutation in POLE mutant tumors is R130Q. The R130G mutation makes a stable protein in cells and both R130G and R130Q show increased p-Akt in glioblastomas and endometrial tumors, suggesting loss of lipid phosphatase activity in POLE mutant tumors (53). However, mutations at R130 have been proposed to act by suppressing the activity of the wild-type partner in a mixed heterodimer in a dominant negative fashion. Since the effects of R130 dominant negative mutation have not yet been tested on nuclear Pten function, it is tempting to speculate that the enriched PTEN-R130Q is specifically helping facilitate POLE mutant tumor development via interfering with a nuclear genome stabilizing activity. Loss of PI3K-AKT regulation would serve as an added unrelated tumorigenic boost.
Our observations that POLE mutant tumor formation in Trp53+/– mice is accelerated, likely via Trp53 LOH, is also consistent with mutant Pole alleles triggering replisome dysfunction. We showed that cells expressing the mutant polymerase enzyme activate p53. Both p53 and Pten could be involved in suppressing possible resulting genome instability. Normally two hits would be required to inactivate p53 and relieve this suppression, while a single hit to Pten would begin to relieve this suppression. The observation that mutant Pol ϵ has hyperpolymerization characteristics (54) is particularly intriguing in light of our results in POLE mutant tumors.
It should be noted that both the PoleP286R and Trp53– mutant alleles used in our mouse studies are germline. While there are human patients with germline mutations in the Pol ϵ exonuclease domain, V411L and L424V/I are overrepresented and P286R has not been observed (55). This is of interest as these are either weak mutators or not mutators at all in yeast, while the yeast P268R ortholog (P310R) is a strong mutator (8). As such, caution is warranted in extrapolating from our results in mice to the situation in humans.
Taken together, we propose that development of tumors with mutations in POLE depends on two features. The elevated mutagenesis is required to supply the loss of function mutations in specific genes, but by itself is insufficient to drive tumor formation. Also required is a loss of genome instability suppression activity, possibly involving replisomes that have become dysfunctional via engaging the mutant Pol ϵ.
DATA AVAILABILITY
The datasets generated during this study are available at the NCBI Sequence Read Archive (SRA), accession code: PRJNA589337.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr Hua Lu for advice on the manuscript.
Author contributions: Conceptualization, V.S.P., J.G.J. and Z.F.P. Investigation, V.S.P., M.J.S.S., W.D.F., L.G.W., K.P.H., J.D.S. and S.J.W. Data curation, L.G.W. Writing—original draft, V.S.P. and Z.F.P. Writing—review and editing, V.S.P., M.J.S.S., L.G.W., J.G.J. and Z.F.P.
Notes
Present address: Vivian S. Park, AbbVie Inc., North Chicago, IL, USA.
Present address: Karl P. Hodel, Foundation Oncology, La Jolla, CA, USA.
Contributor Information
Vivian S Park, Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, USA.
Meijuan J S Sun, Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, USA.
Wesley D Frey, Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, USA.
Leonard G Williams, Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, USA; BioInnovation Program, Tulane University, New Orleans, LA, USA.
Karl P Hodel, Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, USA.
Juliet D Strauss, Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, USA.
Sydney J Wellens, Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, USA.
James G Jackson, Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, USA; Tulane Cancer Center, Tulane University School of Medicine, 1430 Tulane Avenue, New Orleans, LA, USA.
Zachary F Pursell, Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, USA; Tulane Cancer Center, Tulane University School of Medicine, 1430 Tulane Avenue, New Orleans, LA, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
Department of Defense (DoD) Breast Cancer Research Program (BCRP) Breakthrough Award (in part) [to J.G.J.]; National Institute of Environmental Health Sciences [R01ES028271 to Z.F.P.]; Tulane University Carol Lavin Bernick Faculty Grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N.et al.. The repertoire of mutational signatures in human cancer. Nature. 2020; 578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campbell B.B., Light N., Fabrizio D., Zatzman M., Fuligni F., de Borja R., Davidson S., Edwards M., Elvin J.A., Hodel K.P.et al.. Comprehensive analysis of hypermutation in human cancer. Cell. 2017; 171:1042–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Galati M.A., Hodel K.P., Gams M.S., Sudhaman S., Bridge T., Zahurancik W.J., Ungerleider N.A., Park V.S., Ercan A.B., Joksimovic L.et al.. Cancers from novel pole-mutant mouse models provide insights into polymerase-mediated hypermutagenesis and immune checkpoint blockade. Cancer Res. 2020; 80:5606–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haradhvala N.J., Kim J., Maruvka Y.E., Polak P., Rosebrock D., Livitz D., Hess J.M., Leshchiner I., Kamburov A., Mouw K.W.et al.. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun. 2018; 9:1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Church D.N., Briggs S.E.W., Palles C., Domingo E., Kearsey S.J., Grimes J.M., Gorman M., Martin L., Howarth K.M., Hodgson S.V.et al.. DNA polymerase ϵ and δ exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013; 22:2820–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levine D.A.The Cancer Genome Atlas Research Network The Cancer Genome Atlas Research Network Getz G., Gabriel S.B., Cibulskis K., Lander E., Sivachenko A., Sougnez C., Lawrence M., Kandoth C.et al.. Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Palles C., Cazier J.-B., Howarth K.M., Domingo E., Jones A.M., Broderick P., Kemp Z., Spain S.L., Almeida E.G., Salguero I.et al.. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013; 45:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barbari S.R., Kane D.P., Moore E.A., Shcherbakova P.V.. Functional analysis of cancer-associated DNA polymerase epsilon variants in Saccharomyces cerevisiae. G3 (Bethesda). 2018; 8:1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hodel K.P., Sun M.J.S., Ungerleider N., Park V.S., Williams L.G., Bauer D.L., Immethun V.E., Wang J., Suo Z., Lu H.et al.. POLE mutation spectra are shaped by the mutant allele identity, its abundance, and mismatch repair status. Mol. Cell. 2020; 78:1166–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Herzog M., Alonso-Perez E., Salguero I., Warringer J., Adams D.J., Jackson S.P., Puddu F.. Mutagenic mechanisms of cancer-associated DNA polymerase alleles. Nucleic Acids Res. 2021; 49:3919–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jacks T., Remington L., Williams B.O., Schmitt E.M., Halachmi S., Bronson R.T., Weinberg R.A.. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994; 4:1–7. [DOI] [PubMed] [Google Scholar]
- 12. Bellelli R., Borel V., Logan C., Svendsen J., Cox D.E., Nye E., Metcalfe K., O’Connell S.M., Stamp G., Flynn H.R.et al.. Pol epsilon instability drives replication stress, abnormal development, and tumorigenesis. Mol. Cell. 2018; 70:707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang H., Barbour J.A., Poulos R.C., Katainen R., Aaltonen L.A., Wong J.W.H.. Mutational processes of distinct POLE exonuclease domain mutants drive an enrichment of a specific TP53 mutation in colorectal cancer. PLoS Genet. 2020; 16:e1008572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shinbrot E., Henninger E.E., Weinhold N., Covington K.R., Goksenin A.Y., Schultz N., Chao H., Doddapaneni H., Muzny D.M., Gibbs R.A.et al.. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014; 78:1740–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Poulos R.C., Olivier J., Wong J.W.H.. The interaction between cytosine methylation and processes of DNA replication and repair shape the mutational landscape of cancer genomes. Nucleic Acids Res. 2017; 45:7786–7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H.D., Lu C., Zhang H., Hu Q., Zhang J., Cuevas I.C., Sahoo S.S., Aguilar M., Maurais E.G., Zhang S.et al.. A PoleP286R mouse model of endometrial cancer recapitulates high mutational burden and immunotherapy response. JCI Insight. 2020; 5:e138829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hatakeyama K., Ohshima K., Nagashima T., Ohnami S., Ohnami S., Serizawa M., Shimoda Y., Maruyama K., Akiyama Y., Urakami K.et al.. Molecular profiling and sequential somatic mutation shift in hypermutator tumours harbouring POLE mutations. Sci. Rep. 2018; 8:8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kandoth C., Schultz N., Cherniack A.D., Akbani R., Liu Y., Shen H., Robertson A.G., Pashtan I., Shen R., Benz C.C.et al.. Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dou Y., Kawaler E.A., Cui Zhou D., Gritsenko M.A., Huang C., Blumenberg L., Karpova A., Petyuk V.A., Savage S.R., Satpathy S.et al.. Proteogenomic characterization of endometrial carcinoma. Cell. 2020; 180:729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bellelli R., Belan O., Pye V.E., Clement C., Maslen S.L., Skehel J.M., Cherepanov P., Almouzni G., Boulton S.J.. POLE3–POLE4 is a histone H3–H4 chaperone that maintains chromatin integrity during DNA replication. Mol. Cell. 2018; 72:112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li H.D., Cuevas I., Zhang M., Lu C., Alam M.M., Fu Y.X., You M.J., Akbay E.A., Zhang H., Castrillon D.H.. Polymerase-mediated ultramutagenesis in mice produces diverse cancers with high mutational load. J. Clin. Invest. 2018; 128:4179–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Todaro G.J., Green H.. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963; 17:299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aubrey B.J., Janic A., Chen Y., Chang C., Lieschke E.C., Diepstraten S.T., Kueh A.J., Bernardini J.P., Dewson G., O’Reilly L.A.et al.. Mutant TRP53 exerts a target gene-selective dominant-negative effect to drive tumor development. Genes Dev. 2018; 32:1420–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanel W., Marchenko N., Xu S., Yu S.X., Weng W., Moll U.. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013; 20:898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giacomelli A.O., Yang X., Lintner R.E., McFarland J.M., Duby M., Kim J., Howard T.P., Takeda D.Y., Ly S.H., Kim E.et al.. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018; 50:1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marutani M., Tonoki H., Tada M., Takahashi M., Kashiwazaki H., Hida Y., Hamada J., Asaka M., Moriuchi T.. Dominant-negative mutations of the tumor suppressor p53 relating to early onset of glioblastoma multiforme. Cancer Res. 1999; 59:4765–4769. [PubMed] [Google Scholar]
- 27. Pan Y., Haines D.S.. Identification of a tumor-derived p53 mutant with novel transactivating selectivity. Oncogene. 2000; 19:3095–3100. [DOI] [PubMed] [Google Scholar]
- 28. Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L.B., Martin S., Wedge D.C.et al.. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chung J., Maruvka Y.E., Sudhaman S., Kelly J., Haradhvala N.J., Bianchi V., Edwards M., Forster V.J., Nunes N.M., Galati M.A.et al.. DNA polymerase and mismatch repair exert distinct microsatellite instability signatures in normal and malignant human cells. Cancer Discov. 2021; 11:1176–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park V.S., Pursell Z.F.. POLE proofreading defects: contributions to mutagenesis and cancer. DNA Repair. 2019; 76:50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E.et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nemeth E., Lovrics A., Gervai J.Z., Seki M., Rospo G., Bardelli A., Szuts D.. Two main mutational processes operate in the absence of DNA mismatch repair. DNA Repair. 2020; 89:102827. [DOI] [PubMed] [Google Scholar]
- 33. Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E.et al.. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chakravarty D., Gao J., Phillips S.M., Kundra R., Zhang H., Wang J., Rudolph J.E., Yaeger R., Soumerai T., Nissan M.H.et al.. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 2017; 2017:PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang M.T., Bhattarai T.S., Schram A.M., Bielski C.M., Donoghue M.T.A., Jonsson P., Chakravarty D., Phillips S., Kandoth C., Penson A.et al.. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 2018; 8:174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dedes K.J., Wetterskog D., Ashworth A., Kaye S.B., Reis-Filho J.S.. Emerging therapeutic targets in endometrial cancer. Nat. Rev. Clin. Oncol. 2011; 8:261–271. [DOI] [PubMed] [Google Scholar]
- 37. Lee J.O., Yang H., Georgescu M.M., Di Cristofano A., Maehama T., Shi Y., Dixon J.E., Pandolfi P., Pavletich N.P.. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999; 99:323–334. [DOI] [PubMed] [Google Scholar]
- 38. Smith I.N., Briggs J.M.. Structural mutation analysis of PTEN and its genotype–phenotype correlations in endometriosis and cancer. Proteins. 2016; 84:1625–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caserta E., Egriboz O., Wang H., Martin C., Koivisto C., Pecot T., Kladney R.D., Shen C., Shim K.S., Pham T.et al.. Noncatalytic PTEN missense mutation predisposes to organ-selective cancer development in vivo. Genes Dev. 2015; 29:1707–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Denning G., Jean-Joseph B., Prince C., Durden D.L., Vogt P.K.. A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene. 2007; 26:3930–3940. [DOI] [PubMed] [Google Scholar]
- 41. Vogelstein B., Lane D., Levine A.J.. Surfing the p53 network. Nature. 2000; 408:307–310. [DOI] [PubMed] [Google Scholar]
- 42. Wasylishen A.R., Lozano G.. Attenuating the p53 pathway in human cancers: many means to the same end. Cold Spring Harb. Perspect. Med. 2016; 6:a026211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Donehower L.A., Soussi T., Korkut A., Liu Y., Schultz A., Cardenas M., Li X., Babur O., Hsu T.K., Lichtarge O.et al.. Integrated analysis of TP53 gene and pathway alterations in The Cancer Genome Atlas. Cell Rep. 2019; 28:3010. [DOI] [PubMed] [Google Scholar]
- 44. Proctor L., Pradhan M., Leung S., Cheng A., Lee C.H., Soslow R.A., Gilks C.B., Talhouk A., McAlpine J.M., Danielsen H.E.et al.. Assessment of DNA ploidy in the ProMisE molecular subgroups of endometrial cancer. Gynecol. Oncol. 2017; 146:596–602. [DOI] [PubMed] [Google Scholar]
- 45. Leon-Castillo A., Britton H., McConechy M.K., McAlpine J.N., Nout R., Kommoss S., Brucker S.Y., Carlson J.W., Epstein E., Rau T.T.et al.. Interpretation of somatic POLE mutations in endometrial carcinoma. J. Pathol. 2020; 250:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leon-Castillo A., Gilvazquez E., Nout R., Smit V.T., McAlpine J.N., McConechy M., Kommoss S., Brucker S.Y., Carlson J.W., Epstein E.et al.. Clinicopathological and molecular characterisation of ‘multiple-classifier’ endometrial carcinomas. J. Pathol. 2020; 250:312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Post K.L., Belmadani M., Ganguly P., Meili F., Dingwall R., McDiarmid T.A., Meyers W.M., Herrington C., Young B.P., Callaghan D.B.et al.. Multi-model functionalization of disease-associated PTEN missense mutations identifies multiple molecular mechanisms underlying protein dysfunction. Nat. Commun. 2020; 11:2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fan X., Kraynak J., Knisely J.P.S., Formenti S.C., Shen W.H.. PTEN as a guardian of the genome: pathways and targets. Cold Spring Harb. Perspect. Med. 2020; 10:a036194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ho J., Cruise E.S., Dowling R.J.O., Stambolic V.. PTEN nuclear functions. Cold Spring Harb. Perspect. Med. 2020; 10:a036079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Feng J., Liang J., Li J., Li Y., Liang H., Zhao X., McNutt M.A., Yin Y.. PTEN controls the DNA replication process through MCM2 in response to replicative stress. Cell Rep. 2015; 13:1295–1303. [DOI] [PubMed] [Google Scholar]
- 51. He J., Kang X., Yin Y., Chao K.S., Shen W.H.. PTEN regulates DNA replication progression and stalled fork recovery. Nat. Commun. 2015; 6:7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang G., Li Y., Wang P., Liang H., Cui M., Zhu M., Guo L., Su Q., Sun Y., McNutt M.A.et al.. PTEN regulates RPA1 and protects DNA replication forks. Cell Res. 2015; 25:1189–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Papa A., Wan L., Bonora M., Salmena L., Song M.S., Hobbs R.M., Lunardi A., Webster K., Ng C., Newton R.H.et al.. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014; 157:595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xing X., Kane D.P., Bulock C.R., Moore E.A., Sharma S., Chabes A., Shcherbakova P.V.. A recurrent cancer-associated substitution in DNA polymerase epsilon produces a hyperactive enzyme. Nat. Commun. 2019; 10:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Palles C., Cazier J.B., Howarth K.M., Domingo E., Jones A.M., Broderick P., Kemp Z., Spain S.L., Almeida E.G., Salguero I.et al.. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2012; 45:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at the NCBI Sequence Read Archive (SRA), accession code: PRJNA589337.