

Abstract
The actin motor protein myosin VI is a multivalent protein with diverse functions. Here, we identified and characterised a myosin VI ubiquitous interactor, the oral‐facial‐digital syndrome 1 (OFD1) protein, whose mutations cause malformations of the face, oral cavity, digits and polycystic kidney disease. We found that myosin VI regulates the localisation of OFD1 at the centrioles and, as a consequence, the recruitment of the distal appendage protein Cep164. Myosin VI depletion in non‐tumoural cell lines causes an aberrant localisation of OFD1 along the centriolar walls, which is due to a reduction in the OFD1 mobile fraction. Finally, loss of myosin VI triggers a severe defect in ciliogenesis that could be, at least partially, ascribed to an impairment in the autophagic removal of OFD1 from satellites. Altogether, our results highlight an unprecedent layer of regulation of OFD1 and a pivotal role of myosin VI in coordinating the formation of the distal appendages and primary cilium with important implications for the genetic disorders known as ciliopathies.
Keywords: autophagy receptor, centrioles, myosin VI, OFD1, primary cilium
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton
The actin motor myosin VI is critical for the correct localization of the Oral‐facial‐digital syndrome 1 protein (OFD1) at the centrioles distal tip and for OFD1 removal from the centriolar satellites during primary ciliogenesis.

Introduction
Primary cilia are sensory structures extending from the surface of mammalian cells, with a major role in several signalling pathways essential for growth and differentiation, such as the Hedgehog and the Wnt signalling pathways (Berbari et al, 2009; Malicki & Johnson, 2017). The formation of the primary cilium is a highly regulated multi‐step process that occurs in cells that exit the cell cycle and become quiescent. The primary cilium originates from the older centriole, called mother centriole, which docks to the plasma membrane and acts as basal body for the assembly of the microtubule ciliary axoneme (Sanchez & Dynlacht, 2016). Impairment in the formation or function of the primary cilia leads to a variety of severe genetic syndromes, termed ciliopathies (Reiter & Leroux, 2017). This group of disorders includes the oral‐facial‐digital (OFD) type I, the Simpson–Golabi–Behmel type 2 and the Joubert syndromes, which are caused by mutations in the oral‐facial‐digital syndrome 1 (OFD1) gene (Feather et al, 1997; Ferrante et al, 2001, 2006; Budny et al, 2006; Coene et al, 2009; Zullo et al, 2010; Field et al, 2012). The mechanisms underlying many of the disease phenotypes associated with ciliary dysfunction have yet to be fully elucidated. Dissecting the regulatory mechanisms of OFD1 has the potential to offer new therapeutic tools for the treatment of ciliopathies.
OFD1 is a protein component of the centrioles and pericentriolar satellites and acts both as a positive and negative regulator of primary ciliogenesis (Ferrante et al, 2006; Tang et al, 2013; Morleo & Franco, 2020). The localisation of OFD1 is differentially regulated depending on the cell compartment. At the centrioles, OFD1 is recruited to the distal tip through the interaction with the C2 domain containing 3 centriole elongation regulator (C2CD3), a protein required for centriole elongation (Thauvin‐Robinet et al, 2014). OFD1 recruitment is under the control of a subset of proteins that are present only in the daughter centriole (daughter centriole‐specific proteins), but the molecular link is unknown (Wang et al, 2018). OFD1 in turn promotes the recruitment of the distal appendages, structures present on the distal side of the mother centriole. These structures are essential for the docking of the mother centriole to the cellular membrane and its conversion to basal body, required for ciliogenesis (Singla et al, 2010; Wang et al, 2018). Furthermore, OFD1 is required for the recruitment of IFT88, a protein essential for the assembly of the primary cilium (Singla et al, 2010). Despite the growing literature that explores the process of primary cilium formation, the mechanistic details of this sequential recruitment of C2CD3, OFD1 and distal appendages remain unclear.
OFD1 is also localised at the centriolar satellites, which are non‐membrane electron‐dense particles containing regulatory proteins of centrosomes and cilia (Lopes et al, 2011). This localisation depends on the trafficking protein particle complex subunit 8 (TRAPPC8), which mediates the interaction between OFD1 and the main structural component of the centriolar satellites, namely PCM1 (Zhang et al, 2020).
While the centriolar pool of OFD1 is rather stable, the satellite pool shows a higher turnover (Tang et al, 2013). Indeed, upon serum starvation, OFD1 is removed from the satellites by autophagy, and this process is required for ciliogenesis (Tang et al, 2013; Park et al, 2018). Interestingly, the depletion of OFD1 from the satellites is sufficient to induce ciliogenesis also in autophagy‐impaired cells (Tang et al, 2013), indicating that OFD1 turnover at the satellites controls per se the formation of the primary cilium. Moreover, OFD1 appears to regulate autophagosome biogenesis in a feedback loop that aims at limiting autophagy activation (Morleo et al, 2021).
A proteomic study identified OFD1 as a possible interactor of myosin VI (O'Loughlin et al, 2018), a unique motor protein that moves towards the minus end of the actin filaments (Magistrati & Polo, 2020). The molecular pathways and the physiological relevance of this interaction remain unknown. Here, we characterised the interaction between the two proteins and we investigated the role of myosin VI in OFD1 regulation.
Results
OFD1 interacts with both myosin VI isoforms
The functional and phenotypic diversity associated with myosin VI arises from multiple interactors and the existence of alternatively spliced isoforms (Magistrati & Polo, 2020). Indeed, two distinct isoform types, myosin VI short and myosin VI long, differ in an isoform‐specific helix that modifies their conformation and restricts binding to their interactomes (Wollscheid et al, 2016). With the aim of capturing common interactors, we used co‐immunoprecipation assay and analysed by mass spectrometry the interactome of endogenous myosin VI in a set of cell lines of epithelial origin expressing the different isoforms at various level, namely MDA‐MB‐231, HeLa, MCF10A and Caco‐2 cells (Fig EV1A and B and Dataset EV1). Thirteen proteins were found in all four cell lines (Fig 1A) and most of them were identified as myosin VI interactors for the first time. We focused our attention on OFD1 and we validated the interaction between myosin VI and OFD1, using a second anti‐myosin VI antibody (Fig 1B) and by performing a reverse co‐immunoprecipitation experiment (Fig 1C).
Figure EV1. Myosin VI localisation at the centrosome.
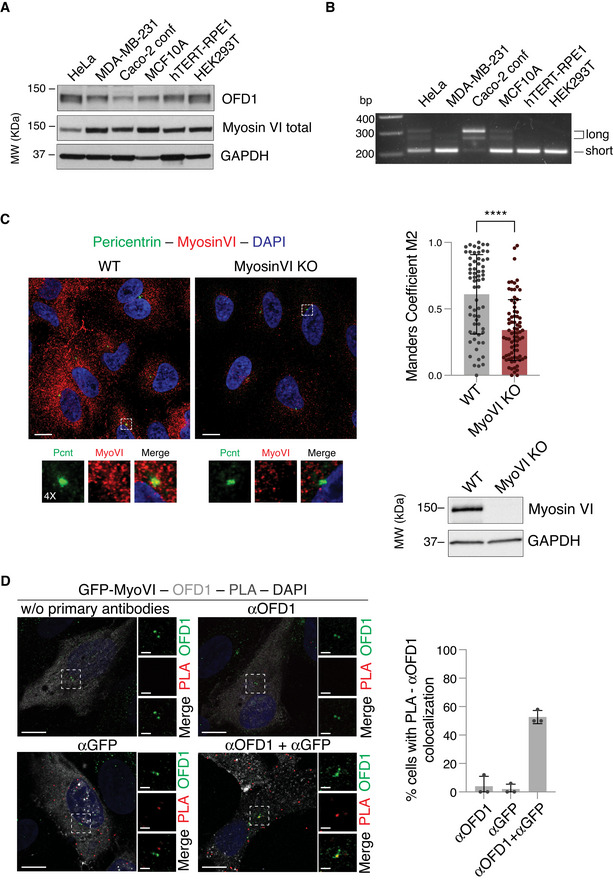
- Total lysates from the indicated cell lines were IB as indicated. No major difference at the level of expression level is visible.
- Expression of myosin VI isoforms in the indicated cell lines was assessed by RT–PCR analysis as previously described (Wollscheid et al, 2016). Caco‐2 cells grown at confluency express mainly myosin VI long isoform.
- A549 wild‐type and myosin VI KO cells were immunostained with anti‐myosin VI and anti‐pericentrin antibodies. Co‐localisation between myosin VI and pericentrin is lost in myosin VI KO cells, as evaluated by Manders’ coefficient M2 that measures the fraction of pericentrin overlapping with myosin VI staining. Bars represent mean ± SD. n = 64‐77 cells from two independent experiments. ****P < 0.0001 by Kruskal–Wallis test. Representative images are shown; scale bar, 10 μm. Bottom panel, total lysates from the indicated cell lines were IB as indicated.
- Proximity ligation assay (PLA) in hTERT‐RPE1 cells overexpressing GFP‐myosin VIshort full‐length, using anti‐OFD1 and anti‐GFP antibodies. After PLA assay, counterstaining with fluorescently labelled secondary antibodies allowed the detection of OFD1 and GFP localisation. Left: representative images of technical controls with single or no primary antibodies are shown. Scale bar, 10 μm (2 μm for the magnification). Right: quantification of the percentage of GFP‐positive cells that show co‐localisation between OFD1 and PLA signal. Bars represent mean ± SD. n = 3 independent experiments (46‐58 cells/condition).
Source data are available online for this figure.
Figure 1. OFD1 interacts with both myosin VI isoforms.
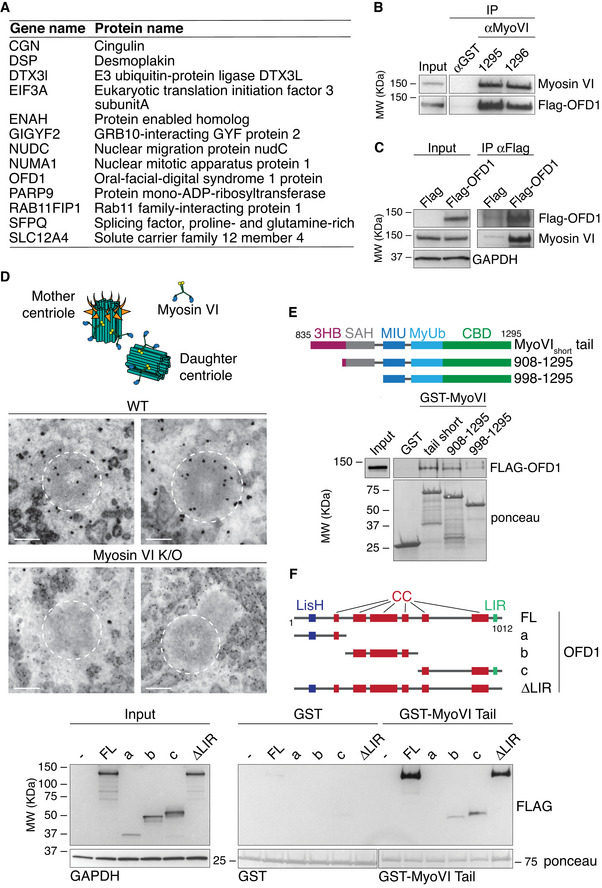
- List of myosin VI interactors commonly found in the four cell lines tested. See Dataset EV1 for the full list of interactors.
- Total lysates from HEK293T transfected with Flag‐OFD1 were IP with anti‐myosin VI antibodies (1295 and 1296) and an unrelated rabbit antibody (anti‐GST, as control). IB was performed with anti‐Flag and anti‐myosin VI antibodies.
- Total lysates from HEK293T transfected with Flag‐OFD1 or Flag (as control) were IP with anti‐Flag antibody‐conjugated beads. IB was performed with anti‐Flag and anti‐myosin VI antibodies.
- Selected sections deriving from TEM analysis of A549 wild‐type versus myosin VI KO cells. The cells were immunogold‐labelled with anti‐myosin VI antibody. The centrioles in the images are indicated with white dashed circles. Scale bar, 200 nm. Top: representation of the estimated localisation of myosin VI at the centrioles.
- GST pull‐down assay using the indicated myosin VI constructs or GST alone (as control) and lysates from HEK293T cells transfected with Flag‐OFD1 construct. IB was performed with anti‐Flag antibody. Ponceau staining as indicated.
- GST pull‐down assay using myosin VI tail (835–1295) construct (or GST alone as control) and lysates from HEK293T cells transfected with the indicated Flag‐OFD1 constructs (or Flag alone, as control). IB was performed with anti‐Flag antibody. Ponceau staining as indicated.
Source data are available online for this figure.
Since OFD1 is a centriolar protein, we assessed the localisation of myosin VI in this organelle. By immunofluorescence (Fig EV1C) and proximity ligation assay (PLA, Fig EV1D), we could demonstrate that myosin VI localises at centrioles in correspondence of OFD1 staining, suggesting that the interaction between the two proteins can occur at least at the level of the centrioles. To further confirm this idea, we performed immunogold electron microscopy analysis of the endogenous myosin VI. As expected, myosin VI shows multiple signals corresponding to the various cytoplasmatic organelles where it performs its function (Magistrati & Polo, 2020). Notably, a specific signal was evident at the level of centriole walls in wild type but not in a myosin VI knock‐out (KO) cell line (Fig 1D).
Next, we examined in detail the interaction between myosin VI and OFD1. Through immunoprecipitation and pull‐down experiments, we confirmed that the short and long myosin VI isoforms can equally bind OFD1 and that the binding is mediated by the tail domain of myosin VI (Fig EV2A and B). In an attempt to characterise the site of the interaction, we focused on the major, well‐characterised myosin VI cargo binding sites, namely the WWY motif (Spudich et al, 2007), the RRL motif (Sahlender et al, 2005) and the MyUb (Myosin VI Ubiquitin‐binding) domain (Penengo et al, 2006; He et al, 2016). Point mutations in key residues did not affect the binding to OFD1 (Fig EV2C and D), prompting us to look for another binding region inside the myosin VI tail. By structure–function analysis we identified the C‐terminal region that starts from the MyUb (1080) as critical but not sufficient to reach the OFD1 binding level of the entire tail domain (Fig EV2E). Addition of the MIU domain is unable to strengthen the interaction (Fig EV2E). Interestingly, the SAH domain, while it does not interact with OFD1 per se (Fig EV2F), appears to be required for the maximum binding (Fig 1E), implying that the conformation of the tail may be important for the interaction (Magistrati & Polo, 2020).
Figure EV2. Characterisation of the myosin VI minimal region of binding to OFD1.
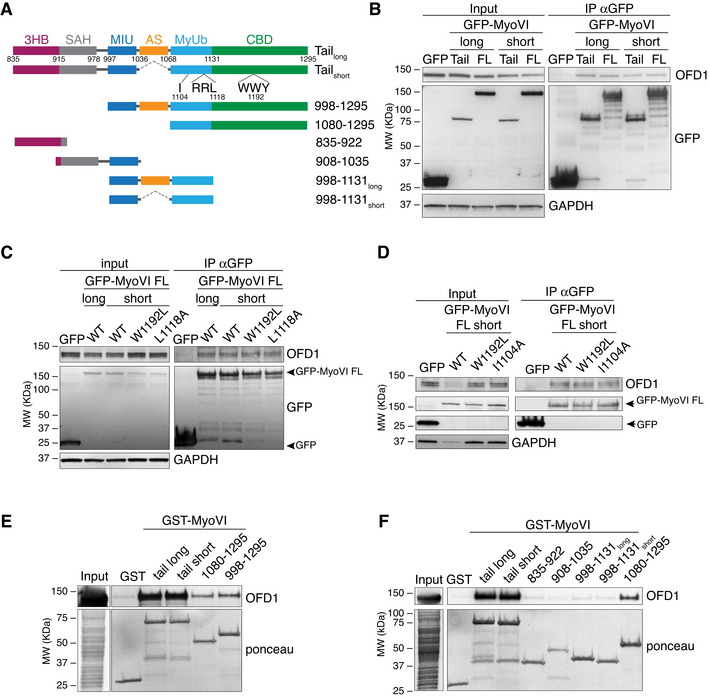
-
AA scheme of the structure and domain organisation of myosin VI. The tail domain is composed of a three‐helix bundle (3HB), a single alpha helix (SAH), two ubiquitin binding regions (MIU, motif interacting with ubiquitin; MyUb, Myosin VI ubiquitin binding domain), and a cargo binding domain (CBD). Between the MIU and the MyUb, an alternative spliced region (AS, in orange) is present in myosin VIlong isoform, while it is absent in the myosin VIshort isoform. RRL and WLY domains and their single amino acid mutations used in (C, D) are indicated. I1104A affects the Ub binding capacity (He et al, 2016), L1118A induces a conformational change that converts a myosin VI long into a myosin VI short‐like protein and abrogates binding to GIPC, optineurin and clathrin, while W1192L abrogates binding to Dab2 (Wollscheid et al, 2016).
-
B–DTotal lysates from HEK293T transfected with Flag‐OFD1 and the indicated GFP‐myosin VI constructs were IP with anti‐GFP antibody‐conjugated beads. IB was performed with anti‐OFD1 and anti‐GFP antibodies. Anti‐GAPDH was used as loading control. FL, full length.
-
E–FTotal lysates from HEK293T cells transfected with Flag‐OFD1 were subjected to pull‐down assay using the indicated GST‐myosin VI constructs or GST alone as control. IB was performed with anti‐Flag antibody. Ponceau staining as indicated.
Source data are available online for this figure.
We then investigated the region of OFD1 that mediates myosin VI binding. Starting from the N terminus, OFD1 is composed of a highly conserved Lis1 homology (LisH) motif that is important for protein–protein interactions, protein stability and intracellular localisation (Singla et al, 2010), followed by six coiled‐coils that are important for centrosomal localisation (Romio et al, 2004; Lopes et al, 2011) and a C‐terminal LIR domain that mediates the binding to the autophagosomes (Morleo et al, 2021). Pull‐down experiments showed that OFD1 binds myosin VI through its coiled‐coils region, while the LIR domain and the N‐terminal region containing the LisH motif appear dispensable for the interaction (Fig 1F). Unfortunately, shorter constructs of the coiled‐coils region of OFD1 were barely soluble, precluding further analysis of the interaction boundaries, as well as possible crystallisation attempts of the complex.
Myosin VI regulates OFD1 levels at the centrioles and Cep164 distal appendage recruitment
We next sought to determine the physiological role of myosin VI‐OFD1 interaction by examining the effects of myosin VI depletion on centriole morphology and activity. To this end, we moved to retinal pigment epithelial (hTERT‐RPE1) cells, diploid immortalised cells that maintain normal checkpoints on cell cycle progression and are commonly used for ciliogenesis assay. First, we confirmed the interaction between the endogenous proteins by co‐immunoprecipitation in RPE1 cells (Fig 2A). Initial characterisation of myosin VI knock‐down (KD) RPE1 cells failed to detect major alterations in the structure of the centrioles, as evident by transmission electron microscopy (TEM) analysis (Fig EV3A). Both TEM and immunofluorescence analysis highlighted that the subcellular localisation of the centrosome, identified by pericentrin staining, is significantly altered upon myosin VI depletion, with an increased centrosome–plasma membrane distance in myosin VI KD cells (Fig EV3B and C).
Figure 2. Myosin VI depletion leads to increased OFD1 and Cep164 recruitment at the centrioles.
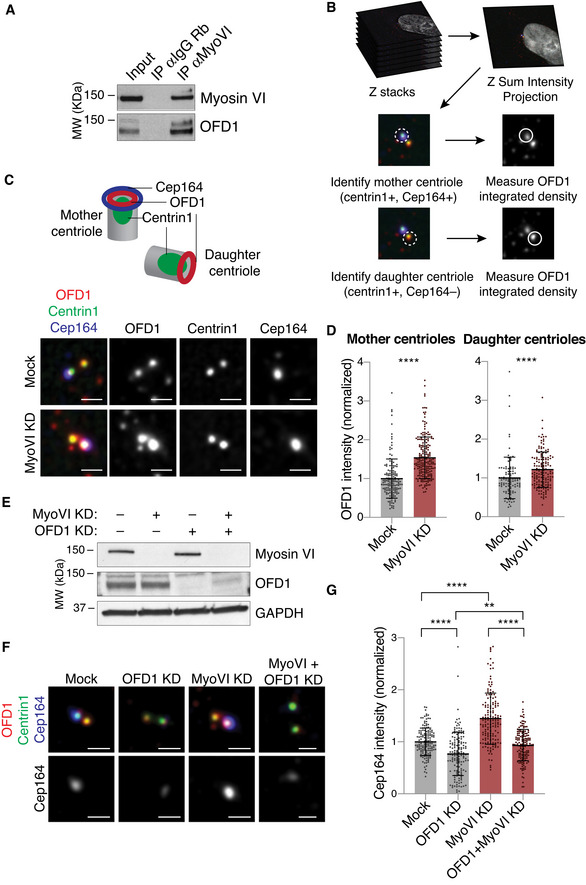
-
ATotal lysates from hTERT‐RPE1 cells were IP with anti‐myosin VI antibody and an anti‐rabbit IgG antibody (as control). IB was performed with anti‐OFD1 and anti‐myosin VI antibodies.
-
BA scheme of the IF analysis performed to calculate the total intensity of OFD1 staining at the mother and daughter centrioles.
-
C, DIF analysis of centriole‐associated OFD1 signal. hTERT‐RPE1 cells were transfected with siRNA against myosin VI. Four days after transfection, cells were treated with nocodazole (1 h, 6 μg/ml) and immunostained with anti‐OFD1, anti‐centrin1 and anti‐Cep164 antibodies. Mother centrioles were identified by the coincident staining of centrin1 and Cep164, while daughter centrioles were centrin1‐only stained. A scheme of the position of the markers used is depicted above. (C) Representative images, scale bar, 1 μm. (D) Quantification of OFD1 intensity at the mother or daughter centrioles. Results are expressed as fold change with respect to mock average intensity. Bars represent mean ± SD. Mother centrioles: Mock, n = 148 cells; MyoVI KD, n = 199 cells, from four independent experiments. Daughter centrioles: Mock, n = 101 cells; MyoVI KD, n = 148 cells, from three independent experiments. ****P < 0.0001 by Mann–Whitney test.
-
EIB analysis of hTERT‐RPE cells treated with the indicated siRNAs with anti‐myosin VI and anti‐OFD1 antibodies. Anti‐GAPDH was used as loading control.
-
F, GIF analysis of Cep164 signal. hTERT‐RPE1 cells were transfected with siRNA against myosin VI and/or OFD1. Four days after transfection, cells were treated with nocodazole (1 h, 6 μg/ml) and immunostained with anti‐OFD1, anti‐centrin1 and anti‐Cep164 antibodies. (F) Representative images. Scale bar, 1 μm. (G) Quantification of Cep164 intensity at the mother centrioles. Results are expressed as fold change with respect to mock average intensity. Bars represent mean ± SD. Mock, n = 147 cells; OFD1 KD, n = 146 cells; MyoVI KD, n = 150 cells; MyoVI + OFD1 KD, n = 151 cells, from four independent experiments. **P < 0.005; ****P < 0.0001 by Kruskal–Wallis test.
Source data are available online for this figure.
Figure EV3. Myosin VI depletion does not affect centrioles ultrastructure.
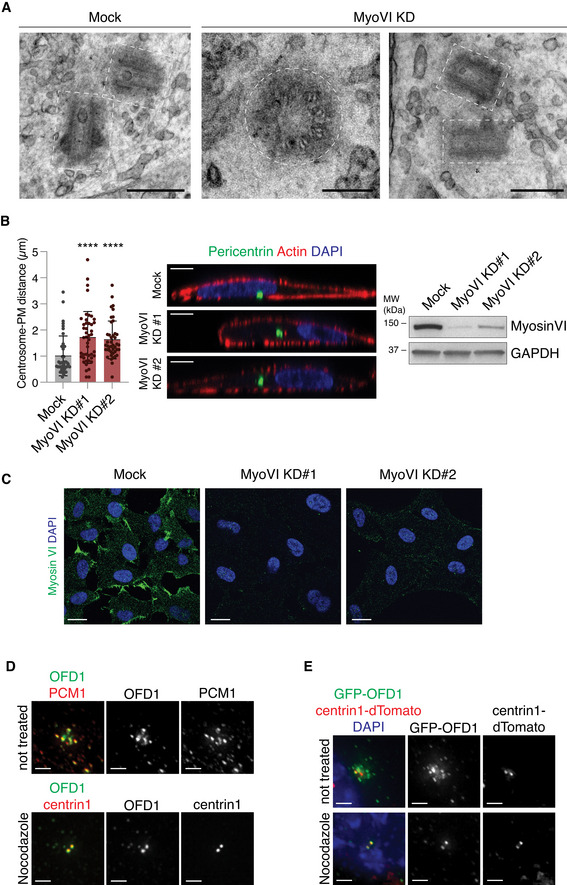
- Transmission electron microscopy (TEM) analysis of centriole structure in hTERT‐RPE1 control and myosin VI‐depleted cells. Representative images are shown, scale bar 500 nm except for the central image that is 200 nm. Centrioles appear morphologically well‐organised.
- Depletion of myosin VI leads to displacement of the centrosomes from the cell cortex. hTERT‐RPE1 cells were transfected with the indicated siRNA oligos and, after 96 h, were stained with anti‐pericentrin to mark centrioles, phalloidin‐TRITC to mark cortical actin, and DAPI to mark the nuclei. Left, the distance of the centrosome from the plasma membrane was calculated using ImageJ from XZ‐axis images of the cells. Bars represent mean ± SD. n = 41–45 cells/condition were counted from three independent experiments. ****P < 0.0001 by Kruskal–Wallis test. Centre, representative images are shown; scale bar, 5 μm. Increased distance between centrioles and nuclear membrane in myosin VI‐depleted cells is evident. Right, IB analysis showing myosin VI depletion in representative samples.
- Representative images of the samples quantified in B showing myosin VI depletion across the different cell populations. Scale bar, 20 μm.
- Nocodazole treatment leads to the dispersion of OFD1 satellite pool. hTERT‐RPE1 cells were treated with nocodazole at 6 μg/ml for 1 h prior fixation to scatter the centriolar satellites and remove OFD1 satellites pool. Cells were immunostained with the anti‐OFD1 antibody and the indicated markers for centriolar satellites (anti‐PCM1) and centrioles (anti‐centrin1). Representative images are shown; Scale bar, 2 μm.
- Effect of nocodazole treatment on GFP‐OFD1 localisation in hTERT‐RPE cells stably expressing GFP‐OFD1 and centrin1‐dTomato, used in FRAP experiments shown in Fig 4. Representative images are shown; scale bar, 2 μm.
Source data are available online for this figure.
Then, we focused on OFD1 behaviour at centrioles. OFD1 localisation in the cytoplasm is exquisitely limited to two specific pools—at the centrioles and at the centriolar satellites—that have different functions and regulations (Lopes et al, 2011; Tang et al, 2013; Wang et al, 2018). We first focused on the centriolar pool using the microtubules depolymerising drug nocodazole (Dammermann & Merdes, 2002) to remove the satellites (Fig EV3D). To assess the amount of OFD1 at the centrioles, we calculated the fluorescence intensity at the centrioles’ spots, identified by the anti‐centrin1 staining. To avoid biases due to the different phases of centriole duplication, we considered only cells with two centrioles, one of which marked by the distal appendage protein Cep164 (Fig 2B). Our analysis revealed that myosin VI depletion causes an accumulation of OFD1 signal both in the mother centriole, identified with Cep164, and in the daughter Cep164‐negative centriole (Fig 2C and D). Interestingly, the increase of OFD1 signal at the centrioles is not accompanied by a parallel increase of the protein level (Fig 2E), indicating a selective increased recruitment of OFD1 at the centrioles.
Previous studies demonstrated that OFD1 is required to constrain centriole elongation and to promote the recruitment of components of the distal appendages at the mother centriole (Singla et al, 2010; Wang et al, 2018). Confirming the aberrant accumulation of OFD1 at centrioles, we found that myosin VI depletion leads to an increased recruitment of the distal appendage protein Cep164 at the mother centriole (Fig 2F and G). Moreover, the effect scored in myosin VI KD cells was rescued by the parallel depletion of OFD1, indicating that the Cep164 accumulation is a secondary effect caused by OFD1 increase at centrioles (Fig 2F and G).
Myosin VI depletion induces cell cycle arrest in non‐tumoural cells through p53 activation
While exploring the effects of myosin VI KD, we unexpectedly observed a severe proliferation impairment in myosin VI‐depleted RPE1 cells (Fig 3A). Similar results were obtained with additional siRNA oligos (Fig EV4A–C) and in BJ‐hTERT cells, another non‐tumoural cell line (Fig 3B). Analysis of the cell cycle by PI staining and FACS showed that myosin VI depletion induces an arrest in the G0/G1 phase, resulting in cellular senescence (Fig 3C and D). This cell cycle arrest was not visible in other cancer cell lines that showed no major defect in cell proliferation (Fig EV4D and E), despite displaying similar myosin VI depletion levels (Fig EV4F).
Figure 3. Myosin VI depletion leads to p53 activation and cell cycle arrest.
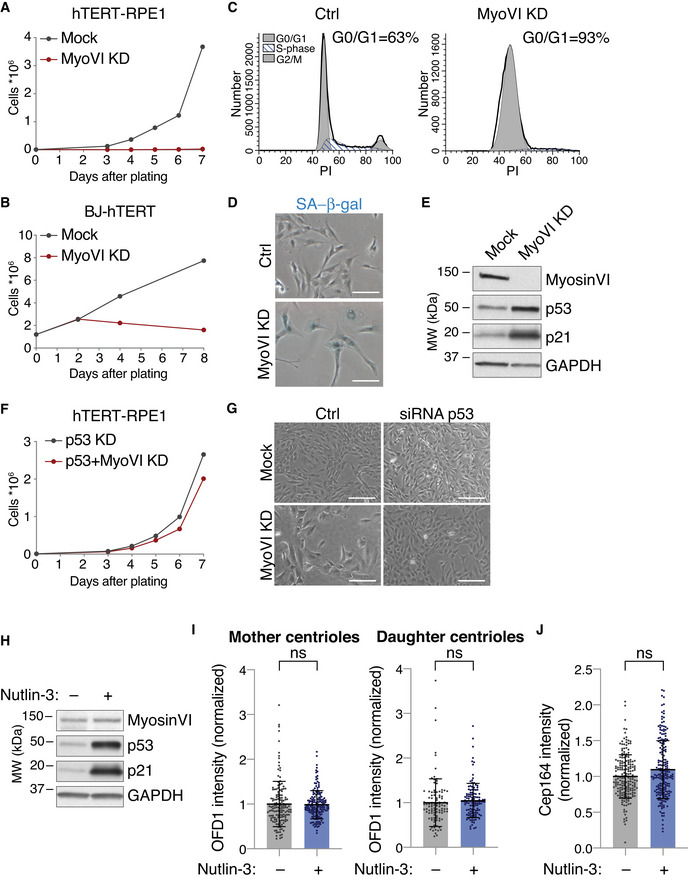
- Growth curve of hTERT‐RPE1 cells transfected with myosin VI siRNA. A representative plot of three independent experiments is shown.
- Growth curve of BJ‐hTERT cells transfected with myosin VI siRNA. A representative plot of two independent experiments is shown.
- Analysis of DNA content in hTERT‐RPE1 cells stably expressing a myosin VI shRNA. After 10 days of doxycycline induction, the cells were stained with propidium iodide (PI) and analysed by FACS. Ctrl, control cells, not induced.
- Senescence‐associated β‐gal assay (SA‐β‐gal) of cells treated as in C. Scale bar, 100 μm.
- IB analysis of hTERT‐RPE1 cells transfected with myosin VI siRNA, with anti‐myosin VI, anti‐p53 and anti‐p21 antibodies. Anti‐GAPDH was used as loading control.
- Growth curve of hTERT‐RPE1 cells transfected with the indicated p53 and myosin VI siRNAs. A representative plot of three independent experiments is shown.
- Representative bright‐field images of cells treated with the indicated p53 and myosin VI siRNAs. Scale bar, 200 μm.
- IB analysis with anti‐myosin VI, anti‐p53 and anti‐p21 antibodies of hTERT‐RPE cells treated with Nutlin‐3, or not treated as control. Anti‐GAPDH was used as loading control.
- Quantification of OFD1 intensity at the mother or daughter centrioles. hTERT‐RPE1 cells treated or not with Nutlin‐3 were incubated with nocodazole for 1 h (6 μg/ml) and immunostained with anti‐OFD1, anti‐centrin1 and anti‐Cep164 antibodies. Mother centrioles were identified by the coincident staining of centrin1 and Cep164, while daughter centrioles were centrin1‐only stained. Results are expressed as fold change with respect to mock average intensity. Bars represent mean ± SD. Mother centrioles: Mock, n = 148 cells; Nutlin‐3, n = 169 cells, from four independent experiments. Daughter centrioles: Mock, n = 101 cells; Nutlin‐3, n = 122 cells, from three independent experiments. ns, not significant by Mann–Whitney test.
- Quantification of Cep164 intensity at the mother centrioles in hTERT‐RPE1 cells treated as in (I). Results are expressed as fold change with respect to mock average intensity. Bars represent mean ± SD. Mock, n = 195 cells; Nutlin‐3, n = 196 cells, from four independent experiments. ns, not significant by Mann–Whitney test.
Source data are available online for this figure.
Figure EV4. Myosin VI ablation causes p53‐dependent cell cycle arrest.
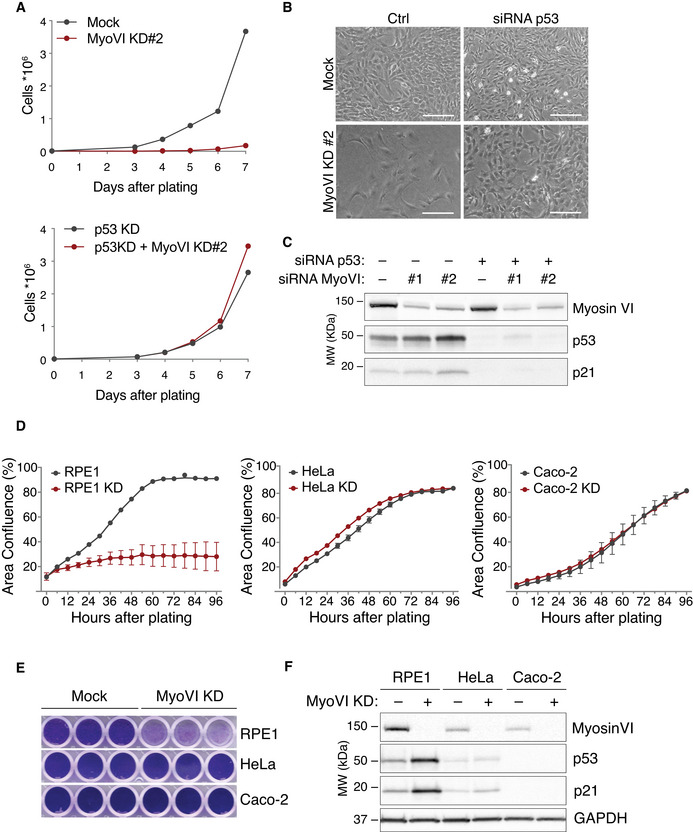
-
A–CSevere proliferation impairment in myosin VI‐depleted RPE1 cells is reproducible using a second siRNA oligo against myosin VI (KD#2). Impairment in cell proliferation was previously shown in rat p53‐proficient PC12 cells (Majewski et al, 2011). (A) Growth curve of hTERT‐RPE1 cells transfected with a second myosin VI siRNA oligo +/− p53 siRNA oligo. A representative plot of three independent experiments is shown. (B) Representative bright‐field images of cells treated with the indicated p53 and myosin VI siRNAs. Scale bar 200 nm. (C) IB analysis of control and myosin VI‐depleted hTERT‐RPE cells with anti‐myosin VI, anti‐p53 and anti‐p21 antibodies.
-
D–FMyosin VI depletion does not affect cell proliferation in p53‐null tumour cells. (D) Growth curve of hTERT‐RPE1, HeLa and Caco‐2 cells transfected with myosin VI siRNA measured by Incucyte. A representative plot of two independent experiments is shown. Data are reported as mean ± SD of three technical replicates. (E) Cell viability of the indicated cell lines measured by crystal violet staining. (F) IB analysis of the indicated cells lines, control and myosin VI‐depleted, with anti‐myosin VI, anti‐p53 and anti‐p21 antibodies. Anti‐GAPDH was used as loading control.
Source data are available online for this figure.
To investigate the cause of the cell cycle arrest, we performed immunoblot analysis and found that myosin VI‐depleted cells display an increase in both p53 and p21 expression levels (Fig 3E). Consistently, double depletion of myosin VI and p53 by siRNA oligos rescued the proliferation of RPE1 cells (Figs 3F and G, and EV4A–C).
To clarify if the increase in OFD1 and Cep164 recruitment to the centrioles upon myosin VI depletion may be due to p53 accumulation and cellular senescence, cells were treated with Nutlin‐3. This drug is a potent inhibitor of the p53 antagonist Mdm2 and induces p53 accumulation with consequent cell cycle arrest (Vassilev et al, 2004). After Nutlin‐3 treatment, cells did not show any changes in the levels of OFD1 or Cep164 at the centrioles (Fig 3H–J), confirming that the increased accumulation of OFD1 at centrioles we scored upon myosin VI depletion is specific to the lack of the myosin VI activity.
We then assessed if the activation of p53 upon myosin VI KD depends on defects occurring at centrioles. Indeed, alterations in the number or structure of the centrioles have been shown to cause cell cycle arrest, which is mediated by the activation of different pathways depending on the type of damage (Mikule et al, 2007; Ganem et al, 2014; Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016; Fava et al, 2017). To assess if the centrioles contribute to myosin VI depletion‐induced p53 activation and cell cycle arrest, we sought to remove the centrioles from the cells and analyse cell proliferation. We used centrinone, a Plk4 inhibitor that blocks the formation of new centrioles (Wong et al, 2015), thus depleting them over a few cell cycles (Fig EV5A). To avoid the activation of p53 due essentially to centriole loss, we performed this experiment in 53BP1 KO RPE1 cells, in which p53 is not activated following centrinone treatment (Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016) (Fig EV5B). Strikingly, in centrinone‐treated 53BP1 KO cells, myosin VI depletion caused p53 activation and cell cycle arrest similar to wild‐type cells, as shown by Western blot and growth analysis (Fig EV5B and C).
Figure EV5. p53 activation in myosin VI‐depleted cells is not due to its effect on centrioles.
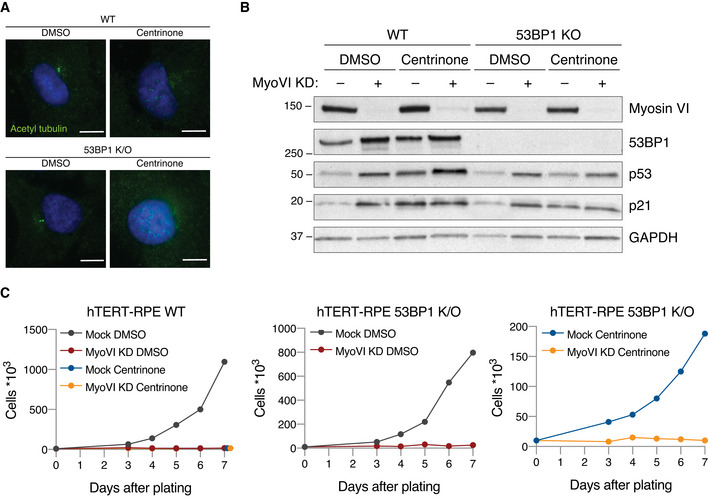
- hTERT‐RPE1 wild‐type or 53BP1 KO cells were treated with centrinone or DMSO and immunostained with anti‐acetylated tubulin antibody and DAPI to verify the presence or absence of centrioles. Representative images are shown, scale bar, 20 μm.
- IB analysis of hTERT‐RPE wild‐type or 53BP1 KO cells transfected with myosin VI siRNA and treated as indicated. Anti‐GAPDH was used as loading control.
- Growth curves of hTERT‐RPE1 wild‐type or 53BP1 KO cells transfected with myosin VI siRNA and treated as indicated. Representative plots of three independent experiments are shown.
Taken together, these results demonstrate that a p53‐dependent cell cycle arrest occurs upon myosin VI depletion in non‐tumoural cell lines and that a general p53 activation does not induce OFD1 and Cep164 accumulation, as myosin VI KD does.
Myosin VI controls the turnover of OFD1 at the centrioles
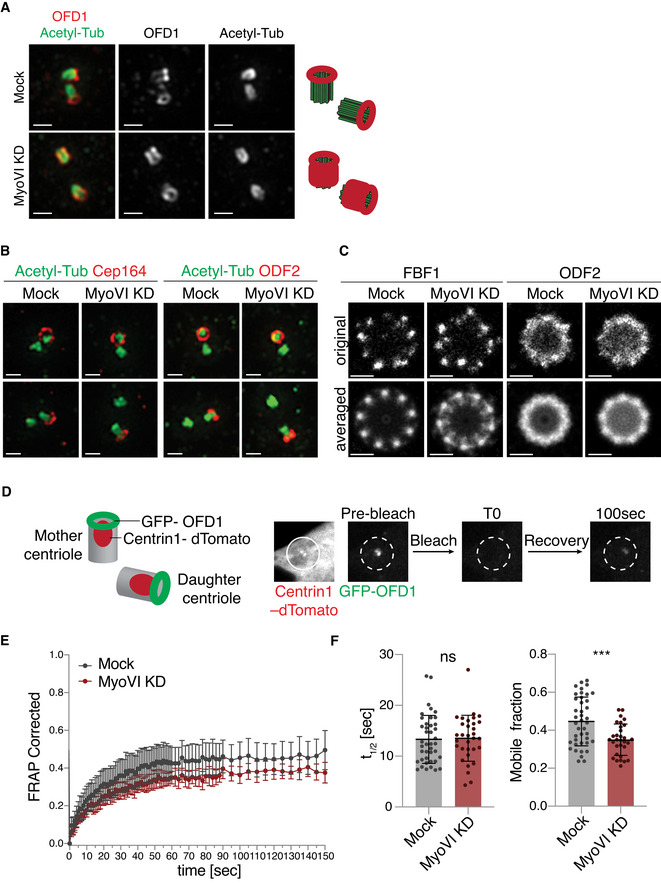
Given the increased recruitment of OFD1 at the centrioles upon myosin VI depletion and the localisation of the motor protein at the centrioles, we hypothesised that myosin VI could have a direct impact on OFD1 at the centrioles. Through super‐resolution structured illumination microscopy (SIM), we confirmed the ring‐like localisation of OFD1 at the distal tip of the mother and daughter centrioles in control cells (Wang et al, 2018). Conversely, we found that the lack of myosin VI caused an aberrant accumulation of OFD1 along the entire centriolar walls (Fig 4A). Centriolar appendage proteins do not show the same behaviour, as both Cep164 and ODF2 correctly localised at the distal end (Fig 4B). Moreover, distal appendage number is not affected by myosin VI depletion, as measured by direct stochastic optical reconstruction microscopy (dSTORM) analysis (Fig 4C).
Figure 4. Lack of myosin VI alters the mobility and localisation of OFD1 at the centrioles.
-
ASuper‐resolution analysis of OFD1 localisation at the centrioles. hTERT‐RPE1 cells were transfected with siRNA against myosin VI, immunostained with anti‐OFD1 and anti‐acetylated tubulin antibodies and visualised using structured illumination microscopy (SIM). Representative images are shown, scale bar, 500 nm. A scheme of the estimated localisation of OFD1 in the two conditions is depicted on the right side.
-
BTwo‐colour SIM images in control and myosin VI‐depleted cells illustrating the localisation and distribution of the distal appendage markers Cep164 and ODF2 at the centrioles, stained with anti‐acetylated tubulin antibody. Representative images are shown; scale bar, 500 nm.
-
CdSTORM super‐resolution analysis of the distal appendage markers FBF1 and ODF2 in control and myosin VI‐depleted cells. To emphasise the symmetry of the structures, the signals from all nine appendages were averaged (bottom). Representative images are shown; scale bar, 200 nm.
-
D–FFRAP analysis of centriole‐associated GFP‐OFD1. hTERT‐RPE cells stably expressing GFP‐OFD1 and centrin1‐dTomato were transfected with siRNA against myosin VI. After four days, cells were treated with nocodazole (1 h, 6 μg/ml) and subjected to live‐cell imaging. (D) Left: a scheme of the localisation of GFP‐OFD1 and the centriole marker centrin1‐dTomato. Right: a scheme of photobleaching and recovery of GFP‐OFD1 at the centrioles. (E) A representative graph of one out of three experiments. For each time point, the fraction of recovery of GFP‐OFD1 is shown. Results are expressed as means with 95% confidence interval. n = 12 cells (Mock), n = 13 cells (MyoVI KD). (F) Quantification of the half‐time of fluorescence recovery (t1/2,) and of the mobile fraction of GFP‐OFD1. Results are expressed as mean ± SD. n = 41 cells (Mock), n = 31 cells (MyoVI KD), from three independent experiments. ns, not significant; ***P < 0.0005 by unpaired t‐test.
Source data are available online for this figure.
These results prompted us to analyse the behaviour of OFD1 by fluorescence recovery after photobleaching (FRAP) to determine the turnover of the protein. Centrioles were identified with centrin1‐dTomato (Fig 4D), while the satellite pool of OFD1 was eliminated with nocodazole treatment during the live cell imaging (Fig EV3E). While the speed of recovery was not affected, myosin VI depletion caused a significant decrease in the mobile fraction of OFD1, indicating the presence of a stable pool of OFD1 at the centrioles that cannot be mobilised (Fig 4E and F).
Collectively, these data indicate that in the absence of myosin VI, OFD1 cannot be removed from the centrioles where it aberrantly accumulates.
Myosin VI removes OFD1 from the satellites
Besides the centrioles, OFD1 also localises at the centriolar satellites in cycling cells. Previous studies showed that serum starvation induces OFD1 removal from the satellites by autophagy, a process that is required for primary ciliogenesis (Tang et al, 2013; Morleo et al, 2021). Interestingly, myosin VI has been previously implicated in autophagosome–lysosome fusion during LC3‐mediated autophagosome maturation (Tumbarello et al, 2012), as well as in mitophagy (Kruppa & Buss, 2018). To determine if myosin VI exerts a role on satellites, we first examined the effect of myosin VI depletion using the satellite marker PCM1, which is virtually absent at the centrioles (Dammermann & Merdes, 2002). Although we could observe a strong reduction of PCM1 intensity around centrioles in myosin VI KD cells (Fig 5A and B), cells treated with Nutlin‐3 showed a similar decrease (Fig 5C), suggesting that the p53 activation induced by myosin VI KD causes satellites loss. Consistently, myosin VI depletion does not affect PCM1 intensity in RPE1 p53 KO cells (Fig 5D).
Figure 5. Myosin VI depletion affects the centriolar satellites via p53.
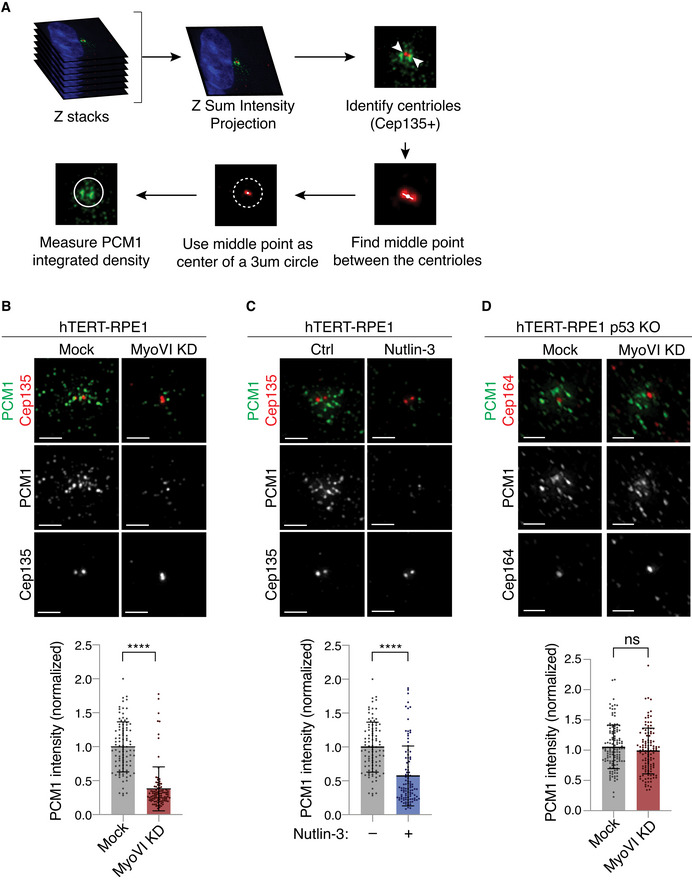
- A scheme of the IF analysis performed to calculate the total intensity of satellite staining that surrounds the centrioles. The centriole marker Cep135 or Cep164 are used to define the centre of a 3 μm circle, in which the intensity of the satellite marker PCM1 was calculated.
- IF analysis of PCM1 signal. hTERT‐RPE1 cells were transfected with siRNA against myosin VI and immunostained with anti‐PCM1 and anti‐Cep135 antibodies. Upper panel, representative images, scale bar, 2 μm. Lower panel, quantification of PCM1 intensity. Results are expressed as fold change with respect to mock average intensity. Bars represent mean ± SD. Mock, n = 96 cell; MyoVI KD, n = 98 cells, from two independent experiments. ****P < 0.0001 by Mann–Whitney test.
- IF analysis of PCM1 signal. hTERT‐RPE1 cells were treated with Nutlin‐3 for 24 h and immunostained with anti‐PCM1 and anti‐Cep135 antibodies. Panels as in B. Mock, n = 96 cells; Nutlin‐3, n = 100 cells, from two independent experiments. ****P < 0.0001 by Mann–Whitney test.
- IF analysis of PCM1 signal. hTERT‐RPE1 p53 KO cells were transfected with siRNA against myosin VI and immunostained with anti‐PCM1 and anti‐Cep164 antibodies. Panels as in (B). Quantification of PCM1 intensity refers to a 3 μm circle around the mother centriole, identified with anti‐Cep164 staining. Mock, n = 128 cells; MyoVI KD, n = 114 cells from three independent experiments. ns, not significant by Mann–Whitney test.
Source data are available online for this figure.
Based on these results, we used RPE1 p53 KO cells to analyse the effect of myosin VI depletion on the satellite pool of OFD1. To eliminate the contribution of the centriolar pool of OFD1, we identified the satellites via PCM1 staining and calculated OFD1 intensity only in the area covered by PCM1 (Fig 6A). As previously shown (Tang et al, 2013; Akhshi & Trimble, 2021), serum starvation induces the removal of OFD1 from the satellites (Fig 6B and C). We could observe that this event is significantly impaired by the parallel depletion of myosin VI (Fig 6B). While serum starvation induces a decrease in OFD1 total protein levels (Akhshi & Trimble, 2021; Morleo et al, 2021), the same effect is weaker in myosin VI‐depleted cells (Fig 6C).
Figure 6. Myosin VI contributes to OFD1 removal from the centriolar satellites required for ciliogenesis.
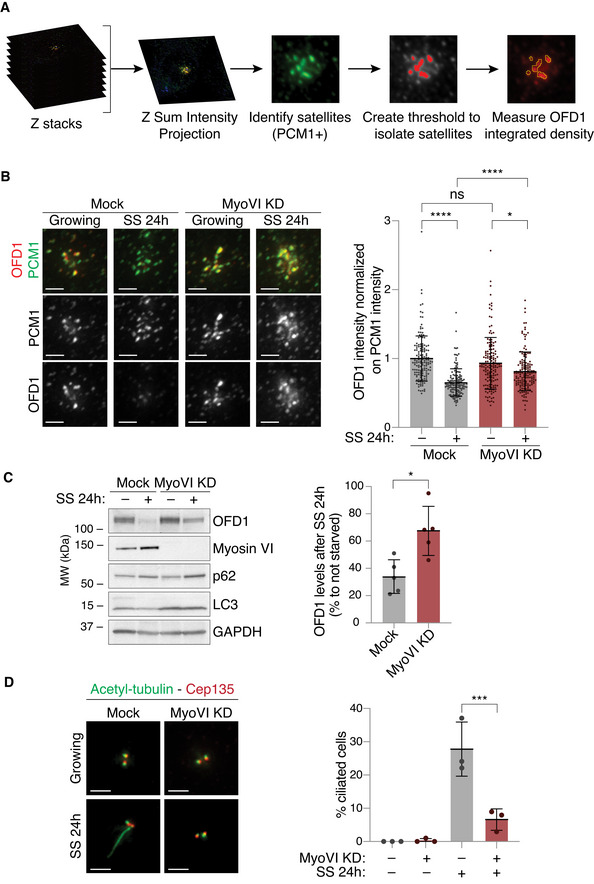
- A scheme of the IF analysis performed to calculate the total intensity of OFD1 staining at the centriolar satellites.
- IF analysis of OFD1 signal at the centriolar satellites upon serum starvation. hTERT‐RPE1 p53 KO cells were transfected with siRNA against myosin VI. After four days, cells were fixed (growing) or serum starved for 24 h (SS). Cells were immunostained with anti‐OFD1 and anti‐PCM1 antibodies. The intensity of OFD1 signal in the area covered by PCM1 was quantified and normalised against the intensity of PCM1 staining in the same area. Left, representative images, scale bar, 2 μm. Right, results are expressed as fold change with respect to mock average intensity. Bars represent mean ± SD. Mock_growing, n = 150 cells; Mock_SS, n = 149 cells; MyoVI KD_growing, n = 149 cells; MyoVI KD_SS, n = 150 cells, from three independent experiments. ns, not significant; *P < 0.05; ****P < 0.0001 by Kruskal–Wallis test.
- IB analysis of OFD1 after serum starvation in control and myosin VI‐depleted cells. hTERT‐RPE1 p53 KO cells were transfected with siRNA against myosin VI. After four days, cells were serum starved for 24 h (SS). Lysates were analysed by IB with anti‐OFD1, anti‐myosin VI, anti‐p62 and anti‐LC3 antibodies. Anti‐GAPDH was used as loading control. The amount of OFD1 protein was normalised against GAPDH signal and is expressed as percentage of OFD1 levels in cells grown in serum‐starved conditions compared to cells grown in media containing serum. Bars represent mean ± SD. n = 5 independent experiments. *P < 0.05 by Kruskal–Wallis test.
- IF analysis of primary cilium upon serum starvation. hTERT‐RPE1 p53 KO cells were transfected with siRNA against myosin VI. After four days, cells were fixed (growing) or serum starved for 24 h (SS). Cells were immunostained with anti‐acetylated tubulin (to identify the cilia), anti‐Cep135 (to identify the centrioles) and DAPI. Left, representative images, scale bar, 2 μm. Right, results are expressed as fold change with respect to mock average intensity. Bars represent mean ± SD. n = 3 independent experiments. 100–200 cells/condition were counted for each experiment. ***P < 0.001 by two‐way ANOVA test.
Source data are available online for this figure.
Ciliogenesis in tissue culture is mainly initiated by serum starvation, which arrests the cell cycle and triggers autophagy (Tang et al, 2013; Morleo et al, 2021). Our data support the idea that myosin VI contributes to OFD1 degradation induced by serum starvation. Thus, we examined the formation of primary cilia in myosin VI‐depleted cells. To avoid confounding effect due to the cell cycle arrest induced by myosin VI depletion, we performed the experiment in RPE p53 KO cells. Remarkably, while around 30% of the p53 KO cells were found ciliated after starvation even in the absence of G1 arrest, myosin VI‐depleted cells completely failed to form cilia (Fig 6D). These data imply a requirement of myosin VI activity for primary cilium formation.
Discussion
Here, we identified and characterised a novel myosin VI interactor, OFD1, whose turnover is modulated by myosin VI both at the centrioles and at the centriolar satellites. Myosin VI activity on OFD1 appears to be critical for maintaining the correct amount of the distal appendages at the mother centriole and for the removal of OFD1 from the centriolar satellites to promote primary ciliogenesis. Thus, OFD1 can be added to the list of specialised cargo‐adaptor proteins that link myosin VI to distinct cellular compartments and processes (Magistrati & Polo, 2020). Importantly, both myosin VI short and long isoforms (Wollscheid et al, 2016) interact with OFD1, indicating that this functional interaction is conserved in all cells and is maintained upon epithelial cell polarisation where a switch between isoforms was observed (Buss et al, 2001; Biancospino et al, 2019).
At the centrioles, OFD1 localisation is restricted to the distal tip (Fig 4A) (Tang et al, 2013). Our FRAP analysis determined that the 45% of OFD1 present at the centrioles is mobile and rapidly exchanges with the cytoplasmic pool. In the absence of myosin VI, this fraction is reduced, allowing accumulation of OFD1 on the entire microtubule wall. At present, the mechanism by which myosin VI controls OFD1 turnover remains unclear. Since the speed of recovery is not affected (t1/2 in FRAP analysis, Fig 4F), it is conceivable that myosin VI is needed for short‐range transport and may exploit the actin filaments that are nucleated by the centrosome to promote the turnover of OFD1 via its motor activity. In this context, it is noteworthy that the SAH domain involved in myosin VI dimerisation and required for the movement (Mukherjea et al, 2014) is critical for the interaction between the two proteins (Fig 1E). At the distal tip, OFD1 could be stabilised by other proteins like C2CD3 (Thauvin‐Robinet et al, 2014) that protect this centriolar protein from myosin VI‐mediated removal.
Recent studies have highlighted a synergistic interplay between actin and microtubule dynamics in several cellular compartments. Actin–microtubule crosstalk is functionally relevant for mitotic spindle positioning (Farina et al, 2019; Inoue et al, 2019) or for cell shape and polarity during cell migration, and many proteins that mediate actin–microtubule interactions have already been identified (Dogterom & Koenderink, 2019). Our data raise the possibility that myosin VI is one of these critical modulators at the centrosomes where it could exploit the actin‐based network surrounding the centrosomes and the microtubule interactors that we have identified with the proteomic approach (i.e. Numa, Dataset EV1). Further investigations are needed to uncover the functional implications of this intriguing hypothesis that is supported by the recent identification of a Gαi‐LGN‐NuMA‐dynein axis activated upon Shh‐Smo induction to promote ciliogenesis (Akhshi & Trimble, 2021) and which is consistent with the aberrant positioning of the centrosomes observed in the absence of myosin VI (Fig EV3B).
Our study also unveils an unprecedent phenotype of cell cycle arrest and senescence following myosin VI depletion in p53‐proficient cells. This effect was not evident in p53‐null cancer cell lines, suggesting that myosin VI contributes to a pathway that sustains proliferation and maintains the cell cycle in check and that becomes deregulated during carcinogenesis. While this possibility is currently under investigation, the cause of this phenotype is certainly unrelated to centriolar biology as demonstrated by the centrinone experiment (Fig EV5).
Another surprising finding of our study is that p53 activation is responsible for the satellite dispersion occurring upon myosin VI depletion, which was established by Nutlin‐3 treatment that phenocopies myosin VI depletion, as well as by the rescue of the phenotype obtained in p53 KO cells (Fig 5). Thus, our study highlights a new intriguing function of p53 at satellites. Determination of how p53 senses the lack of myosin VI is an important area of future work that is predicted to shed light also on this phenotype.
Finally, our study identified for the first time myosin VI as a critical regulator of ciliogenesis. Based on its role in other cellular compartments, it is possible that myosin VI may act as an actin anchor for the basal body, by docking the mother centriole at the plasma membrane, or as an actin‐based motor contributing to the trafficking towards the ciliary pocket (Akhshi & Trimble, 2021). Nonetheless, our evidence suggests that myosin VI depletion affects autophagy‐mediated removal of OFD1 from the satellites (Fig 6A–C), a prerequisite for ciliogenesis (Tang et al, 2013; Akhshi & Trimble, 2021; Morleo et al, 2021). Several findings link myosin VI to autophagy and autophagy receptors (Tumbarello et al, 2012; Magistrati & Polo, 2020), and emerging evidence suggests that autophagy and ciliogenesis influence each other (Pampliega & Cuervo, 2016; Morleo & Franco, 2019). Mechanistically, myosin VI has been shown to act at the damaged mitochondria where it mediates their engulfment and clearance via the formation of F‐actin cages (Kruppa & Buss, 2018). The interaction with OFD1 suggests that myosin VI may be directly involved in the first phases of the autophagosome formation, although implication of F‐actin cages cannot be excluded. Whatever the case, the identification of myosin VI as OFD1 regulator has important implications for a variety of ciliopathy syndromes in which OFD1 mutations compromise ciliogenesis (Singla et al, 2010).
Materials and Methods
Constructs and antibodies
pGEX‐myosin VI tail (835–1295) and pEGFP‐myosin VI full‐length (FL) constructs were previously described (Wollscheid et al, 2016). pcDNA CMV‐10 3xFlag‐OFD1 full‐length (1–1012), a (1–276), b (277–663), c (664–1012) and ΔLIR (mutation of the LIR domain EKYMKI to EKAMKA) were kindly provided by Brunella Franco (TIGEM, Napoli). pEGFP‐OFD1 full‐length (FL) construct was generated by subcloning the OFD1 gene from the pcDNA CMV‐10 3xFlag‐OFD1 construct into the pEGFP_C1 vector with SmaI/BamHI restriction enzymes. pLVX‐GFP‐OFD1 lentiviral construct was generated using the Infusion HD cloning system (Takara Clontech) following manufacturer’s instructions. Briefly, EGFP‐OFD1 sequence was amplified by PCR using primers that anneal on the GFP template and are complementary to pLVX‐Puro vector (forward: 5′‐CTCAAGCTTCGAATTCATGGTGAGCAAGGGCGAG‐3′; reverse: 5′‐TAGAGTCGCGGGATCCATCAGTTATCTAGATCCGGTGG‐3′), followed by EcoRI/BamHI vector linearisation and in‐fusion reaction. pSLIK‐NEO myosin VI shRNA was generated with Gateway™ LR Clonase™ II Enzyme mix (Thermo Fisher Scientific) by subcloning a nucleotide sequence targeting myosin VI (5′‐AGTAATTCAGCACAATATTCCAA‐3′) into a pENTR vector, followed by recombination into pSLIK‐NEO empty vector.
All the other truncated constructs were engineered by site‐directed mutagenesis or recombinant PCR and sequence‐verified. Details are available upon request.
The antibodies with indicated dilutions were as follows:
| Primary antibodies | ||||
|---|---|---|---|---|
| Epitope | Species | Supplier | Identifier | Application |
| 53BP1 | Rabbit | Abcam | ab36823 | WB 1:1,000 |
| Acetylated tubulin | Mouse | Sigma | T7451 | IF 1:1,000 |
| Acetylated tubulin | Mouse | Sigma | 6‐11B‐1 | IF 1:1,000 |
| Centrin1 | Mouse | Millipore | 04‐1624 | IF 1:1,000 |
| Cep135 | Rabbit | Abcam | ab75005 | IF 1:1,000 |
| Cep164 | Goat | Santa‐Cruz Biotechnology | sc‐240226 | IF 1:700 |
| FBF1 | Rabbit | Sigma | HPA023677 | IF 1:50 |
| FLAG | Mouse | Sigma | F3165 | WB 1:5,000 |
| GAPDH | Mouse | Santa‐Cruz Biotechnology | Sc‐32233 | WB 1:5,000 |
| GFP | Rabbit | Sigma | G1544 | WB 1:5,000 |
| GFP | Mouse | Thermo Fisher Scientific | A‐11120 | PLA 1:2,000 |
| GST | Rabbit | Generated in house | IP | |
| LC3 | Rabbit | Cell Signaling | 12741 | WB 1:1,000 |
| Myosin VI | Mouse | Sigma | MUD‐19 | WB 1:1,000 |
| Myosin VI | Rabbit | Generated by EUROGENTEC S.A., purified by Cogentech | 1295 | IP |
| Myosin VI | Rabbit | Generated by EUROGENTEC S.A., purified by Cogentech | 1296 | IP; WB 1:2,000; IF 1:400 |
| ODF2 | Rabbit | Abcam | ab43840 | IF 1:200 |
| OFD1 | Rabbit | Sigma | HPA031103 | IF 1:500; PLA 1:2,000 |
| OFD1 | Rabbit | Kindly provided by Brunella Franco (TIGEM, Napoli) (Giorgio et al, 2007) | WB 1:100 | |
| p21 | Rabbit | Cell Signaling | 2947 | WB 1:5,000 |
| p53 (DO‐1) | Mouse | Cell Signaling | 18032 | WB 1:1,000 |
| p62 | Rabbit | Enzo Life Sciences | BML_PW9860 | WB 1:1,000 |
| PCM1 | Mouse | Sigma | SAB1406228 | IF 1:200 |
| Pericentrin | Mouse | Abcam | 28144 | IF 1:200 |
| IgG from rabbit serum | Sigma | I5006 | IP | |
| Secondary antibodies | ||||
|---|---|---|---|---|
| Antibody | Species | Supplier | Catalogue number | Application |
| Anti‐mouse IgG HRP | Goat | Bio‐Rad | 1721011 | WB 1:5,000 |
| Anti‐rabbit IgG HRP | Goat | Bio‐Rad | 1706515 | WB 1:5,000 |
| Anti‐mouse Alexa488 | Donkey | Thermo Fisher Scientific | A21202 | IF 1:400 |
| Anti‐mouse Cy3 | Donkey | Jackson Lab | 715‐165‐150 | IF 1:400 |
| Anti‐mouse Alexa488 | Goat | Molecular Probes | A11001 | IF 1:500 |
| Anti‐rabbit Alexa555 | Donkey | Molecular Probes | A31572 | IF 1:500 |
| Anti‐rabbit Alexa488 | Donkey | Thermo Fisher Scientific | A21206 | IF 1:400 |
| Anti‐rabbit Cy3 | Donkey | Jackson Lab | 711‐165‐152 | IF 1:400 |
| Anti‐rabbit Alexa647 | Donkey | Thermo Fisher Scientific | A31573 | IF 1:400 |
| Anti‐goat Alexa647 | Donkey | Thermo Fisher Scientific | A21447 | IF 1:400 |
Commercial antibodies were validated by the manufacturer. The home‐made anti‐myosin VI was previously described (Wollscheid et al, 2016).
Cell lines
hTERT‐RPE1 cells (ATCC) were maintained in Dulbecco’s modified Eagle medium: Nutrient Mixture F‐12 (DMEM/F12, Gibco), supplemented with 10% foetal bovine serum (FBS), 2 mM l‐glutamine, 0.5 mM Na‐Pyruvate, 15 mM Hepes pH 7.5. HEK293T (ICLC) and A549 (NCI‐60) cells were maintained in DMEM, supplemented with 10% FBS and 2 mM l‐glutamine. BJ hTERT cells (ATCC) were maintained in DMEM – M199 (4:1), supplemented with 10% FBS and 2 mM l‐glutamine. HeLa cells (ATCC) were maintained in minimum essential medium (MEM), supplemented with 10% FBS, 0.1 mM non‐essential amino acids (NEAA), 2 mM l‐glutamine and 1 mM Na‐Pyruvate. CaCo2 (DSMZ) cells were maintained in MEM, supplemented with 20% FBS, 0.1 mM NEAA and 2 mM l‐glutamine. MDA‐MB‐231(NCI‐60) cells were maintained in RPMI 1640, supplemented with 10% FBS and 2 mM l‐glutamine. MCF‐10A (ATCC) cells were maintained in DMEM‐Ham's F12, supplemented with 5% Horse serum, 10 μg/ml Insulin, 20 ng/ml EGF, 500 ng/ml Hydrocortisone, 100 ng/ml cholera toxin and 2 mM l‐glutamine.
hTERT‐RPE1 p53 knock‐out (KO) and 53BP1 KO were kindly provided by Luca Fava (University of Trento, Trento). In brief, hTERT‐RPE1 cells were transduced with Lenti‐CRISPR‐V2 targeting coding exons of the genes of interest, selected with puromycin, and single clones were characterised through sequencing of the targeted genomic region (Burigotto et al, 2021).
hTERT‐RPE1 with stable expression of centrin1‐dTomato were kindly provided by Francesca Farina (IRTSV, Grenoble). In brief, hTERT‐RPE1 cells were transfected with pdTomato‐centrin1 construct (Farina et al, 2016) and selected with geneticin. dTomato‐positive cells were selected by fluorescence‐activated cell sorting (FACS). To generate the hTERT‐RPE1 centrin1‐dTomato GFP‐OFD1 cell line used for fluorescence recovery after photobleaching (FRAP) experiments, hTERT‐RPE1 centrin1‐dTomato were transduced with the pLVX‐GFP‐OFD1 lentiviral construct. Two weeks after transduction, GFP‐ and dTomato‐positive cells were selected by FACS.
To generate A549 myosin VI KO cell line through CRISPR/Cas9, A549 cells were transiently transfected with pD1301‐AD vector (prepared by ATUM, atum.bio), containing Cas9, a GFP reporter and a guide RNA (gRNA) targeting exon 2 of myosin VI (gRNA sequence: 5′‐GTTCAATTGTTAAGCTGTCG‐3′, designed using ATUM’s design tool). GFP‐positive cells were FACS‐sorted, single cell clones were screened for myosin VI deletion through immunoblot (IB) and IP, and the genomic mutations were characterised by PCR amplification and sequencing of the targeted genomic region. Clone 13S34 that was selected for our experiments have two different mutant alleles: one has an insertion of 2 nucleotides, while the other has a deletion of 19 nucleotides. In both cases, the frameshift caused stop codons, generating truncated proteins spanning the first 30 or 38 amino acids, respectively.
All cell lines were authenticated by STR profiling (StemElite ID System, Promega) and tested for mycoplasma using PCR and biochemical test (MycoAlert, Lonza).
siRNA transfection, shRNA expression
Transient knock‐downs (KD) were performed using Stealth siRNA oligonucleotides from Thermo Fischer Scientific (Waltham, MA, USA). Cells were transfected twice using RNAiMax (Invitrogen), first in suspension and the following day in adhesion. Based on immunoblot analyses, cells were considered myosin VI‐depleted four days after the first transfection.
myosin VI #1 (when not specified, this siRNA is used to deplete myosin VI): 5′‐GAGGCUGCACUAGAUACUUUGCUAA‐3′. myosin VI #2: 5′‐GAGCCTTTGCCATGGTACTTAGGTA‐3′. OFD1: 5′‐GAGAAUGAAGUGUACUGCAAUCCAA‐3′. p53: 5′‐CCAGUGGUAAUCUACUGGGACGGAA‐3′.
hTERT‐RPE1 cells with doxycycline‐inducible shRNA for myosin VI were generated by transducing the cells with pSLIK‐NEO myosin VI shRNA and selection with neomycine. Expression of the shRNA was induced with 0.5 μg/ml doxycycline for 10 days.
Proliferation assays
About 10,000 cells transfected with the indicated siRNAs were plated on a 6‐well plate (day 0). Starting from day 3 after plating, cells were counted every day using Beckman Multisizer 3 Coulter Counter. Cells were split to maintain a confluence < 70%.
For the experiment shown in Fig EV4D, 2,000 cells transfected with the indicated siRNAs were seeded in triplicate in a 96‐well cell culture plate (Corning #3596) and four regions per well were imaged every 6 h over a period of 96 h using Incucyte® SX5 Live‐Cell Analysis System (Sartorius). The surface area occupied by the cells was calculated by Incucyte analysis software and expressed as per cent (%) cell confluency.
To measure cell viability (Fig EV4E), 10,000 cells/well transfected with the indicated siRNAs were seeded in triplicate in 48‐well plates and grown for four days. Cell viability was determined using the 1% crystal violet staining containing 20% methanol (Sigma #1092180500).
Biochemical assays
Cells were lysed in JS buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 5 mM EGTA, 10% glycerol and 1% Triton X‐100) supplemented with 20 mM sodium pyrophosphate, pH 7.5, 250 mM sodium fluoride, 2 mM PMSF, 10 mM sodium orthovanadate and protease inhibitors (Calbiochem) and lysates were cleared by centrifugation at 18,800 g. For immunoblot (IB) analysis, 50 μg of lysates were used, unless otherwise id specified. For co‐immunoprecipitation (co‐IP) analysis, 1 mg of fresh lysates were incubated with GFP‐Trap (Chromotek) or anti‐Flag M2‐conjugated beads (Sigma) for 2 h at 4°C. For pull‐down experiments, 500 µg of transfected HEK293T cellular lysates were incubated with 1 μM of GST‐fusion proteins immobilised onto GSH beads for 2 h at 4°C in JS buffer. For anti‐myosin VI IP, 1 mg lysates were incubated with anti‐myosin VI antibodies (1295 or 1296) or anti‐GST rabbit antibody as negative control. After 2 h of incubation at 4°C, protein A sepharose beads were added to the IP and the mixture was incubated for an additional hour. In all cases, after extensive washes with JS buffer, beads were re‐suspended in Laemmli buffer and proteins were analysed through sodium dodecyl sulphate–polyacrylamide gel electrophoresis (4–20% TGX precast gel, Bio‐Rad). Detection was performed either by staining the gels with Coomassie or by IB using specific antibodies. Ponceau‐stained membranes were used to show equal loading.
Protein expression and purification
GST fusion proteins were expressed in E. coli Bl21 (DE3) Rosetta at 18°C for 16 h after induction with 0.5 mM IPTG at an OD600 of 0.6. Cell pellets were resuspended in lysis buffer (50 mM Na‐HEPES, pH 7.5, 250 mM NaCl, 1 mM EDTA, 5% Glycerol, 0.1% NP40, Protease Inhibitor Cocktail set III, Calbiochem and 0.1 mM PMSF). Sonicated lysates were cleared by centrifugation at 30,000 g for 30 min. Supernatants were incubated with 1 ml of Glutathione Sepharose beads (GE Healthcare) per litre of bacterial culture. After 2 h at 4°C, the beads were washed with lysis buffer, high salt buffer (50 mM Tris, pH 8, 1 M NaCl, 1 mM EDTA, 1 mM DTT and 5% glycerol) and equilibrated in storage buffer (20 mM Tris, pH 8, 150 mM NaCl, 1 mM EDTA, 1 mM DTT and 5% glycerol).
Liquid chromatography–tandem MS (LC–MS/MS) analysis
To identify myosin VI interactors, anti‐myosin VI co‐IP was performed using 3 mg of fresh lysates of HeLa, MDA‐MB‐231, MCF10A and Caco‐2 cells grown in confluent conditions. Parallel co‐IP was performed using anti‐myosin VI antibody (1295) or a rabbit control antibody as negative control. Precipitated immunocomplexes were washed, loaded on a 4–20% TGX precast gel (Bio‐Rad) and stained with colloidal blue (Colloidal Blue Staining Kit, Invitrogen).
Gels were cut in slices and trypsinised as previously described (Shevchenko et al, 1996). Peptides were desalted as described (Rappsilber et al, 2003), dried in a Speed‐Vac and resuspended in 10 µL of solvent A (2% ACN, 0.1% formic acid). About 3 μl was injected on a quadrupole Orbitrap Q‐Exactive mass spectrometer (Thermo Scientific) coupled with an UHPLC Easy‐nLC 1000 (Thermo Scientific), with a 25‐cm fused‐silica emitter of 75 μm inner diameter. Columns were packed in‐house with ReproSil‐Pur C18‐AQ beads (Dr. Maisch Gmbh, Ammerbuch, Germany), 1.9 μm of diameter, using a high‐pressure bomb loader (Proxeon, Odense, Denmark). Peptide separation was achieved with a linear gradient from 95% solvent A (2% ACN, 0.1% formic acid) to 40% solvent B (80% acetonitrile, 0.1% formic acid) over 30 min and from 40% to 100% solvent B for 2 min at a constant flow rate of 0.25 μl/min, with a single run time of 33 min. MS data were acquired using a data‐dependent top 12 method, and the survey full‐scan MS spectra (300–1750 Th) were acquired in the Orbitrap with 70,000 resolution, AGC target 1e6, IT 120 ms. For HCD spectra, the resolution was set to 35,000, AGC target 1e5, IT 120 ms; 25% normalised collision energy and isolation width of 3.0 m/z.
For protein identification, the raw data were processed using Proteome Discoverer (version 1.4.0.288, Thermo Fischer Scientific). MS2 spectra were searched with Mascot engine against uniprot_human_20150401 database (90,411 entries), with the following parameters: enzyme trypsin, maximum missed cleavage 2, fixed modification carbamidomethylation (C), variable modification oxidation (M) and protein N‐terminal acetylation, peptide tolerance 10 ppm, MS/MS tolerance 20 mmu. Peptide Spectral Matches (PSM) were filtered using percolator based on q‐values at a 0.01 FDR (high confidence). Proteins were considered identified with 2 unique high confident peptides (Kall et al, 2007). Scaffold (version Scaffold_4.3.4, Proteome Software Inc., Portland, OR) was used to validate MS/MS‐based peptide and protein identifications. Peptide identifications were accepted if they could be established at a probability greater than 95.0% by the Peptide Prophet algorithm (Keller et al, 2002) with Scaffold delta‐mass correction. Protein identifications were accepted if they could be established at a probability greater than 99.0% and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al, 2003). Proteins that contained similar peptides and that could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters.
Immunofluorescence (IF)
Cells were grown on coverslips and fixed with cold 100% methanol at −20°C for 10 min. The coverslips were incubated in PBS with 10% FBS for 30 min for blocking, followed by incubation with primary antibodies (overnight at 4°C) and then secondary antibodies (1 h at RT) in PBS with 10% FBS. Incubation with DAPI (Sigma‐Aldrich, cat. D9542) for 10 min was performed to stain the nuclei. The coverslips were mounted on glass slides using Mowiol Mounting Medium (Calbiochem) or Dako Faramount Aqueous Mounting Medium (s3025, DAKO). Images were acquired using a GE HealthCare Deltavision OMX system, equipped with 2 PCO Edge 5.5 sCMOS cameras, using a 60× 1.42 NA Oil immersion objective, or using a Deltavision Elite system (GE Healthcare) equipped with a IX71 microscope (Olympus), a sCMOS camera, using a 60× PlanApo 1.42 NA oil immersion objective and driven by softWoRx version 7.0.0. Images were acquired as a z‐series (0.2‐µm z interval in a range of 4 μm) and deconvolution was performed using softWorx software. The images are presented as maximum‐intensity projections and were prepared using ImageJ/Fiji (National Institutes of Health).
For myosin VI and pericentrin co‐staining (Fig EV1C), cells were fixed with 4% PFA for 10 min, permeabilised with 0.2% Triton X‐100 and blocked with 20% donkey serum in PBS. Primary and secondary antibodies were diluted in PBS with 20% donkey serum and incubated at RT for 1 h and 30 min, respectively. After nuclei staining with DAPI for 10 min, the coverslips were mounted on glass slides using Mowiol Mounting Medium (Calbiochem). Confocal microscopy was performed on a Leica TCS SP5 laser confocal scanner mounted on a Leica DMI 6000B inverted microscope equipped with HCX PL APO 63×/1.4 NA oil immersion objective. Leica LAS AF software was used for image acquisitions. For co‐localisation analysis, ROIs were drawn around individual centrosomes and the M2 Manders’ coefficient was obtained using JACoP plugin (ImageJ/Fiji) and processed for statistical analysis with Prism (GraphPad software).
OFD1, Cep164 and PCM1 fluorescence intensity analysis
For OFD1 and Cep164 fluorescence intensity quantifications at the centrioles, hTERT‐RPE1 cells were treated with 6 μg/ml nocodazole (M1404, Sigma) for 1 h to depolymerise the microtubules and thus remove the centriolar satellites. The removal of the satellites is required for the quantification of the centriolar pool of OFD1, which is otherwise indistinguishable from the satellite pool. Z‐stack images (0.2 μm intervals) of 20 sections were acquired as detailed above. Centrioles were considered for quantification when paired signals of centrin and OFD1 were observed. The presence Cep164 staining was used to identify mother centrioles. Centrioles were excluded from the analysis if residual OFD1 satellite staining was visible in the vicinity of the centrioles. The integrated intensity of a circular 1 × 1 μm area around the specific OFD1 and Cep164 signals were recorded in the sum projected images using ImageJ/Fiji (National Institutes of Health). Integrated density was corrected for background intensity signal with the following formula: corrected total fluorescence = integrated density − (area of selected region × mean intensity of background). The number of cells analysed and of experiments performed is detailed in the figure legends.
For centriolar satellite intensity quantifications, staining of PCM1 was performed in combination with a centriole marker (Cep135 or Cep164). The centriole marker was used to determine the centre of a circular 3 × 3 μm area, where the PCM1 signals were recorded in the sum projected images. For quantification of OFD1 intensity at the centriolar satellites, OFD1 staining was performed in combination with PCM1 and Cep164. In order to exclude the contribution of the centriolar pool of OFD1 from the analysis, OFD1 intensity was recorded only in the area covered by PCM1 in the sum projected images. Corrected total fluorescence was calculated as detailed above.
To induce p53 activation, hTERT‐RPE1 cells were incubated with 10 μM Nutlin‐3 (N6287, Sigma) for 24 h before fixation and IF.
Statistical analysis
Statistical analyses were performed with Prism (GraphPad software). Unless differently specified, all the statistical significance calculations were determined using either unpaired Student’s t or ANOVA tests, or the non‐parametric Mann–Whitney or Kruskal–Wallis test, after assessing the normal distribution of the sample with Normal (Gaussian) distribution test. Sample sizes are indicated in the figure legends and were chosen arbitrarily with no inclusion and exclusion criteria. The investigators were not blind to the group allocation during the experiments and data analyses.
Proximity ligation assay (PLA)
hTERT‐RPE1 cells were transfected with pEGFP‐C1 myosinVIshort FL using Lipofectamine 2000 reagent (Invitrogen) and fixed at 48 h after transfection with 100% MeOH at −20°C for 10 min. PLA was performed with the Duolink™ In Situ Orange Starter Kit (Sigma, DUO92102) according to manufacturer's instructions using mouse anti‐GFP (1:2,000; Thermo Fisher Scientific, A11120) and rabbit anti‐OFD1 (1:2,000; Sigma, HPA031103) primary antibodies and secondary anti‐mouse MINUS and anti‐rabbit PLUS probes. As negative controls for the PLA signal, the secondary antibodies were used without previous primary antibody incubation or with the single primary antibody. Counterstaining with anti‐mouse A488 and anti‐rabbit A647 was performed to identify GFP‐positive cells and to localise OFD1. Confocal microscopy was performed on a Leica TCS SP5 laser confocal scanner mounted on a Leica DMI 6000B inverted microscope equipped with motorised stage. The images were acquired with an HCX PL APO 63X/1.4NA oil immersion objective using 405, 488, 568 and 647 nm laser lines. Leica LAS AF software was used for all acquisitions.
Fluorescence recovery after photobleaching (FRAP)
hTERT‐RPE1 centrin1‐dTomato GFP‐OFD1 cells were transfected with myosin VI siRNA and plated on MatTek glass bottom dishes (P35G‐1.5‐14‐C, MatTek Life Sciences). FRAP experiments were performed on the UltraVIEW VoX spinning‐disk confocal system (PerkinElmer) equipped with an EclipseTi inverted microscope (Nikon), provided with an integrated FRAP PhotoKinesis unit (PerkinElmer), a Hamamatsu CCD camera (C9100‐50), a Okolab cage incubator, and driven by Volocity software (Improvision; Perkin Elmer). Photobleaching was achieved on a square region of 3 × 3 μm by using the 488 nm laser at the maximum output to bleach the GFP signal. Initially, 10 images with a 400‐ms time‐frame were acquired to determine the levels of pre‐bleach fluorescence. Images were acquired through a 60× oil‐immersion objective (Nikon Plan Apo VC, NA 1.4) with the following time‐frame: every second for the first 60 s, every 2 s for the following 60 s and every 5 s for the following 120 s.
A custom ImageJ macro and a set of functions written in Matlab software were used to analyse the recovery curves. ImageJ was used to measure the mean intensity value over time in the bleached area and the background over time in an area in the field without cells. StackReg ImageJ Plugin was used to align the bleached area over time. The photobleaching recovery curves were then normalised to the pre‐bleaching mean intensity values after background correction using Matlab. Matlab was then used to fit the first 150 s after photobleaching of the recovery curves with a one phase exponential recovery function with f0 fixed as the first value after photobleaching:
Half maximum time (t1/2) was then evaluated as:
and Mobile Fraction as:
Structured illumination microscopy (SIM)
hTERT‐RPE1 control and myosin VI‐depleted cells were plated on 13‐mm coverslips, thickness 1.5 mm, Boroslicate glass (631‐0150, VWR). Four days after transfection, cells were incubated for 1 h on ice in PBS to induce the depolymerisation of cytoplasmic microtubules and were subsequently stained with anti‐acetylated tubulin and anti‐OFD1, anti‐Cep164 or anti‐ODF2 antibodies (for more details, see the IF staining section). SIM Images were acquired using a GE HealthCare Deltavision OMX system, equipped with 2 PCO Edge 5.5 sCMOS cameras, using a 60× 1.42NA Oil immersion objective. Images were deconvolved and reconstructed with Applied Precision's softWorx software. The images are presented as maximum‐intensity projections and were prepared using ImageJ/Fiji (National Institutes of Health).
Direct stochastic optical reconstruction microscopy (dSTORM)
hTERT‐RPE1 cells were transfected with myosin VI siRNA and plated on MatTek glass bottom dishes (P35G‐1.5‐14‐C, MatTek Life Sciences). Four days after transfection, cells were fixed with methanol and stained with anti‐pericentrin, anti‐ODF2 or anti‐FBF1 antibodies. dSTORM imaging was performed using the Nikon N‐STORM microscope equipped with a 1.49 NA CFI Apochromat TIRF objective, exciting the Alexa Fluor 647 dye with the 647 nm laser light in HILO (highly inclined and laminated optical sheet) mode (Tokunaga et al, 2008). The 405 nm laser light was used to reactivate Alexa Fluor 647 into a fluorescent state. The activation laser power was constantly increased during the acquisitions up to a maximum of 10% of the laser power. Each dSTORM acquisition consisted of 30,000 images recorded with an Orca‐Flash4.0 sCMOS camera (Hamamatsu) with an exposure time of 20 ms, a pixel size of 161.5 nm and a field of view of 64 × 64 pixels. During dSTORM acquisitions, cells were kept in imaging buffer (100 mM MEA, 1% glucose, 560 μg/ml Glucose Oxidase, and 34 μg/ml Catalase in PBS). Prior to dSTORM imaging, the pericentrin staining was imaged in wide‐field mode to identify the centrioles oriented perpendicular to the imaging plane.
dSTORM image sequences were processed using the ImageJ/Fiji plug‐in ThunderSTORM (Ovesny et al, 2014) with a pre‐detection wavelet filter (B‐spline, scale 2, order 3), initial detection by non‐maximum suppression (radius 1, threshold at one standard deviation of the F1 wavelet), and sub‐pixel localisation by integrated Gaussian point‐spread function and maximum likelihood estimator with a fitting radius of 3 pixels. Detected localisations were filtered (intensity > 500 photons, sigma range of 50–500, and localisation uncertainty < uncertainty mean). The filtered dataset was then corrected for sample drift (cross‐correlation of images from five bins at a magnification of 5) and repeated localisations were removed by merging points that reappeared within 3*uncertainty nm. STORM images were visualised using the Average shifted histograms method with 2 lateral shifts. To emphasise the symmetry of the distribution of the distal appendages, the STORM image was rotated for 40 degrees × 8 times around the centre of the centriole, and the average projection from 9 orientations was generated.
Primary cilium assay
hTERT‐RPE1 p53 KO cells were transfected with myosin VI or OFD1 siRNAs and plated on coverslips coated with 0.2% gelatine. Four days after transfection, cells were serum starved in medium with 0% serum (DMEM/F12 supplemented with 2 mM l‐glutamine, 0.5 mM Na‐Pyruvate and 15 mM Hepes, pH 7.5) for 24 h to allow cilium assembly. Microtubule depolymerisation was induced by incubating the cells in PBS at 4°C for 1 h before Me‐OH fixation. Subsequently, cell lines were stained with anti‐acetylated tubulin and anti‐Cep135 primary antibodies and DAPI. Z‐stack images (0.2 μm intervals) of 20 sections were acquired using a Deltavision Elite system (GE Healthcare) equipped with a IX71 microscope (Olympus), a sCMOS camera, using a 60× PlanApo 1.42 NA oil immersion objective and driven by softWoRx version 7.0.0. The number of cells displaying a primary cilium was determined using anti‐acetylated tubulin staining, and centrioles were identified through the double Cep135/acetylated tubulin staining.
Correlative Light Electron Microscopy (CLEM)
hTERT‐RPE1 cells were transfected with the indicated siRNA 96 h before fixation and plated on gridded MatTek glass bottom dishes (P35G‐1.5‐14‐CGRD, MatTek Life Sciences). Cells were fixed and stained as described (Mironov & Beznoussenko, 2013), using anti‐pericentrin antibody to identify the position of the centrosomes. Images were acquired on a XZ plane to calculate the distance between the glass bottom and the centrosome, using a Leica TCS SP5 laser confocal scanner mounted on a Leica DMI 6000B inverted microscope equipped with a HCX PL APO 63X/1.4NA oil immersion objective using 488 nm laser line. Leica LAS AF software was used for all acquisitions. For transmission electron microscopy (TEM), samples were then fixed with 2.5% GA in 0.2 M Cacodylate buffer, pH 7.2, and embedded in resin (Beznoussenko & Mironov, 2015). The samples embedded in resins were sectioned with diamond knife (Diatome, Switzerland) using Leica EM UC7 ultramicrotome. Sections (50‐60 nm) were analysed with a Tecnai 20 High Voltage EM (FEI, The Netherlands) operating at 200 kV.
For immunogold staining of myosin VI, A549 wild‐type and myosin VI KO cells were plated on gridded MatTek glass bottom dishes and treated as detailed/described above for CLEM. Then, cells were stained with anti‐myosin VI 1296 antibody, incubated with Nanogold®‐Fab' Goat anti‐Rabbit IgG (#2004, Nanoprobes). GoldEnhance™ (EM #2113, Nanoprobes) was used to enhance the signal from nanogold particles.
Flow cytometry analysis of cell cycle profile
Myosin VI shRNA expression was induced with 0.5 µg/ml doxycycline for 10 days. Cells were then trypsinised and washed once in PBS. After centrifugation, cell pellets were resuspended in 250 µl of 4°C‐cold PBS, fixed by adding 750 µl of 100% Et‐OH (−20°C) dropwise while vortexing and left 1 h on ice. Cells were then washed in PBS‐1% BSA and resuspended in 1 ml PI (50 µg/ml) and RNAse (250 µg/ml). After incubation at 4°C overnight, flow cytometry was performed for cell cycle analysis. Sample acquisition was performed with FACSCanto II (Beckton Dickinson). Analysis of cell cycle distribution was performed with ModFitLT V3.1 software.
Senescence‐associated β‐galactosidase (SA‐β‐gal) assay
After fixation with 4% PFA for 10 min, cells were incubated with SA‐β‐gal staining solution containing 1 mg/ml 5‐bromo‐4‐chloro‐3‐indolyl beta‐D galactopyranoside (X‐Gal), 40 mM citric acid/sodium phosphate pH 6.0, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl and 2 mM MgCl2. After incubation at 37°C overnight, cells were washed with PBS and mounted on glass slides using Mowiol Mounting Medium. Images were acquired using a digital camera connected to a white‐light microscope. SA‐β‐gal activity was detected in senescent cells as local perinuclear blue precipitate.
Author contributions
EMag and SP conceptualised; EMag performed all the experiments with the help of GM, CN and EMas; MLF aided in the imaging acquisition and SIM experiments; GB and AM performed the EM experiment; MBD contributed to the planning and interpretation of data; SP coordinated the team, designed and supervised the project; EMag and SP wrote the paper with contributions from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Brunella Franco for DNA constructs, and Francesca Farina and Luca Fava for the generation of cell lines. We are grateful to Simona Rodighiero (Imaging Unit at IEO, Milan, Italy) for the dSTORM image acquisition. We thank Paolo Soffientini, Angela Cattaneo, Massimiliano Garrè and Emanuele Martini at Cogentech facilities (Milan, Italy) for support in mass spectrometry and FRAP analysis. We are grateful to Wessen Maruwge for English language editing. This work was supported by the Associazione Italiana per Ricerca sul Cancro, (Investigator grant 2017‐ 19875 to S.P.). Elisa Magistrati’s work was supported by the Associazione Italiana per la Ricerca sul Cancro. E. Mag. was and G.M. is a PhD student at the European School of Molecular Medicine (SEMM).
EMBO reports (2022) 23: e54160.
Contributor Information
Elisa Magistrati, Email: elisa.magistrati@fmi.ch.
Simona Polo, Email: simona.polo@ifom.eu.
Data availability
The mass spectrometry raw datasets were deposited in PRIDE database and can be accessed through ProteomeXchange with the following Project Name: Study of endogenous interactome of Myosin VI, Project accession: PXD026697 (http://www.ebi.ac.uk/pride/archive/projects/PXD026697). Full list of the specific interactors is provided in Dataset EV1.
References
- Akhshi T, Trimble WS (2021) A non‐canonical Hedgehog pathway initiates ciliogenesis and autophagy. J Cell Biol 220: e202004179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK (2009) The primary cilium as a complex signaling center. Curr Biol 19: R526–R535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beznoussenko GV, Mironov AA (2015) Correlative video‐light‐electron microscopy of mobile organelles. Methods Mol Biol 1270: 321–346 [DOI] [PubMed] [Google Scholar]
- Biancospino M, Buel GR, Niño CA, Maspero E, Scotto di Perrotolo R, Raimondi A, Redlingshöfer L, Weber J, Brodsky FM, Walters KJ et al (2019) Clathrin light chain A drives selective myosin VI recruitment to clathrin‐coated pits under membrane tension. Nat Commun 10: 4974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M, Jensen LR, Raynaud M, Shoichet SA, Badura M et al (2006) A novel X‐linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral‐facial‐digital type I syndrome. Hum Genet 120: 171–178 [DOI] [PubMed] [Google Scholar]
- Burigotto M, Mattivi A, Migliorati D, Magnani G, Valentini C, Roccuzzo M, Offterdinger M, Pizzato M, Schmidt A, Villunger A et al (2021) Centriolar distal appendages activate the centrosome‐PIDDosome‐p53 signalling axis via ANKRD26. EMBO J 40: e104844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss F, Arden SD, Lindsay M, Luzio JP, Kendrick‐Jones J (2001) Myosin VI isoform localized to clathrin‐coated vesicles with a role in clathrin‐mediated endocytosis. EMBO J 14: 3676–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coene KLM, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJF, Ngu LH, Budny B, van Wijk E, Gorden NT et al (2009) OFD1 is mutated in X‐linked Joubert syndrome and interacts with LCA5‐encoded lebercilin. Am J Hum Genet 85: 465–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammermann A, Merdes A (2002) Assembly of centrosomal proteins and microtubule organization depends on PCM‐1. J Cell Biol 159: 255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogterom M, Koenderink GH (2019) Actin‐microtubule crosstalk in cell biology. Nat Rev Mol Cell Biol 20: 38–54 [DOI] [PubMed] [Google Scholar]
- Farina F, Gaillard J, Guérin C, Couté Y, Sillibourne J, Blanchoin L, Théry M (2016) The centrosome is an actin‐organizing centre. Nat Cell Biol 18: 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina F, Ramkumar N, Brown L, Samandar Eweis D, Anstatt J, Waring T, Bithell J, Scita G, Thery M, Blanchoin L et al (2019) Local actin nucleation tunes centrosomal microtubule nucleation during passage through mitosis. EMBO J 38: e99843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA et al (2017) The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 31: 34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feather SA, Woolf AS, Donnai D, Malcolm S, Winter RM (1997) The oral‐facial‐digital syndrome type 1 (OFD1), a cause of polycystic kidney disease and associated malformations, maps to Xp22.2‐Xp22.3. Hum Mol Genet 6: 1163–1167 [DOI] [PubMed] [Google Scholar]
- Ferrante MI, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS, Sylvie O et al (2001) Identification of the gene for oral‐facial‐digital type I syndrome. Am J Hum Genet 68: 569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dolle P, Franco B (2006) Oral‐facial‐digital type I protein is required for primary cilia formation and left‐right axis specification. Nat Genet 38: 112–117 [DOI] [PubMed] [Google Scholar]
- Field M, Scheffer IE, Gill D, Wilson M, Christie L, Shaw M, Gardner A, Glubb G, Hobson L, Corbett M et al (2012) Expanding the molecular basis and phenotypic spectrum of X‐linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet 20: 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CS, Mazo G, Das T, Goodman J, Kim M, O'Rourke BP, Izquierdo D, Tsou MF (2016) 53BP1 and USP28 mediate p53‐dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. eLife 5: e16270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D (2014) Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 158: 833–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio G, Alfieri M, Prattichizzo C, Zullo A, Cairo S, Franco B (2007) Functional characterization of the OFD1 protein reveals a nuclear localization and physical interaction with subunits of a chromatin remodeling complex. Mol Biol Cell 18: 4397–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Wollscheid HP, Nowicka U, Biancospino M, Valentini E, Ehlinger A, Acconcia F, Magistrati E, Polo S, Walters KJ (2016) Myosin VI contains a compact structural motif that binds to ubiquitin chains. Cell Rep 14: 2683–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Obino D, Pineau J, Farina F, Gaillard J, Guerin C, Blanchoin L, Lennon‐Dumenil AM, Thery M (2019) Actin filaments regulate microtubule growth at the centrosome. EMBO J 38: e99630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ (2007) Semi‐supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4: 923–925 [DOI] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74: 5383–5392 [DOI] [PubMed] [Google Scholar]
- Kruppa AJ, Buss F (2018) Actin cages isolate damaged mitochondria during mitophagy. Autophagy 14: 1644–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ (2016) A USP28‐53BP1‐p53‐p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 214: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CA, Prosser SL, Romio L, Hirst RA, O'Callaghan C, Woolf AS, Fry AM (2011) Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral‐facial‐digital syndrome 1. J Cell Sci 124: 600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistrati E, Polo S (2020) Myomics: myosin VI structural and functional plasticity. Curr Opin Struct Biol 67: 33–40 [DOI] [PubMed] [Google Scholar]
- Majewski L, Sobczak M, Wasik A, Skowronek K, Redowicz MJ (2011) Myosin VI in PC12 cells plays important roles in cell migration and proliferation but not in catecholamine secretion. J Muscle Res Cell Motil 32: 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malicki JJ, Johnson CA (2017) The Cilium: cellular antenna and central processing unit. Trends Cell Biol 27: 126–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK et al (2016) 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol 214: 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S (2007) Loss of centrosome integrity induces p38—p53—p21‐dependent G1—S arrest. Nat Cell Biol 9: 160–170 [DOI] [PubMed] [Google Scholar]
- Mironov AA, Beznoussenko GV (2013) Correlative microscopy. Methods Cell Biol 113: 209–255 [DOI] [PubMed] [Google Scholar]
- Morleo M, Brillante S, Formisano U, Ferrante L, Carbone F, Iaconis D, Palma A, Buonomo V, Maione AS, Grumati P et al (2021) Regulation of autophagosome biogenesis by OFD1‐mediated selective autophagy. EMBO J 40: e105120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morleo M, Franco B (2019) The Autophagy‐Cilia axis: an intricate relationship. Cells 8: 905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morleo M, Franco B (2020) OFD Type I syndrome: lessons learned from a rare ciliopathy. Biochem Soc Trans 48: 1929–1939 [DOI] [PubMed] [Google Scholar]
- Mukherjea M, Ali MY, Kikuti C, Safer D, Yang Z, Sirkia H, Ropars V, Houdusse A, Warshaw DM, Sweeney HL (2014) Myosin VI must dimerize and deploy its unusual lever arm in order to perform its cellular roles. Cell Rep 8: 1522–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75: 4646–4658 [DOI] [PubMed] [Google Scholar]
- O'Loughlin T, Masters TA, Buss F (2018) The MYO6 interactome reveals adaptor complexes coordinating early endosome and cytoskeletal dynamics. EMBO Rep 19: e44884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovesný M, Křížek P, Borkovec J, Švindrych Z, Hagen GM (2014) ThunderSTORM: a comprehensive ImageJ plug‐in for PALM and STORM data analysis and super‐resolution imaging. Bioinformatics 30: 2389–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampliega O, Cuervo AM (2016) Autophagy and primary cilia: dual interplay. Curr Opin Cell Biol 39: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Lim JS, Ramakrishina S, Kim SH, Kim WK, Lee J, Kang H‐C, Reiter JF, Kim DS, Kim H( et al (2018) Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination Neuron 99: 83–97.e87 [DOI] [PubMed] [Google Scholar]
- Penengo L, Mapelli M, Murachelli AG, Confalonieri S, Magri L, Musacchio A, Di Fiore PP, Polo S, Schneider TR (2006) Crystal structure of the ubiquitin binding domains of rabex‐5 reveals two modes of interaction with ubiquitin. Cell 124: 1183–1195 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- Reiter JF, Leroux MR (2017) Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol 18: 533–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA (2004) OFD1 is a centrosomal/basal body protein expressed during mesenchymal‐epithelial transition in human nephrogenesis. J Am Soc Nephrol 15: 2556–2568 [DOI] [PubMed] [Google Scholar]
- Sahlender DA, Roberts RC, Arden SD, Spudich G, Taylor MJ, Luzio JP, Kendrick‐Jones J, Buss F (2005) Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol 169: 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Dynlacht BD (2016) Cilium assembly and disassembly. Nat Cell Biol 18: 711–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver‐stained polyacrylamide gels. Anal Chem 68: 850–858 [DOI] [PubMed] [Google Scholar]
- Singla V, Romaguera‐Ros M, Garcia‐Verdugo JM, Reiter JF (2010) Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell 18: 410–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich G, Chibalina MV, Au JS, Arden SD, Buss F, Kendrick‐Jones J (2007) Myosin VI targeting to clathrin‐coated structures and dimerization is mediated by binding to Disabled‐2 and PtdIns(4,5)P2. Nat Cell Biol 9: 176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q (2013) Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502: 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin‐Robinet C, Lee JS, Lopez E, Herranz‐Pérez V, Shida T, Franco B, Jego L, Ye F, Pasquier L, Loget P et al (2014) The oral‐facial‐digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat Genet 46: 905–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, Sakata‐Sogawa K (2008) Highly inclined thin illumination enables clear single‐molecule imaging in cells. Nat Methods 5: 159–161 [DOI] [PubMed] [Google Scholar]
- Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick‐Jones J, Buss F (2012) Autophagy receptors link myosin VI to autophagosomes to mediate Tom1‐dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol 14: 1024–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C et al (2004) In vivo activation of the p53 pathway by small‐molecule antagonists of MDM2. Science 303: 844–848 [DOI] [PubMed] [Google Scholar]
- Wang L, Failler M, Fu W, Dynlacht BD (2018) A distal centriolar protein network controls organelle maturation and asymmetry. Nat Commun 9: 3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollscheid HP, Biancospino M, He F, Magistrati E, Molteni E, Lupia M, Soffientini P, Rottner K, Cavallaro U, Pozzoli U et al (2016) Diverse functions of myosin VI elucidated by an isoform‐specific alpha‐helix domain. Nat Struct Mol Biol 23: 300–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW et al (2015) Reversible centriole depletion with an inhibitor of Polo‐like kinase 4. Science 348: 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Li C, Siu GKY, Luo X, Yu S (2020) Distinct roles of TRAPPC8 and TRAPPC12 in ciliogenesis via their interactions with OFD1. Front Cell Dev Biol 8: 148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zullo A, Iaconis D, Barra A, Cantone A, Messaddeq N, Capasso G, Dolle P, Igarashi P, Franco B (2010) Kidney‐specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum Mol Genet 19: 2792–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]