

Summary
Rapid protein degradation enables cells to quickly modulate protein abundance. Dysregulation of short-lived proteins plays essential roles in disease pathogenesis. A focused map of short-lived proteins remains understudied. Cycloheximide, a translational inhibitor, is widely used in targeted studies to measure degradation kinetics for short-lived proteins. Here, we combined cycloheximide chase assays with advanced quantitative proteomics to map short-lived proteins under translational inhibition in four human cell lines. Among 11,747 quantified proteins, we identified 1,017 short-lived proteins (half-lives ≤ 8hr). These short-lived proteins are less abundant, evolutionarily younger, and less thermally stable than other proteins. We quantified 103 proteins with different stabilities among cell lines. We showed that U2OS and HCT116 cells express truncated forms of ATRX and GMDS, respectively, which have lower stability than their full-length counterparts. This study provides a large-scale resource of human short-lived proteins under translational arrest, leading to untapped avenues of protein regulation for therapeutic interventions.
Graphical Abstract
Introduction
Protein turnover encompasses protein synthesis and elimination, occurring continuously to maintain normal cellular functions. Specifically, rapid protein degradation serves vital roles in various dedicated cellular processes, including cell cycle, signal transduction, and differentiation (Meszaros et al., 2017; Teixeira and Reed, 2013). Because protein synthesis is comparatively slow, rapid protein degradation is required to achieve immediate protein abundance changes in response to perturbations. Alterations in the degradation of disease-related proteins often play essential roles in disease pathogenesis. For example, cell cycle phases are tightly controlled by event-driven activators and inhibitors (CCND1, AURKA, etc.). The selective, programmed, and rapid degradation of these regulators provides the direction and appropriate timing of cell cycle phases (Teixeira and Reed, 2013). Mutations in the target recognition subunits of E3 ligase complexes disrupt CCND1 degradation, which is associated with breast cancer and esophageal cancer (Meszaros et al., 2017). These individual examples suggest that systematic investigation of short-lived proteins will uncover proteome-wide regulatory roles for protein degradation across cellular processes.
Cycloheximide, a global translational inhibitor, blocks the eukaryotic ribosome, specifically, the mRNA-tRNA translocation process (Garreau de Loubresse et al., 2014). Combined with Western blotting, the cycloheximide chase assay has been widely used in targeted studies to measure degradation kinetics for short-lived proteins (Dai et al., 2021; Huang et al., 2020; Ross et al., 2020; Schwanhausser et al., 2011; Xu et al., 2017; Yoshida et al., 2020). Besides, a systematic investigation determined the half-lives for over 3,700 yeast proteins with the cycloheximide chase assay and Western blotting (Belle et al., 2006). However, no comparable study has been performed in human cells. Although global translational inhibition introduces stress responses and short treatment times are thus advised, the cycloheximide chase assay has proven effective for interrogating rapidly degraded proteins (Ross et al., 2020). The fast elimination of these proteins pinpoints certain mechanisms for rapid protein removal, which may guide the development of therapeutic interventions via regulated protein degradation.
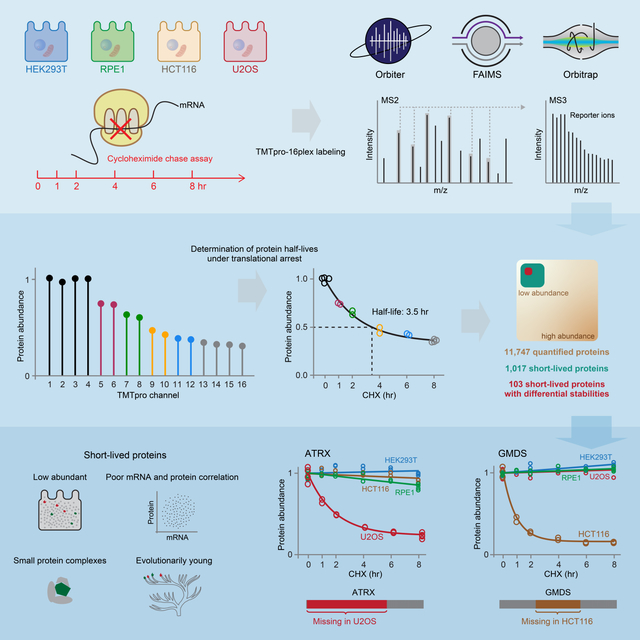
In this work, we combined the cycloheximide chase assay with multiplexed quantitative proteomics to profile short-lived proteins (half-lives ≤ 8 hr) under translational inhibition in widely used human cell lines. We chose four genetically distinct cell lines (U2OS, HEK293T, HCT116, and RPE1) as they provide complementary views of the human proteome. They have different tissues of origin, represent cancer-derived (U2OS and HCT116) and non-cancer derived (HEK293T and RPE1) cells, have different driver mutations and immortalization mechanisms. HCT116 and RPE1 are epithelial cells and HEK293T and U2OS are mesenchymal in origin. Using sample multiplexing, we achieved deep proteome coverage and quantified a total of 11,747 proteins. We identified over 1,000 short-lived proteins under translational inhibition and revealed over 100 proteins showing distinct stabilities between cell lines. The resource represents potential pinch points for therapeutic interventions that could disrupt dedicated biology by controlling protein stability.
Results
The experimental design and quality control of the data
Cells were treated with cycloheximide for different amounts of time (Fig. 1A). Protein abundance was then monitored as a function of time with TMTpro-based quantitative proteomics (Li et al., 2020). Labeled samples were analyzed on an Orbitrap Eclipse mass spectrometer utilizing high-field asymmetric-waveform ion mobility spectrometry (FAIMS) (Schweppe et al., 2019) coupled with real-time search (RTS)-synchronous-precursor-selection (SPS)-MS3 (Schweppe et al., 2020). Protein abundance measurements were used to calculate protein half-lives under translational arrest (hereinafter referred to as “half-lives”) (Fig. 1A). Our expectation was that deep proteome coverage achieved by combining FAIMS and RTS would enable thorough sampling of low abundant short-lived proteins under translational inhibition (hereinafter referred to as “short-lived proteins”) and that the accurate fold change measurements by RTS-SPS-MS3 would facilitate precise half-life calculations. For example, Geminin, an inhibitor of DNA replication, is a known short-lived protein (McGarry and Kirschner, 1998). It showed dramatic degradation and half-lives of 3–4 hr under translational arrest in all four cell lines (Fig. 1B).
Figure 1. An overview of the experimental design.

(A) Cells were treated with cycloheximide, and samples were digested and then labeled with TMTpro 16-plex reagents. Labeled samples were fractionated and concatenated into 24 fractions. Samples were analyzed on an Orbitrap Eclipse mass spectrometer with high-field asymmetric-waveform ion mobility spectrometry (FAIMS) and real-time-search (RTS)-synchronous-precursor-selection (SPS)-MS3. Relative protein abundance was used to calculate protein half-lives under translational inhibition. (B) Relative protein abundance and half-lives of Geminin. Geminin is a known short-lived protein. #PSM is the number of peptide-spectrum-matches. A protein with a PSM count of three means that the protein’s quantification is based on a weighted average of three quantified peptides. (C) Replicate samples grouped together in principal component analysis. Samples collected at different time points were separated mainly on the first principal component (PC1). Relative protein abundance is TMTpro signal-to-noise normalized to the mean of the first time point in panel A and B. See also Figure S1 and Table S1.
Samples collected at the same time point grouped together in principal component analysis (Fig. 1C). Specifically, samples from different time points were separated mainly by the first principal component, which accounted for the majority of the total variance (75%−79%) (Fig. 1C). Coefficients of variance (CV) were excellent between replicates (median CV < 5%) (Fig. S1A). These results indicate that our assay can provide datasets of high quality with excellent reproducibility.
A comprehensive resource of short-lived proteins under translational inhibition in human cells
We performed routine normalization to account for i) small loading differences across TMTpro channels and ii) changes due to small artifacts across time points under the assumption that long-lived proteins would not change in abundance over 8 hr cycloheximide treatment (Mathieson et al., 2018; McShane et al., 2016; Toyama et al., 2013) (Fig. S1B, see Methods for details). Protein half-lives were then determined under the assumption that protein degradation followed first order decay kinetics (Belle et al., 2006) with slight modifications (Fig. S1C, D, see Methods for details).
We achieved excellent proteome coverage, quantifying ~9,000 proteins in each cell line and a total of over 11,000 proteins in all cell lines (Fig. 2, Table S1). The relative expression levels for the majority of proteins remained unchanged, and only a small fraction (~5%) showed dramatic decreases following 8 hr cycloheximide treatment (Fig. 2A, B). Among them, we determined ~500 short-lived proteins in each cell line (Fig. S2A, Table S2). Generally, proteins that were short-lived in one cell line also presented decreasing trends in other cell lines (Fig. S2B). Considering that the criteria for assigning short-lived proteins were arbitrarily based on a half-life cutoff of ≤ 8 hr, we assembled a list of all proteins deemed short-lived in one or more cell lines for many of the subsequent systematic analyses.
Figure 2. Overview of short-lived proteins under translational arrest.

(A) A representative view of quantified proteins and short-lived proteins. Deep proteome coverage was achieved (>9,000 proteins) and only a small fraction of the quantified proteome (~5%) was short-lived in U2OS cells. Six proteins with diverse half-lives are highlighted in panel B. (B) Example degradation curves showing proteins with variable stability. Relative protein abundance is TMTpro signal-to-noise normalized to the mean of the first time point. (C) The number of quantified proteins (upper panel) and short-lived proteins (lower panel). About 4.4%−5.3% of the quantified proteome was short-lived (orange blocks in the upper panel). (D) Distributions of short-lived protein half-lives. Values in parentheses are numbers of short-lived proteins. The eight most short-lived proteins and their half-lives are shown on the left. Bin size is 0.5 hr. The dashed lines indicate medians. See also Figure S2 and Table S2.
In a preliminary analysis, we found that extracellular matrix (ECM) proteins were enriched among short-lived proteins (Fig. S2C). ECM proteins are known long-lived proteins, especially collagen (Toyama and Hetzer, 2013; Toyama et al., 2013) (Fig. S2C). The decreases of ECM proteins were likely caused by secretion without synthesis and not by degradation. Thus, we excluded all ECM proteins in subsequent analyses. After removing ECM proteins, we identified between 403 and 508 short-lived proteins in each cell line (Fig. 2C), representing 4.4%−5.3% of the quantified proteome. In total, 1,017 proteins were short-lived with 90 short-lived in all four cell lines (Fig. S2D, E). The median half-lives of the short-lived proteins were 3–4 hr in each cell line, and about 100 half-lives were as short as 0–2 hr (Fig. 2D). For example, the half-life of PRELID3B was about 0.3 hr (18 min) in U2OS, HEK293T, and HCT116 cells (Fig. 2D). We did not identify PRELID3B in RPE1 cells, but we found a related protein PRELID1 with a half-life of 0.6 hr (36 min) in RPE1 cells (Fig. 2D). PRELID3B and PRELID1 are lipid transfer proteins in mitochondria that shuttle phosphatidic acid and phosphatidylserine across the mitochondrial intermembrane space, respectively (Deshwal et al., 2020). Their homologs in yeast (UPS1 and UPS2) are known short-lived proteins (half-lives ~10 min) (Deshwal et al., 2020). Human PRELID3B and PRELID1 showed half-lives of a similar scale to their yeast homologs despite the dramatically different lifespans of yeast and human cells.
Some phase-specific cell cycle inhibitors and activators are known short-lived proteins (Teixeira and Reed, 2013). Our data recapitulated the immediate degradation of proteins that regulate the transition between cell cycle checkpoints (Fig. 3A). We noted that cleaved NOTCH proteins have short half-lives (Bray, 2016). As bottom-up proteomics cannot routinely differentiate full length and cleaved proteins, NOTCH proteins did not decrease as other proteins (Fig. 3A). Degrons are short linear motifs characterized by specific amino acid sequence patterns. E3 ligases often specifically recognize degrons in substrate proteins for degradation. Some degrons, including PEST sequences, are associated with short protein half-lives (Meszaros et al., 2017). We found that degron-containing proteins were significantly enriched in proteins deemed short-lived (Fig. 3B). These results demonstrated that our data effectively characterize well-recognized short-lived proteins, with half-lives determined under short-term translational arrest.
Figure 3. Known short-lived proteins were captured, and E3 ligases and substrate recognition subunits of E3 ligase complexes were enriched in short-lived proteins under translational inhibition.

(A) Known short-lived cell cycle inhibitors and activators were recapitulated. Replicates are shown individually in the heatmaps. Gray indicates no detection in the plex in the heatmaps. (B) Gene set enrichment analysis (GSEA). Degron-containing proteins were significantly enriched in proteins deemed short-lived. The gene set contains proteins that have at least one known degrons. Log2 fold changes (8 hr vs 0 hr) were used in GSEA. Higher ranks indicate shorter half-lives. (C) InterPro categories enriched among short-lived proteins. DNA-binding proteins, E3 ubiquitin-protein ligases, and substrate recognition subunits of E3 ubiquitin ligase complexes were significantly enriched. (D) Subcellular components enriched among short-lived proteins. See also Figure S3.
Gene Ontology (GO) analyses of short-lived proteins under translational inhibition
Next, we performed GO enrichment analyses using the combined list of short-lived proteins (Fig. 3C, D, Fig. S3). Short-lived proteins were enriched in DNA-binding proteins, E3 ubiquitin-protein ligases, and substrate recognition subunits of E3 ubiquitin ligase complexes (Fig. 3C). Some signaling pathway proteins (JAK-STAT signaling pathway, Hippo signaling pathway, cell cycle, etc.) were also enriched (Fig. S3B). The enrichment of DNA-binding proteins, signaling proteins and cell cycle proteins agreed with previous observations in the subset of unstable proteins (Schwanhausser et al., 2011). Interestingly, we noticed that E3 ubiquitin-protein ligases and substrate recognition subunits of E3 ubiquitin ligase complexes were also enriched (Fig. 3C, D). A few single-unit E3 ligases (e.g. MDM2 (Kubbutat et al., 1999), RNF8 (Ma et al., 2018), RNF138 (Yu et al., 2020)) are known to be short-lived. Three substrate recognition subunits (GRR1, CDC4, and MET30) of the SCF complex are short-lived in yeast (Galan and Peter, 1999). Beyond that, few studies have shown systematically that E3 ligases and substrate recognition subunits of E3 ubiquitin ligase complexes have short half-lives.
The majority of the E3 ligases are RING E3 ligases (Morreale and Walden, 2016). Among RING E3 ligases, the cullin-RING ligase complex and the anaphase promoting complex/cyclosome (APC/C) require substrate recognition subunits to ubiquitylate substrates. We extracted a list of components of the cullin-RING ligase complex and APC/C complex from GO and mapped the log2 fold changes at 8 hr to the list (Fig. 4A, B). We quantified ~90% of all cullin-RING ligase complex members and APC/C complex subunits in GO. Core components of these E3 ligase complexes remained nearly unchanged at 8 hr, while substrate recognition subunits and adaptors underwent fast degradation (Fig. 4). A separate hand-curated list of cullin-RING ligase complex subunits displayed the same results (Fig. S4A). We also noticed that the E3 ligases in these complexes remained largely unchanged. Thus, the enriched E3 ligases in short-lived proteins in GO analysis should be mostly E3 ligases that function as a monomer or dimer and do not require substrate recognition subunits (Morreale and Walden, 2016) (Fig. 3C, Fig. S4B, Fig. S4D).
Figure 4. Substrate recognition subunits of E3 ubiquitin ligase complexes displayed faster degradation than core components under translational inhibition.

About 90% of the E3 ubiquitin ligase complex subunits were found in this work. Core components of cullin-RING ubiquitin ligase complexes (A) and anaphase-promoting complex/cyclosome (B) were stable, and substrate recognition subunits were quickly degraded under 8 hr of translational arrest. The list of E3 ubiquitin ligase complex subunits was from Gene Ontology. Western blotting (C) shows that four adaptor or catalytic proteins (KLHL12, CCNF, CDC20, and ANAPC11) were short-lived, and five core component proteins (CUL3, CUL1, ANAPC1, CDC23, and CDC16) were stable in U2OS cells. See also Figure S4.
It is possible that the enrichment of the substrate recognition subunits of E3 ubiquitin ligase complexes and single-unit ligases was caused by feedback mechanisms when protein synthesis was inhibited. Besides the abovementioned known short-lived single-unit E3 ligases, among all short-lived substrate recognition subunits (Fig. S4A), four were quantified in a pulse-chase metabolic labeling study of RPE1 cells with short half-lives (KLHL9, 6.6 hr; LRRC42, 3.8 hr; DCAF7, 12.5 hr; and LRRC41; 8.9 hr) (McShane et al., 2016). The possibility remains that certain negative feedback mechanisms exist, but evidence from other sources cross-validated that some E3 ligases and substrate recognition subunits are indeed short-lived without translation inhibition. Of note, our results suggested that some working mechanisms were available in cells to rapidly degrade these proteins, which potentially represented entry points for therapeutic interventions via protein stability regulation.
Properties of short-lived proteins under translational inhibition
We next examined if certain protein properties were associated with short-lived proteins. Previous research has studied the relationships between several protein properties and protein turnover rates on a global level, including protein abundance and membership in protein complexes (Cambridge et al., 2011; Mathieson et al., 2018). Besides extending the investigation of these properties into the subset of short-lived proteins, we also explored additional properties, including mRNA abundance and protein expression correlation and evolutionary age.
As expected, we observed significantly fewer peptides for short-lived proteins (Fig. S5A). We did not observe a difference in protein length between short-lived and non-short-lived proteins (Fig. S5B). After normalizing the number of identified peptides by protein length and the fraction of protein intensities at 0 hr, we still observed a significantly lower abundance for short-lived proteins (Fig. 5A). We further retrieved protein copy numbers in U2OS cells from the literature (Beck et al., 2011) and found the same result (Fig. 5B). It is generally recognized that rapidly degraded proteins are largely regulatory and low abundant. A previous protein half-life analysis with pulse-chase metabolic labeling revealed a weak negative association been protein abundance and turnover rates at the proteome level (Cambridge et al., 2011). The conclusion held true in our focused study on short-lived proteins.
Figure 5. Evaluating the properties of short-lived proteins under translational arrest.

(A) Short-lived proteins were less abundant than other proteins. Scaled protein abundance is TMTpro signal-to-noise (SN) normalized by protein length and the fraction of TMTpro SN at 0 hr among summed TMTpro SN. (B) Short-lived proteins displayed lower protein copies/cell than other proteins in U2OS cells. (C) Short-lived proteins showed higher instability indices, higher aliphatic indices and lower melting temperatures than non-short-lived proteins. The list of short-lived proteins contains proteins that were short-lived in at least one cell line. (D) Short-lived proteins tended to reside in smaller protein complexes. (E) ORC1 and ORC6 are loosely attached subunits of the origin recognition complex with shorter half-lives than other subunits. (F) TSEN34 of tRNA splicing endonuclease showed a higher degradation rate than other subunits. (G) Spliceosome components that are assembled or disassembled dynamically between different spliceosome conformational states (transitory subunits) presented greater losses at 8 hr, especially in HCT116 and RPE1 cells. Numbers in the plot are median log2 fold changes. (H) Short-lived proteins had fewer interacting proteins than other proteins in HEK293T cells. Numbers in the plot are median in-degree interactions. (I) Rapidly degraded proteins showed lower mRNA expression and protein abundance correlation across human tissues compared to stable proteins. (J) Short-lived proteins were evolutionarily younger than stable proteins. Numbers in parentheses are bin size. Rank 1 and 8 represent the least and the most stable proteins, respectively in panel I and J. Statistical significance was determined by unpaired Wilcoxon test (two-sided) in panel A-D, G and H. Data are presented as box plots (center line: median; box limits: the first and third quartiles; whiskers: 1.5x interquartile range) in panel A, B, D and G-I. See also Figure S5.
The instability and aliphatic indices calculated from protein primary sequences estimate in vitro protein stabilities (Guruprasad et al., 1990) and protein thermal stabilities (Ikai, 1980), respectively. High instability indices indicate low stability. We found that short-lived proteins tended to have higher instability indices (Fig. 5C, Fig. S5C). The instability index was originally developed by studying the correlation between the stability of a protein in vitro and its dipeptide composition (Guruprasad et al., 1990). Our results suggested that this index could also indicate protein stability in vivo. Low aliphatic indices mean relatively low densities of nonpolar and hydrophobic amino acids (alanine, valine, isoleucine, and leucine) and low melting temperatures (Ikai, 1980). Short-lived proteins had lower aliphatic indexes, and thus presumably low melting temperatures. We further mapped the protein stability data to human protein melting temperature data (Jarzab et al., 2020), and found that short-lived proteins did show lower melting temperatures (Fig. 5C, Fig. S5C).
We next mapped the CORUM database (Giurgiu et al., 2019) onto the list of short-lived proteins under translation inhibition. The largest protein complex was kept if a protein belonged to multiple protein complexes. Short-lived proteins were found preferentially in small complexes (Fig. 5D). Moreover, the half-lives of the members of the same small protein complex varied, and most small protein complexes contained only a minor fraction of short-lived proteins (Fig. S5D). For example, the origin recognition complex (ORC complex) and the tRNA splicing endonuclease have six and five subunits, respectively, but they both harbored just one short-lived component (Fig. 5E, F). Interestingly, ORC1 and ORC6 are loosely attached subunits of the ORC complex (Ghosh et al., 2011; Radichev et al., 2006; Ranjan and Gossen, 2006). They both displayed faster degradation than other subunits. This observation agreed with the abovementioned notion that peripheral subunits generally turned over more rapidly than core subunits of E3 ubiquitin ligase complexes (Fig. 4). Some subunits have been suggested to undergo targeted degradation, controlling the activity of the protein complex and allowing for fast switching between complex assembly and disassembly (Hinkson and Elias, 2011). Many protein complexes, especially small complexes, had subunits of differing stability in our results, which might implicate them as regulatory factors within the complexes. TSEN34, which comprises the active site of the tRNA splicing endonuclease complex (Paushkin et al., 2004), showed a shorter half-life than other subunits (Fig. 5F), suggesting that this catalytic protein may contribute to complex activity regulation via rapid degradation.
Previous studies have shown that members of the long-lived proteasome and nuclear pore complex (NPC) share highly similar turnover rates (Cambridge et al., 2011; Martin-Perez and Villen, 2017; Mathieson et al., 2018). Yet, protein half-lives also can vary within individual complexes. Peripheral subunits of the proteasome and NPC, for example, have faster turnover rates than core components (Mathieson et al., 2018). Although our study was not designed to accurately determine degradation rates for medium- or long-lived protein complexes, we still observed that transitory subunits in the spliceosome (Zhan et al., 2018; Zhang et al., 2018) demonstrated more extensive degradation than core components (Fig. 5G). Mapping the results from cycloheximide-treated HEK293T cells to the global protein-protein interaction data in HEK293T cells (Huttlin et al., 2021) demonstrated a subtle trend that short-lived proteins had fewer interacting proteins than other proteins (Fig. 5H). The trend was consistent with the observation that short-lived proteins tended to reside in smaller protein complexes (Fig. 5D). However, the much smaller effect size suggested that short-lived proteins still maintained comparable numbers of interacting proteins.
Protein degradation partially accounts for the poor correlations between mRNA expression and protein abundance (Liu et al., 2016). We retrieved the correlations between mRNA expression and protein abundance across human tissues (Jiang et al., 2020; Wang et al., 2019) and multiple human cancer cell lines (Nusinow et al., 2020). As expected, we observed a trend that rapidly degraded proteins had lower correlations (Rank 1), while stable proteins had relatively higher correlations (Rank 8) (Fig. 5I, Fig. S5E). Finally, we analyzed the evolutionary ages (Liebeskind et al., 2016) of short-lived proteins. Most of the quantified proteins dated to eukaryotic, eumetazoan, and vertebrate ages (Fig. 5J). Compared to the most stable proteins (Rank 8), the fraction with the least stable proteins (Rank1) increased as protein evolutionary age decreased (Fig. 5J). Short-lived proteins tended to be evolutionarily younger than stable proteins, suggesting that mechanisms underlying rapid protein degradation have evolved to support more precise control over cellular proteome dynamics.
Differential stability of short-lived proteins across cell lines
Although a global view of short-lived proteins reflected common patterns of degradation in all cell lines (Fig. S2B), we found that 103 short-lived proteins showed different half-lives between cell lines using strict criteria (Fig. 6A, Table S3, see Methods for details), and the cell line in which protein half-lives were significantly shorter tended to have overall lower protein abundance (Fig. S6).
Figure 6. Cell line specific differences in the stability of short-lived proteins.

(A) Numbers of short-lived proteins with differential stability between any two cell lines. (B) HEK293T expresses large T-antigen, which binds TP53 and RB1 proteins and renders them non-functional. TP53 is stabilized by its interaction with large T-antigen. (C) Non-functional RB1 results in higher expression of CDKN2A, which competitively binds and stabilizes CDK4. (D) The fast degradation of TP53 in U2OS and HCT116, and the stabilization of TP53 by interaction with large T-antigen in HEK293T were captured by the mass spectrometry data. Values in parentheses are half-lives. TP53 was not identified in RPE1 cells, presumably due to its very low abundance. (E) TP53 showed higher protein abundance in HEK293T due to stabilization by large T-antigen. (F) Western blotting shows that TP53 was long-lived only in HEK293T cells. Cyclin D1 is a known short-lived protein (positive control). (G) The stabilization of CDK4 uniquely in HEK293T cells. (H) CDKN2A and CDK4 have higher overall protein expression levels in HEK293T cells. Relative protein abundance is TMTpro signal-to-noise normalized to the mean of the first time point in panel D and G. Relative protein abundance is TMTpro signal-to-noise normalized to the maximum in panel E and H. Note that relative protein abundance is comparable for the same protein across different cell lines, and not comparable for different proteins in the same cell line in panel H. See also Figure S6 and Table S3.
As an example, a difference among the four cell lines is that HEK293T uniquely express the large T-antigen, which interacts with TP53 and RB1, rendering them dysfunctional (DeCaprio et al., 1988; Deppert and Haug, 1986; Deppert et al., 1987; Hein et al., 2009; Oren et al., 1981) (Fig. 6B, C). TP53 has a short half-life, but it is stabilized by the interaction with large T-antigen (Deppert and Haug, 1986; Deppert et al., 1987; Hein et al., 2009; Oren et al., 1981) (Fig. 6B). We observed that TP53 displayed a significantly longer half-life in HEK293T cells than U2OS and HCT116 cells (Fig. 6D). TP53 also presented higher protein abundance in HEK293T cells (Li et al., 2020) (Fig. 6E). Western blotting confirmed the mass spectrometry data (Fig. 6F). This observation agreed with a previous result that TP53 was more stable in HEK293T than U2OS cells as measured by flow cytometry and microarray (Yen et al., 2008). As another large T-antigen-dependent event, stabilized CDK4 was also found to be HEK293T specific (Fig. 6 B, C, and G). The loss of RB1 function in HEK293T cells leads to high CDKN2A expression (Li et al., 2020; Li et al., 1994) (Fig. 6H), which binds and stabilizes CDK4 (Parry et al., 1995; Parry et al., 1999) (Fig. 6G).
The cell lines analyzed rely on different mechanisms for telomere maintenance. U2OS is an alternative lengthening of telomeres (ALT)-positive cell line (Heaphy et al., 2011; Lovejoy et al., 2012), while the other three are telomerase-positive cell lines (Bodnar et al., 1998; Dilley et al., 2016; Lovejoy et al., 2012) (Fig. 7A). About 85% of human cancers contain telomerase-positive cells and achieve telomere maintenance through telomerase. Most of the remaining 15% maintain telomeres by ALT (Cesare and Reddel, 2010). The loss of ATRX, a SWI/SNF-like chromatin remodeling protein, is a hallmark of ALT (Dyer et al., 2017). It is widely recognized that ATRX protein expression is missing in U2OS cells (Heaphy et al., 2011; Newhart et al., 2012; Yost et al., 2019; Zeineldin et al., 2020). U2OS cells express mutated ATRX mRNA, yet the mutation has been reported as both deletion of exons 2–19 and 2–30 (Heaphy et al., 2011; Lovejoy et al., 2012). Surprisingly, we still detected the ATRX protein with a short half-life (2.1 hr) and a lower expression in U2OS cells (Fig. 7B, C). We first verified that exon 2–19 of ATRX mRNA were missing in U2OS cells using the public RNA-seq data (Ghandi et al., 2019) (Fig. S7A, B). Literature review revealed a 60–80 kDa band in anti-ATRX Western blots in U2OS cells (Newhart et al., 2012; Yost et al., 2019; Zeineldin et al., 2020), which did not match any known ATRX isoforms in the Uniprot database. Subsequent siRNA experiments confirmed the existence of the truncated ATRX (Fig. 7D). Four distinct siRNAs targeting the remaining mRNA sequence abolished the detection of the truncated ATRX, while scramble siRNAs and the siRNA targeting the deletion did not (Fig. 7D). Independent repeat experiments confirmed that the truncated ATRX in U2OS was short-lived, while the full length ATRX in other three cell lines was not (Fig. 7E). An artificially truncated version of ATRX covered by the remaining sequence still interacts with MeCP2 (Nan et al., 2007). It is unknown if the truncated version of ATRX is functional in U2OS cells. In conclusion, a previously unrecognized truncated version of ATRX with a short half-life was identified in U2OS cells. These findings raise questions about established models of telomere maintenance based on expression of ATRX isoforms.
Figure 7. Examples of truncated protein forms unique to one cell line with concomitant short half-lives.

(A) U2OS is an alternative lengthening of telomeres-positive cell line with a mutated ATRX gene. The other three are telomerase-positive cell lines carrying the wild type ATRX gene. (B) ATRX in U2OS cells displayed a significantly shorter half-life than other cell lines. (C) ATRX expression levels in U2OS cells are lower than other cell lines. Only ATRX peptides that were identified in U2OS cells were used for quantification. (D) siRNA experiments verified that U2OS cells expressed a truncated form of ATRX protein. siRNAs (siRNA-1, siRNA-2, siRNA-3, and siRNA-5) targeting the remaining exons abolished the detection of the truncated ATRX, while scramble siRNAs and siRNA (siRNA-4) targeting a deleted exon did not. (E) Western blotting results showed that the truncated form of ATRX was short-lived in U2OS cells, while full length ATRX was not in other cell lines. (F) GMDS has a significantly shorter half-life (0.9 hr) in HCT116 cells. (G) GMDS is expressed at a much lower level in HCT116 cells. Only GMDS peptides that were identified in HCT116 cells were used for quantification. (H) Evidence of GMDS truncation based on peptide sequencing. No GMDS peptides were identified in the deletion region in HCT116 cells in this study or in another large unpublished HCT116 dataset from our lab. However, GMDS peptides were identified throughout the full primary sequence at an equivalent frequency in other cell lines. #PSM means the number of peptide-spectrum-matches (how many times the peptide was detected). Blue blocks indicate the identified peptides and their positions in the full primary sequence. Light gray in the same row indicates no peptide detected in the corresponding position. Relative protein abundance is TMTpro signal-to-noise normalized to the mean of the first time point in panel B and F. Relative protein abundance is TMTpro signal-to-noise normalized to the maximum in panel C and G. See also Figure S7.
The protein GMDS (GDP-mannose 4,6 dehydratase) also displayed a significantly shorter half-life in HCT116 cells than other cell lines. HCT116 cells have a deleted GMDS transcript, are unable to synthesize GDP-fucose, and demonstrate an almost complete absence of fucosylation. Wild type GMDS can restore the cellular fucosylation in HCT116 cells (Moriwaki et al., 2011; Nakayama et al., 2013). No mutant GMDS protein was detected with Western blotting (Moriwaki et al., 2009). However, we identified two unique GMDS peptides in HCT116 cells (Fig. 7F). GMDS displayed a short half-life (0.9 hr) in HCT116 and was relatively long-lived in other cell lines (Fig. 7F). HCT116 also had the lowest expression level of GMDS (Fig. 7G). We then examined all the identified peptides for GMDS in all four cell lines and in another large-scale unpublished HCT116 dataset in our lab. We identified multiple unique peptides in HCT116 cells, yet none fell within the deleted region. However, we identified multiple unique peptides throughout the full primary sequence at an equivalent frequency in other cell lines (Fig. 7H). This finding suggested that the absence of peptides in the deleted region in HCT116 cells was not due to the stochastic precursor selection in bottom-up proteomics. Further inspection of the MS2 spectra, reporter ion quantification quality, and retention times also confirmed the identification and the quantification of the truncated GMDS protein (Fig. S7C, D). This deletion likely contributes to the short half-life of GMDS in HCT116 cells.
Comparison with pulse-chase metabolic labeling studies
We chose to combine TMTpro-based protein quantification and cycloheximide treatment to characterize short-lived proteins under translational arrest in this work. We note that MS-based pulse-chase metabolic labeling has also been broadly used to determine 50% turnover times or half-lives for proteins, especially medium- and long-lived proteins (Martin-Perez and Villen, 2017; Ross et al., 2020). Some short-lived proteins could be detected with short labeling times. However, many factors reduced proteome coverage such that between zero and ~200 short-lived proteins were detected. An overview of these studies is presented in Table S4 and Fig. S8 (Boisvert et al., 2012; Cambridge et al., 2011; Doherty et al., 2009; Jovanovic et al., 2015; Mathieson et al., 2018; McShane et al., 2016; Schwanhausser et al., 2011). Due to stress responses and secondary effects, half-lives determined under translational inhibition may not always represent half-lives determined under more steady-state conditions. We thus compared the half-lives of the short-lived proteins in RPE1 cells in this work to those using RPE1 and pulse-chase metabolic labeling in the literature (McShane et al., 2016). Over 85% of the overlapping proteins presented consistently short half-lives in the literature (Fig. S8A) (McShane et al., 2016). Thus, the short-lived proteins under short-term translational arrest still reflected those under more steady-state conditions to a large extent.
Discussion
We achieved proteome-wide coverage, presenting a dataset of over 11,000 proteins and their 8-hr degradation kinetics following translational arrest in four cell lines whose distinct biological profiles afford complementary views of the human proteome. We identified ~500 short-lived proteins with half-lives between 20 min and 8 hr under translational arrest. For an additional ~1,400 proteins of intermediate stability, half-lives ranging from 8 hr to 34 hr (>15% protein abundance loss at 8 hr) were determined with strong coefficients of determination (R2 > 0.8) (Table S2, Fig. S2F). Caution is advised when interpreting half-lives for the proteins with intermediate stability, as cycloheximide may sufficiently disrupt cellular physiology to result in half-life miscalculation for medium- and long-lived proteins (Darvishi and Woldemichael, 2016; Watanabe-Asano et al., 2014). In addition, quantitative accuracy is essential for protein half-life calculation. Small deviations in the measured protein abundance can have a pronounced effect on half-life measurement. For example, a 50% decrease in protein abundance at 8 hr results in a half-life measurement of 8 hr, while decreases of 60% and 75% equate to half-lives of 6.1 hr and 4 hr, respectively. We combined FAIMS, RTS-SPS-MS3, and TMTpro reagents to improve accuracy and minimize interference. The quantitative accuracy of the datasets should be considered when comparing half-lives between cell lines or studies. Minor differences in half-lives may not be true biological differences and a stringent effect size cutoff is recommended in such comparisons.
Evaluation of differential protein stability under varying conditions can offer insights into the regulatory mechanisms that fine tune proteome remodeling through protein degradation. For example, we observed that TP53 was stabilized in HEK293T cells, but not in other cell lines lacking the large T-antigen (Fig. 6). We also observed how intrinsic protein factors can impact differential stability among protein isoforms, such as truncated versions of ATRX and GMDS that were rapidly degraded and uniquely short-lived in U2OS and HCT116 cells, respectively (Fig. 7). Intrinsic and extrinsic factors that affect protein stability can also be evaluated using this experimental model and in theory converge at the level of interaction between an E3 ligase and a substrate protein of interest.
Pulse-chase metabolic labeling using heavy amino acids has also been used to determine 50% turnover time or half-lives for proteins (Ross et al., 2020). While this method is preferred for measuring protein with medium and long half- lives, there are caveats to its use for short-lived proteins. i) Because sample multiplexing is not used, proteins are not always identified in replicates or time points, and missing values are present. ii) Short-lived proteins are often found at lower abundance levels where detection can be made more difficult by the presence of both heavy and light peaks for every peptide. iii) Early time points are often unreliable since after switching to the media with heavy amino acids the pool of existing charged tRNAs must be depleted and recharged entirely with heavy amino acids. iv) Specifically for short-lived proteins, these studies measure the turnover rates only for newly synthesized proteins where the heavy amino acid has been incorporated just hours previously. If the kinetics for degradation of the newly-synthesized form of a protein are different from those of the mature form (McShane et al., 2016; Schubert et al., 2000), then inaccurate half-lives will be determined. These issues can be largely avoided through translational arrest with cycloheximide. The use of sample multiplexing eliminates missing values within a plex while maintaining proteome-scale depth. In addition, no tRNA re-charging is required. Finally, the half-life of the entire population of a protein (not just the newly synthesized) under translational arrest is captured. Note that this may not be a major issue since comparing our dataset to several others from the literature (Fig. S8 and Table S4) showed that metabolic labeling specifically of newly synthesized proteins using azidohomoalanine and click chemistry returned the best consistency with our work (Fig. S8).
In conclusion, we have employed state of the art isobaric labeling technology in conjunction with translational arrest to identify short-lived proteins within the proteomes of four contrasting human cell lines. These short-lived proteins span myriad cellular processes whose activities they often regulate. While there is some possibility that cycloheximide may have perturbed the half-lives of some proteins, the short half-lives we have observed for these short-lived proteins require mechanisms that govern their rapid degradation. Each of the >1000 short-lived proteins identified in this resource thus has the potential to be targeted for rapid destruction. An understanding of the underlying degradation strategies represents an opportunity to target these proteins with small molecules, causing their degradation for therapeutic interventions.
Limitations of the study
The short-lived proteins and their half-lives were determined under translational inhibition in this work. Although metabolic labeling of proteins using azidohomoalanine returned the best consistency with our work, there is still a likelihood that certain half-lives only occur under cycloheximide treatment. We showed evidence that truncated versions of ATRX and GMDS were rapidly degraded and uniquely short-lived in U2OS and HCT116 cells, respectively. The mechanisms, however, remain unclear and can be further investigated, which may contribute to the understanding of telomere maintenance and protein fucosylation in cancers.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Steven P. Gygi (steven_gygi@hms.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The mass spectrometry data have been deposited in the ProteomeXchange Consortium with the dataset identifier PXD024513.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
U2OS (female human osteosarcoma cells), HEK293T (female human embryonic kidney cells), HCT116 (male human colorectal carcinoma cells), and RPE1 (female human retinal pigment epithelial cells) were purchased from ATCC. Cells were maintained in DMEM with high glucose/pyruvate (Invitrogen) supplemented with 10% FBS (Hyclone). All cell lines were maintained in a 5% CO2 incubator at 37°C.
METHOD DETAILS
Cycloheximide treatment
For cycloheximide treatment, cells were grown under normal conditions to approximately 80% confluence in biological replicate 10 cm (U2OS and RPE1) or 6 cm (HEK293T and HCT116) plates, followed by 0 hr (4 replicates), 1 hr (2 replicates), 2 hr (2 replicates), 4 hr (2 replicates), 6 hr (2 replicates), and 8 hr (4 replicates) treatment with 50 μg/ml cycloheximide. Cells were washed three times with cold PBS, then harvested and lysed by scraping into 8 M urea lysis buffer (8 M urea, 200 mM EPPS, pH 8.5) with protease inhibitors (Complete Mini, Roche). Cell extracts were syringe-lysed and sonicated. Protein concentration was determined with BCA assays for subsequent LC-MS sample preparation.
ATRX siRNA experiment
Two siRNA universal negative controls and five siRNA targeting ATRX mRNA were purchased from Sigma-Aldrich (see key resource table for details). siRNA transfections were carried out with Lipofectamine RNAiMAX (13778–150, Invitrogen) following the standard transfection protocol. Cells were collected for Western blotting 72 hours after transfection.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-ATRX antibody produced in rabbit | Sigma-Aldrich | HPA001906 |
| Monoclonal Anti-Vinculin antibody produced in mouse | Sigma-Aldrich | V9131 |
| p53 Antibody | Cell Signaling Technology | #9282 |
| Cyclin D1 (92G2) Rabbit mAb | Cell Signaling Technology | #2978 |
| Monoclonal ANTI-FLAG® M2-Peroxidase (HRP) antibody produced in mouse | Sigma-Aldrich | A8592 |
| Anti-β-Actin Antibody (C4) | Santa Cruz Biotechnology | sc-47778 |
| CUL3 Antibody | Cell Signaling Technology | #2759 |
| KLHL12 (2G2) Mouse mAb | Cell Signaling Technology | #9406 |
| CUL1 Antibody | Cell Signaling Technology | #4995 |
| Cyclin F (D9K2U) Rabbit mAb | Cell Signaling Technology | #81045 |
| APC1 (D1E9D) Rabbit mAb | Cell Signaling Technology | #13329 |
| APC8 (D5O2D) Rabbit mAb | Cell Signaling Technology | #15100 |
| APC6 (D8D8) Rabbit mAb | Cell Signaling Technology | #9499 |
| CDC20 (D6C2Q) Rabbit mAb | Cell Signaling Technology | #14866 |
| APC11 (D1E7Q) Rabbit mAb | Cell Signaling Technology | #14090 |
| ITCH (D8Q6D) Rabbit mAb | Cell Signaling Technology | #12117 |
| PJA2 Antibody | Cell Signaling Technology | #40180 |
| LRSAM1 (D1O5S) Rabbit mAb | Cell Signaling Technology | #28405 |
| RCHY1 Antibody | Cell Signaling Technology | #5754 |
| Critical commercial assays | ||
| TMTpro™ 16plex Label Reagent Set | ThermoFisher Scientific | A44520 |
| Deposited data | ||
| The mass spectrometry data have been deposited in the ProteomeXchange Consortium. | This paper | PXD024513 |
| Experimental models: Cell lines | ||
| U2OS | ATCC | HTB-96 |
| HEK293T | ATCC | CRL-3216 |
| HCT116 | ATCC | CCL-247 |
| RPE1 | ATCC | CRL-4000 |
| Oligonucleotides | ||
| siRNA Universal Negative Control #1 (siNC_1) | Sigma-Aldrich | SIC001 |
| siRNA Universal Negative Control #2 (siNC_2) | Sigma-Aldrich | SIC002 |
| ATRX, siRNA-1 (5’-3’) GGAUUCAACCUCUUGAGGA[dT][dT] UCCUCAAGAGGUUGAAUCC[dT][dT] | Sigma-Aldrich | SASI_Hs01_00081483 |
| ATRX, siRNA-2 (5’-3’) CUACAAUGAUGUAUUGACA[dT][dT] UGUCAAUACAUCAUUGUAG[dT][dT] | Sigma-Aldrich | SASI_ Hs02_00302645 |
| ATRX, siRNA-3 (5’-3’) CAGAUUCUACCAUGGUAGA[dT][dT] UCUACCAUGGUAGAAUCUG[dT][dT] | Sigma-Aldrich | SASI_ Hs02_00302642 |
| ATRX, siRNA-4 (5’-3’) CAAAGUGGCUCAUCAUCAU[dT][dT] AUGAUGAUGAGCCACUUUG[dT][dT] | Sigma-Aldrich | SASI_Hs02_00302643 |
| ATRX, siRNA-5 (5’-3’) CAUUGACUAUUACCGUUUA[dT][dT] UAAACGGUAAUAGUCAAUG[dT][dT] | Sigma-Aldrich | SASI_Hs01_00081480 |
| Software and algorithms | ||
| R package ClusterProfiler 3.16.1 | (Yu et al., 2012) | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| R package Limma 3.40.6 | (Ritchie et al., 2015) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| R package Peptides 2.4.2 | (Daniel Osorio, 2015) | https://cran.r-project.org/web/packages/Peptides/index.html |
| R 4.0.2 | N/A | https://www.r-project.org/ |
Western blotting
For the detection of ATRX (Fig. 7D, E), samples were loaded into NuPAGE 3–8% Tris-Acetate Gel (EA03785BOX, Invitrogen). HiMark pre-stained protein standard (LC5699, Invitrogen) was loaded to indicate the molecular weights of target proteins. Tris-Acetate SDS running buffer (LA0041, Life Technologies) was used in electrophoresis. Proteins were then transferred to 0.45 μm nitrocellulose membranes (#1620115, BioRad) in NuPAGE transfer buffer (NP0006–1, Life Technologies). For the detection of TP53, Cyclin D1 and Vinculin (Fig. 6F), samples were loaded onto a 10% SDS-PAGE gel and separated with in Tris-Glycine SDS-PAGE running buffer. PageRuler Plus Prestained Protein Ladder (#26619, Thermo Scientific) was used. After electrophoresis, proteins were transferred to 0.45 μm nitrocellulose membranes (#1620115, BioRad) in Towbin transfer buffer. Membranes were blocked in 5% BSA and incubated with primary antibodies. Membranes were then washed with TBST with 0.5% Tween 20 and incubated with IRDye secondary antibodies (926–32211 or 925–68070, LI-COR). Membranes were scanned, and images were collected with ODYSSEY CLx (LI-COR). Membranes were stripped with ReBlot Plus Strong Antibody Stripping Solution (#2504, Millipore) for detection of other targets when needed. Anti-ATRX and anti-Vinculin were from Sigma-Aldrich (HPA001906, V9131). Anti-p53 and anti-Cyclin D1 were from Cell Signaling Technology (#9282, #2978).
For the immunoblot analysis in Fig. S4C, U2OS cells were transduced with c-terminal FLAG-HA lentiviral constructs harboring the cDNA of DCAF15, KLHL9, or FBXO5. Cells were selected with 1 μg/ml puromycin, expanded to 60 mm plates and treated with 50 μg/ml cycloheximide. Cells were harvested in 8 M urea lysis buffer (8 M urea, 200 mM EPPS, pH 8.5) followed by flash frozen. Frozen lysates were thawed, cleared by centrifugation, and subjected to BCA assay to determine protein concentration. Fifty μg of protein per sample was separated by SDS-PAGE using 4–20% acrylamide gradient gels (BioRad). Proteins were transferred to PVDF membranes and immunoblotted with anti-FLAG (M2)-HRP (A8592, Sigma-Aldrich) and anti-β-Actin (sc-47778, Santa Cruz Biotechnology). For the immunoblot analysis in Fig. 4C and Fig. S4D, Western blotting experiments were carried out as described above. Anti-CUL3 (#2759), anti-KLHL12 (#9406), anti-CUL1 (#4995), anti-CCNF (#81045), anti-ANPAC1 (#13329), anti-CDC23 (#15100), anti-CDC16 (#9499), anti-CDC20 (#14866), anti-ANAPC11 (#14090), anti-ITCH (#12117), anti-PJA2 (#40180), anti-LRSAM1 (#28405) and anti-RCHY1 (#5754) were from Cell Signaling Technology. Western blotting membranes were cut for multiple antibodies, thus the blots for the loading control β-Actin were the same in two panels in Fig. 4C and Fig. S4D.
Mass spectrometry sample preparation
Samples were reduced with 5 mM TCEP for 30 min, alkylated with 10 mM iodoacetamide for 30 min in the dark and then quenched with 10 mM DTT for 15 min at room temperature. Streamlined (SL)-TMT protocol was used for sample preparation (Navarrete-Perea et al., 2018). Briefly, 100 μg of protein from whole cell lysate were chloroform-methanol precipitated and reconstituted in 100 μl of 200 mM EPPS (pH 8.5). The samples were digested by Lys-C overnight at room temperature and then trypsin for 6 hr at 37°C, both at a 1:100 protease-to-protein ratio.
Peptides corresponding to 40 μg of protein were then labeled with TMTpro 16-plex reagents (Li et al., 2020) in the presence of 29% acetonitrile. Samples from the same cell line were allocated in the same TMT group (Labels used: 126–0 hr; 127n-0 hr; 127c-0 hr; 128n-0 hr; 128c-1 hr; 129n-1 hr; 129c-2 hr; 130n-2 hr; 130c-4 hr; 131n-4 hr; 131c-6 hr; 132n-6 hr; 132c-8 hr; 133n-8 hr; 133c-8 hr; 134n-8 hr). Samples were labeled for 90 min at room temperature. Two microliters of each sample were pooled, desalted and analyzed on a Q Exactive (ThermoFisher Scientific) to check labeling efficiency. After labeling efficiency check, samples were quenched by adding 5% hydroxylamine and pooled. Pooled samples were then desalted with 100 mg Sep-Pak solid-phase extraction columns, and fractionated with high-pH reversed-phase chromatography. Fractions were collected in a 96-well plate and combined for a total of 24 fractions prior to desalting and subsequent LC-MS/MS analysis.
Mass spectrometry analysis
Data were collected on an Orbitrap Eclipse mass spectrometer (ThermoFisher Scientific) coupled to a Proxeon EASY-nLC 1200 LC pump (ThermoFisher Scientific). Peptides were separated on a 35 cm column (i.d. 100 μm, Accucore, 2.6 μm, 150 Å) packed in-house using a 90 min gradient at 550 nl/min. High-field asymmetric-waveform ion mobility spectrometry (FAIMS) was enabled during data acquisition with compensation voltages (CVs) set as −40V, −60V, and −80V (Schweppe et al., 2019). MS1 data were collected using the Orbitrap (120,000 resolution; maximum injection time 50 ms; AGC 4×105). Determined charge states between 2 and 5 were required for sequencing, and a 120 s dynamic exclusion window was used. MS2 scans were performed in the ion trap with CID fragmentation (isolation window 0.5 Da; Turbo; NCE 35%; maximum injection time 35 ms; AGC 1×104). An on-line real-time search algorithm (Orbiter) was used to trigger MS3 scans for quantification (Schweppe et al., 2020). MS3 scans were collected in the Orbitrap using a resolution of 50,000, NCE of 45%, maximum injection time of 150 ms and AGC of 1.5×105. The close out was set at two peptides per protein per fraction (Schweppe et al., 2020).
Mass spectrometry data analysis
Raw files were converted to mzXML and monoisotopic peaks were re-assigned using Monocle (Rad et al., 2021). Database searching included all human entries from Uniprot (downloaded on 02/15/2020). The database was concatenated with one composed of all protein sequences in reverse. Sequences of common contaminant proteins (e.g. trypsin, keratins, etc.) were appended as well. Searches were performed using the Comet search algorithm and a 50 ppm precursor ion tolerance and 0.9 Da product ion tolerance. TMTpro on lysine residues and peptide N termini (+304.2071 Da) and carbamidomethylation of cysteine residues (+57.0215 Da) were set as static modifications (except when testing for labeling efficiency, in which TMTpro modification was set to variable), while oxidation of methionine residues (+15.9949 Da) was set as a variable modification. MS1 and MS3 masses were re-calibrated as necessary.
Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR) (Elias and Gygi, 2007). PSM filtering was performed using linear discriminant analysis as described previously (Huttlin et al., 2010), while considering the following parameters: XCorr, ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy. Each run was filtered separately. Protein-level FDR was subsequently estimated. For each protein across all samples from each cell line, the posterior probabilities reported by the linear discriminant analysis (LDA) model for each peptide were multiplied to give a protein-level probability estimate. Using the Picked FDR method (Savitski et al., 2015), proteins were filtered to the target 1% FDR level, again using the target-decoy approach to model false positives.
For reporter ion quantification, a 0.003 Da window around the theoretical m/z of each reporter ion was scanned, and the most intense m/z was used. Reporter ion intensities were adjusted to correct for the isotopic impurities of the different TMTpro reagents according to manufacturer specifications. Briefly, the correction factors are applied to the raw peak intensity before calculating signal-to-noise. The correction is done by arranging a system of linear equations where the unknowns are the corrected channel intensities, their coefficients are the correction factors, and the constant terms are the observed peak intensities. A solution is found by solving the system using matrix operations. Peptides were filtered to include only those peptides with a summed signal-to-noise (SN) ≥ 160 across 16 TMTpro channels. For each protein, the filtered peptide TMTpro SN values were summed to create protein quantification values. To control for different total protein loading within a TMTpro experiment, the summed protein quantities of each channel were adjusted to be equal within the experiment.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bioinformatics analysis
TMTpro SN values were scaled to the mean of the first time point (0 hr) so that the average scaled TMTpro SN values were 1.0 at the first time point. Assuming that long-lived proteins did not change over 8 hr cycloheximide treatment, we further normalized TMTpro SN values such that the median of T-complex proteins and Lamin B proteins were equal across time points (Mathieson et al., 2018; McShane et al., 2016; Toyama et al., 2013). Overall, the normalization factors by long-lived proteins were small and fell within the range of 0.9–1.1 (Fig. S1B). We noted that the normalization by T-complex proteins and Lamin B proteins might not be sufficient. We chose to use these proteins because we wanted to ensure the list of short-lived proteins was not inflated by excessive normalization.
Protein half-lives were determined under the assumption that protein degradation follows first order decay kinetics (Belle et al., 2006) with slight modifications (Fig. S1C). The protein abundance of some very short-lived proteins dropped rapidly under translation inhibition (Fig. S1D). The protein abundance could be below the detection limit at the last few time points and the low intensities detected were just basal level signals. This rendered the conventional way of calculating protein half-lives unsuitable for many very short-lived proteins. Thus, we included an asymptote parameter (Asym, the bottom plateau) in the model for proteins of greater than 30% losses at 8 hr. For these proteins, a non-linear model was fitted to the time course of scaled TMTpro SN with R function nls (Fig. S1C). The time point when f(t) was 50% of the R0 (the response when t=0) was designated as the half-life (Fig. S1C). If the coefficient of determination (R2) of the non-linear model was greater than 0.8 and half-live shorter than 8 hr (greater than 50% loss at the last time point), the R2 and half-life were returned. Otherwise, a linear model was fitted to the time course of the natural logarithm transformed scaled TMTpro SN with R function lm. This linear model fitting is the conventional way of calculating protein half-lives. It was essentially the same as the non-linear model fitting except that the linear model did not have the asymptote parameter. The time point when f(t) was 50% of the R0 was designated as the half-life and R2 of the linear model was returned. Proteins with half-lives shorter than 8 hr and R2 greater than 0.8 were defined as short-lived proteins. An extra manual inspection step was carried out for proteins that failed these criteria but showed greater than 50% loss at the last time point (t8). For these proteins, a linear model was fitted to the first two time points (0 hr and 1 hr) of natural logarithm transformed scaled TMTpro SN and half-lives were then derived from the slope (rate constant). Finally, non-linear model fitting captured the majority of the short-lived protein events (1,862 events). Linear model fitting and manual inspection recovered another 39 events and 13 events, respectively (Fig. S1D).
In the removal of extracellular matrix proteins, proteins in the following Gene Ontology categories were excluded (GO: 0062023, collagen-containing extracellular matrix; GO: 0031012, extracellular matrix; GO: 0005604, basement membrane; GO: 0005581, collagen trimer; GO: 0098644, complex of collagen trimers; GO: 0005583, fibrillar collagen trimer; GO: 0098643, banded collagen fibril; GO: 0043256, laminin complex; GO: 0005588, collagen type V trimer; GO: 0001527, microfibril). In the end, 130, 144, 176, and 189 ECM proteins were removed from the ~9,000 quantified proteins in each cell line, and 16, 19, 42, and 47 proteins were excluded from the lists of short-lived proteins in each cell line.
Protein domain (InterPro) enrichment analysis (Fig. 3C) was performed with DAVID (6.8) (Huang da et al., 2009). Other Gene Ontology enrichment analyses (Fig. 3D, Fig. S3) were performed with R package ClusterProfiler (3.16.1) (Yu et al., 2012). With the goal to assemble as comprehensive a list as possible, we used proteins that were short-lived in at least one cell line (a combined list of short-lived proteins in all cell lines) as the foreground. All quantified proteins in the four cell lines were used as the background. Adjusted p-values (Benjamini-Hochberg) were filtered at 0.01 in GO enrichment analyses unless otherwise stated.
Gene set enrichment analysis (GSEA) (Fig. 3B) was performed with R package ClusterProfiler (3.16.1) (Yu et al., 2012). The gene set of proteins containing degrons was from (Meszaros et al., 2017). The log2 fold changes (8 hr vs 0 hr) were used in GSEA (Fig. 3B) and other analyses where log2 fold changes were needed (Fig. 4A, B, Fig. S4A, B, Fig. 5I, J, Fig. S5C, E). For short-lived proteins, theoretical log2 fold changes (8 hr vs 0 hr) assuming no horizontal asymptote (the bottom plateau) were calculated following the linear model (Fig. S1C) and used. The theoretical log2 fold changes reflected the half-lives better, especially for very short-lived proteins whose abundance dropped rapidly (Fig. S1D). For analyses where a combined result from four cell lines was needed (Fig. 4A, B, Fig. S4A, B), the smallest log2 fold change (greatest loss at 8 hr) for a given protein was kept as it was the shortest half-life observed. Binned protein ranks based on theoretical log2 fold change were also used in some analyses (Fig. 5I, J, Fig. S5C, E). Proteins were separated into eight bins of equal size. Rank 1 contained the least stable proteins and Rank 8 contained the most stable proteins.
R package Peptides (2.4.2) (Daniel Osorio, 2015) was used to calculate protein instability index and aliphatic index (Fig. 5C). Cell cycle activator and inhibitor data (Fig. 3A) were from (Teixeira and Reed, 2013). The list of components of anaphase-promoting complex/cyclosome (APC/C) and cullin-RING ubiquitin ligase complex (Fig. 4A, B) was from Gene Ontology (GO:0031466, Cul5-RING ubiquitin ligase complex; GO:0031467, Cul7-RING ubiquitin ligase complex; GO:0080008, Cul4-RING E3 ubiquitin ligase complex; GO:0019005, SCF ubiquitin ligase complex; GO:0005680, anaphase-promoting complex; GO:0035361, Cul8-RING ubiquitin ligase complex; GO:0031462, Cul2-RING ubiquitin ligase complex; GO:0031463, Cul3-RING ubiquitin ligase complex). The hand-curated list of components of APC/C and cullin-RING ubiquitin ligase complex (Fig. S4A) was kindly provided by Wade Harper’s laboratory at Harvard Medical School. The list of E3 ligases was from (Li et al., 2017). E3 ligases that are members of APC/C and cullin-RING ubiquitin ligase complexes were further removed to generate a list of single-unit E3 ligases (Fig. S4B). CORUM database (Giurgiu et al., 2019) was used to retrieve protein complex information unless otherwise stated (Fig. 5D–G). Dynamic components of spliceosome (Fig. 5G) were from (Zhan et al., 2018; Zhang et al., 2018). Protein copy number data in U2OS cells (Fig. 5B) were from (Beck et al., 2011). BioPlex data correspond to interactions detected within HEK 293T cells (Fig. 5H) were taken from (Huttlin et al., 2021). Only in-degree network was used to avoid the potential artifact of bait overexpression. Protein melting temperature data (Fig. 5C) were from (Jarzab et al., 2020). Human protein melting temperatures were used and median was taken when multiple melting temperatures were reported. mRNA expression and protein abundance correlation data (Fig. 5I, Fig. S5E) were from (Jiang et al., 2020; Nusinow et al., 2020; Wang et al., 2019). Protein evolutionary age data (Fig. 5J) were from (Liebeskind et al., 2016). Following the authors’ guidelines, we only accepted protein age estimates with the following parameters: entropy smaller than 1.0, HGT_flag equal to false, NumDBsContributing greater than 3, and Bimodality smaller than 5. Protein abundance data in the four cell lines (Fig. 6E, H, Fig 7C, G, Fig. S6) were from (Li et al., 2020). RNA-seq data for U2OS cells (Fig. S7B) was profiled as part of the CCLE project (Ghandi et al., 2019). Bam files aligned to hg19 was downloaded from GDC Data Portal. Sashimi plot of mapped reads to ATRX mRNA was generated using IGV software.
R package Limma (3.40.6) (Ritchie et al., 2015) was used to determine short-lived proteins of differential stability among cell lines (Fig. 6A, Table S3). TMTpro SN values were normalized to the first time point (0 hr), and then analyzed by fitting a temporal trend using a regression spline. Comparisons were carried out between each pair of cell lines. Proteins that were short-lived in at least one cell line were included in the test. For each protein in each comparison, an effect size (distance) was defined as the average distance of the last two time points between the two cell lines. Proteins of adjusted p-values smaller than 1e−5 and effect sizes greater than 0.4 were designated as proteins of differential stability between cell lines. As we adopted very strict criteria, we did not perform multiple hypothesis testing corrections to account for multiple comparisons of each pair of cell lines.
All data analyses were conducted in R (4.0.2) unless otherwise stated.
ADDITIONAL RESOURCES
The data generated during this study are also available using the viewer on the Gygi lab website (https://gygi.hms.harvard.edu/resources.html).
Supplementary Material
Table S1. All protein quantifications in the four cell lines. Related to Figure 1.
Table S2. Protein half-lives in the four cell lines. Related to Figure 2.
Table S3. Proteins with differential stabilities between cell lines. Related to Figure 6.
Acknowledgements
We thank the members of the Gygi lab at Harvard Medical School for the invaluable discussions. We thank the Wade Harper’s laboratory at Harvard Medical School for the hand-curated list of components of cullin-RING ubiquitin ligase complexes. We thank Dr. Dong Li at Beijing Proteome Research Center for the list of E3 ligases. This work was funded in part by NIH grants GM132129 (J.A.P.) and GM67945 (S.P.G).
Footnotes
Competing interests statement
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, and Aebersold R (2011). The quantitative proteome of a human cell line. Mol Syst Biol 7, 549. 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle A, Tanay A, Bitincka L, Shamir R, and O’Shea EK (2006). Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci U S A 103, 13004–13009. 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, and Wright WE (1998). Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–352. 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Boisvert FM, Ahmad Y, Gierlinski M, Charriere F, Lamont D, Scott M, Barton G, and Lamond AI (2012). A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol Cell Proteomics 11, M111 011429. 10.1074/mcp.M111.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray SJ (2016). Notch signalling in context. Nat Rev Mol Cell Biol 17, 722–735. 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- Cambridge SB, Gnad F, Nguyen C, Bermejo JL, Kruger M, and Mann M (2011). Systems-wide proteomic analysis in mammalian cells reveals conserved, functional protein turnover. Journal of proteome research 10, 5275–5284. 10.1021/pr101183k. [DOI] [PubMed] [Google Scholar]
- Cesare AJ, and Reddel RR (2010). Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11, 319–330. 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- Dai X, Bu X, Gao Y, Guo J, Hu J, Jiang C, Zhang Z, Xu K, Duan J, He S, et al. (2021). Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol Cell. 10.1016/j.molcel.2021.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel Osorio PR-V, Torres Rodrigo (2015). Peptides: A Package for Data Mining of Antimicrobial Peptides. The R Journal 7, 4–14. [Google Scholar]
- Darvishi E, and Woldemichael GM (2016). Cycloheximide Inhibits Actin Cytoskeletal Dynamics by Suppressing Signaling via RhoA. J Cell Biochem 117, 2886–2898. 10.1002/jcb.25601. [DOI] [PubMed] [Google Scholar]
- DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, and Livingston DM (1988). SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54, 275–283. 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- Deppert W, and Haug M (1986). Evidence for free and metabolically stable p53 protein in nuclear subfractions of simian virus 40-transformed cells. Mol Cell Biol 6, 2233–2240. 10.1128/mcb.6.6.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppert W, Haug M, and Steinmayer T (1987). Modulation of p53 protein expression during cellular transformation with simian virus 40. Mol Cell Biol 7, 4453–4463. 10.1128/mcb.7.12.4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshwal S, Fiedler KU, and Langer T (2020). Mitochondrial Proteases: Multifaceted Regulators of Mitochondrial Plasticity. Annu Rev Biochem 89, 501–528. 10.1146/annurev-biochem-062917-012739. [DOI] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, and Greenberg RA (2016). Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58. 10.1038/nature20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty MK, Hammond DE, Clague MJ, Gaskell SJ, and Beynon RJ (2009). Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. Journal of proteome research 8, 104–112. 10.1021/pr800641v. [DOI] [PubMed] [Google Scholar]
- Dyer MA, Qadeer ZA, Valle-Garcia D, and Bernstein E (2017). ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harb Perspect Med 7. 10.1101/cshperspect.a026567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214. 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Galan JM, and Peter M (1999). Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc Natl Acad Sci U S A 96, 9124–9129. 10.1073/pnas.96.16.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, and Yusupov M (2014). Structural basis for the inhibition of the eukaryotic ribosome. Nature 513, 517–522. 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Vassilev AP, Zhang J, Zhao Y, and DePamphilis ML (2011). Assembly of the human origin recognition complex occurs through independent nuclear localization of its components. J Biol Chem 286, 23831–23841. 10.1074/jbc.M110.215988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Ruepp A (2019). CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res 47, D559–D563. 10.1093/nar/gky973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruprasad K, Reddy BV, and Pandit MW (1990). Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng 4, 155–161. 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425. 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS, Roberts TM, and Gjoerup OV (2009). Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J Virol 83, 117–127. 10.1128/JVI.01515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkson IV, and Elias JE (2011). The dynamic state of protein turnover: It’s about time. Trends Cell Biol 21, 293–303. 10.1016/j.tcb.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Huang D, Li Q, Sun X, Tang Y, Qu Y, Liu D, Yu T, Li G, Tong T, and Zhang Y (2020). CRL4(DCAF8) dependent opposing stability control over the chromatin remodeler LSH orchestrates epigenetic dynamics in ferroptosis. Cell Death Differ. 10.1038/s41418-020-00689-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, et al. (2021). Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell. 10.1016/j.cell.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikai A (1980). Thermostability and aliphatic index of globular proteins. J Biochem 88, 1895–1898. [PubMed] [Google Scholar]
- Jarzab A, Kurzawa N, Hopf T, Moerch M, Zecha J, Leijten N, Bian Y, Musiol E, Maschberger M, Stoehr G, et al. (2020). Meltome atlas-thermal proteome stability across the tree of life. Nat Methods 17, 495–503. 10.1038/s41592-020-0801-4. [DOI] [PubMed] [Google Scholar]
- Jiang L, Wang M, Lin S, Jian R, Li X, Chan J, Dong G, Fang H, Robinson AE, and Snyder MP (2020). A Quantitative Proteome Map of the Human Body. Cell 183, 269–283 e219. 10.1016/j.cell.2020.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic M, Rooney MS, Mertins P, Przybylski D, Chevrier N, Satija R, Rodriguez EH, Fields AP, Schwartz S, Raychowdhury R, et al. (2015). Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science 347, 1259038. 10.1126/science.1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Ludwig RL, Levine AJ, and Vousden KH (1999). Analysis of the degradation function of Mdm2. Cell Growth Differ 10, 87–92. [PubMed] [Google Scholar]
- Li J, Van Vranken JG, Pontano Vaites L, Schweppe DK, Huttlin EL, Etienne C, Nandhikonda P, Viner R, Robitaille AM, Thompson AH, et al. (2020). TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat Methods 17, 399–404. 10.1038/s41592-020-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Nichols MA, Shay JW, and Xiong Y (1994). Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Cancer Res 54, 6078–6082. [PubMed] [Google Scholar]
- Li Y, Xie P, Lu L, Wang J, Diao L, Liu Z, Guo F, He Y, Liu Y, Huang Q, et al. (2017). An integrated bioinformatics platform for investigating the human E3 ubiquitin ligase-substrate interaction network. Nat Commun 8, 347. 10.1038/s41467-017-00299-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebeskind BJ, McWhite CD, and Marcotte EM (2016). Towards Consensus Gene Ages. Genome Biol Evol 8, 1812–1823. 10.1093/gbe/evw113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, and Aebersold R (2016). On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550. 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, De S, Petrini JH, Sung PA, Jasin M, et al. (2012). Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet 8, e1002772. 10.1371/journal.pgen.1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Ha K, Kim MS, Noh YW, Lin H, Tang L, Zhu Q, Zhang D, Chen H, Han S, and Zhang P (2018). The anaphase promoting complex promotes NHEJ repair through stabilizing Ku80 at DNA damage sites. Cell Cycle 17, 1138–1145. 10.1080/15384101.2018.1464836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Perez M, and Villen J (2017). Determinants and Regulation of Protein Turnover in Yeast. Cell Syst 5, 283–294 e285. 10.1016/j.cels.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson T, Franken H, Kosinski J, Kurzawa N, Zinn N, Sweetman G, Poeckel D, Ratnu VS, Schramm M, Becher I, et al. (2018). Systematic analysis of protein turnover in primary cells. Nat Commun 9, 689. 10.1038/s41467-018-03106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry TJ, and Kirschner MW (1998). Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93, 1043–1053. 10.1016/s0092-8674(00)81209-x. [DOI] [PubMed] [Google Scholar]
- McShane E, Sin C, Zauber H, Wells JN, Donnelly N, Wang X, Hou J, Chen W, Storchova Z, Marsh JA, et al. (2016). Kinetic Analysis of Protein Stability Reveals Age-Dependent Degradation. Cell 167, 803–815 e821. 10.1016/j.cell.2016.09.015. [DOI] [PubMed] [Google Scholar]
- Meszaros B, Kumar M, Gibson TJ, Uyar B, and Dosztanyi Z (2017). Degrons in cancer. Sci Signal 10. 10.1126/scisignal.aak9982. [DOI] [PubMed] [Google Scholar]
- Moriwaki K, Noda K, Furukawa Y, Ohshima K, Uchiyama A, Nakagawa T, Taniguchi N, Daigo Y, Nakamura Y, Hayashi N, and Miyoshi E (2009). Deficiency of GMDS leads to escape from NK cell-mediated tumor surveillance through modulation of TRAIL signaling. Gastroenterology 137, 188–198, 198 e181–182. 10.1053/j.gastro.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Moriwaki K, Shinzaki S, and Miyoshi E (2011). GDP-mannose-4,6-dehydratase (GMDS) deficiency renders colon cancer cells resistant to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor- and CD95-mediated apoptosis by inhibiting complex II formation. J Biol Chem 286, 43123–43133. 10.1074/jbc.M111.262741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morreale FE, and Walden H (2016). Types of Ubiquitin Ligases. Cell 165, 248–248 e241. 10.1016/j.cell.2016.03.003. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Moriwaki K, Imai T, Shinzaki S, Kamada Y, Murata K, and Miyoshi E (2013). Mutation of GDP-mannose-4,6-dehydratase in colorectal cancer metastasis. PLoS One 8, e70298. 10.1371/journal.pone.0070298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Hou J, Maclean A, Nasir J, Lafuente MJ, Shu X, Kriaucionis S, and Bird A (2007). Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci U S A 104, 2709–2714. 10.1073/pnas.0608056104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete-Perea J, Yu Q, Gygi SP, and Paulo JA (2018). Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J Proteome Res 17, 2226–2236. 10.1021/acs.jproteome.8b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newhart A, Rafalska-Metcalf IU, Yang T, Negorev DG, and Janicki SM (2012). Single-cell analysis of Daxx and ATRX-dependent transcriptional repression. J Cell Sci 125, 5489–5501. 10.1242/jcs.110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER 3rd, Kalocsay M, Jane-Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M, et al. (2020). Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402 e316. 10.1016/j.cell.2019.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren M, Maltzman W, and Levine AJ (1981). Post-translational regulation of the 54K cellular tumor antigen in normal and transformed cells. Mol Cell Biol 1, 101–110. 10.1128/mcb.1.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry D, Bates S, Mann DJ, and Peters G (1995). Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumour suppressor gene product. Embo J 14, 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry D, Mahony D, Wills K, and Lees E (1999). Cyclin D-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol Cell Biol 19, 1775–1783. 10.1128/mcb.19.3.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin SV, Patel M, Furia BS, Peltz SW, and Trotta CR (2004). Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3’ end formation. Cell 117, 311–321. 10.1016/s0092-8674(04)00342-3. [DOI] [PubMed] [Google Scholar]
- Rad R, Li J, Mintseris J, O’Connell J, Gygi SP, and Schweppe DK (2021). Improved Monoisotopic Mass Estimation for Deeper Proteome Coverage. Journal of proteome research 20, 591–598. 10.1021/acs.jproteome.0c00563. [DOI] [PubMed] [Google Scholar]
- Radichev I, Kwon SW, Zhao Y, DePamphilis ML, and Vassilev A (2006). Genetic analysis of human Orc2 reveals specific domains that are required in vivo for assembly and nuclear localization of the origin recognition complex. J Biol Chem 281, 23264–23273. 10.1074/jbc.M603873200. [DOI] [PubMed] [Google Scholar]
- Ranjan A, and Gossen M (2006). A structural role for ATP in the formation and stability of the human origin recognition complex. Proc Natl Acad Sci U S A 103, 4864–4869. 10.1073/pnas.0510305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AB, Langer JD, and Jovanovic M (2020). Proteome Turnover in the Spotlight: Approaches, Applications, and Perspectives. Mol Cell Proteomics 20, 100016. 10.1074/mcp.R120.002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Wilhelm M, Hahne H, Kuster B, and Bantscheff M (2015). A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol Cell Proteomics 14, 2394–2404. 10.1074/mcp.M114.046995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, and Bennink JR (2000). Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404, 770–774. 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Schweppe DK, Eng JK, Yu Q, Bailey D, Rad R, Navarrete-Perea J, Huttlin EL, Erickson BK, Paulo JA, and Gygi SP (2020). Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. Journal of proteome research 19, 2026–2034. 10.1021/acs.jproteome.9b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweppe DK, Prasad S, Belford MW, Navarrete-Perea J, Bailey DJ, Huguet R, Jedrychowski MP, Rad R, McAlister G, Abbatiello SE, et al. (2019). Characterization and Optimization of Multiplexed Quantitative Analyses Using High-Field Asymmetric-Waveform Ion Mobility Mass Spectrometry. Analytical chemistry 91, 4010–4016. 10.1021/acs.analchem.8b05399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira LK, and Reed SI (2013). Ubiquitin ligases and cell cycle control. Annu Rev Biochem 82, 387–414. 10.1146/annurev-biochem-060410-105307. [DOI] [PubMed] [Google Scholar]
- Toyama BH, and Hetzer MW (2013). Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol 14, 55–61. 10.1038/nrm3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR 3rd, and Hetzer MW (2013). Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982. 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Eraslan B, Wieland T, Hallstrom B, Hopf T, Zolg DP, Zecha J, Asplund A, Li LH, Meng C, et al. (2019). A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol Syst Biol 15, e8503. 10.15252/msb.20188503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe-Asano T, Kuma A, and Mizushima N (2014). Cycloheximide inhibits starvation-induced autophagy through mTORC1 activation. Biochem Biophys Res Commun 445, 334–339. 10.1016/j.bbrc.2014.01.180. [DOI] [PubMed] [Google Scholar]
- Xu J, Zhou W, Yang F, Chen G, Li H, Zhao Y, Liu P, Tan M, Xiong X, and Sun Y (2017). The beta-TrCP-FBXW2-SKP2 axis regulates lung cancer cell growth with FBXW2 acting as a tumour suppressor. Nat Commun 8, 14002. 10.1038/ncomms14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, Xu Q, Chou DM, Zhao Z, and Elledge SJ (2008). Global protein stability profiling in mammalian cells. Science 322, 918–923. 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Choi J, Jin HR, Li Y, Bajpai S, Qie S, and Diehl JA (2020). Fbxl8 suppresses lymphoma growth and hematopoietic transformation through degradation of cyclin D3. Oncogene. 10.1038/s41388-020-01532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost KE, Clatterbuck Soper SF, Walker RL, Pineda MA, Zhu YJ, Ester CD, Showman S, Roschke AV, Waterfall JJ, and Meltzer PS (2019). Rapid and reversible suppression of ALT by DAXX in osteosarcoma cells. Sci Rep 9, 4544. 10.1038/s41598-019-41058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Li W, Deng Q, Liu H, Wang X, Hu H, Cao Y, Xu-Monette ZY, Li L, Zhang M, et al. (2020). MYD88 L265P Elicits Mutation-specific Ubiquitination to Drive NF-kappaB Activation and Lymphomagenesis. Blood. 10.1182/blood.2020004918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeineldin M, Federico S, Chen X, Fan Y, Xu B, Stewart E, Zhou X, Jeon J, Griffiths L, Nguyen R, et al. (2020). MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat Commun 11, 913. 10.1038/s41467-020-14682-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Yan C, Zhang X, Lei J, and Shi Y (2018). Structure of a human catalytic step I spliceosome. Science 359, 537–545. 10.1126/science.aar6401. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yan C, Zhan X, Li L, Lei J, and Shi Y (2018). Structure of the human activated spliceosome in three conformational states. Cell Res 28, 307–322. 10.1038/cr.2018.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. All protein quantifications in the four cell lines. Related to Figure 1.
Table S2. Protein half-lives in the four cell lines. Related to Figure 2.
Table S3. Proteins with differential stabilities between cell lines. Related to Figure 6.
Data Availability Statement
The mass spectrometry data have been deposited in the ProteomeXchange Consortium with the dataset identifier PXD024513.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.