

Abstract
Alternative splicing is a potent modifier of protein function. Stromal interaction molecule 1 (Stim1) is the essential activator of store‐operated Ca2+ entry (SOCE) triggering activation of transcription factors. Here, we characterize Stim1A, a splice variant with an additional 31 amino acid domain inserted in frame within its cytosolic domain. Prominent expression of exon A is found in astrocytes, heart, kidney, and testes. Full‐length Stim1A functions as a dominant‐negative regulator of SOCE and ICRAC, facilitating sequence‐specific fast calcium‐dependent inactivation and destabilizing gating of Orai channels. Downregulation or absence of native Stim1A results in increased SOCE. Despite reducing SOCE, Stim1A leads to increased NFAT translocation. Differential proteomics revealed an interference of Stim1A with the cAMP‐SOCE crosstalk by altered modulation of phosphodiesterase 8 (PDE8), resulting in reduced cAMP degradation and increased PIP5K activity, facilitating NFAT activation. Our study uncovers a hitherto unknown mechanism regulating NFAT activation and indicates that cell‐type‐specific splicing of Stim1 is a potent means to regulate the NFAT signalosome and cAMP‐SOCE crosstalk.
Keywords: NFAT, Orai, PDE8, PIP2, PIP5K
Subject Categories: Membranes & Trafficking, Signal Transduction
Alternative splicing of the Ca2+ regulator gene (STIM1) is able to interfere both with ion channel (ORAI) gating and with Ca2+‐cAMP crosstalk to adjust for cell‐type‐specific transcription factor activation.
Introduction
Stromal interaction molecule (STIM) and ORAI proteins are the hallmark constituents of store‐operated Ca2+ entry (SOCE). SOCE is critically important for T‐cell function, but STIM and ORAI proteins have been found in virtually all cell types. SOCE is particularly relevant for relatively slow‐ and long‐lasting processes such as gene expression and cell proliferation (Müller & Rao, 2010; Yeh & Parekh, 2018). It is triggered by receptor‐mediated release of Ca2+ from the endoplasmic reticulum (ER), which is sensed by STIM1 and STIM2 proteins through their respective luminal EF hands that then cluster and trap ORAI channels located in the plasma membrane (PM) into ER‐PM junctions (reviewed in Soboloff et al (2012), Prakriya and Lewis (2015), Zhou et al (2015), Nwokonko et al (2017)). In patients and murine gene‐modified models, loss‐of‐function (LOF) mutations of either STIM1 or ORAI1 lead to severe immune pathologies, myopathies, defects in enamel formation, and anhidrosis (reviewed in Lacruz & Feske, 2015; Michelucci et al, 2018; Feske, 2019). In mice, global deletion of Stim1 results in a severe reduction in the number of offspring and those that are born die within the first weeks (Oh‐hora et al, 2008). The early lethality indicates essential roles for Stim1 that are independent of its immune cell function, a conclusion that is supported by multi‐system phenotypes in gain‐of‐function (GOF) mutations.
Here, we explore whether all of these highly diverse functions of STIM1 are mediated by the same splice variant(s), or whether and how alternative splicing modifies STIM1 structure and function to adapt to cell‐type‐specific requirements. In the past, we and others have identified a splice variant of STIM2 (STIM2.1; STIM2ß), which prevents interaction and gating of Orai channels (Miederer et al, 2015; Rana et al, 2015) and thus acts as a strong dominant‐negative regulator of SOCE by altering the density of activated Orai complexes (Zhou et al, 2018). This splice variant has recently been shown to regulate myogenesis by controlling SOCE‐dependent transcriptional factors (Kim et al, 2019). Darbellay et al (2011) showed that alternative splicing of STIM1 leads to a longer STIM1 protein, STIM1L, which is expressed exclusively in adult human muscle fibers and in in vitro‐differentiated myotubes. We recently identified a mutually exclusive alternate splice event of STIM1 at the same exon 11/12 boundary. This neuron‐specific alternate splice event leads to a shorter STIM1 protein variant (STIM1B) with preferential localization to presynaptic ER where it enables increased synaptic vesicle release at high stimulus frequencies (Ramesh et al, 2021). In this study, we focus on an upstream alternative splice event, which inserts an exon into the exon 10/11 boundary, creating a variant with prominent mRNA expression in cells involved in barrier function such as astrocytes and Sertoli cells and with more ubiquitous expression compared to Stim1B. In contrast to the recently characterized STIM1B, in frame insertion of the A domain leads to ICRAC with altered fast calcium‐dependent inactivation (FCDI). Applying computational molecular docking in conjunction with mutational analysis we show that STIM1A destabilizes gating of Orai channels. Interestingly, despite reduced SOCE, expression of STIM1A resulted in increased NFAT translocation and proteomic analysis revealed differential interaction with a specific phosphodiesterase (PDE8B). We found an unexpected STIM1‐dependent regulation of cAMP levels affecting NFAT translocation, which was altered in the presence of STIM1A and was due to cAMP‐driven changes in phosphoinosite levels, PIP or PIP2. These results point toward an important role of STIM1 splicing in modulating the crosstalk between Gαs/αi‐ and Gq‐mediated cellular signaling cascades.
Results
Stim1A is an evolutionary conserved expressed Stim1 splice variant
Both Stim1 and Stim2 genes contain multiple exons. Database mining predicts that several variants including at least 3 murine variants, several human variants, and EST clones contain an additional exon insertion between conventional exons 10 and 11, named here exon A, encoding 31 amino acids (NM_001374058.1, XM_006507535.4, XM_006507536.2), human variant (NM_001382567.1), and EST clones (BQ068737.1, BU553936.1). The exon 12 and exon 14 boundaries harbor mutually exclusive alternative exons 13 (L) or 13 (B) (Fig 1A). Exon A splicing extends the C‐terminal domain by 93 nucleotides and translates to a 31 amino acid in‐frame insertion (domain A) downstream of residue 491. Using cDNA derived from different murine tissues, we devised both a flanking PCR and qRT–PCR strategies to screen for the presence of the alternative exon A and that of the conventional variant (ØA = wt) (Fig 1B). This strategy enabled us to detect multiple splice events because primers extend over several splice sites and could result in 310 bp for Stim1 wt, 402 bp for exon A, 438 bp for exon AB, 613 bp for exon L, and 346 bp for an exon B splice event. We were able to detect 310‐bp (wt) products in all selected tissues and a prominent additional 402‐bp (exon A) product in testes and astrocytes. Only in skeletal muscle, we detected the 613‐bp product (exon L) (Fig 1C). Using quantitative real‐time PCR either with an exon A‐specific primer or a primer bridging the exon 10/exon 11 boundary (ØA) (Fig 1B), we confirmed significant relative expression of exon A in testes, heart, astrocytes, and kidney, whereas CD8+ T cells did not show detectable expression of exon A (Fig 1D). In addition to the primers indicated in Fig 1, we also devised standard and qRT–PCR primers to check for potential N‐terminal splice events (Fig EV1A). Using primers indicated in Fig 1B, the absolute 2−∆Cq values demonstrate that expression of exon A is seen in most tissues with less variability compared with Stim1ØA expression (Fig EV1B). As database mining also indicated that expression of exon A may occur in conjunction with alternative splicing within the 5’UTR region (putative transcript XM_006507535.4), we confirmed the existence of exon A (reverse primer) with the regular STIM1 N‐term by PCR using forward primers specific for the 5´UTR (wt), 5´UTR (alt 1), or 5´UTR (alt 2) (Figs 1A and EV1A). Both 5´UTR splice events would lead to deletion of exon 1 and result in translation at the first ATG within exon 2. Translation of these variants would lead to proteins lacking the signal peptide and essential parts of the first EF hand to start at M75 of Stim1 and are not functionally investigated here. For testis, Fig EV1C shows a prominent PCR product of the 5´UTR (wt) together with an A‐specific reverse primer, a weaker product of A with the alt1 primer, and only a faint product of A with the alt2 primer, which required a longer exposure time at which other products were saturated. Using the non‐A reverse primer, we also detected dominant 5´UTR (wt) and slightly decreased alt1 (5% of ØA products) PCR products (Fig EV1C). In astrocytes, we did not detect expression of exon A in conjunction with alt 1 or alt 2 N‐terminal variants. To further quantitate the relative amount of N‐terminal splicing, we also used qRT‐PCR primers specific for alternate 5’UTR regions together with a reverse primer within exon 2 (Fig EV1A) to show that 86% and 97% of all Stim1 variants in testes and astrocytes, respectively, contain the wt 5´UTR (Fig EV1D). Having confirmed the existence of exon A within Stim1, we cloned the entire open reading frame of murine Stim1A from cDNA into bicistronic or directly tagged expression vectors and also inserted exon A into human STIM1. The resulting constructs translate to a 716 amino acid protein in which domain A is in close proximity to the negatively charged inhibitory domain (ID) (Fig 1E). 71% of the amino acids encoded by exon A are evolutionarily highly conserved from fish, amphibians, reptiles, and birds to mammals (Fig 1F). Only a 12‐nucleotide intra‐exonic splice event in the middle of domain A appears to have co‐evolved with the subclass Theria as it is absent in Monotremes (egg‐laying mammalia, such as Ornithorhynchus anatinus = platypus), but present in live‐bearing mammals (Theriiformes, such as Phascolarctos cinereus = koala). Insertion of domain A adds 3.55 KDa to the molecular mass of Stim1, a difference that can only be detected by running optimally separating SDS–PAGE as shown after heterologous expression of both variants in STIM‐deficient HEK cells (Fig 1G). Note that insertion of domain A does not significantly alter protein levels as quantified in the right panel of Fig 1G or after quantification of fluorescently tagged proteins (see below). Protein extracts from testes show native protein species, which run at similar masses as Stim1A and as glycosylated and unglycosylated Stim1 (Fig 1H). Importantly, the signal of the molecular mass corresponding to Stim1A is absent in recombinant mice (10ACon) with fused exons 10, 11, and 12 (Yu et al, 2019), thereby disabling splicing at these sites (Fig 1H).
Figure 1. Identification of a novel STIM1 splice variant.
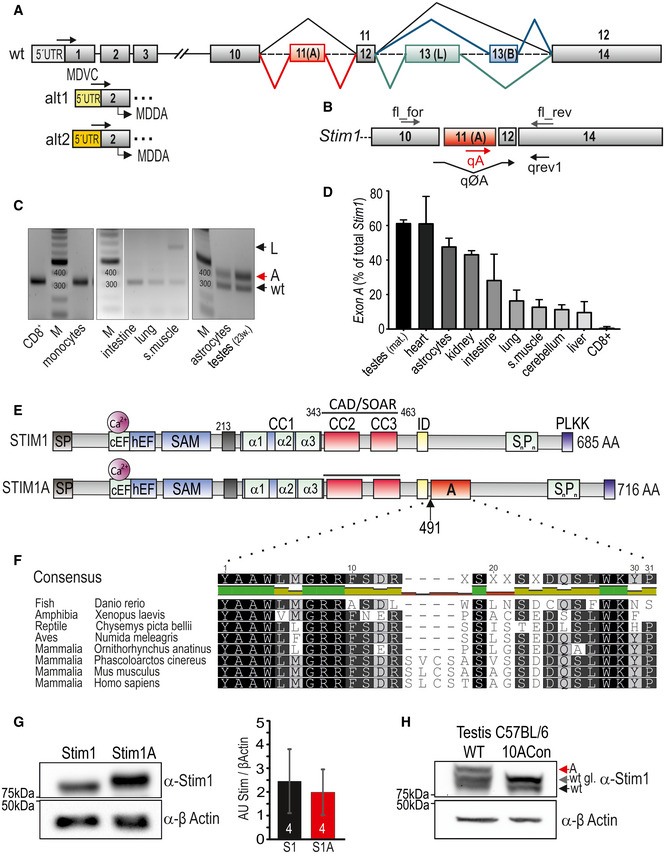
- Schematic exon structure of Stim1 gene with conventional exons depicted in gray. Highlighted in red is the alternative exon 11 (named A), and additional mutually exclusive splicing of exon 13 (named L, green) or exon 13 (named B, blue) is also shown. Alternative usage of 5’UTR regions is indicated with alt1 and alt2. Arrows indicate schematic location of primers, and sequences listed in Table EV2.
- Schematic representation of primer annealing sites on the coding mRNA.
- Analytic PCR with primers flanking the splice site using cDNA’s tissues or cells as indicated. L denotes exon L of STIM1L, and A denotes additional exon A.
- qRT–PCR of exon A‐specific reaction normalized to sum of exon‐specific and wt‐specific (total) Stim1 (± SD) in tissues using cDNA derived from 3 to 4 mice.
- Schematic protein structure of STIM1 displaying functional motives. Domain A (red) is located 8 amino acids downstream of the inhibitory domain (ID, yellow) after aa 491.
- Evolutional conservation of domain A. Identical amino acids within black boxes.
- Western blot showing Stim1 and Stim1A following heterologous expression in HEK293DKO (left); right panel: quantification from 4 transfections, mean ± SD.
- Endogenous levels of Stim1 in testes from C57BL/6 wt and splice‐deficient 10A mice, using an antibody detecting the N‐term of STIM1, detecting STIM1A (A, red arrow), STIM1, and its glycosylated (wt) and unglycosylated (wt gl.) form.
Figure EV1. Alternative splicing and PCR analysis of Stim1 and exon A.
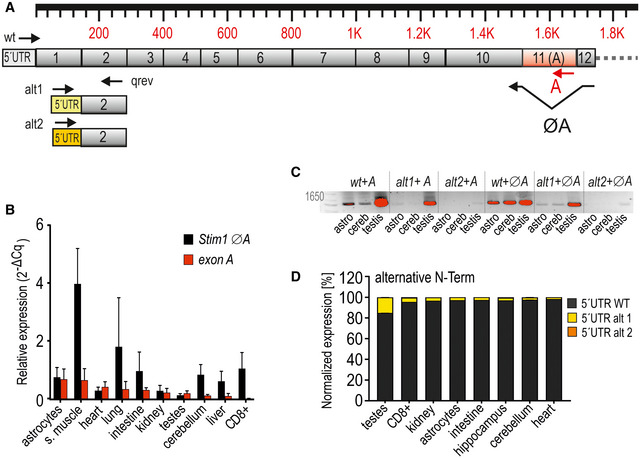
- Schematic exon structure of Stim1 with conventional exons depicted in gray. Highlighted in red is the alternative exon 11 (A). Alternative usage of 5’UTR regions is indicated with alt1 and alt2. Annealing sites of primers are depicted as arrows.
- Expression of exon A and ØA (wt) normalized to TATA box‐binding protein Tbp (mean ± SD) as determined by qRT–PCR from cDNA templates isolated from indicated tissues and cells derived from 3 to 4 mice.
- Analytic PCR with reverse primers flanking the A splice site or within exon A in combination with forward primers specific for 5´URT wt, alt1, or alt2 as also depicted in (A) using cDNA of murine astrocytes, cerebellum, and testis. Abbreviations: astro = astrocytes, cereb = cerebellum. Red indicates saturated pixels (see text).
- Quantification of alternative N‐term splicing via qRT–PCR. Expression levels were normalized to Tbp.
STIM1A is a dominant‐negative regulator of Orai‐mediated Ca2+ entry and ICRAC
To analyze the impact of the inserted residues, we expressed either Stim1 or Stim1A in murine embryonic fibroblast (MEF) cells derived from Stim1 −/− ; Stim2 −/− mice (MEF S1/S2 −/−; Oh‐hora et al, 2008) and performed Fura‐2‐based Ca2+ imaging experiments. Figure 2A shows that re‐expression of Stim1 in this background leads to recovery of the otherwise absent SOCE (see also Ramesh et al, 2021). In comparison with Stim1, Stim1A induces a reduced SOCE with a significantly decreased influx rate, peak, and plateau (Fig 2A and B). We also co‐expressed Stim1A while keeping Stim1 constant to investigate whether Stim1A is able to exert a dominant‐negative effect on SOCE with endogenous Orai channel expression. Influx rate and peak indeed were significantly reduced, while the plateau reached similar levels to single Stim1 expression (Fig 2A and B, blue traces). To investigate the phenotype in co‐overexpression with Orai1, we expressed either Stim variant together with Orai1 in HEK293 cells. Stim1A led to a general decrease in SOCE parameters (Fig 2C and D), despite similar protein levels (Fig 1G; see also Fig EV2 and below). The same outcome was observed upon co‐overexpression of human STIM1 or STIM1A in combination with either ORAI1, ORAI2, or ORAI3 (Fig EV2A–D) in HEK cells deficient of all endogenous ORAI proteins with absent SOCE; (see Alansary et al, 2020). The presence of STIM1A thus reduced SOCE independent of the ORAI homolog, indicating a conserved and not homolog‐specific mechanism responsible for the reduced activity. To facilitate comparison, we quantified the resulting % change of the SOCE parameters and found that Δ peak and Δ plateau were reduced to the same relative extent (Fig EV2D). Using a third expression system, this time calibrating the Fura‐2 signal to yield absolute values of [Ca2+]i, we expressed mCherry‐tagged human constructs (see Materials and Methods) in SH‐SY5Y cells lacking endogenous STIM1 (Ramesh et al, 2021) and confirmed reduced Ca2+ influx despite equal protein expression (Fig EV2E and F). We also checked whether the presence or absence of endogenous STIM2 altered the STIM1A phenotype by conducting Ca2+ imaging experiments in SH‐SY5Y cells either devoid of both endogenous STIM1 and STIM2 with experiments in cells only devoid of endogenous STIM1 (Ramesh et al, 2021). Figure EV3, EV4, EV5A–C demonstrates that STIM1A reduces SOCE parameters to a similar degree in the presence or absence of STIM2. Using BiFC, we did not detect any difference in the ability of STIM1A versus STIM1 to interact either with STIM1 or with STIM2 (Fig EV3D).
Figure 2. Stim1A reduces SOCE.
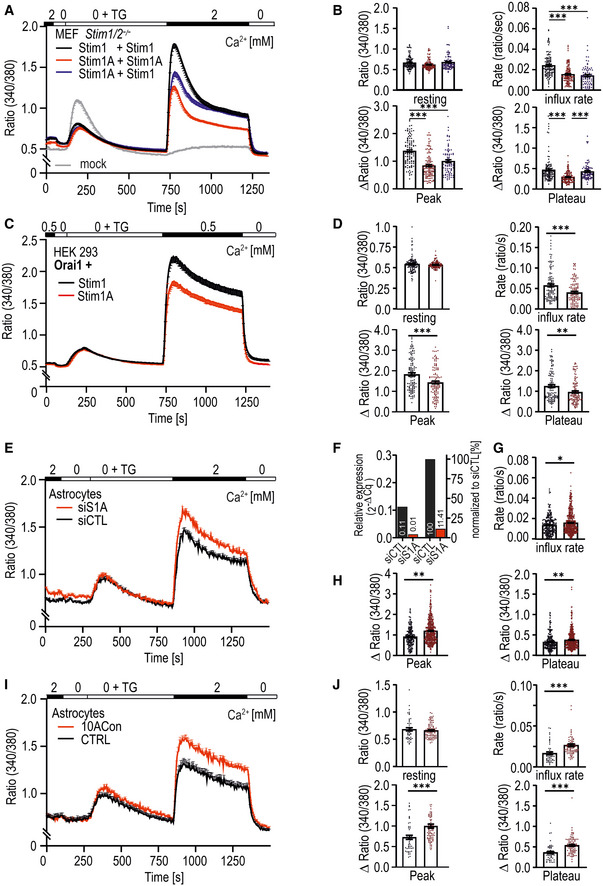
-
ATraces showing average changes (mean ± s.e.m.) in intracellular Ca2+ (Fura‐2 ratio) over time in response to perfusion of different [Ca2+]o as indicated in the upper bar in MEF Stim1/Stim2−/− cells transfected with Stim1 (black trace, n = 111), Stim1A (red trace, n = 116), and the combination of Stim1 with Stim1A (blue trace, n = 86) or with vector only (gray trace, n = 79).
-
BQuantification of changes in ratio of resting, influx rate (Δratio/time), Δpeak, and Δplateau measured in A. ***P < 0.001, Kruskal–Wallis ANOVA.
-
CTraces showing average changes (mean ± s.e.m.) in intracellular Ca2+ (Fura‐2 ratio) in HEK293 cells co‐transfected with Orai1 and either Stim1‐ (black trace, n = 119) or Stim1A‐IRES‐mCherry (red trace, n = 118).
-
DQuantification of changes in ratio of resting, influx rate (Δratio/time), Δpeak, and Δplateau measured in C. **P < 0.01 and ***P < 0.001, Mann–Whitney test.
-
ETraces showing average changes (mean ± s.e.m.) in intracellular Ca2+ (Fura‐2 ratio) in primary astrocytes after transfection with non‐silencing (siCTL, black trace, n = 216) or exon A‐specific siRNA (siS1A, red trace, n = 263).
-
FEfficiency of siRNA as determined by qRT–PCR.
-
G, HQuantification of parameters shown in (E). *P < 0.05 and **P < 0.01, Mann–Whitney test.
-
ITraces showing average changes (mean ± s.e.m.) in intracellular Ca2+ (Fura‐2 ratio) in primary astrocytes from C57BL/6 wt (CTRL, black trace, n = 49) and splice‐deficient C57BL/6 10ACon mice (10ACon, red trace, n = 82).
-
JQuantification of parameters determined in [I]. ***P < 0.001, Mann–Whitney test.
Data information: Data (except F) were obtained from three biological replicates (e.g., transfections) each with three measured dishes (technical replicates) with multiple cells each (yielding a total number n) and are shown as mean ± s.e.m.
Figure EV2. STIM1A reduces SOCE independent of the ORAI homolog.
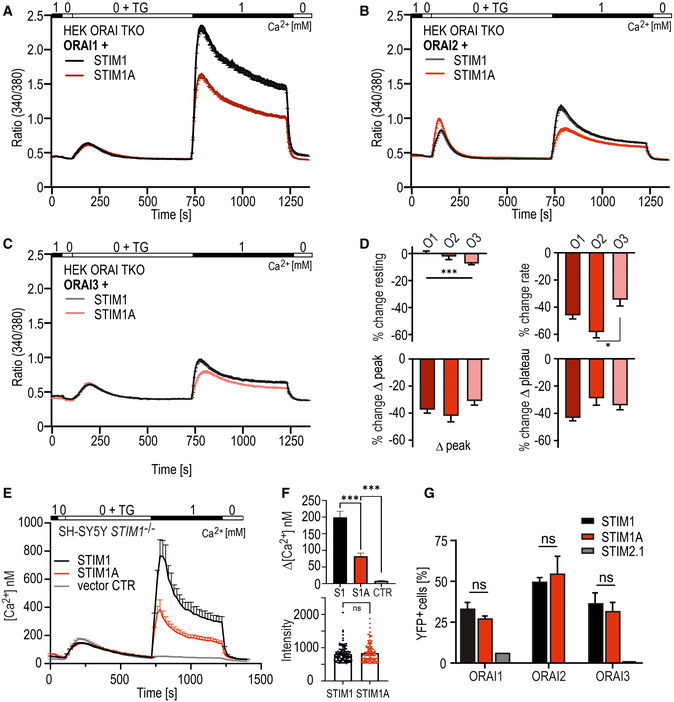
-
A–CTraces showing average changes (mean ± s.e.m.) in intracellular Ca2+ (Fura‐2 ratio) in HEK293 triple ORAI‐deficient cells (HEK ORAI TKO) co‐transfected with either STIM1‐mCherry (black traces, n = 60–98) or Stim1A‐IRES‐mCherry (red traces, n = 51–78), and ORAI1 IRES‐GFP (A) co‐transfected with ORAI2 IRES‐GFP (B) or ORAI3 IRES‐GFP (C).
-
DRelative change compared with the average of each respective control. Statistical analysis: Kruskal–Wallis ANOVA with Dunn's multiple comparisons test with significance indicated (*P < 0.05 and ***P < 0.001, error bars indicate s.e.m., data were obtained from three technical replicates each derived from three biological replicates with multiple cells, total n indicated above).
-
ECalibrated traces showing mean [Ca2+]i (+s.e.m.) after expression of mCherry‐tagged STIM1 (black trace, n = 68), STIM1A (red trace, n = 54) or vector control (gray trace, n = 47 cells) in SH‐SY5Y STIM1−/− cells. Solution exchanges similar as indicated in C, and data were obtained from three technical replicates each (with multiple cells) derived from three biological replicates.
-
FAnalysis of [Ca2+]i plateau (upper panel) and mCherry expression levels (lower panel) (mean ± s.e.m.). ***P < 0.001, Kruskal–Wallis ANOVA with Dunn’s multiple comparisons test (for replicates, see under C).
-
GBimolecular fluorescence complementation (BiFC) assay: Analysis for interaction via flow cytometry. HEKS1−/−/S2−/− were transfected with STIM1‐EYFPN (black), STIM1A‐EYFPN (red), or STIM2.1‐EYFPN (gray) or with ORAI1‐EYFPC, ORAI2‐EYFPC or ORAI3‐EYFPC and screened for YFP+ cells via FACS, and results (mean ± SD) were obtained from 2–3 transfections (1 transfection for STIM2.1) each with 10,000 sorted cells. n.s.: non‐significant, 2‐way ANOVA.
Figure EV3. STIM2 has no impact on STIM1A‐mediated SOCE.
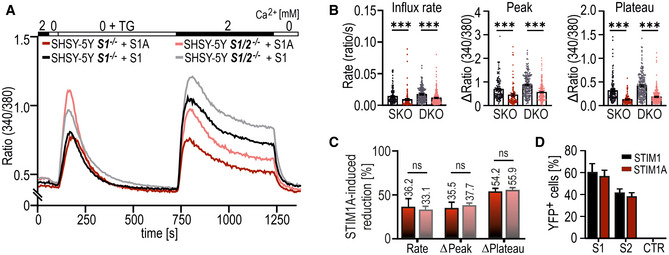
- Traces showing mean changes in Fura‐2 ratio in SH‐SY5Y STIM1−/− or in SH‐SY5Y STIM1−/−;STIM2−/− cells transfected with either STIM1‐mCherry (black traces, n = 136 and gray traces, n = 187) or STIM1A‐IRES‐mCherry (red traces, n = 107 and light red traces, n = 217).
- Quantification of changes in ratio of resting, influx rate (Δratio/time), Δpeak, and Δplateau measured in A. Bars underlying scatter plots depict mean ± s.e.m., ***P < 0.001, Kruskal–Wallis ANOVA with Dunn's multiple comparisons test, and data were obtained from three technical replicates each derived from three biological replicates.
- STIM1A induced reduction in SOCE parameter (B) after expression in single KO (dark red) or in double KO (dark red) in %. ns: not significant, Kruskal–Wallis ANOVA with Dunn's multiple comparisons test.
- Percentage of YFP+ cells from BiFC assay after transfection of HEKS1−/−;S2−/−cells with STIM1(A)‐YFPC with STIM1‐YFPN, STIM2‐YFPN, or negative control. Results (mean ± SD) were obtained from 2‐3 transfections each with 10,000 sorted cells.
Figure EV4. Site‐directed mutagenesis of exon A.
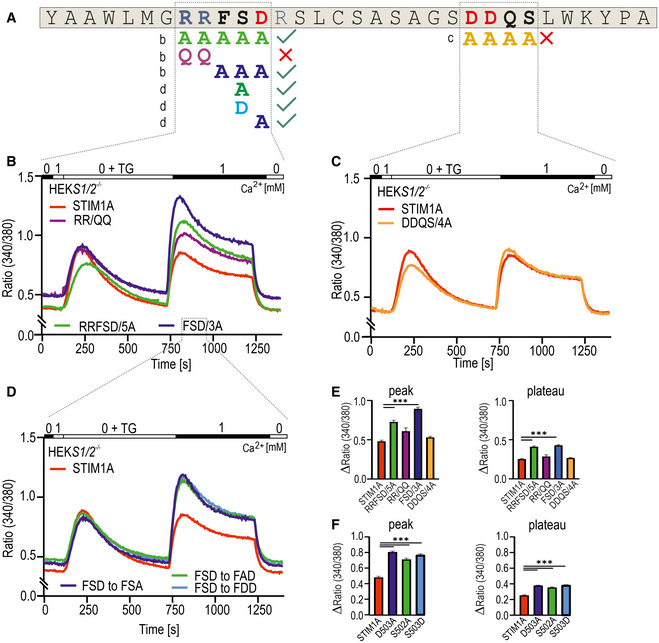
-
AAmino acids of exon A. Positive charged amino acids are marked blue, and negative charged ones are shown in red. Mutated amino acids depicted below are marked in the color code used for B–F.
-
B–DTraces showing average changes (mean ± s.e.m.) in intracellular Ca2+ (Fura‐2 ratio) over time in response to perfusion of different [Ca2+]o as indicated in the upper bar with mutant constructs as indicated expressed in HEKS1−/−;S2−/− cells.
-
EQuantification of changes in ratio measured in B and C.
-
FQuantification of changes in ratio measured in D.
Data information: All constructs were directly tagged with mCherry. ***P < 0.001, Kruskal–Wallis ANOVA with Dunn's multiple comparisons test. Data were obtained from three technical replicates (each with multiple cells) from three biological replicates (129 < total n < 708) and are shown as mean ± s.e.m.
Figure EV5. Ca2+ dependence of FCDI, expression of PDE and ADCY genes, receptor‐mediated Ca2+ entry, and NFAT translocation.
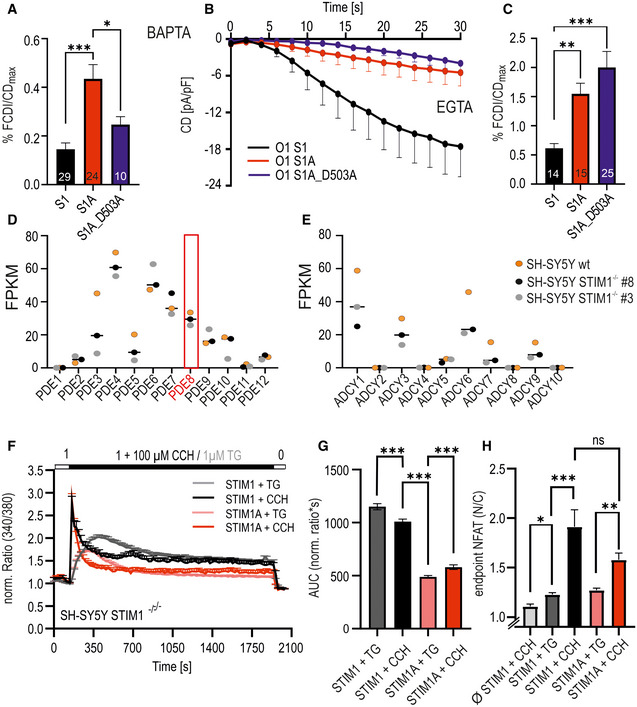
-
ANormalized FCDI of STIM1, STIMA, and STIM1A_D503A with intracellular BAPTA buffering. Number of patch‐clamped cells from > 3 transfections each indicated in bars with mean + s.e.m. *P < 0.05 and ***P < 0.001, ordinary one‐way ANOVA.
-
BCurrent densities over time (data points: mean ± s.e.m.) measured with intracellular EGTA buffering after expression of indicated constructs in HEKS1−/−;S2−/− cells. Number of patch‐clamped cells from > 3 transfections (biological replicates).
-
CNormalized FCDI of STIM1, STIMA, and STIM1A_D503A with intracellular EGTA buffering. Number of patch‐clamped cells from > 3 transfections indicated in bars with mean + s.e.m. **P < 0.01 and ***P < 0.001, Kruskal–Wallis ANOVA.
-
D, EFPKM counts of PDE genes (E) and ADCY genes (F) from RNA‐seq analysis of mock‐transfected SH‐SY5Y cell line and two independent CRISPR/Cas9‐generated STIM1‐deficient cell lines, horizontal lines indicate median, and red box highlights expression of PDE8. Variants of one gene are summed.
-
FTraces showing average changes (mean + s.e.m.) of normalized ratio over time in response to perfusion of different stimuli as indicated in the upper bar with mCherry‐tagged constructs expressed in SH‐SY5Y STIM1−/− cells (clone#8). Darker colors indicate responses to carbachol (CCH) (26 < total n < 105 from 3 transfections with technical replicates).
-
GQuantification of the area under the curve (AUC) of ratios (mean ± s.e.m.) shown in (F). ***P < 0.001, ordinary one‐way ANOVA indicated only for selections (26 < total n < 105 from 3 transfections with technical replicates).
-
HNormalized NFAT ratios (nuclear/cytoplasm), mean + s.e.m, after 30 min with stimulation as shown in (F). *P < 0.05, **P < 0.01, and ***P < 0.001, Kruskal–Wallis ANOVA (13 < total n < 103, 3 transfections).
To confirm the dominant‐negative nature of Stim1A, we performed siRNA experiments targeting only exon A in cultured murine hippocampal astrocytes (Fig 2E–H; see Fig 2F for knockdown efficiency). siRNA‐mediated downregulation (Fig 2G and H) led to increased rate, peak, and plateau levels of SOCE and verified the significance of Stim1A in primary cells. In addition, we also measured SOCE in astrocytes isolated from the 10ACon splice‐deficient mouse model (Yu et al, 2019). Confirming our siRNA results, astrocytes derived from these mice showed significantly increased SOCE compared with their respective WT controls (Fig 2I and J).
STIM1A co‐localizes with STIM1 in HEK cells and interacts with ORAI1
To investigate whether insertion of domain A leads to altered localization of STIM1, we transfected both variants containing mCherry into HEK cells lacking endogenous STIM proteins (HEK S1/S2 −/− cells; Zhou et al, 2018) and investigated STIM1‐STIM1 versus STIM1‐STIM1A co‐localization before and after store depletion. As depicted in Fig 3A, neither overt pre‐clustering nor differential localization of STIM1A when compared to STIM1 was observed before store depletion. After stores were depleted, STIM1 or STIM1A clustered and co‐localized to the same ER‐PM junctions in HEK cells (Fig 3B). Co‐localization coefficient analyses of 9 cells using Mander’s overlap coefficients (M1 and M2) were as follows in the absence of thapsigargin (‐TG/TG) (as in Fig 3A): S1+S1 ± SD: M1 = 0.83 ± 0.09, M2 = 0.87 ± 0.07; S1+S1A ± SD: M1 = 0.79 ± 0.07, M2 = 0.89 ± 0.06; and after addition of TG: S1+S1 ± SD: M1 = 0.84 ± 0.05, M2 = 0.93 ± 0.05; S1+S1A ± SD: M1 = 0.81 ± 0.03, M2 = 0.83 ± 0.08, revealing no differences either (Fig 3B). To investigate whether the reduced SOCE/ICRAC is due to defective interaction or clustering with ORAI1, we analyzed co‐localization and interaction by total internal reflection fluorescence microscopy (TIRFM). Both STIM1 and STIM1A co‐localized with ORAI1 after store depletion at ER/PM junctions with no apparent difference in the Mander’s coefficients of co‐localization (Fig 3C and D). Quantification of FRET efficiencies (E‐FRET) also indicated that interaction between the respective CAD (SOAR) domains of STIM1A and the C‐terminal domain of ORAI1 was unaltered when compared to STIM1 (Fig 3C and E). Comparison of the cluster sizes of STIM1 and STIM1A revealed no significant differences (Fig 3F and G). As a further confirmation for the normal interaction seen with FRET, we checked the interaction between STIM1 and STIM1A and ORAI homologs using bimolecular fluorescence complementation (BiFC) assays and also did not detect a difference between both STIM variants. Using BiFC, both STIM1 and STIM1A showed an increased interaction with ORAI2 (Fig EV2G). As a negative control, we tested the interaction of STIM2.1, which we have previously demonstrated being unable to activate and interact with ORAI channels (Miederer et al, 2015) and also showed absent interaction using BiFC (Fig EV2G).
Figure 3. Co‐localization and interaction analysis.
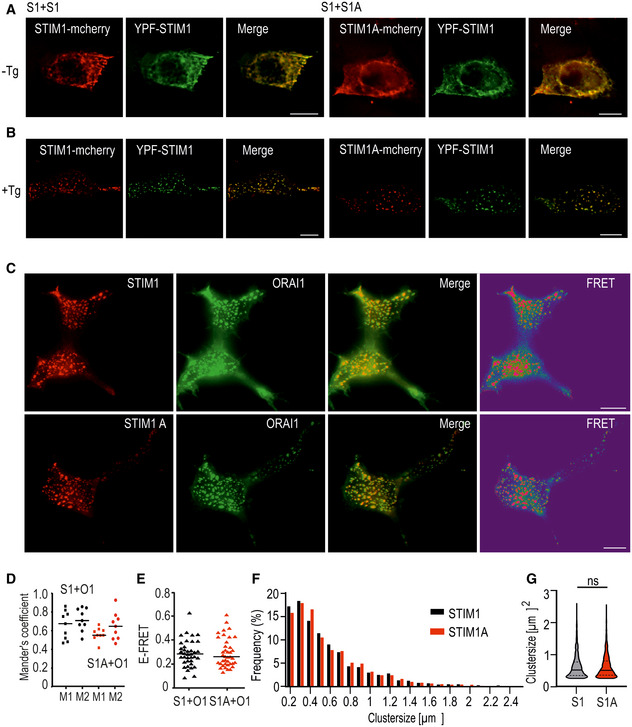
-
A, BImages showing representative HEK S1/S2 −/− cells co‐expressing YFP‐STIM1 (green) or either STIM1‐mCherry or STIM1A‐mCherry (red) and merged images before (A) and after (B) stimulation with TG.
-
CImages showing representative HEK293 cells expressing Orai1‐GFP (green = donor molecule) or either STIM1‐mCherry (upper panel = acceptor molecule) or STIM1A‐mCherry (lower panel = acceptor molecule) after stimulation with TG. Also shown is the overlay of the two channels, as well as the FRET signal.
-
DCo‐localization analysis of cells from (C). For each condition, 9 cells were analyzed using Mander’s overlap coefficients (M1 and M2), S1 M1 = 0.68, M2 = 0.71; S1A M1 = 0.55, M2 = 0.65.
-
EQuantification of FRET signal measured in C (STIM1: 0.28, n = 37; STIM1A: 0.26, n = 39).
-
FDistribution of cluster sizes in HEK293 cells transfected with STIM1‐mCherry (black, n = 2644) or STIM1A‐mCherry (red, n = 4754) at 0.2‐µm2 intervals in %.
-
GViolin blot of the cluster shown in (F), shown as median, and first and third quartile. ns: not significant with the Mann–Whitney test.
Data information: Data were obtained from three independent transfections and, in D and E, are shown as single values with corresponding median. Scale bars, 10 µm.
Specific residues in domain A mediate the reduced function
To characterize the molecular mechanism of reduced SOCE and ICRAC in more detail, we next asked whether the mere insertion of residues shortly downstream of the inhibitory domain (ID) is responsible for the phenotype or whether specific and conserved residues of domain A are required. As shown in Fig 1F, domain A contains several highly conserved and also charged residues. The initial strategy included mutagenesis of the charged RRFSD stretch to alanine residues (AAAAA) or, alternatively, the downstream DDQS motif to alanine residues (AAAA) within the mCherry‐tagged construct utilized for the aforementioned FRET experiments (Figs 4A and EV4A). The RRFSD/AAAAA mutant recovered the STIM1 wt phenotype, whereas mutation of DDQS/AAAA did not rescue the reduced SOCE (Fig EV4B,C,E). These results indicate that, indeed, specific residues and not a sole extension of the C‐terminal domain encode the domain A phenotype. Further mutational analyses show that the charged di‐arginine motif (RR, with mutant QQFSD) is not responsible for reduced SOCE but rather that the FSD motif is a critical determinant of the STIM1A phenotype, as the FSD/AAA within STIM1A is sufficient to restore the STIM1 wt phenotype (Fig EV4B and E). Single‐point mutations of residues S502 and/or D503 both lead to increased SOCE (Fig EV4D and F). As S502 can be potentially modified by phosphorylation, we also created a phospho‐mimetic S502D mutant. As this displayed a similar rescue as the S502A mutation, phosphorylation of S502 is likely not the cause for the reduced function (Fig EV4D and F). The described functional screening was performed in HEKS1/S2 −/− cells with only endogenous ORAI channels mediating Ca2+ entry, thus leading to very small SOCE. We therefore confirmed the effects of STIM1A_D503A in HEK cells stably overexpressing ORAI1 (HEKO1; Kilch et al, 2013). Here, we found a more pronounced difference between SOCE parameters of STIM1 and STIM1A, which were fully reversed in D503A mutant (Fig 4B). Concomitant measurement of mCherry fluorescence of the tagged constructs enabled control for equal protein expression (Fig 4C), with quantitative analysis of all parameters confirming the reversion of the STIM1A phenotype (Fig 4D), proving that the insertion of 31 additional residues does not per se alter initiation or maintenance of SOCE.
Figure 4. Mutation of D503 rescues phenotype of domain A.
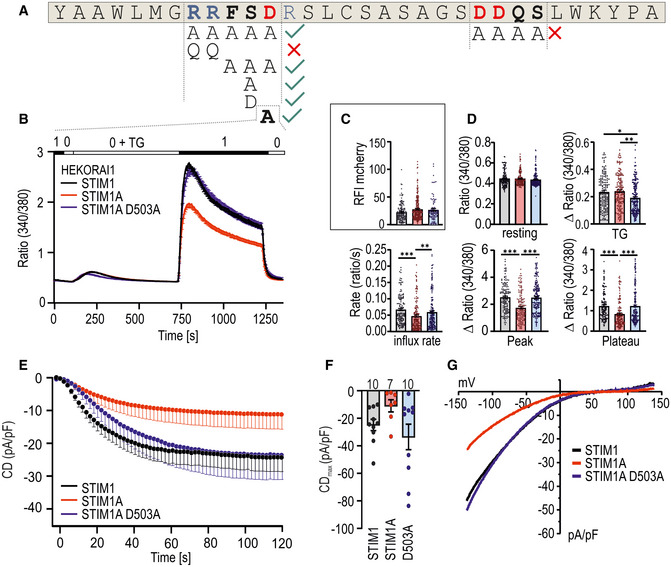
- Amino acids of exon A. Positive charged amino acids are marked blue, and negative charged ones are shown in red. Mutated regions are in bold, and different mutant combinations investigated in Fig EV5 are depicted below.
- Traces showing average changes (mean ± s.e.m.) in intracellular Ca2+ (Fura‐2 ratio) over time in response to perfusion of different [Ca2+]o as indicated in the upper bar with constructs as indicated expressed in HEKO1. STIM1 (black, n = 145), STIM1A (red, n = 157), and STIM1A_D503A (n = 185).
- Relative fluorescence intensities of mCherry‐tagged constructs measured in (B).
- Quantification of changes in resting ratio, TG peak (∆ ratio), and SOCE parameters measured in B.
- Average traces showing whole‐cell current density (CD) over time extracted at −80 mV in HEKO1 cells transfected with STIM1 (black), STIM1A (red), or STIM1A_D503A (blue).
- Average maximum CDs recorded from cells measured in E (n within bars).
- Average current–voltage (I–V) relationship of all cells recorded in E.
Data information: *P < 0.05, **P < 0.01, and ***P < 0.001, Kruskal–Wallis ANOVA with Dunn's multiple comparisons test. Data points (total n) were obtained from three biological replicates each with three technical replicates (each with multiple cells) and are shown as mean ± s.e.m for traces and as scatter plots with the underlying boxes showing the means for individual parameters.
In line with the Ca2+ imaging results, whole‐cell patch‐clamp analysis confirmed that STIM1A reduced ICRAC relative to STIM1 and that STIM1A_D503A fully rescued the reduced current densities seen with STIM1A (Fig 4E–G) in conditions of strong internal buffering. The resulting IV relationships seen upon co‐overexpression with Orai1 display the typical inward rectification (Fig 4G), suggesting no overt alteration of selectivity.
Domain A interferes with stabilized gating of ORAI1
Gating of ORAI1 by STIM1 is initiated by a primary hydrophobic interaction of the STIM1 CAD/SOAR coiled‐coil domain with the C‐terminus of ORAI1 and potentially directly with the “nexus” site (amino acids 261–265) of ORAI1 (Zhou et al, 2016). Alternation of the nexus site elicits a conformational transduction pathway via transmembrane domain TM4 over TM3 to TM1 to open the pore (reviewed recently in Yeung, Yamashita et al, 2020). In the past, several groups have suggested that the CAD domain also interacts with the Extended Transmembrane Orai1 N‐terminal (ETON) region (aa 73–90) (Lis et al, 2010; Mcnally et al, 2013; Derler et al, 2018), which was also recently confirmed biochemically and functionally (Niu et al, 2020). Sequential activation involving interaction with the ETON region is critical for full gating and potentially for fast Ca2+‐dependent inactivation (FCDI) (Mullins & Lewis, 2016). ORAI1 gating may thus be stabilized by a STIM1‐mediated bridging between the cytosolic TM1 and TM4‐extended helices of ORAI1, thereby applying a force to the helical TM1 extension to stabilize the open pore state (Derler et al, 2013, 2018), or alternatively between TM1 and the TM2‐TM3 linker (see discussion). Indeed, when we checked FCDI upon expression of STIM1A, we found a significant increase in fast inactivation (Fig 5A and B), which could explain the reduced currents/SOCE (Figs 2 and 4). As STIM1A_D503A rescued SOCE (Fig 4), we tested whether STIM1A_D503A also rescued FCDI in conditions of strong buffering, which indeed was the case (Fig EV5A). As FCDI is dependent on the capacity and speed of intracellular buffering, we also recorded with EGTA instead of BAPTA in the patch pipette. Strikingly, now, neither the reduced current densities of STIM1A nor the increased FCDI was rescued with STIM1A_D503A (Fig EV5B and C), uncovering a strong and highly localized Ca2+ dependency of STIM1A_D503A. To estimate spatial interactions of STIM1 with the pore residues of ORAI1, we performed two independent approaches of in silico molecular docking of the STIM1 CAD domain with both the closed and open structures of ORAI1. Herein, the first docking study (Fig 5C and D) had also allowed us previously to perform MD simulations to open ORAI1 and to correctly estimate residues in vicinity of ORAI1 C195 (Alansary et al, 2016). The interacting residues leading to channel opening in those MD simulations (Fig 5D) include within ORAI1: R77‐K78, R170, K265, D284 and D287 and with E173 and H264 making contact after 50‐ns simulation and H264 and K265 being most important and in good agreement with experimental data concerning gating (Zhou et al, 2016; Yeung et al, 2020; Tiffner et al, 2021). Interacting gating residues in STIM1 included K386, R387, N388, T393, F394, H395, and H398 with F391 in addition within 50‐ns simulation. We also conducted an independent docking approach with the constraint that the CAD/SOAR domain should interact with the “nexus” region of ORAI1 (261LVSHK265) (Zhou et al, 2016; Fig 5E). Both approaches demonstrated tight spatial restrictions of the interacting surfaces between ORAI1 and the CAD domain. In both models, we observed a close proximity of the pore‐outward facing residue ORAI1R77 with CAD residues (i.e., T393) and with the apex of the STIM1 CAD domain, with any downstream domain clearly not being able to directly interact with residues close to the ORAI1 pore region (Fig 5C and E). However, the model (Fig 5C) also shows that the second helix within the STIM1 CAD domain moves toward R77/K78 in the open state (green). Because STIM1A showed normal FRET with ORAI1, we hypothesized that domain A might interfere with the secondary stabilizing interaction between STIM1 CAD residues and the ORAI1 ETON region by slightly altering the conformation of the CAD apex. Therefore, an N‐terminal gate‐modified ORAI1 such as ORAI1_R77E (Lis et al, 2010; Niu et al, 2020) might mask the STIM1A phenotype. Using Ca2+ imaging, we observed a 35‐40% reduced STIM1‐mediated SOCE with ORAI1_R77E (Fig 5F and G). This difference indeed was absent when STIM1A was co‐expressed with ORAI1_R77E, but could be restored with the single D503A mutation within domain A (Fig 5F and G). Patch‐clamp analysis in HEKS1/S2 −/− cells confirmed the reduced currents with STIM1A (Fig 5H and J) similar to those observed with Stim1A (murine) in HEKwt cells (Fig 4E). Co‐expression with ORAI1_R77E leads to a strong current reduction compared with ORAI1 (Fig 5I and J; see Lis et al, 2010). However, STIM1A did not cause further reduction in the background of R77E (Fig 5H–J). These results suggest that insertion of domain A could affect the interaction between STIM1 CAD residues and R77 of ORAI1, thus either reducing or destabilizing full gating of ORAI1. The initial hydrophobic coiled‐coil interaction between STIM1 CAD and the C‐terminus of ORAI1 likely guides the CAD apex toward the nexus region and underlies the strong FRET signal (Frischauf et al, 2009). Interestingly, the single‐point mutation D503A within domain A is able to revert the reduced gating phenotype and FCDI, but only in a highly buffer‐dependent manner as further discussed below.
Figure 5. Mutation within the ORAI1 ETON region masks the STIM1A phenotype.
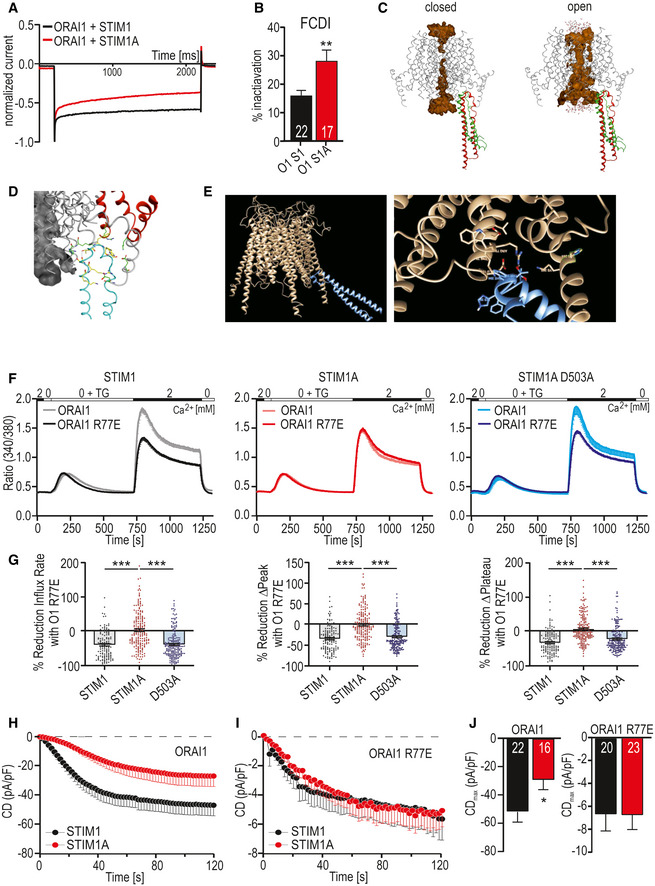
-
AExemplary normalized current responses at −120 mV pulse showing fast Ca2+‐dependent inactivation (FCDI).
-
BPercentage of FCDI (plateau/peak, mean ± s.e.m.), for HEKS1/S2−/− cells transfected with STIM1+ORAI1 (O1 S1), 22 patch‐clamped cells or transfected with STIM1A and ORAI1 (O1 S1A, 17 cells) from three independent transfections.
-
CDocking model of the STIM1 CAD domain to ORAI at the beginning (red) and at the end (green) of a 50‐ns MD simulation.
-
DClose‐up of interacting residues.
-
EAlternate docking approach.
-
FTraces showing average changes (mean ± s.e.m.) in intracellular Ca2+ (Fura‐2 ratio) over time in response to perfusion of different [Ca2+]o as indicated in the upper bar with constructs as indicated expressed in HEKS1/S2−/−.
-
GQuantification of the relative reduction of ratio/time or ratio for cells (135 < n < 202) measured in (F).
-
H, IAverage traces (mean ± s.e.m.) showing whole‐cell current density (CD) over time extracted at −80 mV in HEKS1/S2−/− cells co‐transfected either with STIM1 (black) or STIM1A (red) and ORAI1 (H) or with ORAI1 R77E (I) and recorded using extracellular solution containing 2 mM Ca2+.
-
JAverage maximum CDs recorded from cells measured in [H,I] (n within bars).
Data information: In G, ***P < 0.001, Kruskal–Wallis ANOVA with Dunn’s multiple comparisons test; in B, J, *P < 0.05 and **P < 0.01, unpaired t‐test with Welch’s correction. Data points (total n) were obtained from three biological replicates each with three technical replicates (with multiple cells) and are shown as mean ± s.e.m for traces (F) and as scatter plots with the underlying boxes showing the means for individual parameters (G). For patch‐clamp experiments, numbers in bars (B, J) indicate measured cells, which were from three independent transfections.
Stim1A alters SOCE‐cAMP crosstalk and affects NFAT translocation
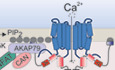
In addition to its potential effects on channel gating, insertion of 31 additional residues could either code for an additional protein binding interface or interfere with known or unknown interaction partners of STIM1. We screened for differentially interacting proteins using pull‐down of HA‐tagged variants of STIM1‐mCherry, STIM1A‐mCherry, and STIM1A_D503A‐mCherry in HEK STIM1/2 −/− cells with subsequent analysis by mass spectrometry and identified several new potential interaction partners. Selected hits showing significant differential label‐free quantification (LFQ) intensities (Fig 6A) between STIM1 and STIM1A were tested for interaction using bimolecular fluorescence complementation (BiFC, Fig 6B) and plotted against mass spectrometry scores to identify interaction partners with a significant discrepancy between STIM1 and STIM1A (Fig 6C). For all hits shown in Fig 6A, we found that STIM1A_D503A yielded average LFQ intensities that were not different to STIM1A; thus these data are not included in the table (Fig 6A). One of the strongest differential candidates was phosphodiesterase 8B (PDE8B), which showed a significant difference in mean LFQ intensities between STIM1 and STIM1A or between STIM1 and STIM1A_D503A pull‐down (STIM1: 37627, STIM1A: 7064 and STIM1A_D503A: 64118; see Data availability); however, only a moderate difference was found in the BiFC experiments, potentially due to the C‐terminal tags required for BiFC or due to an indirect interaction requiring a mediator protein (see Discussion). Because a signaling complex between STIM, ORAI1, and A‐kinase anchoring protein (AKAP79) is critical and essential for efficient activation of the transcription factor NFAT (Kar et al, 2014, 2021a, 2021b; Samanta et al, 2015) and as Son et al (2020) showed that the polybasic domains of STIM proteins are necessary for the assembly of this signaling complex, we asked whether PDE8B might also be involved in NFAT translocation by altering cAMP levels. Indeed, forskolin, an activator of adenylate cyclase, has been shown to increase NFAT translocation in osteoblasts (Huang et al, 2010) and to increase bisperoxovanadium phosphotyrosyl‐phosphatase inhibitor‐mediated NFAT translocation in Jurkat T cells (Barat & Tremblay, 2003). We expressed STIM1 and STIM1A in STIM1‐deficient SH‐SY5Y cells (STIM2 present; Ramesh et al, 2021) together with NFATc1‐GFP and quantified translocation. We observed little TG‐induced translocation in the absence of STIM1 re‐expression and relatively inefficient NFATc1 translocation upon expression of STIM1 (Fig 6D and E); however, despite reduced SOCE, increased STIM1A‐mediated NFAT translocation was observed. Stimulation with 100 µM carbachol leads to more efficient Ca2+ release from internal stores and resulted in an increased NFAT translocation despite a slight reduction in total Ca2+ entry compared with TG for STIM1 (Fig EV5F–H). STIM1A‐mediated Ca2+ entry stimulated by carbachol was reduced but did not result in reduced NFAT translocation (Fig EV5F and G). STIM1A_D503A, showing similar Ca2+ entry compared with STIM1wt (Fig 4), also slightly increased TG‐induced NFAT translocation (Fig 6D and E). To test a potential role for differential effects of the variants on PDE8B activity, we applied the PDE8‐specific inhibitor PF‐04957325 (Vang et al, 2016), after confirming that PDE8 is expressed in these cell lines (Fig EV5D), with expression levels of adenylate cyclases shown in Fig EV5E. In the absence of STIM1 expression, PF‐04957325 did not alter NFAT translocation (Fig 7A and C). Strikingly, inhibition of PDE8B in the presence of STIM1 led to a significant increase in NFAT translocation (Fig 7A and C). Expression of STIM1A showed a differential effect upon PF‐04957325 treatment: PF‐04957325 had a significantly stronger impact on STIM1 compared with STIM1A‐mediated NFAT translocation (Fig 7B), suggesting that PDE8 is more active in the presence of STIM1 compared with STIM1A. In parallel to NFAT translocation, we measured SOCE over the entire time of the NFAT translocation assay with lower sampling rates and found that PF‐04957325 did not alter the amount of Ca2+ influx and also confirmed a slightly lower amount of total Ca2+ entry upon expression of STIM1A with rescue of STIM1A_D503A (Fig 7D and E). To exclude potential unspecific effects of PF‐04957325 and to address whether increased cAMP as expected by inhibition of PDE8 triggers altered NFAT translocation via activation of PKA or via alternate activation of the Exchange protein activated by cAMP (Epac), we applied membrane‐permeant analogs of cAMP that either activate PKA specifically (6‐Bnz‐cAMP) or signal via activation of Epac (8‐pCPT‐2’‐O‐methyl‐cAMP) in the presence or absence of STIM1. Quantifying NFAT translocation, we observed a similar increase in TG‐triggered NFAT translocation as with the PDE8B inhibitor but only in the presence of a STIM1 isoform (Fig 7C); however, we could not ascribe this effect to either the PKA or the Epac pathway. Flooding the cells with cAMP also obliterated differences seen between STIM1 and STIM1A. Related to the reported Orai‐independent effects of STIM1 on the activity of ADCY6 resulting in altered melanogenesis (Motiani et al, 2018), we find that inhibition of PDE8B leads to a strong and differential STIM1 isoform‐dependent effect on NFAT translocation, which did not scale with the total amount of global change in Ca2+ levels. A reduced activation of PDE8B by STIM1A likely explains the reduced effect of the inhibitor PF‐04957325; thus, we expect expression of STIM1A to cause less degradation of cAMP when compared to STIM1. Using the novel and sensitive (K d = 0.3 μM) genetically encoded cAMP sensor RflincA (Ohta et al, 2018; Fig 7F), we indeed find a slowed degradation of cAMP level 5 min after addition of TG in STIM1A‐expressing cells when compared to STIM1, with the cAMP‐insensitive mutant of RflincA as control for effects of bleaching (Fig 7F). However, after 30 min endogenous cAMP levels drop to a similar endpoint value, suggesting that ambient local cAMP may facilitate early activation of NFAT translocation. Because STIM‐regulated NFAT translocation requires the presence of the C‐terminal polybasic domain (PBD; Son et al, 2020), we also asked whether deletion of the PBD from both STIM1 and STIM1A could obliterate the effect of PF‐04957325. Figure 8A and B shows that deletion of the PBD decreased total Ca2+ entry with STIM1 by about 30%, but did not reduce Ca2+ entry of STIM1A. NFAT translocation, however, was obliterated with deletion of the PBD for both STIM1 and STIM1A, despite the presence of endogenous STIM2, and, in contrast to full‐length proteins, could not be increased with incubation with PF‐04957325 (Fig 8C). These data prompted us to speculate whether the effects of increasing cAMP (either by PF‐04957325 or by applying membrane‐permeant cAMP analogs) were due to a cAMP‐mediated increase in plasma membrane PIP2 levels, facilitating the assembly of the STIM/ORAI/AKAP signalosome (see Discussion). Indeed, in S. cerevisiae, the phosphatidylinositol‐4‐phosphate 5‐kinase (PIP5K)‐mediated PIP2 production is regulated by cAMP (Kato et al, 1989). We therefore tested whether the PIP5K inhibitor ISA‐2011B, which also impaired CD28‐dependent costimulatory effects in T cells (Kunkl et al, 2017), had an effect on cAMP‐facilitated NFAT translocation. Indeed, inhibition of PIP5K abolished the cAMP‐dependent enhancement of NFAT translocation (Fig 8D) and uncovered a hitherto unknown target (PIP5K) of Ca2+‐triggered and cAMP‐dependent regulation of NFAT translocation, which is independent of PKA activity.
Figure 6. Domain A interferes with an interaction of STIM1 with PDE8B and increases NFAT translocation.
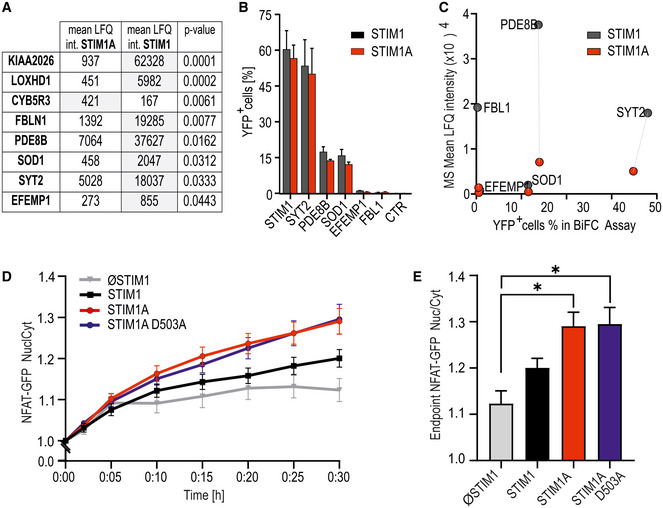
- Mean LFQ intensities of interaction candidates of three replicates each with pull‐down experiments of HA‐tagged STIM1A‐mCherry and STIM1‐mCherry after expression and TG activation in HEKS1/S2−/− cells. Eluates were analyzed via mass spectrometry, and only hits with significant differential scores (P < 0.05, Student’s t‐test) are shown.
- Hits from (A) tested for interaction with commercially available constructs quantified with bimolecular fluorescence complementation (BiFC) via flow cytometry. HEKS1/S2−/− were transfected with STIM1‐YFPC (black) or STIM1A‐YFPC (red) in combination with POI‐YFPN and screened for YFP+ cells via FACS; results (mean ± SD) were obtained from three transfections each with 10,000 sorted cells.
- Correlation of the mean LFQ intensities from (A) and YFP+ cells from BiFC assay from (B). Dotted lines show differences in interaction of the POIs with STIM1 in comparison with STIM1A.
- Ratio of nuclear NFAT‐GFP vs. cytosolic GFP intensity normalized to t = 0 (mean ± s.e.m.) after transfection of SH‐SY5Y S1−/− cells with STIM (n = 42), STIM1A (n = 82), STIM1A_D503A (n = 49), or vector‐only (ØSTIM, n = 15) IRES‐mCherry and induction of SOCE after stimulation with 1 µM TG. Data (total n as indicated) were obtained from at least three independent transfections.
- Normalized endpoint ratios at 30 min (mean ± s.e.m.) from numbers indicated in (D). *P < 0.05, Kruskal–Wallis ANOVA with Dunn’s multiple comparisons test.
Figure 7. STIM1A alters PDE8B‐ and cAMP‐dependent NFAT translocation.
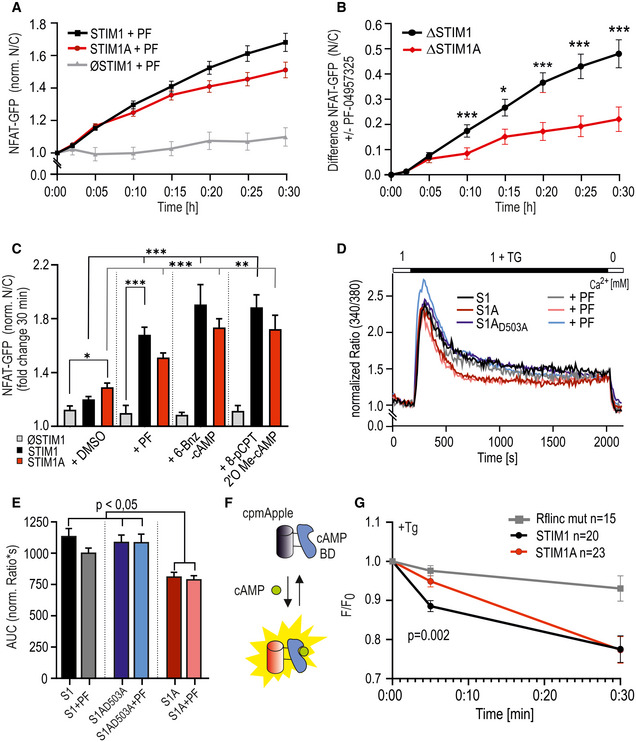
- Time course of translocation (nuclear NFAT‐GFP signal/cytosolic signal normalized to TG addition at t = 0, mean ± s.e.m., for each time point) after transfection of indicated constructs and preincubation with 1 µM PF‐04957325 (analyzed cells: STIM1 + PF, n = 75; STIM1A + PF, n = 50; and ØSTIM + PF, n = 9) Data (total n as indicated) were obtained from at least three independent transfections. *P < 0.05, **P < 0.01, and ***P < 0.001, Kruskal–Wallis ANOVA with Dunn’s multiple comparisons test.
- PF‐04957325 induced difference in the nuclear NFAT‐GFP signal vs. cytosolic signal from (A) to mean of control without the blocker (see Fig 6D). *P < 0.05, **P < 0.01, and ***P < 0.001, Mann–Whitney test of differences.
- Endpoint value with addition of PF or indicated cAMP analogs loaded for 30 min as AM esters before the experiment, shown as mean ± s.e.m., ØSTIM + 6‐Bnz‐cAMP/8‐pCPT‐cAMP: n = 15/14 cells; STIM1 + 6‐Bnz‐cAMP/8‐pCPT‐cAMP: n = 87/66; and STIM1A + 6‐Bnz‐cAMP: n = 78/67, from three independent transfections.
- Traces showing average changes (mean) in intracellular Ca2+ (Fura‐2 ratio) over time in response to perfusion of TG as indicated in the upper bar in SH‐SY5Y S1 −/− cells transfected with STIM1, STIMA, or STIM1A_D503A‐IRES‐mCherry and measured with and without incubation with 1 µM PF‐04957325 (n = 82‐134).
- Area under the curve calculated from individual cells as measured in (D). Bars indicate mean ± s.e.m.
- Schematic representation of the RflincA, modified after Ohta et al (2018).
- Normalized fluorescence signal change (mean ± s.e.m.) of R‐FlincA, SH‐SY5Y S1−/− cells expressing STIM1 (n = 20) or STIM1A (n = 22) IRES‐GFP variants, and the indicator were stimulated by TG (1 μM). To avoid photobleaching, values were only recorded after 5 and 30 min, respectively. The mutant R‐Flinc (n = 15) was measured in SH‐SY5Y wt cells after TG stimulation.
Data information: *P < 0.05, **P < 0.01, and ***P < 0.001, 2‐way ANOVA (B) and Kruskal–Wallis ANOVA with Dunn’s multiple comparisons test (C, E, F, G). Data (total number of cells, n) were obtained from at least three independent transfections with several technical replicates each.
Figure 8. cAMP modulates STIM‐ORAI‐AKAP signalosome by alteration of PIP2.
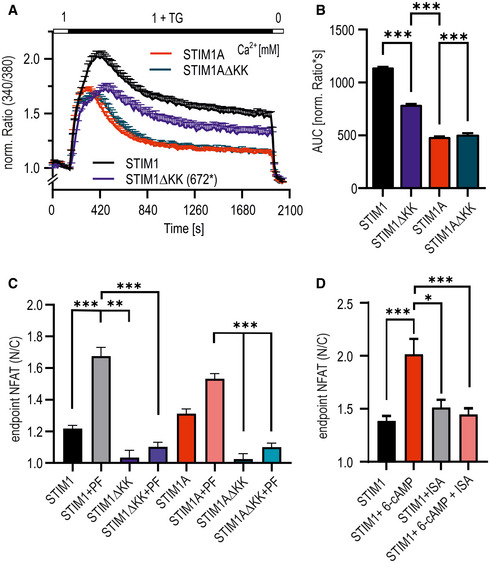
- Traces showing average changes (mean ± sem) in intracellular Ca2+ (Fura‐2 ratio) over time in response to perfusion of TG as indicated in the upper bar in SH‐SY5Y S1−/− cells transfected with mCherry‐tagged constructs as indicated (n = 46–105).
- Quantification of the area under the curve (AUC, mean ± SD), calculated from traces shown in (A) with STIM1 (n = 105), STIM1∆KK (n = 56), STIM1A (n = 75), and STIM1A∆KK (n = 46). ***P < 0.001, Kruskal–Wallis ANOVA with Dunn's multiple comparisons test. Total n were obtained from three independent transfections with 2–3 technical replicates each.
- Normalized endpoint NFAT translocation (mean ± s.e.m.) of control constructs and STIM variants with deleted PBD (∆KK) with or without addition of 1 µM PF‐04957325, Kruskal–Wallis ANOVA, n = 18–19 for ∆KK constructs ±PF, and 56–120 for STIM1 and STIM1A controls obtained from three independent transfections. **P < 0.01 and ***P < 0.001, Kruskal–Wallis ANOVA with Dunn's multiple comparisons test. For clarity, not all differences are shown.
- Normalized endpoint NFAT translocation (mean ± s.e.m.) of cells transfected with indicated constructs that were treated ON with 1 µM ISA with or without addition of 1 µM 6‐Bnz‐cAMP 30 min before measurement. *P < 0.05, **P < 0.01, and ***P < 0.001, Kruskal–Wallis ANOVA, n = 26‐50 from 3 biological replicates.
Discussion
In our study, we report the identification and functional characterization of a novel STIM1 splice variant, STIM1A (α), that contains 31 additional amino acids in its cytosolic domain downstream of the CAD/SOAR domain and immediately adjacent to the highly charged acidic inhibitory domain (ID). While exon A is expressed in many tissues at low levels, its relative expression varies among tissues and cell types and is particularly high in cells with barrier functions, i.e., astrocytes and Sertoli cells. Based on the genomic structure of both the murine and human STIM1 gene, additional splice variants are predicted involving both alternative and mutually exclusive splice events. Alternative 5’UTR regions can be spliced in before exon 2 and may shift the translation start to an internal methionine; therefore, it is important to confirm which splice combinations are present in given tissue. Splice insertion of exon A within full‐length Stim1 translates to a protein detected within testes (Fig 1H) and astrocytes. Testes, but not astrocytes, also express splice combinations leading to N‐terminally truncated Stim1 and Stim1A, although at low levels (Fig EV1C and D). Currently, the specificities of available Stim1 antibodies are re‐evaluated by breeding testis‐specific Stim1‐deficient mice. Therefore, the differential immunohistochemical localization of STIM1 and STIM1A as published in the associated preprint Knapp et al (preprint: Knapp et al, 2020) has been excluded and will be published separately. While the relative amount of N‐terminal spliced variants in several investigated tissues is low, these variants would lack the signal peptide and ER localization and thus might represent a class of STIM1 proteins activating ORAI channels independent of ER [Ca2+] content. We recently identified a neuronally expressed variant (STIM1B) with splice insertion ~60 aa downstream from the insertion of domain A. While STIM1B also decreased SOCE, it did not affect FCDI, but rather led to slower activation and inactivation (SCDI) and caused altered synaptic signaling at high‐frequency stimulations (Ramesh et al, 2021). Functionally, STIM1A reduces SOCE and ICRAC in a sequence‐specific dominant‐negative manner, with the conserved 501FSD503 motif within domain A being essential for its SOCE phenotype and either S502A or D503A point mutants sufficient for SOCE phenotype reversal. However, while current density and FCDI analysis in conditions of strong buffering (BAPTA) demonstrated rescue of the STIM1A phenotype with D503A, weaker buffering (EGTA) uncovered that neither currents nor FCDI was rescued by the point mutant, pointing to a strong Ca2+‐dependent modulation of the effect of the added protein domain. In contrast to the splice insertion (STIM2.1) investigated for STIM2 (Miederer et al, 2015; Rana et al, 2015), we found no overt defect in interaction with ORAI1 as measured by E‐FRET or BiFC nor differences in cluster sizes between STIM1A and ORAI1 after store depletion (Fig 2). Because STIM1A showed increased FCDI and as docking simulations suggest that the CAD/SOAR domain is in very close spatial proximity to both the C‐terminal “nexus” region of ORAI1 (261–265) and its extended N‐terminal pore loop helix, it is highly unlikely that domain A interferes directly with the initial hydrophobic coiled‐coil interactions between STIM1 and ORAI1 and subsequent initial gating residues in ORAI1, but might affect a stabilizing interaction with the ETON region (Lis et al, 2010; Derler et al, 2013; Mcnally et al, 2013), confirmed biochemically and functionally by Niu et al (2020). Indeed, mutating R77 within the ORAI1 ETON region prevented a further reduction by STIM1A. While many of the ETON residue side chains are facing into the pore lumen (Yamashita et al, 2020), the side chain of R77 points outwards (Fig 5D and E). The docking studies that we performed earlier (Alansary et al, 2016) and now repeated with docking constraints of STIM1 interacting with the ORAI1 nexus region revealed STIM1 T393 as a putative amino acid directly interacting with ORAI1 R77. T393 was shown to be highly important for STIM1 activation, since several point mutations led to impaired activation properties. This effect strongly depends on the particular amino acid chosen to replace T393 (Ma et al, 2020). One possible explanation for the observed effect of STIM1A on channel gating is that domain A adds intramolecular bending forces to the CAD domain to weaken a stabilizing interaction between the ETON region (R77) and CAD/SOAR, necessary for full and stabilized gating as modeled by molecular docking (Fig 5C). Our finding that normal function can be restored by mutating the FSD motif or with single residue mutations (S502A or D503A) could be due to a structural change resulting in regained flexibility and accessibility of the CAD/SOAR domain to ORAI1R77; however, additional interacting interfaces are also possible. The close vicinity of domain A (inserted after aa 491) to the inhibitory domain (ID, aa 475–483) in STIM1 and the strong Ca2+ dependency of the point mutant STIM1A_D503A strengthen the hypothesis that gating and FCDI are altered. The reduced SOCE seen upon overexpression is matched by increased SOCE upon downregulation or knockout of endogenous STIM1A in primary astrocytic cultures. Although increased translocation of NFAT is known to be enhanced upon increasing calcium concentration (Rao et al, 1997; Kar et al, 2011), NFAT translocation does not scale with the global amount of Ca2+ entry (Son et al, 2020). We found that STIM1A, despite leading to reduced Ca2+‐entry, caused a slightly increased TG‐induced translocation of NFAT and, despite a clearly reduced Ca2+‐entry upon carbachol stimulation, also did not show reduced NFAT translocation when compared to STIM1. The TG‐triggered difference was still seen upon expression of the D503A mutant, proving that the differential NFAT translocation is likely independent of STIM1A’s effect on gating (Fig 6). However, the difference in NFAT translocation was reverted in a STIM‐dependent manner when cells were treated with a specific inhibitor of the highly cAMP‐specific PDE8B, the phosphodiesterase that we differentially pulled down in IP experiments (Fig 6A). This suggests that STIM1A is a less efficient activator of PDE8B, thereby reducing the effect of the inhibitor. Surprisingly, the PDE8 inhibitor led to a strong increase in NFAT translocation in conditions of submaximal stimulation (TG). PKA‐mediated phosphorylation of ORAI1 serine 34 had been postulated to increase FCDI leading to reduced nuclear NFAT (Zhang et al, 2019), a finding that has recently been challenged (Kar et al, 2021b). While we find that current densities and FCDI are altered upon expression of STIM1A, reduced SOCE is also seen with ORAI2 and ORAI3, which lack serine 34. In addition, the strong and differential effects observed with the PDE8B inhibitor, which did not alter the amount of Ca2+ entry (Fig 7D and E) but increased NFAT translocation, point toward a positive and unexpected cAMP‐dependent modulation of NFAT translocation. Indeed, acute treatment of cells with membrane‐permeant cAMP analogs (both PKA‐specific and exchange protein activated by cAMP (Epac)‐specific) increased NFAT translocation in SH‐SY5Y cells to a similar degree as PF‐04957325. As with the PDE8 inhibitor, the cAMP analogs had no effect on NFAT translocation in the absence of STIM1, pointing toward a dominant requirement of the presence of STIM1 or STIM1‐mediated Ca2+ entry (Fig 7C). However, deletion of the PBD in both variants obliterated NFAT translocation and PF‐04957325 effects, despite the presence of endogenous levels of STIM2 and substantial Ca2+ entry (Fig 8). Together with published effects of PIP2 depletion on NFAT translocation (Son et al, 2020), we speculated that local cAMP at membrane contact sites may lead to increased PIP2 production by stimulation of PIP5K, an effect reported before for PIP5K from yeast (Kato et al, 1989). Our experiments with the PIP5K blocker ISA‐2011B, which prevented cAMP‐induced increases in NFAT translocation, demonstrate that it is also highly likely that mammalian PI4K/PIP5K are cAMP‐regulated enzymes. However, these cAMP effects only become significant, if recruitment/activation of PDE8 (or potentially of other PDEs) to the STIM1/ORAI1/AKAP79 signalosome are held in check, for example, by expressing the splice variant STIM1A, which hinders PDE8 recruitment (Fig 9). The cAMP‐mediated differences might be masked by strong Gαq stimulation (Fig EV5F–H), as carbachol efficiently stimulates phosphoinositide turnover in the time frame of the translocation experiments (Quist & Satumtira, 1987) Higher steady‐state levels of PIP2 facilitate recruitment of STIM proteins and AKAP79 to the plasma membrane (Dell'Acqua et al, 1998), likely stabilizing AKAP79’s interaction with the ORAI1 AKAR region (aa 39–59; Kar et al, 2021b). This stabilized STIM(A)‐ORAI1‐AKAP signalosome then efficiently mobilizes NFAT dephosphorylation and translocation to the nucleus.
Figure 9. Model summarizing altered functions of STIM1A.
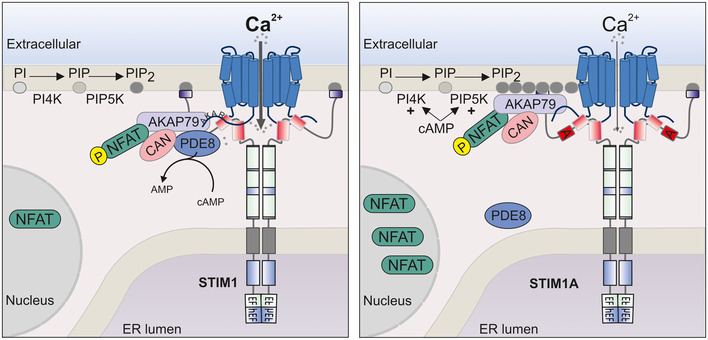
Left image: SOCE mediated by STIM1 and ORAI1 leads to Ca2+ influx, and recruitment of the AKAP79/NFAT signaling complex and the phosphodiesterase PDE8. Locally recruited PDE8 reduces cAMP at ER‐PM junctions. The AKAP79 signalosome recruitment is instable due to limited membrane PIP2 levels. CAN: calcineurin. Right image: SOCE mediated by STIM1A and ORAI1 leads to reduced Ca2+ influx due to destabilized gating and hampers recruitment of PDE8. Reduced recruitment leads to reduced local cAMP degradation, which enhances PIP2 levels by activating PIP kinases. Increased levels of PIP2 stabilize the AKAP79 signalosome, enabling more efficient NFAT translocation.
In summary, our results offer an intriguing insight how altered STIM1 splicing can affect local SOCE and cAMP‐modulated activation of NFAT translocation (Fig 9) and point to a role of STIM1A in altering Gαs/adenylate cyclase‐modulated effects on initiation of NFAT translocation and gene expression. Through alternate splicing, domain A can interfere with STIM1‐mediated recruitment and activation of PDE8, thereby maintaining higher local cAMP and PIP2 levels, stabilizing the AKAP79/NFAT complex and facilitating NFAT translocation. Therefore, the presence of STIM1A may contribute to hyperproliferative phenotypes induced by Gαs and SOCE pathways in cells with high expression of STIM1A, potentially in neonatal cardiomyocytes (Rosenberg et al, 2021), astrocytes, and Sertoli cells. Whether regulation of NFAT translocation via PDE8 (showing only very high expression in selected cells) is a universal phenomenon or restricted to specified cell types, different NFAT‐ or PDE isoforms will require further investigation.
Materials and Methods
Cell lines and transfection
All cell lines were cultivated at 37°C, 5% CO2 in a humidified incubator in respective medium (HEK293 minimum essential medium, HEK293STIM1/2 −/− Dulbecco’s modified Eagle’s medium, MEFStim1/2 −/− Dulbecco’s modified Eagle’s medium, HEKO1 minimum essential medium, and SH‐SY5Y STIM1−/− and SH‐SY5Y STIM1 −/− ;STIM2 −/− Dulbecco’s modified Eagle’s medium with 1% non‐essential amino acids; NEAA, Gibco). All culture media were supplemented with 10% fetal calf serum. For passaging, cells were detached using trypsin. For transfections, HEK cells were transfected via electroporation using the Amaxa® Nucleofector II® (Lonza) according to the user manual. Amount of DNA used was 1µg/1.000.000 cells. MEF/SH‐SY5Y cells were transfected using jetPRIME® (Polyplus transfections) transfection reagent following the manufacturer’s protocol. siRNA (#1+#2, 20 nM total) or siCTL (20 nM) were transfected 24h before recording using INTERFERin® (Polyplus transfections). Cells were analyzed 20‐24 h after transfection of siRNA or with specified DNA constructs as listed in Table EV1; primers and siRNA are listed in Table EV2.
Hippocampal astrocyte cultures
Hippocampal astrocytes were prepared from C57BL/6 or 10ACon mice of either sex at postnatal days P0–P1. Hippocampi were dispersed using a cell strainer in DMEM (Invitrogen). After centrifugation for 10 min at 702 g at RT, cells were resuspended and plated in collagen (0.5 mg/ml; Bioscience)‐coated culture flasks and cultured at 37°C in culture medium (DMEM + 10% FCS + 0.1% P/S + 0.1% Mito Serum Extender) at 8% CO2 until cultures became confluent (5–7 DIC). Cells were subsequently trypsinized, plated on collagen‐coated coverslips, and grown until confluent (5–7 DIC).
PCR and quantitative real‐time PCR
For cDNA synthesis, cells were first harvested in TRIzol (Life Technologies) and RNA was isolated according to the manufacturer’s instructions. The cDNA transcription was obtained using SuperScriptTM II Reverse Transcriptase (Life Technologies). For analytic PCR, DreamTaq Green PCR Master Mix (Thermo Fisher Scientific) was used. Amplifications were analyzed in GTQ‐agarose gels with Safe‐Red DNA dye. qRT–PCRs were performed using QuantiTect SYBR Green Kit (Qiagen) and a CFX96 Real‐Time System (Bio‐Rad). All primers are listed in Table EV2. Using the ΔC q (quantification cycle) method, results were normalized to TBP, whereas normalization with HPRT1 showed comparable findings. Values are shown as 2−(ΔCq).
Western blot/antibodies
Transfected cells were washed with PBS, detached with a cell scraper, and transferred in ice‐cold RIPA buffer containing 150 mM NaCl, 50 mM Tris–HCl, 1% Non‐Idet P 40, 1% Triton and Protease Inhibitor Complete™ (Sigma‐Aldrich). For lysis, the cells were frozen for 10 min at −80°C and vortexed (3 × 20 s). Murine tissues were washed in PBS and lysed using a microhomogenizer (neoLab). Lysed cells were spun down at 21,380 g (4°C) for 30 min. Proteins were linearized in Tris–glycine buffer at 95°C for 5 min, electrophoresed in 7% SDS–PAGE, and blotted onto a nitrocellulose membrane (GE Healthcare). Membranes were blocked with 5% skimmed milk in TBST buffer. Primary and secondary antibodies used for Western blot are listed in Table EV3.
Fluorescent‐based Ca2+ Imaging
Cells were loaded with 1 µM Fura‐2‐AM for 30 min at RT. For perfusion, a fully automated perfusion system (ALA Scientific Instruments) was used. Cells were flushed with 1 ml of Ca2+ Ringer of different concentrations. The Ca2+ Ringer solution contained (in mM) the following: 155 NaCl, 2 MgCl2, 10 glucose, 5 HEPES, and 0.5–2 CaCl2 (= 0.5–2 Ca2+ Ringer) or no CaCl2, but 1 EGTA plus 3 MgCl2 instead (= 0 Ca2+ Ringer) (pH 7.4 with NaOH). To block the SERCA, 1 µm TG in 0 Ca2+ Ringer was used. Images were taken at 340 and 380 nm every 5 s at RT and analyzed with VisiView® software. To quantify 340/380 nm signal ratio, IGOR Pro was used. Parameters analyzed were the average basal signal, the maximal TG‐induced peak, maximal and plateau after readdition of 0.5–2 mM [Ca2+]o, and influx rate. The minimal signal before addition of TG or 0.5 mM [Ca2+]o was subtracted respectively from TG peak or Ca peak/plateau to determine the Δ values.
TIRF microscopy for FRET analysis
HEK293 cells were transfected with 3 µg STIM1‐mCherry‐pmax (containing mCherry after position L599 (Alansary et al, 2016) or STIM1A‐mCherry‐pmax (acceptor) and 1µg Orai1‐GFP‐pmax (donor) 24 h before measurements). Prior to the measurements, TG‐sensitive stores were depleted using 1 µm TG in 0 [Ca2+]o Ringer. For recording fluorescence images, the Leica AM TIRF MC system was used. Images were taken with a 100 × 1.47 oil HCX Plan Apo objective. The GFP‐Donor signal was used to determine the TIRF focal plane. For each cell, three images were taken: I. GFP excited at 488 nm (suppression filter BP 525/50); II. mCherry excitation wavelength at 561 nm (suppression filter BP 600/40); and III. FRET excitation with a 488‐nm laser and suppression filter BP 600/40. To calibrate laser intensities and excitation durations for each day of experiments, single transfected cells were used, only expressing the donor or acceptor construct. Calibrated parameters were constant for all channels and images taken. For image acquisition and analysis, the LAS (Leica Application Suite) FRET module was used. To calculate FRET efficiency according to van Rheenen et al (2004), all images were corrected for background signal, bleed‐through, and crosstalk factors.
Live cell imaging
HEK STIM1/2−/− cells were transfected with SP‐YFP‐STIM1 and STIM1‐mCherry or STIM1A‐mCherry. Fluorescence was detected using the wide‐field epifluorescence microscope cell observer A1 (Zeiss) with the Fluor 40×/1.3 M27 oil objective. To detect YFP, fluorescence filter cube 54HE and LED 470 (470/40) were used, and filter cube 56HE and LED N‐White + Ex (556/20) were used to detect mCherry. Images were taken as Z‐stacks and processed as a maximum intensity projection (MIP) for visualization. Co‐localization was analyzed using the Fiji plugin JACoP from a single stack. Threshold was set as mean plus three times SD.
Patch‐clamp electrophysiology
Recordings were performed at room temperature in the tight‐seal whole‐cell configuration as in Kilch et al (2013) . Briefly, the indicated cells were transfected as above and recordings were done 24 h later with an EPC‐10 patch‐clamp amplifier controlled by Patchmaster software (HEKA). Series resistance was compensated to 85% for the transfected HEK293 cells. Immediately after establishing whole‐cell configuration, linear voltage ramps from −150 mV to +150 mV (50‐ms duration) were applied every 2 s from a holding potential of 0 mV for the indicated time period. Currents were analyzed at −80 mV and 120 s after break‐in. The pipette solution contained the following (in mM): 120 Cs‐glutamate, 3 MgCl2, 20 Cs‐BAPTA or 10 Cs‐EGTA, 10 HEPES, and 0.05 IP3 (pH 7.2 with CsOH). Bath solution contained the following (in mM): 120 NaCl, 10 TEA‐Cl, 2 CaCl2 (or 10 CaCl2), 2 MgCl2, 10 HEPES, and glucose (pH 7.2 with NaOH).
Mass spectrometry and Data processing
For sample preparation, HEK STIM1/2 −/− cells were transfected with SP‐HA‐STIM1‐mCherry or SP‐HA‐STIM1A‐mCherry. 24 h post‐transfection, cells were stimulated adding 1 µM thapsigargin to the medium. After lysate generation and concentration determination, 5 mg of whole‐cell lysate was used to precipitate STIM1(A) using Pierce™ anti‐HA agarose (Thermo Fisher Scientific). Bound protein was eluted using 2% SDS containing 50mM Tris–HCl (pH 6.8). Protein samples were loaded on 12% SDS–PAGE and allowed to migrate approximately 8 mm into the separating gel. Protein bands from three independent replicates were cut out, followed by tryptic in‐gel protein digestion and peptide desalting. Peptides were resuspended in a solution of 2% acetonitrile, and 1% formic acid just before the LC‐MS/MS run. The LC‐MS/MS system (Eksigent nanoLC 425 coupled to a TripleTOF 6600, AB Sciex) was operated as described in Hammel et al (2018). Quantitative analysis of MS measurements was performed using MaxQuant 1.6.0.1 (Cox & Mann, 2008). The library used for peptide spectrum matching was constructed based on the Homo sapiens reference proteome UP000005640 (Bateman, 2019) including the sequences of hSTIM1A and hSTIM1A_D503A. The peptide library was extended by methionine oxidation and acetylation of protein N‐termini as variable modifications. False discovery rate (FDR) thresholds for peptide spectrum matching and protein identification were set to 0.01. Proteins were quantified using the label‐free quantification (LFQ) algorithm (Cox et al, 2014). Protein abundances were normalized according to the detected amount of bait protein. Missing values were imputed on protein level by mean value imputation, adding random Gaussian noise estimated using sample mean and variance. If both parameters could not be estimated from the data, the random value imputation was based on the 0.3 quantile of the empirical distribution of the experiment, simulating the detection limit for lowly abundant proteins. Statistical significance was determined by one‐way ANOVA adjusted for multiple testing and the Hays post hoc test.
Bimolecular fluorescence complementation
HEK STIM1−/−;STIM2−/− cells (Zhou et al, 2018) were transfected in a 6‐well scale with STIM1 or STIM1A fused to AA 156–720 of YFP in pmax as bait in combination with POI fused to AA1–155 of YFP in pBabe as prey. Transfection was performed at equimolar ratios of DNA using a total amount of 2 µg DNA. To induce SOCE, 1 µM TG was added to the media and incubated for 10 min 24 h post‐transfection. After detaching cells in PBS containing 1 µM TG, cells were centrifuged at 320 g in FACS tubes at 4°C for 10 min and resuspended in 400 µl ice‐cold FACS buffer (5% FCS, 0.5% BSA and 0.07% NaN3 in PBS) with additional Zombie Aqua™ (1:1,000 Fixable Viability Kit, BioLegend) to stain for vital cells. To screen for YFP signal in vital cells FACSVerse system (BD Biosciences) was used. BD FACSuite™ and FloJo 10.0.7 (BD Biosciences) were used for determination of the percentages of YFP‐positive cells.
NFAT assay
SH‐SY5Y STIM1 −/− cells were transfected with STIM1‐mCherry, STIM1A‐mCherry, or noSTIM‐mCherry in combination with NFAT1‐GFP. 24 h post‐transfection, the GFP signal in the cytoplasm vs. in the nucleus was detected using the Zeiss AXIO observer. Immediately before the measurement, DNA was stained using 1 mg/ml Hoechst 33342 dye (Thermo Fisher Scientific). For inhibition of PDE8B, the specific blocker PF‐04957325 (1 µM, MCE) was added 2 h prior to the experiment and kept in all reagents during the measurement. To test the effect of cAMP analogs on NFAT translocation, cells were loaded 30 min prior to the measurement with 1 μM of 6‐Bnz‐cAMP‐AM (BioLog) or 8‐pCPT‐2'‐O‐Me‐cAMP‐AM (BioLog). The ratio of GFP signal in the cytoplasm vs. in the nucleus was determined using Fiji (Rueden et al, 2017).
Homology modeling and molecular docking
At the time of writing, crystal structures for Orai proteins exist only for Drosophila melanogaster and not for humans. Since we were interested in the specific interaction of human Orai1 (hOrai1) with human STIM1 (hSTIM1), we performed homology modeling using the SWISS‐MODEL webserver (Waterhouse et al, 2018). We inserted the hOrai1 sequence obtained from the entry in the UniProt database (Bateman et al, 2021) with entry name CRCM1_HUMAN as a target sequence. We were interested in the open conformation of hOrai1 and thus used a structure of Orai from Drosophila melanogaster with the H206A gain‐of‐function mutation (Hou et al, 2018) stored in the Protein Data Bank (PDB; Berman et al, 2000) with PDB‐ID 6BBF as a template. The obtained homology model was afterward used to perform solvated protein‐protein docking of hOrai1 and the SOAR domain of STIM1 (chain A from crystal structure stored at PDB‐ID 3TEQ; Yang et al, 2012) using the expert mode of HADDOCK2.2 webserver (Dominguez et al, 2003; Van Zundert et al, 2016). Unambiguous distance restraints with a potential of zero between a lower limit of 0 Å and an upper limit of 8 Å distance between the residues and increasing potential outside of this range were applied between residues 76–80 of hOrai1 and residues 383–403 of hSTIM1. Default parameters for the force constants and clustering parameters were used. 2000 structures were generated during rigid‐body docking stage with 10 trials for rigid‐body minimization, 400 structures were refined during semi‐flexible refinement, and 400 structures were refined during explicit solvent refinement (in water). The second docking approach was executed as described in Alansary et al (2016).
Author contributions
BAN designed experiments and wrote the manuscript with the help of MLK and input from DA and AL. MLK and VP performed imaging, PCR, microscopy, and biochemical experiments. KF performed qRT–PCR experiments. YS provided astrocytic cultures. DA and AL performed patch‐clamp experiments. FS and MS performed the mass spectrometry (MS) analyses. DZ and TM performed MS data evaluation and statistical tests. NK, VH, and AK performed docking models. KM provided splice‐deficient mice. All authors conducted data analysis and provided input into writing of the manuscript.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Acknowledgments
We thank Phillip Knapp for help with the Cell Observer, and Alina Gilson and Maik Konrad for initial experiments. We are very thankful to Don Gill for the gift of STIM‐deficient HEK cells and to Yubin Zhou for providing us with STIM1 BiFC constructs. We also thank members of the Hoth’ and Niemeyer’ laboratories for critical input, reading of the manuscript, and discussions. The research was funded by the DFG SFB894 (A2), SFB1027 (C4), and TRR219 (322900939) to BAN. Mona L. Knapp (née Schoeppe) was a graduate student supported by IRTG1830 (to BN and MH). Open Access funding enabled and organized by Projekt DEAL.
Conflict of interest
The authors declare that they have no conflict of interest.
EMBO reports (2022) 23: e53135.
Data availability
RNA‐Seq data: European Nucleotide Archive (ENA) submission: (https://www.ebi.ac.uk/ena/browser/view/PRJNA768993?show=reads).
BioProject ID: PRJNA768993 (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA768993).
Protein interaction AP‐MS data: The mass spectrometric proteomic data are available via the ProteomeXchange Consortium partner repository, PRIDE (Perez‐Riverol et al, 2019) with the dataset identifier PXD028974 (http://www.ebi.ac.uk/pride/archive/projects/PXD028974).
References
- Alansary D, Peckys DB, Niemeyer BA, de Jonge N (2020) Detecting single ORAI1 proteins within the plasma membrane reveals higher‐order channel complexes. J Cell Sci 133: jcs240358 [DOI] [PubMed] [Google Scholar]
- Alansary D, Schmidt B, Dörr K, Bogeski I, Rieger H, Kless A, Niemeyer BA (2016) Thiol dependent intramolecular locking of Orai1 channels. Sci Rep 6: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barat C, Tremblay MJ (2003) Treatment of human T cells with bisperoxovanadium phosphotyrosyl phosphatase inhibitors leads to activation of cyclooxygenase‐2 gene. J Biol Chem 278: 6992–7000 [DOI] [PubMed] [Google Scholar]
- Bateman A (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47: D506–D515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Martin MJ, Orchard S, Magrane M, Agivetova R, Ahmad S, Alpi E, Bowler‐Barnett EH, Britto R, Bursteinas B et al (2021) UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res 49: D480–D489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Darbellay B, Arnaudeau S, Bader CR, Konig S, Bernheim L (2011) STIM1L is a new actin‐binding splice variant involved in fast repetitive Ca2+ release. J Cell Biol 194: 335–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD (1998) Membrane‐targeting sequences on AKAP79 bind phosphatidylinositol‐4,5‐bisphosphate. EMBO J 17: 2246–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Butorac C, Krizova A, Stadlbauer M, Muik M, Fahrner M, Frischauf I, Romanin C (2018) Authentic CRAC channel activity requires STIM1 and the conserved portion of the Orai N terminus. J Biol Chem 293: 1259–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Plenk P, Fahrner M, Muik M, Jardin I, Schindl R, Gruber HJ, Groschner K, Romanin C (2013) The extended transmembrane orai1 N‐terminal (ETON) region combines binding interface and gate for orai1 activation by STIM1. J Biol Chem 288: 29025–29034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C, Boelens R, Bonvin AMJJ (2003) HADDOCK: A protein‐protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125: 1731–1737 [DOI] [PubMed] [Google Scholar]
- Feske S (2019) CRAC channels and disease – From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 80: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischauf I, Muik M, Derler I, Bergsmann J, Fahmer M, Schindl R, Groschner K, Romanin C (2009) Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1‐3 channels by a STIM1 coiled‐coil mutant. J Biol Chem 284: 21696–21706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel A, Zimmer D, Sommer F, Muhlhaus T, Schroda M (2018) Absolute quantification of major photosynthetic protein complexes in Chlamydomonas reinhardtii using quantification concatamers (QconCATs). Front Plant Sci 9: 1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Burstein SR, Long SB (2018) Structures reveal opening of the store‐operated calcium channel Orai. eLife 7: e36758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Chikazu D, Voznesensky OS, Herschman HR, Kream BE, Drissi H, Pilbeam CC (2010) Parathyroid hormone induction of cyclooxygenase‐2 in murine osteoblasts: role of the calcium‐calcineurin‐NFAT pathway. J Bone Miner Res 25: 819–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P, Barak P, Zerio A, Lin Y‐P, Parekh AJ, Watts VJ, Cooper DMF, Zaccolo M, Kramer H, Parekh AB (2021a) AKAP79 orchestrates a cyclic AMP signalosome adjacent to orai1 Ca2+ channels. Function 2: zqab036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P, Lin YP, Bhardwaj R, Tucker CJ, Bird GS, Hediger MA, Monico C, Amin N, Parekh AB (2021b) The N terminus of Orai1 couples to the AKAP79 signaling complex to drive NFAT1 activation by local Ca2+ entry. Proc Natl Acad Sci USA 118: e2012908118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P, Nelson C, Parekh AB (2011) Selective activation of the transcription factor NFAT1 by calcium microdomains near Ca2+ release‐activated Ca2+ (CRAC) channels. J Biol Chem 286: 14795–14803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P, Samanta K, Kramer H, Morris O, Bakowski D, Parekh AB (2014) Dynamic assembly of a membrane signaling complex enables selective activation of NFAT by orai1. Curr Biol 24: 1361–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Uno I, Ishikawa T, Takenawa T (1989) Activation of phosphatidylinositol kinase and phosphatidylinositol‐4‐phosphate kinase by cAMP in Saccharomyces cerevisiae. J Biol Chem 264: 3116–3121 [PubMed] [Google Scholar]
- Kilch T, Alansary D, Peglow M, Dorr K, Rychkov G, Rieger H, Peinelt C, Niemeyer BA (2013) Mutations of the ca2+‐sensing stromal interaction molecule stim1 regulate Ca2+ influx by altered oligomerization of stim1 and by destabilization of the ca2+ channel orai1. J Biol Chem 288: 1653–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM, Rana A, Park CY (2019) Orai1 inhibitor STIM2β regulates myogenesis by controlling SOCE dependent transcriptional factors. Sci Rep 9: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp ML, Förderer K, Alansary D, Jung M, Schwarz Y, Lis A, Niemeyer BA (2020) Alternative splicing switches STIM1 targeting to specialized membrane contact sites and modifies SOCE. bioRxiv 10.1101/2020.03.25.005199v1 [PREPRINT] [DOI] [Google Scholar]
- Kunkl M, Porciello N, Mastrogiovanni M, Capuano C, Lucantoni F, Moretti C, Persson JL, Galandrini R, Buzzetti R, Tuosto L (2017) ISA‐2011B, a phosphatidylinositol 4‐phosphate 5‐kinase a inhibitor, impairs CD28‐dependent costimulatory and pro‐inflammatory signals in human T lymphocytes. Front Immunol 8: 502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS, Feske S (2015) Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci 1356: 45–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis A, Zierler S, Peinelt C, Fleig A, Penner R (2010) A single lysine in the N‐terminal region of store‐operated channels is critical for STIM1‐mediated gating. J Gen Physiol 136: 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma G, He L, Liu S, Xie J, Huang Z, Jing J, Lee YT, Wang R, Luo H, Han W et al (2020) Optogenetic engineering to probe the molecular choreography of STIM1‐mediated cell signaling. Nat Commun 11: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcnally BA, Somasundaram A, Jairaman A, Yamashita M, Prakriya M (2013) The C‐ and N‐terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J Physiol 591: 2833–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A, García‐Castañeda M, Boncompagni S, Dirksen RT (2018) Role of STIM1/ORAI1‐mediated store‐operated Ca2+ entry in skeletal muscle physiology and disease. Cell Calcium 76: 101–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miederer AM, Alansary D, Schwär G, Lee PH, Jung M, Helms V, Niemeyer BA (2015) A STIM2 splice variant negatively regulates store‐operated calcium entry. Nat Commun 6: 6899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motiani RK, Tanwar JK, Raja DA, Vashisht A, Khanna S, Sharma S, Srivastava So, Sivasubbu S, Natarajan VT, Gokhale RS (2018) STIM 1 activation of adenylyl cyclase 6 connects Ca2+ and cAMP signaling during melanogenesis. EMBO J 37: e97597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller MR, Rao A (2010) NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol 10: 645–656. [DOI] [PubMed] [Google Scholar]
- Mullins FM, Lewis RS (2016) The inactivation domain of STIM1 is functionally coupled with the Orai1 pore to enable Ca2+‐dependent inactivation. J Gen Physiol 147: 153–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L, Wu F, Li K, Li J, Zhang SL, Hu J, Wang Q (2020) STIM1 interacts with termini of Orai channels in a sequential manner. J Cell Sci 133: jcs239491 [DOI] [PubMed] [Google Scholar]
- Nwokonko RM, Cai X, Loktionova NA, Wang Y, Zhou Y, Gill DL (2017) The STIM‐Orai pathway: Conformational coupling between STIM and Orai in the activation of store‐operated Ca2+ entry. In Store‐Operated Ca2+ Entry (SOCE) Pathways. Advances in Experimental Medicine and Biology, Groschner K, Graier W, Romanin C (eds), Vol. 993, pp 83–98. Cham: Springer; 10.1007/978-3-319-57732-6_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh‐hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A (2008) Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 9: 432–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Furuta T, Nagai T, Horikawa K (2018) Red fluorescent cAMP indicator with increased affinity and expanded dynamic range. Sci Rep 8: 1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS (2015) Store‐operated calcium channels. Physiol Rev 95: 1383–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quist EE, Satumtira N (1987) Muscarinic receptor stimulated phosphoinositide turnover in cardiac atrial tissue. Biochem Pharmacol 36: 499–505 [DOI] [PubMed] [Google Scholar]
- Ramesh G, Jarzembowski L, Schwarz Y, Poth V, Konrad M, Knapp ML, Schwar G, Lauer AA, Grimm MOW, Alansary D et al (2021) A short isoform of STIM1 confers frequency‐dependent synaptic enhancement. Cell Rep 34: 108844 [DOI] [PubMed] [Google Scholar]
- Rana A, Yen M, Sadaghiani AM, Malmersjö S, Park CY, Dolmetsch RE, Lewis RS (2015) Alternative splicing converts STIM2 from an activator to an inhibitor of store‐operated calcium channels. J Cell Biol 209: 653–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG (1997) Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15: 707–747 [DOI] [PubMed] [Google Scholar]
- Rosenberg P, Zhang H, Bryson VG, Wang C (2021) SOCE in the cardiomyocyte: the secret is in the chambers. Pflügers Archiv – Eur J Physiol 473: 417–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW (2017) ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform 18: 529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta K, Kar P, Mirams GR, Parekh AB (2015) Ca2+ Channel re‐localization to plasma‐membrane microdomains strengthens activation of Ca2+‐dependent nuclear gene expression. Cell Rep 12: 203–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, Gill DL (2012) STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol 13: 549–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son GY, Subedi KP, Ong HL, Noyer L, Saadi H, Zheng C, Bhardwaj R, Feske S, Ambudkar IS (2020) STIM2 targets Orai1/STIM1 to the AKAP79 signaling complex and confers coupling of Ca2+ entry with NFAT1 activation. Proc Natl Acad Sci USA 117: 16638–16648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffner A, Maltan L, Weiß S, Derler I (2021) The orai pore opening mechanism. Int J Mol Sci 22: 533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen J, Langeslag M, Jalink K (2004) Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys J 86: 2517–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, Van Dijk M, De Vries SJ, Bonvin AMJJ (2016) The HADDOCK2.2 Web server: user‐friendly integrative modeling of biomolecular complexes. J Mol Biol 428: 720–725 [DOI] [PubMed] [Google Scholar]
- Vang AG, Basole C, Dong H, Nguyen RK, Housley W, Guernsey L, Adami AJ, Thrall RS, Clark RB, Epstein PM et al (2016) Differential expression and function of PDE8 and PDE4 in effector T cells: implications for PDE8 as a drug target in inflammation. Front Pharmacol 7: 259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, De Beer TAP, Rempfer C, Bordoli L et al (2018) SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Ing CE, Yeung PSW, Maneshi MM, Pomès R, Prakriya M (2020) The basic residues in the Orai1 channel inner pore promote opening of the outer hydrophobic gate. J Gen Physiol 152: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Jin H, Cai X, Li S, Shen Y (2012) Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc Natl Acad Sci USA 109: 5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh Y‐C, Parekh AB (2018) CRAC Channels and Ca2+‐dependent gene expression. In Calcium Entry Channels in Non‐Excitable Cells, Kozak JA, Putney JW Jr (eds), pp 93–106. Boca Raton, FL: CRC Press/Taylor & Francis; [PubMed] [Google Scholar]
- Yeung PSW, Yamashita M, Prakriya M Molecular basis of allosteric Orai1 channel activation by STIM1. J Physiol 598: 1707–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Hubrack SZ, Chakraborty S, Sun L, Alcantara‐Adap E, Kulkarni R, Billing AM, Graumann J, Taylor CW, Machaca K (2019) Remodeling of ER‐plasma membrane contact sites but not STIM1 phosphorylation inhibits Ca(2+) influx in mitosis. Proc Natl Acad Sci USA 116: 10392–10401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Pathak T, Yoast R, Emrich S, Xin P, Nwokonko RM, Johnson M, Wu S, Delierneux C, Gueguinou M et al (2019) A calcium/cAMP signaling loop at the ORAI1 mouth drives channel inactivation to shape NFAT induction. Nat Commun 10: 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cai X, Loktionova NA, Wang X, Nwokonko RM, Wang X, Wang Y, Rothberg BS, Trebak M, Gill DL (2016) The STIM1‐binding site nexus remotely controls Orai1 channel gating. Nat Commun 7: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Nwokonko RM, Cai X, Loktionova NA, Abdulqadir R, Xin P, Niemeyer BA, Wang Y, Trebak M, Gill DL (2018) Cross‐linking of Orai1 channels by STIM proteins. Proc Natl Acad Sci USA 115: E3398–E3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang X, Wang X, Loktionova NA, Cai X, Nwokonko RM, Vrana E, Wang Y, Rothberg BS, Gill DL (2015) STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat Commun 6: 8395 [DOI] [PMC free article] [PubMed] [Google Scholar]